

Abstract
Rare species are vital members of a microbial community, but retrieving their genomes is difficult because of their low abundance. The ReadUntil (RU) approach allows nanopore devices to sequence specific DNA molecules selectively in real time, which provides an opportunity for enriching rare species. Despite the robustness of enriching rare species by reducing the sequencing depth of known host sequences, such as the human genome, there is still a gap in RU-based enriching of rare species in environmental samples whose community composition is unclear, and many rare species have poor or incomplete reference genomes in public databases. Therefore, here we present metaRUpore to overcome this challenge. When we applied metaRUpore to a thermophilic anaerobic digester (TAD) community and human gut microbial community, it reduced coverage of the high-abundance populations and modestly increased (∼2×) the genome coverage of the rare taxa, facilitating successful recovery of near-finished metagenome-assembled genomes (nf-MAGs) of rare species. The simplicity and robustness of the approach make it accessible for laboratories with moderate computational resources, and hold the potential to become the standard practice in future metagenomic sequencing of complicated microbiomes.
Microbial communities are composed of a high number of rare species (Jousset et al. 2017). Rare species play a vital role in ecosystem health and stability (Shade et al. 2014). For example, the slow-growing autotrophic microbes of ammonia-oxidizing bacteria or archaea (AOB/AOA) and anammox enable the rate-limiting step for natural nitrogen turnover (Kartal et al. 2010; Zhang et al. 2015). Rare species are also known as blind spots in biodiversity, like the extremely slow-growing “Asgard” archaea which provide crucial insights into the evolutionary emergence of eukaryotic cells in the history of life on our planet (Zaremba-Niedzwiedzka et al. 2017). Therefore, identifying the functional capacities of these rare species (a species with a relative abundance of <1% in the community) is essential to understanding the community dynamics and ecological function of a natural microbiome (Shade et al. 2014; Xiong et al. 2021).
The recovery of draft genomes (referred to as metagenome-assembled genomes, MAGs) from high-throughput metagenomic whole-genome sequencing (thereafter short as metagenomic) data sets ushered in a new era for understanding the ecological and evolutionary traits of the unculturable majority of natural microbiomes. However, high-quality (HQ, defined as >90% single copy gene [SCG]-completeness with <5% contamination and the presence of the 23S, 16S, and 5S ribosomal RNA [rRNA] genes and at least 18 tRNAs [Bowers et al. 2017]) MAGs recovery for low-abundance species is always difficult. Because the abundance distribution of the natural microbiome tends to follow a power law (Matthews and Whittaker 2015), the rare species are often missed or simply neglected in metagenomic sequencing. To get sufficient genome coverage of low-abundance species, extremely deep sequencing will be required. For example, Liu et al. sequenced 111.2 Gbytes paired-end short reads and 69.4 Gbytes long reads of a partial-nitrification anammox reactor to retrieved MAGs with low coverage (Liu et al. 2020). Also, based on regression analysis, a minimum of 186 Gbytes 454 reads (averagely 500 bp) are required for a full overlap-based assembly (without unassembled singleton) of soil community (Delmont et al. 2012). It would be a great waste of sequencing throughput as well as money if the study aims were to focus on rare species. Bioinformatic analysis can become more intractable during the data analyses and recovering unknown genomes from hundreds of gigabytes to terabytes of data is a massive computational challenge (Pop 2009). Furthermore, MAGs of rare taxa assembled from short-read data are often too fragmented to meet the necessity for understanding the effect of genome structure on function.
To effectively raise coverage of rare taxa from a high-abundance background, molecular biology-based methods including hybrid capture or CRISPR-Cas9 enrichment are adapted in library preparation to enrich the target (Gu et al. 2016; Gilpatrick et al. 2020). On the other hand, depletion of high-abundance species like saponin-based depletion (Charalampous et al. 2019) and osmotic lysis (Marotz et al. 2018) may serve the same purpose. What is evident, however, is that these approaches require the use of extra reagents and preparatory procedures. This is compounded by the fact that they require known information about the enrichment or depletion targets to design the experiment, which does not appear to work for enriching low-abundance species in natural communities with unknown compositions.
Unlike the endeavors made before sequencing, Nanopore sequencing (Oxford Nanopore Technology, ONT) users can program their system to reverse the voltage polarity of the sequencing pore to eject reads identified as not of interest, which provides a potential solution to enrich rare species in metagenomic samples. ONT supplied the ReadUntil (RU) programming interface and delegated the task of figuring out how to examine the raw pore signal to decide if a read was on-target to third-party developers. In the first published RU implementation, Loose et al. used the dynamic time warping (DTW) algorithms to compare the signal from the pore with precomputed reference signals for sequences of interest (Loose et al. 2016). However, this algorithm could not scale to references larger than millions of bases, which limits its widespread usage (Loose et al. 2016). With a similar goal of mapping streaming raw signal to DNA reference, UNCALLED has a lighter computational footprint than DTW (Kovaka et al. 2021). Still, it requires abundant computational resources. The newly designed Readfish toolkit eliminates the need for complex signal mapping algorithms and exploits existing ONT tools to provide a robust toolkit for designing and controlling selective sequencing experiments (Payne et al. 2021). Until now, the application of RU is principally limited to the elimination of known host species or the enrichment of known targets such as mitogenomes of blood-feeding insects (Gan et al. 2021; Martin et al. 2022; Kipp et al. 2023).
By ejecting dominant species while accepting low-abundance species, selective sequencing provides a potential solution to enrich rare species in metagenomic samples (Martin et al. 2022). However, enrichment for low-abundance species in real metagenomic samples by selective sequencing remains challenging because the community composition is never known. BOSS-RUNS, a Bayesian algorithm-based algorithm recently proposed by Weilguny et al., has only been applied in mock communities for the enrichment of low-abundance species without a prior reference, but it lacks practical applications in real complex communities (Weilguny et al. 2023). Readfish's native adaptive sampling keeps track of the most abundant species in a metagenomic sample and then depletes them to enrich rare taxa, but this strategy necessitates an Internet connection to retrieve references and is constrained by the reference database's composition (Payne et al. 2021). In fact, a large proportion of the species in a natural microbiome lacks a corresponding reference in public databases. To specifically address such a metagenomic issue and to realize effective targeted enrichment of rare species within a complicated environment microbiome, we introduced metaRUpore, a protocol for configuring selective nanopore sequencing and necessary bioinformatic scripts to achieve efficient enrichment of rare species within a complicated environment microbiome. By implementing metaRUpore, we aimed to show a novel approach for enriching low-abundance microorganisms in diverse environmental samples, which could advance our understanding of the microbial diversity and ecological dynamics in complex ecosystems.
