

Abstract
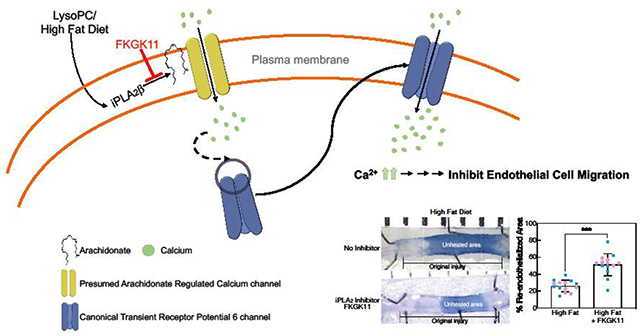
Lipid oxidation products, including lysophosphatidylcholine (lysoPC), accumulate at the site of arterial injury after vascular interventions and hinder re-endothelization. LysoPC activates calcium-permeable channels, specifically canonical transient receptor potential 6 (TRPC6) channels that induce a sustained increase in intracellular calcium ion concentration [Ca2+]i and contribute to dysregulation of the endothelial cell (EC) cytoskeleton. Activation of TRPC6 leads to inhibition of EC migration in vitro and delayed re-endothelization of arterial injuries in vivo. Previously, we demonstrated the role of phospholipase A2 (PLA2), specifically calcium-independent PLA2 (iPLA2), in lysoPC-induced TRPC6 externalization and inhibition of EC migration in vitro. The ability of FKGK11, an iPLA2-specific pharmacological inhibitor, to block TRPC6 externalization and preserve EC migration was assessed in vitro and in a mouse model of carotid injury. Our data suggest that FKGK11 prevents lysoPC-induced PLA2 activity, blocks TRPC6 externalization, attenuates calcium influx, and partially preserves EC migration in vitro. Furthermore, FKGK11 promotes re-endothelization of an electrocautery carotid injury in hypercholesterolemic mice. FKGK11 has similar arterial healing effects in male and female mice on a high-fat diet. This study suggests that iPLA2 is a potential therapeutic target to attenuate calcium influx through TRPC6 channels and promote EC healing in cardiovascular patients undergoing angioplasty.
Keywords: phospholipase A2, canonical transient receptor potential 6 channel, calcium, iPLA2 inhibitor FKGK11, endothelial migration, lysophosphatidylcholine, hypercholesterolemia
Graphical Abstract
1. INTRODUCTION1
Dyslipidemia contributes to atherosclerosis and the eventual need for vascular intervention. Angioplasty is one of the leading interventions to treat arterial disease. This intervention denudes the endothelial layer [1], requiring the migration of endothelial cells (EC) from the healthy artery [2, 3]. However, lipid accumulation and subsequent lipid oxidation at the site of injury are triggers that set-in motion a cascade of events that inhibit arterial re-endothelialization [4–8]. Delayed re-endothelialization prolongs the presence of a thrombogenic surface and promotes the development of intimal hyperplasia [9, 10], increasing the risk of complications.
Lipid oxidation products, including lysophosphatidylcholine (lysoPC), inhibit EC migration, in part, by activating canonical transient receptor potential (TRPC) channels, specifically, TRPC6 and TRPC5 [11, 12]. LysoPC triggers an increase in calcium [5], possibly through arachidonic acid (ArA)-regulated calcium (ARC) channels, that leads to the translocation of TRPC6 to the plasma membrane. LysoPC-induced TRPC6 externalization initiates the cascade of events that induces TRPC5 activation leading to a prolonged increase in intracellular calcium ion concentration ([Ca2+]i) that activates calpains, disrupts EC focal adhesion and cytoskeleton that regulate EC movement resulting in the inhibition of EC migration [5, 7, 11, 12]. In vivo, a high fat diet and associated increase in plasma lysoPC markedly decreases EC healing of carotid injuries in wild-type (WT) mice but not in TRPC6−/− mice [13]. This suggests that blocking TRPC6 externalization and activation could potentially preserve EC migration and promote re-endothelialization of the angioplasty site.
Although small molecule TRPC6 modulators have led to the advancements in understanding channel function and regulation, targeted in vivo inhibition remains challenging and no specific TRPC6 inhibitor is available for clinical use [14–16]. Our recent work suggests that phospholipase A2 (PLA2) plays a critical role in TRPC6 externalization [17]. PLA2 enzymes hydrolyze membrane phospholipids to release ArA and lysolipids. While there are three major PLA2 subfamilies including cytosolic calcium independent PLA2 (iPLA2), cytosolic calcium-dependent PLA2 (cPLA2), and secretory PLA2 (sPLA2), iPLA2, specifically the β isoform of iPLA2 (iPLA2β), is a primary mediator of lysoPC-induced release of ArA, TRPC6 externalization, increase in [Ca2+]i, and subsequent inhibition of EC migration [17]. This suggests that iPLA2 is an alternative target to attenuate lysoPC-induced TRPC6 externalization and preserve EC migration.
The purpose of this study is to evaluate the potential of pharmacological inhibition of iPLA2 to attenuate lysoPC-induced TRPC6 externalization in vitro and improve re-endothelialization of arterial injuries in hypercholesterolemic mice. We use FKGK11, a potent and reversible iPLA2 inhibitor belonging to the family of polyfluoroketones, for our studies [18, 19]. FKGK11 has shown promising results in various disease models [20–24]; however, the use of this inhibitor in EC migration and arterial healing has not been explored. Our data show that FKGK11 prevents lysoPC-induced ArA release from EC membranes, prevents TRPC6 externalization, attenuates the increase in [Ca2+]i, and preserves EC migration in vitro. Importantly, in hypercholestrolemic mice FKGK11 restores re-endothelization of carotid injuries to levels equivalent to that in normocholesterolemic (chow-fed) mice. Our results identify iPLA2 as a potential new target to promote arterial healing in cardiovascular patients undergoing vascular interventions.
2. MATERIALS AND METHODS
2.1. Reagents
1-palmitol-2-hydroxy-sn-glycero-3-phosphocholine (16:0 LysoPC) (catalog number (Cat. No.): 855675p) and ArA (Cat. No. 861810) were obtained from Avanti Polar Lipids, Inc., Alabaster, AL. Pharmacological inhibitor, PLA2 assay kits (Cat. No. 765021), and FKGK11 (Cat. No. 13179) were purchased from Cayman Chemical, Ann Arbor, MI. FKGK11 was received in powder form and readily soluble in PBS. Aliquots were made in PBS and stored at −20 °C until use for in vitro studies. FKGK11 for in vivo studies was received in ethanol. The FKGK11 was aliquoted and ethanol evaporated. FKGK11 was stored at −20 °C until use in in vivo studies. FKGK11 was resuspended in PBS immediately prior to intraperitoneal injections (i.p.), consistent with other studies [20, 21, 24, 25]. The ArA ELISA kits (Cat. No. MBS2608709) were purchased from MyBioSource, San Diego, CA. Antibodies for immunoblot analysis were purchased from Cell Signaling and Santa Cruz as indicated below. Evans Blue dye was purchased form Sigma-Aldrich (Cat. No. E2129). Paraformaldehyde (Cat. No. 416780250) and Total cholesterol assay kits (Cat. No. TR13421) were purchased from ThermoFisher Scientific.
2.2. EC Harvest and Culture
Primary bovine aortic endothelial cells (BAECs) were used in our studies. BAECs were isolated from adult bovine aortas (abattoir, unknown sex) by scraping after collagenase treatment [12]. Assays involving BAECs were performed in replicates using cells from at least three different bovine aortas. BAECs were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% (vol/vol) Fetal Bovine Serum (FBS, HyClone Laboratories, Cat. No. SH30541.03) and 1% antibiotic mixture (penicillin/streptomycin). BAECs between passage 4 and 9 were used for the assays.
2.3. Measurement of PLA2 Activity
PLA2 enzyme activity was measured using PLA2 assay kit (Cayman) as described previously [17]. Briefly, BAECs cultured in 60 mm dishes and serum-starved for 18 h were preincubated with medium with PBS referred to hereafter as medium (control) or PLA2 inhibitor (FKGK11, 20 μmol/L) for 1 h, followed by addition of lysoPC (12.5 μmol/L) for 15 min. Cells were then lysed in lysis buffer without EDTA (50 mmol/L HEPES, 150 mmol/L NaCl, 200 μmol/L Na3VO4, 100 mmol/L NaF, 1% Triton X-100, pH 7.4) containing protease inhibitors (Complete, Roche) for 30 min at 4 °C. Lysates were passed through needles, 20 gauge (20x) and 25 gauge (15x), and cleared by centrifugation at 12,000 g for 15 min. The sample (lysates), negative control (PBS), and positive control (bee venom, 10 μmol/L) were added to triplicate wells of a 96-well plate. Arachidonoyl Thio-PC (200 μmol/L) substrate was added to each well to initiate the reaction, and the plate was incubated for 60 min at room temperature. DNTB/EGTA was added to stop the reaction, and the absorbance was read at 405 nm using a plate reader (SpectraMAX 190).
