

Abstract

Semisynthetic modifications of natural products have bestowed us with many anticancer drugs. In the present work, a natural product, eugenol, has been modified synthetically to generate new anticancer agents. The final compounds were structurally confirmed by NMR, IR, and mass techniques. From the cytotoxicity results, compound 17 bearing morpholine was found to be the most active cytotoxic agent with IC50 1.71 (MCF-7), 1.84 (SKOV3), and 1.1 μM (PC-3) and a thymidylate synthase (TS) inhibitor with an IC50 of 0.81 μM. Further cellular studies showed that compound 17 could induce apoptosis and arrest the cell cycle at the S phase in PC-3 carcinoma. The docking study strongly favors compound 17 to be a TS inhibitor as it displayed a similar interaction to 5-fluorouracil. The in silico pharmacokinetics and DFT computational studies support the results obtained from docking and biological evaluation and displayed favorable pharmacokinetic profile for a drug to be orally available. Compound 17 was found to be a promising TS inhibitor which could suppress DNA synthesis and consequently DNA damage in prostate cancer cells.
Introduction
Natural products are reservoir of bioactive molecules with diverse and remarkable pharmacological potential.1 Natural products and their semisynthetic molecules have played a fascinating role in the advancement of cancer therapy.2,3 The semisynthesized natural products derivatives has been an advantage in terms of a better pharmacokinetics profile, diminishing toxicity with improved antiproliferative potential.4,5 Eugenol (4-allyl-2-methoxyphenol) is a natural aromatic pale yellow phenol with moderate water solubility and complete solubility in organic solvents.6,7 It possesses important antioxidant, anticancer, antiinflammatory, and antiviral potential.8,9 It is a nonmalignant and nonmutagenic molecule that exerts anticancer activity through β-catenin/E2F1/surviving downregulation, DNA synthesis inhibition, increased reactive oxygen species production, decreased mitochondrial membrane potential, and triggering apoptosis with cell cycle arrests.10−13
Cancer is the second leading cause of deaths globally and increasing tremendously with lung cancer at the top followed by breast and colorectal cancers.14 The most common treatments employed to fight the disease are chemotherapy, radiotherapy, and surgery while targeted therapies are receiving great attention currently. Among the targets, thymidylate synthase (TS), a folate-dependent enzyme required for DNA replication is attracting medicinal chemists in the field of oncology. This enzyme catalyzes methylation of deoxyuridine monophosphate (dUMP) to thymine monophosphate (dTMP) using CH2THF cofactor, which after phosphorylation results in thymidine triphosphate (dTTP) formation, a precursor for DNA synthesis.15 Inhibition of TS causes thymine deprivation resulting in thymidine triphosphate (dTTP) exhaustion, eventually leading to cell death, induction of apoptosis, and antiproliferation.16 Also, TS enzyme regulates various proteins involved in the apoptosis process.17,18 Owing to resistance, insensitivity, and toxicity of the available TS inhibitors,19,20 new chemotherapeutic agents with better efficacy and safety is an emerging area in the cancer therapy.
During the last decades, the design of 1,3,4-oxadiazole based derivatives as anticancer agents has increased remarkably in medicinal chemistry due to their wide mode of actions.21 For instance, compound I showed a telomerase inhibitory activity with IC50 2.30 μM,22 compound II as HDAC1 inhibitor with IC50 = 0.017 μM,23 compound III was found to be 8-fold (IC50 2.1 μM) more effective as FAK inhibitor than Cisplatin (IC50 8.6 μM),24 while compound IV was 15-fold more potent TS inhibitor with IC50 0.62 μM than pemetrexed.25 Moreover, conjugation of oxadiazole scaffold with bioactive natural products also represents a promising approach.26−28 Oxadiazole fused thymol (V),29 furanolabdane,30 and isosteviol derivatives31 have shown significant inhibitory activities with IC50 1.95 μM, GI50 0.08–0.34 μM, and IC50 0.95–3.36 μM, respectively, toward tested cancer cells (Figure 1). These abovementioned reports of eugenol and 1,3,4-oxadiazole with broad biological targets for antiproliferative effect encouraged us to conjugate natural product eugenol with 1,3,4 oxadiazole scaffold. The present work reports the synthesis of eugenol-based 1,3,4-oxadiazole-hybrids (5–17) and their cytotoxicity, docking, and computational studies as TS inhibitors.
Figure 1.

Rationale for the present work.
Results and Discussion
Synthesis and Characterization of Compounds
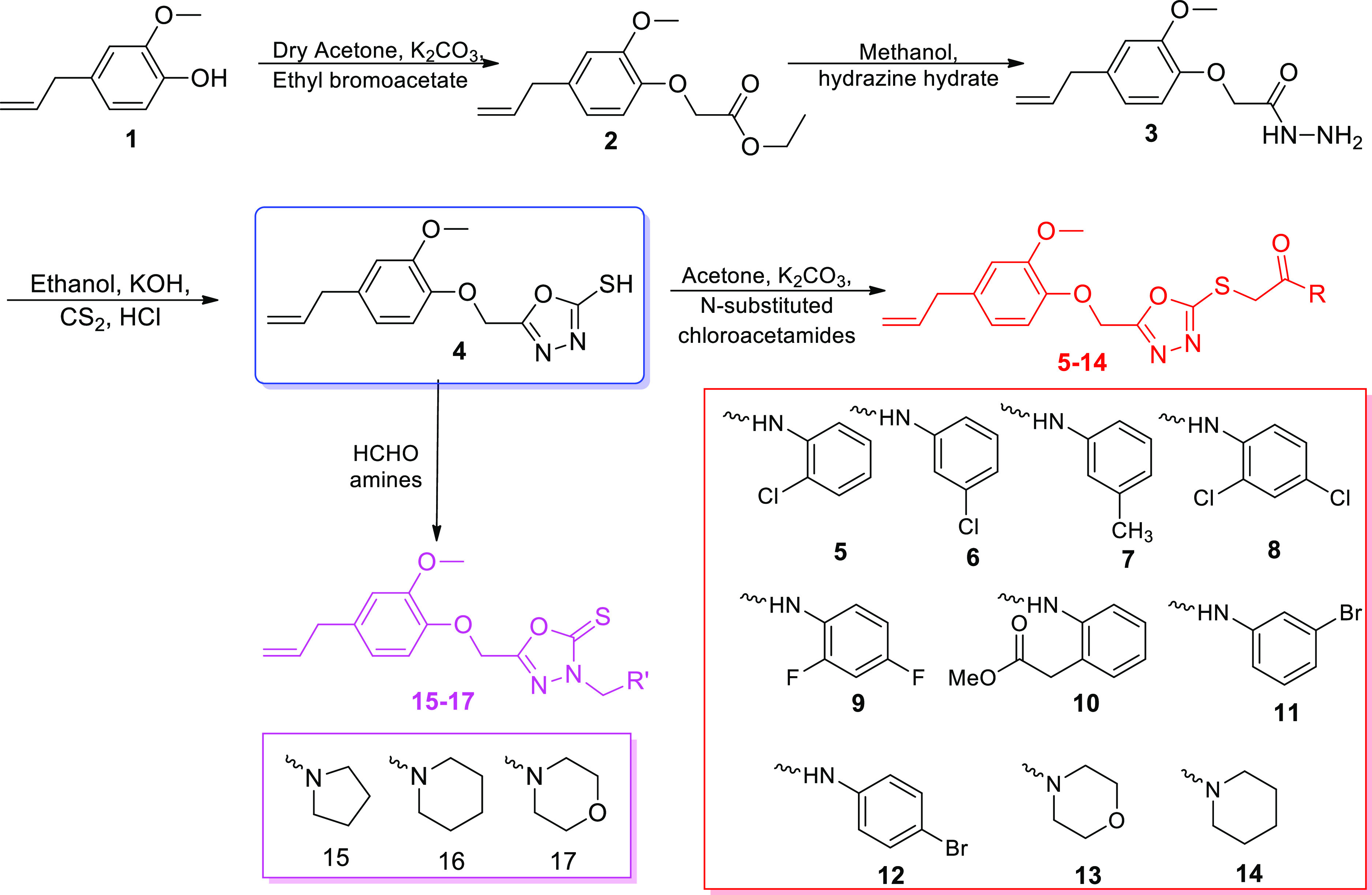
A natural product, eugenol 1, was used as the starting material for the synthesis target compounds 5–17 (Scheme 1). All the intermediates 2–4 were prepared and confirmed by comparing their melting point in the literature.8 Compound 1 was reacted with ethyl bromo acetate in the presence of potassium carbonate and anhydrous acetone to yield compound 2, which was treated with hydrazine hydrate in methanol to yield compound 3. Then, after the addition of carbon disulfide drop by drop in the basic alcoholic mixture of compound 3 at 0–5 °C, the mixture was refluxed for 12 h, and acidification using HCl yielded the key intermediate 4 in 86% yield, which was utilized for the obtainment of final compounds 5–17. Treatment of 4 with freshly prepared different N-substituted chloroacetamides in dry propanone afforded final hybrids 5–14 (68–90% yield) and with formaldehyde and different aliphatic amines in ethanol gave compounds 15–17 (70–85% yield).