Results
H. mediterrane enrichment in a mock community
To evaluate the performance of enriching low-abundance species with RU, we first constructed a mock community. The Haloferax mediterranei strain, which accounts for 1% of the mock community, was the target of our enrichment, whereas the other seven bacteria species were targets to be depleted during the RU run. In the mock run, a MinION flow cell was configured into two parts, in which the first half of the channels performed selective sequencing, and the other half performed normal sequencing as a control. In the RU channels, the reads were basecalled and then mapped to a 33-Mbyte reference (Supplemental Table S1) of all these eight microorganisms when they were being sequenced. A DNA molecule would be first sequenced for 0.4 sec before the obtained sequence was aligned to decide if it should be sequenced continually or ejected. The average length of rejected reads was 537 bases, which showed that the entire process of basecalling, mapping, and rejection decision could be completed in about 1.3 sec, based on the average nanopore sequencing speed of 400 bp/sec with R9.4.1 chemistry (Supplemental Fig. S1; Payne et al. 2021). In the RU-delivered data set, >99.9% of archaeal reads were kept, whereas >99% of bacterial reads were ejected (Supplemental Fig. S2). H. mediterranei was enriched to the absolute dominant population within the community with a proportion of accepted reads number of 62% in kept reads (Fig. 1A,B) with the coverage increased twice to 21.19× in RU data (Fig. 1C).
Figure 1.

Enriching low-abundance species in mock community with RU. (A) Bar plot of accepted reads number of the eight microbial species in RU and control runs. (B) Bar plot of the proportion of accepted reads number of the eight microbial species in RU and control runs. (C) Bar plot of the coverage of the eight microbial species’ genome in RU and control runs. (D) Heatmap of data yield per channel in RU and control runs, and (E) plot of the number of sequencing channels over the course of the sequencing run.
Despite the high rejection precision and fairly ideal enrichment result, it must be noted that the yield of selective sequencing channels was ∼60% lower than that of normal sequencing channels during the mock run (Fig. 1D). This reduction in throughput can be partly attributed to the increased idle time of each nanopore caused by a large number of ejections (Kovaka et al. 2021). At an enriched target prevalence of 1% within a community, each nanopore ejected an average of 2430 short fragments, whereas 267 contiguous long fragments were sequenced in a 7-h run. In addition, a rapid drop in active channels happed after 1-h sequencing in RU channels (Fig. 1E) and the effective pores were depleted after a 6-h runtime which was four times shorter than normal run whose pores could normally last for 24 h (Fig. 1E). This downward trend becomes slower as the proportion of enriched targets increases (Payne et al. 2021).
In situ metagenomic selective sequencing protocol and performance on TAD community
We introduced a pipeline, metaRUpore (https://github.com/sustc-xylab/metaRUpore), to selectively sequence rare populations in complex microbiome samples. The protocol consists of three consecutive steps (Fig. 2A): (1) a short-time normal sequencing (1 h for the community tested) to obtain an overall picture of the community structure and the genomic profile of the dominant populations, (2) bioinformatics analysis to determine the reference and target data set for optimized RU configuration, and (3) finally a 40-h selective sequencing for enriching rare populations in the sample. The pore control of the nanopore device was implemented by Readfish (Payne et al. 2021), which combines Guppy with minimap2 (Li 2018) to determine the eject/keep action for a pore.
Figure 2.

The workflow of metaRUpore and bioinformatic analysis. (A) The workflow of metaRUpore. (B) A MinION flow cell in metaRUpore is configured into two parts, 1/8th of the channels for normal sequencing and the remaining channels for selective sequencing. (C) The bioinformatic workflow for HQ-MAGs retrieval based on data sets derived from nanopore selective sequencing and Illumina sequencing.
Here we show our results in applying the metaRUpore protocol to facilitate the genome recovery of rare populations within the thermophilic anaerobic digester (TAD) community, which consists of 2977 OTUs with a Shannon index of 8.74, representing a typical diversity level of bioreactor systems (Supplemental Fig. S3). Genomic DNA of the TAD community was extracted by QIAGEN DNeasy PowerSoil Kit (100) and the libraries were constructed using the SQK-LSK109 kit. In order to improve the enrichment efficiency, we aim to retain longer DNA segments during the library construction process by removing short fragments using 0.4× beads at each step (Martin et al. 2022). ONT MinION flow cells v.R9.4.1 were used for all sequencing on an ONT GridION X5; see methods for more details. Rarefaction analysis showed that nanopore reads sequenced in the first 1 h normal sequencing already covered 90% of the overall diversity in the TAD community (Supplemental Fig. S4). Among the 125,606 reads sequenced in the first-hour normal sequencing, 66% of them could be assigned to a known reference by Centrifuge (Kim et al. 2016). All of these classified reads obtained in the first 1-h run were set as the target for ejection in subsequent RU run as it mostly consisted of the known and abundant populations within the community. It is worth noting that the classified reads obtained in the first 1-h normal sequencing predictably contain genomic fragments from the rare and unknown populations we intend to enrich, which will result in incomplete genome coverage of rare populations in the sequences obtained in the RU channels. Therefore, a small fraction of the channels still need to be set to normal sequencing in the subsequent 40-h RU run and the delivered data set needs to be assembled together with the RU-derived data sets. For our RU sequencing of the TAD community, we set 1/8 channels to normal sequencing (‐‐channels 1 448) (Fig. 2B). Our subsequent data analysis revealed that 29 HQ-MAGs would be missed if reads derived from selective sequencing were assembled alone. To further manipulate the selection, the users can manually select which taxa to keep during subsequent RU run; reads belonging to these taxa will be subtracted from the eject target data set based on their taxonomic affiliations determined by ARGpore2 (Wu et al. 2022). For example, in our TAD community, we intended to keep all the archaea reads, so we eliminated them from the ejection target data set. The entire aforementioned bioinformatic analysis can be completed in <5 min (Supplemental Text S4; Supplemental Table S2); such a short suspension of experimental work will not affect the flow cell chemistry and the subsequent RU run may directly start without refreshing the sequencing library.