2.4. Measurement of Membrane ArA Content
BAECs were serum-starved for 18 h followed by the addition of lysoPC (12.5 μmol/L) for 15 min. Using Mem-PER Plus membrane extraction kit (ThermoFisher, Cat no. 89842), the membrane fraction was isolated. Briefly, BAECs were rinsed with PBS x 2 and collected by centrifugation at 1000 g for 5 min. The pellet was resuspended in the permeabilization buffer and incubated at 4 °C for 10 min. The cells were then centrifuged at 16,000 g for 15 min at 4°C. The upper cytosolic fraction was removed carefully and discarded. The pellet containing the membrane fraction was resuspended in solubilization buffer and incubated at 4°C for 30 min. The sample was then centrifuged at 16,000 g for 15 min. ArA content in the membrane fraction was assessed using an ELISA assay following the manufacturer’s protocol, and the absorbance read at 450 nm using a plate reader (SpectraMAX 190).
2.5. TRPC6 Externalization by Biotinylation Assay
TRPC6 was measured by biotinylation assay as previously described [26]. Briefly, BAECs were cultured in 60 mm dishes to 80% confluency, serum-starved for 18 h, then lysoPC (12.5 μmol/L) added for 15 min. The ECs were then treated with Sulfo-NHS-Biotin (2 mg/ml, ThermoFisher, Cat. No. 21217), cells were then lysed and incubated with streptavidin-agarose beads (ThermoFisher, Cat. No. 20349) after separating an aliquot for total TRPC6 determination. Externalized (biotin-TRPC6) and total TRPC6 samples (30 μg) were run on 4-20% Tris-Glycine gels (ThermoFisher, Cat. No. XP04200BOX) and detected by immunoblot analysis as previously described [12], using a rabbit anti-TRPC6 antibody (1:1000, Cell Signaling, Cat. No.16716S-RRID:AB_2798768) and an anti-rabbit IgG (1:1000, Antibodies-online, Cat. No. ABIN102010-RRID:AB_10762386) as the secondary antibody. An anti-β-actin antibody (1:2000, Santa Cruz, Cat. No. sc47778 HRP-RRID:AB_2714189) was used to detect actin that served as the loading control.
2.6. Measurement of [Ca2+]i
When 80-90% confluent, BAECs were loaded with the FITC-conjugated fluorophore Calbryte™ 520 AM dye (AAT Bioquest, Cat. No. 36310) following the manufacturer’s protocol and allowed to incubate for 35 min. ECs were loaded into the sort chamber of a BD FACS Melody™ cell sorter (BD Biosciences, San Jose, CA) maintained at 37 °C. After a stable baseline was obtained, lysoPC (12.5 μmol/L) was added, and the relative change in [Ca2+]i was read using the kinetic reading mode at Ex/Em 490/525 nm. Data were analyzed using the FlowJo™ v10 software (BD Biosciences).
2.7. EC Migration
A razor-scrape assay was used to study EC migration as previously described [27]. Briefly, confluent BAECs were serum-starved for 18 h and pre-incubated with medium (control) or FKGK11 (20 μmol/L) for 1 h. The razor scrape was performed, then cells were washed and allowed to migrate with or without FKGK11(20 μmol/L) and lysoPC (12.5 μmol/L) for 24 h. Images were acquired using a digital CCD camera mounted on a phase-contrast microscope. Images were processed using NIH ImageJ analysis software (NIH, Bethesda, MD), and migration was quantitated by an observer blinded to the experimental conditions.
2.8. Animal Use
WT C57Bl/6 male and female mice were used in this study. Mice between the ages of six and eight weeks were randomly assigned to four weeks of chow diet (Envigo, Cat. No. 2920X) or high fat (HF) diet (Envigo, Cat. No. TD. 150869) containing 21% hydrogenated vegetable oil and 0.2% cholesterol by weight. Half the mice on each diet were then randomly assigned to receive daily i.p. injections of either PBS (vehicle control) or freshly prepared FKGK11 in PBS (5.6 mg/kg/day, each mouse received approximately 200 μL of 2 mmol/L FKGK11 in PBS) [20, 21, 24]. Daily i.p. injections were started two days prior to carotid injury and continued until the end of the study. The Institutional Animal Care and Use Committee approved the study protocol. The animal protocol and care complied with the American Association of Laboratory Care Guidelines and the Guide for the Care and Use of Laboratory Animals, Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council, Washington, DC: National Academies Press, 2011.
2.9. Mouse Carotid Injury
When mice were 10 to 12 weeks of age, the right common carotid artery (RCCA) was injured using electrocautery as described previously [28]. Briefly, mice were anesthetized by i.p. injection of ketamine (80 mg/kg, West-Ward Pharmaceuticals, Eatontown, NJ) and xylazine (5 mg/kg, Akorn, Inc., Lake Forest, IL). Electrocautery at 2 watts of power was applied to the RCCA for 3 seconds using custom bipolar forceps (Elmed, Inc., Addison, IL; Cat. No. 5000SP-M) to produce a 5-mm injury. Prior studies documented that this perivascular electrical injury denuded the endothelium and injured the smooth muscle layer of the vessel without damaging the adventitial layer [29]. The injury caused via electrocautery induced changes in the arterial wall similar to changes after arterial angioplasty [30].
2.10. Mouse Carotid Artery Harvest and Analysis for EC Migration
At 120 h after injury when approximately 50% of the injured carotid is re-endothelialized in mice on chow diet [28], mice were anesthetized as described above. Blood was drawn from inferior vena cava and 5% Evans Blue in PBS was injected for 10 minutes to stain arteries that lacked intact EC surface [28]. The carotids were perfusion-fixed for 10 min with freshly prepared 4% paraformaldehyde and then removed. The arteries were pinned flat and imaged with a SPOT Insight camera (SPOT Imaging, Sterling Heights, MI). The images were analyzed using NIH Image software, and the area of artery that stained blue was measured. An observer blinded to the experimental conditions verified the measurements of endothelial healing. In our prior studies, the ability of the Evans Blue method to accurately quantify the area lacking endothelial cells was verified by scanning electron microscopy [13, 28].
2.11. Plasma Assay
Plasma levels of cholesterol and lysoPC were assessed at the conclusion of the study. Plasma was prepared from whole blood and stored at −80 °C until analyzed. Total cholesterol was determined by a cholesterol oxidase method as previously described [31]. Briefly, cholesterol standards (ThermoFisher, Chemistry calibrator, Cat. No. 23-666-189/203) were prepared (range of 0-24 μg/well), and plasma samples were diluted 1:1 with PBS. Standards and samples were added to wells in duplicate and incubated with Thermotrace cholesterol reagent (ThermoFisher, Cat. No.TR13421) for 15 min at room temperature. The absorbance was read at 500 nm using a plate reader (SpectraMAX 190).
Plasma lysoPC concentration was measured by an enzymatic method as previously described [32]. Briefly, lysoPC standards were freshly prepared (range of 0-2000 μg/mL) in sterile distilled water. Standards and plasma samples were placed in wells of a 96-well plate in duplicate, followed by addition of reagent cocktail A (0.1 mol/L Tris-HCl, pH 8.0, 0.01% Triton X-100, 1 mmol/L CaCl2, 3 mmol/L N-ethyl-N-(2-hydroxy-3-sulfopropyl)-3-methylaniline sodium dehydrate, 10 kU/L peroxidase, 0.1 kU/L glycerophosphoryl-choline phosphodiesterase, 1 kU/L choline oxidase). The mixture was incubated for 30 min, and initial absorbance at 600 nm was recorded. The reaction was initiated by the addition of reagent cocktail B (0.1 mol/L Tris-HCl, pH 8.0, 0.01% Triton X-100, 5 mmol/L 4-aminoantipyrine, and 1 kU/L phospholipase B), and the mixture was incubated for 30 min. Final absorbance was read at 600 nm using a plate reader (SpectraMAX 190). The lysoPC concentration was evaluated by subtracting the background from final absorbance.
2.12. Statistical Analysis
All experiments were performed at least in triplicate with cells cultured from three different animals or independent biological samples. Experimental results were represented as means ± SD. Data were analyzed by Student’s t-test or one-way ANOVA with Tukey’s multiple comparison test post hoc analysis, and p < 0.05 was considered statistically significant.
