

Oxidative stress in Alzheimer’s disease: Oxidative stress refers to an imbalance between the pro-oxidative and anti-oxidative states that are part of normal cell metabolism that results in the elevated production of reactive oxygen species (ROS) and free radicals. These products oxidize lipids, proteins, and DNA, significantly threatening the integrity of neurons (Praticò, 2008; Padurariu et al., 2013). Specifically, free radicals such as the superoxide anion (O2•–), hydroxyl radical (OH•), and non-radical molecules including hydrogen peroxide (H2O2) and singlet oxygen (1O2), are often produced in the mitochondria during oxidative phosphorylation and can enter the cytoplasm of the cells and causes damage outside of the mitochondria (Guo et al., 2013; Padurariu et al., 2013). After this step, there is a subsequent reduction in the cell’s endogenous-antioxidant potential along with inhibition of mitochondrial respiration, reduction of adenosine triphosphate (ATP) levels, mitochondrial bursting, Ca2+ dyshomeostasis, membrane damage, and subsequent production of insoluble aggregate which results in proteotoxicity and protein aggregation (Praticò, 2008; Guo et al., 2013; Padurariu et al., 2013). The brain is particularly vulnerable to the oxidative stress due to its high oxygen requirement and lipid-rich composition. Therefore, damage to the brain caused by oxidative stress has a high chance of impairing normal central nervous system processes and may result in progressive cell damage, neurodegeneration, and cell death (Guo et al., 2013). The term antioxidant often refers to a group of vitamins and synthetic, plant-derived, or inorganic substances that can inhibit the production of, promote the detoxification, or scavenge reactive oxidant species (Praticò, 2008).
Alzheimer’s disease (AD) is the most common form of dementia in older adults, and amyloid-β (Aβ) is thought to play a key role in its etiology (Allan Butterfield and Boyd-Kimball, 2018). Free-radical-mediated oxidative stress of neuronal lipids, proteins, DNA, and RNA is extensive in brain areas such as the hippocampus, frontal cortex, cerebellum, globus pallidus, and locus coeruleus in patients with AD; Aβ is abundant in these areas and is a known cause of this neurodegeneration (Wang and Michaelis, 2010; Padurariu et al., 2013; Allan Butterfield and Boyd-Kimball, 2018). An intriguing and complex relationship has been shown between Aβ and mitochondrial dysfunction, redox metal imbalance, failure of protein clearance mechanism, advanced glycation end-products, and calcium excitotoxicity that may further contribute to the oxidative stress in the brains of AD patients (Praticò, 2008; Padurariu et al., 2013; Ganguly et al., 2017; Allan Butterfield and Boyd-Kimball, 2018). The high number of mitochondria found in human neurons makes the brain particularly more vulnerable to ROS and free radical generation than other organs and tissues. As byproducts of normal cellular metabolism, ROS in low to moderate concentrations are thought to be necessary for brain development and function; however, excessive levels are threatening. This narrow range for safe ROS concentrations renders especially the brain vulnerable to oxidative stress and the neurodegeneration and cognitive decline that results from too much exposure. However, clinical trials for the use of antioxidants to protect against this damage have revealed only a slightly favorable benefit or no effect despite epidemiological studies showing that dietary intake of antioxidants lowers the incidence of AD (Ashok et al., 2022; Pritam et al., 2022).
Oxidative stress: a culprit in Alzheimer’s amyloidopathy: In addition to the brain, the sections of the central nervous system are also exposed to free radicals. The entorhinal cortex and CA1 region of the hippocampus are the most vulnerable areas of the brain in patients with AD, expressing high concentrations of antioxidants as well as genes involved in oxidative stress (Praticò, 2008; Wang and Michaelis, 2010). Thus, oxidative damage to these areas may result in reduced cell proliferation, impaired cellular remodeling capacity, altered neuronal plasticity, disrupted neurogenesis, impaired neurotransmission, and accumulation of toxic proteins such as Aβ due to the failure of proteasomal and lysosomal pathways (Wang and Michaelis, 2010). Free radicals have been found to oxidize prosthetic groups, residual amino acids, cross-links, and protein clumps, and to engage in the proteolysis of structural and enzymatic proteins. Polyunsaturated fatty acids undergo a process known as lipid peroxidation or auto-oxidation as a result of the presence of several double bonds in their structure, resulting in the formation of peroxides, ROS, and other reactive organic free radicals that lead to the creation of Aβ (Padurariu et al., 2013). Altogether, this chain of events offers a convincing explanation for oxidative stress-induced initiation of AD amyloidopathy.