Scheme 1. Synthetic Pathway for the Eugenol Derivatives (5–17).
Biological Evaluation
Antiproliferative Activity
The final eugenol compounds (5–17) were screened for antiproliferative activity against three human adenocarcinomas, namely, breast (MCF-7), prostate (PC-3), and ovarian (SKOV3) using the MTT method as previously described.32 The results are illustrated in Table 1. Among all the final compounds, 1,3,4-oxadiazole-Mannich base bearing morpholine heterocycle (17) was the most active cytotoxic agent with IC50 1.71, 1.84, and 1.1 μM, while doxorubicin exhibited IC50 1.74, 2.88, and 2.61 μM, against MCF-7, SKOV3, and PC-3, respectively. Also, compound 9 having fluoro-substituted thioacetamide group exhibited promising cytotoxicity with IC50 in the range 2.09–3.36 μM toward the tested cell lines. Against the breast cancer cells, compounds 8, 12, and 15 displayed significant cytotoxicity with IC50 < 10 μM, compounds 5, 6, and 11 were moderately cytotoxic with IC50 less than 25 μM and other compounds were found to be mild and inactive. Against ovarian and prostate cancer cells, compound 10 with the COOMe group displayed good sensitivity with IC50 8.74 and 7.07 μM, respectively; however, it was mildly cytotoxic against breast cancer cells. Besides, compound 15 possessing pyrrolidine ring in the Mannich base skeleton displayed good antiproliferation toward prostate carcinoma with IC50 10.01 μM. Compounds 5, 11, 12, 15, and 16 with IC50 in the range 14.09–26.33 μM and compounds 5, 6, 7, 8, 12, 13, and 16 with IC50 in the range 11.18–29.96 μM were found to be moderate in killing the ovarian and prostate cancer cells, respectively. The remaining compounds were either mild (IC50 > 50) or less active (IC50 > 100). From these data, it is clear that most of the eugenol derivatives (except 14) have the ability to hinder cancer cell proliferation.
Table 1. In Vitro Cytotoxicity of Eugenol Derivatives (5–17)a.
| half
maximal inhibitory concentration (IC50, μM) |
|||
|---|---|---|---|
| compounds | breast MCF7 | ovarian SKOV3 | prostate PC3 |
| 5 | 25.74 ± 3.52 | 15.73 ± 3.43 | 11.18 ± 2.72 |
| 6 | 16.33 ± 2.80 | 41.06 ± 5.27 | 16.98± 1.08 |
| 7 | 45.89 ± 3.80 | 65.4 ± 2.81 | 24.30 ± 1.53 |
| 8 | 10.74 ± 2.76 | 70.01 ± 2.42 | 29.96 ± 0.91 |
| 9 | 2.87±0.56 | 3.36±0.80 | 2.09±0.68 |
| 10 | 45.01 ± 2.92 | 8.74 ± 1.06 | 7.07 ± 3.90 |
| 11 | 15.21 ± 3.77 | 26.33 ± 3.09 | 51.09 ± 2.92 |
| 12 | 6.54 ± 1.42 | 14.09 ± 2.17 | 16.72 ± 4.10 |
| 13 | 38.92 ± 3.20 | 61.09 ± 3.84 | 20.27 ± 0.75 |
| 14 | 172.86 ± 1.36 | 181.46 ± 3.58 | 124.90 ± 0.36 |
| 15 | 9.55 ± 0.71 | 16.86 ± 1.64 | 10.01 ± 0.99 |
| 16 | 33.18 ± 2.04 | 19.54 ± 2.48 | 26.65 ± 0.86 |
| 17 | 1.71±0.95 | 1.84±0.27 | 1.1±0.07 |
| doxorubicin | 1.74 ± 0.34 | 2.88 ± 0.68 | 2.61 ± 0.23 |
Data represent the mean values ± standard deviation of three independent experiments.
In Vitro Thymidylate Synthase Activity
The cytotoxic hybrids (8, 9, 10, 12, 15, and 17) with IC50 ≤ 10 μM against the tested cell lines, were selected for the TS inhibition activity in order to recognize their mechanism as TS inhibitors. These compounds as shown in Table 2 significantly inhibited TS with IC50 1.01, 1.21, and 0.81 μM for compounds 9, 10, and 17, respectively, which supports the promising cytotoxicity of these compounds whereas pemetrexed inhibited TS with IC50 2.81 μM. Other compounds 12 and 15 displayed comparable TS repression to pemetrexed, while compound 8 was found to be a moderate TS inhibitor. These data suggest that the synthesized eugenol derivatives have the potential to suppress TS enzyme leading to cancer cell antiproliferation.
Table 2. In Vitro TS Activitya.
| compounds | IC50 (μM) |
|---|---|
| 8 | 3.67 ± 0.58 |
| 9 | 1.01±0.41 |
| 10 | 1.25 ± 0.35 |
| 12 | 2.93 ± 0.71 |
| 15 | 2.57 ± 0.31 |
| 17 | 0.81±0.21 |
| pemetrexed | 2.81 ± 0.31 |
IC50 values are the mean ± S.D. of three separate experiments.
Compound 17 Arrest Cell Cycle at S Phase
Cell cycle dysregulation is an important cause of cancer cells proliferation, therefore blocking the cell cycle is an effective strategy to prevent cell proliferation. Compound 17, the most potent cytotoxic compound on PC-3 cells and elicited highest TS inhibitory activity was selected to explore its cellular mechanism responsible for cancer cell antiproliferation. Exposure of PC-3 cells with compound 17 and the control were stained with propidium iodide at their pre calculated, IC50 for 48 h and their effects on cell cycle profile and apoptosis were analyzed by flow cytometry.33 As illustrated in Figure 2, compound 17 increased the percentage of cell population at the S phase of the cell cycle from 37.69 to 62.07%, compared to vehicle control. Such increase was accompanied by the significant reduction of cell population at G1 and G2 phases from 37.69 to 29.30 and 24.60 to 8.61%, respectively. These results suggested that compound 17 could arrest cell cycle at the S phase.
Figure 2.

Cell cycle distribution of compound 17 and control at their pre calculated IC50 for 48 h in prostate PC3 cells by PI staining using flow cytometry. One-way ANOVA was used to test for statistical difference (*p < 0.05).
Compound 17 Induced Cancer Cell Apoptosis in PC-3 Cells
To further assure the apoptotic ability of compound 17, a flow cytometric assay utilizing Annexin V-propidium iodide dual staining was performed, which differentiates between live, apoptotic (early and late), and necrotic cells. As shown in Figure 3, after 48 h of treatment of PC-3 cells with compound 17, a decrease in the percentage of live cells in green (R1) were noticed. Moreover, a significant increase in total apoptotic (early and late in purple and blue) cells was observed from 1.08 to 47.58% (R2 and R4) as a slight increase in necrotic cells to 3.56% (R1, red color) from 1.62%. This result indicates that compound 17 induces apoptosis in PC-3 cells.
Figure 3.

