

Abstract
The receptor for advanced glycation end-products (RAGE) is a pattern recognition receptor that regulates inflammation, cell migration, and cell fate. Systemic lupus erythematosus (SLE) is a chronic multiorgan autoimmune disease. To understand the function of RAGE in SLE, we generated RAGE-deficient (Ager−/−) lupus-prone mice by backcrossing MRL/MpJ-Faslpr/J (MRL-lpr) mice with Ager−/− C57BL/6 mice. In 18-week-old Ager−/− MRL-lpr, the weights of the spleen and lymph nodes, as well as the frequency of CD3+CD4−CD8− cells, were significantly decreased. Ager−/− MRL-lpr mice had significantly reduced urine albumin/creatinine ratios and markedly improved renal pathological scores. Moreover, neutrophil infiltration and neutrophil extracellular trap formation in the glomerulus were significantly reduced in Ager−/− MRL-lpr. Our study is the first to reveal that RAGE can have a pathologic role in immune cells, particularly neutrophils and T cells, in inflammatory tissues and suggests that the inhibition of RAGE may be a potential therapeutic strategy for SLE.
Keywords: RAGE, lupus nephritis, systemic lupus erythematosus, neutrophil extracellular traps
1. Introduction
The receptor for advanced glycation end-products (RAGE) is a pattern recognition receptor belonging to the immunoglobulin superfamily[1]. Advanced glycation end-products (AGEs), members of the S100/calgranulin family, high mobility group box-1 (HMGB1), Mac-1, amyloid β sheet fibrils, complement C3a and CpG DNA oligonucleotides bind to RAGE[2]. By engaging with its ligands, RAGE activates mitogen-activated protein kinase and NF-kB, leading to the upregulation of inflammatory cytokines, cell migration, and proliferation[3–6]. RAGE is expressed in tissues, such as the lung, central nervous system, kidney, heart, vasculature, and immune cells[7, 8]. A soluble form of RAGE (sRAGE) acts as a decoy receptor that blocks RAGE and prevents its ligand binding[9–11]. RAGE is upregulated under inflammation and mediates proinflammatory responses upon alarmin recognition.
HMGB1 is included in damage-associated molecular patterns (DAMPs) and promotes an inflammatory immune response by actively or passively translocating from the nucleus to the extracellular space and binding with DNA[12, 13]. RAGE recognizes nucleic acids and HMGB1 and delivers them to intracellular endosomes where Toll-like receptors (TLRs) 7 and 9 are activated[14, 15]. The HMGB1-RAGE axis promotes proinflammatory cytokines, including type I interferon (IFN)[16]. Type 1 IFN is known as a crucial cytokine in systemic lupus erythematosus (SLE), a chronic multiorgan autoimmune disease characterized by antinuclear antibodies, especially anti–double-stranded DNA (anti-dsDNA) antibodies[17]. Lupus nephritis (LN) is a refractory complication of SLE that causes end-stage renal disease, resulting in lower survival rates and quality of life[18, 19].
Neutrophils are polymorphonuclear granulocytes that play an essential role in host defense against infection and maintain tissue homeostasis[20]. They are terminally differentiated within the bone marrow to yield short-lived cytotoxic cells[21], and hydrogen peroxide, chemokines, and lipid mediators at a tissue injury site direct the recruitment of neutrophils[22]. Neutrophils express RAGE, and the many DAMPs that are released from damaged and necrotic cells also function to directly modulate neutrophils[23–25]. Recent transcriptome analysis of lupus patients has revealed that a gradual enrichment of a neutrophil signature in whole blood was associated with subsequent active nephritis development within a few years[26]. The formation of neutrophil extracellular traps (NETs) is one of the significant antimicrobial mechanisms employed by neutrophils[27]. NETs are large, extracellular, web-like structures composed of cytosolic and granule proteins built on a scaffold of decondensed chromatin[27]. A pathological role for NET formation exists in conditions including autoimmunity such as vasculitis, rheumatoid arthritis, and lupus[28–30].
Circulating HMGB1 forms complexes with nucleosomes as well as immune complexes[16], and RAGE chaperones nucleic acid and immune complexes to endosomal TLR 7 and 9[15]. However, studies on the HMGB1-RAGE axis in murine lupus models and their biological effects are still contradictory. While some studies reported a pathophysiologic role of RAGE, other studies reported that RAGE deficiency did not significantly impact clinical severity in murine models of pristane-induced lupus[31]. Treatment with sRAGE, which sequesters ligands from RAGE, significantly improved LN in (NZB/NZW)F1 mice[32], while RAGE deficiency in B6. MRL-Faslpr/J (B6-lpr) mice exhibited aggravated lymphoproliferation and an exacerbated lupus phenotype[33].
To elucidate the discrepancy between those studies, we examined lupus-prone MRL/MpJ-Faslpr/J (MRL-lpr) mice deficient for RAGE and determined whether it ameliorates lupus-like features, including nephritis and serological abnormalities. MRL-lpr mice are characterized by the production of anti-DNA antibodies, lymphoproliferation, and glomerulonephritis. MRL-lpr mice have a loss-of-function mutation in Fas/CD95 that contributes to defective cell death[34, 35]. Since RAGE deficiency alone does not significantly affect lupus-like autoimmunity, including the pristane-induced model and B6-lpr[31, 33], we aimed to clarify the relevance of RAGE signaling in LN pathogenesis based on MRL-lpr lupus-prone animals.
In this study, we demonstrated that lymphoid organomegaly was alleviated by the deletion of RAGE, and a reduction in CD3+CD4−CD8− cells was prominent in RAGE-deficient MRL-lpr mice, contrary to a previous report[33]. Notably, we further revealed that the deletion of RAGE improved nephritis in this lupus model mouse through the inhibition of neutrophil infiltration and NET formation.
2. Materials and Methods
2.1. Generation of Ager−/− MRL-lpr mice
C57BL/6 mice deficient in RAGE (Ager−/−) were generated as described[36]. We then generated Ager−/− MRL-lpr mice by mating Ager−/− female C57BL/6 mice with male MRL-lpr mice (Jackson Laboratory, Bar Harbor, ME, USA) to yield heterozygous F1 offspring. RAGE+/− female F1 mice were further backcrossed with male MRL-lpr mice for 9–10 generations. Finally, RAGE+/− F9 or F10 mice were intercrossed to obtain Ager−/− female MRL-lpr littermates and Ager+/+ MRL-lpr littermates. Genotyping for RAGE deletion was conducted as previously described[36], and we performed PCR to determine the region of the Ager gene. PCR primer sequences were: 5’-CCAGAGTGACAACAGAGCAGAC-3’, 5’-GGTCAGAACATCACAGCCCGGA-3’, and 5’-CCTCGCCTGTTAGTTGCCCGAC-3’. Genomic DNAs from tails were extracted using DirectPCR Lysis Reagent (Mouse Tail) according to the manufacturer’s instruction (Viagen Biotech Inc, Los Angeles, CA, USA). These mice were housed under specific pathogen–free conditions in a 12-hour light/dark cycle, with free access to water and standard rodent chow (MF, Oriental Yeast, Tokyo, Japan), and further monitored until they were 18 weeks old. Plasma and twenty-four-hour urine samples were collected at 10, 14, and 18 weeks of age. Mice were sacrificed at 18 weeks of age, and tissue samples, including those from the spleen, lymph nodes, thymus, and kidneys, were obtained.
These methods were carried out under the approved guidelines. All experimental protocols were approved by the Animal Care and Use Committee of the Department of Animal Resources, Advanced Science Research Center, Okayama University (OKU-2017429).
