

Abstract
In this issue of Developmental Cell, Murthy et al. identify AP-1 as a driver of oncogenic KRAS early tumor progression and demonstrate the distinct paths of transformation from two different cells of origin.
The best outcomes for lung cancer patients occur with early diagnosis and treatment with surgical resection or definitive radiation with or without adjuvant chemotherapy (Nasim et al., 2019). Recently, treatment with osimertinib, an EGFR antagonist, after surgical resection of mutant EGFR lung adenocarcinoma (LUAD) led to a substantial improvement in disease-free survival (Wu et al., 2020). Such studies highlight the need to understand the mechanisms that drive epithelial transformation so as to establish more precise, targeted therapies in early stage lung cancer settings.
In this context, the efforts to identify the cells of origin of lung cancers will be critical (Ferone et al., 2020). Candidates for cell of origin for LUAD include club cells in terminal bronchioles, alveolar type 2 pneumocytes (AEC2) in alveoli, and bronchioalveolar stem cells (BASCs) in bronchioalveolar duct junctions (Ferone et al., 2020). Key markers for these cells include Secretoglobin 1a1 (SCGB1a1, also known as Clara cell 10 kD protein [CC10] and Clara cell secretory protein [CCSP]) for club cells, surfactant protein C (SFTPC) for AEC2 cells and a combination of SFTPC and CC10 in a rare population of AEC2 cells, and a combination of SFTPC and CC10 for BASCs. Club cells and AEC2 cells have been demonstrated to generate adenocarcinoma (Sutherland et al., 2014; Xu et al., 2012). BASCs (Kim et al., 2005) as a cell of origin has been debated (Ferone et al., 2020). In this issue of Developmental Cell, Murthy and colleagues examine the epigenetic basis of club cell and AEC2 cell transformation by oncogenic KRAS-G12D (Murthy et al., 2022). Tamoxifen was administered to Sftpc-CreER;KrasLSL-G12D/+ and Scgb1a1-CreER;KrasLSL-G12D/+ mice to induce mutant KRAS-G12D expression and initiate transformation of AEC2 and club cells, respectively. Epigenetic states of developing tumors were assessed by ATAC-seq. In both club cell and AEC2 transformed cells, chromatin accessible sites were enriched in AP-1 transcription factor complex motif sites and AP-1 complexes containing FOSL1 were critical for the proliferation of transformed cells.
The authors then examined the accessible chromatin regions of fluorescence-activated cell sorting-isolated transformed club cells at 4, 10, 16, and 22 weeks after tamoxifen administration. A linear increase in open chromatin regions enriched for AP-1 binding motifs and decreased nucleosome occupancy of AP-1 motif centers correlated with increasing time after KRAS-G12D induction, suggesting that the activity of FOSL1 containing AP-1 complexes may be driving early tumor progression. Interestingly, as time increased, the general accessibility of chromatin regions bearing AP-1 motifs more closely those of 4-week-transformed AEC2-derived cells. scRNA-seq of 6-week-transformed club cells and normal lung cells identified several subpopulations of transformed club cells including club-like, three AEC2-like, and one pre-AEC2-like-1 cell population(s). Club-like cells were strongly enriched for “surfactant metabolism” genes with many of the cells expressing high levels of Sftpc mRNA. RNA velocity analysis suggested that club-like cells become AEC2-like cells. Principle component analysis of open chromatin regions containing AP-1 motifs demonstrated that normal and transformed club cells (4 weeks to 22 weeks after mutant KRAS induction) clustered together, distinct from AEC2, CC10+ AEC2 cells, and AEC2 transformed cells. Strikingly, despite the apparent transdifferentiation of transformed club cells toward AEC-like cells, they still maintained their club cell identity (Figure 1A). Transformed club cells were enriched for open chromatin sites common to club cells such as FOX and SOX family of transcription factors and Notch pathway target genes, HES1 and HEY1. Transformed AEC2 cells were enriched for sites common to AEC2 cells.
Figure 1. Tumor progression from cells of origin.
