

Abstract
The catalytic function of lysyl hydroxylase-2 (LH2), a member of the Fe(II)/αKG-dependent oxygenase superfamily, is to catalyze the hydroxylation of lysine to hydroxylysine in collagen, resulting in stable hydroxylysine aldehyde-derived collagen cross-links (HLCCs). Reports show that high amounts of LH2 lead to the accumulation of HLCCs, causing fibrosis and specific types of cancer metastasis. Some members of the Fe(II)/αKG-dependent family have also been reported to have intramolecular O2 tunnels, which aid in transporting one of the required cosubstrates into the active site. While LH2 can be a promising target to combat these diseases, efficacious inhibitors are still lacking. We have used computational simulations to investigate a series of 44 small molecules as lead compounds for LH2 inhibition. Tunneling analyses indicate the existence of several intramolecular tunnels. The lengths of the calculated O2-transporting tunnels in holoenzymes are relatively longer than those in the apoenzyme, suggesting that the ligands may affect the enzyme’s structure and possibly block (at least partially) the tunnels. The sequence alignment analysis between LH enzymes from different organisms shows that all of the amino acid residues with the highest occurrence rate in the oxygen tunnels are conserved. Our results suggest that the enolate form of diketone compounds establishes stronger interactions with the Fe(II) in the active site. Branching the enolate compounds with functional groups such as phenyl and pyridinyl enhances the interaction with various residues around the active site. Our results provide information about possible leads for further LH2 inhibition design and development.
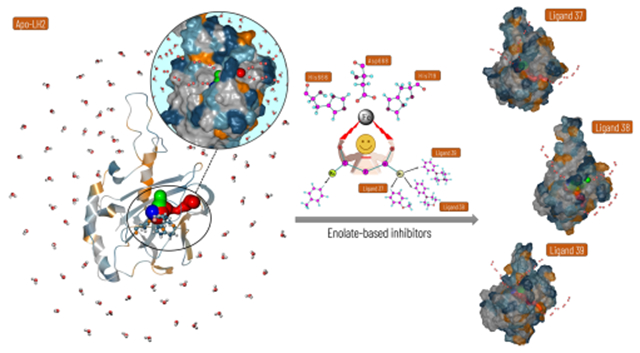
Graphical Abstract
INTRODUCTION
Lysyl hydroxylases (LHs) belong to the iron(II)/α-ketoglutarate-dependent (Fe(II)/αKG-dependent) enzyme superfamily, with more than 60 oxygenases, using Fe(II) and 2-oxoglutarate (αKG) as cofactor and cosubstrate, respectively (Figure 1A).1 The enzymatic oxidation reaction catalyzed in the active site of these enzymes occurs by activating a nonheme Fe with the concomitant oxidation of αKG into succinate and carbon dioxide, followed by a H abstraction and hydroxyl rebound to hydroxylate the substrate (Figure 1B).1–3 The LH subfamily consists of LH1, LH2, and LH3, encoded by the procollagenlysine, 2-oxoglutarate 5-dioxygenase genes PLOD1, PLOD2, and PLOD3, respectively.4–6 All three enzymes catalyze the hydroxylation of lysine to hydroxylysine on collagen (Figure 1C), with LH2 being the only modifier for telopeptidyl lysine residues.7
Figure 1.
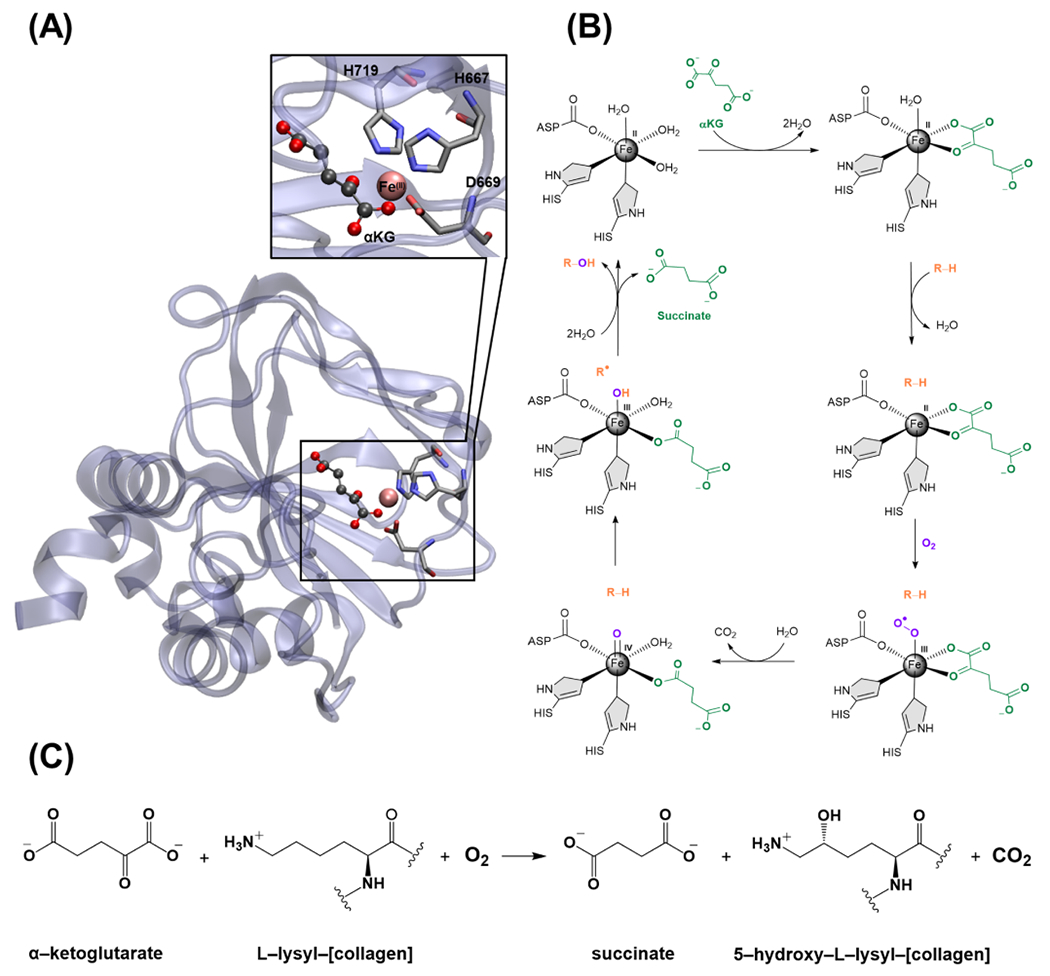
(A) Crystal structure of full-length human lysyl hydroxylase LH3 and its active site complexed with αKG (PDB ID: 6FXK). 24 (B) The general catalytic mechanism of the αKG-dependent hydroxylase superfamily. (C) Hydroxylation of lysine to hydroxylysine by the LH2 in the presence of molecular oxygen.
The role of LH2 in the human body is the formation of highly stable hydroxylysine aldehyde-derived collagen cross-links (HLCCs) mediated by the telopeptidyl lysine residues.8,9 These cross-links are more durable than lysine-derived collagen cross-links (LCCs) that form in the absence of LH2.10 The large stabilization and resistance of HLCCs to collagenase cleavage are necessary to stabilize the extracellular matrix,8 as the deficiency of HLCCs is seen in patients with Bruck syndrome suffering from deformed and fragile bones.11 On the other hand, the excessive accumulation of HLCCs by high levels of LH2 expression can cause fibrosis.12,13 Moreover, pathological studies on sarcoma14 and metastasis in lung and breast cancers15–17 show that high levels of LH2 directly contribute to these conditions. Thus, LH2 is a promising target for potential inhibitors to cope with these diseases. Various assays have been developed to determine LH2 activity, some of which are currently employed to develop possible LH2 inhibitors.18–22 However, as of yet, no LH2 inhibitors are available. A recent study by Scietti et al. on self-inhibition of the lysyl hydroxylase catalytic site induced by the binding of in a noncatalytic site showed a dual role of iron as a simultaneous cofactor and inhibitor of the lysyl hydroxylase activity.23 They realized that the LH/PLOD enzyme activity is extremely sensitive to the balance of concentration. The authors believe that developing specific inhibitors of LH/PLOD is a challenging effort with probable additional obstacles for which extra care is needed.
The hydroxylation reaction performed by LH2 is carried out via a reaction mechanism consistent with other enzymes in the Fe/αKG-dependent superfamily. This mechanism involves molecular oxygen to activate the nonheme Fe to carry out the required oxidations. Experimental and computational studies have shown that various enzymes have intramolecular tunnels formed by flexible hydrophobic residues to transport molecular oxygen into the active site.25–31 This feature has also been observed in some Fe/αKG-dependent superfamily enzymes such as AlkB.32–36 Yu et al. proposed the possibility of oxygen diffusion by intramolecular tunnels based on the original AlkB crystal structure.35 Subsequently, a computational investigation showed the likelihood of two O2-transporting tunnels in this enzyme by various computational approaches.36
Classical atomistic molecular dynamics (MD) simulations have become a useful tool to aid in lead development and inhibitor design.37–40 Many applications use classical nonpolarizable force fields implemented in various software packages such as OPLS,41 CHARMM,42,43 AMBER,44,45 and GROMOS.46 Polarizable force fields such as the atomic multipole optimized energetics for biomolecular application (AMOEBA) can also be employed.47–49 In cases where transition metals are involved, an accurate description of the electronic structure around the metal may be required. One approach that can be used in these cases is quantum mechanics/molecular mechanics (QM/MM). This method combines two levels of theory and can be used to study the interaction energies, reaction energies, and reaction mechanisms in chemical and biochemical systems.50–56
A wide variety of combinations can be used to treat the QM subsystem and the MM region, such as empirical valence bond, semiempirical or ab initio Hamiltonians for the QM, and nonpolarizable or polarizable force fields for the MM region. It has been shown that, similar to MD, considering polarization in QM/MM simulations is important and can improve the description of the inter- and intramolecular interactions.57–63 Thus, combining high-level QM and polarizable/anisotropic force fields can help achieve more reliable results.64–67
Classical molecular dynamics simulations have been employed to study the structural features of other members of the αKG-dependent superfamily, such as AlkB and TET,68–73 where the effects of mutagenesis and different types of DNA/RNA are investigated. However, the presence of Fe(II) in the active site requires a highly accurate description of intermolecular interactions due to the electronic state of the cation.74 Therefore, high-level QM combined with polarizable/anisotropic MM force fields has been employed in tandem with other tools and techniques, such as noncovalent interaction (NCI) analysis to investigate the interaction of all ligands in the active site in detail.