3. RESULTS
3.1. FKGK11 Blocks LysoPC-induced PLA2 Activity and Prevents Membrane ArA Release
We previously showed that lysoPC increases iPLAβ activity in BAECs [17]. To verify that the pharmacological inhibitor, FKGK11, blocks lysoPC-induced PLA2 activity, BAECs were serum-starved for 18 h, then incubated with medium (control) or FKGK11 (20 μmol/L) for 1 h followed by lysoPC (12.5 μmol/L) for 15 min. The lysoPC-induced change in PLA2 activity in control BAECs was 0.166 ± 0.0007 μmol/min/mig (Fig. 1A, Medium). In cells pretreated with FKGK11, the lysoPC-induced change in PLA2 activity decreased to 0.089 ± 0.0118 μmol/min/mg (Fig. 1A, FKGK11). The iPLA2 inhibitor FKGK11 (20 μmol/L) reduced lysoPC-induced PLA2 activity by approximately 50% in BAECs (n= 3 independent biological samples, p < 0.001, Fig. 1A).
Figure 1. FKGK11, an iPLA2 inhibitor, blocks lysoPC-induced PLA2 activity and prevents ArA release in BAECs.
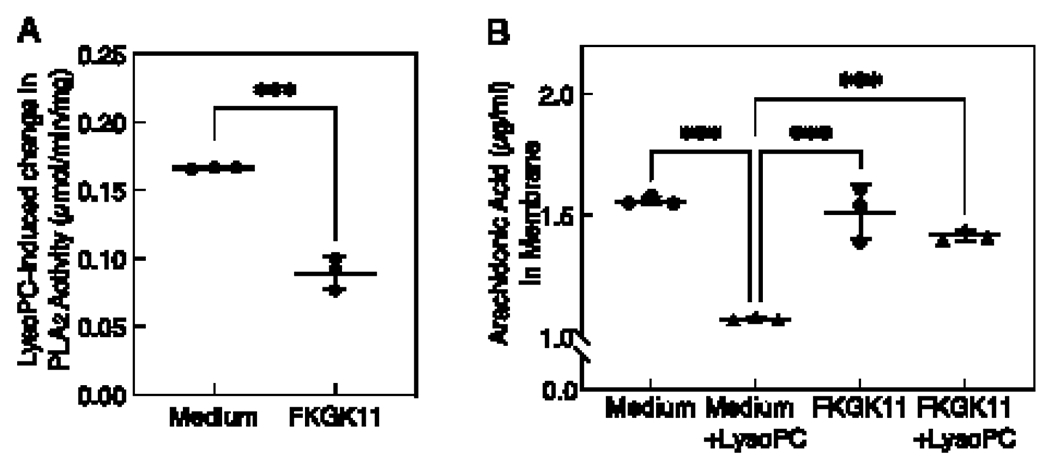
Confluent ECs made quiescent for 18 h were preincubated with medium or PLA2 inhibitor (FKGK11, 20 μmol/L) for 1 h, followed with or without the addition of lysoPC (12.5 μmol/L) for 15 min. (A) Total PLA2 enzyme activity was measured using colorimetric assay. LysoPC-induced change in PLA2 activity was calculated by subtracting activity under basal conditions and is presented as a dot and whisker plot. Values shown are the means ± SD, n= 3 independent biological samples. Statistical analysis was performed using Student’s t-test and p values calculated, ***p < 0.001. (B) ArA content of membrane fractions was measured by ELISA assay and presented as a dot and whisker plot. Values shown are the means ± SD, n= 3 independent biological samples. Statistical analysis was performed using one-way ANOVA with Tukey’s multiple comparison test and p values calculated, ***p < 0.001. iPLA2: calcium-independent phospholipase A2; LysoPC: lysophosphatidylcholine; ArA: arachidonic acid; BAEC: bovine aortic endothelial cell; EC: endothelial cell.
The effect of FKGK11 on lysoPC-induced ArA release from BAEC membranes was assessed. ECs were serum-starved for 18 h, then incubated with medium (control) or FKGK11 (20 μmol/L) for 1 h, followed by lysoPC (12.5 μmol/L) for 15 min. Basal level of membrane ArA content in control cells was 1.56 ± 0.10 μg/mL (n= 3 independent biological samples, Fig. 1B). LysoPC induced ArA release from BAEC membranes, and the ArA content was reduced to 1.07 ± 0.04 μg/mL (n= 3 independent biological samples, p < 0.001 compared with the basal level, Fig. 1B). In BAECs pretreated with FKGK11 (20 μmol/L) for 1 h, membrane ArA content was 1.51 ± 0.14 μg/mL, similar to medium (control) (n= 3 independent biological samples, Fig. 1B). FKGK11 reduced lysoPC-induced ArA release from the membrane, with membrane ArA content being 1.41 ± 0.06 μg/mL in treated cells incubated with lysoPC (n= 3 independent biological samples, Fig. 1B). Cumulatively, this confirmed the ability of FKGK11 to block lysoPC-induced PLA2 activity and prevent ArA release from EC membranes.
3.2. FKGK11 Prevents LysoPC-induced TRPC6 Externalization and Attenuates the Increase in [Ca2+]i
The effect of the iPLA2 inhibitor on TRPC6 externalization was evaluated using a biotinylation assay. At baseline, TRPC6 externalization was comparable between medium (control) and FKGK11 (20 μmol/L) treated cells (n= 3 independent biological samples, Fig. 2A). Incubation with lysoPC (12.5 μmol/L) for 15 min significantly increased TRPC6 externalization in control cells (n= 3 independent biological samples, p < 0.001 compared with medium alone, Fig. 2A). However, lysoPC-induced TRPC6 externalization was significantly attenuated in cells pretreated with FKGK11 and was comparable to control levels (n= 3 independent biological samples, p < 0.001 compared with lysoPC, Fig. 2B). These data suggested that FKGK11 prevented iPLA2 mediated ArA release and in turn blocked TRPC6 externalization. To determine if addition of ArA exogenously could induce TRPC6 externalization in FKGK11-treated cells, ArA (10 μmol/L) was added to FKGK11-pretreated cells for 15 min (Supplemental Fig. S1). FKGK11 blocked lysoPC-induced TRPC6 externalization, but addition of exogenous ArA (10 μmol/L) induced TRPC6 externalization despite PLA2 inhibition (Supplemental Fig. S1A and B). Cumulatively, this data suggested that iPLA2 inhibition blocked lysoPC-induced TRPC6 externalization in BAECs by preventing ArA release, but this could be circumvented by adding exogenous ArA.
Figure 2. FKGK11, an iPLA2 inhibitor, prevents lysoPC-induced TRPC6 externalization and attenuates the rise in [Ca2+]i.