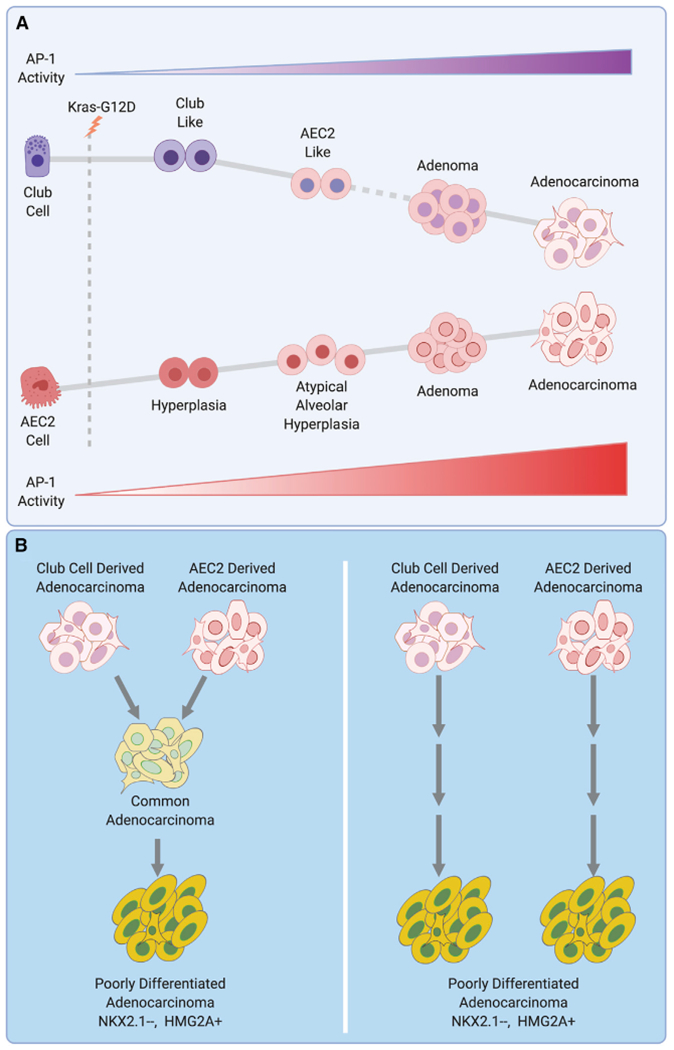
(A) A model of mutant Kras-induced tumor progression from club cell and alveolar type 2 pneumocyte (AEC2) is shown. Dashed line represents an assumption that the AEC2-like cells progress to adenomas. Cell states maintain the overall identity of their cell of origin. Activity of AP-1 transcription factor complex is low in normal club and AEC2 cells but progressively increases as cells transform toward adenocarcinoma. AP-1 activity is higher in AEC2-derived tumors.
(B) Two models of progression from differentiated adenocarcinomas to poorly differentiated adenocarcinoma. Left: adenocarcinomas derived from club and AEC2 cells converge on a common cell state that progresses to aggressive poorly differentiated adenocarcinoma with NKX2.1 loss and HMGA2 gain. Right: differentiated adenocarcinomas from club and AEC2 cells progress to poorly differentiated adenocarcinoma in parallel. The resulting aggressive tumors may still maintain characteristics of their respective cell of origin. Figure created with BioRender.com.
The progression of transformed club cells into AEC2-like states may account for the increased latency of tumor formation from club cells compared to tumors from AEC2 cells. The accessible chromatin states of club cell-derived tumors at 22 weeks resembled those of AEC2-derived lesions at 4 weeks. Late stage, aggressive, poorly differentiated club cell-derived tumors in KrasLSL-G12D/+; Trp53f/f mice with p53 loss gain the expression of a developmental master regulator, HMGA2, and lose the lung lineage marker, NKX2.1 (Sutherland et al., 2014), analogous to AEC2-derived poorly differentiated adenocarcinomas (Winslow et al., 2011). These observations raise the question whether tumor progression from club and AEC2 cells converge upon a common cell population that ultimately gives rise to a poorly differentiated adenocarcinoma or if tumors from the two lineages progress in parallel and maintain a semblance of their cell of origin identity (Figure 1B). If the former is true, then poorly differentiated adenocarcinomas from the two lineages may share common behaviors and therapeutic vulnerabilities. If the latter is true, then tumors from the two lineages would have divergent behaviors and therapeutic vulnerabilities. Most likely, the reality will be more complex than the binary paths hypothesized here. scATAC-seq and scRNA-seq at different time points from club cell and AEC2 cell-derived hyperplasia to poorly differentiated tumors may resolve this issue analogous to recent studies performed in KrasLSL-G12D/+ and KrasLSL-G12D/+;Trp53f/f mice treated with adenovirus-expressing Cre recombinase (LaFave et al., 2020; Marjanovic et al., 2020). Consistent with the trends of this study, regions of accessible chromatin regions with FOSL1 motifs were highest in late-stage and metastatic tumors (LaFave et al., 2020). These studies identified a highly plastic cell population (HPCS) from which other cell states were derived to increase intratumoral heterogeneity. The HPCS population most likely were derived from AEC2 cells, as AEC2 cells are the dominant cell of origin in this model (Sutherland et al., 2014), although a club cell origin cannot be excluded.