This contribution presents a combined polarizable MD and QM/MM investigation of a series of 44 diketone-, enol-, and enolate-based ligands in three sets as potential inhibitors of LH2. Tunnel analyses are also presented for LH2 to investigate the possibility of the existence of molecular oxygen transport tunnels and whether the various ligands may affect them. Several candidates of this study, in addition to other designed compounds based on the findings of this investigation, are used as lead compounds in another hybrid computational/experimental contribution, in which we developed a series of antagonists to find competitive inhibitors of αKG.75 The remainder of the manuscript is organized as follows: the Methodology section describes the various computational approaches used. Subsequently, results for MD, QM/MM, and tunneling simulations are presented and discussed, followed by concluding remarks.
METHODOLOGY
Molecular Docking and MD Simulations.
The homology model for LH2 was constructed by using the SWISS-MODEL server.76 The PLOD2 sequence (residues 598–737) was submitted. The LH3 PDB structure 6fxm.1.A with a 64.29% sequence identity was used as the template. Next, the protonation states of the output structure at pH 7 were predicted by using propka77 via the PDB2PQR server78 with H atoms added/removed on the ionizable residues accordingly. The initial structures of LH2–ligand complexes were prepared using GOLD molecular docking software (version 5.8.1)79. Initially, 16 ligands were considered in this work, followed by 28 modified compounds based on the initial results. A visual check of each docked structure was performed to confirm that the ligands were in the proper position of the LH2 binding pocket. All systems were solvated within an AMOEBA water box and neutralized.80 NaCl ions were added to reach a physiological concentration of 0.15 M. The resulting simulation boxes are about 70 × 90 × 70 Å3 with total atoms of ~44 000. After 1 ns equilibration, the production MD simulations were carried out under an NPT ensemble (298 K and 1 bar) for 5 ns for each protein–ligand complex. The RESPA integrator,81,82 Monte Carlo barostat,83 and BUSSI thermostat84 were employed in the simulations. To accelerate the simulation, the integration time step was chosen to be 2 fs, and the induced dipole moment was converged to 0.0001 D. Following the conventional settings used by AMOEBA simulations, the cutoff distances for van der Waals (vdW) interactions and real-space electrostatics are 12 and 7 Å, respectively. The long-range interactions were treated with the Ewald summation method, as implemented in the Tinker software package. MD trajectories were saved every 50 ps for a total simulation time of 5 ns for each system. The generated trajectories were subjected to a clustering analysis and further investigation using QM/MM calculations described below. All MD simulations were performed using the Tinker software implemented via the OpenMM plugin 85 on NVIDIA GPU cards (GTX 1070 or RTX 2070).
Clustering and QM/MM Calculations.
The k-means clustering analysis86 was performed on MD trajectories of the apoenzyme and selected lead compounds based on the distances between the and the donor atoms of the coordinated residues (H666, D668, and H718), ligand (N/A for apoenzyme), and water molecules (whenever applicable). The Layered Interacting CHEmical Model (LICHEM)87,88 program was combined with Gaussian1689 and TINKER790,91 for all energy and optimization calculations. The ωB97X-D/6-31G-(d,p)92,93 level of theory and AMOEBAbio1894 force field were employed for the QM region and the MM environment, respectively. The QM/MM long-range electrostatic correction (QM/MM-LREC) method95 was applied with a 25 Å cutoff for the QM subsystem coupled with the particle mesh Ewald (PME) method96 for the MM calculations. The QM subsystem includes the , ligand molecule, H666, D668, H718, and one or two water molecules depending on the ligand to complete the octahedral coordination sphere around the divalent cation. In contrast, the remaining residues and solvent molecules were assigned to the MM subsystem. The MM environment consists of two subregions: the active region, in which all of the protein atoms and the solvent are flexible inside a sphere of a 30 Å radius, and the frozen subregion, where the remaining MM atoms are kept fixed. Boundary conditions across the QM and MM subsystems, where covalent bonds were cut, were described using the pseudobond approach.97 Interaction energies () were calculated using the following approach
| (1) |
| (2) |
| (3) |
Equations 1 and 2 give the interaction energy between the ligand, the solvated protein, and water. Equation 3 reports the difference between the two calculated interaction energies. The terms and correspond to the gas-phase energies of the respective systems including the basis set superposition error (BSSE) correction using the counterpoise approach.98,99
Noncovalent Interaction and Thermal Fluctuation Index (TFI) Analyses.
Noncovalent interaction (NCI), average noncovalent interaction (aNCI), and thermal fluctuation index (TFI) analyses were used as implemented in the Multiwfn3.7100 software, employing promolecular densities.101 This analysis has a default RGB color code for plotting isosurfaces. Blue surfaces represent strong-attractive interactions; red surfaces refer to strong repulsive interactions, and green ones show weak attractive/repulsive interactions. The thermal fluctuation index (TFI) visually indicates the variations between NCI surfaces and is color-coded like the NCI. Blue (red) surfaces in this analysis mean that the TFI is smaller (larger), and the noncovalent interactions do not fluctuate that much (fluctuate a lot). NCI analysis was studied on QM/MM-optimized structures, while aNCI and TFI analyses were used for the last 5 ns of MD trajectories. A cubic grid of 200 au and an isovalue of 0.4 au with a color scale of were used for all surfaces. The VMD102 software package was used to render images and visualize MD trajectories and surfaces.
Sequence Alignment.
Forty-one lysyl hydroxylase (LH) enzymes coded by PLOD1, PLOD2, and PLOD3 genes from 14 different organisms were aligned with the Human LH2 (UniProtKB: O00469) via the Clustal Omega program accessed via UniProtKB.103 A specific portion of the studied enzymes (V538 to P737 in human LH2), formed by a sequence of 199 residues corresponding to the catalytically active region for the hydroxylase reaction,24 was selected for further multiple alignments by the Expresso mode of the T-Coffee server.104–107 Lastly, conserved residues on the target systems from these multiple alignments were assessed by visual inspection to determine their proximity and positions toward the active site.
O2-Transporting Tunnels.
Intramolecular tunnel analysis was done with the CAVER 3.01 algorithm108 implemented in Caver Analyst 2.0.109 The k-means clustering analysis was used on the MD trajectories of selected ligand systems and apo-LH2 to identify representative structures to perform the tunneling analysis. The was considered the starting point of the tunnels in all cases. The minimal radius of the computed tunnels (min. probe radius) was set to 0.9 Å, the maximum depth of the surface region (shell depth) was set to 4.0 Å, and the radius of the shell probe (shell radius), defining which parts of the Voronoi diagram represent the bulk solvent, was set to 3.0 Å.110
RESULTS AND DISCUSSION
The natural cosubstrate of the LH2 enzyme, αKG, which is the deprotonated form of 2-oxoglutaric acid, coordinates with the iron cation of the active site via its carbonyl oxygens in a bidentate form (see Figure 1A). Thus, we started with a series of structures having diketone, enol, or enolate ligand skeletons branched with aromatic rings and various functional groups (see Scheme 1A). We also considered aromatic compounds with two N: donor atoms (cpd3 and cpd4 in Scheme 1A) and picolinamide-based compounds with C═O: and N: donors (cpd12 and cpd13 in Scheme 1A) to study the interaction tendencies of different combinations of donor atoms and negative charges. We analyzed the QM/MM interaction energies between each lead compound (ligand) and its environment () in three configurations: first, the docking configuration with the best score; second, the last frame of a 5 ns of MD simulation (MD1) with AMOEBAbio18 using NPT ensemble at 298 K and 1 bar, with 1 kcal mol−1 Å−2 of positional restraints on all protein atoms (excluding solvent, ions, and ligand); and third, the last frame of a 5 ns MD simulation without restraints (MD2) with the same conditions as the second configuration.
Scheme 1. (A) First Set of Test Compoundsa and (B) Possible Coordination Geometries of the Ligands to Fe(II) in the Active Site.
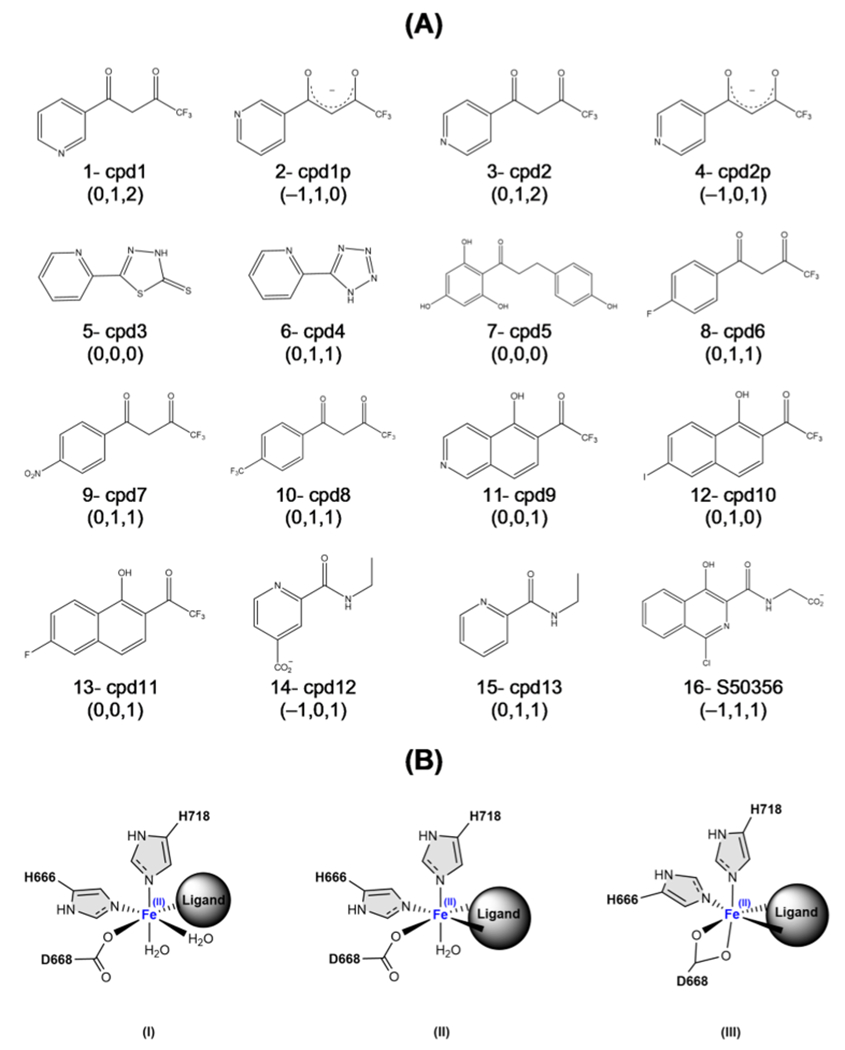
aThe numbers in parentheses show the ligand’s charge and number of coordinated waters to the Fe cation in the active site in MD1 and MD2, respectively.