Apoptosis analysis of compound 17 and control at their pre calculated IC50 for 48 h in prostate PC3 cells by PI staining using flow cytometry. One-way ANOVA was used to test for statistical difference (*p < 0.05).
The outcomes of cell cycle and apoptosis studies were found to be in agreement with the reported literature.34,35 5-fluorouracil and other TS inhibitors arrest the S stage of the cycle in different cancer cells. In the present work also, compound 17 displayed significant TS suppression via arresting S stage and apoptosis, indicating that compound 17 could block thymidine triphosphate (dTTP) leading to controlled antiproliferation.
In Silico Physicochemical and Pharmacokinetics Studies
The development of new drugs has been a costly, risky and challenging venture with a low victory success rate. Most of the molecules, due to undesirable drawbacks and low efficacy in clinical trials do not reach the market. Therefore, in silico pharmacokinetic and toxicity prediction in early stages of drug development provides an idea about the effectiveness and success of the molecule therapeutically. The emergence of computational studies has led to optimization of pharmacokinetic and toxicity parameters resulting in drug discovery in an efficient manner. The physicochemical and pharmacokinetic properties of the newly synthesized hybrids (5–17) have been evaluated by Swiss ADME software.36 The fate of the synthesized molecules for successful drug depends upon certain rules such as Lipinski and Veber. Lipinski rules are hall mark in the innovation and discovery of a drug which states that a molecule must have MW < 500, lipophilicity (i log Po/w) < 5, hydrogen bond acceptor (HBA) below 10 and hydrogen bond donor (HBD) below 5. These rules further included molecule flexibility (nROTB) and polar surface area (PSA) less than 10 and 140 Å2, respectively. It can be observed from Table 3 that the synthesized molecules possess promising oral absorption and permeability as they did not show any Lipinski violations in respect of molecular weight, HBA, HBD, and lipophilicity. Also all the compounds except 10 were found to be flexible, suggesting that they did not have bioavailability problem. All the compounds possess high gastrointestinal absorption in the range 70.43–79.76%, except compound 10 which displayed low gastrointestinal absorption of 61.36%. All the molecules were found to be nonpermeable to the brain. These results suggested that the synthesized molecules have acceptable physicochemical and pharmacokinetics features, which are required for a molecule to be orally available.
Table 3. In Silico Physicochemical/Pharmacokinetic Properties of Eugenol Hybrids (5–17)d.
| Lipinski
parameters |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| compd. no. | MWa | HBAb | HBDc | i log Po/w | violations | nROTBe | TPSAf | % ABSg | BBBh | GI ABSi |
| 5 | 445.92 | 6 | 1 | 4.23 | 0 | 10 | 111.78 | 70.43 | no | high |
| 6 | 445.92 | 6 | 1 | 3.53 | 0 | 10 | 111.78 | 70.43 | no | high |
| 7 | 425.50 | 6 | 1 | 3.55 | 0 | 10 | 111.78 | 70.43 | no | high |
| 8 | 480.36 | 6 | 1 | 4.45 | 0 | 10 | 111.78 | 70.43 | no | high |
| 9 | 447.46 | 8 | 1 | 3.95 | 0 | 10 | 111.78 | 70.43 | no | high |
| 10 | 469.51 | 8 | 1 | 4.17 | 0 | 11 | 138.08 | 61.36 | no | low |
| 11 | 490.37 | 6 | 1 | 3.12 | 0 | 10 | 111.78 | 70.43 | no | high |
| 12 | 490.37 | 6 | 1 | 3.20 | 0 | 10 | 111.78 | 70.43 | no | high |
| 13 | 405.47 | 7 | 0 | 3.62 | 0 | 9 | 112.22 | 70.28 | no | high |
| 14 | 403.50 | 6 | 0 | 3.95 | 0 | 9 | 102.99 | 73.46 | no | high |
| 15 | 361.46 | 5 | 0 | 3.97 | 0 | 8 | 84.75 | 79.76 | no | high |
| 16 | 375.49 | 5 | 0 | 4.16 | 0 | 8 | 84.75 | 79.76 | no | high |
| 17 | 377.46 | 6 | 0 | 3.97 | 0 | 8 | 93.98 | 76.57 | no | high |
Molecular weight.
Hydrogen bond acceptor.
Hydrogen bond donor.
Partition coefficient.
Number of rotable bonds.
Topological PSA.
Absorption %.
Blood–brain barrier.
Gastrointestinal absorption.
The toxicity prediction of the synthesized hybrids (5–17) was done by available software tools (http://biosig.unimelb.edu.au/pkcsm/prediction) and is shown in Table 4. All the molecules were found to be non-mutagenic, noncarcinogenic, and safe with LD50 in the acceptable range of 2.46–2.82 mol/kg. Chronic toxicity was also found to be in the safer zone with no skin sensitization effect. However, these compounds were found to be hepatotoxic.
Table 4. Toxicity Studies of the Eugenol Hybrids (5–17).
| compds | AMES toxicity | LD50 (mol/kg) | oral rat chronic toxicity (log mg/kg b.w/day) | hepatotoxicity | skin sensitization |
|---|---|---|---|---|---|
| 5 | no | 2.52 | 0.987 | yes | no |
| 6 | no | 2.52 | 0.987 | yes | no |
| 7 | no | 2.46 | 1.21 | yes | no |
| 8 | no | 2.63 | 0.785 | yes | no |
| 9 | no | 2.50 | 0.916 | yes | no |
| 10 | no | 2.46 | 1.04 | yes | no |
| 11 | no | 2.54 | 0.977 | yes | no |
| 12 | no | 2.54 | 0.977 | yes | no |
| 13 | no | 2.71 | 1.834 | yes | no |
| 14 | no | 2.88 | 1.294 | yes | no |
| 15 | no | 2.79 | 1.08 | yes | no |
| 16 | no | 2.82 | 1.007 | yes | no |
| 17 | no | 2.622 | 1.273 | yes | no |
DFT Physicochemical Properties
The quantum theory approximation is one of the reliable computational methods in drug discovery. DFT/B3LYP/6-311G**(d,p) with TD-DFT measurements were done for eugenol derivatives 5–17. The difference between the charge separation in HOMO and LUMO of the molecules designates Fermi molecular orbitals, as depicted in Figure 4.
Figure 4.

HOMO and LUMO molecular orbital for compounds 5–17.
The electronic envelop was cumulated on the donor fragment, 4-allyl-1,2-dimethoxyphenyl fragments in HOMO orbital which was switched to LUMO orbital characterized by thiomethyl triazole and the substituted aromatic moieties for compounds 1–10, 13, 14, thiomethyl triazole and oxadiazole ring in compounds 11, 15–17; however, in compound 12, localization of the LUMO zone was distributed in similar manner to HOMO. DFT calculations were applied to calculate HOMO, LUMO, and energy gap (Eg) illustrated in Table 5.
Table 5. DFT Calculations of Eugenol Derivatives 5–17 Computed by the B3LYP and 6-311G** (d,p) Basis Set.
