

Abstract

Q203 (telacebec) is an imidazopyridine amide (IPA) targeting the respiratory CIII2CIV2 supercomplex of the mycobacterial electron transport chain (ETC). Aiming for a better understanding of the molecular mechanism of action of IPA, 27 analogues were prepared through a seven-step synthetic scheme. Oxygen consumption assay was designed to test the inhibition of purified Mycobacterium smegmatis CIII2CIV2 by these compounds. The assay results generally supported structure–activity relationship information obtained from the structure of M. smegmatis CIII2CIV2 bound to Q203. The IC50 of Q203 and compound 27 was 99 ± 32 and 441 ± 138 nM, respectively. All IPAs including Q203 showed no inhibition of mitochondrial ETC, proving their selectivity against mycobacteria. In vitro Mycobacterium tuberculosis growth inhibition and M. smegmatis CIII2CIV2 binding did not correlate perfectly. These observations suggest that further investigation into the mechanisms of resistance in different mycobacterial species is needed to understand the lack of the correlation pattern between CIII2CIV2 inhibition and cellular activity.
Introduction
Tuberculosis (TB) is one of the oldest and most pervasive respiratory diseases and remains one of the leading causes of death worldwide.1−4 In 2021, 10.6 million new TB infections and 1.6 million deaths were reported.1 The disease is caused by Mycobacterium tuberculosis (Mtb), a slow-growing acid-fast bacterium that can be transmitted by air droplets. TB bacilli can adapt to the high oxidative stress levels in human macrophages and survive for years by downregulating their metabolism and entering a state of dormancy until the host’s immune status weakens, and they become active.5 Long-term combination therapy, most commonly a combination of isoniazid, rifampicin, pyrazinamide, and ethambutol, and problems with patient compliance with the prescribed regimen, lead to the continuous emergence of multidrug-resistant and extensively drug-resistant Mtb. The emergence of drug resistance emphasizes the need to better understand the mechanism of action of existing anti-TB drugs to optimize their activity, as well as discover new and better TB targets.6 Among the challenges that face TB drug development are the slow growth and pathogenicity of Mtb, the latter requiring handling of the pathogen in BSL-3 facilities. To overcome these challenges, the fast-growing non-pathogenic Mycobacterium smegmatis (Msmeg) is often used as a model due to structural similarities with Mtb for known and putative drug targets.7 Handling of Msmeg requires lower biosafety laboratories (BSL-2), easing assay conduction.8,9
Mycobacterial cellular respiration is a promising target for TB treatment.6,10 Cellular respiration results in the production of adenosine triphosphate (ATP), the chemical energy currency in cells, using energy from the oxidation of nutrients. Cellular respiration needs the combined activities of the electron transport chain (ETC) and ATP synthase. The ETC is a series of membrane-embedded protein complexes that establish a transmembrane proton motive force, using energy from a series of redox reactions. The proton motive force drives ATP synthesis by the ATP synthase. Structural differences between mycobacterial ETC complexes compared to ETC complexes from eukaryotic mitochondria and other bacteria allow selective inhibition of mycobacterial respiration.
Electron Transport Chain (ETC)
The transfer of electrons from complex III to IV is different in mycobacteria than that in mitochondria. Complexes III and IV in mycobacteria form an obligatory supercomplex (CIII2CIV2), rather than existing as two separate entities (Figure 1).11,12 Instead of ubiquinone, mycobacteria use menaquinone (MK) as a membrane-embedded electron carrier. Complex III has three subunits: QcrA, QcrB, and an anchored QcrC domain with two c-type hemes that transfer electrons to complex IV. There are two MK-binding sites in the QcrB subunit of complex III, one where MKH2 is oxidized and the other where MK is reduced in a process known as the Q cycle. The oxidation site for MKH2 (Qo) is the target of Q203 (telacebec). Telacebec was involved and is present in clinical studies: a first-in-human trial (clinicaltrials.gov identifier NCT02530710); a phase 1 ascending multiple-dose study (NCT02858973); and a phase 2a multiple-dose trial for the evaluation of early bactericidal activity (NCT03563599) where it showed a dose-dependent reduction in the load of viable mycobacteria in the sputum. It was also tested for its effect on an inflammation biomarker in coronavirus patients (NCT04847583).11,13−15
Figure 1.

Mycobacterial ETC. Mycobacteria have two types of NADH dehydrogenases, NDH-1 (complex I) and NDH-2. Electrons from redox reactions in NDH-1, NDH-2, and complex II (succinate dehydrogenase, SDH) are transported via menaquinone (MK, electron carrier) to the mycobacterial respiratory supercomplex (CIII2CIV2). Menaquinol (MKH2) is oxidized in the QcrB subunit of complex III, and the resulting electrons are transported via an anchored domain (QcrC) to complex IV where oxygen is reduced to water. The QcrB subunit has two MK-binding sites: Qo, where MKH2 is oxidized to MK and Qi where MK is reduced to MKH2 in a process known as the Q cycle. Q203 binds at the Qo site, preventing the oxidation of MKH2 to MK. The cytochrome bd complex16 can also oxidize MKH2 to MK and reduce O2 to H2O, functioning as an alternative pathway when CIII2CIV2 is inhibited.
Imidazopyridine Amides (IPA)
Q203 belongs to the chemical class of imidazopyridine amides (IPAs). The imidazopyridine system is synthetically relatively easy to construct and modify. IPA analogues have been designed by many research groups. For example, Marvin Miller’s group reported several IPA analogues (Figure 2A–C) with good activity (micromolar to nanomolar) and selectivity against mycobacteria as well as good PK parameters.17−19 Q203 resulted from the modifications of an IPA hit compound identified from screening a library of ∼120,000 compounds.11,20 It targets the mycobacterial respiratory CIII2CIV2, also known as cytochrome bcc-aa3. It was reported that a long hydrophobic tail increases the potency of IPAs, suggesting that it helps in penetrating the cell membrane.21 In vitro potency is high indeed, for example, Q203 has an MIC50 of 2.7 nM. However, its high lipophilicity and poor solubility,22 leading to an extended half-life of 321.12 ± 227.29 h,23 necessitate searching for IPAs with good potency and better pharmacokinetic (PK) parameters. Despite its low minimum inhibitory concentration (MIC) and extended half-life, Q203 is given in doses up to 300 mg/d in clinical trials (NCT03563599).
Figure 2.

Examples of imidazopyridine amide analogues. A, B, and C from.18,25,26 D has approximately the same MIC as Q203; an analogue to compound 24 later in this study.22
To solve the high lipophilicity problem of Q203, a series of analogues with fused ring systems in the tail part were synthesized by different research groups.22,24 Fused ring systems helped greatly reduce the log P value and thus the overall lipophilicity of Q203.
Kang et al. 2017 synthesized a series of analogues with different heterocycles and compared them to Q203 in terms of their metabolic stability and both extracellular and intracellular antimycobacterial activity against Mtb. An IPA analogue with a benzoxazole ring in the tail (Figure 2D) showed comparable activity and PK parameters to Q203 but a much lower log P value.
Design Based on the Target–Ligand Structure
The recently published model of Q203 bound to the Msmeg respiratory CIII2CIV2 gave insights into the drug–target interactions.15 It showed three main interactions in the head region (IP) of Q203, namely, a halogen bond between the chlorine at C-6 and the carbonyl of Leu 166, a hydrogen bond between the N of the IPA and His 368, and van der Waals interactions between the ethyl group and Ile 178. Although not detected, a hydrogen bond between Asp 309 in Msmeg and N1 of IPA is also plausible due to their distance of ∼3.5 Å. In the neck region of Q203, a hydrogen bond between the carbonyl oxygen and Thr 308 was found. Finally, a π–π interaction was observed between Phe 156 and the benzyl group in the tail.15
The IPAs reported here were devised for an improved understanding of the molecular mechanism of action, making use of the molecular assay facilities in our laboratories and in particular trying to approach the long-term goal of targeting problematic mycobacteria that are not sensitive to Q203. These mycobacteria include M. abscessus and the M. avium complex, non-tuberculous mycobacteria of emerging importance.20 The insensitivity may be due to PK differences IPAs display in different mycobacteria or differences in target binding and inhibition. We decided to synthesize IPA analogues with a shorter tail than Q203 and variations of substituents in the head to explore the details of binding to CIII2CIV2. Compound D (Figure 2) was chosen as the lead compound. The benzoxazole ring system has the advantage of decreasing the high lipophilicity of Q203 and being hydrophobic enough to allow the membrane penetration.22 We designed and synthesized a set of analogues with changes in the R1, R2, and R3 groups (Figure 3). In total, 28 compounds were synthesized and tested biochemically against the Msmeg CIII2CIV2, as well as in vitro against Mtb, Msmeg, andM. abscessus for growth inhibition.
Figure 3.

Q203 is shown on the left. The tail was replaced with a benzoxazole that is less lipophilic compared to the tail of Q203.
Results and Discussion
Synthesis and Physicochemical Characterization
Through a seven-step synthetic scheme, a series of 27 analogues were synthesized, 25 of which had the benzoxazole heterocycle in their side chain. The general scheme was divided into three major parts; synthesis of the imidazopyridine scaffold (Scheme 1A), synthesis of the benzoxazole side chain (Scheme 1B), and amide coupling (Schemes 1C and 2). The synthesis of compound 27 (Scheme 3) was slightly different from the general scheme.
Scheme 1. General Synthetic Scheme, (A) Synthesis of imidazo[1,2-a]pyridine-3-carboxylic Acids, (B) Synthesis of 2-substituted benzo[d]oxazole-5-methylamine, and (C) Synthesis of 1–24 and 26.

Reaction conditions: (A) (i) 2-aminopyridine, EtOH, reflux overnight; overnight. (ii) N-Bromosuccinimide (NBS), H2O, 30 min, 80 °C; 2-aminopyridine, 30 min–1 h, 80 °C. (iii) Ester hydrolysis: LiOH, EtOH/H2O (3:1, v/v), reflux overnight. (B) (iv) thionylchloride, DCM/DMF, room temperature, 1 h; (v) 3-amino-4 hydroxybenzonitrile, methanesulfonic acid, dioxane, 100 °C. (vi) di-tert-butyl-dicarbonate, NiCl2.6H2O, NaBH4, MeOH, 0 °C to room temperature, overnight; (vii) 4 M HCl in dioxane, 2 h. n = 1 or 3. (C) (viii) Amide coupling: HATU, DMF, DIPEA, room temperature, 1 h.
Scheme 2. Synthesis of 25.

Reaction conditions: (i) HATU, DIPEA, DMF, room temperature, 1 h. This compound was previously described in the literature.18
Scheme 3. Synthesis of 27.

Reaction conditions: (i) phthalimide, DIAD, Ph3P in dry THF, room temperature, 4 h. (ii) Hydrazine (50% w/w in H2O), MeOH, overnight, room temperature. (iii) (1) 7c, DIPEA, dry DMF, 10 min; (2) HATU, stir 20 min; and (3) 1g, room temperature, 1 h.
Scheme 1A: the imidazopyridine scaffold was synthesized through a one-pot reaction in which halogenation and cyclization occurred in the same reaction flask. The advantage of this method is that the highly reactive brominated product can react directly with the nucleophile (aminopyridine) once it is formed.
Scheme 1B: different synthetic methods of benzoxazoles have been reported due to their importance as a compound class with various pharmacological activities.27 They are usually synthesized through the condensation of o-aminophenol and benzoic acids or benzoic acid derivatives. After several trials, we were able to obtain the benzoxazole side chain (1d and 2d) with ∼30–50% yield by using methane sulfonic acid as a catalyst.
The selective reduction of the benzonitrile to the primary benzylamine was a challenging step. Borane dimethylsulfide and LiAlH4 reduced the imine functional group of the benzoxazole ring together with the nitrile group, leading to ring opening. Starting with the reduction of the educt, 3-amino-4-hydroxybenzonitrile, to avoid ring opening, followed by amide coupling between the formed amine and the imidazopyridine-3-carboxylic acid was not successful. To selectively reduce the nitrile without affecting the benzoxazole ring, NaBH4 was used as the reducing agent with NiCl2.6H2O as a catalyst.28,29 However, the primary amine could not be isolated from the reaction mixture. The masses of a secondary amine and primary amide were also detected in ESI-MS. To solve this problem, di-tert-butyl-dicarbonate was added to the reaction mixture. The resulting boc-protected primary amine was easily isolated from the reaction mixture through column chromatography. Stirring in 4 M HCl in dioxane for 1 h was sufficient to precipitate the primary amine as a hydrochloride salt (1e and 2e).
Scheme 1C: coupling of the imidazopyridine carboxylic acids and the benzoxazole side chain was achieved by using either HATU or PyBOP. With HATU, amide formation was faster than that with PyBOP, leading to complete conversion after 1 h. It is noteworthy that the order of adding the reactants in this reaction made a difference in the yield. First, the acid and DIPEA were dissolved in DMF. Then, HATU was added, and the mixture was left to stir for 20 min before adding the amine. This order allowed enough time for the formation of the activated ester before the amine was added. Afterward, brine was added to precipitate the product. Washing thoroughly with brine helped remove DMF and DIPEA. Finally, washing with a small amount of MeOH removed the remaining impurities.
Scheme 2: coupling of 15c to the benzylamine was performed using HATU as described above.
Scheme 3: the tail of compound 27 consists of three isoprene units rather than a 2-substituted benzoxazole heterocycle. It was synthesized via coupling of an imidazopyridine carboxylic acid with E-farnesyl amine, which was synthesized from E-farnesol in two steps. The first step was the synthesis of N-farnesylphthalimide (1f) from farnesol. This reaction is a variation of the Mitsunobu reaction. The second step was the deprotection of the N-farnesylpthalimide to farnesyl amine (1g) with hydrazine hydrate.30 The conversion of farnesyl bromide to farnesyl amine following Gabriel’s synthesis was also successful but with much lower yields.
Biochemical Assays
To quantify the inhibition of CIII2CIV2, we employed CIII2CIV2 activity assay using a Clark-type oxygen electrode. In contrast to our recent study where the electron donor 2,3-dimethyl[1,4]naphthoquinone (DMW) was reduced chemically,15 here we used an assay where DMW was reduced to DMWH2 by adding the NADH dehydrogenase enzyme from Caldalkalibacillus thermarum (NDH-2) to the reaction mixture (Figure 4A).31 The enzymatic reduction of DMW was found to be more reliable than chemical reduction. In assay, NADH is added in excess to the chamber of the oxygen electrode to initiate the rapid reduction of all the DMW to DMWH2, which then allows a slower reduction of O2 to H2O by CIII2CIV2 (Figure 4B, orange curve). This reduction of oxygen proceeds until all oxygen in the chamber is consumed (Figure 4B, orange curve). The specific activity of CIII2CIV2 in this assay was found to be 340 ± 24 e–/s (±s.e. from five separate measurements each from a different batch of protein), ∼3-fold higher than measurements with chemically reduced DMW.15 As in our previous studies with chemically reduced DMW,15 there was residual CIII2CIV2 activity even at high concentrations of Q203 (Figure 4B, green curve vs blue curve). All compounds, including Q203, were tested for binding and inhibition of the purified Msmeg CIII2CIV2 via this assay.
Figure 4.

Oxygen belongs to biochemical assays. (A) 2,3-dimethyl[1,4]naphthoquinone (DMW) was reduced by NDH-2 using electrons from the oxidation of NADH. The reduced DMWH2 is re-oxidized to DMW by the Msmeg CIII2CIV2 (SC) catalyzing the reduction of oxygen to H2O. (B) Rate of oxygen consumption in case of no inhibitor “-Q203” (orange), with inhibitor “+Q203” (green), and baseline auto-oxidation in the absence of CIII2CIV2, “-SC-Q203” (blue). The red arrow represents the point where NADH was injected.
None of the Compounds Tested Inhibited Caldalkalibacillus thermarum NDH-2 nor Bos tausus Complex I
NADH oxidation assays confirmed that all the compounds tested including Q203 did not inhibit NDH-2, supporting the validity of the oxygen consumption assay results. The oxidation of NADH was monitored spectrophotometrically at 340 nm (Figure S2). By monitoring the NADH oxidation at 340 nm in the presence of sub-mitochondrial particles (SMPs), it was confirmed that at 10 μM, 27 and Q203 do not inhibit mitochondrial complex I (Figure S3).