Figure 2A compares the calculated interaction energies for the first set of ligands. The interaction energy differences () indicate that the neutral compounds have the weakest interaction energies (detailed results in Table S1). Overall, represents ~20–50% of . The negatively charged ligands, cpd1p, cpd2p, cpd12, and S50356 (IDs: 2, 4, 14, and 16), have the most negative interaction energies, and their corresponding () is 0.42, 0.50, 0.48, and 0.44, respectively. Two compounds, cpd3 and cpd13, show positive , as shown in Figure 2A. This is because these neutral compounds are the only two that coordinate the metal cation in a monodentate fashion, and the interaction with the rest of the protein environment is insufficient to stabilize the repulsive energy between the ligand and the active site, resulting in an overall unfavorable interaction.
Figure 2.
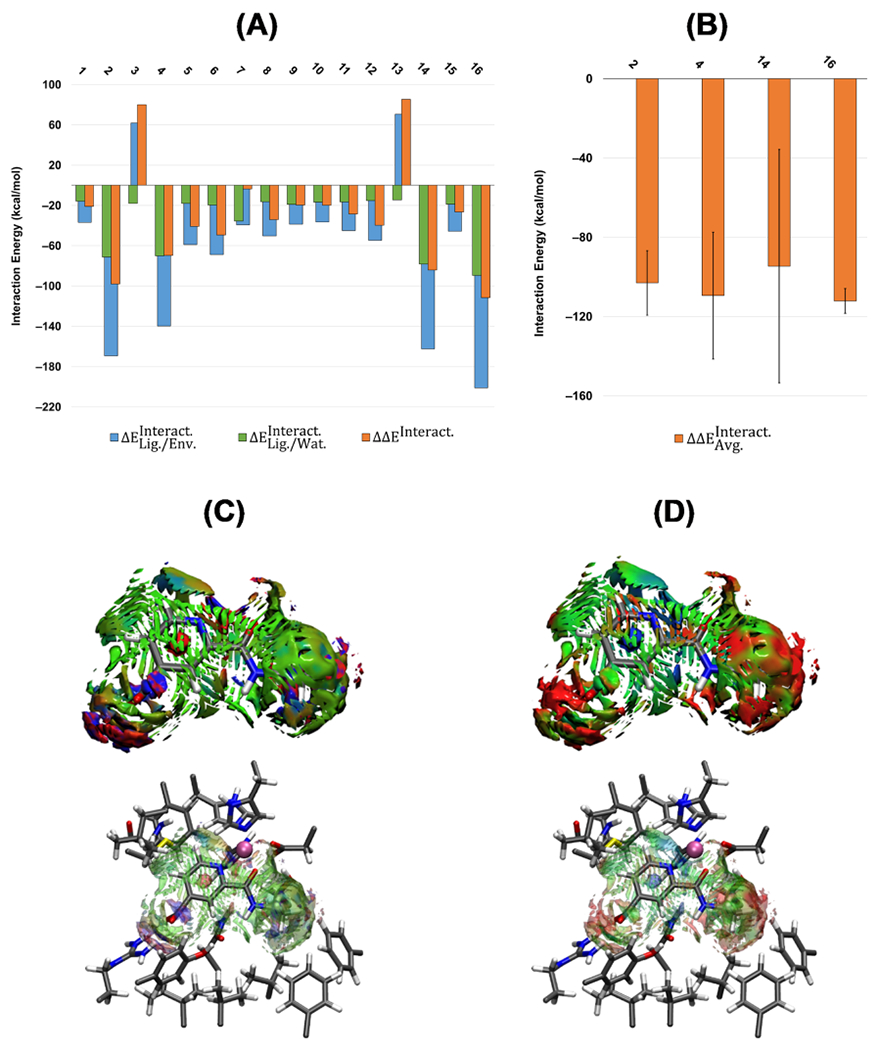
(A) Calculated QM/MM interaction energies (kcal mol−1) for the first set of test compounds (ID: 1–16). (B) Average QM/MM interaction energies () and the standard deviation for the compounds with the most negative interaction energies. All of the calculations are at the ωB97X-D/6-31G(d,p) level of theory and AMOEBAbio18 force field. (C) Averaged noncovalent index (aNCI) and (D) thermal fluctuation index (TFI) analyses for cpd12 (ID: 14). The isosurface cutoff for NCI and TFI is 0.4 au, and the data is plotted in the color range of .
Clustering analysis was performed on the ensemble generated from the MD trajectory (without restraints) for the ligands with the largest interaction energy differences, compounds cpd1p, cpd2p, cpd12, and S50356. The calculated averaged QM/MM interaction energies () on the selected representatives of the clusters for ligands cpd1p, cpd2p, and cpd12 shown in Figure 2B are similar (−103.1, −109.5, and −94.6 kcal mol−1, respectively), while this value for S50356 is slightly larger (−112.2 kcal mol−1). Calculated standard deviations for cpd12 and S50356 show the largest and smallest values of ±58.91 and ±6.26 kcal mol−1, respectively. Although compounds cpd1p and cpd2p are similar, the standard deviation of cpd1p is smaller than that of cpd2p (±16.27 and ±31.98 kcal mol−1, respectively).
To understand the nature of these differences in the calculated QM/MM interaction energies, we employed averaged noncovalent interaction index (aNCI) and thermal fluctuation index (TFI) analyses. The noncovalent interaction index (NCI) is a qualitative analysis that visually identifies the noncovalent interactions between the molecule of interest and the surrounding residues. The thermal fluctuation index is another qualitative analysis showing fluctuation and instability in the noncovalent interactions between the compound and the surrounding residues. aNCI in Figure 2C shows continuous and blue-green surfaces among cpd12 and the surrounding residues, which corresponds to van der Waals interactions, and red surfaces with well-defined blue spots for the strong repulsive–attractive interactions (cpd12-CO2 group and LH2-Arg residue). This visual analysis shows that overall, cpd12 has many and consistent interactions with the neighboring residues. Based on the TFI surfaces for cpd12 in Figure 2D, the high standard deviation is due to the high fluctuations (red surfaces) around the carboxylate and ethyl groups. In the case of ligand S50356 (ID: 16), despite the carboxylate group, more stability is seen due to a polycyclic aromatic hydrocarbon (see Figure S3). For cpd1p and cpd2p (ID: 2 and 4), the nitrogen atom’s position in the pyridine group affects the enolate’s binding to the ion (see Figures S1 and S2).
Based on these results, it became evident that the negative charge significantly increases the interaction tendency, but the values of among the ligands with the most robust interactions were relatively similar. Thus, the specific effect of the negative charge was still unclear. In other words, it is unclear whether the enolate-based compounds with a negative charge on the donor oxygen or the anionic compounds with a negative charge far from the donor atoms are better ligands. To better understand this issue, we broadened the combinations of the second series to have diketone-, enolate-, and picolinamide-based structures branched with more aromatic rings and various functional groups (see Scheme 2). Compounds 2- cpd1p, 4- cpd2p, 8- cpd6 (enolate form), and 9- cpd7 (enolate form) of this set were used in a parallel in vitro investigation to study their selectivity and potency to inhibit LH2.75
Scheme 2.
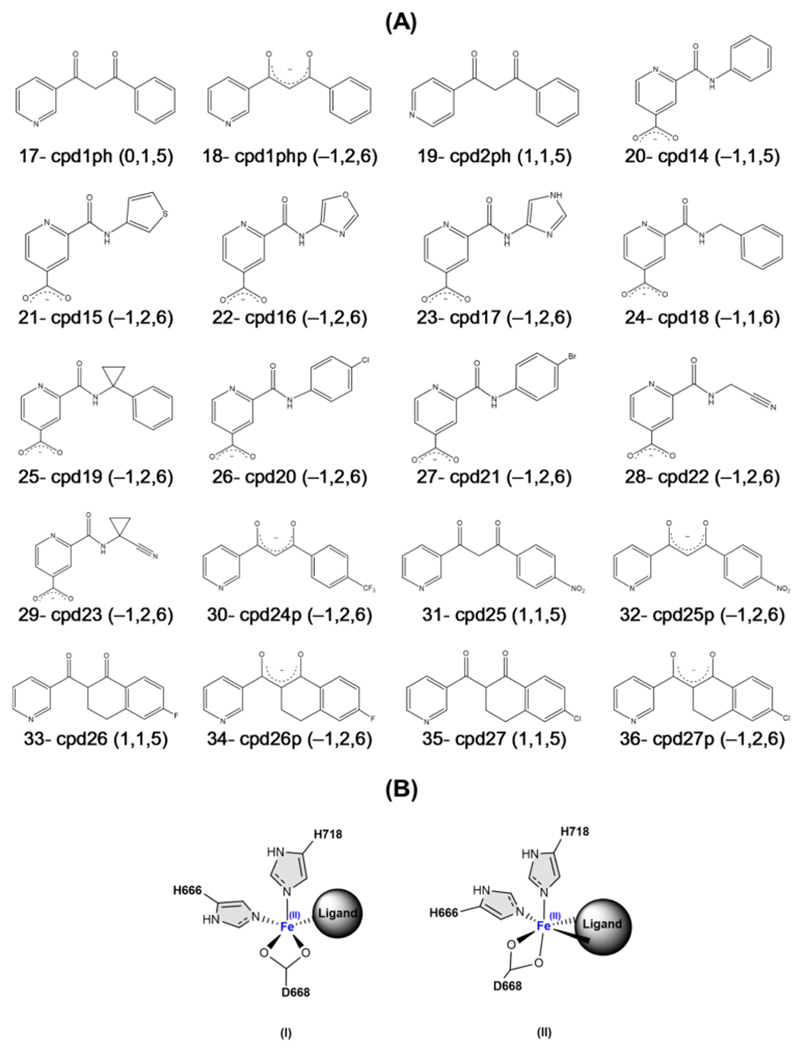
(A) Second Set of the Studied Ligandsa and (B) Different Geometries of the Active Site in Coordination with the Lead Compounds
a Numbers in parentheses are ligand’s charge, ligand’s coordination mode (monodentate or bidentate), and coordination number of the complex, respectively.
In the second set of the lead compounds listed in Scheme 2A, an equilibrated LH2 system with one of the selected ligands from the first set (cpd1p, cpd2p, cpd12, or S50356) was used, and the ligand was replaced with a new one, followed by QM/MM optimization. The results of the interaction energies are shown in Figure 3A. Our QM/MM optimizations showed distorted square pyramidal or octahedral geometries based on the ligand’s coordination type (see Scheme 2B). All of the neutral diketone compounds (IDs: 17, 19, 31, 33, and 35) are coordinated in a monodentate mode, while all of the picolinamide- and enolate-based compounds are bidentate-coordinated (except IDs: 20 and 24).
Figure 3.