2.2. Flow cytometry analysis
The spleen, mesenteric, and axillary lymph nodes from mice at 18 weeks of age were finely minced, and single-cell suspensions were obtained by filtering through a 70 μm mesh. For splenocytes, red blood cells were removed using red cell lysis buffer (BD Biosciences, San Jose, CA, USA). The cells (1×106) derived from the spleen and lymph nodes were incubated at 4 °C for 30 min in staining buffer (HBSS plus 3% horse serum and 0.02% sodium azide) with the relevant optimized amount of fluorochrome-conjugated antibodies: FITC–conjugated anti–CD4 (GK1.5), allophycocyanin (APC)–conjugated anti–CD8a (53–6.7), APCVio770–conjugated anti–CD8a (53–6.7), APC–conjugated anti–CD62L (MEL14-H2.100), phycoerythrin (PE)–conjugated anti–CD44 (IM7.8.1). All antibodies were purchased from Miltenyi Biotec (Bergisch Gladbach, Germany) except BV421–conjugated anti–CD3 (SK7) and PE–conjugated anti–CD8a (53–6.7), which were purchased from BD Biosciences; APC-Cy7–conjugated anti–CD11c (N418), which was purchased from Tonbo Biosciences (San Diego, CA, USA); and eFluor 450-conjugated anti–CD11b (M1/70) and PerCP-Cyanine5.5–conjugated anti-CD19 (eBio1D3), which were purchased from Thermo Scientific (Waltham, MA, USA). Apoptotic cells were detected using 7-AAD (BD Biosciences) and APC–conjugated Annexin V/binding buffer (BD Biosciences). Flow cytometry data were acquired on a MACSQuant Analyzer (Miltenyi Biotec) and analyzed using FlowJo software (FlowJo, LLC, Ashland, OR, USA). All procedures were performed according to the manufacturer’s instructions.
2.3. Measurement of urinary albumin excretions, plasma anti-dsDNA antibodies, HMGB1, RAGE, and cytokines
Urinary albumin excretions were measured using the turbidimetric immunoassay (FUJIFILM Wako Shibayagi Co., Shibukawa, Japan) and standardized by urine creatinine concentration. Plasma anti-dsDNA IgG titers, HMGB1 levels, and RAGE levels were measured with commercially available ELISA kits (anti-dsDNA IgG: FUJIFILM Wako Shibayagi, HMGB1: Shino-Test Co., Sagamihara, Japan, and RAGE: R&D Systems, Inc. Minneapolis, MN, USA, respectively). Cytokine analyses were performed using a Bio-Plex Pro™ Mouse Cytokine Th17 Panel A 6-Plex kit (Bio-Rad Laboratories, Inc., Hercules, CA, USA) with a Bio-Plex 200 Luminex machine according to the manufacturer’s protocol.
2.4. Histopathology and scoring
Kidney specimens were fixed with 10% buffered formalin and embedded in paraffin. Serial 2-μm sections were stained with hematoxylin and eosin (H&E) or periodic acid-Schiff (PAS) for histological examination by light microscopy. Activity and chronicity indices were scored according to previously described criteria[37]. Ten renal glomeruli and 10 selected tubular areas were examined in each mouse by CMIC Pharma Science Co., Ltd. (Hokuto, Japan).
2.5. Immunofluorescence staining
Kidney specimens were embedded in an optimum cutting temperature compound (Sakura Finetek Japan Co. Ltd., Tokyo, Japan) and immediately frozen in acetone cooled on dry ice. Frozen sections (4 μm thick) were stained with fluorescein-conjugated anti-mouse IgG (MP Biomedicals LLC., Santa Ana, CA, USA) or anti-mouse C3 (MP Biomedicals). The images were captured using an Olympus microscope. The brightness of each image file was analyzed using ImageJ[38]. Image files were converted to 16-bit grayscale. IgG or C3 indices were calculated using the following formula: [X (density) × positive area]/glomerular total area, where staining density is indicated by a number from 0 to 65535 in grayscale.
To evaluate neutrophil infiltration, the kidney sections were washed with PBS and immersed in 10% normal goat serum (Nichirei, Tokyo, Japan) containing 1% BSA for 30 min to block nonspecific binding. The sections were incubated overnight with a rat anti-Ly-6G antibody (1A8; BioLegend, San Diego, CA, USA) as the primary antibody at 4 °C and then with Alexa-568-labeled anti-rat IgG as the secondary antibody at room temperature for 30 min. Following staining with DAPI, the slides were mounted using a fluorescence mounting medium (Agilent, Santa Clara, CA, USA). Images were captured using an Olympus microscope, and Ly-6G-positive cells were counted using ImageJ. The TIFF file of each glomerulus was split into each RGB channel, a Gaussian filter was applied, and then the background was corrected using a rolling ball algorithm. Watershed separation was applied for the DAPI-positive area. The analyze particles algorithm counted the DAPI and Ly-6G double-positive area that was 30 μm2 or more and was recognized as Ly-6G-positive cells. The number of Ly-6G-positive cells was calculated in ten glomeruli per animal, and the mean number of positive cells per glomerulus was used for estimation.
NETs were stained as previously described[39]. In brief, the kidney sections were incubated overnight with an anti-Ly-6G antibody (1A8) in combination with an anti-Histone H3 (citrulline R2 + R8 + R17) (cit-Histone H3) antibody (Abcam plc. Cambridge, UK) as the primary antibodies at 4 °C and then with Alexa-568-labeled anti-rat IgG (Invitrogen) and Alexa-488-labeled anti-rabbit IgG (Invitrogen) as the secondary antibodies at room temperature. Following image capture using an LSM 780 confocal imaging system (Carl Zeiss, Oberkochen, Germany), the area of NETs in each glomerulus was evaluated using ImageJ. After measuring the area of each glomerulus, TIFF files were split into each RGB channel, a Gaussian filter was applied, and then the background was corrected using a rolling ball algorithm. Then, the area positive for both cit-Histone-H3 and Ly-6G in each glomerulus was measured, and the area percentages of cit-Histone-H3 and Ly6G double-positive sites to the glomerular area were calculated. We evaluated ten glomeruli per animal. The mean area percentages were used for estimation.
2.6. Immunoperoxidase Staining
Macrophage infiltration was analyzed as previously described[39]. In brief, the kidney sections were incubated with a monoclonal murine monocyte/macrophage antibody (F4/80; Abcam) as the primary antibody, followed by an HRP-conjugated goat anti-rat IgG antibody (Merck KGaA, Darmstadt, Germany) as the secondary antibody. The number of F4/80-positive cells was calculated in ten glomeruli per animal, and the mean number of positive cells per glomerulus was used for estimation.
2.7. Statistical analysis
Data were analyzed by JMP for Windows, version 9.0.2 (SAS Institute Inc., Cary, NC, USA). The data presented are the median (interquartile). For comparisons between two groups, the Mann-Whitney U test was employed if the distribution was non-normal; if it had a normal distribution, the Student’s t-test or Welch’s t-test (in case of unequal variances) would have been used instead. For plasma RAGE or cytokines analysis using the 6-plex Bio-plex kit, concentrations less than the limit of detection (LOD) were given a value of LOD/√2 for statistical analysis and plotting[40, 41]. The urine albumin/Cr ratios or anti-dsDNA IgG titers were compared between 14 and 18 weeks in each group using the Wilcoxon signed-rank sum test. The tests were two-tailed, and the threshold for significance was p<0.05. For the 6-plex Bio-Plex kit, statistical significance was determined at 0.05/6 using the Bonferroni correction to adjust for multiple testing. Each figure legend describes the statistical analysis method used.
3. Results
3.1. RAGE deletion attenuates splenomegaly and lymph node enlargement and alters the composition of immune cells in lymphoid tissue.
To determine the role of RAGE in lupus, we generated Ager−/− MRL-lpr mice and Ager+/+ MRL-lpr control mice. The complete deletion of RAGE was confirmed at the protein level (Supplementary Figure 1). It is well known that MRL-lpr mice develop lymphadenopathy and splenomegaly[42]; Ager−/− MRL-lpr mice showed a significantly smaller spleen and lymph nodes than Ager+/+ MRL-lpr mice at 18 weeks of age; an approximately 50% reduction was observed (Figure 1A, Table 1). The ratios of the spleen to body weight showed consistent results (Table 1). The number of splenocytes also decreased in Ager−/− MRL-lpr mice compared to Ager+/+ MRL-lpr mice at 18 weeks of age (Figure 1B).