| no. | EHOMO (eV) | ELOMO (eV) | Eg (eV) | H (eV) | S (eV) | χ (eV) | IP (eV) | ω (eV) |
|---|---|---|---|---|---|---|---|---|
| 5 | –5.959 | –1.061 | 4.898 | 2.449 | 302.318 | –3.510 | 3.510 | 2.503 |
| 6 | –6.095 | –1.524 | 4.572 | 2.286 | 323.815 | –3.810 | 3.810 | 3.175 |
| 7 | –5.905 | –1.225 | 4.680 | 2.340 | 316.196 | –3.565 | 3.565 | 2.715 |
| 8 | –8.003 | –0.767 | 7.236 | 3.618 | 204.650 | –0.017 | 0.017 | 0.000 |
| 9 | –5.905 | –1.687 | 4.218 | 2.109 | 351.114 | –3.796 | 3.796 | 3.416 |
| 10 | –5.905 | –0.871 | 5.034 | 2.517 | 294.176 | –3.388 | 3.388 | 2.280 |
| 11 | –5.823 | –2.068 | 3.755 | 1.878 | 394.367 | –3.946 | 3.946 | 4.146 |
| 12 | –5.986 | –1.497 | 4.490 | 2.245 | 329.834 | –3.742 | 3.742 | 3.118 |
| 13 | –5.769 | –1.551 | 4.218 | 2.209 | 351.114 | –3.660 | 3.660 | 3.176 |
| 14 | –5.986 | –1.551 | 4.435 | 2.218 | 333.881 | –3.769 | 3.769 | 3.202 |
| 15 | –5.959 | –1.279 | 4.680 | 2.340 | 316.411 | –3.619 | 3.619 | 2.798 |
| 16 | –5.905 | –0.980 | 4.925 | 2.463 | 300.678 | –3.442 | 3.442 | 2.406 |
| 17 | –5.905 | –0.980 | 4.925 | 2.463 | 300.678 | –3.442 | 3.442 | 2.406 |
It was found that strong relationship exists between Eg and nature of the substituents such as 2–Cl–C6H5; 3–Cl–C6H5; 2–CH3–C6H5; 2,4-dichlore–C6H4; 2,4-difoloro–C6H4; morpholineyl; C6H11; 2–COOH–C6H4–; 2–Br–OCH3–C6H4; 4–Br–C6H4; pyrrolidinyl; and piperidinyl and morpholinomethyl and the Eg was found in the order, 3.755 (11) < 4.218 (9, 13) < 4.435 (14) < 4.490 (12) < 4.572 (6) < 4.680 (7, 15) < 4.898 (5) < 4.925 (16) < 4.925 (17) < 5.034 (10) < 7.23 (8). For chemical reactivity, parameters such as electronegativity (χ), chemical potential (IP), and electrophilicity index (ω) were calculated which correspond to more electrophilic (HOMO) and nucleophilic (LUMO) sites. The antioxidant potential of the molecules is inversely proportional to ionization potential,16 and we observed that all the synthesized eugenol derivatives possess low IP indicating that these molecules have good potential to scavenge free radicals.
Molecular Potential Maps
The presence of electron density and chemical reactivity of synthesized molecules toward nucleophilic or electrophilic sites of biological media can be distinguished by molecular electrostatic potential (MEP). The MEP of the synthesized molecules was generated by the same DFT parameters as discussed before. As shown in Figure 5, the red color signifies the electrophilic zone with negative potential, blue color as the nucleophilic zone and positive potential, while the green zone for a neutral site. It can be observed that the negative potential (red surfaces) surrounds the oxadiazole ring and positive potential (blue surfaces) was distributed around the molecular skeletons. These zones in MEPs were responsible in detection of the active site for the receptors.
Figure 5.

MEP for the eugenol derivatives (5–17).
Molecular Docking Study
The synthesized eugenol derivatives (5–17) were docked against TS protein (PDB 6QXG) to support out biological results. It has been reported that 5-fluorouracil (5-FU) interacts with the active site through GLY222, SER216, ASN226, ARG50, HIS256, ARG175, CYS195, GLY217, ASP218, ARG215, and ARG176 residues.30 The induce-fit-docking was applied to generate final poses by OPLS force field. The final pose was selected based on both lowest binding free energy (ΔG) and RMSD. The inhibition-constant (Ki) was generated for all the compounds 5–17, which must be in the range 0.1–1.0 μM, and inversely proportional to binding energy efficiency (Table 6). Our docking findings revealed that the compounds 5–17 can perceive the key amino acids in diverse ways such as hydrogen bonding, arene cations, and arene–arene interactions.
Table 6. Binding-Affinity Energies (kcal/mol) of Eugenol Derivatives (5–17) against TSa.
| no. | ΔG | RMSD | H. B | EInt. | Eele | Ki |
|---|---|---|---|---|---|---|
| 5FU | –8.10 | 2.19 | 435.10 | –16.66 | –12.39 | 0.94 |
| 5 | –7.52 | 1.53 | 26.24 | –17.01 | –7.86 | 1.98 |
| 6 | –7.48 | 1.84 | 6.83 | –16.56 | –8.75 | 1.54 |
| 7 | –7.56 | 3.52 | 13.84 | –13.42 | –8.03 | 2.16 |
| 8 | –7.68 | 2.61 | 21.41 | –16.67 | –7.21 | 1.73 |
| 9 | –7.75 | 1.71 | 38.28 | –16.21 | –7.32 | 1.94 |
| 10 | –7.82 | 3.69 | 37.42 | –6.71 | –7.25 | 1.81 |
| 11 | –7.59 | 2.16 | 17.01 | –13.90 | –8.19 | 1.43 |
| 12 | –7.76 | 2.66 | 13.18 | –12.18 | –9.71 | 1.49 |
| 13 | –7.52 | 2.87 | 65.71 | –14.14 | –8.31 | 1.56 |
| 14 | –7.74 | 1.69 | 10.64 | –16.29 | –7.34 | 0.16 |
| 15 | –7.20 | 1.38 | 57.63 | –16.47 | –9.53 | 0.94 |
| 16 | –6.90 | 2.41 | 53.95 | –9.61 | –8.42 | 1.02 |
| 17 | –7.89 | 4.26 | 95.13 | –10.23 | –7.41 | 1.98 |
Where ΔG: free binding energy of the ligand; RMSD: root-mean-square deviation; H.·B: H-bonding energy between protein and ligand; EInt.: binding affinity of H-bond interaction with receptor; and Eele: electrostatic interaction over the receptor.
As shown in Table 6 and Figure 6, all the compounds except 16 have nearly the same binding energy (ΔG in the range −7.20 to −7.89 kcal/mol), but lower than reference drug, 5-FU (ΔG −8.10 kcal/mol), and displayed interactions with the active site similar to 5FU, suggestion that the synthesized molecules interacted with TS protein analogues to 5FU as TS inhibitor. The most active compounds 9 and 17 were found to bind with the TS pocket via H-bond with Arg50 amino acid. Furthermore, compound 9 also formed other H-bond with Arg176 and π–π interaction with Trp109, whereas C=S of compound 17 formed strong H-bond with Arg50, Arg175, and π–π interaction with Arg215. Other compounds were stabilized in binding pocket of the vital TS backbone through binding with ASN226, CYS195, ASP218, ARG50, ARG175, ARG215, SER216, and ARG176. The docking interaction of compounds 9 and 17 strongly support their in vitro TS inhibitory activity with IC50 1.01 and 0.81 μM, respectively. From the above results, compound 17 emerged as a promising lead for a TS inhibitor.
Figure 6.

Docking poses of compounds 9, 17, and 5-FU
Conclusions
Eugenol-based new 1,3,4-oxadiazole incorporated N-substituted acetamide and Mannich bases as anticancer agents were prepared with moderate to good yield. In silico physicochemical and toxicity studies of the molecules showed that most of the molecules have drug likeness properties and found to be non-mutagenic, noncarcinogenic, and safe with LD50 in the acceptable range of 2.46–2.82 mol/kg. Cytotoxicity results concluded that compound 17 bearing a morpholine ring was found to be the most active which blocks proliferation of breast, ovarian, and prostate cancer cells with IC50 1.71, 1.84, and 1.1 μM, respectively, and TS inhibition effectively with IC50 of 0.81 μM. It induces S phase arrest due to S phase checkpoint activation and triggers apoptosis by irreparable DNA damage in PC3 cells.
Experimental Section
Materials and Methods
The solvents and other reagents required for the synthesis of target molecules were either purchased from Sigma-Aldrich (Merck Germany), Across (USA) or freshly prepared in the lab. NMR spectra, FT-IR, and mass spectra were recorded on a Bruker spectrometer in CDCl3 at 80 MHz, Thermo Scientific spectrophotometer (ATR method), and Thermo Scientific LCQ Fleet (LCF10605), respectively, while melting points were performed on a SMP40 machine which were uncorrected. Target molecules were analyzed for their elemental composition on LEECO Elementar Analyzer. Compounds 2–4 were prepared according to our previous reported work.8
General Procedure for Synthesis of Compounds 5–14
Compound 4 (2 mmol), anhydrous acetone (50 mL), and potassium carbonate (1.5 mmol) were refluxed for an hour and then cooled to 40–45 °C and added by different N-substituted chloroacetamides (1.1 mmol). Refluxing was continued for 6–12 h, while reaction monitoring was carried out by TLC silica gel 60 WF254S aluminum sheet. After consumption of the starting reactants, it was filtered, and the filtrate was concentrated and poured in water (50 mL) and extracted with ethyl acetate (50 mL). Recrystallization was employed to purify the compounds using petroleum ether/ethyl acetate or isopropyl alcohol or dichloromethane/cyclohexane.