According to the theory of free radicals, physiological aging can be viewed as a gradual and inevitable process that is at least partially generated through the accumulation of specific types of oxidative damage, in turn causing protein misfolding or unfolding (Ganguly et al., 2017). Interestingly, the simultaneous processing of ROS and aging promote the accumulation of advanced glycation end products, which in turn serve not only as proinflammatory molecules but also as potent neurotoxins (Padurariu et al., 2013). Other exogenous factors such as excessive caloric intake from high-calorie, high-fat, and processed food; infection and subsequent inflammatory states; environmental toxins, radiation exposure; consumption of tobacco or certain drugs; stress; smoking; alcohol; and unbalanced nutrition can disrupt the oxidant-antioxidant balance in the brain, resulting in over-production of ROS and free radicals leading to oxidative stress (Ganguly et al., 2017; Sharma and Kim, 2021). These ROS and free radicals can then connect to and alter the structure of neuronal fat molecules, changing the fluidity and permeability of membranes and interfering with membrane activities related to transport as well as barrier-like functions. These elevated levels of oxidant species can influence Aβ formation by increasing amyloid precursor protein (APP) levels, as well as Aβ processing by altering the activity and levels of key enzymes such as β-secretase and γ-secretase, furthering the intra-lysosomal induction of Aβ and supporting amyloid development (Padurariu et al., 2013; Ganguly et al., 2017; Allan Butterfield and Boyd-Kimball, 2018; Sharma and Kim, 2021; Tamagno et al., 2021). In fact, according to previous reports, the central nervous system and peripheral tissues of patients with AD exhibit higher levels of lipid peroxidation reacions that result in oxidation of the extracellular sites of glutamatergic N-methyl-D-aspartate receptor and Aβ-induced impairment of long-term potentiation and synaptic neurotransmission (Praticò, 2008; Guo et al., 2013; Padurariu et al., 2013). Specifically, the reactive lipid peroxidation byproduct 4-hydroxynonenal was reported to significantly alter the APP structure and function and decrease the viability of hippocampal neurons by disrupting Ca2+ homeostasis, reducing Na+/K+-ATPase activity, impairing the glucose transport system, and inhibiting choline acetyltransferase activity in vivo (Sharma and Kim, 2021). This results in the accumulation of free radicals and respiratory chain defects that consequently participate in the formation of excess free radicals, as well as extracellular Aβ deposits that induce local inflammatory processes and activate microglia, another potential source of ROS, and reduction in antioxidant defense mechanism, thus creating a vicious cycle (Allan Butterfield and Boyd-Kimball, 2018; Cheignon et al., 2018; Tamagno et al., 2021).
Oxidative stress: a consequence of Alzheimer’s amyloidopathy: Aβ has a pro-oxidant role that has been demonstrated both in vitro and in vivo, causing oxidative stress and creating positive feedback for levels of APP levels and its proteolytic pathway (Ganguly et al., 2017; Allan Butterfield and Boyd-Kimball, 2018). This in turn causes an increase in lipid peroxidation, alteration of proteins by the reactive lipid peroxidation byproducts such as 4-hydroxynonenal and acrolein, increases in carbonylic proteins, and mitochondrial oxidative damage from elevated ROS levels, resulting in reduced neuronal survival (Guo et al., 2013). Aβ also oxidizes and inhibits several neuronal and glial transporters including glutamine transporters, glucose transporters, GTP-coupled transmembrane signaling proteins, and polyamine transporters (Ganguly et al., 2017; Sharma and Kim, 2021). Loss of function in each of these can produce deleterious effects on neurons that include loss of cell potential, accumulation of excitotoxic glutamate, reduced glucose availability, deteriorated intracellular communication, increased neurotoxicity, and reduced cognitive function (Allan Butterfield and Boyd-Kimball, 2018; Sharma and Kim, 2021). Release of free fatty acids, protein oxidation, protein misfolding, Ca2+ dyshomeostasis, impaired mitochondrial function, autophagy failure, induction of apoptosis, formation of peroxynitrite, and neuroinflammatory response are additional effects of Aβ-associated oxidative stress that ultimately contribute to impaired neurotransmission, neurodegeneration, and cognitive failure resulting in the neuronal loss (Guo et al., 2013; Ganguly et al., 2017; Allan Butterfield and Boyd-Kimball, 2018; Simpson and Oliver, 2020; Sharma and Kim, 2021; Tamagno et al., 2021).