Figure 1. Lymphoid tissue organomegaly and the composition of inflammatory cells at 18 weeks of age.
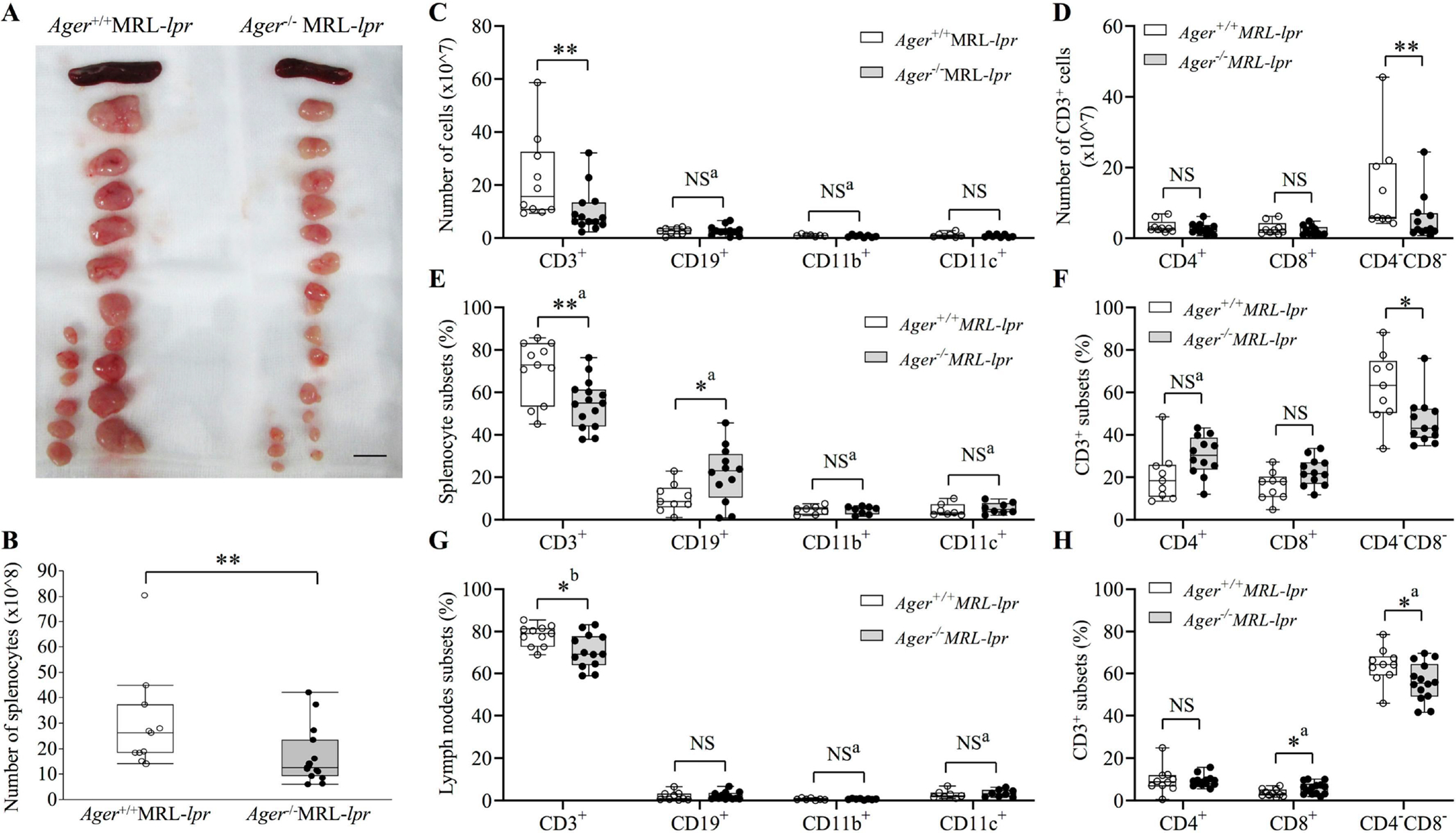
(A) Representative image of spleens and lymph nodes. The bar indicates 1 cm. (B) The total number of splenocytes Ager+/+ MRL-lpr (n = 11), and Ager−/− MRL-lpr (n = 15). Student’s t-test. **p < 0.01. (C-F) Comparisons of the absolute numbers or proportions of splenocyte subsets (C, E) and the absolute numbers or proportions of CD3+ cells in the spleen (D, F). The proportions indicate the percentages in total splenocytes. (G) Comparisons of the proportions of lymph node subsets. (H) Comparisons of the proportions of CD3+ cells in lymph nodes. The proportions indicate the percentages in total lymph node cells. (C-H) CD3+ cells were evaluated in 10 Ager+/+ MRL-lpr and 14 Ager−/− MRL-lpr mice. CD19+ cells were evaluated in 9 Ager+/+ MRL-lpr and 12 Ager−/− MRL-lpr mice. CD11b+ or CD11c+ populations were compared between 7 Ager+/+ MRL-lpr and 8 Ager−/− MRL-lpr. aStudent t-test; bWelch’s t-test. The Mann‒Whitney U test was used if not stated otherwise. *P < 0.05; **P < 0.01. NS, not significant.
Table 1.
Body and organ weights at 18 weeks of age
| Ager+/+ MRL-lpr | Ager −/− MRL-lpr | |
|---|---|---|
| (n = 9) | (n = 9) | |
|
| ||
| Body weighta | 41 (38–43) | 41 (37–43) |
| Rt. Kidney**a, b | 0.24 (0.23–0.3) | 0.2 (0.19–0.22) |
| Lt. kidney**c | 0.24 (0.23–0.29) | 0.2 (0.2–0.22) |
| Spleen***d | 0.6 (0.45–0.65) | 0.3 (0.25–0.32) |
| Spleen (%body weight)**d | 1.4 (0.99–1.8) | 0.77 (0.6–0.84) |
| Thymusc | 0.09 (0.07–0.12) | 0.08 (0.05–0.1) |
| Axially LN**d | 0.94 (0.63–1.2) | 0.4 (0.25–0.53) |
| Neck LN***a | 1.2 (1–1.4) | 0.58 (0.32–0.61) |
| Abdominal LN***c | 2.8 (2.7–3.1) | 1.3 (1–1.5) |
| Total LN***c | 4.9 (4.5–5.8) | 2.1 (1.7–2.6) |
Nine animals in each group were evaluated. Total lymph nodes (LNs) are a combination of peritoneal, cervical, and axillary LNs. The units are expressed in grams.
The MannWhitney U test
One value is missing in Ager −/−
Student’s t-test
Welch’s t-test.
p < 0.05
p < 0.01
p < 0.001.
We next investigated the changes in the composition of immune cells in lymphoid organs to elucidate the effect of RAGE deletion on the immunophenotype. Flow cytometry analysis of the spleen revealed that the absolute number and proportion of CD3+ T cells significantly decreased in Ager−/− MRL-lpr mice compared to Ager+/+ MRL-lpr mice in the spleen at 18 weeks of age (Figure 1C, E). The proportion of CD19+ B cells increased in Ager−/− MRL-lpr mice, and the absolute numbers were comparable between the two groups (Figure 1C, E). Among CD3+ T cells, only CD3+CD4−CD8− T cells were decreased by the deletion of RAGE in terms of both absolute number and proportion in the spleen (Figure 1D, F). We further examined memory T-cell subsets in CD4-positive or CD8-positive T cells, but there were no significant differences in the proportions of naïve, central memory, and effector memory T cells between the two groups (Supplementary Figure 2A–B). In lymph nodes, the proportions of CD3+ T cells decreased, and CD3+CD4−CD8− T cells decreased significantly in Ager−/− MRL-lpr mice compared with Ager+/+ MRL-lpr mice (Figure 1G–H). Naïve, central memory, and effector memory T cells were not altered by RAGE deletion (Supplementary Figure 2C–D).