Compound 27 Showed Inhibition of CIII2CIV2 that was Comparable to Q203
Compound 27 was synthesized to test the effect of a tail that resembles MK on binding to the active site and electron transfer. This compound had the same head and neck as Q203 and a short isoprene tail, similar to the natural substrate (MK). At the tested concentration (10 μM), compound 27 showed 88% inhibition of CIII2CIV2 activity, which is similar to inhibition by Q203 (85%). Unexpectedly, the substitution of the 6-Cl in the IP with a hydrogen atom showed better percentage inhibition despite the possibility of a halogen bond formation by the chlorine atom. Compound 24(22) (R1=Cl) showed only 20% inhibition, while compound 26 (R1=H) showed 50% inhibition. These differences imply that the main binding interactions between the IPAs and CIII2CIV2 are between N1 of the imidazopyridine head with His 368 and between the carbonyl O-atom in the neck with Thr 308 (Figure 3). No significant inhibition was detected in compounds with methyl18 (R1=CH3), trifluoromethyl (R1=CF3), or trifluoromethoxy (R1=OCF3) in the head region (Table 1). Bromo-substituted analogues (R1=Br), however, showed approximately 50% inhibition (Table 1 and Figure S1).
Table 1. Average Percent Inhibition at Particular Concentrations of Msmeg CIII2CIV2 by Q203 and IPA Analoguesa.

Experiments were performed in triplicates to calculate the standard deviation (mean ± s.e., n = 3 independent assays).
54 μM has been previously reported as the nephelometric detection limit for compounds with a molecular weight of 500.38
The structure–activity relationship (SAR) was based on electron cryomicroscopy data of Q203 bound to the active site, and the results of CIII2CIV2 activity assays supported the hypothesis that the main interactions are in the head and not in the tail. A single concentration (10 μM) was chosen to screen all analogues, then the compound which showed high percentage inhibition was then tested at different concentrations. 10 μM is a very high concentration considering the reported MIC50 of Q203 (2.7 nM) and MIC90 of compound 24 (<40 nM) in growth inhibition assays.
Q203 and Compound 27 IC50 Values
The IC50 against Msmeg CIII2CIV2 of both Q203 and 27 was determined through the repetition of the oxygen consumption assay with different concentrations of test compounds (10, 1 μM, 100, and 10 nM). The IC50 values for Q203 and compound 27 were calculated as 99 ± 32 and 441 ± 138 nM, respectively (mean ± s.e., n = 4 independent assays from biological triplicates, Figure 5). These values show that Q203 and 27 have relatively similar potencies on the enzyme, supporting the hypothesis that the tail has no significant effect on the binding.
Figure 5.

IC50 of Q203 (A) and compound 27 (B). The rate of oxygen consumption was measured after incubating the Msmeg CIII2CIV2 with different concentrations of Q203 and 27 to calculate their IC50. The IC50 for Q203 and 27 were 99 ± 32 and 441 ± 138 nM, respectively (mean ± s.e., n = 4 independent assays). The assays were repeated on different days and using different batches of CIII2CIV2, which could explain the high s.e. values. Nevertheless, these values show that Q203 and 27 have relatively similar potencies on the enzyme, supporting the hypothesis that the tail has no significant effect on the binding.
Q203, 24, and 27 Do Not Inhibit Mitochondrial Cytochrome bc1 (CIII)
Compounds (Q203, 24,22 and 27) that showed activity against Msmeg did not show activity against bovine mitochondrial cytochrome bc1 (complex III) nor Candida albicans mitochondrial cytochrome bc1, confirming their specificity against the mycobacterial CIII2CIV2. Although 24 did not show activity in Msmeg inhibition assay, it showed high activity in the in vitro Mtb growth inhibition assays. Oxygen consumption assays, where Q203, 24,22 or 27 were incubated with SMPs at concentrations of 10 and 1 μM, were performed. The results showed some inhibition at 10 μM, while at 1 μM no inhibition was detected (Figure 6). To further confirm the specificity of Q203, 24,22 and 27, the reduction of cytochrome c was monitored spectrophotometrically at 550 nm to measure the activity of purified mitochondrial CIII2 from C. albicans. No inhibition was observed at the concentrations tested (10 and 1 μM), confirming the specificity of Q203 and analogues to mycobacteria (Figure S4).
Figure 6.

Comparison of the activity in SMPs. Rotenone, KCN, and antimycin A were used as positive controls. The graph shows that Q203, 24, and 27 inhibit complex III at 10 μM; 80, 30, and 50% inhibition respectively, while at 1 μM, they show no inhibition (mean ± s.d., n = 3 independent assays).
Growth Inhibition Assays
In Mtb in vitro growth inhibition assays, Compounds 1–4 showed an MIC90 of 0.2 μM, while 24(22) showed ∼5× higher potency (MIC90 ≤ 0.04 μM). This finding is consistent with the previously reported MIC80 of compound 24 (0.027 μM).22 Br-substitution at C-7 in compounds 5–8 led to a 50-fold decrease in activity (≥10 μM). CF3 and OCF3 instead of bromine or chlorine led to a ∼125-fold decrease in activity (Table S1). These results were rather consistent with the results of the CIII2CIV2 activity assays, suggesting that a group bigger than bromine at C6 or C7 is not well tolerated. Contradictory to the results of the oxygen consumption assays described above, replacing the halogen at C6 or C7 with a hydrogen atom led to a decrease in activity. For instance, compound 26 (R1=H) showed a ∼250-fold decrease in activity compared to 24(22) (R1=Cl). In addition, compound 24 in Msmeg CIII2CIV2 assays did not show strong inhibition (Table 1), unlike its high in vitro Mtb activity. Compound 27 showed MIC90 > 10 μM contrary to its high activity in the Msmeg CIII2CIV2 assay. One explanation for this observation could be that the efflux mechanisms could also contribute to the high in vitro MIC of 27.
Msmeg and M. abscessus were not susceptible to all compounds analyzed with MIC90 values >25, 50, or 100 μM (Table S1). It was reported that combining Verapamil with Q203 increases its potency in vitro and ex vivo.32 However, no change in activity was detected when we combined verapamil with the test compounds in Msmeg assays. Polymorphisms in the QcrB subunit of M. abscessus were previously reported to be the reason for its insensitivity to Q203.33 However, superimposing the QcrB subunit of Mtb (PDB:7e1w),34Msmeg (PDB:7rh7),15 and an AlphaFold model (UniProt: A0A0U1AIY2)35,36 of M. abscessus showed that these mutations do not affect the binding site. The models were superimposed using UCSF Chimera software (Figure S5).37
The in vitro inactivity against Msmeg and M. abscessus could be related to the compensatory mechanism by the cytochrome bd complex, other unknown compensatory mechanisms, or different binding affinities in different species. The Q203-bound to Mtb CIII model34 showed that Q203 forms three hydrogen bonds compared to the two shown in Msmeg. The halogen in Mtb forms a halogen bond with a water molecule, which in turn forms a hydrogen bond with a Tyr 164.34 In Msmeg, the halogen directly forms a halogen bond with the carbonyl of Leu 166 in QcrB.15
Conclusions
With the panel of IPAs prepared and tested for this investigation, data were generated that allow some generalizations on the IPA-CIII2CIV2 interaction in Msmeg, tentative correlation with in vitro activities, and extrapolation to other mycobacterial species.
As for SAR of the IPAs, a halogen substituent at C6/C7 of the IP moiety is important for CIII2CIV2 inhibitory activity. Substitution at C6 leads to a stronger effect than that at C7. Electron cryomicroscopy data, corroborated by in silico models, show that a halogen bond39 can be formed with Tyr16434 in Mtb and the carbonyl of Leu166 in Msmeg.15 A substituent bigger than bromine at C6/C7 is not tolerated. More generally, the NADH oxidation assays for both the NDH-2 enzyme and the SMPs support that the head of the IPA, not the tail, affected specificity. None of the compounds tested showed activity against bovine mitochondrial complex I or Caldalalibacillus thermarum NDH-2 despite both of these enzymes having a quinone-binding site. The inactivity in these assays for compound 27, which has an isoprene tail similar to the natural quinone substrate, thus allowing binding through the tail, further supports the hypothesis that binding and in particular specificity of binding to a mycobacterial CIII2CIV2 mainly rests in the head region.
Mostly, no generally applicable correlation could be made between the results in the Msmeg CIII2CIV2 oxygen consumption assay and the Msmeg and Mtb growth inhibition assays. For example, the strong inhibition by compound 27 in the Msmeg CIII2CIV2 binding assays did not translate to high potency in the Mtb assays. The opposite was observed in the case of compound 24: weak inhibition of CIII2CIV2 and strong growth inhibition of Mtb. However, for Msmeg, all compounds did not inhibit or only weakly inhibited growth, regardless of any inhibition in the CIII2CIV2 oxygen consumption or Mtb growth inhibition assays.
This lack of a general correlation pattern between assays implies that inhibition of CIII2CIV2 activity is not sufficient to translate into growth inhibition. This finding may be due to either efflux or alternative resistance mechanisms the mycobacteria can resort to, or the activity of the cytochrome bd terminal oxidase as a rescue mechanism when CIII2CIV2 is inhibited. The cytochrome bd oxidase in Mtb has previously been shown to prevent bactericidal activity of Q203 despite its low MIC (nM). Bactericidal activity against Mtb can only be achieved through the combination of Q203 with a cytochrome bd inhibitor.40 In M. abscessus, the cytochrome bd complex has been reported to have no effect on susceptibility to Q203 and other IPAs. Therefore, other yet unknown mechanisms must be the reason for the insensitivity of M. abscessus to Q203.33 In addition, PK parameters such as solubility, permeation through human and bacterial cell membranes, and bacterial metabolism of the IPAs could play a role in limiting or preventing growth inhibitory activity. The similarity of the binding sites of the Msmeg and Mtb CIII2CIV2 electron cryostructures supports the hypothesis that the resistance could be due to one of the above reasons (Figure S5A). Furthermore, the Msmeg CIII2CIV2 electron cryomicroscopy structure and M. abscessus AlphaFold QcrB model (Figure S5B) displayed even higher similarity, but we did not observe growth inhibition of M. abscessus with any of the IPAs tested.
The findings reported here will be investigated further to assess the role of the tail in IPA activity, evaluate the role of cytochrome bd and expression level of CIII2CIV2 for bacteriostatic or bactericidal activity, and to improve PK parameters of novel IPAs compared to Q203. Activity against other mycobacterial species will also be studied.
Experimental Procedures
General Procedures
Reagents obtained from Sigma-Aldrich, abcr, TCI, and enamine were used without further purification. All organic solvents used were of pure analytical grade. Dioxane and THF were dried over molecular sieves (4 Å), while DCM was dried over 3 Å molecular sieves. Column chromatography was carried out using silica-gel 40–60 μm mesh with Heptane: EtOAc: Heptane and CHCl3: MeOH. Flash chromatography was performed on puriFlash 430 (Interchim, Montluçon, France). Prepacked columns with silica gel of 30 μm pore size were used. Thin-layer chromatography (TLC) was performed using TLC silica gel 60 F254 plates (Merck). Mass spectrometry was performed on APCI-MS (Advion expression CMS; Ithaca, NY, USA). The flow rate used was 10 μL/min, and the super soft method was used to avoid fragmentation. m/z range 10 to 1000 with an acquisition speed was 10,000 m/z units/sec. The ESI-MS spectra were recorded on LCQ-Classic, Thermo Finnigan; direct injection). For the high-resolution mass spectrometry (HRMS), a Q ExactiveTM Plus Orbitrap mass spectrometer (Thermo Scientific, Bremen, Germany) was used. Melting points (mp) were measured on a Kofler bench apparatus. 1H NMR and 13C NMR spectra were recorded at 400, 500 and 126, 101 MHz, respectively, using a lampe-vnmrs400 spectrometer. 1H shifts are referenced to the residual protonated solvent signal [δ 7.26 for CDCl3, δ 3.31, 4.87 for CD3OD, and δ 2.5 for 11.5 trifluoroacetic acid (TFA)/dimethyl sulfoxide (DMSO)], and 13C shifts are referenced to the deuterated solvent signal (δ 77.0 for CDCl3, δ 49.0 for CD3OD, and δ 164.2 for TFA/DMSO). Abbreviations: s: singlet, d = doublet, dd = double doublet t = triplet, dt = doublet of triplets, q = quartet, p = pentet, and m = multiplet. Assignments were proven by HSQC. Chemical shifts are given in parts per million (ppm), and all coupling constants (J) are given in Hz. In most 13C NMR, some quaternary carbon atoms were covered by noise due to low concentrations and solubility in organic solvents. The purities of the tested compounds were determined by high-performance liquid chromatography (HPLC). The purity of the final compounds was 95% or higher. The instrument used was from Shimadzu (Kyoto, Japan). Pump: two LC-10AD pumps, Detector: SPD-M10A VP PDA detector, and Sampler: SIL-HT autosampler. Analytical HPLC: LiChrospher column RP-18, 5 μm particle size, and 10 cm length was used, and the solvent system was MeOH: H2O, 5 to 95 + 0.05% TFA over 30 min. Preparative HPLC: NUCLEODUR 100–5 C18 ec column was used and, the used mobile phase was acetonitrile (ACN): water, 5 to 95% ACN +0.1% formic acid over 30 min. Samples were dissolved in CHCl3: MeOH, 1:1. Peaks were detected at λ = 254 nm.
Synthesis of the Imidazo[1,2-a]pyridine-3-carboxylic Acid Scaffold
2-Ethylimidazo[1,2-a]pyridine-3-carboxylic Acids
A solution of NBS (3.46 mmol, 1.2 equiv) dissolved in H2O (5 mL) was heated to 80 °C, and then, ethyl-3-oxo-pentanoate (3.03 mmol, 1.05 equiv) was added with a syringe and left to stir for 30 min. After complete addition, the color of the solution changes from yellow to colorless. Afterward 1 equiv 2-aminopyridine (2.89 mmol) was added and stirred for 30 min to 1 h at 80 °C. To stop the reaction, saturated Na2CO3 solution was added, then the mixture was extracted three times with EtOAc. The organic phase was dried over anhydrous MgSO4. After evaporation of the solvent, the product was purified by flash column chromatography (gradient mobile phase, Heptane: EtOAc from 90:1 to 66:34) to give a pale-yellow solid (1–8b, Scheme 1(A)).41 The yield ranged from 15 to 30% depending on the substituents on the pyridine.
The purified ester was dissolved in absolute ethanol (30 mL) followed by the addition of an aqueous solution of LiOH (10 mL). The ratio of EtOH/H2O was 3:1 v/v. The reaction mixture was left to reflux overnight. After evaporating EtOH on the Rotavap, 1 N HCl was added dropwise until pH dropped to 4. The formed pale solid residue was filtered and washed with water and then dried in the desiccator to give 1–8c.42 The yield obtained was up to 90%.
2-Methylimidazo[1,2-a]pyridine-3-carboxylic Acids
Ethyl-2-chloroacetoacetate (6.075 mmol) was added to 25 mL of EtOH, followed by 2-aminopyridine (1 equiv). The reaction was left to reflux (80 °C) overnight. The solvent was evaporated, and 20 mL of EtOAc was added. The organic phase was extracted 3× with H2O and dried over anhydrous MgSO4. The product was purified by flash column chromatography (Heptane: EtOAc, 4:1) to give a solid (9–15b), Scheme 1(A)).25 The ester hydrolysis was performed in the same way as mentioned above (9–15c).(42)
Ethyl-6-bromo-2-ethylimidazo[1,2-a]pyridine-3-carboxylate (1b)
C12H13BrN2O2, yellow solid, 17% yield, ESI-MS: m/z 295.06 [M – H]−, Rf = 0.26 (EtOAc: Heptane, 1:2), 1H NMR (400 MHz, Chloroform-d) δ 9.51 (dd, J = 1.9, 0.9 Hz, 1H), 7.52 (dd, J = 9.4, 0.9 Hz, 1H), 7.44 (dd, J = 9.4, 1.9 Hz, 1H), 4.44 (q, J = 7.1 Hz, 2H), 3.10 (q, J = 7.5 Hz, 2H), 1.44 (t, J = 7.1 Hz, 3H), 1.35 (t, J = 7.5 Hz, 3H).