The 40-h RU run on one flow cell delivered 6.84 Gbytes of effective long reads with an average read length of 3.46 kbp, whereas the normal sequencing channels produced 1.71 Gbytes reads with an average read length of 3.60 kbp (Supplemental Figs. S5–S7). To ensure adequate genome coverage, we sequenced the TAD community following metaRUpore protocol using three flow cells on GridION X5 (flow cells’ yield could be found in Supplemental Table S3). A marked change in the community structure was observed: As shown in the three-dimensional (3D) density plot of the phylogeny distribution of the overall TAD community (Fig. 3A), several density peaks of the original TAD community were depleted in the RU-run delivered data sets, indicating DNA of the high-abundance populations of the TAD community was effectively ejected during RU sequencing and the community became homogeneous with coverage of different populations becoming much more unified. Such unified coverage of different populations will help to minimize the disparity of k-mer frequency in the data set, preventing k-mers of the rare species from being filtered out as error-containing k-mers because of coverage drop during the k-mer-counting step of a de novo assembly algorithm (Nurk et al. 2017; Kolmogorov et al. 2020).
Figure 3.
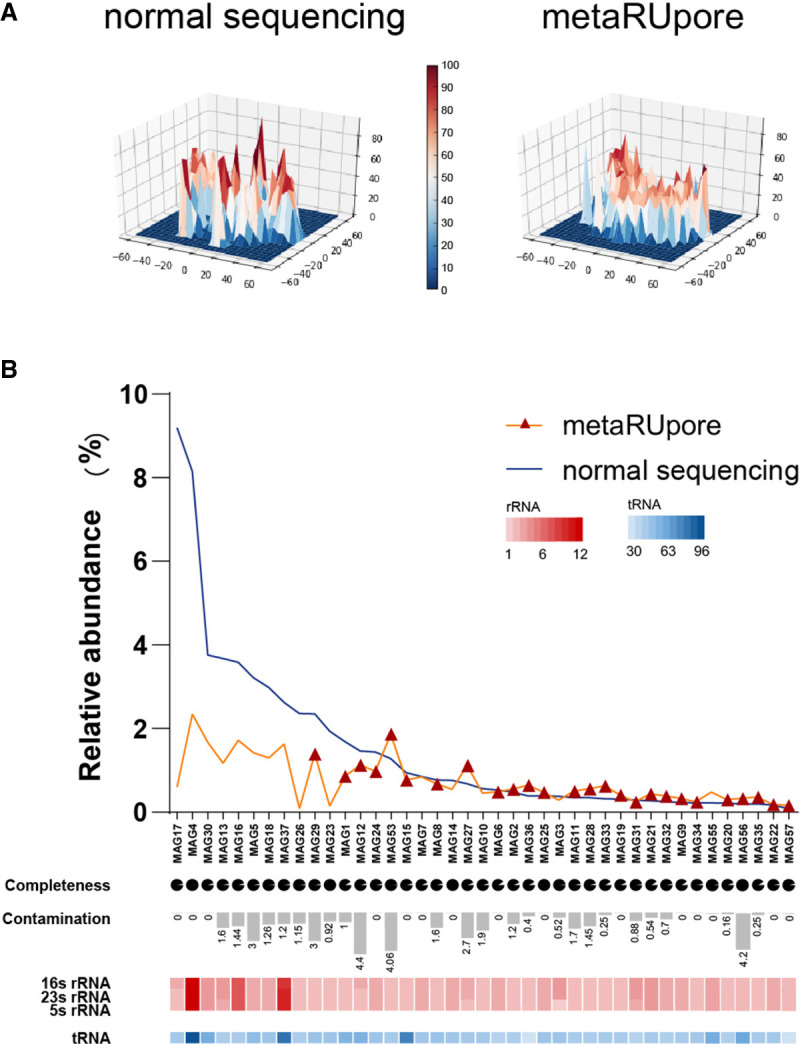
Performance of metaRUpore on recovery of high-quality MAGs in TAD community. (A) 3D density plots of t-SNE downscaling results for normal sequencing data sets and selective sequencing data sets by metaRUpore at four base frequencies, showing that metaRUpore renders the TAD community structure homogenous. (B) The distribution of 41 retrieved HQ MAGs in normal and RU sequencing data set. The red triangles indicate MAGs that could only be assembled in the metaRUpore data set. The pie chart and bar chart represent the level of genomic SCG-completeness and contamination by CheckM. The copy numbers of 16S rRNA, 23S rRNA, and 5S rRNA ribosomal RNA genes are represented by the red heatmap, whereas the copy number of tRNA is represented by the blue heatmap.
Bioinformatics pipeline for de novo metagenomic assembly and genome recovery
As illustrated in the assembly pipeline (Fig. 2C), the 25 Gbytes data generated by metaRUpore were assembled, respectively, using three different assemblers, namely Canu (Koren et al. 2017), Unicycler (Wick et al. 2017), and metaFlye (Kolmogorov et al. 2020). The basic statistics of assembled contigs were summarized in Supplemental Table S4. We defined HQ MAGs as having multiple rRNA ribosomal genes (23S/16S/5S), SCG-completeness >90%, and contamination <5%. Draft-quality (DQ) MAGs mean MAGs having >70% SCG-completeness, <10% contamination, and the presence of 16S rRNA genes, whereas if a MAG meets all of the DQ criteria but misses 16S rRNA, it was regarded as a low-quality (LQ) genome (Bowers et al. 2017). To improve the robustness of the binning, 139 > 1 Mbp contigs were first picked, as the candidate of the HQ genomes (Arumugam et al. 2021). The rest of the shorter contigs derived by the three assemblers were, respectively, binned by MetaBAT2 (Kang et al. 2019). Only contigs longer than 100 kbp were kept for subsequent binning (Supplemental Fig. S8). The MAGs retrieved above were subject to consensus correction by Medaka with nanopore data and polished by Pilon (Walker et al. 2014) with Illumina short reads (SRs). Next, polished MAGs were further corrected for frame-shift errors using MEGAN-LR (Huson et al. 2018) based on DIAMOND (Buchfink et al. 2015) alignment against the nr database (O'Leary et al. 2016). Finally, MAGs obtained by the different assemblers were deduplicated using dRep (Olm et al. 2017) with a relatedness threshold of ANI > 0.95 to obtain species-level representative MAGs (Supplemental Fig. S9). This pipeline is less time-consuming than the hybrid assembly and results in less fragmented MAGs. Totally, we obtained 46 draft-quality MAGs after dereplication. Among them, 41 MAGs including six complete circular genomes were HQ (Supplemental Fig. S10; Supplemental Table S5). All of these MAGs contained less than 13 contigs with an average N50 > 2 Mbp, demonstrating that they are highly continuous and meet the near-finished (nf) standard. In comparison, the normal nanopore sequencing data set yielded 29 draft-quality MAGs, including 16 HQ MAGs; 15 of them were included in the 41 HQ MAGs retrieved by metaRUpore strategy (Supplemental Fig. S10). The 26 HQ MAGs that are additionally obtained by RU-based selective sequencing were mainly from the rare populations of the TAD community (Fig. 3B); this proves that metaRUpore facilitates the recovery of more rare species genomes. Additionally, evident coverage reduction was observed in the dominant populations, such that the coverage of MAG17, MAG4, and MAG30, which together accounted for 21% of the TAD community, was reduced by 78% after RU-based selective sequencing (Fig. 3B; Supplemental Table S6), demonstrating the effectiveness of metaRUpore protocol in eliminating dominant populations. Despite the lowered overall throughput, coverage of the rare species MAG33, MAG35, MAG57, and MAG56 was doubled compared to normal sequencing at the current sequencing effort.