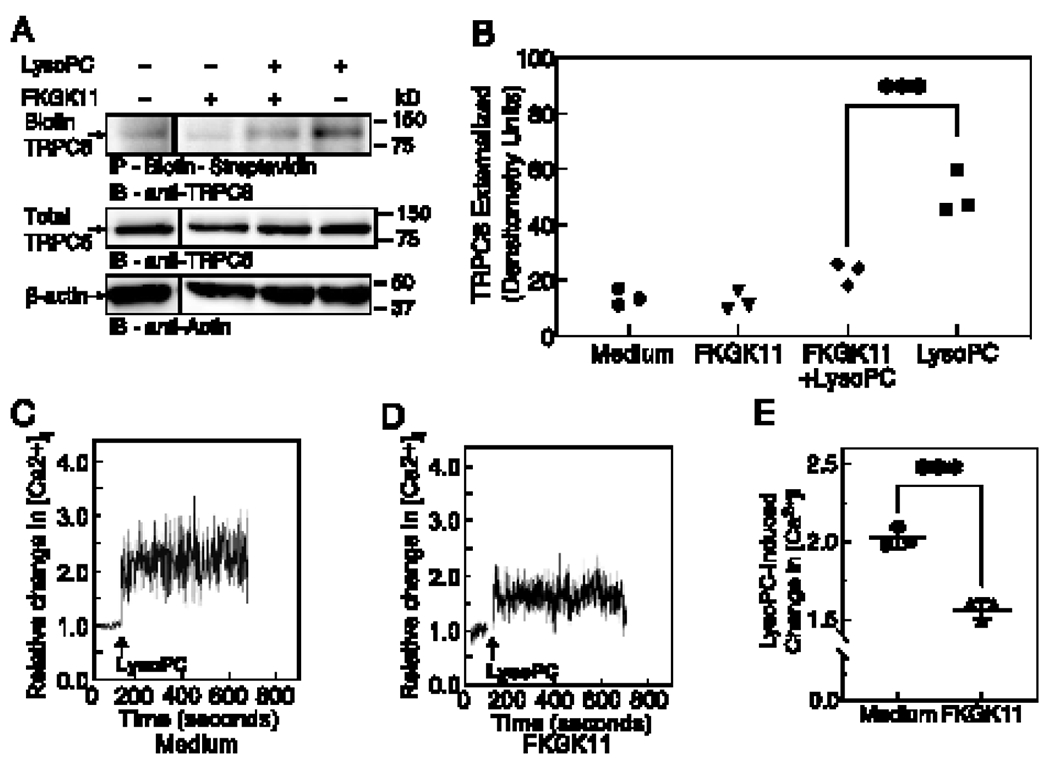
(A-B) ECs were pre-incubated with medium or iPLA2 inhibitor (FKGK11, 20 μmol/L) for 1 h, followed with or without the addition of lysoPC (12.5 μmol/L) for 15 min. (A) Externalized TRPC6 was detected by biotinylation assay. Total TRPC6 was detected in an aliquot of cell lysate removed prior to biotinylation, and actin served as a loading control. A representative blot is shown of n= 3 independent biological samples, lines indicate lanes rearranged from the same gel. (B) Densitometric measurements of externalized TRPC6 is presented as a scatter plot; ●, medium control; ▼, FKGK11; ◆, FKGK11 + lysoPC; ■, medium control + lysoPC. Statistical analysis using one-way ANOVA with Tukey’s multiple comparison test was performed and p values calculated, ***p < 0.001. (C-E) LysoPC-induced change in [Ca2+]i was determined by fluorometric assay. After adjusting the baseline, lysoPC (12.5 μmol/L) was added (arrow) and relative change in [Ca2+]i in medium control cells (C) and FKGK11-treated cells (D) was determined. Representative images of n= 3 independent biological samples are shown. (E) LysoPC-induced change in [Ca2+]i measured by difference in mean [Ca2+]i at baseline and after addition of lysoPC is presented as a dot and whisker plot. Values shown are the means ± SD, n= 3 independent biological samples. Statistical analysis was performed using Student’s t-test and p value calculated, ***p < 0.001. iPLA2: calcium-independent phospholipase A2; LysoPC: lysophosphatidylcholine; TRPC6: canonical transient receptor potential 6 channel; EC: endothelial cell.
Previous studies in our lab showed that lysoPC caused an increase in [Ca2+]i in EC [12]. Furthermore, lysoPC induced a significant rise in [Ca2+]i in mouse aorta EC (MAEC) isolated from WT mice, but not in TRPC6−/− MAEC [12]. This provided strong evidence that lysoPC-induced TRPC6 externalization triggered an increase in [Ca2+]i. We recently showed that lysoPC activates iPLA2 to release ArA from EC membrane leading to TRPC6 externalization and an increase in [Ca2+]i [17]. To verify that FKGK11-dependent attenuation of lysoPC-induced TRPC6 externalization was accompanied by reduced [Ca2+]i, [Ca2+]i was measured by fluorometric assay. LysoPC (12.5 μmol/L) increased the relative [Ca2+]i in control cells to 2.02 ± 0.05 times the baseline (representative graph, Fig. 2C). In cells pretreated with FKGK11 (20 μmol/L), lysoPC increased the relative [Ca2+]i to 1.56 ± 0.05 times baseline (representative graph, Fig. 2D). FKGK11 significantly attenuated lysoPC-induced increase in [Ca2+]i in BAECs (n=3 independent biological samples, p < 0.001 compared with untreated cells with lysoPC, Fig. 2E). Cumulatively, the data showed that the iPLA2 inhibitor, FKGK11, inhibited lysoPC-induced TRPC6 externalization and attenuated the increase in [Ca2+]i.
3.3. FKGK11 Preserves EC Migration in Presence of LysoPC
LysoPC inhibits EC migration in WT MAEC but has little effect on migration of MAEC [12]. To evaluate pharmacological inhibition of iPLA2 on EC migration, a razor scrape EC migration assay was performed. Basal migration of medium control BAECs and FKGK11-pretreated BAECs was similar (n= 3 independent biological samples, Fig. 3). In the presence of lysoPC, EC migration was reduced by 78% (n= 3 independent biological samples, Fig. 3). However, for EC pretreated with FKGK11, lysoPC reduced migration by only 32% (n= 3 independent biological samples, p < 0.001, compared with untreated ECs with lysoPC, Fig. 3). The iPLA2 inhibitor partially preserved EC migration in presence of lysoPC.
Figure 3. FKGK11, an iPLA2 inhibitor, preserves EC migration in presence of lysoPC.
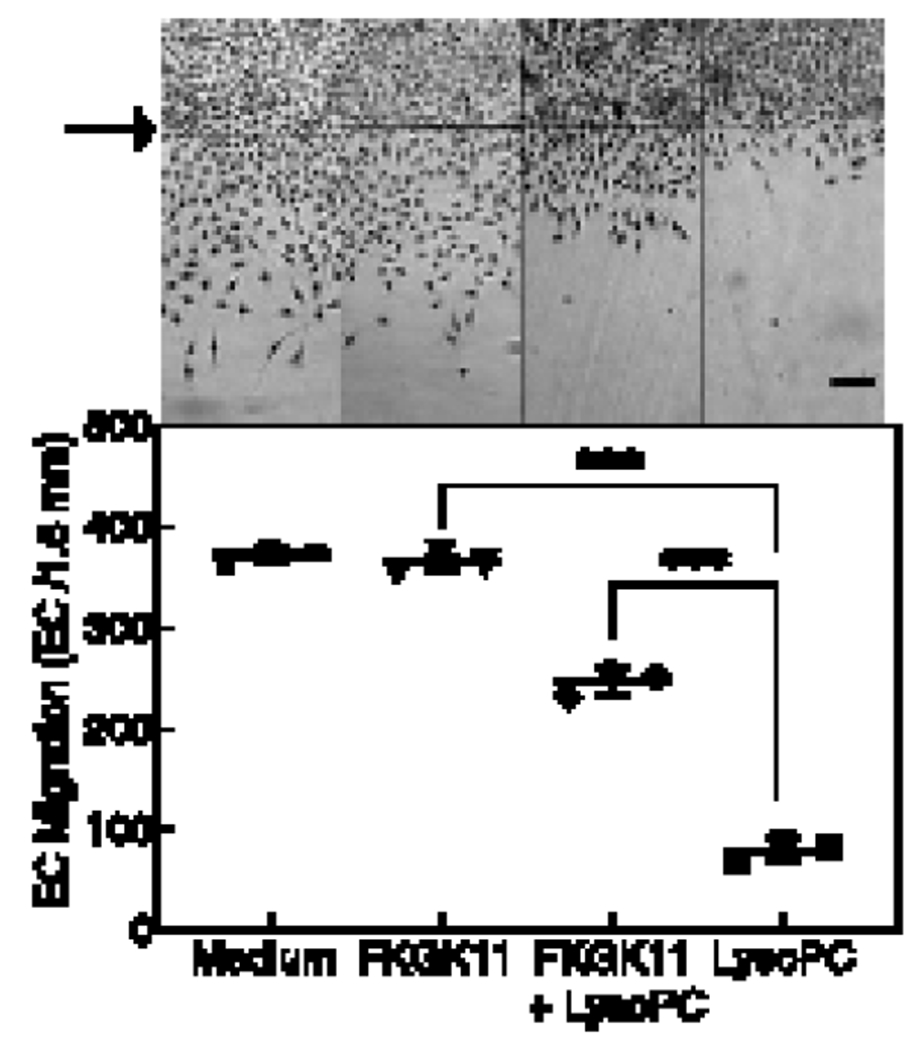
BAECs were serum-starved for 18 h, incubated with medium or FKGK11 (20 μmol/L) for 1 h, then migration assay was initiated. Migration in the presence or absence of lysoPC (12.5 μmol/L) was assessed at 24 h. Top: Representative images of migration at 40x magnification, scale bar 100 μm. The arrow indicates starting line of cell migration. Bottom: Dot and whisker plot of EC migration represented as means ± SD, n= 3 independent biological samples. Statistical analysis using one-way ANOVA with Tukey’s multiple comparison test was performed and p values calculated, ***p < 0.001. iPLA2: calcium-independent phospholipase A2; EC: endothelial cell; lysoPC: lysophosphatidylcholine; BAEC: bovine aortic endothelial cell.