The ultimate validation of the importance of the findings by Murthy and colleagues will come when they lead to clinical benefits. Thus, a series of clinically relevant questions arise. Does transformation by other oncogenic drivers depend on the same mechanisms as reported here for mutant KRAS? Does the cell of origin influence metastatic behavior, treatment, prevention vulnerabilities, mechanisms of immune escape, or gender/racial susceptibilities? Can lung cancers be categorized by cell of origin along with histology? This study provides new therapeutic directions including the development of AP-1 complex inhibitors. Can AP-1 complex antagonists prevent recurrence after surgery or definitive radiation of early-stage mutant KRAS LUAD? Do AP-1 inhibitors have utility for AEC2 cell-derived atypical alveolar hyperplasia or adenocarcinoma in situ that often present as multifocal disease with poor response to chemotherapy? Can Notch pathway antagonists be utilized for early-stage mutant KRAS papillary tumors derived from club cells? The results of recent single cell sequencing studies (LaFave et al., 2020; Marjanovic et al., 2020) corroborate the low complexity of cell populations within preneoplastic and early neoplastic lesions identified in this study, in contrast to more complex cell populations in advanced cancers. These observations further reinforce what we already know–the greatest opportunity to cure lung cancer is still in its early stages.
Footnotes
DECLARATION OF INTERESTS
J.K. is on the Scientific Advisory Board for Sanofi Genzyme and had consulted for COTA, Inc. J.D.M. receives licensing fees from the National Institutes of Health and University of Texas Southwestern for distribution of human tumor and normal cell lines.
REFERENCES
- Ferone G, Lee MC, Sage J, and Berns A (2020). Cells of origin of lung cancers: lessons from mouse studies. Genes Dev. 34, 1017–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, Crowley D, Bronson RT, and Jacks T (2005). Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 121, 823–835. [DOI] [PubMed] [Google Scholar]
- LaFave LM, Kartha VK, Ma S, Meli K, Del Priore I, Lareau C, Naranjo S, Westcott PMK, Duarte FM, Sankar V, et al. (2020). Epigenomic State Transitions Characterize Tumor Progression in Mouse Lung Adenocarcinoma. Cancer Cell 38, 212–228 e213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marjanovic ND, Hofree M, Chan JE, Canner D, Wu K, Trakala M, Hartmann GG, Smith OC, Kim JY, Evans KV, et al. (2020). Emergence of a High-Plasticity Cell State during Lung Cancer Evolution. Cancer Cell 38, 229–246 e213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy PKL, Xi R, Arguijo D, Everitt JI, Kocak DD, Kobayashi Y, Bozec A, Vicent S, Ding S, Crawford GE, et al. (2022). Epigenetic basis of oncogenic-Kras mediated epithelialcellular proliferation and plasticity. Dev. Cell 57, 310–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasim F, Sabath BF, and Eapen GA (2019). Lung Cancer. Med. Clin. North Am 103, 463–473. [DOI] [PubMed] [Google Scholar]
- Sutherland KD, Song JY, Kwon MC, Proost N, Zevenhoven J, and Berns A (2014). Multiple cells-of-origin of mutant K-Ras-induced mouse lung adenocarcinoma. Proc. Natl. Acad. Sci. USA 111, 4952–4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winslow MM, Dayton TL, Verhaak RG, Kim-Kiselak C, Snyder EL, Feldser DM, Hubbard DD, DuPage MJ, Whittaker CA, Hoersch S, et al. (2011). Suppression of lung adenocarcinoma progression by Nkx2-1. Nature 473, 101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YL, Tsuboi M, He J, John T, Grohe C, Majem M, Goldman JW, Laktionov K, Kim SW, Kato T, et al. ; ADAURA Investigators (2020). Osimertinib in Resected EGFR-Mutated Non-Small-Cell Lung Cancer. N. Engl. J. Med 383, 1711–1723. [DOI] [PubMed] [Google Scholar]
- Xu X, Rock JR, Lu Y, Futtner C, Schwab B, Guinney J, Hogan BL, and Onaitis MW (2012). Evidence for type II cells as cells of origin of K-Ras-induced distal lung adenocarcinoma. Proc. Natl. Acad. Sci. USA 109, 4910–4915. [DOI] [PMC free article] [PubMed] [Google Scholar]