For nf-MAGs of rare species, a total of 562,883 reads were sequenced with the metaRUpore pipeline, which accounted for 15.5% of the total number of RU-accepted reads, whereas, in the normal sequencing, a total of 297,778 reads were detected for these species which accounted for 8.2% of the total number of sequencing reads. These results showed that metaRUpore effectively redirected the sequencing throughput to rare species. Notably, when applying metaRUpore, rare species could be identified based on their prevalence in the normal sequencing data set derived from the 1/8 normal sequencing channels or in the total reads (both ejected and received reads) as recommended by other researchers (Weilguny et al. 2023). Despite the largely unaltered community composition (Supplemental Fig. S11), the total reads obtained by selective sequencing might detect different rare taxa as compared to the normal sequencing data set. For the TAD community, compared to normal sequencing, the total reads derived by selective sequencing detected 903 extra species with the highest community prevalence reached to 0.23%, whereas 20 species cannot be covered (Supplemental Fig. S11). Owing to the unavoidable bias introduced by the substantial disparity in read length between ejected and received reads in the total reads, we used normal sequencing data set to define rare species in this study. Nevertheless, the normal sequencing-based quantification cannot quantify rare species only detected by RU methods. In this case, these species can only be regarded as extremely rare species in the consortia.
The 41 HQ MAGs introduce five new phyla, namely WOR-3, OLB16, Omnitrophota, Gemmatimonadota, and Deferribacterota, into the global HQ genome collection of the anaerobic digester (AD) microbiome (Campanaro et al. 2020). They also represent the first nf-MAGs for 21 phyla in the AD microbiome (Fig. 4; Campanaro et al. 2020). Furthermore, these MAGs show much better integrity and continuity than those in the previous collection assembled with SRs in terms of N50, number of contigs as well as the integrity of rRNA operon (Supplemental Table S7). Evolutional traits analysis reveals a much more conservative scale of gene flow based on nf-MAGs we assembled than that based on the previous collection (Supplemental Fig. S12; Supplemental Table S8; Alvarez-Ponce et al. 2013). An archaeal HQ-MAG retrieved from the TAD community revealed a giant (112 kbp) function-related genomic island, extending the evolutionary traits of the important Bathyarchaeota phylum (Supplemental Text S3; Supplemental Fig. S13).
Figure 4.

Phylogenomics of MAGs in anerobic reactor. A phylogenetic tree was constructed from 41 HQ-MAGs derived by metaRUpore (red branches) and 1108 HQ-MAGs collection derived from other AD systems (black branches). External circles represent, respectively: (1) taxonomic assignment at phylum level, (2) genome size (heatmap), (3) bar plot representing the genome continuity, which is calculated as the reciprocal of the number of contigs. The gray shaded areas indicate phyla with near-finished genomes obtained by metaRUpore, and the name of each phylum is in the legend on the left.
Enriching the rare flora of the human gut with metaRUpore
To testify to the generality of metaRUpore, we further applied metaRUpore in human gut consortia. The vast majority of reads in this human gut microbiota tested belong to bacteria, with only 1% classified as human hosts. Our aim is to enrich rare bacteria in the human gut flora. The same metaRUpore sequencing strategy that applied to the TAD community was used to sequence a human gut flora. The 40-h RU run on one flow cell delivered 1.17 Gbytes of effective long reads with an average read length of 1.57 kbp, whereas the normal sequencing channels produced 2.03 Gbytes reads with an average read length of 2.78 kbp (Supplemental Fig. S14). Notably, during selective sequencing of the human gut community with metaRUpore, we observed a high ejection ratio of around 98.0%, which is substantially higher than that of the TAD community (about 68.0%). And this high level of ejection rate is irrelevant to the duration of the pre-RU normal sequencing which neither shortening normal sequencing to 15 min nor extending it to 6 h showed a minimum effect in reducing ejection ratio (Supplemental Fig. S15). Therefore, we speculate such a high ejection rate was mainly associated with the exhaustive ejection of the dominant populations during RU sequencing. Given the extensive culturomics and metaomics studies of the human gut flora (Franzosa et al. 2014, 2019; Vatanen et al. 2019), nearly all the dominant populations in human gut flora were well documented in the public databases; thus, they were categorized as “classified” (defined as classifiable at phylum by Centrifuge) and included in the ejection target data set by metaRUpore. 88.6% of pre-RU normal sequencing data set of human gut flora was classified at phylum level, whereas the unknown proportion for TAD consortia could reach 32.2%. Despite the high ejection rate, comparable sequencing performance data was observed during RU sequencing of the human gut community. A similar effect of community homogenization (Supplemental Fig. S16) facilitated the recovery of 36.7% more nf-MAGs from the human gut community, with 2 MAGs showing community prevalence as low as 0.4% being recovered as circular MAGs.