6-Bromo-2-ethylimidazo[1,2-a]pyridine-3-carboxylic Acid (1c)
C10H9BrN2O2, beige powder, 70%, ESI-MS: m/z 271.23 [M + H] +, 1H NMR (400 MHz, Methanol-d4): δ 9.60 (dd, J = 1.9, 0.9 Hz, 1H), 7.68 (dd, J = 9.4, 1.9 Hz, 1H), 7.59 (dd, J = 9.4, 0.9 Hz, 1H), 3.16 (q, J = 7.6 Hz, 2H), 1.36 (t, J = 7.6 Hz, 3H).
Ethyl-7-bromo-2-ethylimidazo[1,2-a]pyridine-3-carboxylate (2b)
C12H13BrN2O2, yellow solid, APCI-MS: m/z 297, 299 (79Br,81Br) [M + H] +, this compound was directly taken to the next step without further purification.
7-Bromo-2-ethylimidazo[1,2-a]pyridine-3-carboxylic Acid (2c)
C10H9BrN2O2, white powder, 74% yield, APCI-MS: m/z 269.1, 271.1 (79Br,81Br) [M + H]+, 1H NMR (500 MHz, Methanol-d4): δ 9.31 (d, J = 7.4 Hz, 1H), 7.85 (dd, J = 2.0, 0.8 Hz, 1H), 7.30 (dd, J = 7.4, 2.0 Hz, 1H), 3.13 (q, J = 7.6 Hz, 2H), 1.33 (t, J = 7.6 Hz, 3H).
Ethyl-6-(trifluoromethyl)-2-ethylimidazo[1,2-a]pyridine-3-carboxylate (3b)
C13H13F3N2O2, white solid, 27% yield, APCI-MS: m/z 287.0 [M + H] +, 1H NMR (400 MHz, Chloroform-d): δ 9.81 (dt, J = 2.1, 1.1 Hz, 1H), 7.93 (d, J = 9.3 Hz, 1H), 7.64 (dd, J = 9.1, 1.7 Hz, 1H), 4.49 (q, J = 7.1 Hz, 2H), 3.20 (q, J = 7.6 Hz, 2H), 1.47 (t, J = 7.1 Hz, 3H), 1.41 (t, J = 7.6 Hz, 3H).
2-Ethyl-6-(trifluoromethyl)imidazo[1,2-a]pyridine-3-carboxylic Acid (3c)
C11H9F3N2O2, white solid, 88% yield, APCI-MS: m/z 259.1 [M + H]+, 1H NMR (400 MHz, cd3od): δ 9.82 (dp, J = 2.3, 1.2 Hz, 1H), 7.78 (dt, J = 9.4, 0.9 Hz, 1H), 7.72 (dd, J = 9.4, 1.9 Hz, 1H), 3.16 (q, J = 7.6 Hz, 2H), 1.34 (t, J = 7.6 Hz, 3H).
Ethyl-7-(trifluoromethyl)-2-ethylimidazo[1,2-a]pyridine-3-carboxylate (4b)
C13H13F3N2O2, white solid, 28% yield, APCI-MS: m/z 287.0 [M + H] +, 1H NMR (400 MHz, Chloroform-d): δ 9.44 (dt, J = 7.3, 0.8 Hz, 1H), 7.93 (dp, J = 1.9, 1.0 Hz, 1H), 7.14 (dd, J = 7.3, 1.9 Hz, 1H), 4.46 (q, J = 7.1 Hz, 2H), 3.15 (q, J = 7.5 Hz, 2H), 1.45 (t, J = 7.1 Hz, 3H), 1.36 (t, J = 7.5 Hz, 3H).
7-(Trifluoromethyl)-2-ethylimidazo[1,2-a]pyridine-3-carboxylic Acid (4c)
C11H9F3N2O2, white powder, 93% yield, APCI-MS: m/z 259.1 [M + H] +, 1H NMR (400 MHz, Methanol-d4): δ 9.55 (dt, J = 7.2, 0.9 Hz, 1H), 7.93 (dt, J = 2.1, 1.0 Hz, 1H), 7.33 (dd, J = 7.3, 1.9 Hz, 1H), 3.16 (q, J = 7.5 Hz, 2H), 1.34 (t, J = 7.5 Hz, 3H).
Ethyl 2-Ethyl-6-(trifluoromethoxy)imidazo[1,2-a]pyridine-3-carboxylate (5b)
C13H13F3N2O3, pale yellow, 22% yield, APCI-MS: m/z 303.0 [M + H] +, 1H NMR (400 MHz, Chloroform-d): δ 9.44 (dt, J = 2.3, 0.8 Hz, 1H), 7.65 (dd, J = 9.7, 0.8 Hz, 1H), 7.34 (ddd, J = 9.7, 2.3, 1.0 Hz, 1H), 4.45 (q, J = 7.1 Hz, 2H), 3.13 (q, J = 7.5 Hz, 2H), 1.44 (t, J = 7.1 Hz, 3H), 1.36 (t, J = 7.5 Hz, 3H).
2-Ethyl-6-(trifluoromethoxy)imidazo[1,2-a]pyridine-3-carboxylic Acid (5c)
C11H9F3N2O3, white powder, 73% yield, APCI-MS: m/z 275.1 [M + H] +, 1H NMR (400 MHz, Methanol-d4): δ 9.53 (dt, J = 2.4, 0.9 Hz, 1H), 7.71 (dd, J = 9.7, 0.8 Hz, 1H), 7.58 (ddd, J = 9.7, 2.3, 1.0 Hz, 1H), 3.15 (q, J = 7.6 Hz, 2H), 1.34 (t, J = 7.6 Hz, 3H).
Ethyl 2-Ethyl-7-(trifluoromethoxy)imidazo[1,2-a]pyridine-3-carboxylate (6b)
C13H13F3N2O3, pale yellow, 17% yield, APCI-MS: m/z 303.0 [M + H] +, 1H NMR (400 MHz, Chloroform-d): δ 9.36 (dd, J = 7.6, 0.8 Hz, 1H), 7.48 (d, J = 2.2 Hz, 1H), 6.89 (dd, J = 7.4, 2.4 Hz, 1H), 4.45 (q, J = 7.1 Hz, 2H), 3.12 (q, J = 7.5 Hz, 2H), 1.44 (t, J = 7.1 Hz, 3H), 1.36 (t, J = 7.6 Hz, 3H).
2-Ethyl-7-(trifluoromethoxy)imidazo[1,2-a]pyridine-3-carboxylic Acid (6c)
C11H9F3N2O3, white powder, 42% yield, APCI-MS: m/z 275.1 [M + H] +, 1H NMR (400 MHz, Methanol-d4): δ 9.47 (dd, J = 7.6, 0.8 Hz, 1H), 7.48 (tt, J = 2.4, 1.2 Hz, 1H), 7.12 (ddd, J = 7.6, 2.6, 0.8 Hz, 1H), 3.13 (q, J = 7.6 Hz, 2H), 1.33 (t, J = 7.6 Hz, 3H).
Ethyl-6-chloro-2-ethylimidazo[1,2-a]pyridine-3-carboxylate (7b)
C12H13ClN2O2, pale yellow solid, 28% yield, ESI-MS: m/z 253.2 [M + H]+, 1H NMR (400 MHz, Chloroform-d): δ 9.41 (dd, J = 2.1, 0.8 Hz, 1H), 7.57 (dd, J = 9.5, 0.8 Hz, 1H), 7.34 (dd, J = 9.5, 2.1 Hz, 1H), 4.44 (q, J = 7.1 Hz, 2H), 3.10 (q, J = 7.5 Hz, 2H), 1.44 (t, J = 7.1 Hz, 3H), 1.35 (t, J = 7.5 Hz, 3H).
6-Chloro-2-ethylimidazo[1,2-a]pyridine-3-carboxylic Acid (7c)
C10H9ClN2O2, white powder, 42% yield, ESI-MS: m/z 223.11 [M – H]−, 1H NMR (500 MHz, Methanol-d4) δ 9.51 (dd, J = 2.1, 0.8 Hz, 1H), 7.61 (dd, J = 9.5, 0.8 Hz, 1H), 7.54 (dd, J = 9.5, 2.0 Hz, 1H), 3.15 (q, J = 7.6 Hz, 2H), 1.34 (t, J = 7.6 Hz, 3H).
Ethyl 2-Ethylimidazo[1,2-a]pyridine-3-carboxylate (8b)
C12H14N2O2, orange solid, 5% yield, APCI-MS: m/z 219.0 [M + H]+, 1H NMR (400 MHz, Chloroform-d): δ 9.33 (dt, J = 7.0, 1.2 Hz, 1H), 7.66 (dt, J = 8.9, 1.2 Hz, 1H), 7.37 (ddd, J = 8.9, 6.8, 1.3 Hz, 1H), 6.97 (td, J = 6.9, 1.3 Hz, 1H), 4.43 (q, J = 7.1 Hz, 2H), 3.13 (q, J = 7.5 Hz, 2H), 1.44 (t, J = 7.1 Hz, 3H), 1.36 (t, J = 7.5 Hz, 3H).
2-Ethylimidazo[1,2-a]pyridine-3-carboxylic Acid (8c)
C10H10N2O2, orange solid, 90% yield, ESI-MS: m/z 189.17 [M – H]−, 1H NMR (400 MHz, Methanol-d4): δ 9.65 (d, J = 6.9 Hz, 2H), 8.13 (ddd, J = 8.5, 7.0, 1.1 Hz, 1H), 8.06 (d, J = 8.9 Hz, 1H), 7.67 (td, J = 7.0, 1.3 Hz, 1H), 3.34–3.26 (m, 2H), 1.43 (t, J = 7.6 Hz, 4H).
Ethyl-6-bromo-2-methylimidazo[1,2-a]pyridine-3-carboxylate (9b)
C11H11BrN2O2, pale yellow solid, 39% yield, APCI-MS: m/z 283.1 [M + H] +, 1H NMR (400 MHz, Chloroform-d): δ 9.49 (dd, J = 1.9, 0.9 Hz, 1H), 7.52–7.46 (m, 1H), 7.44 (dd, J = 9.4, 1.9 Hz, 1H), 4.44 (q, J = 7.1 Hz, 2H), 2.70 (s, 3H), 1.44 (t, J = 7.1 Hz, 3H).
6-Bromo-2-methylimidazo[1,2-a]pyridine-3-carboxylic Acid (9c). C9H7BrN2O2
Beige solid, 65% yield, APCI-MS: m/z 255.1 [M + H]+, 1H NMR (400 MHz, Methanol-d4) δ 9.60 (dd, J = 2.0, 0.8 Hz, 1H), 7.61 (dd, J = 9.5, 2.0 Hz, 1H), 7.52 (dd, J = 9.4, 0.8 Hz, 1H), 2.68 (s, 3H).
Ethyl-7-bromo-2-methylimidazo[1,2-a]pyridine-3-carboxylate (10b)
C11H11BrN2O3, pale yellow solid, 18% yield, APCI-MS: m/z 283.0, 285.0 (79Br,81Br) [M + H]+. This compound was taken directly to the next step without further purification.
7-Bromo-2-methylimidazo[1,2-a]pyridine-3-carboxylic Acid (10c)
C10H7BrN2O2, white solid, 70% yield, APCI-MS: m/z 255.1, 257.1(79Br,81Br) [M]+, 1H NMR (400 MHz, Methanol-d4): δ 9.29 (dd, J = 7.4, 0.8 Hz, 1H), 7.81 (dd, J = 2.0, 0.8 Hz, 1H), 7.27 (dd, J = 7.4, 2.0 Hz, 1H), 2.68 (s, 3H).
Ethyl-6-(trifluoromethyl)-2-methylimidazo[1,2-a]pyridine-3-carboxylate (11b)
C12H11F3N2O2, beige powder, 22%, APCI-MS: m/z 273.4 [M + H]+, 1H NMR (400 MHz, Methanol-d4): δ 9.79–9.70 (m, 1H), 7.84–7.75 (m, 1H), 7.74 (dd, J = 9.4, 1.8 Hz, 1H), 4.47 (q, J = 7.1 Hz, 2H), 2.72 (s, 3H), 1.45 (t, J = 7.1 Hz, 3H).
6-(Trifluoromethyl)-2-methylimidazo[1,2-a]pyridine-3-carboxylic Acid (11c)
C12H11BrN2O2F3, light orange solid, 82% yield, APCI-MS: m/z 273.4 [M + H]+, 1H NMR (400 MHz, Methanol-d4): δ 9.91–9.74 (m, 1H), 7.82–7.78 (m, 1H), 7.76 (dd, J = 9.4, 1.8 Hz, 1H), 2.76 (s, 3H).
Ethyl-7-(trifluoromethyl)-2-methylimidazo[1,2-a]pyridine-3-carboxylate (12b)
C12H11F3N2O2, white solid,17% yield, APCI-MS: m/z 272.9 [M]+,1H NMR (400 MHz, Chloroform-d): δ 9.42 (dt, J = 7.3, 0.9 Hz, 1H), 7.89 (dt, J = 1.9, 1.0 Hz, 1H), 7.14 (dd, J = 7.3, 1.9 Hz, 1H), 4.45 (q, J = 7.1 Hz, 2H), 2.74 (s, 3H), 1.45 (t, J = 7.1 Hz, 3H).
7-(Trifluoromethyl)-2-methylimidazo[1,2-a]pyridine-3-carboxylate (12c)
C10H7F3N2O2, pale yellow, 65% yield, APCI-MS: m/z 272.9 [M]+, 1H NMR (400 MHz, Methanol-d4): δ 9.69 (d, J = 7.3 Hz, 1H), 8.10 (s, 1H), 7.56 (dd, J = 7.3, 2.0 Hz, 1H), 2.82 (s, 3H).
Ethyl-6-(trifluoromethoxy)-2-methylimidazo[1,2-a]pyridine-3-carboxylate (13b)
C12H11F3N2O3, white powder, 21% yield, APCI-MS: m/z 289.1[M + H]+. This compound was taken directly to the next step without further purification.
6-(Trifluoromethoxy)-2-methylimidazo[1,2-a]pyridine-3-carboxylic Acid (13c)
C10H7F3N2O3, white solid, 85% yield, APCI-MS: m/z 261.1 [M + H]+, 1H NMR (400 MHz, Methanol-d4): δ 9.51 (dt, J = 2.5, 0.8 Hz, 1H), 7.69 (dd, J = 9.7, 0.8 Hz, 1H), 7.62–7.53 (m, 1H), 2.71 (s, 3H).
Ethyl-2-methyl-7-(trifluoromethoxy)imidazo[1,2-a]pyridine-3-carboxylate (14b)
C12H11F3N2O3, white powder, 22% yield, APCI-MS: m/z 288.9 [M + H]+. This compound was taken directly to the next step without further purification.
2-Methyl-7-(trifluoromethoxy)imidazo[1,2-a]pyridine-3-carboxylic Acid (14c)
C10H7F3N2O3, white solid, 43% yield, APCI-MS: m/z 261.1 [M + H]+,1H NMR (500 MHz, Methanol-d4): δ 9.52 (d, J = 7.7 Hz, 1H), 7.55 (s, 1H), 7.23 (d, J = 7.6 Hz, 1H), 2.73 (s, 3H).
Ethyl-2,7-dimethylimidazo[1,2-a]pyridine-3-carboxylate (15b)
C12H14N2O2, pale yellow solid, 17%, APCI-MS: m/z 219.0 [M + H]+, 1H NMR (400 MHz, Chloroform-d): δ 9.14 (dd, J = 7.0, 0.9 Hz, 1H), 7.34 (dt, J = 2.0, 1.0 Hz, 1H), 6.78 (dd, J = 7.1, 1.8 Hz, 1H), 4.40 (q, J = 7.1 Hz, 2H), 2.67 (s, 3H), 2.44–2.39 (m, 6H), 1.41 (t, J = 7.1 Hz, 3H).
2,7-Dimethylimidazo[1,2-a]pyridine-3-carboxylic Acid (15c)
C12H14N2O2, white solid, 39% yield, ESI-MS: m/z 191.28 [M + H]+, 1H NMR (400 MHz, Methanol-d4): δ 9.45 (dd, J = 7.1, 0.8 Hz, 1H), 7.63 (dt, J = 1.9, 1.0 Hz, 1H), 7.33 (dd, J = 7.1, 1.7 Hz, 1H), 2.76 (s, 3H), 2.66–2.49 (m, 3H).