2-((5-((4-Allyl-2-methoxyphenoxy)methyl)-1,3,4-oxadiazol-2-yl)thio)-N-(2-chlorophenyl) Acetamide (5)
Recryst. solvent: petroleum ether-ethylacetate; yield: 82%; mp 90–92 °C; FT-IR (cm–1): 2927.47, 1699, 1607, 1588, 1510, 1367, 1248, 1139, 1027, 753; 1H NMR: 3.36 (d, J = 6.4 Hz, 2H, Ar–CH2−), 3.89 (s, 3H, Ar–O–CH3), 4.08 (s, 2H, −S–CH2−), 4.61 (s, 2H, −O–CH2−), 5.00–5.25 (m, 2H, CH2=CH−), 5.73–6.23 (m, 1H, CH2=CH−), 6.66–6.83 (m, 3H, Ar–H), 7.40–7.50 (m, 4H, Ar–H), 9.14 (s, 1H, Ar–N–H); 13C NMR: 32.98 (S–CH2−), 39.84 (−CH2–Ar), 55.85 (Ar–O–CH3), 69.97 (Ar–O–CH2−), 112.54, 116.02, 120.96, 128.16, 130.48, 131.13, 132.45, 135.58, 137.25, 145.53, 149.68, 158.29, 164.56, 169.76 (−C=O). ESI (+ve): 446.50 [M + H]+. Elemental analyses for C21H20ClN3O4S (calcd): C, 56.56; H, 4.52; N, 9.42; S, 7.19. Found: 56.44; H, 4.54; N, 9.47; S, 7.17.
2-((5-((4-Allyl-2-methoxyphenoxy)methyl)-1,3,4-oxadiazol-2-yl)thio)-N-(3-chlorophenyl) Acetamide (6)
Recryst. solvent: petroleum ether-ethylacetate; yield: 69%, mp 88–90 °C, FT-IR (cm–1): 3287, 3009, 2929, 1673, 1596, 1535, 1510, 1480, 1458, 1418, 1392, 1282, 1247, 1220, 1161, 1024, 824, 803, 749, 731. 1H NMR: 3.34 (d, J = 6.4 Hz, 2H, Ar–CH2−), 3.84 (s, 3H, Ar–O–CH3), 4.09 (s, 2H, S–CH2−), 4.99–5.25 (m, 4H, CH2=CH–, O–CH2−), 5.72–6.13 (m, 1H, CH2=CH−), 6.64–7.01 (m, 6H, Ar–H), 8.25–8.36 (m, 1H, Ar–H), 9.14 (s, 1H, Ar–NH−); 13C NMR: 36.50 (S–CH2−), 39.92 (−CH2–Ar), 55.85 (Ar–OCH3), 61.81 (Ar–O–CH2–oxadiazole), 110.20, 112.78, 116.04, 116.66, 120.08, 120.70, 121.03, 124.43, 127.44, 135.94, 137.31, 144.52, 148.42, 164.93, 175.05 (C=O). ESI (+ve): 446.33 [M + H]+. Elemental analyses for C21H20ClN3O4S (calcd): C, 56.56; H, 4.52; N, 9.42; S, 7.19. Found: C, 56.48; H, 4.54; N, 9.39; S, 7.14.
2-((5-((4-Allyl-2-methoxyphenoxy)methyl)-1,3,4-oxadiazol-2-yl)thio)-N-(m-tolyl)acetamide (7)
Recryst. solvent: dichloromethane-cyclohexane; yield: 69%, mp 72–74 °C, 1H NMR: 2.29 (s, 3H, Ar–CH3), 3.38 (d, J = 6.4 Hz, Ar–CH2−), 3.90 (s, 3H, Ar–O–CH3), 4.09 (s, 2H, S–CH2), 4.63 (s, 2H, O–CH2−), 5.03–5.19 (m, 2H, CH2=CH−), 5.75–6.14 (m, 1H, CH2=CH−), 6.79–6.85 (m, 3H, Ar–H), 7.31–7.37 (m, 4H, Ar–H), 9.13 (s, 1H, Ar–NH−); 13C NMR: 17.71, 33.02, 39.91, 55.91, 69.92, 112.65, 115.93, 116.09, 121.01, 127.36, 128.35, 130.06, 131.55, 135.62, 137.33, 145.55, 149.72, 164.61, 170.37. ESI (+ve): 426.33 [M + H]+; elemental analyses for C22H23N3O4S (calcd): C, 62.10; H, 5.45; N, 9.88; S, 7.54. Found: C, 62.21; H, 5.48; N, 9.85; S, 7.56.
2-((5-((4-Allyl-2-methoxyphenoxy)methyl)-1,3,4-oxadiazol-2-yl)thio)-N-(2,4-di-chlorophenyl) Acetamide (8)
Recryst. solvent: isopropyl alcohol; yield: 84%, mp 96–98 °C, FT-IR (cm–1): 3264, 3064, 1689, 1645, 1596, 1506, 1481, 1211, 829, 800. 1H NMR: 3.33 (d, J = 6.4 Hz, 2H, Ar–CH2−), 3.87 (s, 3H, Ar–O–CH3), 4.07 (s, 2H, S–CH2−), 4.59 (s, 2H, −O–CH2−), 4.99–5.14 (m, 2H, CH2=CH−), 5.71–6.00 (m, 1H, CH2=CH=), 6.75 (brd, s, 3H, Ar–H), 7.35–7.54 (m, 3H, Ar–H), 9.15 (s, 1H, Ar–N–H); 13C NMR: 32.98, 39.93, 55.91, 70.08, 112.55, 116.09, 121.05, 128.65, 130.56, 131.35, 133.48, 137.27, 145.57, 150.69, 155.00, 161.37, 164.59, 169.51. ESI (+ve): 481.17 [M + H]+; 483.17 [M + 2 + H]+; elemental analyses for C21H19Cl2N3O4S (calcd): C, 52.15; H, 3.99; N, 8.75; S, 6.68. Found: C, 52.26; H, 4.02; N, 8.77; S, 6.65.
2-((5-((4-Allyl-2-methoxyphenoxy)methyl)-1,3,4-oxadiazol-2-yl)thio)-N-(2,4-diflorophenyl) Acetamide (9)
Recryst. solvent: petroleum ether-ethylacetate; yield: 72%, mp 124–128 °C, FT-IR (cm–1): 3341, 3063, 1689, 1505, 1446, 1399, 1293, 1210, 830, 800. 1H NMR: 3.41 (brs, 2H, Ar–CH2−), 3.92 (s, 3H, Ar–O–CH3), 6.72–7.33 (m, 6H, Ar–H), 8.82 (s, 1H, Ar–N–H); 13C NMR: 32.98, 38.07, 54.34, 63.74, 112.79, 114.74, 116.84, 121.16, 122.44, 127.43, 128.90, 130.15, 141.85, 145.03, 154.70, 158.53, 163.37. ESI (−ve): 446.08 [M – H]+; elemental analyses for C21H19F2N3O4S (calcd): C, 56.37; H, 4.28; N, 9.39; S, 7.17. Found: C, 56.45; H, 4.30; N, 9.36; S, 7.15.