Consistent with this postulate, research on post-mortem brains of Alzheimer’s patients and AD transgenic mice has shown that Aβ and APP impair the mitochondrial transport of proteins and disrupt electron transport chains resulting in permanent and irreversible neuronal damage (Guo et al., 2013). Aβ not only promotes ROS production at the mitochondrial level but also limits the scavenging of ROS molecules by inhibiting the activity of mitochondrial superoxide dismutase. Aβ also binds and inhibits the activity of Aβ-binding alcohol dehydrogenase, contributing to a compromised free radical and ROS detoxification process. The ability of Aβ to bind iron is the most likely route by which it may induce oxidative stress by increasing BACE1 activity, which in turn promotes Aβ production in vitro (Tamagno et al., 2021). Fibrillar Aβ can bind to receptors causing advanced glycation end-products to induce oxidative stress and also can initiate an oxidative inflammatory response from activated microglia and astrocytes via induction of NLRP3 (NOD-, LRR- and pyrin domain-containing 3) and nuclear factor-κB pathway (Praticò, 2008; Padurariu et al., 2013; Allan Butterfield and Boyd-Kimball, 2018; Cheignon et al., 2018; Simpson and Oliver, 2020; Tamagno et al., 2021; Li et al., 2022). Additionally, the phosphorylation of tau protein is favored by the high amounts of Aβ oligomers. Senile plaques are created over time by the accumulation of Aβ oligomers in the extracellular space, whereas neuronal neurofibrillary tangles are created by hyperphosphorylated tau (Ganguly et al., 2017). Both lesions cause persistent inflammation and further oxidative stress reactions that ultimately result in irreversible damage, gradual cellular aging, and cell death (Simpson and Oliver, 2020; Li et al., 2022). Clinical manifestations of these cell-biologic events include gradual cognitive deterioration, dementia-like symptoms in the early stages, and full-blown AD (Praticò, 2008; Ashok et al., 2022; Pritam et al., 2022).
Antioxidants and Alzheimer’s disease: It is evident that Aβ production and accumulation are influenced by ROS, the generation of which also contributes to the self-perpetuation of Aβ. As a result, numerous promising techniques that are currently being evaluated in clinical trials involve the inhibition of Aβ-oligomerization or reducing ROS production through the design of multitargeted agents such as antioxidants (Praticò, 2008). The behavioral and amyloidotic phenotypes of animals have been shown to consistently positively be influenced by antioxidants such as vitamins and natural polyphenols by reducing oxidative stress, inhibiting Aβ aggregation and the associated inflammatory processes, and preventing neuronal loss both in vitro and in vivo (Praticò, 2008; Padurariu et al., 2013; Ganguly et al., 2017; Allan Butterfield and Boyd-Kimball, 2018; Simpson and Oliver, 2020). Reductions in Aβ levels, phosphorylated tau, mitochondrial dysfunction, and microglial activation, along with increased synaptic activity and improved cognition are important outcomes of these investigations, suggesting that antioxidant therapy may be beneficial for reducing and/or preventing AD progression (Praticò, 2008). However, these studies have called for future research (such as antioxidant therapy for elderly patients without AD) and clinical trials (such as antioxidant treatment for patients with AD) as results from clinical trials with antioxidants have only revealed slight favorable benefit or no effect, despite epidemiological studies showing that dietary intake of antioxidants lowers the incidence of AD (Ashok et al., 2022; Pritam et al., 2022). So far, clinical trials with curcumin and vitamin E have shown changes in pathological markers and improvement in cognitive decline in AD patients (Ashok et al., 2022). The main drawback is that administering a single oxidant at a higher concentration can be more harmful since it acts as a pro-oxidant. Less bioavailability, instability, co-dependency on another antioxidant, time-span of consumption, dose-dependent toxicity, age of people and associated cognitive decline, mode of administration, movement across the blood-brain barrier, hormone replacement therapy are some of the other shortcomings of antioxidant therapy in clinical trials (Ashok et al., 2022; Pritam et al., 2022).