Synthesis of 2-Substituted 1,3-Benzoxazole-5-methylamine
2-(4-Chlorophenyl)-1,3-benzoxazole-5-carbonitrile (1d)
To a suspension of 3-amino-4-hydroxybenzonitrile (5.7 mmol) in dioxane, 4-chlorobenzoylchloride (5.7 mmol) was added. The mixture was stirred at 100 °C, and then, methanesulfonic acid (17.1 mmol, 3 equiv) was added carefully. The reaction was stirred at 100 °C overnight. After the evaporation of dioxane, a saturated solution of NaHCO3 was added carefully (∼5 mL). Effervescence was observed due to the release of CO2. The formed brown precipitate was filtered and washed thoroughly with H2O. This precipitate was recrystallized using hot isopropanol (70%) to get 1y in the form of reddish-brown crystals.43 Yield = 54%, APCI-MS: m/z 255.3 [M + H]+, 1H NMR (400 MHz, Chloroform-d): δ 8.19 (d, J = 8.6 Hz, 2H), 8.08 (d, J = 1.1 Hz, 1H), 7.67 (d, J = 1.4 Hz, 2H), 7.58–7.49 (m, 2H). 13C NMR (101 MHz, Chloroform-d): δ 164.2, 153.1, 142.5, 138.9, 129.5, 129.3, 129.2, 124.6, 124.5, 118.6, 111.9, 108.8.
2-(4-Chlorophenyl)-1,3-benzoxazole-5-methylamine (1e)
1y (0.208 mmol) was suspended in dry MeOH (10 mL). Di-tert-butyl-dicarbonate (0.416, 2 equiv) and NiCl2.6H2O (0.02 mmol, 0.1 equiv) were added. After cooling the mixture to 0 °C, NaBH4 (0.93 mmol, 4.5 equiv) was added portionwise and left to stir vigorously at room temperature overnight. 300 μL of diethylenetriamine was added before the evaporation of the solvent. The mixture was then diluted with ethyl acetate and washed with 10% citric acid, saturated NaHCO3, and brine. The organic phase was dried over anhydrous MgSO4, concentrated, and then purified with column chromatography using EtOAc/heptane (1:2) as the mobile phase. To the purified 1eBoc, 4 M HCl in dioxane was added (3–5 mL). The mixture was stirred for 2 h. As a workup, diethyl ether was added, and the formed precipitate was filtered and dried to give a grayish-white solid.28 90% yield, ESI-MS: m/z 259.16 [M + H]+, 1H NMR (400 MHz, Methanol-d4): δ 8.32–8.18 (m, 2H), 7.88 (dd, J = 1.8, 0.6 Hz, 1H), 7.78 (dd, J = 8.4, 0.6 Hz, 1H), 7.74–7.56 (m, 3H), 7.54 (dd, J = 8.4, 1.8 Hz, 1H), 4.28 (s, 3H).
2-(4-(Trifluoromethoxy)phenyl)-1,3-benzoxazole-5-carbonitrile (2d)
Under argon, 4-(trifluoromethoxy)benzoic acid (4.85 mmol) was suspended in dry DCM (20 mL), then a catalytic amount of DMF (3 drops) was added followed by thionyl chloride (2 equiv). The colorless suspension was left to stir at room temperature for 2 h. The solvent was co-evaporated with toluene to get rid of excess remaining thionyl chloride.44 The produced 4-(trifluoromethoxy)benzoyl chloride oil was directly added to a suspension of 3-amino-4-hydroxybenzonitrile (4.85 mmol, 1 equiv) in dioxane. The procedure continues in the same manner as mentioned above for (1d); however, 3y crystals were paler and gave a lower yield (∼30%). APCI-MS: m/z 305 [M + H]+, 1H NMR (400 MHz, Chloroform-d): δ 8.42–8.19 (m, 2H), 8.09 (t, J = 1.1 Hz, 1H), 7.73–7.64 (m, 2H), 7.45–7.26 (m, 2H). 13C NMR (101 MHz, Chloroform-d): δ 163.8, 153.1, 152.2 (q, C-OCF3, 1JCF = 1.7 Hz), 142.5, 129.8, 129.35, 124.7, 124.5, 121.6, 120.2 (q, OCF3, 1JCF = 265 Hz), 118.8, 111.9, 108.8.
2-(4-(Trifluoromethoxy)phenyl)-1,3-benzoxazole-5-methylamine (2e)
3y (0.208 mmol) was suspended in dry MeOH (10 mL). Di-tert-butyl-dicarbonate (0.416, 2 equiv) and NiCl2.6H2O (0.02 mmol, 0.1 equiv) were added. After cooling the mixture to 0 °C, NaBH4 (0.93 mmol, 4.5 equiv) was added portionwise and left to stir vigorously at room temperature overnight. 300 μL of diethylenetriamine was added before the evaporation of the solvent. The mixture was then diluted with ethyl acetate and washed with 10% citric acid, saturated NaHCO3, and brine. The organic phase was dried over anhydrous MgSO4, concentrated, and then 4 M HCl in dioxane was added (3–5 mL). The mixture was stirred for 2 h. As a workup, diethyl ether was added, and the formed precipitate was filtered and dried to give a gray precipitate.28 73% yield, ESI-MS: m/z 309.32 [M + H] +, 1H NMR (400 MHz, Methanol-d4): δ 8.43–8.27 (m, 2H), 7.90 (dd, J = 1.8, 0.6 Hz, 1H), 7.79 (dd, J = 8.4, 0.6 Hz, 1H), 7.56 (d, J = 1.8 Hz, 1H), 7.54–7.51 (m, 2H), 4.29 (s, 2H).
General Procedure for the Synthesis of Farnesyl Amine30 (Scheme 3)
2-((2E,6E)-3,7,11-trimethyldodeca-2,6,10-trien-1-yl)isoindoline-1,3-dione (1f)
In a 100 mL round-bottom flask, E-farnesol, phthalimide (1.2 equiv), and Ph3P (1.3 equiv) were stirred in anhydrous THF (25 mL) in the dark. 1.3 equiv DIAD was added dropwise, and then, the mixture was left stirring at room temperature for 4 h. H2O (100 mL) was added, and the aqueous phase was extracted with heptane (3 × 50 mL). The collected organic phases were washed with brine before drying over anhydrous Na2SO4 and then filtered. N-farnesylphthalimide (1f) was isolated by flash column chromatography (gradient elution, heptane/EtOAc, 100:0 to 50:50). Colorless oil, 72% yield, APCI-MS: m/z 352.1 [M + H]+, 1H NMR (500 MHz, Chloroform-d): δ 7.83 (dd, J = 5.4, 3.0 Hz, 2H), 7.72–7.62 (m, 2H), 5.28 (tq, J = 7.1, 1.3 Hz, 1H), 5.09–5.02 (m, 2H), 4.28 (d, J = 7.1 Hz, 2H), 2.11–1.87 (m, 8H), 1.84 (d, J = 1.3 Hz, 3H), 1.66 (d, J = 1.3 Hz, 3H), 1.57 (dd, J = 2.7, 1.3 Hz, 6H). 13C NMR (126 MHz, Chloroform-d): δ 168.1, 140.6, 135.3, 133.7, 132.3, 131.2, 124.3, 123.6, 123.1, 118.0, 39.6, 39.5, 35.8, 26.7, 26.2, 25.6, 17.6, 16.3, 15.9.
(2E,6E)-3,7,11-trimethyldodeca-2,6,10-trien-1-amine (1g)
The purified 1f was dissolved in MeOH, and then, hydrazine hydrate (50% w/w) was added. The solution was left to stir at room temperature overnight. The reaction mixture was then diluted with an equal amount of H2O, acidified with conc. HCl (pH < 2), and washed with diethyl ether. The organic phase was discarded. The aqueous phase was basified with solid KOH to pH > 10 and extracted with diethyl ether (3×). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, and concentrated under vacuum. The resulting oil was passed through a silica gel plug with 3% MeOH/DCM to yield 4z as a colorless oil, APCI-MS: m/z 222.1 [M + H]+, 1H NMR (500 MHz, Chloroform-d): δ 5.25 (tp, J = 6.8, 1.3 Hz, 1H), 5.13–5.05 (m, 2H), 3.27 (d, J = 6.8 Hz, 2H), 2.10–1.92 (m, 8H), 1.67 (d, J = 1.3 Hz, 3H), 1.62 (d, J = 1.3 Hz, 3H), 1.59 (d, J = 1.3 Hz, 6H).
Synthesis of Imidazopyridine Amide Analogues (1–27)
Method A
In a round-bottom flask covered with aluminum foil (for light exclusion), nc, 1z, or 3z (1 equiv) and DIPEA (5 equiv) were dissolved in DMF (∼10 mL) under argon. PyBOP (1.1 equiv), dissolved in DMF, was added through a septum over 30 min, and the reaction was left to stir overnight at room temperature. The mixture was diluted with H2O to stop the reaction. The desired product crushed out and was filtered and washed with H2O several times to get rid of DMF (1–5).
Method B
Under an inert atmosphere, nc and DIPEA (5 equiv) were dissolved in anhydrous DMF. The yellow solution was left to stir for 10 min before adding HATU (1.1 equiv). The solution got darker in color as it stirred for 30 min. 1z or 3z (1 equiv) was added, and the reaction was stirred at room temperature for 1 h. Brine (50 mL) was added to precipitate the product. The filtered product was washed several times with water and EtOAc and one time with MeOH to get rid of DMF and impurities (6–27).
6-Bromo-2-ethyl-N-((2-(4-chlorophenyl)-1,3-benzoxazol-5-yl)methyl)imidazo[1,2a]pyridine-3-carboxamide (1)
C24H18BrClN4O2, beige solid, 29% yield, mp 260–262 °C, ESI-HRMS: m/z 509.0379, 511.0354 (79BrCl, 81BrCl) [M + H]+, 1H NMR (500 MHz, Chloroform-d): δ 9.63 (dd, CH (IPA), J = 1.9, 0.9 Hz, 1H), 8.22–8.08 (m, 2 CH, 2H), 7.75 (dd, CH, J = 1.7, 0.7 Hz, 1H), 7.57 (dd, CH, J = 8.3, 0.6 Hz, 1H), 7.53–7.47 (m, 2CH, 2H), 7.48 (dd, CH (IPA), J = 9.5, 0.9 Hz, 1H), 7.41 (dd, CH, J = 8.5, 1.8 Hz, 1H), 7.38 (dd, CH (IPA), J = 9.5, 1.8 Hz, 1H), 6.18 (t, NH, J = 5.8 Hz, 1H), 4.81 (d, CH2NH, J = 5.8 Hz, 2H), 2.98 (q, CH2CH3, J = 7.6 Hz, 2H), 1.40 (t, CH3, J = 7.5 Hz, 3H). 13C NMR (126 MHz, Chloroform-d): δ 162.8, 161.1, 151.29, 150.31, 144.6, 142.6, 137.9, 135.1, 130.4, 129.3, 128.9, 128.4, 125.5, 125.1, 119.1, 117.2, 114.8, 110.9, 108.1, 43.6, 23.6, 13.2. Rf = 0.44 (DCM: 3% MeOH).
6-Bromo-2-methyl-N-((2-(4-chlorophenyl)-1,3-benzoxazol-5-yl)methyl)imidazo[1,2a]pyridine-3-carboxamide (2)
C23H16BrClN4O2, beige solid, 40% yield, mp 285–288 °C, ESI-HRMS: m/z 495.52, 497.019 (79BrCl, 81BrCl) [M + H] +, 1H NMR (400 MHz, Chloroform-d): δ 9.67 (dd, CH (IPA), J = 1.9, 0.9 Hz, 1H), 8.23–8.14 (m, 2CH, 2H), 7.77 (dd, CH, J = 1.6, 0.7 Hz, 1H), 7.58 (d, CH, J = 8.3 Hz, 1H), 7.55–7.49 (m, 2CH, 2H), 7.47 (dd, CH (IPA), J = 9.4, 0.9 Hz, 1H), 7.45–7.37 (m, 2CH, 2H), 6.17 (t, NH J = 5.7 Hz, 1H), 4.83 (d, CH2NH, J = 5.8 Hz, 2H), 2.69 (s, CH3, 3H). 13C NMR (101 MHz, Chloroform-d): δ 162.8, 161.1, 150.3, 144.5, 142.6, 137.9, 135.1, 130.5, 129.3, 128.8, 128.4, 125.5, 125.1, 119.04, 117.01, 110.9, 108.2, 43.5, 16.8. Rf = 0.22 (DCM: 2% MeOH).
6-Bromo-2-ethyl-N-((2-(4-(trifluoromethoxy)phenyl)-1,3-benzoxazol-5yl)methyl)imidazo[1,2a]pyridine-3-carboxamide (3)
C25H18BrF3N4O3, pale pink solid, 70% yield, mp 228–232 °C, ESI-HRMS: m/z 559.0577, 561.0559 (79Br, 81Br) [M + H]+, 1H NMR (400 MHz, Chloroform-d): δ 9.65 (dd, CH (IPA), J = 1.9, 0.9 Hz, 1H), 8.34–8.25 (m, 2CH, 2H), 7.78 (dd, CH, J = 1.8, 0.7 Hz, 1H), 7.59 (dd, CH, J = 8.4, 0.6 Hz, 1H), 7.50 (dd, CH (IPA), J = 9.5, 0.9 Hz, 1H), 7.46–7.41 (m, 2CH, 2H), 7.40–7.35 (m, 2CH, 2H), 6.2 (t, NH, J = 5.8 Hz, 1H), 4.83 (d, CH2NH, J = 5.8 Hz, 2H), 3.00 (q, CH2CH3, J = 7.6 Hz, 2H), 1.42 (t, CH3, J = 7.6 Hz, 3H). 13C NMR (101 MHz, Chloroform-d): δ 162.5, 161.1, 151.3, 150.3, 144.6, 142.5, 137.8, 135.1, 130.4, 129.4, 128.4, 124.2 (q, OCF3, 1JCF = 267 Hz), 125.2, 122.4, 121.1, 119.1, 117.2, 116.5, 110.9, 108.1, 43.5, 23.6, 13.2. Rf = 0.23 (CHCl3: 1% MeOH).
6-Bromo-2-methyl-N-((2-(4-(trifluoromethoxy)phenyl)-1,3-benzoxazol-5-yl)methyl)imidazo[1,2a]pyridine-3-carboxamide (4)
C24H16BrF3N4O3, pale pink solid, 20% yield, mp 251–255 °C, ESI-HRMS: m/z 545.32, 547.0411 (79Br, 81Br) [M + H] +, 1H NMR (500 MHz, Chloroform-d): δ 9.77–9.48 (m, CH (IPA), 1H), 8.37–8.19 (m, 2CH, 2H), 7.82–7.75 (m, CH, 1H), 7.60 (d, CH, J = 8.3 Hz, 1H), 7.53 (d, CH (IPA), J = 9.3 Hz, 1H), 7.47 (d, CH (IPA), J = 9.3 Hz, 1H), 1H), 7.43 (dd, CH, J = 8.3, 1.8 Hz, 1H), 7.41–7.31 (m, 2H), 6.28 (s, NH, 1H), 4.84 (d, J = 5.7 Hz, 2H), 2.72 (s, 3H). 13C NMR (101 MHz, Chloroform-d): δ 162.5, 161.1, 151.6 (q, J = 1.8 Hz), 150.3, 145.8, 144.5, 142.5, 135.2, 130.6, 129.4, 128.4, 125.5, 125.2, 121.6, 121.1, 119.1, 116.9, 115.5, 110.9, 108.2, 43.5, 16.8. Rf = 0.28 (CHCl3: 2% MeOH).
7-Bromo-2-ethyl-N-((2-(4-chlorophenyl)-1,3-benzoxazol-5-yl)methyl)imidazo[1,2a]pyridine-3-carboxamide (5)
C24H18BrClN4O2, white solid, 50% yield, mp 294–296 °C, ESI-HRMS: m/z 509.0374, 511.0351, 513.0325(79BrCl, 81BrCl, 81Br37Cl) [M + H]+, 1H NMR (500 MHz, Chloroform-d): δ 9.33 (dd, J = 7.4, 0.8 Hz, 1H), 8.24–8.18 (m, 2H), 7.80 (dd, J = 2.0, 0.8 Hz, 1H), 7.78 (dd, J = 1.7, 0.7 Hz, 1H), 7.59 (dd, J = 8.3, 0.6 Hz, 1H), 7.55–7.50 (m, 2H), 7.42 (dd, J = 8.4, 1.7 Hz, 1H), 7.04 (dd, J = 7.4, 2.0 Hz, 1H), 6.19 (s, 1H), 4.83 (d, J = 5.7 Hz, 2H), 3.00 (q, J = 7.5 Hz, 2H), 1.41 (t, J = 7.6 Hz, 3H). 13C NMR (126 MHz, Chloroform-d): δ 162.3, 161.1, 142.6, 135.1, 129.3, 128.9, 128.4, 128.1, 125.5, 125.1, 121.2, 119.1, 117.0, 110.9, 43.6, 23.6, 13.2. Rf = 0.1 (EtOAc: Heptane: DCM, 1:1:1).