(A) Calculated QM/MM interaction energies (kcal mol−1) for the second set of the studied ligands (ID: 17–36) at the ωB97X-D/6-31G(d,p) level of theory and AMOEBAbio 18 force field. The plot of the noncovalent interactions (NCIs) between the ligand and surrounding amino acid residues for (B) cpd27p, (C) cpd26p, and (D) cpd2ph. The isosurface cutoff for the NCI is 0.35 au, and the data is plotted in the color range of . Residues H666, D668, and H718 are shown in thin sticks, ligand atoms are in ball and sticks (with different color codes), is in pink vdW sphere, and the surrounding residues with the noncovalent interactions are in thick sticks. Hydrogen atoms are not shown for more clarity.
Based on the results of the interaction energies, calculated values of are ~30–50% of , similar to the first set of ligands. Most of the ligands have considerable negative values of , except for cpd1ph, cpd2ph, cpd25, cpd26, and cpd27 with the corresponding values of −43.3, −33.6, −21.6, −34.9, and −45.2 kcal mol−1, respectively. All of the mentioned correspond to the neutral, monodentate ligands. Moreover, once the enolate form of cpd1ph, cpd25, cpd26, and cpd27, which are cpd1php, cpd25p, cpd26p, and cpd27p, coordinates to the active site, the significantly improves to −115.0, −124.5, −143.9, and −144.7 kcal mol−1, respectively. These results are consistent with our previous observation for the first set, which showed that the enolate ligands have stronger interactions than the neutral ligands. We also realized that the negative charge effect on the interaction energy is significantly higher when closer to the active site. In other words, the enolate-based compounds having a negative charge on their donor oxygen have a stronger interaction with the active site than the picolinamide-based ligands in which a group is far from the active site.
Noncovalent interactions between the second set’s ligands and their surrounding residues were another important factor that needed to be considered. Interaction energies showed that enolate-based compounds are more energetically favorable, while the surrounding residues should stabilize a good candidate structurally. The main reason to branch the second series of the compounds with more aromatic rings, substituted with electron-donating and electron-withdrawing groups, was to see the effect of expanding the ligand’s size on the noncovalent interactions. The residues that show the most interactions with the ligands are F651, V653, Y655, L663, N675, C690, I730, and V732 (Table S2 and Figure S4). As mentioned before, based on our values in Figure 3A, compounds cpd27p and cpd26p exhibit the strongest interactions, while cpd2ph had one of the weakest interactions. However, the NCI plots for these compounds in Figure 3B–D show negligible differences in the numbers of interacting amino acid residues among these compounds. This observation may come from the fact that the second series of designed compounds are made from an equilibrated LH2 system with one of the selected ligands of the first set, subjected to QM/MM optimization without MD simulations.
Our observations from the first and the second series of the compounds indicate that the enolate-based ligands have stronger interaction with the active site. Additionally, several residues around the active site have attractive noncovalent interactions with compounds of the second set, which means all of the designed ligands of this series are stabilized by the environment. Previous studies have reported O2-transporting tunnels in other αKG-dependent superfamily members;32–36 based on this, we hypothesize that O2-transporting tunnels might also exist in LH2. Therefore, before designing the third set of enolate-based compounds, we studied the existence of O2-transporting tunnels in the apoenzyme. We performed a 5 ns MD simulation on the apo structure and applied k-means clustering on the trajectories. The geometry of the active site was considered octahedral at the starting point, in which H666, D668, H718, and two water molecules were coordinated to the iron (see clusters 2 and 4 in Figure S5). However, in agreement with experimental observations, a water molecule was replaced by one of the aspartate’s oxygen after 1 ns of MD.
Figure 4A shows the three major tunnels that are accessible for the apoenzyme with the priority/availability of 0.92/63% (blue), 0.83/51% (green), and 0.52/32% (red). There were some other accessible tunnels, but their availability and priority were much less than the three mentioned tunnels (detailed results in Table S11). It should be noted that, in addition to the tunnel’s availability, two other factors are essential when considering a calculated tunnel as a major tunnel: the cost and priority of the tunnel.110,111 Here, the cost of each tunnel, which is defined as the balance between the width and length of the tunnel (Å), was considered the priority of the tunnels. The tunnels with a cost of less than ~0.7 (wide and short) and a higher priority than ~0.5 were considered the threshold for selecting the tunnels.
Figure 4.
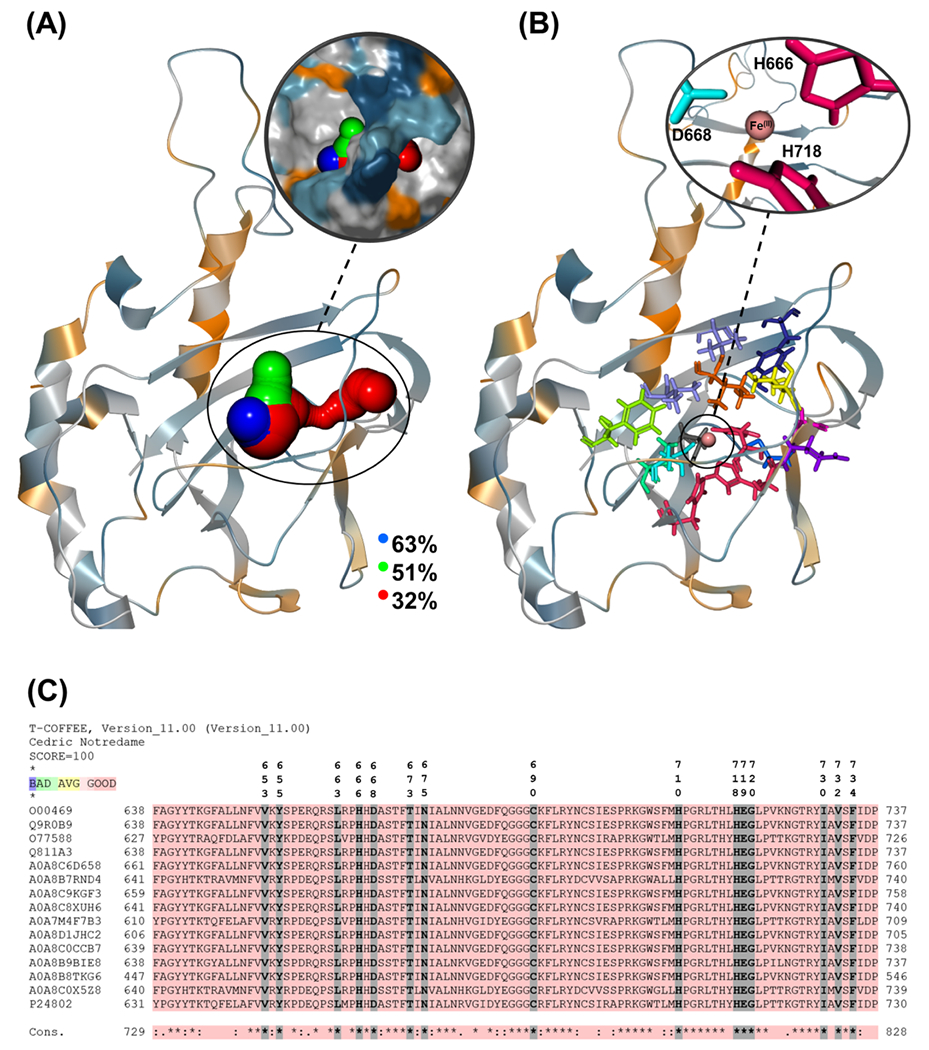
(A) O2-transporting tunnels with the largest calculated percentages along the trajectory observed in the apoenzyme. Calculated tunnels are colored in blue, green, and red based on the tunnel’s length (blue: shortest, red: longest). The intersection of the tunnels inside the protein is the active site, which is not shown for clarity. The molecular surface of the protein is shown in magnification to give a better view of the enzyme’s cavities. (B) Close-up of the approximate positions of the tunnel-lining residues with more than ~80% presence in all of the calculated tunnels/representatives. (C) Condensed sequence alignment between human LH2 (UniProtKB: O00469) and selected members of the LH family. Key: “*” indicates residues conserved in all of the sequences, while “:” and “.” indicate highly and weakly conserved residues.
Several common residues are present in most of the representatives’ tunnels.Table S3 shows that the tunnel-defining residues present in more than ~80% of the tunnels are V653, Y655, L663, H666, D668, T673, N675, C690, H710, H718, E719, G720, I730, V732, and F734. These residues are shown in Figure 4B. A sequence alignment between all of the 3 LH enzymes of Homo sapiens (LH1, LH2, and LH3) and 14 other organisms shows that all of the tunnel-defining residues are conserved (see Figure 4C and Tables S5 and S6). Similar to the previous set, selected compounds of the second set (IDs: 18, 19 (enolate form), 32, 34, and 36) were studied experimentally to investigate their inhibition properties.75
Based on our observations from the last two series of compounds and the presence of the oxygen-transporting tunnels in the apoenzyme with conserved amino acids, we designed a third set of lead compounds consisting of eight enolate-based structures with more aromatic rings substituted with electron-donating and electron-withdrawing groups (see Figure 5). One of the goals for the third set was to see if the new ligands could affect oxygen transport by blocking the putative tunnels and establishing further interactions with conserved residues around the active site. Furthermore, we calculated the interaction energies between each compound and the active site and studied their noncovalent interactions with the surrounding amino acids. All of the ligands were parameterized, and holo-structures were subjected to MD simulations. The stability of the whole structure during the MD simulations has been illustrated by the root-mean-square deviation (RMSD) of the core part of LH2 bound with the compounds of this set (see Figure S6). At the same time, the main interactions between the Fe ion and the surrounding active site (H666, D668, and H718) in those complexes have been kept stable. The specific data for those distances have been recorded in Table S7. The 5 ns of each structure’s MD trajectories was taken for further clustering analysis to select the best representative structures. The k-means clustering results and the related QM/MM optimization energies for each representative are given in Table S8. The geometries of the active site in all of the representatives for each compound are also shown in Figures S7–S14.
Figure 5.