3.4. FKGK11 Did Not Affect Weight, Plasma Cholesterol or Plasma LysoPC in Mice.
The effect of FKGK11 on EC healing in vivo was evaluated in C57Bl/6J mice. A total of 29 male and 30 female mice underwent an electrocautery injury to the RCCA. One male and two female mice were excluded because these mice died before completion of the study period. Mice at the age of six to eight weeks were placed on chow or high fat (HF) diet for four weeks. Half of the mice on each of the diets were treated with FKGK11 (5.6 mg/kg/day) starting 48 h prior to the injury and continued until 120 h post injury. Mice on the HF diet tended to gain more weight compared with mice on chow diet, however, weight of control mice and mice treated with FKGK11 was not significantly different (Table 1).
Table 1. Mouse data.
FKGK11, an iPLA2 inhibitor, does not affect diet-induced change in cholesterol or lysoPC levels in mice. C57Bl/6J mice on chow or HF diet for 4 weeks underwent electrical injury to the RCCA. Half the mice on each diet were treated with FKGK11 (5.6 mg/kg/day) intraperitoneally from 48 h prior to injury until 120 h postinjury. Values for weight (at the time of injury), cholesterol and lysoPC (at the time of sacrifice) are shown as means ± SD, n=14 mice for each study group.
| Group | # Mice, n | Weight, g | Cholesterol, mmol/L | LysoPC, μmol/L |
|---|---|---|---|---|
| Chow | 14 | 22.65 ± 4.2 | 2.12 ± 0.37 | 870.58 ± 175.2 |
| Chow + FKGK11 | 14 | 22.30 ± 4.1 | 2.15 ± 0.43 | 892.71 ± 242.8 |
| HFat | 14 | 25.64 ± 3.3 | 3.4 ± 0.65† | 1067.70 ± 115.0‡ |
| HFat + FKGK11 | 14 | 24.50 ± 2.6 | 3.2 ± 0.58†† | 1139.08 ± 187.7‡‡ |
Statistical analysis using one-way ANOVA with Tukey’s multiple comparison test was performed and p ≤ 0.05 was considered significantly different and shown in the table: for cholesterol
p < 0.001 Chow vs HF− and
p < 0.001 Chow + FKGK11 vs HF + FKGK11; for lysoPC
p = 0.01 Chow vs HF− and
p = 0.038 Chow + FKGK11 vs HF + FKGK11 Differences between Chow and Chow + FKGK11 or HF and HF + FKGK11 were not significant. iPLA2: calcium-independent phospholipase A2; lysoPC: lysophosphatidylcholine; RCCA: right common carotid artery; Chow: chow-fed mice; HF high fat-fed mice; Chow + FKGK11: chow-fed mice treated with FKGK11; HF + FKGK11: high fat-fed mice treated with FKGK11.
Plasma cholesterol was increased with the HF diet, and FKGK11 did not affect cholesterol levels. Cholesterol was 2.12 ± 0.37 mmol/L in chow-fed control mice and 2.15 ± 0.43 mmol/L in chow-fed mice treated with FKGK11 (n=14/group, Table 1). Plasma cholesterol in HF-fed control mice was 3.4 ± 0.65 mmol/L (n=14, p < 0.001, compared with chow-fed mice, Table 1). In HF-fed mice treated with FKGK11 plasma cholesterol was 3.2 ± 0.58 mmol/L (n=14, p < 0.001, compared with chow-fed mice treated with FKGK11, Table 1). These data indicated that FKGK11 did not have a significant effect on cholesterol levels in mice on a chow diet, and a high-fat diet increased plasma cholesterol in both control and FKGK11-treated mice.
Plasma lysoPC levels were measured as a marker of oxidative stress. FKGK11 treatment did not have a significant effect on plasma lysoPC. LysoPC was 870.58 ± 175.2 μmol/L in chow-fed mice and 892.71 ± 242.8 μmol/L in chow-fed mice treated with FKGK11 (Table 1). The lysoPC level was 1067.70 ± 115.0 μmol/L in mice on the HF diet (n=14, p =0.01, compared with chow-fed mice, Table 1). The lysoPC level in mice on the HF diet and treated with FKGK11 was 1139.08 ± 187.7 μmol/L (n=14, p = 0.038, HF + FKGK11 compared with chow-fed mice treated with FKGK11, Table 1). FKGK11 did not significantly affect the cholesterol or lysoPC values in mice on chow or HF diet.
3.4. FKGK11 Promotes Arterial Injury Healing in Hypercholesterolemic Mice
A HF diet decreases EC healing of arterial injuries in WT mice, but not in TRPC6−/− mice [13]. To determine if inhibition of iPLA2 and the subsequent blocking of TRPC6 externalization improved EC healing in vivo, arterial healing was quantitated at 120 h after electrical injury of the carotid artery in male and female mice on a chow or HF diet with or without FKGK11 treatment (n= 7/group). No significant difference in arterial healing between the two sexes was observed across different groups, therefore data from male and female mice were pooled for interpretation (Supplemental Table 1). Figure 4A, shows a representative image of carotid arteries at 120 h after electrical injury in control and FKGK11-treated mice on a chow or HF diet. Percent re-endothelialized area of the 5 mm injury at 120 h after injury was calculated and graphically represented in Fig. 4B. Arterial healing in chow-fed control mice was 50 ±9.3 % at 120 h (n=14, Fig. 4B). Arterial healing in chow-fed mice treated with FKGK11 was 49 ± 8.4 %, comparable to chow-fed control mice (n=14, Fig. 4B). HF diet significantly reduced arterial healing to 26 ± 6.9 % in control mice (n=14, p < 0.001, compared with chow-fed control mice, Fig. 4B). Arterial healing in HF mice treated with FKGK11 was 51 ± 13.1 %, similar to mice on a chow diet but significantly increased from HF-fed control mice (n=14, p < 0.001 compared with HF-fed control mice, Fig. 4B). These results indicated that the iPLA2 inhibitor, FKGK11, preserved EC healing in hypercholesterolemic mice.
Figure 4. FKGK11, an iPLA2 inhibitor, promotes re-endothelization of injured carotid artery in hypercholesterolemic mice.
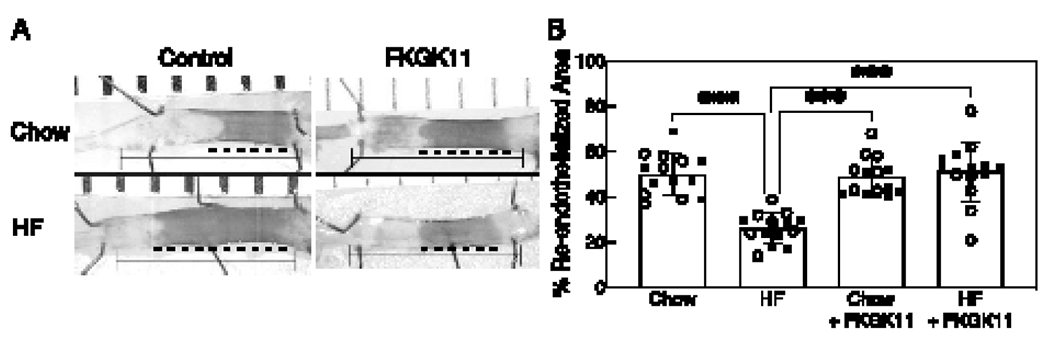
C57Bl/6J mice on chow or HF diet for 4 weeks underwent electrical injury to the RCCA. Half the mice on each diet were treated with FKGK11 (5.6 mg/kg/day) intraperitoneally from 48 h prior to injury until 120 h postinjury. At 120 h after injury, Evans blue dye was injected intravenously to identify the area lacking an intact endothelial monolayer. (A) Representative images of carotid arteries 120 h after electrical injury are shown, the line identifies the length of the initial 5 mm injury and the darker area and dotted line represents the unhealed portion of the injury; left: Control (PBS) and right: FKGK11 (FKGK11 in PBS). (B) Reendothelialization results are shown as the percent of re-endothelialized area relative to the total injured area. Results are represented as means ± SD, n=14 mice (7 male and 7 female) for each study group. Values for individual mice are also depicted (○, male; ●, female). Statistical analysis using one-way ANOVA with Tukey’s multiple comparison test was performed and p values calculated, ***p < 0.001. iPLA2: calcium-independent phospholipase A2; RCCA: right common carotid artery; Chow: chow diet; HF: high fat diet; Chow + FKGK11: chow diet with FKGK11 treatment; HF + FKGK11: high fat diet with FKGK11 treatment.