7-Bromo-2-methyl-N-((2-(4-chlorophenyl)-1,3-benzoxazol-5-yl)methyl)imidazo[1,2a]pyridine-3-carboxamide (6)
C23H16BrClN4O2, white solid, 7% yield, mp 281–285 °C, ESI-HRMS: m/z 495.0227, 497.0202 (79BrCl, 81BrCl) [M + H]+, 1H NMR (400 MHz, Chloroform-d): δ 9.35 (d, J = 7.4 Hz, 1H), 8.22–8.17 (m, 2H), 7.81–7.75 (m, 1H), 7.59 (d, J = 8.1 Hz, 1H), 7.56–7.51 (m, 2H), 7.44–7.39 (m, 2H), 7.06 (dd, J = 7.4, 1.9 Hz, 1H), 6.16 (t, J = 5.7 Hz, 1H), 4.82 (d, J = 5.8 Hz, 2H), 2.69 (s, 3H). 13C NMR (126 MHz, Chloroform-d): δ 162.8, 161.1, 150.3, 142.6, 138.00, 135.1, 129.3, 128.9, 128.4, 125.5, 125.1, 121.4, 119.1, 118.8, 117.1, 110.9, 43.5, 16.8. (purified by preparative HPLC), Rf = 0.15 (DCM: 2% MeOH).
7-Bromo-2-ethyl-N-((2-(4-(trifluoromethoxy)phenyl)-1,3-benzoxazol-5-yl)methyl)imidazo[1,2a]pyridine-3-carboxamide (7)
C25H18BrF3N4O3, white solid, 77% yield, mp 266–270 °C, ESI-HRMS: m/z 559.0582, 561.0561 (79Br, 81Br) [M + H] +, 1H NMR (500 MHz, Chloroform-d): δ 9.33 (dd, J = 7.4, 0.8 Hz, 1H), 8.33–8.26 (m, 2H), 7.80 (dd, J = 2.1, 0.8 Hz, 1H), 7.79 (dd, J = 1.7, 0.7 Hz, 1H), 7.59 (dd, J = 8.3, 0.6 Hz, 1H), 7.42 (dd, J = 8.4, 1.8 Hz, 1H), 7.40–7.35 (m, 2H), 7.04 (dd, J = 7.4, 2.1 Hz, 1H), 6.19 (s, 1H), 4.83 (d, J = 5.7 Hz, 2H), 2.99 (q, J = 7.6 Hz, 2H), 1.41 (t, J = 7.6 Hz, 3H). 13C NMR (126 MHz, Chloroform-d): δ 161.1, 142.5, 135.1, 129.4, 128.4, 125.2, 121.1, 119.1, 119.1, 117.1, 110.9, 43.5, 23.5, 13.2. (purified by preparative HPLC). Rf = 0.3 (DCM:1% MeOH).
7-Bromo-2-methyl-N-((2-(4-(trifluoromethoxy)phenyl)-1,3-benzoxazol-5-yl)methyl)imidazo[1,2a]pyridine-3-carboxamide (8)
C24H16BrF3N4O3, white solid, 22% yield, mp 272–275 °C, ESI-HRMS: m/z 545.0422, 547.0401 (79Br, 81Br) [M + H]+, 1H NMR (500 MHz, Chloroform-d): δ 9.33 (d, J = 7.4 Hz, 1H), 8.38–8.23 (m, 2H), 7.88 (br s, 1H), 7.81 (d, J = 1.6 Hz, 1H), 7.59 (d, J = 8.3 Hz, 1H), 7.44 (dd, J = 8.3, 1.7 Hz, 1H), 7.42–7.38 (m, 2H), 7.20–7.06 (m, 1H), 6.58 (s, 1H), 4.83 (d, J = 5.5 Hz, 2H), 2.74 (s, 3H). 13C NMR (126 MHz, Chloroform-d): δ 162.5, 151.6 (q, J = 1.8 Hz), 150.4, 142.5, 135.0, 129.4, 128.5, 125.5, 125.3, 124.5 (q, OCF3, 1JCF = 271.1 Hz), 121.4, 121.1, 119.3, 110.9, 43.6, 11.5.92% purity (HPLC), Rf = 0.1 (CHCl3:1% MeOH). For the purification of this compound, a column was prepared using the mobile phase CHCl3/MeOH (99:1). This compound was not further purified as it did not show activity in biochemical and in vitro assays.
6-Trifluoromethyl-2-ethyl-N-((2-(4-chlorophenyl)-1,3-benzoxazol-5-yl)methyl)imidazo[1,2a]pyridine-3-carboxamide (9)
C25H18ClF3N4O2, white solid, 6% yield, mp 255–257 °C, ESI-HRMS: m/z 499.1144 [M + H], 1H NMR (500 MHz, Chloroform-d): δ 9.89 (dq, J = 2.1, 1.2 Hz, 1H), 8.27–8.11 (m, 2H), 7.78 (dd, J = 1.8, 0.6 Hz, 1H), 7.71 (dt, J = 9.4, 0.9 Hz, 1H), 7.59 (dd, J = 8.4, 0.6 Hz, 1H), 7.54–7.50 (m, 2H), 7.50–7.46 (m, 1H), 7.42 (dd, J = 8.4, 1.8 Hz, 1H), 6.24 (t, J = 5.4 Hz, 1H), 4.85 (d, J = 5.8 Hz, 2H), 3.03 (q, J = 7.6 Hz, 2H), 1.43 (t, J = 7.5 Hz, 3H). 13C NMR (126 MHz, Chloroform-d): δ 162.8, 160.9, 152.4, 150.3, 146.0, 142.6, 138.01, 134.8, 129.3, 128.9, 127.4 (q, 3JCF = 5.7 Hz), 125.4, 125.1, 123.5 (q, 1JCF = 271.4 Hz), 122.9 (q, 4JCF = 2.6 Hz), 119.1, 117.6 (q, 2JCF = 34.2 Hz), 117.3, 115.6, 110.9, 43.6, 23.6, 13.1.
6-Trifluoromethyl-2-methyl-N-((2-(4-chlorophenyl)-1,3-benzoxazol-5-yl)methyl)imidazo[1,2a]pyridine-3-carboxamide (10)
C24H16ClF3N4O2, white solid, 39% yield, mp 251–254 °C, ESI-HRMS: m/z 485.0989 [M + H]+, 1H NMR (500 MHz, Chloroform-d): δ 10.01–9.84 (m, 1H), 8.25–8.16 (m, 2H), 7.79 (dd, J = 1.7, 0.7 Hz, 1H), 7.69 (d, J = 9.3 Hz, 1H), 7.59 (dd, J = 8.4, 0.6 Hz, 1H), 7.55–7.51 (m, 2H), 7.51–7.48 (m, 1H), 7.43 (dd, J = 8.4, 1.7 Hz, 1H), 6.28–6.16 (m, 1H), 4.85 (d, J = 5.8 Hz, 2H), 2.74 (s, 3H).13C NMR (126 MHz, Chloroform-d): δ 162.8, 160.9, 150.3, 146.9, 142.6, 138.0, 134.9, 129.3, 128.9, 125.4, 125.1, 123.4 (q, 1JCF = 274.6 Hz), 123.1 (q, 4JCF= 2.7 Hz), 119.1, 117.6 (q, 2JCF = 34.2 Hz), 117.1, 110.9, 43.6, 16.8.
6-Trifluoromethyl-2-ethyl-N-((2-(4-(trifluoromethoxy)phenyl)-1,3-benzoxazol-5-yl)methyl)imidazo[1,2a]pyridine-3-carboxamide (11)
C26H18F6N4O3, beige solid, 73% yield, mp 234–236 °C, ESI-HRMS: m/z 549.1363 [M + H], 1H NMR (400 MHz, Chloroform-d): δ 9.88 (dt, CH (IPA) J = 2.2, 1.1 Hz, 1H), 8.38–8.25 (m, 2CH, 2H), 7.82–7.75 (m, CH, 1H), 7.71 (d, CH (IPA), J = 9.4 Hz, 1H), 7.60 (d, CH, J = 8.4 Hz, 1H), 7.49 (dd, CH (IPA), J = 9.4, 1.9 Hz, 1H), 7.43 (dd, CH, J = 8.4, 1.7 Hz, 1H), 7.40–7.31 (m, 2CH, 2H), 6.25 (t, NH, J = 5.6 Hz, 1H), 4.85 (d, CH2NH, J = 5.8 Hz, 2H), 3.03 (q, CH2CH3, J = 7.6 Hz, 3H), 1.43 (t, CH3, J = 7.6 Hz, 3H). 13C NMR (101 MHz, Chloroform-d): δ 162.5, 160.9, 152.4, 151.6 (q, J = 1.6 Hz), 150.4, 146.0, 142.6, 134.9, 129.4, 127.5 (q, 3JCF = 5.7 Hz), 125.5, 125.2, 123.5 (q, 1JCF = 272.6 Hz), 122.9 (q, 4JCF = 2.6 Hz), 121.1, 119.1, 117.6 (q, 2JCF = 34.6 Hz), 117.3, 115.6, 111.0, 43.6, 23.6, 13.1. Rf = 0.25 (DCM: 2% MeOH).
6-Trifluoromethyl-2-methyl-N-((2-(4-(trifluoromethoxy)phenyl)-1,3-benzoxazol-5-yl)methyl)imidazo[1,2a]pyridine-3-carboxamide (12)
C25H16F6N4O3, pale pink solid, 71% yield, mp 199–202 °C, ESI-HRMS: m/z 535.1191 [M + H]+, 1H NMR (400 MHz, Chloroform-d): δ 9.89 (br s, CH (IPA), 1H), 8.34–8.22 (m, 2H), 7.79 (br s, 1H), 7.70 (d, CH (IPA) J = 9.3 Hz, 1H), 7.59 (d, J = 8.3 Hz, 1H), 7.52 (d, CH (IPA), J = 9.0 Hz, 1H), 7.43 (d, J = 8.4 Hz, 1H), 7.40–7.32 (m, 2H), 6.40 (s, NH, 1H), 4.84 (d, J = 4.3 Hz, 2H), 2.74 (s, 3H). 13C NMR (101 MHz, Chloroform-d): δ 162.5, 160.7, 151.6 (q, J = 1.7 Hz), 150.4, 142.5, 134.9, 129.4, 127.4 (q, 3JCF = 5.7 Hz), 125.5, 125.2, 123.3 (q, 1JCF = 273.5 Hz), 123.5 (q, 1JCF = 4.1 Hz), 120.5 (q, OCF3, 1JCF = 261.2 Hz), 121.1, 119.2, 118.0 (q, 2JCF = 35.0 Hz), 116.9, 110.9, 43.6, 16.6. Rf = 0.12 (CHCl3: 1% MeOH).
7-Trifluoromethyl-2-ethyl-N-((2-(4-chlorophenyl)-1,3-benzoxazol-5-yl)methyl)imidazo[1,2a]pyridine-3-carboxamide (13)
This compound was synthesized following method A. C25H18ClF3N4O2, beige solid, 70% yield, mp 265–268 °C, ESI-HRMS: m/z 499.1134 [M + H]+, 1H NMR (400 MHz, Chloroform-d): δ 9.55 (dt, CH (IPA), J = 7.4, 2.0, 1.2 Hz, 1H), 8.28–8.10 (m, 2CH, 2H), 7.92 (dt, CH (IPA), J = 2.0, 1.1 Hz, 1H), 7.81–7.68 (m, CH, 1H), 7.59 (d, CH, J = 8.2 Hz, 1H), 7.55–7.49 (m, 2CH, 2H), 7.41 (dd, CH, J = 8.4, 1.8 Hz, 1H), 7.10 (dd, CH (IPA) J = 7.4, 1.9 Hz, 1H), 6.23 (t, NH, J = 4.6 Hz, 1H), 4.84 (d, CH2NH, J = 5.7 Hz, 2H), 3.03 (q, CH2CH3, J = 7.5 Hz, 2H), 1.43 (t, CH3, J = 7.6 Hz, 3H). 13C NMR (101 MHz, Chloroform-d): δ 171.1, 160.9, 160.7, 152.5, 150.3, 149.8, 142.6, 138.0, 129.3, 129.0, 128.9, 125.4, 125.1, 119.1, 117.5 (q, 1JCF = 279.2 Hz), 117.4, 116.1, 114.5 (q, 3JCF = 3.7 Hz),112.9, 110.9, 108.9 (q, 4JCF = 1.9 Hz), 43.6, 23.5, 13.1. Rf = 0.25 (DCM: 1% MeOH).
7-Trifluoromethyl-2-methyl-N-((2-(4-chlorophenyl)-1,3-benzoxazol-5-yl)methyl)imidazo[1,2a]pyridine-3-carboxamide (14)
C24H16ClF3N4O2, white solid, 3% yield, mp 257–260 °C, ESI-HRMS: m/z 485.0978 [M + H]+, 1H NMR (400 MHz, Chloroform-d): δ 9.58 (d, J = 7.3 Hz, 1H), 8.19 (d, J = 8.2 Hz, 2H), 7.88 (s, 1H), 7.78 (s, 1H), 7.59 (d, J = 8.4 Hz, 1H), 7.51 (d, J = 8.3 Hz, 2H), 7.42 (d, J = 8.4 Hz, 1H), 7.11 (d, J = 7.3 Hz, 1H), 6.23 (s, 1H), 4.84 (d, J = 5.8 Hz, 2H), 2.73 (s, 3H). 13C NMR (126 MHz, Chloroform-d): δ 162.9, 160.9, 150.3, 146.9, 142.6, 138.0, 134.9, 129.3, 128.95, 128.9, 125.4, 125.1, 119.1, 117.6 (q, 1JCF = 280.0 Hz), 114.3 (q, 3JCF = 4.9 Hz), 110.9, 109.0 (q, 3JCF = 3.1 Hz), 43.6, 16.8. Rf = 0.24 (DCM: 1% MeOH).
7-Trifluoromethyl-2-ethyl-N-((2-(4-(trifluoromethoxy)phenyl)-1,3-benzoxazol-5-yl)methyl)imidazo[1,2a]pyridine-3-carboxamide (15)
C26H18F6N4O3, white solid, 47% yield, mp 258–261 °C, ESI-HRMS: m/z 549.1348 [M + H]+, 1H NMR (400 MHz, TFA/DMSO): δ 9.39 (d, J = 7.2 Hz, 1H), 8.50–8.40 (m, 2H), 8.27–8.18 (m, 1H), 8.04 (d, J = 1.6 Hz, 1H), 7.94 (d, J = 8.7 Hz, 1H), 7.86 (dd, J = 8.8, 1.6 Hz, 1H), 7.74 (dd, J = 7.4, 1.7 Hz, 1H), 7.62–7.54 (m, 2H), 5.01 (s, 1H), 3.27 (q, J = 7.6 Hz, 2H), 1.47 (t, J = 7.6 Hz, 3H). 13C NMR (126 MHz, TFA/DMSO): δ 166.5, 161.9, 158.9 (q, J = 1.8 Hz), 150.8, 145.8, 141.2, 140.6, 133.7, 132.5, 131.6, 131.1, 123.4, 119.5, 118.3 (q, 2JCF = 34.6 Hz), 117.1, 117.1, 116.7, 115.7, 115.6 (q, 1JCF = 282.8 Hz), 115.24, 45.91, 21.12, 12.85. Some signals are covered by the solvent signal. Rf = 0.21 (DCM: 1% MeOH).