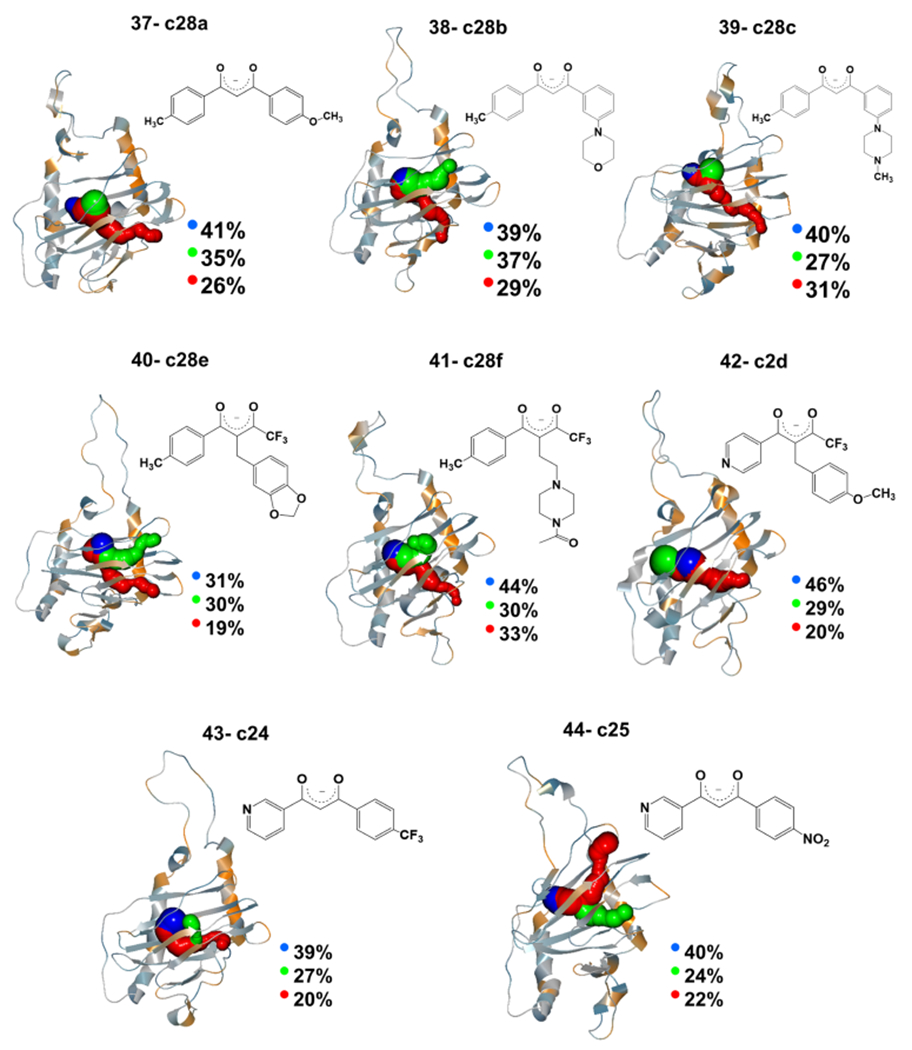
O2-transporting tunnels with the largest calculated percentages along the trajectory observed in the third set of lead compounds. The twodimensional (2D) structure of each ligand is also given next to its tunnel graph. Calculated tunnels are colored in blue, green, and red based on the increase in the tunnel’s length. The intersection of the tunnels inside the protein is the active site of the inhibition reaction, but the active site’s residues are not shown for more clarity.
Like the apo structure, we used each compound’s six representatives to calculate the O2-transporting tunnels. The close-ups of the major calculated tunnels for each compound are given in Figure 5. The length/width of the three major tunnels in the apo structure—selected by the criteria as mentioned earlier—was 2.2/1.9 (blue), 5.8/1.7 (green), and 10.2/0.9 Å (red) (Table S4). Additionally, it is observed that the holo-structures have relatively longer tunnels. This may be due to the structural effects of the incoming ligands after binding to the active site and elongating the tunnel. Comparing the tunnel availability between apo and holo-structures indicates a decrease in tunnel availability by all of the compounds. This reduction is 27–51 and 31–53% for the blue and green tunnels, respectively, with the highest availability in the apo structure. Analysis of the conserved residues lining the tunnels for the holo systems is consistent with the apoenzyme tunnel analysis (Tables S12–S19).
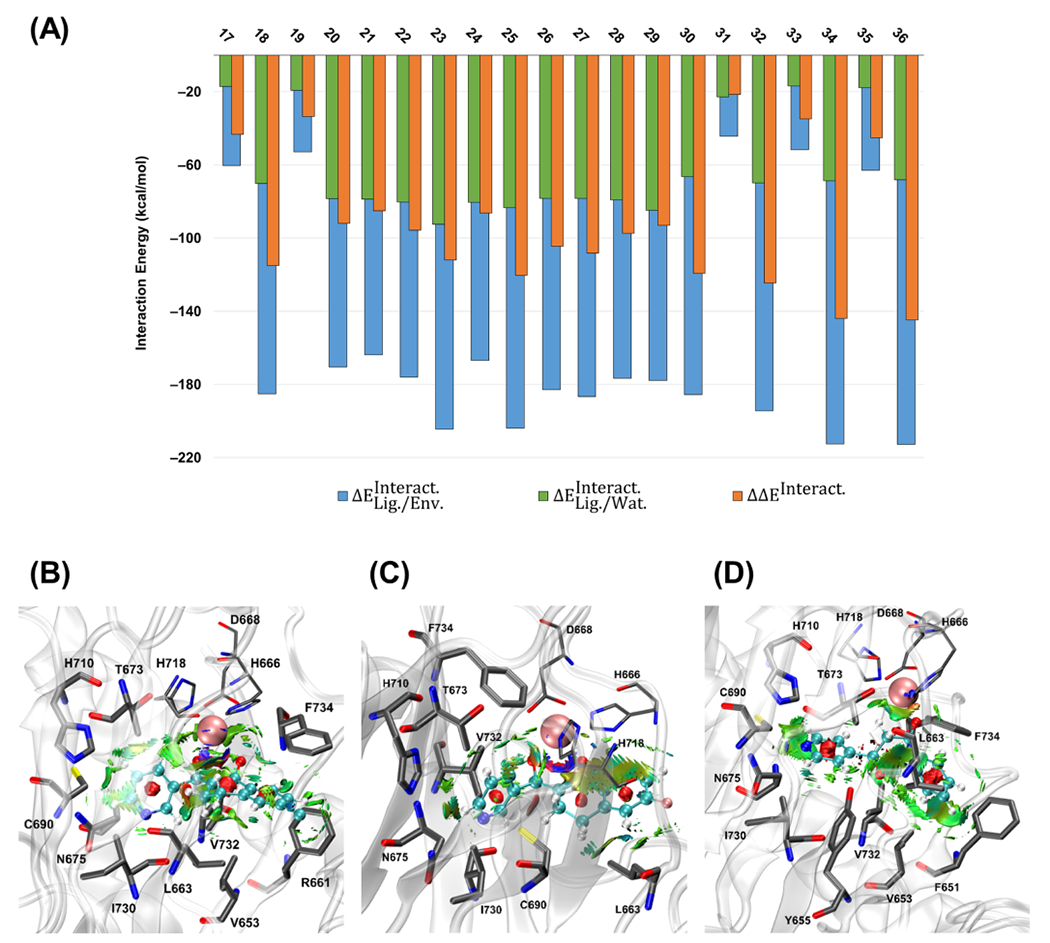
The interaction energies between the most stable representative of each ligand (the representative with the zero value of the relative optimization energy in Table S8) and the active site were calculated for each ligand in the third set. Calculated interaction energies in Figure 6A show that 33–63% of the is due to . Based on the calculated values of , ligands c2d and c24 have the weakest interaction with the active site (−37.9 and −48.2 kcal mol−1, respectively), while c28b and c28a have the strongest interactions, −199.3 and −147.4 kcal mol−1, respectively (Table S1).
Figure 6.
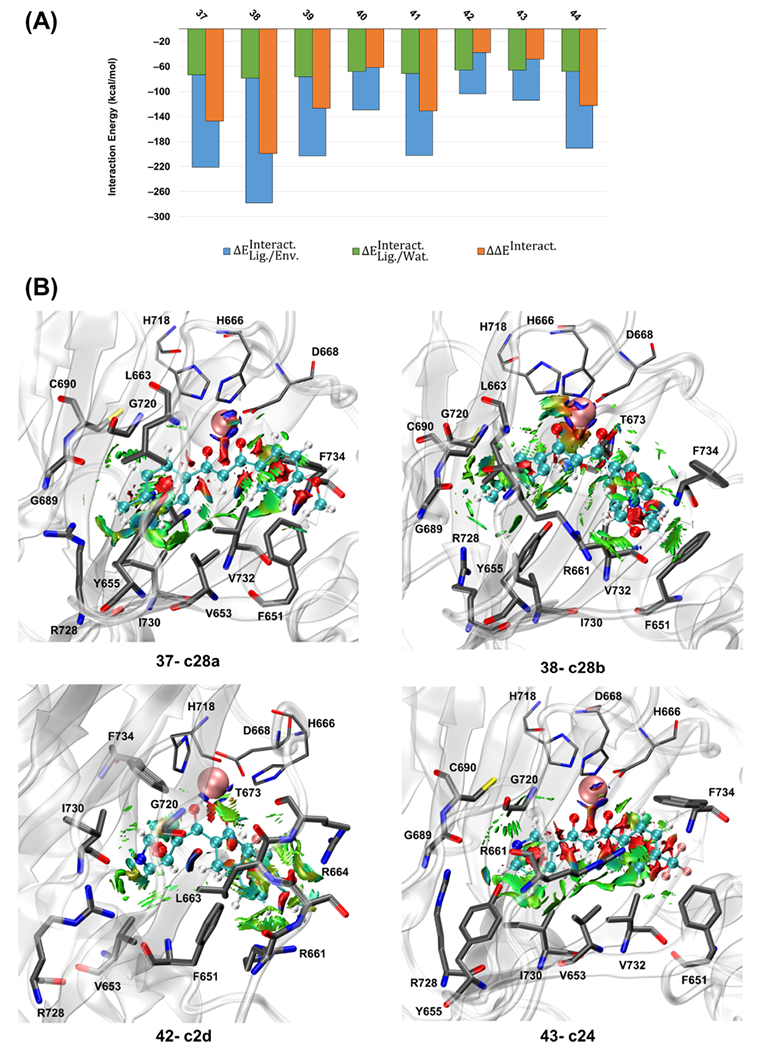
(A) Calculated QM/MM interaction energies (kcal mol−1) for the most stable representative of the studied ligands of the third set (ID: 37–44) at the ωB97X-D/6-31G(d,p) level of theory and AMOEBAbio 18 force field. (B) Averaged noncovalent interactions (aNCIs) between the ligand and its surrounding residues for c28a, c28b, c2d, and c24. The isosurface cutoff for the NCI is 0.35 au, and the data is plotted in the color range of au. Residues H666, D668, and H718 are shown in thin sticks, ligand atoms are in ball and sticks (with different color codes), is in pink vdW sphere, and the surrounding residues with the noncovalent interactions are in thick sticks. Hydrogen atoms are not shown for more clarity.