4. DISCUSSION
iPLA2-mediated TRPC6 externalization plays an important role in the inhibition of EC migration by lysoPC in vitro [17]. Specifically, siRNA mediated transient knockdown of iPLA2, but not cPLA, blocks lysoPC-induced TRPC6 externalization and inhibition of EC migration. Furthermore, we have shown that knockdown of the iPLA2β isoform, but not the iPLA2γ isoform, preserves EC migration in the presence of lysoPC. Based on these findings, the focus of the current study is to evaluate the potential of pharmacological inhibition of iPLA2 to prevent lysoPC-induced TRPC6 externalization and to preserve EC migration in vitro and to test the efficacy of this inhibitor to promote arterial healing in hypercholesterolemic mice. Our data show that FKGK11 attenuates lysoPC-induced PLA2 activity and ArA release from EC membranes in vitro. Blocking ArA release prevents lysoPC-induced TRPC6 externalization and limits the increase in [Ca2+]i, thus preserving EC migration in the presence of lysoPC in vitro. Importantly, FKGK11 promotes re-endothelialization of an arterial injury in hypercholesterolemic mice.
PLA2 enzymes can hydrolyze membrane phospholipids to release ArA, which can potentially activate ARC channels [33]. iPLA2β has been shown to play a critical role in the release and regulation of ArA in ECs [34–36]. Our previous study shows that siRNA-mediated iPLA2β downregulation in a human EC line (EAhy.926) reduces lysoPC-induced ArA release from EC membranes [17]. Oxidized LDL induces PLA2 activity in ECs, and inhibition of iPLA2 using BEL attenuates this PLA2 activity [37]. In our studies we use lysoPC, the major lysophospholipid component of oxidized LDL that contributes to the inhibition of EC migration [38]. which also induces PLA2 activity [17]. We show that pharmacological inhibition of iPLA2 using FKGK11 attenuates lysoPC-induced PLA2 activity and ArA release from BAEC membranes (Fig. 1). Furthermore, blocking ArA release with FKGK11 prevents lysoPC-induced TRPC6 externalization (Fig. 2) which is circumvented by the addition of exogenous ArA (Fig. S1). TRPC6 externalization by exogenous ArA with FKGK11+ lysoPC is lower than by ArA alone, but this may be a dose effect of ArA or FKGK11‘s effect on other pathways of TRPC6 activation. TRPC6 activation requires calcium [7], and restricting release of membrane ArA by iPLA2 prevents lysoPC-induced TRPC6 channel externalization, potentially by limiting calcium influx through ARC channels [17, 33, 39].
We selected FKGK11, a iPLA2-specific inhibitor, for our studies from the variety of potent PLA2 pharmacological inhibitors developed over the years [40]. One of the most potent iPLA2 inhibitors, bromoenol lactone (BEL) and its enantiomers S- and R-BEL [41], have been widely used in both in vitro and in vivo research studies [42]. Although selective for iPLA2 (a serine lipase), BEL inhibits other serine enzymes and has high toxicity owing to its interaction with cysteines [43]. Furthermore, BEL is not ideal for in vivo use due to its irreversible nature [44]. To mitigate the off-target effects of BEL, the next generation of iPLA2 inhibitors developed include the polyfluoroketone (FK) family, which is a group of highly specific and reversible iPLA2 inhibitors [25].
FKGK11 is a potent iPLA2β inhibitor, the mole fraction required for 50 % inhibition (Xi(50)) being 0.0073. FKGK11 has higher selectivity for iPLA2 than other subfamilies, with inhibition of > 95 % at 0.091 mole fraction, compared with 29 % inhibition of sPLA2 and 17 % inhibition of cPLA2 [25]. Thus, FKGK11 is a good candidate to inhibit iPLA2β mediated lysoPC-induced TRPC6 externalization. For our in vitro studies, we tested FKGK11 concentrations from 0.2- 200 μmol/L (data not shown) and found that FKGK11 at 20 μmol/L was sufficient to preserve EC migration in presence of lysoPC. In vitro cytotoxic effects were seen at 200 μmol/L (data not shown). In the literature, FKGK11has been used in vivo in mouse models of brain inflammation and epithelial ovarian cancer [20, 21, 24]. In these studies, FKGK11 was used at 2 mmol/L in 200 μL for each i.p injection which is equal to 5.6 mg/kg/day. This dose was found to be effective in inhibiting iPLA2 without significant cytotoxic effects. Based on literature we used the same concentration in our in vivo studies; however, we did not measure the plasma concentration of FKGK11. We find that i.p. injections of FKGK11 (5.6 mg/kg/day) in C57BI/6J mice over a period of seven days significantly improves re-endothelialization of injured carotid arteries in mice on HF diet with no overt toxicity or side effects.
Previously, we have shown in vitro that lysoPC significantly inhibits EC migration in WT MAEC but has little effect on TRPC6−/− MAEC migration [12]. Hypercholesterolemia and oxidative stress severely inhibit re-endothelialization of injured carotid arteries in WT mice [28], and this is in part due to TRPC6 activation [13]. A HF diet reduces 5-day EC healing of an electrical injury in mice by about 50 % in WT mice, but does not affect re-endothelialization of arteries in TRPC6−/− mice [13]. Our results show that FKGK11 prevents the inhibitory effect of the HF diet on EC healing in the carotid injury model. In fact, the results with FKGK11 are essentially identical to those seen in TRPC6−/− mice [13]. On average, there is no difference in healing between mice on a chow-fed diet and those on a HF-fed diet when the mice are treated with FKGK11. This suggests that an iPLA2β inhibitor could be a potential therapeutic agent to promote EC healing of vascular wounds in the clinical arena. Importantly, our studies incorporating and comparing male and female mice highlights that FKGK11 treatment is not sex dependent (Table S1 and Fig. 4), and may have therapeutic potential in male and female patients.
This study shows that administration of FKGK11 to C57Bl/6J mice promotes EC healing without affecting plasma cholesterol or lysoPC levels. The effect is assumed to be due to iPLA2 inhibition because FKGK11 is highly specific for iPLA2β, but off-target effects cannot be eliminated and certain limitations must be considered. The electrocautery injury model is a controlled electrical injury that denudes the endothelial layer of the carotid for a defined distance of 5 mm. EC from the adjacent healthy artery migrate into the denuded area to re-endothelialize the area of injury. Although it is unlikely that the iPLA2 activity or function of EC from the healthy artery is affected, given the current technological limitations, direct assessment of iPLA2 activity in vivo in the re-endothelialized layer was not measured. Also, the plasma concentration of FKGK11 was not measured in this study. C57Bl/6J mice have a naturally occurring null mutation of sPLA2 [46]. Therefore, results should be confirmed in other mouse strains that express sPLA2. Transient knockdown of cPLA2 suggests that cPLA2 plays a negligible role in lysoPC-induced TRPC6 externalization and inhibition of EC migration in vitro [17], but future studies should address the role of cPLA2 in lysoPC-induced TRPC6 externalization in vivo.
5. CONCLUSION
In conclusion, we show that the iPLA2 inhibitor, FKGK11, attenuates the lysoPC-induced release of ArA, rise in [Ca2+]i, TRPC6 externalization, and subsequent inhibition of EC migration in vitro. FKGK11 administration promotes endothelial healing of a carotid injury in mice on a HF diet. Together these results suggest that iPLA2 represents a novel target to prevent TRPC6 activation by lipid oxidation products in vivo and promote EC migration to restore arterial surface integrity after injury. For cardiovascular patients requiring angioplasty, short-term administration of an iPLA2 inhibitor could promote endothelial healing in hypercholesterolemic conditions and in atherosclerotic arteries with elevated levels of lipid oxidation products.
Supplementary Material
Figure S1. FKGK11, an iPLA2 inhibitor, does not prevent lysoPC-induced TRPC6 externalization in presence of exogenous ArA. (A-B) ECs were pre-incubated with medium or iPLA2 inhibitor (FKGK11, 20 μmol/L) for 1 h, followed with or without the addition of lysoPC (12.5 μmol/L) for 15 min and/or ArA (10 μmol/L) for 15 min as indicated. (A) Externalized TRPC6 was detected by biotinylation assay. Total TRPC6 was detected in an aliquot of cell lysate removed prior to biotinylation, and actin served as a loading control. A representative blot is shown of n= 3 independent biological samples. (B) Densitometric measurements of externalized TRPC6 is presented as a scatter plot; ●, medium; ◆; FKGK11 + lysoPC; ■, medium + lysoPC; ▼, FKGK11 + lysoPC + ArA; ▲, ArA. Statistical analysis using one-way ANOVA with Tukey’s multiple comparison test was performed and p values calculated, #p = 0.014, *p = 0.023 and **p = 0.004. iPLA2: calcium-independent phospholipase A2; lysoPC: lysophosphatidylcholine; TRPC6: canonical transient receptor potential 6 channel; ArA: arachidonic acid; EC: endothelial cell.