Discussion
We have developed and tested the effectiveness of metaRUpore, a method for enriching low-abundance and undiscovered microorganisms in complex microbial communities based on nanopore selective sequencing. metaRUpore heuristically determines populations to reject through initial short-term de novo sequencing of the dominant populations. It overcomes the constraints imposed by the absence of reference genomes for complex natural communities and lowers the relative abundance limit for assembling HQ-MAGs (Fig. 5B). We elaborated the principle and process of metaRUpore and applied it to a TAD community as well as the human gut microbial community. MetaRUpore effectively increases the sequencing throughput of rare species, although the increase in the number of reads from rare taxa over baseline is modest (∼2×). Meanwhile, we show a robust and effective assembling and binning procedure for RU-based nanopore data sets, which facilitates the recovery of 41 nf-MAGs (defined as HQ-MAGs with <15 contigs) in the TAD community and 19 nf-MAGs in the human gut microbial community. Even though the structure and composition of communities changed before and after selective sequencing, metaRUpore is unlikely to introduce systematic biases in the genomic content of MAGs retrieved for rare species (Supplemental Fig. S17). Because environmental microbiomes typically contain a high proportion of genetic fragments that are distinct from all the sequences deposited in a whole-genome collection, this strategy will undoubtedly outcompete the whole-genome retrieval approach in the default metagenomic mode of Readfish in terms of ejecting dominant populations. MetaRUpore could unify the sequenced community structure and increase the genome coverage of low-abundance species, facilitating the assembly of additional nf-MAGs of rare species within a natural microbiota. 1-h normal sequencing is recommended as it achieved the best tradeoff between enrichment effectiveness and throughput loss for the TAD community tested, but this duration is fully adjustable according to the microbiome complexity of the user's sample. Noteworthy is that because the ejection target data set derived based on the first-hour normal sequencing predictably contains genomic fragments from the rare and unknown populations we intend to enrich, a small proportion of channels (1/8 is recommended) need to be set to normal sequencing during the 40-h RU-run to get a full genome coverage. And subsequent assembly procedure of metaRUpore protocol is based on the combined data set of RU-accepted and normal sequencing reads. Furthermore, if the ejection rate is getting too high (higher than 90%) for the selective sequencing channels, to reduce overall throughput loss, the user might consider enlarging the proportion of normal sequencing channels, which could be reconfigured by shortly suspending the current run.
Figure 5.

Performance and simulation of metaRUpore in retrieving MAGs of the rare species within the TAD microbiome at increasing sequencing depths. (A) The number of DQ MAGs retrieved for rare species by metaRUpore and normal nanopore sequencing. (B) Significant promotion in the minimum relative abundance to get HQ MAGs by metaRUpore as compared to normal nanopore sequencing (P-value is shown). The theoretical minimum value at a given sequencing data size was calculated as the lowest relative abundance of a species with a genome size of 3 Mb to reach 30× genome coverage.
Despite the fact that metaRUpore workflow is designed to encounter the challenge of lacking a reference genome when selectively enriching rare species in a metagenome without prior knowledge of community composition and available reference genome collections (e.g., environmental microbiomes), it could be configured to facilitate enriching microbial signals from a high-abundance mammalian DNA (human host) background. Because the host genome is available, the RU sequencing could be applied with the host genome merged into the established eject target set, facilitating a more effective host depletion by providing a more comprehensive reference set for ejection decision making. We tested the effect of this combine-reference approach on a playback selective sequencing of 10 clinical bronchoalveolar lavage fluid (BALF) samples which originally contains 99.2% human DNA background (Cheng et al. 2022). As shown in Supplemental Figure S18, the proportion of human reads retained after selective sequencing was significantly reduced by 38.7%–58.8% using the combined approach, demonstrating the robustness of metaRUpore in increasing reject sensitivity during selective sequencing. We used the most recent GRCh38 assembled version of the human genome as the target, and the eject efficiency is not significantly affected by the reference version of the human genome used, as both the hg19 and GRCh38 versions show consistent ejection efficiency (Supplemental Fig. S18). Noteworthy is that metaRUpore alone is not suitable to deplete DNA molecules of a mammalian host because the host genome is too big to be fully captured during the short-term normal sequencing step for ejection target data set establishment.
Because selective sequencing for the rare species is associated with a reduction in per-flow cell data yield, it is critical to establish an appropriate target proportion for selective sequencing to achieve the best tradeoff between enrichment effectiveness and throughput loss considering the total cost. We conducted a simulation of metaRUpore's MAGs retrieval capability at a different sequencing depth of the TAD microbiome to show its effectiveness as compared to regular nanopore sequencing. As shown in Figure 5A, with 15 Gbytes of nanopore reads respectively derived by metaRUpore and normal sequencing, metaRUpore data set can recover roughly twice as many MAGs of rare species than normal sequencing. Given normal nanopore sequencing by R 9.4 flow cell of a natural microbiome will produce roughly 9.36 Gbytes data according to previous reports (Supplemental Table S9), a roughly 17% per flowcell throughput reduction could be estimated for applying metaRUpore protocol on the TAD community. Therefore, the advantages of metaRUpore can outweigh the loss of data volume in terms of retrieving rare species’ MAGs. Moreover, this advantage will further enlarge with increasing sequencing depth based on the trend of the regression curve (Fig. 5A). PromethION flow cells, having around 1.5× the cost of MinION flow cells and around 5× the yield, may further improve rare species recovery through metaRUpore, as long as the basecaller can keep up with the throughput increase. Additionally, a greater proportion of rare species in more complicated natural microbiomes, plus a dedicated designed and adjustable rejection target during real-time selective sequencing, would all serve to ensure the efficacy of selective rare species enrichment by metaRUpore. Contrarily, if the computational cost is not taken into account, because of high ejection rate and considerable data loss, metaRUpore protocol may not provide a cost-saving advantage over directly increasing normal nanopore sequencing depth of human gut and alike samples whose dominant populations’ genomes have been extensively documented in public databases.
In conclusion, we have showed the effectiveness of metaRUpore which normalizes sequenced genome coverage and increases coverage of low-abundance species, facilitating the assembly of additional nf-MAGs of rare species within a complicated natural microbiota. It could be expected that by enhancing sequencing effort, HQ-MAGs could be obtained for populations with even lower prevalence using the metaRUpore protocol. Furthermore, metaRUpore protocol is robust and requires minimal modification to the experimental procedure of nanopore library construction and sequencing, making it easily applicable to metagenomic investigations of other environmental microbiomes to improve the time-to-answer in terms of sequencing costs and computational requirements.