Since RAGE limits apoptosis in particular conditions[43–45], we then investigated apoptosis of CD3+CD4−CD8− T cells and whether programmed cell death alteration was induced by RAGE deficiency. The proportions of annexin V-positive and 7-AAD-negative apoptotic CD3+CD4−CD8− cells in the spleen and lymph nodes were not different between Ager−/− and Ager+/+ MRL-lpr mice (Supplementary Figure 3A–B). Necrotic and all dead cells among CD3+CD4−CD8− cells were also not altered by RAGE deletion (Supplementary Figure 3 C–F). In Ager−/− MRL-lpr mice, the reduction in CD3+ cells, especially CD3+CD4−CD8− T cells, may significantly improve lymphoid organomegaly. The reduction in this population in RAGE-deficient animals was not suggested to be induced by the alteration in apoptosis.
Anti-dsDNA antibody is a hallmark of human lupus pathogenesis and was detected in MRL-lpr lupus-prone mice at around 4–6 weeks of age[46, 47]. While Ager+/+ MRL-lpr control mice develop anti-dsDNA starting from 14 weeks, the deletion of RAGE did not lead to lower anti-dsDNA antibody titers till 18 weeks of age (Supplementary Figure 4A). While the plasma levels of several cytokines, such as IFN-γ and IL-1β, tended to increase in the Ager−/− MRL-lpr group, the plasma levels of HMGB1 and IL-10 tended to decrease; however, there were no statistically significant differences (Supplementary Figure 4B and 4C).
3.2. Attenuation of nephritis in RAGE-deficient MRL-lpr mice
LN is an example of an immune complex-mediated manifestation in SLE and is also characterized by cell proliferation in the kidney[19]. LN occurs in MRL-lpr mice, which exhibit urinary protein from approximately 3 months of age[48]. In our analysis, urine albumin excretion significantly increased from 14 to 18 weeks of age in Ager+/+ MRL-lpr mice, and the amount of urine albumin was significantly lower in Ager−/− MRL-lpr mice than in Ager+/+ MRL-lpr mice at 18 weeks of age (Figure 2A). The weights of both kidneys at 18 weeks of age were significantly heavier in Ager+/+ MRL-lpr mice than in Ager−/− MRL-lpr mice (Table 1). Although the National Institutes of Health scoring system for LN was developed many years ago[37], the latest classification criteria for LN are still based on this system[49], and we also utilized the activity index and the chronicity index to report the extent of the overall activity of nephritis in this study. At 18 weeks of age, the activity index was attenuated in Ager−/− MRL-lpr mice compared to Ager+/+ MRL-lpr mice (Figure 2B). Among the components of the activity index, glomerular cell proliferation, karyorrhexis, and fibrinoid necrosis were especially alleviated, although no significant differences in cellular crescents and interstitial inflammation were found (Supplementary Figure 5A). For the chronicity index, both groups had a score of 0. Representative images of light and electron microscopy are shown in Figure 2C and Supplementary Figure 5B. These results indicate that RAGE deletion improved proteinuria via the attenuation of glomerular pathologies such as cell proliferation and necrosis.
Figure 2. Attenuation of nephritis in Ager−/− MRL-lpr mice at 18 weeks of age.
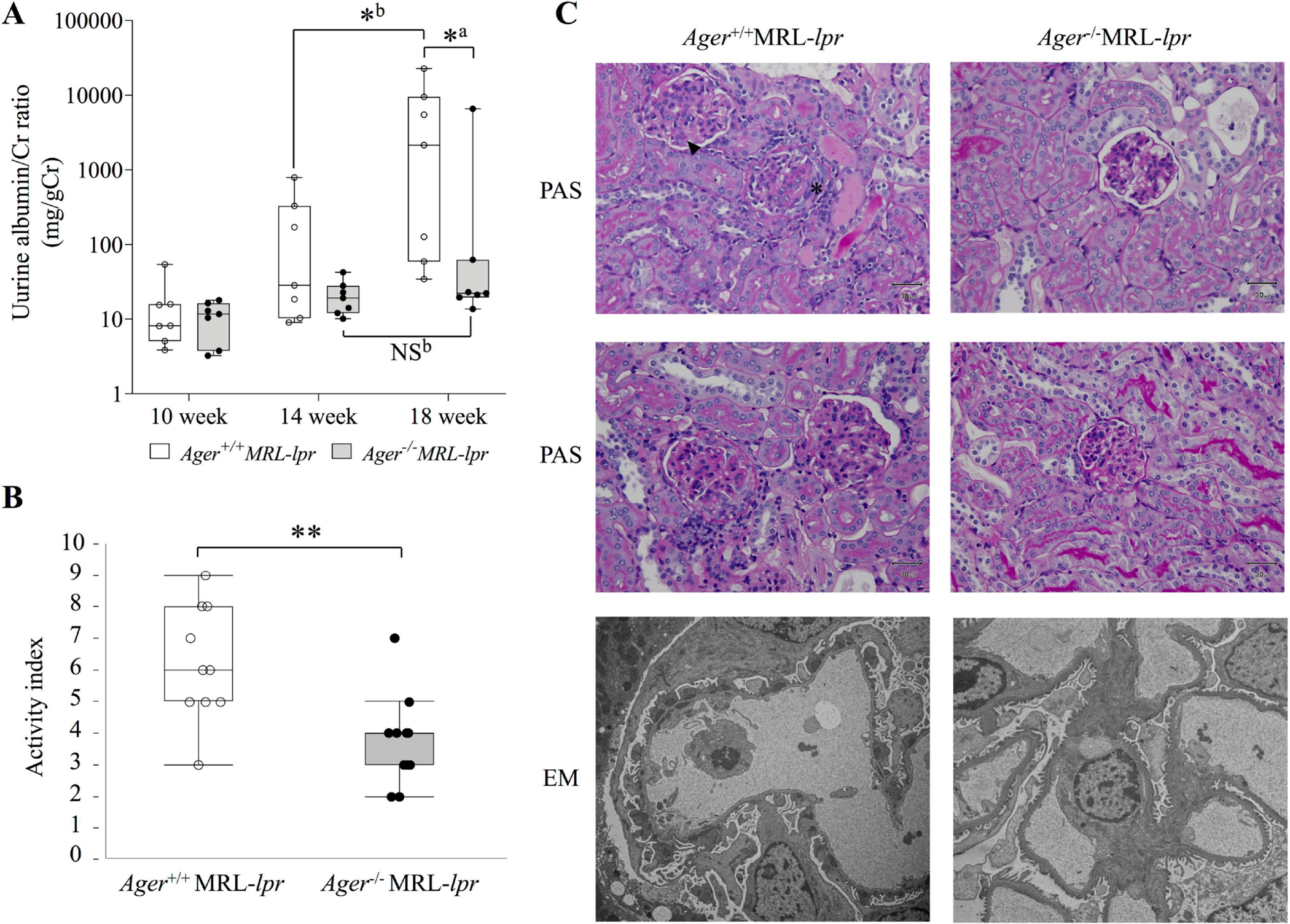
(A) The transition of the urine albumin/Cr ratio (mg/gCr). Ager+/+ MRL-lpr (n = 7), and Ager−/− MRL-lpr (n = 7). aThe Mann‒Whitney U test for between-group comparisons; bWilcoxon signed-rank sum test for comparisons between 14 and 18 weeks. *P < 0.05. (B) Activity index. Ager+/+ MRL-lpr (n = 10), and Ager−/− MRL-lpr (n = 11). Student’s t-test. **P < 0.01. (C) Pathological findings of the kidney. Ager+/+ MRL-lpr shows cellular crescent (*) and karyorrhexis (arrowhead) of glomeruli (top: PAS staining). Moderate glomerular cell proliferation was found in Ager+/+ MRL-lpr mice (middle: PAS staining). By electron microscopy, subepithelial and subendothelial electron-dense deposits and podocyte foot process effacement were observed in Ager+/+ MRL-lpr mice (bottom). Representative images of Ager+/+ MRL-lpr (n = 10), and Ager−/− MRL-lpr (n = 11). NS, not significant, EM: electron microscopy; PAS, periodic acid-Schiff.