Table S1. FKGK11, an iPLA2 inhibitor, promotes arterial healing in male and female mice. Male and female C57Bl/6J mice on chow or high fat diet for 4 weeks underwent electrical injury to the RCCA. Half the mice on each diet were treated with FKGK11 (5.6 mg/kg/day) intraperitoneally from 48 h prior to injury until 120 h postinjury. Mean arterial healing (%) was calculated and statistical analysis using one way ANOVA with Tukey’s multiple comparison test was performed, n=7 mice for each study group. iPLA2: calcium-independent phospholipase A2; RCCA: right common carotid artery; M: male; F: female; Chow (M): chow-fed male; Chow (F): chow-fed female; HF (M): high fat-fed male; HF (F): high fat-fed female; Chow + FKGK11 (M): chow-fed male treated with FKGK11; Chow + FKGK11 (F): chow-fed female treated with FKGK11; HF + FKGK11 (M): high fat-fed male treated with FKGK11; HF + FKGK11 (F): high fat-fed female treated with FKGK11.
HIGHLIGHTS.
Calcium-independent phospholipase A2 (iPLA2) is selectively inhibited by FKGK11.
Calcium influx induced by lysophosphatidylcholine (lysoPC) is blocked by FKGK11.
FKGK11 blocks canonical transient receptor potential 6 channel externalization.
Endothelial cell migration in the presence of lysoPC is preserved by FKGK11.
Reendothelialization of arterial injury in high fat-fed mice is promoted by FKGK11.
Endothelial healing after angioplasty in patients could be promoted by FKGK11
ACKNOWLEDGMENTS
The authors would like acknowledge the Cleveland Clinic Flow Cytometry Core, an ISAC Accredited Shared Resources Laboratory with a special thanks to Kewal Asosingh, PhD, SCYM (ASCP), Scientific Director, and Amy Graham, MS, SCYM (ASCP), Principal Flow Cytometry Technologist for her technical assistance with calcium studies. The authors also thank Parag Joshi for his input during brainstorming and discussion sessions.
GRANTS
This work was supported by NIH National Heart, Lung, and Blood Institute Grant R01-HL-064357 (to L.M.G.) and by Career Development Award #IK2BX003628 and Merit Review #I01BX005823 from the United States (U.S.) Department of Veterans Affairs Biomedical Laboratory Research and Development Service (to M.A.R).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Abbreviations: LysoPC: lysophosphatidylcholine, TRPC6: canonical transient receptor potential 6, [Ca2+]i: intracellular calcium ion concentration, EC: endothelial cell, PLA2: phospholipase A2, iPLA2: calcium-independent PLA2, cPLA2: calcium-dependent PLA2, sPLA2: secretory PLA2, ArA: arachidonic acid, ARC: arachidonic acid regulated calcium channel, WT- wild type, DMEM: Dulbecco’s Modified Eagle’s Medium, FBS: Fetal Bovine Serum, HF: high fat, i.p.: intraperitoneal, RCCA: right common carotid artery, EAhy.926: human EC line, BEL: bromoenol lactone.
DISCLAIMERS
The contents of this article are solely the responsibility of the authors and do not represent the views of the National Institutes of Health, U.S. Department of Veterans Affairs or the United States Government.
DISCOLOSUERS
The authors declare that they have no conflicts of interest with the contents of this article.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Reference:
- 1.Pasternak RC, Baughman KL, Fallon JT, Block PC. Scanning electron microscopy after coronary transluminal angioplasty of normal canine coronary arteries. Am J Cardiol. 1980;45(3):591–8. [DOI] [PubMed] [Google Scholar]
- 2.Cornelissen A, Vogt FJ. The effects of stenting on coronary endothelium from a molecular biological view: Time for improvement? J Cell Mol Med. 2019;23(1):39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fishman JA, Ryan GB, Karnovsky MJ. Endothelial regeneration in the rat carotid artery and the significance of endothelial denudation in the pathogenesis of myointimal thickening. Lab Invest. 1975;32(3):339–51. [PubMed] [Google Scholar]
- 4.Frosen J, Tulamo R, Heikura T, Sammalkorpi S, Niemela M, Hernesniemi J, et al. Lipid accumulation, lipid oxidation, and low plasma levels of acquired antibodies against oxidized lipids associate with degeneration and rupture of the intracranial aneurysm wall. Acta Neuropathol Commun. 2013;1:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chaudhuri P, Colles SM, Damron DS, Graham LM. Lysophosphatidylcholine inhibits endothelial cell migration by increasing intracellular calcium and activating calpain. Arterioscler Thromb Vasc Biol. 2003;23(2):218–23. [DOI] [PubMed] [Google Scholar]
- 6.Chaudhuri P, Colles SM, Fox PL, Graham LM. Protein kinase Cdelta-dependent phosphorylation of syndecan-4 regulates cell migration. Circ Res. 2005;97(7):674–81. [DOI] [PubMed] [Google Scholar]
- 7.Chaudhuri P, Rosenbaum MA, Sinharoy P, Damron DS, Birnbaumer L, Graham LM. Membrane translocation of TRPC6 channels and endothelial migration are regulated by calmodulin and PI3 kinase activation. Proc Natl Acad Sci U S A. 2016;113(8):2110–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Aalst JA, Burmeister W, Fox PL, Graham LM. Alpha-tocopherol preserves endothelial cell migration in the presence of cell-oxidized low-density lipoprotein by inhibiting changes in cell membrane fluidity. J Vasc Surg. 2004;39(1):229–37. [DOI] [PubMed] [Google Scholar]
- 9.Kijani S, Vazquez AM, Levin M, Boren J, Fogelstrand P. Intimal hyperplasia induced by vascular intervention causes lipoprotein retention and accelerated atherosclerosis. Physiol Rep. 2017;5(14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Libby P, Schwartz D, Brogi E, Tanaka H, Clinton SK. A cascade model for restenosis. A special case of atherosclerosis progression. Circulation. 1992;86(6 Suppl):III47–52. [PubMed] [Google Scholar]
- 11.Chaudhuri P, Rosenbaum MA, Birnbaumer L, Graham LM. Integration of TRPC6 and NADPH oxidase activation in lysophosphatidylcholine-induced TRPC5 externalization. Am J Physiol Cell Physiol. 2017;313(5):C541–C55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaudhuri P CS, Bhat M, Birnbaumer VW, Graham LM. Elucidation of a TRPC6-TRPC5 channel cascade that restricts endothelial cell movement. Mol Biol Cell. 2008;19(8):3203–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosenbaum MA, Chaudhuri P, Graham LM. Hypercholesterolemia inhibits re-endothelialization of arterial injuries by TRPC channel activation. J Vasc Surg. 2015;62(4):1040–7 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hofmann T, Schaefer M, Schultz G, Gudermann T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci U S A. 2002;99(11):7461–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Earley S, Brayden JE. Transient receptor potential channels in the vasculature. Physiol Rev. 2015;95(2):645–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bon RS, Wright DJ, Beech DJ, Sukumar P. Pharmacology of TRPC Channels and Its Potential in Cardiovascular and Metabolic Medicine. Annu Rev Pharmacol Toxicol. 2022;62:427–46. [DOI] [PubMed] [Google Scholar]
- 17.Putta P, Smith AH, Chaudhuri P, Guardia-Wolff R, Rosenbaum MA, Graham LM. Activation of the cytosolic calcium-independent phospholipase A2 beta isoform contributes to TRPC6 externalization via release of arachidonic acid. J Biol Chem. 2021;297(4):101180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nikolaou A, Kokotou MG, Vasilakaki S, Kokotos G. Small-molecule inhibitors as potential therapeutics and as tools to understand the role of phospholipases A2. Biochim Biophys Acta Mol Cell Biol Lipids. 2019;1864(6):941–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramanadham S, Ali T, Ashley JW, Bone RN, Hancock WD, Lei X. Calcium-independent phospholipases A2 and their roles in biological processes and diseases. J Lipid Res. 2015;56(9):1643–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lopez-Vales R, Navarro X, Shimizu T, Baskakis C, Kokotos G, Constantinou-Kokotou V, et al. Intracellular phospholipase A(2) group IVA and group VIA play important roles in Wallerian degeneration and axon regeneration after peripheral nerve injury. Brain. 2008;131(Pt 10):2620–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kalyvas A, Baskakis C, Magrioti V, Constantinou-Kokotou V, Stephens D, Lopez-Vales R, et al. Differing roles for members of the phospholipase A2 superfamily in experimental autoimmune encephalomyelitis. Brain. 2009;132(Pt 5):1221–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paliege A, Roeschel T, Neymeyer H, Seidel S, Kahl T, Daigeler AL, et al. Group VIA phospholipase A2 is a target for vasopressin signaling in the thick ascending limb. Am J Physiol Renal Physiol. 2012;302(7):F865–74. [DOI] [PubMed] [Google Scholar]