Methods
Construction of the synthetic mock community
We synthesized a mock community of eight microorganisms, of which archaea accounted for 1% and the other seven bacteria species shared the rest equally based on DNA concentration determined from qubit average measurements. The archaeal species is H. mediterranei and these seven bacteria are Acinetobacter baumannii, Enterococcus faecalis, Escherichia coli, Klebsiella pneumoniae, Pseudomonas aeruginosa, Serratia marcescens, and Staphylococcus aureus. The seven bacterial strains and one archaeal strain were generously given by Professors L. Yang of the School of Medicine and Chuanlun Z. of the School of Oceanography at Southern University of Science and Technology. H. mediterranei was cultivated at 37°C overnight in nutrient-rich AS-168 L medium (per liter, 150 g NaCl, 20 g MgSO4·7H2O, 2 g KCl, 3 g trisodium citrate, 1 g sodium glutamate, 50 mg FeSO4·7H2 O, 0.36 mg MnCl2·4H2O, 5 g Bacto Casamino Acids, 5 g yeast extract and pH 7.2). Meanwhile, the seven bacteria were individually grown overnight in 3 mL of Luria-Bertani broth in a 12 mL tube at 37°C shaking at 200 rpm.
Sampling and DNA extraction
Genomic DNA of the eight microorganisms of the mock community was extracted by QIAamp DNA Micro Kit (50). Samples for the TAD community were taken when the methanogenic bacteria were at their highest activity. Human gut microbial community samples were taken from healthy adults. Genomic DNA of the TAD community and human gut microbial community were extracted by QIAGEN DNeasy PowerSoil Kit (100). DNA concentration was determined using the Thermo Fisher Scientific Qubit high-sensitivity assay kits. The quality of the DNA was measured by Thermo Fisher Scientific NanoDrop to assure that it all met the requirements for nanopore library construction. Short reads DNA Sequencing was performed at Novogene Co., Ltd. (Beijing, China) on the Illumina HiSeq platform with the PE150 strategy. Additionally, 16S rRNA amplicon sequencing was applied to evaluate the community diversity of the TAD microbiome (detailed procedure could be found in Supplemental Text S1).
Library construction and nanopore sequencing
All sequencing libraries were constructed using the ONT Ligation Sequencing Kit (no. SQK-LSK109) according to the manufacturer's instructions. When preparing the reactor sample libraries, to remove as many very short DNA fragments as possible, 0.4× beads (Beckman AMPure XP) were used for each step of the cleanup, and therefore the initial amount of genomic DNA was increased to 2 µg to ensure a sufficient amount of DNA of the final library.
Selective sequencing via metaRUpore
The execution of metaRUpore to enrich unknown low-abundance taxa is divided into the following three steps: firstly, a period (in this case 60 min) of normal sequencing is performed to generate reference file for selective sequencing. Next, the sequenced data is fed into metaRUpore to obtain the reference and target needed to configure Readfish TOML for selective sequencing. We put the reference and target paths into the TOML file and set config_name=“dna_r9.4.1_450 bps_fast”, single_on=unblock, multi_on=unblock, single_off=stop_receiving, multi_off=stop_receiving, no_seq=proceed, no_map=proceed. As recommended by the author of Readfish, we deactivated adapter scaling by editing the config files (dna_r9.4.1_450 bps_fast.cfg) in the guppy data directory. Next, selective sequencing was started. The configuration on MinKNOW was the same as for normal sequencing. Readfish runs at the same time as the sequencing starts. To use adaptive sampling directly in MinKNOW, enter the ejection target generated by metaRUpore into the “reference”, select the “deplete” option. Next, set channels by entering the channels you want to deplete and finally perform selective sequencing of depleting high-abundance species.
Analysis of long-read sequence data
Sequencing-derived FASTQ reads were adapter-trimmed using Porechop (https://github.com/rrwick/Porechop) (version 0.2.2) with default settings. These reads were subsequently assembled by the three tools: Canu (Koren et al. 2017) (version 2.2, default setting except -nanopore, genomeSize = 3 m, maxInputCoverage = 10,000, corOutCoverage = 10,000, corMhapSensitivity = high, corMinCoverage = 0, redMemory = 32, oeaMemory = 32, batMemory = 200 useGrid = false), Unicycler (Wick et al. 2017) (version 0.4.9b, default setting except -t 40, ‐‐keep 3), and metaFlye (Kolmogorov et al. 2020) (version 2.8.3, default setting except –nano-raw, ‐‐threads 50, ‐‐plasmids, ‐‐meta, ‐‐debug). Generated contigs that were at least 1 Mbp in length were regarded as potential whole-chromosome sequence. Among the remaining contigs that are <1 Mbp, we did metagenomic binning for the contigs that are >100 kbp in length. MetaBAT2 (Kang et al. 2019) (version 2.12.1 with default setting) is used to, respectively, bin the contigs assembled by above three assemblers.
Next, we took multiple steps to correct the >1 Mbp potential chromosome and bins we obtained. Firstly, we used nanopore data to perform consensus correction on them using Medaka (https://github.com/nanoporetech/medaka) (version 1.4.3, default setting except -t 20, -m r941_min_high_g360). They were then further corrected with the short reads data using Pilon (Walker et al. 2014) (version 1.24 with default setting except ‐‐fix all, ‐‐vcf). We used DIAMOND (Buchfink et al. 2015) (version 0.9.24) to align the Pilon polished potential chromosome (with default settings except -f 100 -p 40 -v ‐‐log ‐‐long-reads -c1 -b12) against the NCBI nr database (O'Leary et al. 2016) (download on July 2021). We used daa-meganizer in MEGAN Community Edition suite (Huson et al. 2016) (version 6.21.7, run with default settings except ‐‐longReads, ‐‐lcaAlgorithm longReads, ‐‐lcaCoveragePercent 51, ‐‐readAssignmentMode alignedBases) to format the .daa output file and receive frame-shift corrected sequence with “Export Frame-Shift Corrected Reads” option.