Neutrophils are essential components of the innate immune system, and a relationship to the development of LN has been suggested[26]. In our study, glomerular Ly-6G-positive neutrophils were significantly reduced in Ager−/− MRL-lpr mice compared to their Ager+/+ MRL-lpr counterparts (Figure 3A). We further evaluated the change in NET formation in the kidney. Immunofluorescence staining revealed that the deletion of RAGE suppressed the manifestations of NETs in the glomerulus (Figure 3B). We also evaluated glomerular immunoglobulin and complement deposition, and immunofluorescence revealed that the deletion of RAGE did not affect the intensity of the immune complex in MRL-lpr mice at 18 weeks of age (Supplementary Figure 6A). Intrarenal macrophages have been considered important in the development of LN[50], but no significant differences in the glomerular infiltration of F4/80-positive macrophages were apparent (Supplementary Figure 6B). Overall, we demonstrated that the deletion of RAGE improved nephritis in MRL-lpr lupus model mice by inhibiting neutrophil infiltration and NET formation.
Figure 3. Reduction of glomerular neutrophil infiltrations and neutrophil extracellular traps in Ager−/− MRL-lpr mice at 18 weeks of age.
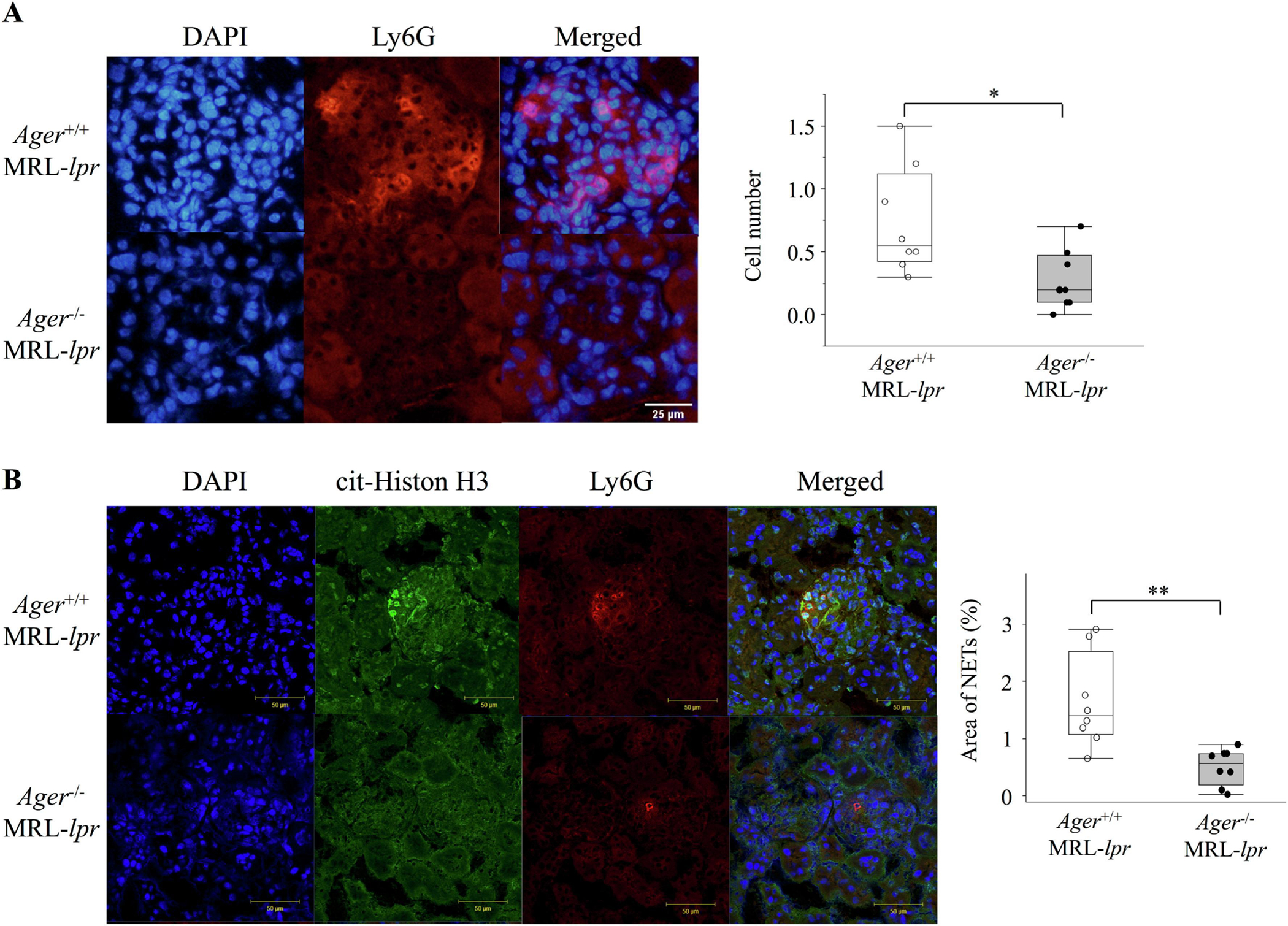
(A) Glomerular neutrophil infiltration. The number of Ly-6G-positive cells was counted in 10 glomeruli per animal, and the mean number of positive cells per glomerulus was used for estimation (n = 8). Student’s t-test. *p < 0.05. (B) The area percentage of NETs in each glomerulus was calculated in 10 glomeruli per animal, and the mean values per glomerulus were used for estimation (n=8). Welch’s t-test. **p < 0.01. NETs, neutrophil extracellular traps, cit-Histone H3, Histone H3 (citrulline R2 + R8 + R17).
4. Discussion
DAMPs from damaged and necrotic cells after tissue injury may be responsible for early neutrophil recruitment through pattern recognition receptors[51]. Recruited neutrophils are then activated, which directly and indirectly promote further secretion of DAMPs to amplify neutrophil recruitment from the circulation[22, 52]. Previous studies have demonstrated that HMGB1 activates neutrophil recruitment toward acetaminophen-induced liver necrosis through RAGE[53, 54]. On the other hand, neither ablation of RAGE signaling nor impaired oligomerization of RAGE has any impact on LPS-induced neutrophil infiltration in the air pouch model[55]. These results are consistent with our findings that RAGE-knockout animals had decreased neutrophil infiltration in the glomeruli. Recruited neutrophils play an important role in autoimmune diseases through formation of NETs[17, 23, 56–58]. NETs are a source of self-antigens in autoimmune disorders with autoantibodies. The formation of NETs has been reported to be increased in lupus[59]. Further studies have revealed an interaction between NETs and DAMPs, including HMGB1[60, 61], and neutralizing antibodies against HMGB1 suppressed the formation of NETs through TLR4[60].
Neutrophil heterogeneity and the association of RAGE and NETs have been described in the context of autoimmunity. Lupus peripheral mononuclear cells collected using density gradient centrifugation contain low-density granulocytes (LDGs), which are correlated with lupus disease activity or organ damage through vascular inflammation[62–64]. Several pathways are suggested to induce NETs, and RAGE ligand binding seems to be an important endogenous factor mediating NET formation[65]. HMGB1-RAGE signaling in neutrophils, with HMGB1 derived from platelets, was reported to be involved in NET formation[66, 67]. These previously proposed mechanisms are consistent with our results of less induction of NETs in glomeruli of the RAGE-knockout lupus model. A previous report further demonstrated that immune complexes containing autoantibodies and self-antigens are potent NET inducers in children with SLE[68]. The induction of NETs by immune complexes seems to be mediated by Fc receptor-binding mechanisms[69]. The Fc receptor-mediated and RAGE-mediated internalization of immune complexes is important for downstream signaling activation[15], and further study is needed to test whether RAGE-mediated internalization is also involved in NET formation in lupus pathogenesis.