The aNCI analysis for the third set of ligands in Table S10 shows that the amino acid residues interacting with most of the ligands from this third set are F651, Y655, C690, R728, and F734. The aNCI graphs in Figure 6B qualitatively show that c28a and c28b, which have the strongest interaction with the active site, also have noncovalent interactions with more surrounding residues than c2d and c24, with the weakest interaction with the active site (supplementary aNCI graphs in Figure S15). The QM/MM interaction energies and the aNCI show that c28a and c28b are energetically and structurally more favorable than the other ligands. These two compounds also had a significant blocking effect on the O2-transporting tunnel. Selected compounds of the third set (IDs: 38, 39, and their pyridine analogs) were used in the parallel experimental investigation.75
CONCLUSIONS
Forty-four small molecules were investigated as candidate compounds for LH2 inhibition in three sequential sets. The first set of compounds was designed to have one or two oxygen, nitrogen, or a combination of both to coordinate with the iron cation of the active site. Results showed that diketone-based compounds do not interact strongly with the active site, while the enolate form has considerably large negative interaction energies. We designed compounds with various functional groups for the second set but with the main skeleton of diketone-, enol-, enolate- (to see the effect of the negative charge and the structural change), and picolinamide-based compounds having oxygen and nitrogen donor atoms. Results showed that enolate-based compounds have the most potent interactions with the active site, similar to the first set. Intramolecular O2-transport tunnel analysis was carried out on apo-LH2 to determine the feasibility of the existence of this feature in LH2. We found three major tunnels with the availability of 63, 51, and 32%, in which a series of tunnel-defining residues had more than 80% of repetition. Further investigation on the evolutionary conservation of amino acid residues via the multiple sequence alignment on LH enzymes (LH1, LH2, and LH3) in 41 organisms showed that all residues showing more than 80% presence in the tunnels are conserved. For the third set of ligands, new enolate-based compounds branched with more rings and functional groups were designed to block the O2-transport tunnels and enhance the interactions with the active site and the surrounding residues. Results showed that the availability of the tunnels is relatively higher in the apo structure than in the holo-structures, with the third set and some of the compounds in this third set blocking the tunnels. The interaction energies and the noncovalent interactions showed that compounds 37-C28a and 38-C28b have the strongest interactions with the active site and noncovalent interactions with most of the surrounding amino acid residues. Selected compounds of this study—in addition to some other candidates based on our findings—were tested in parallel in another computational/experimental contribution to identifying competitive inhibitors of αKG with nanomolar inhibition property.75
Supplementary Material
ACKNOWLEDGMENTS
This work was partially supported by the National Institutes of Health (R01GM108583, R01GM106137, R01GM114237, R01CA105155, R00CA225633, and P50CA070907) and National Science Foundation (CHE-2217856 and CHE-1856173), the Cancer Prevention and Research Institute of Texas (award numbers RP160657, RP210088, and RP160652), and the Welch Foundation (award number F-1390). Computational time was provided by the University of Texas at Dallas CyberInfrastructure Facilities and the University of North Texas CASCaM CRUNTCh3 high-performance cluster, which was partially supported by NSF grant Nos. CHE-1531468 and OAC-2117247. Additional computing time from XSEDE Project TG-CHE160044 is gratefully acknowledged. E.A.V.-M. thanks CONACyT for funding.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.2c01448.
Additional details of molecular dynamics, k-means clustering, interaction energies, NCIs, oxygen-transporting tunnels, and multiple sequence alignments (PDF)
Initial coordinates and parameters for all of the studied systems (ZIP)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jcim.2c01448
The authors declare the following competing financial interest(s): PR is the co-founder of Qubit Pharmaceuticals.
Contributor Information
Yazdan Maghsoud, Department of Chemistry and Biochemistry, The University of Texas at Dallas, Richardson, Texas 75080, United States.
Erik Antonio Vázquez-Montelongo, Department of Physical Medicine and Rehabilitation, University of Texas Southwestern Medical Center, Dallas, Texas 75390, United States.
Xudong Yang, Department of Biomedical Engineering, The University of Texas at Austin, Austin, Texas 78712, United States.
Chengwen Liu, Department of Biomedical Engineering, The University of Texas at Austin, Austin, Texas 78712, United States.
Zhifeng Jing, Department of Biomedical Engineering, The University of Texas at Austin, Austin, Texas 78712, United States.
Juhoon Lee, Division of Chemical Biology and Medicinal Chemistry, College of Pharmacy, The University of Texas at Austin, Austin, Texas 78712, United States.
Matthew Harger, Department of Biomedical Engineering, The University of Texas at Austin, Austin, Texas 78712, United States.
Ally K. Smith, Department of Chemistry, University of North Texas, Denton, Texas 76201, United States
Miguel Espinoza, Department of Chemistry, University of North Texas, Denton, Texas 76201, United States.
Hou-Fu Guo, Department of Molecular and Cellular Biochemistry, College of Medicine, The University of Kentucky, Lexington, Kentucky 40536, United States.
Jonathan M. Kurie, Department of Thoracic/Head and Neck Medical Oncology, The University of Texas MD Anderson Cancer Center, Houston, Texas 77005, United States
Kevin N. Dalby, Division of Chemical Biology and Medicinal Chemistry, College of Pharmacy, The University of Texas at Austin, Austin, Texas 78712, United States
Pengyu Ren, Department of Biomedical Engineering, The University of Texas at Austin, Austin, Texas 78712, United States.
G. Andrés Cisneros, Department of Chemistry and Biochemistry, The University of Texas at Dallas, Richardson, Texas 75080, United States; Department of Physics, The University of Texas at Dallas, Richardson, Texas 75080, United States.
Data Availability Statement
All simulations and analyses employed via third-party software are described and referenced in the Methodology section. The LICHEM software program is available at the Cisneros Research Group GitHub: https://github.com/CisnerosResearch/LICHEM.
REFERENCES
- (1).Loenarz C; Schofield CJ Expanding chemical biology of 2-oxoglutarate oxygenases. Nat. Chem. Biol 2008, 4, 152–156. [DOI] [PubMed] [Google Scholar]
- (2).Hausinger RP Fe (II)/α-ketoglutarate-dependent hydroxylases and related enzymes. Crit. Rev. Biochem. Mol. Biol 2004, 39, 21–68. [DOI] [PubMed] [Google Scholar]
- (3).Flashman E; Schofield CJ The most versatile of all reactive intermediates? Nat. Chem. Biol 2007, 3, 86–87. [DOI] [PubMed] [Google Scholar]
- (4).Hautala T; Byers MG; Eddy RL; Shows TB; Kivirikko KI; Myllyla R Cloning of human lysyl hydroxylase: Complete cDNA-derived amino acid sequence and assignment of the gene (PLOD) to chromosome 1p36. 3→ p36. 2. Genomics 1992, 13, 62–69. [DOI] [PubMed] [Google Scholar]
- (5).Xu Y; Baldassare M; Fisher P; Rathbun G; Oltz EM; Yancopoulos GD; Jessell TM; Alt FW LH-2: a LIM/homeodomain gene expressed in developing lymphocytes and neural cells. Proc. Natl. Acad. Sci. U.S.A 1993, 90, 227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Passoja K; Rautavuoma K; Ala-Kokko L; Kosonen T; Kivirikko KI Cloning and characterization of a third human lysyl hydroxylase isoform. Proc. Natl. Acad. Sci. U.S.A 1998, 95, 10482–10486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Takaluoma K; Lantto J; Myllyharju J Lysyl hydroxylase 2 is a specific telopeptide hydroxylase, while all three isoenzymes hydroxylate collagenous sequences. Matrix Biol. 2007, 26, 396–403. [DOI] [PubMed] [Google Scholar]
- (8).Yamauchi M; Sricholpech M Lysine post-translational modifications of collagen. Essays Biochem. 2012, 52, 113–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Pornprasertsuk S; Duarte WR; Mochida Y; Yamauchi M Lysyl hydroxylase-2b directs collagen cross-linking pathways in MC3T3-E1 cells. J. Bone Miner. Res 2004, 19, 1349–1355. [DOI] [PubMed] [Google Scholar]
- (10).Bailey AJ; Peach CM; Fowler L Chemistry of the collagen cross-links. Isolation and characterization of two intermediate intermolecular cross-links in collagen. Biochem. J 1970, 117, 819–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ha-Vinh R; Alanay Y; Bank RA; Campos-Xavier AB; Zankl A; Superti-Furga A; Bonafé L Phenotypic and molecular characterization of Bruck syndrome (osteogenesis imperfecta with contractures of the large joints) caused by a recessive mutation in PLOD2. Am. J. Med. Genet., Part A 2004, 131A, 115–120. [DOI] [PubMed] [Google Scholar]
- (12).Van Der Slot AJ; Zuurmond A-M; Van Den Bogaerdt AJ; Ulrich MM; Middelkoop E; Boers W; Ronday HK; DeGroot J; Huizinga TW; Bank RA Increased formation of pyridinoline cross-links due to higher telopeptide lysyl hydroxylase levels is a general fibrotic phenomenon. Matrix Biol. 2004, 23, 251–257. [DOI] [PubMed] [Google Scholar]
- (13).van der Slot AJ; Zuurmond A-M; Bardoel AF; Wijmenga C; Pruijs HE; Sillence DO; Brinckmann Jr.; Abraham DJ; Black CM; Verzijl N; et al. Identification of PLOD2 as telopeptide lysyl hydroxylase, an important enzyme in fibrosis. J. Biol. Chem 2003, 278, 40967–40972. [DOI] [PubMed] [Google Scholar]
- (14).Eisinger-Mathason TK; Zhang M; Qiu Q; Skuli N; Nakazawa MS; Karakasheva T; Mucaj V; Shay JE; Stangenberg L; Sadri N; et al. Hypoxia-dependent modification of collagen networks promotes sarcoma metastasis. Cancer Discovery 2013, 3, 1190–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Gilkes DM; Bajpai S; Chaturvedi P; Wirtz D; Semenza GL Hypoxia-inducible factor 1 (HIF-1) promotes extracellular matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. J. Biol. Chem 2013, 288, 10819–10829. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (16).Gilkes DM; Bajpai S; Wong CC; Chaturvedi P; Hubbi ME; Wirtz D; Semenza GL Procollagen lysyl hydroxylase 2 is essential for hypoxia-induced breast cancer metastasis. Mol. Cancer Res 2013, 11, 456–466. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (17).Chen Y; Terajima M; Yang Y; Sun L; Ahn Y-H; Pankova D; Puperi DS; Watanabe T; Kim MP; Blackmon SH; et al. Lysyl hydroxylase 2 induces a collagen cross-link switch in tumor stroma. J. Clin. Invest 2015, 125, 1147–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Luo L; Pappalardi MB; Tummino PJ; Copeland RA; Fraser ME; Grzyska PK; Hausinger RP An assay for Fe (II)/2-oxoglutarate-dependent dioxygenases by enzyme-coupled detection of succinate formation. Anal. Biochem 2006, 353, 69–74. [DOI] [PubMed] [Google Scholar]