All the putative genomes were dereplicated using the dRep (Olm et al. 2017) (version 3.2.2, run with default setting except -p 40 -sa 0.95 –genomeInfo) to get species-level unique MAGs. We checked the SCG-completeness and contamination of these potential genomes with CheckM (Parks et al. 2015) (version v1.0.12, run with default setting except lineage_wf, -t 20). Next, gene annotations were obtained using Prokka (Seemann 2014) (version 1.13). Microbial taxonomic classifications were assigned using GTDB-Tk (Chaumeil et al. 2020) (version 1.3.0, GTDB-Tk reference data version r89). The abundance of each MAG is calculated by dividing the number of reads bases mapped to it by the total bases of selectively sequencing or normally sequencing, then normalizing by the size of the MAG (detailed methods could be found in Supplemental Text S2).
Ethics statement
The research was approved by the Ethical Committee of Southern University of Science and Technology (Approval Number: 20220237).
Data access
The raw nucleotide sequencing data (both Illumina and Nanopore) generated in this study have been submitted to the NCBI BioProject database (http://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA794848. MetaRUpore is available as free software from GitHub (https://github.com/sustc-xylab/metaRUpore) and as Supplemental Code.
Supplementary Material
Acknowledgments
This work was supported by the National Key Research and Development Program of China (Grant No. 2022YFE0103200) and the National Natural Science Foundation of China (Grants No. 42007216 and No. 42277103). We thank the Center for Computational Science and Engineering at Southern University of Science and Technology (SUSTech) and core research facilities at SUSTech for providing quality resources and services.
Author contributions: Y.X. and Y.S. designed the study. Y.S., Q.Y., and Z.W. performed experiments. Y.S., Z.C., and B.Z. analyzed and visualized the data. Y.S., X.L., and Y.X. prepared the manuscript. All authors read and approved the final manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277266.122.
Competing interest statement
The authors declare no competing interests.
References
- Alvarez-Ponce D, Lopez P, Bapteste E, McInerney JO. 2013. Gene similarity networks provide tools for understanding eukaryote origins and evolution. Proc Nat Acad Sci 110: E1594–E1603. 10.1073/pnas.1211371110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugam K, Bessarab I, Haryono MA, Liu X, Zuniga-Montanez RE, Roy S, Qiu G, Drautz-Moses DI, Law YY, Wuertz S, et al. 2021. Recovery of complete genomes and non-chromosomal replicons from activated sludge enrichment microbial communities with long read metagenome sequencing. NPJ Biofilms Microbiomes 7: 23. 10.1038/s41522-021-00196-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers RM, Kyrpides NC, Stepanauskas R, Harmon-Smith M, Doud D, Reddy TBK, Schulz F, Jarett J, Rivers AR, Eloe-Fadrosh EA, et al. 2017. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol 35: 725–731. 10.1038/nbt.3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B, Xie C, Huson DH. 2015. Fast and sensitive protein alignment using DIAMOND. Nat Method 12: 59–60. 10.1038/nmeth.3176 [DOI] [PubMed] [Google Scholar]
- Campanaro S, Treu L, Rodriguez-R LM, Kovalovszki A, Ziels RM, Maus I, Zhu X, Kougias PG, Basile A, Luo G, et al. 2020. New insights from the biogas microbiome by comprehensive genome-resolved metagenomics of nearly 1600 species originating from multiple anaerobic digesters. Biotechnol Biofuels 13: 25. 10.1186/s13068-020-01679-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charalampous T, Kay G, Richardson H, Aydin A, Baldan R, Jeanes C, Rae D, Grundy S, Turner DJ, Wain J, et al. 2019. Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nat Biotechnol 37: 783–792. 10.1038/s41587-019-0156-5 [DOI] [PubMed] [Google Scholar]
- Chaumeil PA, Mussig AJ, Hugenholtz P, Parks DH. 2020. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36: 1925–1927. 10.1093/bioinformatics/btz848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Sun YH, Yang Q, Deng MG, Yu ZJ, Zhu G, Qu JX, Liu L, Yang L, Xia Y. 2022. A rapid bacterial pathogen and antimicrobial resistance diagnosis workflow using Oxford nanopore adaptive sequencing method. Brief Bioinform 23: bbac453. 10.1093/bib/bbac453 [DOI] [PubMed] [Google Scholar]
- Delmont T, Prestat E, Keegan K, Faubladier M, Robe P, Clark IM, Pelletier E, Hirsch PR, Meyer F, Gilbert JA, et al. 2012. Structure, fluctuation and magnitude of a natural grassland soil metagenome. ISME J 6: 1677–1687. 10.1038/ismej.2011.197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzosa EA, Morgan XC, Segata N, Waldron L, Reyes J, Earl AM, Giannoukos G, Boylan MR, Ciulla D, Gevers D, et al. 2014. Relating the metatranscriptome and metagenome of the human gut. Proc Natl Acad Sci 111: E2329–E2338. 10.1073/pnas.1319284111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzosa EA, Sirota-Madi A, Avila-Pacheco J, Fornelos N, Haiser HJ, Reinker S, Vatanen T, Hall AB, Mallick H, Mclver LJ, et al. 2019. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol 4: 293–305. 10.1038/s41564-018-0306-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan M, Wu B, Yan G, Li G, Sun L, Lu G, Zhou W. 2021. Combined nanopore adaptive sequencing and enzyme-based host depletion efficiently enriched microbial sequences and identified missing respiratory pathogens. BMC Genom 22: 732. 10.1186/s12864-021-08023-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilpatrick T, Lee I, Graham JE, Raimondeau E, Bowen R, Heron A, Downs B, Sukmar S, Sedlazeck FJ, Timp W. 2020. Targeted nanopore sequencing with Cas9-guided adapter ligation. Nat Biotechnol 38: 433–438. 10.1038/s41587-020-0407-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Crawford E, O'Donovan B, Wilson M, Chow E, Retallack H, DeRisi JL. 2016. Depletion of Abundant Sequences by Hybridization (DASH): using Cas9 to remove unwanted high-abundance species in sequencing libraries and molecular counting applications. Genome Biol 17: 41. 10.1186/s13059-016-0904-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, Beier S, Flade I, Górska A, El-Hadidi M, Mitra S, Ruscheweyh HJ, Tappu R. 2016. MEGAN community edition-interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput Biol 12: e1004957. 10.1371/journal.pcbi.1004957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, Albrecht B, Bağcı C, Bessarab I, Górska A, Jolic D, Williams RB. 2018. MEGAN-LR: new algorithms allow accurate binning and easy interactive exploration of metagenomic long reads and contigs. Biol Direct 13: 6. 10.1186/s13062-018-0208-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jousset A, Bienhold C, Chatzinotas A, Gallien L, Gobet A, Kurm V, Kirsten Küsel K, Rillig MC, Rivett DW, Salles JF, et al. 2017. Where less may be more: how the rare biosphere pulls ecosystems strings. ISME J 11: 853–862. 10.1038/ismej.2016.174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang DD, Li F, Kirton E, Thomas A, Egan R, An H, Wang Z. 2019. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7: e7359. 10.7717/peerj.7359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kartal B, Kuenen JV, Van Loosdrecht MCM. 2010. Sewage treatment with anammox. Science 328: 702–703. 10.1126/science.1185941 [DOI] [PubMed] [Google Scholar]