Contradictory effects of RAGE inhibition have been reported in a murine lupus model. Lee and colleagues treated (NZB/NZW)F1 lupus-prone mice with sRAGE, which can bind to RAGE ligands and thus competitively inhibit the binding of ligands to membranous RAGE and downstream signaling[32]. They found that sRAGE alleviated the severity of LN pathology and immune complex deposition and decreased serum autoantibody titers. In addition, sRAGE interrupted the nuclear translocation of NF-κB in the kidney and effectively modified T-cell populations. On the other hand, Goury et al. reported that the deletion of RAGE in B6-lpr, lupus model mice, exacerbated lymphoproliferative syndrome, production of anti-dsDNA antibodies, LN, and accumulation of autoreactive CD3+B220+CD4−CD8− T cells compared with wild-type B6-lpr mice[33]. They further showed that autoreactive T cells in the spleen of B6-lpr Ager−/− mice exhibited a delay in apoptosis more than B6-lpr mice. RAGE itself has inherent contradictory pro- and anti-inflammatory roles. sRAGE, which is generated by alternative splicing or the cleavage of membranous RAGE by matrix metalloproteinases, lacks the membrane and cytoplasmic signaling domains and thereby inhibits RAGE signaling[6, 70]. The balance between the pro- and anti-inflammatory roles of RAGE might be different among different lupus models. B6-lpr mice exhibit a mild phenotype of lupus, as indicated by a longer lifetime and less severe nephritis than MRL-lpr mice[71, 72], and the difference in disease severity might be associated with the different effects of RAGE ablation. Indeed, RAGE deficiency did not alter clinical outcomes in the pristane-induced model of C57BL/6 mice, which showed a mild lupus phenotype[31]. Furthermore, it has been reported that NET induction is accelerated in MRL-lpr mice compared to B6-lpr mice[73], and the difference in neutrophil inflammation among the different strains may be the reason for the paradoxical effects of RAGE deficiency.
Several limitations of this study should be acknowledged. First, control animals without mutations in Fas/CD95, such as MRL/MpJ mice, were unavailable; nevertheless, the comparison of Ager−/− with Ager+/+ MRL-lpr littermates enabled us to evaluate the role of RAGE in lupus pathogenesis. Second, we could not elucidate whether infiltrated neutrophils or the origin of NETs were LDGs or normal-density neutrophils because of the lack of specific markers for LDGs[74]. Further study is needed to evaluate their spatial distribution and role in lupus autoimmunity as well as the function of RAGE in this cell subset. Third, the inducer of NETs in this study was also unknown, and it is necessary to develop specific therapeutics targeted against NETs.
5. Conclusions
We found that lymphoid organomegaly was alleviated by the deletion of RAGE, and the reduction in CD3+CD4−CD8− T cells was prominent in RAGE-deficient MRL-lpr lupus mice. Although the deletion of RAGE did not alleviate glomerular immune complex deposition, it suppressed neutrophil infiltration and NET formation. Thus, RAGE may be a promising target to treat LN.
Supplementary Material
Highlights.
MRL/MpJ-Faslpr/J (MRL-lpr) mice develop severe SLE phenotypes, including the high titer of the autoantibody, lymphoid organomegaly, and lupus nephritis.
Ablation of RAGE alleviated lymphoid organomegaly and reduced the frequency of pathogenic CD3+CD4−CD8− T cells in these lupus model mice.
The deletion of RAGE improved lupus nephritis, especially glomerular cell proliferation, karyorrhexis, and fibrinoid necrosis.
Neutrophil infiltration and the formation of neutrophil extracellular traps in the glomerulus were markedly reduced in RAGE-deficient MRL-lpr mice.
6. Acknowledgments
This work was supported by JSPS KAKENHI Grant Number 16K19600, 19K17907, research fellowship from the Uehara Memorial Foundation, the Mochida Memorial Foundation for Medical and Pharmaceutical Research, the Japan Rheumatism Foundation, and grant from the NIH/National Institute of Allergy and Infectious Diseases (R01AI135063 [M.S.]).
We gratefully acknowledge the excellent technical assistance of Ms. Kiyomi Maitani. We wish to thank Dr. Betty Diamond of Feinstein Institutes for Medical Research for critical review of the manuscript.
Footnotes
Conflict of Interest
J.W. receives grant support from Astellas, Bayer, Chugai, Daiichi Sankyo, Kissei, Kyowa Hakko Kirin, MSD, Otsuka, Teijin, Torii, Pfizer, Takeda, and Taisho Toyama. Other authors have no conflict of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
8. References
- 1.Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, Elliston K, Stern D, Shaw A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. The Journal of biological chemistry. 1992;267(21):14998–5004. [PubMed] [Google Scholar]
- 2.Ruan BH, Li X, Winkler AR, Cunningham KM, Kuai J, Greco RM, Nocka KH, Fitz LJ, Wright JF, Pittman DD, Tan XY, Paulsen JE, Lin LL, Winkler DG. Complement C3a, CpG oligos, and DNA/C3a complex stimulate IFN-α production in a receptor for advanced glycation end product-dependent manner. J Immunol. 2010;185(7):4213–22. [DOI] [PubMed] [Google Scholar]
- 3.Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97(7):889–901. [DOI] [PubMed] [Google Scholar]
- 4.Schmidt AM, Yan SD, Yan SF, Stern DM. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest. 2001;108(7):949–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmidt AM. 2016 ATVB Plenary Lecture: Receptor for Advanced Glycation Endproducts and Implications for the Pathogenesis an Treatment of Cardiometabolic Disorders: Spotlight on the Macrophage. Arterioscler Thromb Vasc Biol. 2017;37(4):613–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watanabe H, Son M. The Immune Tolerance Role of the HMGB1-RAGE Axis. Cells. 2021;10(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brett J, Schmidt AM, Yan SD, Zou YS, Weidman E, Pinsky D, Nowygrod R, Neeper M, Przysiecki C, Shaw A, et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol. 1993;143(6):1699–712. [PMC free article] [PubMed] [Google Scholar]
- 8.Tanji N, Markowitz GS, Fu C, Kislinger T, Taguchi A, Pischetsrieder M, Stern D, Schmidt AM, D’Agati VD. Expression of advanced glycation end products and their cellular receptor RAGE in diabetic nephropathy and nondiabetic renal disease. J Am Soc Nephrol. 2000;11(9):1656–66. [DOI] [PubMed] [Google Scholar]
- 9.Hudson BI, Carter AM, Harja E, Kalea AZ, Arriero M, Yang H, Grant PJ, Schmidt AM. Identification, classification, and expression of RAGE gene splice variants. FASEB J. 2008;22(5):1572–80. [DOI] [PubMed] [Google Scholar]
- 10.Hanford LE, Enghild JJ, Valnickova Z, Petersen SV, Schaefer LM, Schaefer TM, Reinhart TA, Oury TD. Purification and characterization of mouse soluble receptor for advanced glycation end products (sRAGE). The Journal of biological chemistry. 2004;279(48):50019–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raucci A, Cugusi S, Antonelli A, Barabino SM, Monti L, Bierhaus A, Reiss K, Saftig P, Bianchi ME. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). FASEB J. 2008;22(10):3716–27. [DOI] [PubMed] [Google Scholar]
- 12.Yanai H, Ban T, Wang Z, Choi MK, Kawamura T, Negishi H, Nakasato M, Lu Y, Hangai S, Koshiba R, Savitsky D, Ronfani L, Akira S, Bianchi ME, Honda K, Tamura T, Kodama T, Taniguchi T. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature. 2009;462(7269):99–103. [DOI] [PubMed] [Google Scholar]
- 13.Harris HE, Andersson U, Pisetsky DS. HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nature reviews Rheumatology. 2012;8(4):195–202. [DOI] [PubMed] [Google Scholar]