- (19).McNeill L; Bethge L; Hewitson K; Schofield C A fluorescence-based assay for 2-oxoglutarate-dependent oxygenases. Anal. Biochem 2005, 336, 125–131. [DOI] [PubMed] [Google Scholar]
- (20).Guo H-F; Cho EJ; Devkota AK; Chen Y; Russell W; Phillips GN Jr.; Yamauchi M; Dalby KN; Kurie JM A scalable lysyl hydroxylase 2 expression system and luciferase-based enzymatic activity assay. Arch. Biochem. Biophys 2017, 618, 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Devkota AK; Veloria JR; Guo H-F; Kurie JM; Cho EJ; Dalby KN Development of a high-throughput lysyl hydroxylase (LH) assay and identification of small-molecule inhibitors against LH2. SLAS Discovery 2019, 24, 484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Rhoads RE; Udenfriend S Decarboxylation of alpha-ketoglutarate coupled to collagen proline hydroxylase. Proc. Natl. Acad. Sci. U.S.A 1968, 60, No. 1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Scietti L; Moroni E; Mattoteia D; Fumagalli M; De Marco M; Negro L; Chiapparino A; Serapian S; De Giorgi F; Faravelli S; et al. A Fe2+-dependent self-inhibited state influences the druggability of human collagen lysyl hydroxylase (LH/PLOD) enzymes. Front. Mol. Biosci 2022, 9, No. 876352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Scietti L; Chiapparino A; De Giorgi F; Fumagalli M; Khoriauli L; Nergadze S; Basu S; Olieric V; Cucca L; Banushi B; et al. Molecular architecture of the multifunctional collagen lysyl hydroxylase and glycosyltransferase LH3. Nat. Commun 2018, 9, No. 3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Ruscio JZ; Kumar D; Shukla M; Prisant MG; Murali T; Onufriev AV Atomic level computational identification of ligand migration pathways between solvent and binding site in myoglobin. Proc. Natl. Acad. Sci. U.S.A 2008, 105, 9204–9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Tomita A; Kreutzer U; Adachi S.-i.; Koshihara S.-y.; Jue T ‘It’s hollow’: the function of pores within myoglobin. J. Exp. Biol 2010, 213, 2748–2754. [DOI] [PubMed] [Google Scholar]
- (27).Colloc’h N; Gabison L; Monard G; Altarsha M; Chiadmi M; Marassio G; Sopkova-de Oliveira Santos J; El Hajji M; Castro B; Abraini JH; Prangé T Oxygen pressurized X-ray crystallography: probing the dioxygen binding site in cofactorless urate oxidase and implications for its catalytic mechanism. Biophys. J 2008, 95, 2415–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Pesce A; Nardini M; Dewilde S; Capece L; Martí MA; Congia S; Salter MD; Blouin GC; Estrin DA; Ascenzi P; et al. Ligand migration in the apolar tunnel of Cerebratulus lacteus minihemoglobin. J. Biol. Chem 2011, 286, 5347–5358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Elber R Ligand diffusion in globins: simulations versus experiment. Curr. Opin. Struct. Biol 2010, 20, 162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Di Russo NV; Condurso HL; Li K; Bruner SD; Roitberg AE Oxygen diffusion pathways in a cofactor-independent dioxygenase. Chem. Sci 2015, 6, 6341–6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Shadrina MS; English AM; Peslherbe GH Effective simulations of gas diffusion through kinetically accessible tunnels in multisubunit proteins: O2 pathways and escape routes in T-state deoxyhemoglobin. J. Am. Chem. Soc 2012, 134, 11177–11184. [DOI] [PubMed] [Google Scholar]
- (32).Li C; Junaid M; Almuqri EA; Hao S; Zhang H Structural analysis of a phosphonate hydroxylase with an access tunnel at the back of the active site. Acta Crystallogr., Sect. F: Struct. Biol. Commun 2016, 72, 362–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Chaplin VD; Valliere MA; Hangasky JA; Knapp MJ Investigations on the role of a solvent tunnel in the α-ketoglutarate dependent oxygenase factor inhibiting HIF (FIH). J. Inorg. Biochem 2018, 178, 63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Yu B; Hunt JF Enzymological and structural studies of the mechanism of promiscuous substrate recognition by the oxidative DNA repair enzyme AlkB. Proc. Natl. Acad. Sci. U.S.A 2009, 106, 14315–14320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Yu B; Edstrom WC; Benach J; Hamuro Y; Weber PC; Gibney BR; Hunt JF Crystal structures of catalytic complexes of the oxidative DNA/RNA repair enzyme AlkB. Nature 2006, 439, 879–884. [DOI] [PubMed] [Google Scholar]
- (36).Torabifard H; Cisneros GA Computational investigation of O2 diffusion through an intra-molecular tunnel in AlkB; influence of polarization on O 2 transport. Chem. Sci 2017, 8, 6230–6238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Buch I; Giorgino T; De Fabritiis G Complete reconstruction of an enzyme-inhibitor binding process by molecular dynamics simulations. Proc. Natl. Acad. Sci. U.S.A 2011, 108, 10184–10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Costa RA; Cruz JN; Nascimento FC; Silva SG; Silva SO; Martelli MC; Carvalho SM; Santos CB; Neto AM; Brasil DS Studies of NMR, molecular docking, and molecular dynamics simulation of new promising inhibitors of cruzaine from the parasite Trypanosoma cruzi. Med. Chem. Res 2019, 28, 246–259. [Google Scholar]
- (39).Śledź P; Caflisch A Protein structure-based drug design: from docking to molecular dynamics. Curr. Opin. Struct. Biol 2018, 48, 93–102. [DOI] [PubMed] [Google Scholar]
- (40).Zhao H; Caflisch A Molecular dynamics in drug design. Eur. J. Med. Chem 2015, 91, 4–14. [DOI] [PubMed] [Google Scholar]
- (41).Jorgensen WL; Maxwell DS; Tirado-Rives J Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc 1996, 118, 11225–11236. [Google Scholar]
- (42).Vanommeslaeghe K; Hatcher E; Acharya C; Kundu S; Zhong S; Shim J; Darian E; Guvench O; Lopes P; Vorobyov I CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem 2010, 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Brooks BR; Bruccoleri RE; Olafson BD; States DJ; Swaminathan S; Karplus M CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem 1983, 4, 187–217. [Google Scholar]
- (44).Lindorff-Larsen K; Piana S; Palmo K; Maragakis P; Klepeis JL; Dror RO; Shaw DE Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins: Struct., Funct., Bioinf 2010, 78, 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Pearlman DA; Case DA; Caldwell JW; Ross WS; Cheatham TE III; DeBolt S; Ferguson D; Seibel G; Kollman P AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun 1995, 91, 1–41. [Google Scholar]
- (46).Oostenbrink C; Villa A; Mark AE; Van Gunsteren WF A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem 2004, 25, 1656–1676. [DOI] [PubMed] [Google Scholar]
- (47).Wu JC; Chattree G; Ren P Automation of AMOEBA polarizable force field parameterization for small molecules. Theor. Chem. Acc 2012, 131, No. 1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Ponder JW; Wu C; Ren P; Pande VS; Chodera JD; Schnieders MJ; Haque I; Mobley DL; Lambrecht DS; DiStasio RA Jr.; et al. Current status of the AMOEBA polarizable force field. J. Phys. Chem. B 2010, 114, 2549–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Ren P; Ponder JW Consistent treatment of inter-and intramolecular polarization in molecular mechanics calculations. J. Comput. Chem 2002, 23, 1497–1506. [DOI] [PubMed] [Google Scholar]
- (50).Magalhães RP; Fernandes HS; Sousa SF Modelling enzymatic mechanisms with QM/MM approaches: current status and future challenges. Isr. J. Chem 2020, 60, 655–666. [Google Scholar]
- (51).Ahmadi S; Barrios Herrera L; Chehelamirani M; Hostaš J; Jalife S; Salahub DR Multiscale modeling of enzymes: QM-cluster, QM/MM, and QM/MM/MD: a tutorial review. Int. J. Quantum Chem 2018, 118, No. e25558. [Google Scholar]
- (52).Duarte F; Amrein BA; Blaha-Nelson D; Kamerlin SC Recent advances in QM/MM free energy calculations using reference potentials. Biochim. Biophys. Acta, Gen. Subj 2015, 1850, 954–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Brunk E; Rothlisberger U Mixed quantum mechanical/molecular mechanical molecular dynamics simulations of biological systems in ground and electronically excited states. Chem. Rev 2015, 115, 6217–6263. [DOI] [PubMed] [Google Scholar]
- (54).van der Kamp MW; Mulholland AJ Combined quantum mechanics/molecular mechanics (QM/MM) methods in computational enzymology. Biochemistry 2013, 52, 2708–2728. [DOI] [PubMed] [Google Scholar]
- (55).Senn HM; Thiel W QM/MM methods for biomolecular systems. Angew. Chem., Int. Ed 2009, 48, 1198–1229. [DOI] [PubMed] [Google Scholar]
- (56).Lin H; Truhlar DG QM/MM: what have we learned, where are we, and where do we go from here? Theor. Chem. Acc 2007, 117, 185–199. [Google Scholar]
- (57).Nakano H; Yamamoto T Variational calculation of quantum mechanical/molecular mechanical free energy with electronic polarization of solvent. J. Chem. Phys 2012, 136, No. 134107. [DOI] [PubMed] [Google Scholar]
- (58).Boulanger E; Thiel W Solvent boundary potentials for hybrid QM/MM computations using classical drude oscillators: a fully polarizable model. J. Chem. Theory Comput 2012, 8, 4527–4538. [DOI] [PubMed] [Google Scholar]
- (59).Illingworth CJR; Parkes K; Snell C; Marti S; Moliner V; Reynolds C The effect of MM polarization on the QM/MM transition state stabilization: application to chorismate mutase. Mol. Phys 2008, 106, 1511–1515. [Google Scholar]
- (60).Giese TJ; York DM Charge-dependent model for manybody polarization, exchange, and dispersion interactions in hybrid quantum mechanical/ molecular mechanical calculations. J. Chem. Phys 2007, 127, No. 194101. [DOI] [PubMed] [Google Scholar]
- (61).Gao J; Alhambra C A hybrid semiempirical quantum mechanical and lattice-sum method for electrostatic interactions in fluid simulations. J. Chem. Phys 1997, 107, 1212–1217. [Google Scholar]
- (62).Slipchenko LV Solvation of the excited states of chromophores in polarizable environment: Orbital relaxation versus polarization. J. Phys. Chem. A 2010, 114, 8824–8830. [DOI] [PubMed] [Google Scholar]
- (63).Gao J Energy components of aqueous solution: Insight from hybrid QM/MM simulations using a polarizable solvent model. J. Comput. Chem 1997, 18, 1061–1071. [Google Scholar]