- Kim D, Song L, Breitwieser FP, Salzberg SL. 2016. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res 26: 1721–1729. 10.1101/gr.210641.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipp EJ, Lindsey LL, Milstein MS, Blanco CM, Baker JP, Faulk C, Oliver JD, Larsen PA. 2023. Nanopore adaptive sampling for targeted mitochondrial genome sequencing and bloodmeal identification in hematophagous insects. Parasit Vectors 16: 68. 10.1186/s13071-023-05679-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolmogorov M, Bickhart DM, Behsaz B, Gurevich A, Rayko M, Shin SB, Kuhn K, Yuan J, Polevikov E, Smith TPL, et al. 2020. metaFlye: scalable long-read metagenome assembly using repeat graphs. Nat Methods 17: 1103–1110. 10.1038/s41592-020-00971-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. 2017. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27: 722–736. 10.1101/gr.215087.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovaka S, Fan Y, Ni B, Timp W, Schatz MC. 2021. Targeted nanopore sequencing by real-time mapping of raw electrical signal with UNCALLED. Nat Biotechnol 39: 431–441. 10.1038/s41587-020-0731-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100. 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Wang Y, Che Y, Chen YQ, Xia Y, Luo RB, Cheng SH, Zheng CM, Zhang T. 2020. High-quality bacterial genomes of a partial-nitritation/anammox system by an iterative hybrid assembly method. Microbiome 8: 155. 10.1186/s40168-020-00937-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loose M, Malla S, Stout M. 2016. Real-time selective sequencing using nanopore technology. Nat Methods 13: 751–754. 10.1038/nmeth.3930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marotz C, Sanders J, Zuniga C, Zaramela L, Knight R, Zengler K. 2018. Improving saliva shotgun metagenomics by chemical host DNA depletion. Microbiome 6: 42. 10.1186/s40168-018-0426-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S, Heavens D, Lan Y, Horsfield S, Clark MD, Leggett RM. 2022. Nanopore adaptive sampling: a tool for enrichment of low abundance species in metagenomic samples. Genome Biol 23: 11. 10.1186/s13059-021-02582-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews TJ, Whittaker RJ. 2015. On the species abundance distribution in applied ecology and biodiversity management. J Appl Ecol 52: 443–454. 10.1111/1365-2664.12380 [DOI] [Google Scholar]
- Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. 2017. metaSPAdes: a new versatile metagenomic assembler. Genome Res 27: 824–834. 10.1101/gr.213959.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, et al. 2016. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44: D733–D745. 10.1093/nar/gkv1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olm MR, Brown CT, Brooks B, Banfield JF. 2017. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J 11: 2864–2868. 10.1038/ismej.2017.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25: 1043–1055. 10.1101/gr.186072.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne A, Holmes N, Clarke T, Munro R, Debebe BJ, Loose M. 2021. Readfish enables targeted nanopore sequencing of gigabase-sized genomes. Nat Biotechnol 39: 442–450. 10.1038/s41587-020-00746-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop M. 2009. Genome assembly reborn: recent computational challenges. Brief Bioinform 10: 354–366. 10.1093/bib/bbp026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30: 2068–2069. 10.1093/bioinformatics/btu153 [DOI] [PubMed] [Google Scholar]
- Shade A, Jones SE, Caporaso JG, Handelsman J, Knight R, Fierer N, Gilbert JA. 2014. Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity. mBio 5: 01371-14. 10.1128/mBio.01371-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatanen T, Plichta DR, Somani J, Münch PC, Arthur TD, Hall AB, Rudolf S, Oakeley EJ, Ke X, Young RA, et al. 2019. Genomic variation and strain-specific functional adaptation in the human gut microbiome during early life. Nat Microbiol 4: 470–479. 10.1038/s41564-018-0321-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, et al. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9: e112963. 10.1371/journal.pone.0112963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weilguny L, De Maio N, Munro R, Manser C, Birney E, Loose M, Goldman N. 2023. Dynamic, adaptive sampling during nanopore sequencing using Bayesian experimental design. Nat Biotechnol 10.1038/s41587-022-01580-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick RR, Judd LM, Gorrie CL, Holt KE. 2017. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol 13: e1005595. 10.1371/journal.pcbi.1005595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Che Y, Dang CY, Zhang M, Zhang XY, Sun YH, Li X, Zhang T, Xia Y. 2022. Nanopore-based long-read metagenomics uncover the resistome intrusion by antibiotic resistant bacteria from treated wastewater in receiving water body. Water Res 226: 119282. 10.1016/j.watres.2022.119282 [DOI] [PubMed] [Google Scholar]
- Xiong C, He J, Singh BK, Zhu Y, Wang J, Li P, Zhang Q, Han L, Shen J, Ge A, et al. 2021. Rare taxa maintain the stability of crop mycobiomes and ecosystem functions. Environ Microbiol 23: 1907–1924. 10.1111/1462-2920.15262 [DOI] [PubMed] [Google Scholar]
- Zaremba-Niedzwiedzka K, Caceres EF, Saw JH, Bäckström D, Juzokaite L, Vancaester E, Seitz KW, Anantharaman K, Starnawski P, Kjeldsen KU, et al. 2017. Asgard archaea illuminate the origin of eukaryotic cellular complexity. Nature 541: 353–358. 10.1038/nature21031 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen L, Dai T, Tian J, Wen D. 2015. The influence of salinity on the abundance, transcriptional activity, and diversity of AOA and AOB in an estuarine sediment: a microcosm study. Appl Microbiol Biotechnol 99: 9825–9833. 10.1007/s00253-015-6804-x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.