7-Trifluoromethyl-2-methyl-N-((2-(4-(trifluoromethoxy)phenyl)-1,3-benzoxazol-yl)methyl)imidazo[1,2a]pyridine-3-carboxamide (16)
C25H16F6N4O3, white solid, 80% yield, mp 276–277 °C, ESI-HRMS: m/z 535.1192 [M + H]+, 1H NMR (500 MHz, Chloroform-d): δ 9.59 (dt, J = 7.3, 0.8 Hz, 1H), 8.34–8.26 (m, 2H), 7.88 (dt, J = 1.8, 1.0 Hz, 1H), 7.80 (dd, J = 1.7, 0.7 Hz, 1H), 7.62–7.57 (m, 1H), 7.43 (dd, J = 8.4, 1.7 Hz, 1H), 7.41–7.35 (m, 2H), 7.12 (dd, J = 7.4, 1.9 Hz, 1H), 6.24 (s, 1H), 4.85 (d, J = 5.8 Hz, 2H), 2.74 (s, 3H). 13C NMR (126 MHz, Chloroform-d): δ 162.5, 160.9, 150.4, 146.9, 144.3, 142.6, 134.9, 129.4, 128.9, 125.2, 121.9, 121.1 (q, 4JCF = 1.2 Hz), 119.2, 117.6 (q, 1JCF = 275.3 Hz), 116.5, 114.3 (q, 3JCF = 5.0 Hz), 111.0, 109.0 (q, 3JCF = 3.0 Hz), 43.6, 16.8.100% purity (HPLC, the compound was purified using preparative HPLC). Rf = 0.2 (DCM: 2% MeOH).
6-Trifluoromethoxy-2-ethyl-N-((2-(4-chlorophenyl)-1,3-benzoxazol-5-yl)methyl)imidazo[1,2a]pyridine-3-carboxamide (17)
C25H18ClF3N4O3, white solid, 10% yield, mp 264–266 °C, ESI-HRMS: m/z 515.1093 [M + H]+, 1H NMR (400 MHz, Chloroform-d): δ 9.64–9.58 (m, 1H), 8.21–8.13 (m, 2H), 7.77 (dd, J = 1.7, 0.6 Hz, 1H), 7.65 (d, J = 9.7 Hz, 1H), 7.58 (dd, J = 8.3, 0.6 Hz, 1H), 7.53–7.48 (m, 2H), 7.42 (dd, J = 8.4, 1.7 Hz, 1H), 7.33 (dd, J = 9.6, 2.5 Hz, 1H), 6.30–6.13 (m, 1H), 4.83 (d, J = 5.7 Hz, 2H), 3.01 (q, J = 7.6 Hz, 2H), 1.42 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, Chloroform-d): δ 162.8, 160.9, 150.3, 142.6, 138.0, 134.9, 129.3, 128.9, 125.4, 125.1, 124.1(q, 1JCF = 260.8 Hz), 122.3, 119.1, 116.8, 110.9, 43.6, 23.6, 13.1. Rf = 0.23 (CHCl3: 1% MeOH).
6-Trifluoromethoxy-2-methyl-N-((2-(4-chlorophenyl)-1,3-benzoxazol-5-yl)methyl)imidazo[1,2a]pyridine-3-carboxamide (18)
C24H16ClF3N4O3, white solid, 42% yield, mp 256–260 °C, ESI-HRMS: m/z 501.0937 [M + H]+, 1H NMR (500 MHz, Chloroform-d): δ 9.71–9.53 (m, 1H), 8.23–8.09 (m, 2H), 7.84–7.71 (m, 1H), 7.60 (dd, J = 9.6, 0.8 Hz, 1H), 7.58 (dd, J = 8.4, 0.5 Hz, 1H), 7.54–7.48 (m, 2H), 7.42 (dd, J = 8.4, 1.8 Hz, 1H), 7.33 (ddd, J = 9.6, 2.4, 1.0 Hz, 1H), 6.20 (s, 1H), 4.84 (d, J = 5.7 Hz, 2H), 2.72 (s, 3H). 13C NMR (126 MHz, Chloroform-d): δ 162.9, 161.0, 150.3, 146.7, 144.3, 142.6, 138.6 (q, J = 2.5 Hz), 138.0, 135.0, 129.3, 128.9, 127.8, 125.4, 125.1, 122.8, 122.2, 120.5 (q, 1JCF = 263.1 Hz), 119.06, 116.74, 110.95, 43.51, 16.89. Rf = 0.12 (CHCl3: 1% MeOH).
6-Trifluoromethoxy-2-ethyl-N-((2-(4-(trifluoromethoxy)phenyl)-1,3-benzoxazol-5-yl)methyl)imidazo[1,2a]pyridine-3-carboxamide (19)
C26H18F6N4O4, white solid, 60% yield, mp 248–251 °C, ESI-HRMS: m/z 565.13 [M + H]+, 1H NMR (400 MHz, Chloroform-d): δ 9.61 (s, 1H), 8.29 (d, J = 8.4 Hz, 2H), 7.78 (s, 1H), 7.62 (d, J = 9.7 Hz, 1H), 7.59 (d, J = 8.3 Hz, 1H), 7.42 (d, J = 8.4 Hz, 1H), 7.38 (d, J = 8.2 Hz, 2H), 7.32 (d, J = 9.7 Hz, 1H), 6.22 (s, 1H), 4.84 (d, J = 5.7 Hz, 2H), 3.01 (q, J = 7.5 Hz, 2H), 1.42 (t, J = 7.5 Hz, 3H). 13C NMR (126 MHz, Chloroform-d): δ 162.5, 161.0, 152.2, 151.6 (q, J = 1.8 Hz), 150.4, 144.5, 142.6, 138.6 (q, J = 2.6 Hz), 135.0, 129.4, 125.5, 125.2, 122.7, 122.3, 121.6 (q, 1JCF = 261.4 Hz), 121.4 (q, 1JCF = 258.5 Hz), 121.1, 119.1, 116.9, 115.7, 110.9, 43.5, 23.7, 13.2. Rf = 0.37 (CHCl3: 2% MeOH).
6-Trifluoromethoxy-2-methyl-N-((2-(4-(trifluoromethoxy)phenyl)-1,3-benzoxazol-5-yl)methyl)imidazo[1,2a]pyridine-3-carboxamide (20)
C25H16F6N4O4, white solid, 55% yield, mp 239–240 °C, ESI-HRMS: m/z 551.1141 [M + H]+, 1H NMR (400 MHz, Chloroform-d): δ 9.62 (s, CH (IPA), 1H), 8.35–8.18 (m, 2CH, 2H), 7.80 (s, CH, 1H), 7.69 (d, CH (IPA), J = 9.6 Hz, 1H), 7.59 (d, CH, J = 8.2 Hz, 1H), 7.44 (d, CH, J = 8.2 Hz, 1H), 7.40 (d, CH (IPA), J = 9.6 Hz, 1H), 7.38–7.33 (m, 2CH, 2H), 6.47 (s, NH, 1H), 4.84 (s, CH2NH, 2H), 2.76 (s, CH3, 3H). 13C NMR (101 MHz, Chloroform-d): δ 162.5, 160.6, 150.4, 142.6, 134.9, 129.4, 125.5, 125.3, 123.7, 122.3, 121.1, 119.2, 116.2, 114.1, 110.9, 43.6, 16.4. Rf = 0.3 (CHCl3: 1% MeOH).
7-Trifluoromethoxy-2-methyl-N_((2-(4_chlorophenyl)-1,3-benzoxazol-5-yl)methyl)imidazo[1,2a]pyridine-3-carboxamide (21)
C24H16ClF3N4O3, beige solid, 40% yield, mp 261–263 °C, ESI-HRMS: m/z 501.0926 [M + H]+, 1H NMR (500 MHz, Chloroform-d): δ 9.51 (dd, J = 7.6, 0.8 Hz, 1H), 8.26–8.11 (m, 2H), 7.78 (dd, J = 1.7, 0.7 Hz, 1H), 7.59 (dd, J = 8.3, 0.6 Hz, 1H), 7.55–7.48 (m, 2H), 7.44–7.38 (m, 2H), 6.86 (dd, J = 7.5, 2.5 Hz, 1H), 6.17 (t, J = 5.5, 5.1 Hz, 1H), 4.84 (d, J = 5.8 Hz, 2H), 2.70 (s, 3H). 13C NMR (126 MHz, Chloroform-d): δ 162.9, 161.1, 150.3, 148.0 (q, J = 1.8 Hz), 146.6, 146.0, 138.0, 135.1, 129.6, 129.3, 128.9, 125.5, 125.1, 119.1, 110.9, 107.9, 106.1, 43.5, 16.8. Rf = 0.086 (CHCl3: 1% MeOH).
7-Trifluoromethoxy-2-ethyl-N-((2-(4-(trifluoromethoxy)phenyl)-1,3-benzoxazol-5-yl)methyl)imidazo[1,2a]pyridine-3-carboxamide (22)
C26H18F6N4O4, white solid, 50% yield, mp 251–255 °C, ESI-HRMS: m/z 565.1303 [M + H]+, 1H NMR (400 MHz, Chloroform-d): δ 9.48 (d, CH (IPA), J = 7.6 Hz, 1H), 8.29 (d, 2CH, J = 8.5 Hz, 2H), 7.78 (s, CH, 1H), 7.59 (d, CH, J = 8.4 Hz, 1H), 7.44 (d, CH, J = 8.5 Hz, 1H), 7.40 (d, CH (IPA), J = 7.6 Hz, 1H), 7.37 (d, 2CH, J = 8.4 Hz, 2H), 6.85 (d, CH (IPA), J = 7.9 Hz, 1H), 6.20 (t, NH, J = 6.3 Hz, 1H), 4.83 (d, CH2NH, J = 5.7 Hz, 2H), 3.00 (q, CH2CH3, J = 7.5 Hz, 2H), 1.42 (t, CH3, J = 7.5 Hz, 3H).13C NMR (101 MHz, Chloroform-d): δ 161.1, 151.6 (q, J = 1.6 Hz), 150.3, 148.0 (q, J = 2.9 Hz), 146.0, 142.6, 135.1, 129.6, 129.4, 125.5, 125.2, 121.5, 121.1, 119.1, 114.9, 110.9, 107.9, 106.1, 43.5, 23.5, 13.1. Rf = 0.24 (CHCl3: 1% MeOH).
7-Trifluoromethoxy-2-methyl-N-((2-(4-(trifluoromethoxy)phenyl)-1,3-benzoxazol-5-yl)methyl)imidazo[1,2a]pyridine-3-carboxamide (23)
C25H16F6N4O4, white solid, 25% yield, mp 255–259 °C, ESI-HRMS: m/z 551.1152 [M + H]+, 1H NMR (400 MHz, Chloroform-d): δ 9.51 (d, J = 7.6 Hz, 1H), 8.32–8.25 (m, 2H), 7.79 (s, 1H), 7.59 (d, J = 8.3 Hz, 1H), 7.47–7.40 (m, 2H), 7.37 (d, J = 8.4 Hz, 2H), 6.89 (d, J = 7.6 Hz, 1H), 6.28 (s, 1H), 4.83 (d, J = 5.4 Hz, 2H), 2.72 (s, 3H). 13C NMR (126 MHz, Chloroform-d) δ 162.5, 161.1, 151.6 (q, J = 1.8 Hz), 150.4, 142.6, 135.1, 129.6, 129.4, 125.5, 125.2, 121.4 (q, 1JCF = 255.9 Hz), 121.3 (q, 1JCF = 259.4 Hz), 121.1, 119.1, 110.9, 107.9, 106.0, 43.5, 16.8.
6-Chloro-2-ethyl-N-((2-(4-(trifluoromethoxy)phenyl)-1,3-benzoxazol-5-yl)methyl)imidazo[1,2a]pyridine-3-carboxamide (24)
(22) C25H18ClF3N4O3, pale pink solid, 29% yield, mp 240–242 °C, ESI-HRMS: m/z 515.1089 [M + H] +, 1H NMR (400 MHz, Chloroform-d): δ 9.56 (dd, J = 2.1, 0.9 Hz, 1H), 8.32–8.26 (m, 2H), 7.78 (t, J = 2.8 Hz, 1H), 7.61–7.49 (m, 2H), 7.42 (dt, J = 8.3, 2.5 Hz, 1H), 7.39–7.34 (m, 2H), 7.31 (dd, J = 9.5, 2.1 Hz, 1H), 6.19 (s, 1H), 4.83 (d, J = 5.8 Hz, 2H), 3.08–2.92 (m, 2H), 1.41 (td, J = 7.6, 1.5 Hz, 3H). 13C NMR (13C NMR (101 MHz, Chloroform-d): δ 161.1, 150.3, 144.5, 142.5, 135.3, 133.9, 133.0, 129.4, 128.9, 128.3, 126.2, 125.5, 125.2, 121.6, 121.1, 119.1, 116.9, 111.0, 43.5, 23.6, 13.1.
N-benzyl-2,7-dimethylimidazo[1,2a]pyridine-3-carboxamide (25)
(18) C17H17N3O, white solid, mp 169–170 °C, ESI-HRMS: m/z 280.144 [M + H]+, 1H NMR (400 MHz, Chloroform-d): δ 9.31 (dd, J = 7.1, 0.9 Hz, 1H), 7.40–7.36 (m, 4H), 7.34–7.30 (m, 2H), 6.76 (dd, J = 7.2, 1.8 Hz, 1H), 4.70 (d, J = 5.7 Hz, 2H), 2.66 (s, 3H), 2.42 (d, J = 1.0 Hz, 3H). 13C NMR (101 MHz, Chloroform-d): δ 161.5, 146.5, 145.3, 138.3, 128.8, 127.6, 127.3, 115.7, 115.0, 43.4, 21.3, 16.8. Rf = 0.17 (DCM: 2% MeOH).
2-Ethyl-N-((2-(4-(trifluoromethoxy)phenyl)-1,3-benzoxazol-5-yl)methyl)imidazo[1,2a]pyridine-3-carboxamide (26)
C25H19F3N4O3, white solid, 25% yield, mp 229–233 °C, ESI-HRMS: m/z 481.1476 [M + H] +, 1H NMR (500 MHz, Chloroform-d): δ 9.43 (dt, J = 7.0, 1.2 Hz, 1H), 8.32–8.24 (m, 2H), 7.81–7.76 (m, 1H), 7.61 (dt, J = 8.9, 1.2 Hz, 1H), 7.58 (d, J = 8.3 Hz, 1H), 7.43 (dd, J = 8.4, 1.8 Hz, 1H), 7.39–7.34 (m, 2H), 7.33 (ddd, J = 9.0, 6.9, 1.4 Hz, 1H), 6.93 (td, J = 6.9, 1.2 Hz, 1H), 6.18 (d, J = 6.2 Hz, 1H), 4.84 (d, J = 5.7 Hz, 2H), 3.01 (q, J = 7.6 Hz, 2H), 1.42 (t, J = 7.6 Hz, 3H). 13C NMR (126 MHz, Chloroform-d): δ 162.4, 161.5, 151.6 (q, J = 1.8 Hz), 150.9, 150.3, 146.3, 142.5, 135.3, 129.4, 128.2, 126.9, 125.5, 125.2, 121.1, 120.3 (q, 1JCF = 259.0 Hz), 119.1, 116.7, 114.6, 113.2, 110.9, 43.5, 23.6, 13.3. Rf = 0.33 (CHCl3: 2% MeOH).
6-Chloro-2-ethyl-N-((2E,6E)-3,7,11-trimethyldodeca-2,6,10-trien-1-yl)imidazo[1,2a]pyridine-3-carboxamide (27)
C25H34ClN3O, white solid, 8% yield, mp 101–106 °C, ESI-HRMS: m/z 428.2459 [M + H] +, 1H NMR (500 MHz, Chloroform-d): δ 9.48 (dd, J = 2.1, 0.9 Hz, 1H), 7.52 (dd, J = 9.4, 0.9 Hz, 1H), 7.28 (dd, J = 9.5, 2.1 Hz, 1H), 5.70 (t, J = 5.2 Hz, 1H), 5.33 (tq, J = 7.0, 1.3 Hz, 1H), 5.13–5.04 (m, 2H), 4.10 (t, J = 5.8 Hz, 2H), 2.98 (q, J = 7.6 Hz, 2H), 2.15–1.95 (m, 8H), 1.76 (d, J = 1.3 Hz, 3H), 1.67 (d, J = 1.3 Hz, 3H), 1.60 (d, J = 1.3 Hz, 3H), 1.58 (d, J = 1.3 Hz, 3H), 1.43 (t, J = 7.6 Hz, 3H). 13C NMR (126 MHz, Chloroform-d): δ 160.9, 151.0, 144.3, 140.9, 135.5, 131.3, 127.9, 126.1, 124.2, 123.6, 121.4, 119.5, 116.8, 115.3, 39.7, 39.5, 37.4, 26.7, 26.3, 25.6, 23.4, 17.6, 16.4, 16.0, 13.2. Rf = 0.35 (CHCl3: 1% MeOH).