Methyl-2-(2-((5-((4-allyl-2-methoxyphenoxy)methyl)-1,3,4-oxadiazol-2-yl)thio)acetamido) Benzoate (10)
Recryst. solvent: petroleum ether-ethylacetate; yield: 74%, mp 86–88 °C, FT-IR (cm–1): 3285, 2981, 1683, 1646, 1590, 1506, 1487, 1229, 1154, 996, 788; 1H NMR: 3.36 (d, J = 6.4 Hz, Ar–CH2−), 3.87–3.94 (m, 6H, Ar–O–CH3), 4.24 (s, 2H, S–CH2−), 5.02–5.27 (m, 4H, CH2=CH–, O–CH2−), 5.83–6.04 (m, 1H, CH2=CH−), 6.66–7.31 (m, 5H, Ar–H), 7.59 (t, J = 6.4 Hz, 1H), 8.50 (d, J = 8.0 Hz, 1H), 8.64 (s, 1H, ArNH); 13C NMR: 37.24, 39.81, 52.45, 55.79, 61.69, 112.65, 115.91, 116.52, 120.61, 123.27, 130.84, 134.57, 137.23, 140.57, 145.09, 150.15, 164.91, 168.24. ESI (+ve): 470.25 [M + H]+; elemental analyses for C23H23N3O6S (calcd): C, 58.84; H, 4.94; N, 8.95; S, 6.83. Found: C, 58.75; H, 4.96; N, 8.99; S, 6.85.
2-((5-((4-Allyl-2-methoxyphenoxy)methyl)-1,3,4-oxadiazol-2-yl)thio)-N-(3-bromophenyl) Acetamide (11)
Recryst. solvent: isopropyl alcohol; yield: 64%, mp 114–116 °C; FT-IR (cm–1): 3185, 2987, 1730, 1673, 1607, 1592, 1509, 1374, 1250, 1141, 1026, 965, 965, 800; 1H NMR: 3.33 (d, J = 6.4 Hz, Ar–CH2−), 3.83 (s, 3H, Ar–O–CH3), 4.03 (s, 2H, S–CH2−), 4.62–5.14 (m, 4H, CH2=CH–, O–CH2−), 5.71–5.97 (m, 1H, CH2=CH−), 6.57–6.81 (m, 3H, Ar–H), 7.18–7.79 (m, 4H, Ar–H), 9.5 (s, 1H, Ar–NH−); 13C NMR: 32.98 (S–CH2−), 39.83 (−CH2–Ar), 55.77 (Ar–OCH3), 61.45 (Ar–O–CH2–oxadiazole), 112.57, 115.87, 116.01, 120.97, 122.64, 126.63, 130.66, 130.94, 132.94, 135.60, 137.39, 145.11, 145.43, 149.02, 164.75. ESI (+ve): 490 [M + H]+; 492 [M + 2 + H]+; elemental analyses for C21H20BrN3O4S (calcd): C, 51.44; H, 4.11; N, 8.57; S, 6.54. Found: C, 51.51; H, 4.12; N, 8.54; S, 6.53.
2-((5-((4-Allyl-2-methoxyphenoxy)methyl)-1,3,4-oxadiazol-2-yl)thio)-N-(4-bromophenyl) Acetamide (12)
Recryst. solvent: isopropyl alcohol; yield: 90%, mp 128–130 °C; FT-IR (cm–1):3188, 2985, 2930, 1729, 1670, 1602, 1509, 1488, 1375, 1251, 1208, 1141, 1030, 964, 913, 798; 1H NMR: 3.42 (d, J = 6.4 Hz, 2H, Ar–CH2−), 3.95 (s, 3H, Ar–O–CH3), 4.12 (s, 2H, S–CH2−), 4.69 (s, 2H, O–CH2−), 5.07–5.31 (m, 2H, CH2=CH−), 5.86–6.08 (s, 1H, CH2=CH−), 6.75–6.96 (m, 3H, Ar–H), 7.26–7.74 (m, 4H, Ar–H), 9.19 (s, 1H, Ar–NH−); 13C NMR: 32.26 (S–CH2−), 39.96 (−CH2–Ar), 55.80 (Ar–OCH3), 61.64 (Ar–O–CH2–oxadiazole), 105.93, 112.70, 116.09, 116.69, 121.08, 123.42, 129.42, 123.81, 136.40, 137.32, 147.92, 150.10, 164.40. ESI (+ve): 490 [M + H]+; 492 [M+2 + H]+; elemental analyses for C21H20BrN3O4S (calcd): C, 51.44; H, 4.11; N, 8.57; S, 6.54. Found: C, 51.46; H, 4.11; N, 8.52; S, 6.52.
2-((5-((4-Allyl-2-methoxyphenoxy)methyl)-1,3,4-oxadiazol-2-yl)thio)-1-morpholinoethanone (13)
Recryst. solvent: dichloromethane-cyclohexane; yield: 70%, mp 84–86 °C, FT-IR (cm–1): 2970, 2928, 1627, 1589, 1514, 1464, 1385, 1279, 1220, 1167, 1069, 1025, 843, 802, 1H NMR: 3.38 (d, J = 6.4 Hz, Ar–CH2−), 3.72–3.90 (m, 11H, morpholine ring protons, Ar–O–CH3), 4.36 (s, 2H, S–CH2−), 5.03–5.27 (m, 4H, CH2=CH–, O–CH2−), 5.76–6.25 (m, 1H, CH2=CH−), 6.69–7.04 (m, 3H, Ar–H). ESI (+ve): 406.83 [M + H]+; C19H23N3O5S (calcd): C, 56.28; H, 5.72; N, 10.36; S, 7.91. Obsd: 56.20; H, 5.75; N, 10.33; S, 7.90.
2-((5-((4-Allyl-2-methoxyphenoxy)methyl)-1,3,4-oxadiazol-2-yl)thio)-1-(piperidin-1-yl)ethanone (14)
Recryst. solvent: dichloromethane-cyclohexane; yield: 64%, mp 82–84 °C, FT-IR (cm–1): 3280, 2984, 2930, 1670, 1602, 1509, 1486, 1376, 1251, 1210, 1141, 1030, 964, 914. 1H NMR: 1.18–2.89 (m, 10H, piperidine ring), 3.32 (d, J = 6.4 Hz, Ar–CH2−), 3.84 (brds, 5H, Ar–O–CH3, S–CH2−), 4.98–5.22 (m, 4H, CH2=CH–, O–CH2−), 5.70–6.19 (m, 1H, CH2=CH−), 6.64–6.98 (m, 3H, Ar–H). ESI (+ve): 404.25 [M + H]+; elemental analyses for C20H25N3O4S (calcd): C, 59.53; H, 6.25; N, 10.41; S, 7.95. Found: C, 59.41; H, 6.20; N, 10.35; S, 7.92.
General Procedure for the Synthesis of Compounds 15–17
Compound 4 (0.005 mol) was taken in a 100 mL clean round-bottom flask and then added 50 mL of ethanol. To the mixture, suitable secondary amine (0.006 mol) and formaldehyde (0.006 mol) were added and the reaction was refluxed for 6–8 h. When the reaction completed, the mixture was concentrated under vacuum and poured on to the cold ice water (50 mL) and products were isolated by extracting with ethyl acetate (25 mL × 2). Finally, crude products were crystallized by ethanol.
5-((4-Allyl-2-methoxyphenoxy)methyl)-3-(pyrrolidin-1-ylmethyl)-1,3,4-oxadiazole-2(3H) Thione (15)
Recryst. solvent: ethanol; yield: 65.46%, mp 134–136 °C, FT-IR (cm–1): 2933, 1507, 1417, 1259, 1216, 1140, 1028, 912, 847, 804. 1H NMR: 1.50–2.22 (m, 4H, pyrrolidine ring −CH2–CH2−), 2.35–2.59 (m, 4H, pyrrolidine ring, CH2–N–CH2), 3.30–3.70 (m, 4H, Ar–CH2–, N–CH2–N−), 3.88 (s, 3H, Ar–O–CH3), 5.07–5.18 (m, 4H, CH2=CH–, O–CH2−), 5.76–6.14 (m, 1H, CH2=CH−), 6.77–7.01 (m, 3H, Ar–H); 13C NMR: 24.42, 39.81, 44.97, 55.85, 61.96, 112.85, 116.03, 117.44, 120.71, 137.17, 144.95, 146.06, 150.31, 158.29, 160.80, 177.80. ESI (+ve): 362.25 [M + H]+; elemental analyses for C18H23N3O3S (calcd): C, 59.81; H, 6.41; N, 11.63; S, 8.87. Found: C, 59.78; H, 6.38; N, 11.60; S, 8.90.