- (64).Giovannini T; Riso RR; Ambrosetti M; Puglisi A; Cappelli C Electronic transitions for a fully polarizable qm/mm approach based on fluctuating charges and fluctuating dipoles: linear and corrected linear response regimes. J. Chem. Phys 2019, 151, No. 174104. [DOI] [PubMed] [Google Scholar]
- (65).Giovannini T; Puglisi A; Ambrosetti M; Cappelli C Polarizable QM/MM approach with fluctuating charges and fluctuating dipoles: the QM/FQFμ model. J. Chem. Theory Comput 2019, 15, 2233–2245. [DOI] [PubMed] [Google Scholar]
- (66).Vázquez-Montelongo E; Vázquez-Cervantes JE; Cisneros GA Polarizable ab initio QM/MM Study of the Reaction Mechanism of N-tert-Butyloxycarbonylation of Aniline in [EMIm][BF4]. Molecules 2018, 23, No. 2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Hagras MA; Glover WJ Polarizable embedding for excited-state reactions: dynamically weighted polarizable QM/MM. J. Chem. Theory Comput 2018, 14, 2137–2144. [DOI] [PubMed] [Google Scholar]
- (68).DeNizio JE; Liu MY; Leddin EM; Cisneros GA; Kohli RM Selectivity and Promiscuity in TET-Mediated Oxidation of 5-Methylcytosine in DNA and RNA. Biochemistry 2019, 58, 411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Liu MY; Torabifard H; Crawford DJ; DeNizio JE; Cao X-J; Garcia BA; Cisneros GA; Kohli RM Mutations along a TET2 active site scaffold stall oxidation at 5-hydroxymethylcytosine. Nat. Chem. Biol 2017, 13, 181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Waheed SO; Chaturvedi SS; Karabencheva-Christova TG; Christov CZ Catalytic mechanism of human ten-eleven translocation-2 (tet2) enzyme: Effects of conformational changes, electric field, and mutations. ACS Catal. 2021, 11, 3877–3890. [Google Scholar]
- (71).Walker AR; Silvestrov P; Müller TA; Podolsky RH; Dyson G; Hausinger RP; Cisneros GA ALKBH7 variant related to prostate cancer exhibits altered substrate binding. PLoS Comput. Biol 2017, 13, No. e 1005345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Silvestrov P; Müller TA; Clark KN; Hausinger RP; Cisneros GA Homology modeling, molecular dynamics, and sitedirected mutagenesis study of AlkB human homolog 1 (ALKBH1). J. Mol. Graphics Modell 2014, 54, 123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Lenz SA; Li D; Wetmore SD Insights into the Direct Oxidative Repair of Etheno Lesions: MD and QM/MM Study on the Substrate Scope of ALKBH2 and AlkB. DNA Repair 2020, 96, No. 102944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Berger MB; Walker AR; Vázquez-Montelongo EA; Cisneros GA Computational investigations of selected enzymes from two iron and α-ketoglutarate-dependent families. Phys. Chem. Chem. Phys 2021, 23, 22227–22240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Lee J; Guo H-F; Maghsoud Y; Vázquez-Montelongo EA; Jing Z; Wang S; Sammons RM; Cho EJ; Ren P; Cisneros GA; Kurie JM; Dalby KN 1,3-Diketone Analogs as Selective Lysyl Hydroxylase 2 (LH2) Antagonists, ChemRxiv 2022. [Google Scholar]
- (76).Waterhouse A; Bertoni M; Bienert S; Studer G; Tauriello G; Gumienny R; Heer FT; de Beer TAP; Rempfer C; Bordoli L; et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Olsson MHM; Søndergaard CR; Rostkowski M; Jensen JH PROPKA3: consistent treatment of internal and surface residues in empirical p K a predictions. J. Chem. Theory Comput 2011, 7, 525–537. [DOI] [PubMed] [Google Scholar]
- (78).Jurrus E; Engel D; Star K; Monson K; Brandi J; Felberg LE; Brookes DH; Wilson L; Chen J; Liles K; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Jones G; Willett P; Glen RC; Leach AR; Taylor R Development and validation of a genetic algorithm for flexible docking11Edited by F. E. Cohen. J. Mol. Biol 1997, 267, 727–748. [DOI] [PubMed] [Google Scholar]
- (80).Ren P; Ponder JW Polarizable atomic multipole water model for molecular mechanics simulation. J. Phys. Chem. B 2003, 107, 5933–5947. [Google Scholar]
- (81).Humphreys DD; Friesner RA; Berne BJ A Multiple-Time-Step Molecular Dynamics Algorithm for Macromolecules. J. Phys. Chem. A 1994, 98, 6885–6892. [Google Scholar]
- (82).Qian X; Schlick T Efficient multiple-time-step integrators with distance-based force splitting for particle-mesh-Ewald molecular dynamics simulations. J. Chem. Phys 2002, 116, 5971–5983. [Google Scholar]
- (83).Frenkel D; Smit B; Ratner MA Understanding Molecular Simulation: From Algorithms to Applications; Academic Press: San Diego, 1996; Vol. 2. [Google Scholar]
- (84).Bussi G; Zykova-Timan T; Parrinello M Isothermal-isobaric molecular dynamics using stochastic velocity rescaling. J. Chem. Phys 2009, 130, No. 074101. [DOI] [PubMed] [Google Scholar]
- (85).Harger M; Li D; Wang Z; Dalby K; Lagardère L; Piquemal J-P; Ponder J; Ren P Tinker-OpenMM: Absolute and relative alchemical free energies using AMOEBA on GPUs. J. Comput. Chem 2017, 38, 2047–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Likas A; Vlassis N; Verbeek JJ The global k-means clustering algorithm. Pattern Recognit. 2003, 36, 451–461. [Google Scholar]
- (87).Gökcan H; Vázquez-Montelongo EA; Cisneros GA LICHEM 1.1: recent improvements and new capabilities. J. Chem. Theory Comput 2019, 15, 3056–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Kratz EG; Walker AR; Lagardère L; Lipparini F; Piquemal JP; Andrés Cisneros G LICHEM: A QM/MM program for simulations with multipolar and polarizable force fields. J. Comput. Chem 2016, 37, 1019–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich AV; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg JL; Williams-Young D; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Henderson T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark MJ; Heyd JJ; Brothers EN; Kudin KN; Staroverov VN; Keith TA; Kobayashi R; Normand J; Raghavachari K; Rendell AP; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas O; Foresman JB; Fox DJ Gaussian 16, revision C.01; Gaussian, Inc.: Wallingford, CT, 2016. [Google Scholar]
- (90).Ponder JW TINKER: Software Tools for Molecular Design; Washington University School of Medicine: Saint Louis, MO, 2004; Vol. 3. [Google Scholar]
- (91).Rackers JA; Wang Z; Lu C; Laury ML; Lagardère L; Schnieders MJ; Piquemal J-P; Ren P; Ponder JW Tinker 8: software tools for molecular design. J. Chem. Theory Comput 2018, 14, 5273–5289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Chai J-D; Head-Gordon M Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys 2008, 10, 6615–6620. [DOI] [PubMed] [Google Scholar]
- (93).Chai J-D; Head-Gordon M Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys 2008, 128, No. 084106 [DOI] [PubMed] [Google Scholar]
- (94).Zhang C; Lu C; Jing Z; Wu C; Piquemal J-P; Ponder JW; Ren P AMOEBA polarizable atomic multipole force field for nucleic acids. J. Chem. Theory Comput 2018, 14, 2084–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Kratz EG; Duke RE; Cisneros GA Long-range electrostatic corrections in multipolar/polarizable QM/MM simulations. Theor. Chem. Acc 2016, 135, No. 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Essmann U; Perera L; Berkowitz ML; Darden T; Lee H; Pedersen LG A smooth particle mesh Ewald method. J. Chem. Phys 1995, 103, 8577–8593. [Google Scholar]
- (97).Fang D; Chaudret R; Piquemal J-P; Cisneros GA Toward a deeper understanding of enzyme reactions using the coupled ELF/NCI analysis: application to DNA repair enzymes. J. Chem. Theory Comput 2013, 9, 2156–2160. [DOI] [PubMed] [Google Scholar]
- (98).Simon S; Duran M; Dannenberg J How does basis set superposition error change the potential surfaces for hydrogen-bonded dimers? J. Chem. Phys 1996, 105, 11024–11031. [Google Scholar]
- (99).Galano A; Alvarez–Idaboy JR A new approach to counterpoise correction to BSSE. J. Comput. Chem 2006, 27, 1203–1210. [DOI] [PubMed] [Google Scholar]
- (100).Lu T; Chen F Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem 2012, 33, 580–592. [DOI] [PubMed] [Google Scholar]
- (101).Johnson ER; Keinan S; Mori-Sánchez P; Contreras-García J; Cohen AJ; Yang W Revealing noncovalent interactions. J. Am. Chem. Soc 2010, 132, 6498–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Humphrey W; Dalke A; Schulten K VMD: visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- (103).The UniProt Consortium. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Armougom F; Moretti S; Poirot O; Audic S; Dumas P; Schaeli B; Keduas V; Notredame C Expresso: automatic incorporation of structural information in multiple sequence alignments using 3D-Coffee. Nucleic Acids Res. 2006, 34, W604–W608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Di Tommaso P; Moretti S; Xenarios I; Orobitg M; Montanyola A; Chang J-M; Taly J-F; Notredame C T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 2011, 39, W13–W17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Notredame C; Higgins DJ; Heringa J T-Coffee: A novel method for multiple sequence alignments. J. Mol. Biol 2000, 302, 205–217. [DOI] [PubMed] [Google Scholar]
- (107).Zhulidov PA; Bogdanova EA; Shcheglov AS; Vagner LL; Khaspekov GL; Kozhemyako VB; Matz MV; Meleshkevitch E; Moroz LL; Lukyanov SA Simple cDNA normalization using kamchatka crab duplex-specific nuclease. Nucleic Acids Res. 2004, 32, No. e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Petřek M; Otyepka M; Banáš P; Košinová P; Koča J; Damborský J CAVER: a new tool to explore routes from protein clefts, pockets and cavities. BMC Bioinf. 2006, 7, No. 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Jurcik A; Bednar D; Byska J; Marques SM; Furmanova K; Daniel L; Kokkonen P; Brezovsky J; Strnad O; Stourac J; et al. CAVER Analyst 2.0: analysis and visualization of channels and tunnels in protein structures and molecular dynamics trajectories. Bioinformatics 2018, 34, 3586–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Chovancova E; Pavelka A; Benes P; Strnad O; Brezovsky J; Kozlikova B; Gora A; Sustr V; Klvana M; Medek P et al. CAVER 3.0: A Tool for the Analysis of Transport Pathways in Dynamic Protein Structures, PLoS Comput. Biol, 2012, e1002708, CAVER 3.0: A Tool for the Analysis of Transport Pathways in Dynamic Protein Structures. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Pavelka A; Sebestova E; Kozlikova B; Brezovsky J; Sochor J; Damborsky J CAVER: algorithms for analyzing dynamics of tunnels in macromolecules. IEEE/ACM Trans. Comput. Biol. Bioinform 2016, 13, 505–517. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All simulations and analyses employed via third-party software are described and referenced in the Methodology section. The LICHEM software program is available at the Cisneros Research Group GitHub: https://github.com/CisnerosResearch/LICHEM.