Nephelometric Solubility Assay
First, equidistant and logarithmic pre-dilutions were prepared in a 96-well plate with round bottoms, using 10 mM stock solutions of the compounds and DMSO. Then, 5 μL of each pre-dilution was added to 245 μL of PBS buffer in a 96 well-plate with straight bottoms. Quadruplets were prepared for the blank (DMSO + PBS), and final concentrations (one plate/compound) were tested. The final concentrations measured started from 200 μM to 0.3125 μM. A nephelometer (NEPHELOstarplus) was used to measure solubilities, and omega-data analysis software was used to evaluate the results.
Biochemical Assays
M. smegmatis Growth, Cell Lysis, and Protein Purification
The protocol followed was the same as described previously.15 An M. smegmatis strain with a 3× FLAG tag at the C terminus of subunit QcrB was grown on 7H9+hygromycin plates for 2 days at 30 °C. A colony from the plate was transferred to a 20 mL preculture of 7H9 (Sigma) supplemented with TDS (10 g/L, tryptone, 2 g/L dextrose, 0.8 g/L NaCl) and grown for 2 days in the dark at 30 °C and 180 rpm before inoculating a 6 L culture and growing under the same conditions for 48 h. Cells were harvested by centrifugation for 20 min at 4 °C, and then, cell pellets were frozen in liquid nitrogen and stored at −80 °C. Thawed cell pellets were resuspended in ∼150 mL of lysis buffer (50 mM Tris–HCl pH 7.5, 100 mM NaCl, and 0.5 mM EDTA) and homogenized first with a Dounce homogenizer, then filtered with cheesecloth and passed through an Avestin homogenizer three times at 20 kpsi. Lysed cells were centrifuged at 39,000g for 30 min to remove cell debris. The supernatant was then centrifuged at 149,000g for 60 min (Beckmann 70Ti rotor) to isolate the membranes. Membrane pellets were resuspended in lysis buffer (12 mL/g membranes) before aliquoting in falcon tubes, freezing, and storing at −80 °C. To isolate and purify CIII2CIV2, thawed membranes were solubilized with 1% (w/v) dodecyl maltoside detergent (DDM), stirring for 45 min at 4 °C. Following addition of detergent, the solution was centrifuged at 149,000g for 50 min to remove insoluble material. The supernatant was filtered (0.45 μm) before loading onto a column of 1.5 mL of Anti-FLAG M2 Affinity gel (Sigma). The column was washed with DTBS buffer (50 mM Tris–HCl pH 7.4, 100 mM NaCl, and 0.02% DDM) and eluted with 6 × 500 μL of 150 μg/mL 3× FLAG peptide. The purified protein was exchanged into 50 mM Tris–HCl pH 7.4, 100 mM NaCl, and 0.003% (w/v) glyco-diosgenin (GDN) using a 100 kDa molecular weight cut-off concentrator (Sigma).15
NDH-2 Purification from Caldalalibacillus thermarum Strain TA2.A1
Escherichia coli C41 (DE3) cells were transformed with the ppTRC99A-ndh vector31 and grown on LB + 100 μg/mL ampicillin plates overnight. A single colony was selected and grown in a 20 mL preculture in LB + 100 μg/mL ampicillin at 220 rpm overnight which was used to inoculate a 1 L growth. The 1 L culture was grown under the same conditions as the preculture until the OD600 ∼ 0.6 when the cells were induced with 1 mM isopropyl ß-d-thiogalactopyranoside and grown for 4 h before being harvested by centrifugation at 65,000g for 20 min and then flash frozen in liquid nitrogen. Cells were thawed and resuspended in 20 mL of NDH-2 lysis buffer (50 mM Tris–HCl pH 7.5, 2 mM MgCl2, 0.001% PMSF) and sonicated (Q Sonica Q500) for 10 min with 2 s pulses and 2 s pause between pulses and an amplitude of 30%. After lysis, cell debris was spun down at 10,000g for 15 min. Membranes from the supernatant were then harvested at 125,000g for 1 h and resuspended in buffer A [50 mM Tris–HCl pH 8.0, 20 mM imidazole, 150 mM NaCl, 0.05% w/v DDM, and 0.001% (w/v) PMSF]. Membranes were solubilized with 1% DDM at 4 °C for 2 h. This solubilization was followed by ultracentrifugation at 125,000g for 1 h to remove insoluble material. The supernatant was filtered (0.45 μm) before loading on a HisTrap column (5 mL) previously equilibrated with 50 mL of buffer A before loading the sample at 1 mL/min and then washed with 25 mL of buffer A to remove the unbound sample. To elute, 55% buffer B [50 mM Tris–HCl pH 8.0, 500 mM imidazole, 150 mM NaCl, 0.05% (w/v) DDM, and 0.001% (w/v) PMSF] was applied over 10 column volumes. Fractions with high absorbance at 280 nm were pooled and dialyzed against 50 mM Tris–HCl pH 8.0, 150 mM NaCl, and 0.05% w/v DDM to remove imidazole.
Sub-Mitochondrial Particle (SMP) Preparation
Bovine hearts were kept on ice, and all subsequent steps were carried out at 4 °C. Fat, blood vessels, and connective tissues were removed from the hearts, and the remaining material was cut into ∼2 cm3 pieces before being ground with a meat grinder. For each minced heart, 1400 mL of buffer A (250 mM sucrose, 10 mM Tris–HCl pH 7.8, 2-mercaptoethanol) was added. The buffer was squeezed out of the mince through muslin. Buffer B (1600 mL per heart; 250 mM sucrose, 10 mM Tris–HCl pH 7.8, 5 mM 2-mercaptoethanol, 0.2 mM EDTA, and 1 mM Tris-succinate) was added, and 25 mL of Tris (2 M not pH adjusted) was added per heart. The suspension was blended for 30 s on high setting. Cell debris was removed by centrifugation at 1600g for 15 min. The supernatant was filtered through muslin cloth, and mitochondria from the filtrate were harvested at 18,500g for 27 min. The mitochondria were resuspended in 3600 mL of buffer B per heart and harvested again at 18,500g for 27 min. A total of ∼30 mL of mitochondria obtained per heart were flash frozen in liquid nitrogen and stored at −80 °C. In a falcon tube, 0.5 mL of bovine heart mitochondria was added to 4 mL of the isotonic buffer [0.25 M sucrose, 10 mM MOPS, and 2 mM EDTA pH 8]. To form SMPs, the sample was sonicated while keeping the tube in a beaker of ice mixed with NaCl. The sonicator (Q Sonica Q500) was adjusted to provide ten 5 s pulses with a 30 s pause between pulses and an amplitude of 20%. SMPs were then centrifuged at 16,000g for 10 min at 4 °C to remove mitochondrial debris.45,46
Oxygen Consumption Assays (Binding Assay)
All oxygen consumption assays were performed with an Oxygraph Clark-type electrode (Hansatech). For assay of M. smegmatis-purified proteins with inhibitors, 500 nM superoxide dismutase (SOD, Sigma), 25 nM purified CIII2CIV2 (SC), 114 nM NADH-dehydrogenase type II (NDH-2, purification described above), 100 μM DMW (Sigma), and 10 μM of inhibitor in DMSO or DMSO alone were incubated in a 500 μL reaction [GTBS: 50 mM Tris–HCl pH 7.4, 100 mM NaCl, and 0.003% (w/v) GDN] at room temperature for 1 h. The reaction was initiated by injecting 1 mM NADH. To account for the effect of auto-oxidation of DMWH2, a baseline oxygen consumption was measured by monitoring oxygen consumption without the addition of the CIII2CIV2. The rate of oxygen consumption was calculated with a Python script where the baseline was subtracted.
To calculate the MICs (IC50) of Q203 and compound 27, different concentrations of the
two compounds (10, 1, 0 0.1, 0.01 μM, and blank) were tested
using the above protocol. The assays were repeated four times on different
days and using different batches of purified CIII2CIV2. The IC50s were calculated with a Python script,
and errors were estimated using Monte Carlo simulations.47 Briefly, the overall standard deviation σdata of the data was estimated by taking the average standard
deviation of all the inhibitor concentrations. The best fit curve
was calculated, and simulated data sets were created using the best
fit parameters. Random errors 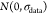 (normal
random numbers with standard deviation
= σdata) were added to each data point, and the simulated
data with errors were fit to extract an IC50. The addition
of random errors and fitting was repeated 10,000 times, and the standard
deviation of the 10,000 best fit parameters was taken to be the standard
deviation of the IC50. The Python matplotlib library was
used to generate plots for figures.
(normal
random numbers with standard deviation
= σdata) were added to each data point, and the simulated
data with errors were fit to extract an IC50. The addition
of random errors and fitting was repeated 10,000 times, and the standard
deviation of the 10,000 best fit parameters was taken to be the standard
deviation of the IC50. The Python matplotlib library was
used to generate plots for figures.
To establish the specificity of compound 27 and Q203, oxygen consumption was measured with bovine heart SMPs. In 500 μL of reaction buffer [0.25 M sucrose, 10 mM 3-(N-morpholino) propanesulfonic acid (MOPS) pH 8.0, and 2 mM EDTA], 200 μL of SMPs was incubated at room temperature with different concentrations (10 and 1 μM) for 1 h. The reaction was initiated by adding 10 mM NADH. Rotenone, KCN, and antimycin A were used as positive controls for inhibition.
NDH-2 Activity Assay
To confirm that Q203 and the analogues that were synthesized do not inhibit the NDH-2 used in oxygen consumption assays, the oxidation of NADH to NAD+ was monitored spectrophotometrically at λ = 340 nm for 1.5 h. In a 96-well plate, 500 nM SOD, 3.15 nM NDH-2, 100 μM DMW, and 10 μM of one of the putative inhibitors (28 in total) were added to each well. The plate was loaded into a Synergy neo2 multi-mode plate reader, and 100 μM NADH (100 μL) was injected to start the reaction (total volume = 200 μL).
Bos taurus Complex I Activity Assay
In a 96-well plate, SMPs (1/5 of the total volume of the reaction) and 10 μM of positive control (Rotenone, antimycin A, or KCN) or inhibitor (Q203 or 27) were added to each well. 200 μM NADH was injected to start the reaction (total volume = 200 μL). The oxidation of NADH was monitored spectrophotometrically for 8 min at λ = 340 nm.
C. albicans Complex III Activity Assay
The isolation and purification of CIII2 followed the protocol described previously.48 The reduction of cytochrome c was followed spectrophotometrically at λ = 550 nm to measure the activity of C. albicans complex III. In a 96-well plate, 50 μL of reaction mixture containing ∼25 nM purified cytochrome bc1 and 150 μM equine cytochrome c in reaction buffer (50 mM KPi pH 7.4, 100 mM KCl, 0.1 mM EDTA, 0.01% GDN, and 0.5 mM KCN) was added to each well. Q203, 24, and 27 were each dispensed from 376 μM DMSO stock to their final concentrations (10 and 1 μM). The final DMSO concentration was 2.66%. After 10 min of incubation at room temperature, 100 μL of 120 μM DBH2 (decylubiquinol) in reaction buffer was added column by column in the well plate to start the reaction. After shaking for 5 min, absorbance was recorded every 15 s for 15 min. Inz-5, a cytochrome bc1 inhibitor, was used as a positive control for complex III inhibition.48
In Vitro Mycobacterial Growth Assays
M. tuberculosis
M. tuberculosis strain H37Rv, harboring the red fluorescent protein (RFP)-expressing plasmid pTEC27, was used for measuring growth. The plasmid confers resistance to hygromycin. M. tuberculosis was grown in 7H9 broth (Difco Middlebrook) supplemented with 10% (v/v) OADC (5% bovine albumin fraction, 2% dextrose, 0.004% catalase, 0.05% oleic acid, and 0.8% NaCl) and 0.05% (v/v) Tween-80 at 37 °C in standing cultures. Hygromycin B was added to the medium at a final concentration of 50 μg/mL to suppress/inhibit the growth of non-transformed (non-plasmid containing) Mtb strains.
MIC90s were determined by the broth microdilution method using flat-bottom 96-well Corning Costar plates. In the first well in each row, two times the desired highest concentration (50 μM) of each compound was added in growth medium 7H9 supplemented with 10% OADS, 0.05% Tween 80, and hygromycin (50 μg/mL). Each well was then diluted two-fold in a 10-point serial dilution. Subsequently, 100 μL of the bacterial inoculum was added to each well to give a final volume of 200 μL. The concentration of the inoculum of 5 × 105 cells/mL (OD600, 0.1 = 0.33 × 108 cfu/mL) was prepared from a starting inoculum that was diluted from a preculture at the mid-log phase (OD600, 0.3 to 0.7). In each plate, a negative control (1% DMSO) and a positive control (4 μM bedaquiline) were included. The plates were sealed with parafilm, placed in a container with moist tissue, and incubated for 6 days at 37 °C. After incubation, the fluorescence intensity (signal) of each well was measured [Synergy H4 plate reader (BioTek), excitation at 530 nm, and emission at 590 nm], and the growth inhibition was calculated with the following equation
MIC90 was calculated as the concentration of the compound that caused more than 90% growth reduction. Each test was performed in duplicate.
M. smegmatis and M. abscessus
MIC90s were determined against M. smegmatis mc2 155 pTEC27 and M. abscessus ATCC 19977(pTEC27) by the broth microdilution method. 96-well flat-bottom tissue culture plates (Sarstedt, 83.3924.500) were used.49 In the third well of each row, two times the desired highest concentration of the tested compound was added in 7H9 medium supplemented with 10% ADS and 0.05% Tween 80. Each compound was diluted two-fold in a nine-point serial dilution. The concentration of the starting inoculum was 5 × 105 cells mL–1. The starting inoculum was diluted from a preculture at the mid-log phase (OD600 0.3 to 0.7), and an OD600 of 0.1 was correlated to 1 × 108 cfu mL–1. The plates were sealed with parafilm, placed in a container with moist tissue, and incubated for 3 days at 37 °C. Each plate had eight negative controls (1% DMSO) and eight positive controls (100 μM amikacin). After incubation, the plates were monitored by the RFP measurement (λex = 544 nm and λem = 590 nm, BMG labtech Fluostar Optima). The assay was performed in duplicate, and the results were validated by the OD measurement at 550 nm.
Every assay plate contained eight wells with 1% DMSO as the negative control, which corresponds to 100% bacterial growth and eight wells with amikacin (100 μM) as a positive control in which 100% inhibition of bacterial growth was reached. Controls were used to monitor the assay quality through the determination of the Z′ score. The Z′ factor was calculated as follows
(SD = standard deviation, M = mean).
The percentage of growth inhibition was calculated by the equation:
Acknowledgments
R.A. research stay in The Hospital for Sick Children’s Research Institute was partially funded by the German Academic Exchange Service (DAAD, Project ID: 57556281) and the Stiftung für Kanada-Studien (Germany, Project no.: T0191/38098/2021). J.D.T. was supported by a postdoctoral fellowship from the Canadian Institutes of Health Research. J.L.R. and L.E.C. were supported by the Canada Research Chairs program. Z.L. was supported by a Precision Medicine Initiative (PRiME) internal fellowship (PRMF2020-005). Research was supported by the Canadian Institutes of Health Research grant PJT162186 (J.L.R.), Foundation grant FDN-154288 (L.E.C.), and the Canadian Institute for Advanced Research Fungal Kingdom research program (L.E.C.). We acknowledge the support of the CL-3 facility, Facility for Infectious Disease and Epidemic Research (FINDER), within the Life Sciences Institute at the University of British Columbia.