Conclusion: It is clear that oxidative stress is a self-propagating phenomenon. Conversely, Aβ interacts with and exacerbates the various sources of oxidative stress including mitochondrial dysfunction, redox metal imbalances, advanced glycation end-products, and calcium excitotoxicity. However, uncertainty remains regarding whether Aβ-associated oxidative stress or oxidative stress-mediated accumulation of Aβ are primary or secondary to each other in the AD brain. Undoubtedly, oxidative stress and AD are intertwined and the intricate pathophysiology of AD points towards oxidative damage to neurons (Figure 1). A reduction in the protein’s breakdown rate owing to impaired or compromised proteasomal or lysosomal pathways, transcriptional activation, or rapid translation of a specific mRNA may result in the accumulation of a specific hazardous protein such as Aβ. A similar event may occur in post-translationally modified proteins resulting from changes in the cellular environment such as those observed in the redox status and kinase activity that may precede other conformational alterations, facilitating oligomerization and self-aggregation, and can be altered by ROS-responsive transcription factors and triggering various cell death pathways. Hence, the task of determining which process occurs first remains complicated. Overall, the association between ROS and AD raises the possibility that ROS plays a crucial role in the pathological process and that precisely targeted antioxidant therapy may be effective for the treatment of AD. Synergistic administration of multiple antioxidants can combat ROS-mediated oxidative stress and neuroinflammation simultaneously and can also improve bioavailability. Additionally, the use of nanotechnology for developing smaller antioxidant molecules can offer target-specific delivery and movement through blood-brain barrier.
Figure 1.
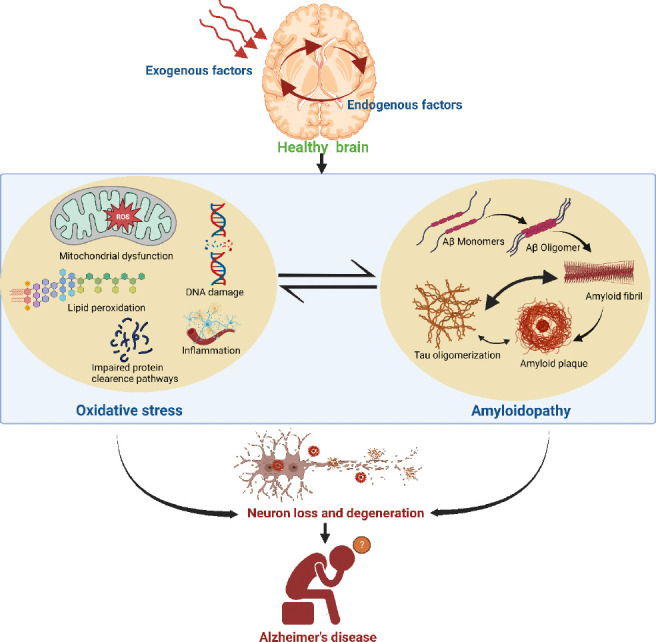
Schematic showing interdependency of oxidative stress and Alzheimer’s amyloidopathy in the brain of a patient with Alzheimer’s disease (AD).