5-((4-Allyl-2-methoxyphenoxy)methyl)-3-(piperidin-1-ylmethyl)-1,3,4-oxadiazole-2(3H)thione (16)
Recryst. solvent: ethanol; yield: 63.50%, mp 138–140 °C, FT-IR (cm–1): 2936, 1516, 1439, 1413, 1316, 1215, 1174, 1142, 1051, 805. 1H NMR: 0.95–1.28 (m, 6H, piperidine protons), 3.24–3.40 (m, 6H, piperidine protons, Ar–CH2−), 3.79–3.89 (brs, 3H (Ar–O–CH3) + 2H (−N–CH2−)), 5.0–5.19 (m, 4H, CH2=CH–, O–CH2−), 5.73–6.22 (m, 1H, CH2=CH−), 6.68–6.99 (m, 3H, Ar–H); ESI (+ve): 376.92 [M + H]+; elemental analyses for C19H25N3O3S (calcd): C, 60.78; H, 6.71; N, 11.19; S, 8.54. Found: C, 60.65; H, 6.73; N, 11.16; S, 8.52.
5-((4-Allyl-2-methoxyphenoxy)methyl)-3-(morpholinomethyl)-1,3,4-oxadiazole-2(3H)-thione (17)
Recryst. solvent: ethanol; yield: 68%, mp 144–146 °C, FT-IR (cm–1): 2907, 1512, 1465, 1417, 1342, 1260, 1225, 1142, 1058, 1027, 923, 808. 1H NMR: 3.35–3.43 (m, 2H, −Ar–CH2−), 3.72–3.92 (m, 11H, Ar–O–CH3, morpholine protons), 5.11–5.21 (m, 4H, CH2=CH–, O–CH2−), 5.77–6.27 (m, 1H, CH2=CH−), 6.71–7.03 (m, 3H, Ar–H), 13C NMR: 39.87, 47.35, 48.09, 55.93, 61.84, 62.04, 112.87, 114.79, 116.07, 116.74, 120.79, 123.63, 137.19, 139.08, 147.43, 160.00, 166.22, 178.78. ESI (+ve): 378.05 [M + H]+; elemental analyses for C18H23N3O4S (calcd): C, 57.28; H, 6.14; N, 11.13; S, 8.49. Found: 57.33; H, 6.16; N, 11.10; S, 8.48.
Antiproliferative Activity
The cytotoxicity study was performed on MCF-7, SKOV3, and PC-3 cancerous cells according to our published work by the MTT method. Doxorubicin was used as a positive control for comparison.30
In Vitro Thymidylate Synthase Enzymatic Assay
It was performed as per previously published work.30 Pemetrexed was used as a reference drug.
Cell Cycle Analysis
It was performed as per previously published work.30 The experimental method has been provided in the Supporting Information.
Apoptosis Analysis
It was performed as per previously published work.30 The experimental method has been provided in the Supporting Information.
Statistical Analysis
Results are presented as mean ± SD, performed in triplicate. One-way ANOVA was used to determine statistical significance (*p < 0.05).
DFT and Docking Studies
They were performed as previously published work.16 The experimental protocol is available in the Supporting Information.
Acknowledgments
The authors thank the Deanship of Scientific Research at King Khalid University for funding this work through large Groups (Project under grant number R.G.P.2/213/44). M.M.A., S.N., N.A., and N.M.A. are greatly thankful to Al-Baha University.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c00933.
NMR (1H and 13C) and mass spectra of final compounds and methods for biological, docking, and computational studies (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Huang M.; Lu J. J.; Ding J. Natural Products in Cancer Therapy: Past, Present and Future. Nat. Prod. Bioprospect. 2021, 11, 5–13. 10.1007/s13659-020-00293-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demain A. L.; Vaishnav P. Natural products for cancer chemotherapy. Microb. Biotechnol. 2011, 4, 687–699. 10.1111/j.1751-7915.2010.00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzobo K. The Role of Natural Products as Sources of Therapeutic Agents for Innovative Drug Discovery. Compr. Pharmacol. 2022, 2, 408–422. 10.1016/B978-0-12-820472-6.00041-4. [DOI] [Google Scholar]
- Lee K. H. Discovery and development of natural product-derived chemotherapeutic agents based on a medicinal chemistry approach. J. Nat. Prod. 2010, 73, 500–516. 10.1021/np900821e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaou K. C.; Pfefferkorn J. A.; Roecker A. J.; Cao G. Q.; Barluenga S.; Mitchell H. J. Natural Product-like Combinatorial Libraries Based on Privileged Structures. 1. General Principles and Solid-Phase Synthesis of Benzopyrans. J. Am. Chem. Soc. 2000, 122, 9939–9953. 10.1021/ja002033k. [DOI] [Google Scholar]
- Alam M. M. Synthesis and anticancer activity of novel Eugenol derivatives against breast cancer cells. Nat. Prod. Res. 2023, 37, 1632–1640. 10.1080/14786419.2022.2103809. [DOI] [PubMed] [Google Scholar]
- Al-Sharif I.; Remmal A.; Aboussekhra A. Eugenol triggers apoptosis in breast cancer cells through E2F1/survivin down-regulation. BMC Cancer 2013, 13, 600–610. 10.1186/1471-2407-13-600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohane S. H.; Chauhan A. J.; Fuloria N. K.; Fuloria S. Synthesis and in vitro antimycobacterial potential of novel hydrazones of Eugenol. Arabian J. Chem. 2020, 13, 4495–4504. 10.1016/j.arabjc.2019.09.004. [DOI] [Google Scholar]
- Kamatou G. P.; Vermaak I.; Viljoen A. M. Eugenol—From the Remote Maluku Islands to the International Market Place: A Review of a Remarkable and Versatile Molecule. Molecules 2012, 17, 6953–6981. 10.3390/molecules17066953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury P.; Barua A.; Roy A.; Pattanayak R.; Bhattacharyya M.; Saha P. Eugenol restricts Cancer Stem Cell population by degradation of β-catenin via N-terminal Ser37 phosphorylation-an in vivo and in vitro experimental evaluation. Chem.-Biol. Interact. 2020, 316, 108938. 10.1016/j.cbi.2020.108938. [DOI] [PubMed] [Google Scholar]
- Yoo C. B.; Han K. T.; Cho K. S.; Ha J.; Park H. J.; Nam J. H.; Kil U. H.; Lee K. T. Eugenol isolated from the essential oil of Eugenia caryophyllata induces a reactive oxygen species-mediated apoptosis in HL-60 human promyelocytic leukemia cells. Cancer Lett. 2005, 225, 41–52. 10.1016/j.canlet.2004.11.018. [DOI] [PubMed] [Google Scholar]
- Shin S. H.; Park J. H.; Kim G. C. The mechanism of apoptosis induced by Eugenol in human osteosarcoma cells. J. Korean Assoc. Oral Maxillofac. Surg. 2007, 33, 20–27. [Google Scholar]
- Abdullah M. L.; Hafez M. M.; Al-Hoshani A.; Al-Shabanah O. Anti-metastatic and anti-proliferative activity of Eugenol against triple negative and HER2 positive breast cancer cells. BMC Complementary Altern. Med. 2018, 18, 321–332. 10.1186/s12906-018-2392-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R. L.; Miller K. D.; Wagle N. S.; Jemal A. Cancer statistics, 2023. Ca-Cancer J. Clin. 2023, 73, 17–48. 10.3322/caac.21763. [DOI] [PubMed] [Google Scholar]
- Chon J.; Stover P. J.; Field M. S. Targeting nuclear thymidylate biosynthesis. Mol. Aspects Med. 2017, 53, 48–56. 10.1016/j.mam.2016.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam M. M.; Malebari A. M.; Nazreen S.; Neamatallah T.; Almalki A. S. A.; Elhenawy A. A.; Obaid R. J.; Alsherif M. A. Design, synthesis and molecular docking studies of thymol based 1,2,3-triazole hybrids as thymidylate synthase inhibitors and apoptosis inducers against breast cancer cells. Bioorg. Med. Chem. 2021, 38, 116136. 10.1016/j.bmc.2021.116136. [DOI] [PubMed] [Google Scholar]