- 14.Sirois CM, Jin T, Miller AL, Bertheloot D, Nakamura H, Horvath GL, Mian A, Jiang J, Schrum J, Bossaller L, Pelka K, Garbi N, Brewah Y, Tian J, Chang C, Chowdhury PS, Sims GP, Kolbeck R, Coyle AJ, Humbles AA, Xiao TS, Latz E. RAGE is a nucleic acid receptor that promotes inflammatory responses to DNA. J Exp Med. 2013;210(11):2447–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Porat A, Giat E, Kowal C, He M, Son M, Latz E, Ben-Zvi I, Al-Abed Y, Diamond B. DNA-Mediated Interferon Signature Induction by SLE Serum Occurs in Monocytes Through Two Pathways: A Mechanism to Inhibit Both Pathways. Front Immunol. 2018;9:2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, Hua J, An LL, Audoly L, La Rosa G, Bierhaus A, Naworth P, Marshak-Rothstein A, Crow MK, Fitzgerald KA, Latz E, Kiener PA, Coyle AJ. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8(5):487–96. [DOI] [PubMed] [Google Scholar]
- 17.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197(6):711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mok CC. Towards new avenues in the management of lupus glomerulonephritis. Nature reviews Rheumatology. 2016;12(4):221–34. [DOI] [PubMed] [Google Scholar]
- 19.Tsokos GC, Lo MS, Costa Reis P, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nature reviews Rheumatology. 2016;12(12):716–30. [DOI] [PubMed] [Google Scholar]
- 20.Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. 2011;11(8):519–31. [DOI] [PubMed] [Google Scholar]
- 21.Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172(5):2731–8. [DOI] [PubMed] [Google Scholar]
- 22.de Oliveira S, Rosowski EE, Huttenlocher A. Neutrophil migration in infection and wound repair: going forward in reverse. Nat Rev Immunol. 2016;16(6):378–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, Kubes P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330(6002):362–6. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Collison KS, Parhar RS, Saleh SS, Meyer BF, Kwaasi AA, Hammami MM, Schmidt AM, Stern DM, Al-Mohanna FA. RAGE-mediated neutrophil dysfunction is evoked by advanced glycation end products (AGEs). Journal of leukocyte biology. 2002;71(3):433–44. [PubMed] [Google Scholar]
- 26.Banchereau R, Hong S, Cantarel B, Baldwin N, Baisch J, Edens M, Cepika AM, Acs P, Turner J, Anguiano E, Vinod P, Kahn S, Obermoser G, Blankenship D, Wakeland E, Nassi L, Gotte A, Punaro M, Liu YJ, Banchereau J, Rossello-Urgell J, Wright T, Pascual V. Personalized Immunomonitoring Uncovers Molecular Networks that Stratify Lupus Patients. Cell. 2016;165(3):551–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–5. [DOI] [PubMed] [Google Scholar]
- 28.Kessenbrock K, Krumbholz M, Schönermarck U, Back W, Gross WL, Werb Z, Gröne HJ, Brinkmann V, Jenne DE. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. 2009;15(6):623–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, Malech HL, Ledbetter JA, Elkon KB, Kaplan MJ. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. 2016;22(2):146–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, Friday S, Li S, Patel RM, Subramanian V, Thompson P, Chen P, Fox DA, Pennathur S, Kaplan MJ. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med. 2013;5(178):178ra40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eichhorst A, Daniel C, Rzepka R, Sehnert B, Nimmerjahn F, Voll RE, Chevalier N. Relevance of Receptor for Advanced Glycation end Products (RAGE) in Murine Antibody-Mediated Autoimmune Diseases. International journal of molecular sciences. 2019;20(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee SW, Park KH, Park S, Kim JH, Hong SY, Lee SK, Choi D, Park YB. Soluble receptor for advanced glycation end products alleviates nephritis in (NZB/NZW)F1 mice. Arthritis and rheumatism. 2013;65(7):1902–12. [DOI] [PubMed] [Google Scholar]
- 33.Goury A, Meghraoui-Kheddar A, Belmokhtar K, Vuiblet V, Ortillon J, Jaisson S, Devy J, Le Naour R, Tabary T, Cohen JH, Schmidt AM, Rieu P, Toure F. Deletion of receptor for advanced glycation end products exacerbates lymphoproliferative syndrome and lupus nephritis in B6-MRL Fas lpr/j mice. J Immunol. 2015;194(8):3612–22. [DOI] [PubMed] [Google Scholar]
- 34.Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356(6367):314–7. [DOI] [PubMed] [Google Scholar]
- 35.Nagata S, Golstein P. The Fas death factor. Science. 1995;267(5203):1449–56. [DOI] [PubMed] [Google Scholar]
- 36.Myint KM, Yamamoto Y, Doi T, Kato I, Harashima A, Yonekura H, Watanabe T, Shinohara H, Takeuchi M, Tsuneyama K, Hashimoto N, Asano M, Takasawa S, Okamoto H, Yamamoto H. RAGE control of diabetic nephropathy in a mouse model: effects of RAGE gene disruption and administration of low-molecular weight heparin. Diabetes. 2006;55(9):2510–22. [DOI] [PubMed] [Google Scholar]
- 37.Austin HA, Muenz LR, Joyce KM, Antonovych TT, Balow JE. Diffuse proliferative lupus nephritis: identification of specific pathologic features affecting renal outcome. Kidney international. 1984;25(4):689–95. [DOI] [PubMed] [Google Scholar]
- 38.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature methods. 2012;9(7):671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Watanabe H, Watanabe KS, Liu K, Hiramatsu S, Zeggar S, Katsuyama E, Tatebe N, Akahoshi A, Takenaka F, Hanada T, Akehi M, Sasaki T, Sada KE, Matsuura E, Nishibori M, Wada J. Anti-high Mobility Group Box 1 Antibody Ameliorates Albuminuria in MRL/lpr Lupus-Prone Mice. Molecular therapy Methods & clinical development. 2017;6:31–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kezic S, O’Regan GM, Lutter R, Jakasa I, Koster ES, Saunders S, Caspers P, Kemperman PM, Puppels GJ, Sandilands A, Chen H, Campbell LE, Kroboth K, Watson R, Fallon PG, McLean WH, Irvine AD. Filaggrin loss-of-function mutations are associated with enhanced expression of IL-1 cytokines in the stratum corneum of patients with atopic dermatitis and in a murine model of filaggrin deficiency. The Journal of allergy and clinical immunology. 2012;129(4):1031–9 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gardner RM, Nyland JF, Silva IA, Ventura AM, de Souza JM, Silbergeld EK. Mercury exposure, serum antinuclear/antinucleolar antibodies, and serum cytokine levels in mining populations in Amazonian Brazil: a cross-sectional study. Environmental research. 2010;110(4):345–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ueno Y, Ishii M, Yahagi K, Mano Y, Kisara N, Nakamura N, Shimosegawa T, Toyota T, Nagata S. Fas-mediated cholangiopathy in the murine model of graft versus host disease. Hepatology. 2000;31(4):966–74. [DOI] [PubMed] [Google Scholar]
- 43.Kang R, Tang D, Schapiro NE, Livesey KM, Farkas A, Loughran P, Bierhaus A, Lotze MT, Zeh HJ. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell death and differentiation. 2010;17(4):666–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mou K, Liu W, Han D, Li P. HMGB1/RAGE axis promotes autophagy and protects keratinocytes from ultraviolet radiation-induced cell death. Journal of dermatological science. 2017;85(3):162–9. [DOI] [PubMed] [Google Scholar]
- 45.Su Z, Wang T, Zhu H, Zhang P, Han R, Liu Y, Ni P, Shen H, Xu W, Xu H. HMGB1 modulates Lewis cell autophagy and promotes cell survival via RAGE-HMGB1-Erk1/2 positive feedback during nutrient depletion. Immunobiology. 2015;220(5):539–44. [DOI] [PubMed] [Google Scholar]
- 46.Elouaai F, Lulé J, Benoist H, Appolinaire-Pilipenko S, Atanassov C, Muller S, Fournié GJ. Autoimmunity to histones, ubiquitin, and ubiquitinated histone H2A in NZB x NZW and MRL-lpr/lpr mice. Anti-histone antibodies are concentrated in glomerular eluates of lupus mice. Nephrol Dial Transplant. 1994;9(4):362–6. [PubMed] [Google Scholar]