The onset of close linkages between oxidative stress and amyloidopathy in the AD brain is marked by numerous exogenous (such as environmental changes, exposure to chemicals and radiation, and dietary habits) or endogenous (intracellular) causes. Oxidative stress is manifested by reactive oxygen species (ROS) formation, lipid peroxidation, and subsequent modification of proteins by reactive lipid peroxidation products which results in faulty protein clearance and accumulation of toxic proteins, inflammation, and DNA damage. However, amyloid-β (Aβ)-induced oxidative stress also results in the accumulation of Aβ monomers to create amyloid fibrils and plaque which may cause tau polymerization, protein oxidation, and subsequent alterations in mitochondrial function that further progressive neuronal degeneration and memory impairment. Created with BioRender.com.
The present work was supported by the National Research Foundation of Korea (NRF-2020R1A2C2007954) and the Korea Healthcare Technology R&D (HI21C1795) grants funded by the Korean government (to SRK).
Footnotes
C-Editors: Zhao M, Sun Y, Qiu Y; T-Editor: Jia Y
References
- 1.Allan Butterfield D, Boyd-Kimball D. Oxidative stress, amyloid-βpeptide, and altered key molecular pathways in the pathogenesis and progression of Alzheimer's disease. J Alzheimer's Dis. 2018;62:1345–1367. doi: 10.3233/JAD-170543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ashok A, Andrabi SS, Mansoor S, Kuang Y, Kwon BK, Labhasetwar V. Antioxidant therapy in oxidative stress-induced neurodegenerative diseases:role of nanoparticle-based drug delivery systems in clinical translation. Antioxidants. 2022;11:408. doi: 10.3390/antiox11020408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheignon C, Tomas M, Bonnefont-Rousselot D, Faller P, Hureau C, Collin F. Oxidative stress and the amyloid beta peptide in Alzheimer's disease. Redox Biol. 2018;14:450–464. doi: 10.1016/j.redox.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ganguly G, Chakrabarti S, Chatterjee U, Saso L. Proteinopathy, oxidative stress and mitochondrial dysfunction:cross talk in Alzheimer's disease and Parkinson's disease. Drug Des Devel Ther. 2017;11:797–810. doi: 10.2147/DDDT.S130514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo CY, Sun L, Chen XP, Zhang DS. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen Res. 2013;8:2003–2014. doi: 10.3969/j.issn.1673-5374.2013.21.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Y, Xia X, Wang Y, Zheng JC. Mitochondrial dysfunction in microglia:a novel perspective for pathogenesis of Alzheimer's disease. J Neuroinflammation. 2022;19:248. doi: 10.1186/s12974-022-02613-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Padurariu M, Ciobica A, Lefter R, Serban IL, Stefanescu C, Chirita R. The oxidative stress hypothesis in Alzheimer's disease. Psychiatr Danub. 2013;25:401–409. [PubMed] [Google Scholar]
- 8.Praticò D. Oxidative stress hypothesis in Alzheimer's disease:a reappraisal. Trends Pharmacol Sci. 2008;29:609–615. doi: 10.1016/j.tips.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 9.Pritam P, Deka R, Bhardwaj A, Srivastava R, Kumar D, Jha AK, Jha NK, Villa C, Jha SK. Antioxidants in Alzheimer's disease:current therapeutic significance and future prospects. Biology (Basel) 2022;11:212. doi: 10.3390/biology11020212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma C, Kim SR. Linking oxidative stress and proteinopathy in Alzheimer's disease. Antioxidants (Basel) 2021;10:1231. doi: 10.3390/antiox10081231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simpson DSA, Oliver PL. ROS generation in microglia:understanding oxidative stress and inflammation in neurodegenerative disease. Antioxidants (Basel) 2020;9:743. doi: 10.3390/antiox9080743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tamagno E, Guglielmotto M, Vasciaveo V, Tabaton M. Oxidative stress and beta amyloid in Alzheimer's disease. Which comes first:the chicken or the egg?Antioxidants (Basel) 2021;10:1479. doi: 10.3390/antiox10091479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X, Michaelis EK. Selective neuronal vulnerability to oxidative stress in the brain. Front Aging Neurosci. 2010;2:12. doi: 10.3389/fnagi.2010.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]