Glossary
Abbreviations
- ACN
acetonitrile
- BSL-2
biosafety level 2
- CIII
complex III
- CIV
complex IV
- cyt.
cytochrome
- CIII2CIV2
supercomplex
- DIAD
diisopropyl azodicarboxylate
- DIPEA
N,N-diisopropylethylamine
- DMW
2,3-dimethyl[1,4]naphthoquinone
- DMWH2
2,3-dimethyl[1,4]naphthoquinol
- ETC
electron transport chain
- EMB
ethambutol
- EtOH
ethanol
- HATU
hexafluorophosphate azabenzotriazole tetramethyl uronium
- IC50
half-maximal inhibitory concentration
- INH
isoniazid
- IPA
imidazopyridine amide
- IP
imidazopyridine
- MeOH
methanol
- MDR
multidrug-resistant
- MIC
minimum inhibitory concentration
- MK
menaquinone
- MKH2
menaquinol
- Msmeg
M. smegmatis
- Mtb
Mycobacterium tuberculosis
- NDH
NADH dehydrogenases
- Ph3P
triphenylphosphine
- PK
pharmacokinetic
- PyBOP
benzotriazole-1-yloxytripyrrolidinophosphonium hexafluorophosphate
- PZA
pyrazinamide
- Q
ubiquinone
- RIF
rifampicin
- SAR
structure–activity relationship
- SDH
succinate dehydrogenase
- s.e.
standard error
- s.d.
standard deviation
- SC
supercomplex
- TB
tuberculosis
- XDR
extensively drug-resistant
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c02259.
Author Contributions
R.A.: synthesis of the IPAs, biochemical assays, and manuscript writing, J.D.T.: biochemical assays and writing editing, H.A.S.: in vitro Mtb assay, L.M. and A.R.: in vitro M. smegmatis and M. abscessus assays, Z.L. and L.E.C.: Candida albicans complex III activity assay, S.A.B.: initial development of the coupled assay, and J.L.R. and P.I.: supervision, writing, and editing.
The authors declare the following competing financial interest(s): LEC is a co-founder and shareholder in Bright Angel Therapeutics, a platform company for development of antifungal therapeutics and is Science Advisor for Kapoose Creek, a company that harnesses the therapeutic potential of fungi. The other authors declare no competing interests.
Supplementary Material
References
- Global Tuberculosis Report 2021. Geneva: World Health Organization; 2021. Licence: CC BY-NC-SA 3.0 IGO.: Geneva, Switzerland.
- United States; Department of Health and Human Services , C for D. C. and P. Tuberculosis (TB): Data and Statistics. https://www.cdc.gov/tb/statistics/tbcases.htm.
- RKI. Report on the Epidemiology of Tuberculosis in Germany 2020. .
- World Bank . Incidence of tuberculosis. https://data.worldbank.org/indicator/SH.TBS.INCD.
- Vergne I.; Chua J. Cell biology of Mycobacterium tuberculosis phagosome. Annu. Rev. Cell Dev. Biol. 2004, 20, 367. [DOI] [PubMed] [Google Scholar]
- Shetye G. S.; Franzblau S. G.; Cho S. New Tuberculosis Drug Targets, Their Inhibitors, and Potential Therapeutic Impact. Transl. Res. 2020, 220, 68–97. 10.1016/j.trsl.2020.03.007. [DOI] [PubMed] [Google Scholar]
- Rajan A.; Shankar V. Features of the Biochemistry of Mycobacterium Smegmatis, as a Possible Model for Mycobacterium Tuberculosis. J. Infect. Public Health 2020, 13, 1255–1264. 10.1016/j.jiph.2020.06.023. [DOI] [PubMed] [Google Scholar]
- Namouchi A.; Cimino M.; Favre-Rochex S.; Charles P.; Gicquel B. Phenotypic and Genomic Comparison of Mycobacterium Aurum and Surrogate Model Species to Mycobacterium Tuberculosis: Implications for Drug Discovery. BMC Genomics 2017, 18, 530–628. 10.1186/s12864-017-3924-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelovic N.; Mitachi K.; Yang J.; Lemieux M. R.; Ji Y.; Kurosu M.; Sciences B.; Avenue C.; States U. Application of Mycobacterium smegmatis as a surrogate to evaluate drug leads against Mycobacterium tuberculosis. HHS Public Access 2020, 73, 780–789. 10.1038/s41429-020-0320-7.Application. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo C. S. Y.; Pethe K.; Lupien A. Oxidative Phosphorylation-an Update on a New, Essential Target Space for Drug Discovery in Mycobacterium Tuberculosis. Appl. Sci. 2020, 10, 2339. 10.3390/app10072339. [DOI] [Google Scholar]
- Wiseman B.; Nitharwal R. G.; Fedotovskaya O.; Schäfer J.; Guo H.; Kuang Q.; Benlekbir S.; Sjöstrand D.; Ädelroth P.; Rubinstein J. L.; et al. Structure of a Functional Obligate Complex III2IV2 Respiratory Supercomplex from Mycobacterium Smegmatis. Nat. Struct. Mol. Biol. 2018, 25, 1128–1136. 10.1038/s41594-018-0160-3. [DOI] [PubMed] [Google Scholar]
- Datta S.; Krishna R.; Ganesh N.; Chandra N. R.; Muniyappa K.; Vijayan M. Crystal Structures of Mycobacterium Smegmatis RecA and Its Nucleotide Complexes. J. Bacteriol. 2003, 185, 4280–4284. 10.1128/JB.185.14.4280-4284.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B. S.; Pethe K. Telacebec: An Investigational Antibacterial for the Treatment of Tuberculosis (TB). Expert Opin. Investig. Drugs 2022, 31, 139–144. 10.1080/13543784.2022.2030309. [DOI] [PubMed] [Google Scholar]
- Malík I.; Čižmárik J.; Kováč G.; Pecháčová M.; Hudecova L. Telacebec (Q203): Is There a Novel Effective and Safe Anti-Tuberculosis Drug on the Horizon?. Ces. a Slov. Farm. Cas. Ces. Farm. Spol. a Slov. Farm. Spol. 2021, 70, 164–171. 10.5817/CSF2021-5-164. [DOI] [PubMed] [Google Scholar]
- Yanofsky D. J.; Di Trani J. M.; Król S.; Abdelaziz R.; Bueler S. A.; Imming P.; Brzezinski P.; Rubinstein J. L.. Structure of Mycobacterial CIII2CIV2 Respiratory Supercomplex Bound to the Tuberculosis Drug Candidate Telacebec (Q203). Elife 2021, 10. DOI: 10.7554/eLife.71959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascolo L.; Bald D. Cytochrome Bd in Mycobacterium Tuberculosis: A Respiratory Chain Protein Involved in the Defense against Antibacterials. Prog. Biophys. Mol. Biol. 2020, 152, 55–63. 10.1016/j.pbiomolbio.2019.11.002. [DOI] [PubMed] [Google Scholar]
- Moraski G. C.; Markley L. D.; Hipskind P. A.; Boshoff H.; Cho S.; Franzblau S. G.; Miller M. J. Advent of Imidazo[1,2-a]Pyridine-3-Carboxamides with Potent Multi- and Extended Drug Resistant Antituberculosis Activity. ACS Med. Chem. Lett. 2011, 2, 466–470. 10.1021/ml200036r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Malley T.; Alling T.; Early J. V.; Wescott H. A.; Kumar A.; Moraski G. C.; Miller M. J.; Masquelin T.; Hipskind P. A.; Parish T. Imidazopyridine Compounds Inhibit Mycobacterial Growth by Depleting ATP Levels. Antimicrob. Agents Chemother. 2018, 62, 1–8. 10.1128/AAC.02439-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glutaminsynthetase H.. Antimykobakterielle Imidazopyridine 2011, 1–2. [Google Scholar]
- Pethe K.; Bifani P.; Jang J.; Kang S.; Park S.; Ahn S.; Jiricek J.; Jung J.; Jeon H. K.; Cechetto J.; et al. Discovery of Q203, a Potent Clinical Candidate for the Treatment of Tuberculosis. Nat. Med. 2013, 19, 1157–1160. 10.1038/nm.3262. [DOI] [PubMed] [Google Scholar]
- Kang S.; Kim Y. M.; Kim R. Y.; Seo M. J.; No Z.; Nam K.; Kim S.; Kim J. Synthesis and Structure-Activity Studies of Side Chain Analogues of the Anti-Tubercular Agent, Q203. Eur. J. Med. Chem. 2017, 125, 807–815. 10.1016/j.ejmech.2016.09.082. [DOI] [PubMed] [Google Scholar]
- Kang S.; Kim Y. M.; Jeon H.; Park S.; Seo M. J.; Lee S.; Park D.; Nam J.; Lee S.; Nam K.; et al. Synthesis and Structure-Activity Relationships of Novel Fused Ring Analogues of Q203 as Antitubercular Agents. Eur. J. Med. Chem. 2017, 136, 420–427. 10.1016/j.ejmech.2017.05.021. [DOI] [PubMed] [Google Scholar]
- Kim J.; Choi J.; Kang H.; Ahn J.; Hutchings J.; van Niekerk C.; Park D.; Kim J.; Jeon Y.; Nam K.; et al. Safety, Tolerability, and Pharmacokinetics of Telacebec (Q203), a New Antituberculosis Agent, in Healthy Subjects. Antimicrob. Agents Chemother. 2022, 66 (). DOI: 10.1128/AAC.01436-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A.; Wang H.; Geng Y.; Fu L.; Gu J.; Wang B.; Lv K.; Liu M.; Tao Z.; Ma C.; Lu Y. Design, Synthesis and Antimycobacterial Activity of Less Lipophilic Q203 Derivatives Containing Alkaline Fused Ring Moieties. Bioorganic Med. Chem. 2019, 27, 813–821. 10.1016/j.bmc.2019.01.022. [DOI] [PubMed] [Google Scholar]
- Moraski G. C.; Markley L. D.; Cramer J.; Hipskind P. A.; Boshoff H.; Bailey M. A.; Alling T.; Ollinger J.; Parish T.; Miller M. J. Advancement of Imidazo[1,2-a]Pyridines with Improved Pharmacokinetics and NM Activity vs. Mycobacterium Tuberculosis. ACS Med. Chem. Lett. 2013, 4, 675–679. 10.1021/ml400088y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- No Z.; Kim J.; Brodin P. B.; Seo M. J.; Kim Y. M.; Cechetto J.; Jeon H.; Genovesio A.; Lee S.; Kang S.; et al. Preparation of Imidazopyridine Derivatives as Anti-Infective Compounds. WO 2011113606 A1, 2011.
- Kumar R. V. Synthetic Strategies towards Benzoxazole Ring Systems: A Review. Asian J. Chem. 2005, 36, 1241–1260. 10.1002/chin.200528243. [DOI] [Google Scholar]
- Caddick S.; Judd D. B.; Lewis A. K. K.; Reich M. T.; Williams M. R. V. A Generic Approach for the Catalytic Reduction of Nitriles. Tetrahedron 2003, 59, 5417–5423. 10.1016/S0040-4020(03)00858-5. [DOI] [Google Scholar]
- Paquette L. A.; Sin N.. Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons, 1995; Vol. 6. [Google Scholar]
- Sen S. E.; Roach S. L. A Convenient Two-Step Procedure for the Synthesis of Substituted Allylic Amines from Allylic Alcohols. Thieme E-Journals 1995, 1995, 756–758. 10.1055/s-1995-4012. [DOI] [Google Scholar]
- Heikal A.; Nakatani Y.; Dunn E.; Weimar M. R.; Day C. L.; Baker E. N.; Lott J. S.; Sazanov L. A.; Cook G. M. Structure of the Bacterial Type II NADH Dehydrogenase: A Monotopic Membrane Protein with an Essential Role in Energy Generation. Mol. Microbiol. 2014, 91, 950–964. 10.1111/mmi.12507. [DOI] [PubMed] [Google Scholar]
- Jang J.; Kim R.; Woo M.; Jeong J.; Park D. E.; Kim G.; Delorme V. Efflux Attenuates the Antibacterial Activity of Q203 in Mycobacterium Tuberculosis. Antimicrob. Agents Chemother. 2017, 61, 1–8. 10.1128/aac.02637-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorayah R.; Manimekalai M. S. S.; Shin S. J.; Koh W. J.; Grüber G.; Pethe K. Naturally-Occurring Polymorphisms in QcrB Are Responsible for Resistance to Telacebec in Mycobacterium Abscessus. ACS Infect. Dis. 2019, 5, 2055–2060. 10.1021/acsinfecdis.9b00322. [DOI] [PubMed] [Google Scholar]
- Zhou S.; Wang W.; Zhou X.; Zhang Y.; Lai Y.; Tang Y.; Xu J.; Yang X.; Guddat L. W.; Wang Q.; et al. Structure of Mycobacterial Cytochrome Bcc in Complex with Q203 and TB47, Two Anti-TB. Drug Candidates 2 3. bioriv 2021, 6, 448498. 10.1101/2021.06.15.448498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J.; Evans R.; Pritzel A.; Green T.; Figurnov M.; Ronneberger O.; Tunyasuvunakool K.; Bates R.; Žídek A.; Potapenko A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadi M.; Anyango S.; Deshpande M.; Nair S.; Natassia C.; Yordanova G.; Yuan D.; Stroe O.; Wood G.; Laydon A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50, D439–D444. 10.1093/nar/gkab1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera--a Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Bevan C. D.; Lloyd R. S. A High-Throughput Screening Method for the Determination of Aqueous Drug Solubility Using Laser Nephelometry in Microtiter Plates. Anal. Chem. 2000, 72, 1781–1787. 10.1021/ac9912247. [DOI] [PubMed] [Google Scholar]
- Dammann M.; Stahlecker J.; Zimmermann M. O.; Klett T.; Rotzinger K.; Kramer M.; Coles M.; Stehle T.; Boeckler F. M. Screening of a Halogen-Enriched Fragment Library Leads to Unconventional Binding Modes. J. Med. Chem. 2022, 65, 14539–14552. 10.1021/acs.jmedchem.2c00951. [DOI] [PubMed] [Google Scholar]
- Lu P.; Asseri A. H.; Kremer M.; Maaskant J.; Ummels R.; Lill H.; Bald D. The Anti-Mycobacterial Activity of the Cytochrome Bcc Inhibitor Q203 Can Be Enhanced by Small-Molecule Inhibition of Cytochrome Bd. Sci. Rep. 2018, 8, 2625–2627. 10.1038/s41598-018-20989-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhagat S. B.; Telvekar V. N. NBS Mediated Protocol for the Synthesis of N-Bridged Fused Heterocycles in Water. Tetrahedron Lett. 2017, 58, 3662–3666. 10.1016/j.tetlet.2017.08.017. [DOI] [Google Scholar]
- Kang S.; Kim R. Y.; Seo M. J.; Lee S.; Kim Y. M.; Seo M.; Seo J. J.; Ko Y.; Choi I.; Jang J.; et al. Lead Optimization of a Novel Series of Imidazo[1,2-a]Pyridine Amides Leading to a Clinical Candidate (Q203) as a Multi- and Extensively-Drug- Resistant Anti-Tuberculosis Agent. J. Med. Chem. 2014, 57, 5293–5305. 10.1021/jm5003606. [DOI] [PubMed] [Google Scholar]
- Kumar D.; Rudrawar S.; Chakraborti A. K. One-Pot Synthesis of 2-Substituted Benzoxazoles Directly from Carboxylic Acids. Aust. J. Chem. 2008, 61, 881–887. 10.1071/CH08193. [DOI] [Google Scholar]
- Gao X.; Liu L.; Liu H.; Tang J.; Kang L.; Wu H.; Cui P.; Yan J. Structure–Activity Relationship Investigation of Benzamide and Picolinamide Derivatives Containing Dimethylamine Side Chain as Acetylcholinesterase Inhibitors. J. Enzyme Inhib. Med. Chem. 2018, 33, 110–114. 10.1080/14756366.2017.1399885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orynbayeva, Zulfiya; Drexel University College of Medicine; PhD Thesis, 2018. [Google Scholar]
- Panov A.Practical Mitochondriology. Pitfalls and Problems in Studies of Mitochondria, 2013. Vol 10, pp 1483963853. [Google Scholar]
- Koehler E.; Brown E.; Haneuse S. J. P. A. On the Assessment of Monte Carlo Error in Simulation-Based Statistical Analyses. Am. Stat. 2009, 63, 155–162. 10.1198/tast.2009.0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Trani J. M.; Liu Z.; Whitesell L.; Brzezinski P.; Cowen L. E.; Rubinstein J. L. Rieske Head Domain Dynamics and Indazole-Derivative Inhibition of Candida Albicans Complex III. Structure 2022, 30, 129–138.e4. 10.1016/j.str.2021.08.006. [DOI] [PubMed] [Google Scholar]
- Richter A.; Strauch A.; Chao J.; Ko M.; Av-Gay Y. Screening of Preselected Libraries Targeting Mycobacterium Abscessus for Drug Discovery. Antimicrob. Agents Chemother. 2018, 62, 1–11. 10.1128/AAC.00828-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.