- Hanauske A. R.; Eismann U.; Oberschmidt O.; Pospisil H.; Hoffmann S.; Hanauske-Abel H.; Ma D.; Chen V.; Paoletti P.; Niyikiza C. In vitro chemosensitivity of freshly explanted tumor cells to Pemetrexed is correlated with target gene expression. Invest. New Drugs 2007, 25, 417–423. 10.1007/s10637-007-9060-9. [DOI] [PubMed] [Google Scholar]
- Roukos D. H.; Katsios C.; Liakakos T. Genotype-phenotype map and molecular networks: a promising solution in overcoming colorectal cancer resistance to targeted treatment. Expert Rev. Mol. Diagn. 2010, 10, 541–545. 10.1586/erm.10.49. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Mitchell-Ryan S.; Raghavan S.; George C.; Orr S.; Hou Z.; Matherly L. H.; Gangjee A. Novel 5-Substituted Pyrrolo[2,3-d]pyrimidines as Dual Inhibitors of Glycinamide Ribonucleotide Formyltransferase and 5-Aminoimidazole-4-carboxamide Ribonucleotide Formyltransferase and as Potential Antitumor Agents. J. Med. Chem. 2015, 58, 1479–1493. 10.1021/jm501787c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson P. M.; Fazzone W.; LaBonte M. J.; Deng J.; Neamati N.; Ladner R. D. Novel opportunities for thymidylate metabolism as a therapeutic target. Mol. Cancer Ther. 2008, 7, 3029–3037. 10.1158/1535-7163.mct-08-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benassi A.; Doria F.; Pirota V. Groundbreaking Anticancer Activity of Highly Diversified Oxadiazole Scaffolds. Int. J. Mol. Sci. 2020, 21, 8692–8720. 10.3390/ijms21228692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q.-Z.; Zhang X.-M.; Xu Y.; Cheng K.; Jiao Q.-C.; Zhu H.-L. Synthesis, biological evaluation, and molecular docking studies of 2-chloropyridine derivatives possessing 1,3,4-oxadiazole moiety as potential antitumor agents. Bioorg. Med. Chem. 2010, 18, 7836–7841. 10.1016/j.bmc.2010.09.051. [DOI] [PubMed] [Google Scholar]
- Rajak H.; Agarawal A.; Parmar P.; Thakur B. S.; Veerasamy R.; Sharma P. C.; Kharya M. D. 2,5-Disubstituted-1,3,4-oxadiazoles/thiadiazole as surface recognition moiety: Design and synthesis of novel hydroxamic acid based histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 5735–5738. 10.1016/j.bmcl.2011.08.022. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Luo Y.; He L.-Q.; Liu Z.-J.; Jiang A.-Q.; Yang Y.-H.; Zhu H.-L. Synthesis, biological evaluation, and molecular docking studies of novel 1,3,4-oxadiazole derivatives possessing benzotriazole moiety as FAK inhibitors with anticancer activity. Bioorg. Med. Chem. 2013, 21, 3723–3729. 10.1016/j.bmc.2013.04.043. [DOI] [PubMed] [Google Scholar]
- Du Q. R.; Li D. D.; Pi Y. Z.; Li J. R.; Sun J.; Fang F.; Zhong W. Q.; Gong H. B.; Zhu H. L. Novel 1,3,4-oxadiazole thioether derivatives targeting thymidylate synthase as dual anticancer/antimicrobial agents. Bioorg. Chem. 2013, 21, 2286–2297. 10.1016/j.bmc.2013.02.008. [DOI] [PubMed] [Google Scholar]
- Popov S.; Semenova M. D.; Baev D. S.; Frolova T. S.; Shestopalov M. A.; Wang C.; Qi Z.; Shults E. E.; Turks M. Synthesis and cytotoxicity of hybrids of 1,3,4- or 1,2,5-oxadiazoles tethered from ursane and lupane core with 1,2,3-triazole. Steroids 2020, 162, 108698. 10.1016/j.steroids.2020.108698. [DOI] [PubMed] [Google Scholar]
- Ren J.; Wu L.; Xin W. Q.; Chen X.; Hu K. Synthesis and biological evaluation of novel 4β-(1,3,4-oxadiazole-2-amino)-podophyllotoxin derivatives. Bioorg. Med. Chem. Lett. 2012, 22, 4778–4782. 10.1016/j.bmcl.2012.05.059. [DOI] [PubMed] [Google Scholar]
- Alam M. M.; Elbehairi S. E. I.; Shati A. A.; Hussien R. A.; Alfaifi M. Y.; Malebari A. M.; Asad M.; Elhenawy A. A.; Asiri A. M.; Mahzari A. M.; Alshehri R. F.; Nazreen S. Design, synthesis and biological evaluation of new eugenol derivatives containing 1,3,4-oxadiazole as novel inhibitors of thymidylate synthase. New J. Chem. 2023, 47, 5021–5032. 10.1039/d2nj05711e. [DOI] [Google Scholar]
- Almalki A. S. A.; Nazreen S.; Malebari A. M.; Ali N.; Elhenawy A. A.; Alghamdi A. A.; Ahmad A.; Alfaifi S. Y. M.; Alsharif M. A.; Alam M. M. Synthesis and Biological Evaluation of 1,2,3-Triazole Tethered Thymol-1,3,4-Oxadiazole Derivatives as Anticancer and Antimicrobial Agents. Pharmaceuticals 2021, 14, 866–886. 10.3390/ph14090866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov M. E.; Pokrovsky M. A.; Kharitonov Y. V.; Shakirov M. M.; Pokrovsky A. G.; Shults E. E. Furanolabdanoid–based 1,2,4-oxadiazoles: Synthesis and cytotoxic activity. ChemistrySelect 2016, 1, 417–424. 10.1002/slct.201600042. [DOI] [Google Scholar]
- Liu C.-J.; Zhang T.; Yu S.-L.; Dai X.-J.; Wu Y.; Tao J.-C. Synthesis, cytotoxic activity, and 2D- and 3D-QSAR studies of 19-carboxyl-modified novel isosteviol derivatives as potential anticancer agents. Chem. Biol. Drug Des. 2017, 89, 870–887. 10.1111/cbdd.12910. [DOI] [PubMed] [Google Scholar]
- Alzahrani H. A.; Alam M. M.; Elhenawy A. A.; Malebari A. M.; Nazreen S. Synthesis, antiproliferative, docking and DFT studies of benzimidazole derivatives as EGFR inhibitors. J. Mol. Struct. 2022, 1253, 132265–132313. 10.1016/j.molstruc.2021.132265. [DOI] [Google Scholar]
- Nazreen S.; Almalki A. S. A.; Elbehairi S. E. I.; Shati A. A.; Alfaifi M. Y.; Elhenawy A. A.; Alsenani N. I.; Alfarsi A.; Alhadhrami A.; Alqurashi E. A.; Alam M. M. Cell Cycle Arrest and Apoptosis-Inducing Ability of Benzimidazole Derivatives: Design, Synthesis, Docking, and Biological Evaluation. Molecules 2022, 27, 6899–6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligabue A.; Marverti G.; Liebl U.; Myllykallio H. Transcriptional Activation and Cell Cycle Block Are the Keys for 5-Fluorouracil Induced Up-Regulation of Human Thymidylate Synthase Expression. PLoS One 2012, 7, e47318 10.1371/journal.pone.0047318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. Y.; Zhang T. J.; Kamara M. O.; Lu G. Q.; Xu H. L.; Wang D. P.; Meng F. H. Discovery of N-phenyl-(2,4-dihydroxypyrimidine-5-sulfonamido) phenylurea-based thymidylate synthase (TS) inhibitor as a novel multi-effects antitumor drugs with minimal toxicity. Cell Death Dis. 2019, 10, 532–547. 10.1038/s41419-019-1773-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- http://www.swissadme.ch/ (accessed on Jan 18, 2023) .
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.