- 47.Lu Q, Kanai Y, Kubota T. The emergence of anti-dsDNA antibodies precedes nucleosome-specific antibodies in MRL/lpr and MRL/+ mice. Journal of medical and dental sciences. 2003;50(1):9–15. [PubMed] [Google Scholar]
- 48.Andrews BS, Eisenberg RA, Theofilopoulos AN, Izui S, Wilson CB, McConahey PJ, Murphy ED, Roths JB, Dixon FJ. Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. J Exp Med. 1978;148(5):1198–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bajema IM, Wilhelmus S, Alpers CE, Bruijn JA, Colvin RB, Cook HT, D’Agati VD, Ferrario F, Haas M, Jennette JC, Joh K, Nast CC, Noël LH, Rijnink EC, Roberts ISD, Seshan SV, Sethi S, Fogo AB. Revision of the International Society of Nephrology/Renal Pathology Society classification for lupus nephritis: clarification of definitions, and modified National Institutes of Health activity and chronicity indices. Kidney international. 2018;93(4):789–96. [DOI] [PubMed] [Google Scholar]
- 50.Davidson A, Aranow C, Mackay M. Lupus nephritis: challenges and progress. Curr Opin Rheumatol. 2019;31(6):682–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pittman K, Kubes P. Damage-associated molecular patterns control neutrophil recruitment. Journal of innate immunity. 2013;5(4):315–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eash KJ, Greenbaum AM, Gopalan PK, Link DC. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest. 2010;120(7):2423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huebener P, Pradere JP, Hernandez C, Gwak GY, Caviglia JM, Mu X, Loike JD, Jenkins RE, Antoine DJ, Schwabe RF. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J Clin Invest. 2015;125(2):539–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arnold K, Xu Y, Sparkenbaugh EM, Li M, Han X, Zhang X, Xia K, Piegore M, Zhang F, Zhang X, Henderson M, Pagadala V, Su G, Tan L, Park PW, Stravitz RT, Key NS, Linhardt RJ, Pawlinski R, Xu D, Liu J. Design of anti-inflammatory heparan sulfate to protect against acetaminophen-induced acute liver failure. Sci Transl Med. 2020;12(535). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li M, Ong CY, Langouët-Astrié CJ, Tan L, Verma A, Yang Y, Zhang X, Shah DK, Schmidt EP, Xu D. Heparan sulfate-dependent RAGE oligomerization is indispensable for pathophysiological functions of RAGE. eLife. 2022;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ferguson PJ, Lokuta MA, El-Shanti HI, Muhle L, Bing X, Huttenlocher A. Neutrophil dysfunction in a family with a SAPHO syndrome-like phenotype. Arthritis and rheumatism. 2008;58(10):3264–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Glennon-Alty L, Hackett AP, Chapman EA, Wright HL. Neutrophils and redox stress in the pathogenesis of autoimmune disease. Free radical biology & medicine. 2018;125:25–35. [DOI] [PubMed] [Google Scholar]
- 58.Hacbarth E, Kajdacsy-Balla A. Low density neutrophils in patients with systemic lupus erythematosus, rheumatoid arthritis, and acute rheumatic fever. Arthritis and rheumatism. 1986;29(11):1334–42. [DOI] [PubMed] [Google Scholar]
- 59.Hakkim A, Fürnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, Herrmann M, Voll RE, Zychlinsky A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A. 2010;107(21):9813–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tadie JM, Bae HB, Jiang S, Park DW, Bell CP, Yang H, Pittet JF, Tracey K, Thannickal VJ, Abraham E, Zmijewski JW. HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. Am J Physiol Lung Cell Mol Physiol. 2013;304(5):L342–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, Wang S, Kim J, Billiar T, Wang Y, Tsung A. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology. 2015;62(2):600–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, McCune WJ, Kaplan MJ. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J Immunol. 2010;184(6):3284–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Midgley A, Beresford MW. Increased expression of low density granulocytes in juvenile-onset systemic lupus erythematosus patients correlates with disease activity. Lupus. 2016;25(4):407–11. [DOI] [PubMed] [Google Scholar]
- 64.Mistry P, Nakabo S, O’Neil L, Goel RR, Jiang K, Carmona-Rivera C, Gupta S, Chan DW, Carlucci PM, Wang X, Naz F, Manna Z, Dey A, Mehta NN, Hasni S, Dell’Orso S, Gutierrez-Cruz G, Sun HW, Kaplan MJ. Transcriptomic, epigenetic, and functional analyses implicate neutrophil diversity in the pathogenesis of systemic lupus erythematosus. Proc Natl Acad Sci U S A. 2019;116(50):25222–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tatsiy O, de Carvalho Oliveira V, Mosha HT, McDonald PP. Early and Late Processes Driving NET Formation, and the Autocrine/Paracrine Role of Endogenous RAGE Ligands. Front Immunol. 2021;12:675315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, Maiuri L, Maseri A, D’Angelo A, Bianchi ME, Rovere-Querini P, Manfredi AA. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. Journal of thrombosis and haemostasis : JTH. 2014;12(12):2074–88. [DOI] [PubMed] [Google Scholar]
- 67.Stark K, Philippi V, Stockhausen S, Busse J, Antonelli A, Miller M, Schubert I, Hoseinpour P, Chandraratne S, von Brühl ML, Gaertner F, Lorenz M, Agresti A, Coletti R, Antoine DJ, Heermann R, Jung K, Reese S, Laitinen I, Schwaiger M, Walch A, Sperandio M, Nawroth PP, Reinhardt C, Jäckel S, Bianchi ME, Massberg S. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood. 2016;128(20):2435–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, Punaro M, Baisch J, Guiducci C, Coffman RL, Barrat FJ, Banchereau J, Pascual V. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. 2011;3(73):73ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Patiño-Trives AM, Pérez-Sánchez C, Pérez-Sánchez L, Luque-Tévar M, Ábalos-Aguilera MC, Alcaide-Ruggiero L, Arias-de la Rosa I, Román-Rodríguez C, Seguí P, Espinosa M, Font P, Barbarroja N, Escudero-Contreras A, Antonio González-Reyes J, Manuel Villalba J, Collantes-Estévez E, Aguirre-Zamorano M, López-Pedrera C. Anti-dsDNA Antibodies Increase the Cardiovascular Risk in Systemic Lupus Erythematosus Promoting a Distinctive Immune and Vascular Activation. Arterioscler Thromb Vasc Biol. 2021;41(9):2417–30. [DOI] [PubMed] [Google Scholar]
- 70.Oshima Y, Harashima A, Munesue S, Kimura K, Leerach N, Goto H, Tanaka M, Niimura A, Hayashi K, Yamamoto H, Higashida H, Yamamoto Y. Dual Nature of RAGE in Host Reaction and Nurturing the Mother-Infant Bond. International journal of molecular sciences. 2022;23(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Izui S, Kelley VE, Masuda K, Yoshida H, Roths JB, Murphy ED. Induction of various autoantibodies by mutant gene lpr in several strains of mice. J Immunol. 1984;133(1):227–33. [PubMed] [Google Scholar]
- 72.Kelley VE, Roths JB. Interaction of mutant lpr gene with background strain influences renal disease. Clinical immunology and immunopathology. 1985;37(2):220–9. [DOI] [PubMed] [Google Scholar]
- 73.Knight JS, Subramanian V, O’Dell AA, Yalavarthi S, Zhao W, Smith CK, Hodgin JB, Thompson PR, Kaplan MJ. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Annals of the rheumatic diseases. 2015;74(12):2199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nakazawa D, Kudo T. Novel Therapeutic Strategy Based on Neutrophil Subset and Its Function in Autoimmune Disease. Frontiers in pharmacology. 2021;12:684886. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.