- 23.Lopez-Vales R, Ghasemlou N, Redensek A, Kerr BJ, Barbayianni E, Antonopoulou G, et al. Phospholipase A2 superfamily members play divergent roles after spinal cord injury. FASEB J. 2011;25(12):4240–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, Zhao Z, Antalis C, Zhao Z, Emerson R, Wei G, et al. Combination therapy of an inhibitor of group VIA phospholipase A2 with paclitaxel is highly effective in blocking ovarian cancer development. Am J Pathol. 2011;179(1):452–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baskakis C, Magrioti V, Cotton N, Stephens D, Constantinou-Kokotou V, Dennis EA, et al. Synthesis of polyfluoro ketones for selective inhibition of human phospholipase A2 enzymes. J Med Chem. 2008;51(24):8027–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cayouette S, Lussier MP, Mathieu EL, Bousquet SM, Boulay G. Exocytotic insertion of TRPC6 channel into the plasma membrane upon Gq protein-coupled receptor activation. J Biol Chem. 2004;279(8):7241–6. [DOI] [PubMed] [Google Scholar]
- 27.Murugesan G, Chisolm GM, Fox PL. Oxidized low density lipoprotein inhibits the migration of aortic endothelial cells in vitro. J Cell Biol. 1993;120(4):1011–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosenbaum MA, Miyazaki K, Graham LM. Hypercholesterolemia and oxidative stress inhibit endothelial cell healing after arterial injury. J Vasc Surg. 2012;55(2):489–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carmeliet P, Moons L, Stassen JM, De Mol M, Bouche A, van den Oord JJ, et al. Vascular wound healing and neointima formation induced by perivascular electric injury in mice. Am J Pathol. 1997;150(2):761–76. [PMC free article] [PubMed] [Google Scholar]
- 30.D’Alessandro D, Neri E, Moscato S, Dolfi A, Bartolozzi C, Calderazzi A, et al. Immediate structural changes of porcine renal arteries after angioplasty: a histological and morphometric study. Micron. 2006;37(3):255–61. [DOI] [PubMed] [Google Scholar]
- 31.Rosenbaum MA, Miyazaki K, Colles SM, Graham LM. Antioxidant therapy reverses impaired graft healing in hypercholesterolemic rabbits. J Vasc Surg. 2010;51(1):184–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kodvawala A, Ghering AB, Davidson WS, Hui DY. Carboxyl ester lipase expression in macrophages increases cholesteryl ester accumulation and promotes atherosclerosis. J Biol Chem. 2005;280(46):38592–8. [DOI] [PubMed] [Google Scholar]
- 33.Shuttleworth TJ. Arachidonic acid, ARC channels, and Orai proteins. Cell Calcium. 2009;45(6):602–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rastogi P, McHowat J. Inhibition of calcium-independent phospholipase A2 prevents inflammatory mediator production in pulmonary microvascular endothelium. Respir Physiol Neurobiol. 2009;165(2-3):167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herbert SP, Walker JH. Group VIA calcium-independent phospholipase A2 mediates endothelial cell S phase progression. J Biol Chem. 2006;281(47):35709–16. [DOI] [PubMed] [Google Scholar]
- 36.Anfuso CD, Giurdanella G, Motta C, Muriana S, Lupo G, Ragusa N, et al. PKCalpha-MAPK/ERK-phospholipase A2 signaling is required for human melanoma-enhanced brain endothelial cell proliferation and motility. Microvasc Res. 2009;78(3):338–57. [DOI] [PubMed] [Google Scholar]
- 37.Lupo G, Nicotra A, Giurdanella G, Anfuso CD, Romeo L, Biondi G, et al. Activation of phospholipase A(2) and MAP kinases by oxidized low-density lipoproteins in immortalized GP8.39 endothelial cells. Biochim Biophys Acta. 2005;1735(2):135–50. [DOI] [PubMed] [Google Scholar]
- 38.Murugesan G, Fox PL. Role of lysophosphatidylcholine in the inhibition of endothelial cell motility by oxidized low density lipoprotein. J Clin Invest. 1996;97(12):2736–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wong JT TK, Pierce GN, Chan AC, O K, Choy PC. . Lysophosphatidylcholine stimulates the release of arachidonic acid in human endothelial cells. J Biol Chem. 1998;273(12):6830–6. [DOI] [PubMed] [Google Scholar]
- 40.Batsika CS, Gerogiannopoulou AD, Mantzourani C, Vasilakaki S, Kokotos G. The design and discovery of phospholipase A2 inhibitors for the treatment of inflammatory diseases. Expert Opin Drug Discov. 2021;16(11):1287–305. [DOI] [PubMed] [Google Scholar]
- 41.Jenkins CM, Han X, Mancuso DJ, Gross RW. Identification of calcium-independent phospholipase A2 (iPLA2) beta, and not iPLA2gamma, as the mediator of arginine vasopressin-induced arachidonic acid release in A-10 smooth muscle cells. Enantioselective mechanism-based discrimination of mammalian iPLA2s. J Biol Chem. 2002;277(36):32807–14. [DOI] [PubMed] [Google Scholar]
- 42.Ong WY, Farooqui T, Kokotos G, Farooqui AA. Synthetic and natural inhibitors of phospholipases A2: their importance for understanding and treatment of neurological disorders. ACS Chem Neurosci. 2015;6(6):814–31. [DOI] [PubMed] [Google Scholar]
- 43.Balsinde J, Dennis EA. Bromoenol lactone inhibits magnesium-dependent phosphatidate phosphohydrolase and blocks triacylglycerol biosynthesis in mouse P388D1 macrophages. J Biol Chem. 1996;271(50):31937–41. [DOI] [PubMed] [Google Scholar]
- 44.Song H, Ramanadham S, Bao S, Hsu FF, Turk J. A bromoenol lactone suicide substrate inactivates group VIA phospholipase A2 by generating a diffusible bromomethyl keto acid that alkylates cysteine thiols. Biochemistry. 2006;45(3):1061–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bao S, Miller DJ, Ma Z, Wohltmann M, Eng G, Ramanadham S, et al. Male mice that do not express group VIA phospholipase A2 produce spermatozoa with impaired motility and have greatly reduced fertility. J Biol Chem. 2004;279(37):38194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kennedy BP, Payette P, Mudgett J, Vadas P, Pruzanski W, Kwan M, et al. A natural disruption of the secretory group II phospholipase A2 gene in inbred mouse strains. J Biol Chem. 1995;270(38):22378–85. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. FKGK11, an iPLA2 inhibitor, does not prevent lysoPC-induced TRPC6 externalization in presence of exogenous ArA. (A-B) ECs were pre-incubated with medium or iPLA2 inhibitor (FKGK11, 20 μmol/L) for 1 h, followed with or without the addition of lysoPC (12.5 μmol/L) for 15 min and/or ArA (10 μmol/L) for 15 min as indicated. (A) Externalized TRPC6 was detected by biotinylation assay. Total TRPC6 was detected in an aliquot of cell lysate removed prior to biotinylation, and actin served as a loading control. A representative blot is shown of n= 3 independent biological samples. (B) Densitometric measurements of externalized TRPC6 is presented as a scatter plot; ●, medium; ◆; FKGK11 + lysoPC; ■, medium + lysoPC; ▼, FKGK11 + lysoPC + ArA; ▲, ArA. Statistical analysis using one-way ANOVA with Tukey’s multiple comparison test was performed and p values calculated, #p = 0.014, *p = 0.023 and **p = 0.004. iPLA2: calcium-independent phospholipase A2; lysoPC: lysophosphatidylcholine; TRPC6: canonical transient receptor potential 6 channel; ArA: arachidonic acid; EC: endothelial cell.
Table S1. FKGK11, an iPLA2 inhibitor, promotes arterial healing in male and female mice. Male and female C57Bl/6J mice on chow or high fat diet for 4 weeks underwent electrical injury to the RCCA. Half the mice on each diet were treated with FKGK11 (5.6 mg/kg/day) intraperitoneally from 48 h prior to injury until 120 h postinjury. Mean arterial healing (%) was calculated and statistical analysis using one way ANOVA with Tukey’s multiple comparison test was performed, n=7 mice for each study group. iPLA2: calcium-independent phospholipase A2; RCCA: right common carotid artery; M: male; F: female; Chow (M): chow-fed male; Chow (F): chow-fed female; HF (M): high fat-fed male; HF (F): high fat-fed female; Chow + FKGK11 (M): chow-fed male treated with FKGK11; Chow + FKGK11 (F): chow-fed female treated with FKGK11; HF + FKGK11 (M): high fat-fed male treated with FKGK11; HF + FKGK11 (F): high fat-fed female treated with FKGK11.