

Abstract
Fragmentation ion spectral analysis of chemically crosslinked proteins is an established technology in the proteomics research repertoire for determining protein interactions, spatial orientation, and structure. Here we present Kojak version 2.0, a major update to the original Kojak algorithm, which was developed to identify crosslinked peptides from fragment ion spectra using a database search approach. A substantially improved algorithm with updated scoring metrics, support for cleavable crosslinkers, and identification of crosslinks between 15N-labeled homomultimers are among the newest features of Kojak 2.0 presented here. Kojak 2.0 is now integrated into the Trans-Proteomic Pipeline, enabling access to dozens of additional tools within that suite. In particular, the PeptideProphet and iProphet tools for validation of crosslinks improve the sensitivity and accuracy of correct crosslink identifications at user-defined thresholds. These new features improve the versatility of the algorithm, enabling its use in a wider range of experimental designs and analysis pipelines. Kojak 2.0 remains open-source and multi-platform.
Keywords: crosslinking mass spectrometry, XL-MS, Kojak, Trans-Proteomic Pipeline, PeptideProphet, iProphet, proteomics, computational proteomics, software tools, protein interaction
Graphical Abstract:
Introduction
Shotgun mass spectrometry (MS/MS) analysis of chemically crosslinked proteins (XL-MS) has become a versatile tool in the field of proteomics.1,2 Data analysis of crosslinked proteins has unique challenges; database and spectral library searching algorithms for standard shotgun analyses are not readily extensible to the analysis of chemically crosslinked proteins. In response to this shortcoming, many analytical tools have been developed to meet the unique challenges of crosslinking spectral analysis and have been recently reviewed and evaluated.3 These tools are diverse in their functionality, capable of analyzing data from static4–7 and cleavable crosslinkers8,9, and can incorporate isotope labeling into the crosslinkers, e.g. as shown in10. Alternatively, the tools must also be able to analyze isotopically labeled proteins.11 Additional tools incorporate such information into larger analysis pipelines to visualize structure12–14 and interaction networks15.
Kojak was first developed as a modern implementation of a database search algorithm for shotgun MS analysis of chemically crosslinked proteins16. At its core, Kojak emulated some of the key features of the Comet algorithm for standard shotgun proteomics analyses17, and applying these features to the identification of crosslinked peptide sequences from MS/MS spectra. Foremost, Kojak was designed to be computationally efficient, capable of analysis with many different crosslinkers on both small and large datasets. The simple interface of the Kojak software, combined with adherence to open data standards, enabled its use with a diverse set of experimental conditions, analytical platforms, and pre- and post-analysis pipelines.
Development of Kojak has continued since its publication. These developments have culminated in Kojak version 2.0, which has several improvements and new features. These improvements include support for additional open formats and standards, further refinement to the search algorithm for efficiency, E-values to normalize the scores of the results, support for cleavable crosslinkers, and methods to identify crosslinks between homomultimer subunits. Kojak 2.0 is integrated into the Trans-Proteomic Pipeline (TPP), and we use PeptideProphet and iProphet from the TPP to assign a probability value to each crosslinked-spectrum match (CSM) returned from Kojak. These probability values are then used to estimate error rates and determine which Kojak results are accepted at a user-defined error threshold. We show that this new pipeline developed around Kojak is both sensitive and accurate in the crosslinks identified, an area of the field that has recently received additional scrutiny in an effort to improve XL-MS technology.1,18–21 Here we present results highlighting the latest features of Kojak 2.0, and describe their use in the analysis of chemically crosslinked proteins.
Methods
Search database
Kojak 2.0 has multiple features for the creation and use of tailored protein sequence databases in the search analysis. Users provide a FASTA file containing presumed protein sequences to search. If validation after Kojak analysis requires decoy protein sequences to be searched, the user can opt to provide those sequences with a label in their FASTA file, or request Kojak generate decoys for them. Kojak’s decoy generation algorithm fixes in place all digestion enzyme sites and reverses the amino acid sequences between them. This approach produces an equal number of decoy peptide sequences as target peptide sequences and identical masses. Thus, for every target peptide sequence searched against a spectrum, an equivalent decoy sequence is also searched against that spectrum. The decoy generation algorithm is dynamic to any enzyme digestion rule provided to Kojak, so that it can be used regardless of the digestion enzyme used. Palindromic sequences are mitigated through additional amino acid swaps. Kojak also recognizes protein name labels for isotopically-labeled proteins, to distinguish these protein sequences from unlabeled protein sequences during the search process. The details of this feature are described in the methods below.
Identification of linked peptides
A two-pass approach is used to identify linked peptide pairs, similar to a previously described approach22. In the first pass, candidate peptides for the larger peptide mass are obtained through fragment ion matching the peptide sequence to the observed spectrum. In this pass, a candidate peptide is searched against a spectrum if it contains a site for binding to the crosslinker and its mass satisfies the following equation:
| eq. 1 |
Where is the mass of the peptide in consideration, is the predicted precursor mass, is the mass of the crosslinker, and is the user-defined smallest allowed peptide mass in the analysis. This equation is applicable to all spectra for which is greater than or equal to .
The mass of the unknown complementary peptide linked to the candidate peptide is computed as:
| eq.2 |
where the complementary peptide mass () is the difference between the precursor mass () and the mass of the peptide in consideration (). is treated as a modification mass on the potential site of linkage of the peptide. This mass can be moved to other sites on the peptide if it contains additional potential sites of linkage. Additionally, any differential modification masses arising from post-translational modifications (PTMs) or chemical modifications are considered. A dynamic tally of the best scoring peptides (i.e. those peptides with the highest cross-correlation score) in this first pass are maintained for each spectrum at a user-defined size, with recommendations between 5 and 15 peptides.
In the second pass, only peptide sequences whose masses sum to the predicted precursor mass with any of the short list of sequences from the first pass are searched. Additionally, only peptides containing a valid crosslinker binding site are considered. This second stage greatly reduces the number of peptide combinations that must be considered, and further prioritizes those combinations to the most likely candidate sequences based on fragment ion match information obtained in the first pass. This approach differs significantly from previous versions of Kojak, which would search all peptides and attempt to find two peptides among a list of hundreds of candidates that sum to the precursor ion mass. This approach is also algorithmically faster than the previous method used in Kojak. This improvement, combined with other improvements in software engineering has reduced computation time to less than 25% of the previously published version of Kojak.
Improved scoring metrics for candidate crosslinked peptides
Kojak uses a modified form of the Comet fast cross-correlation algorithm17,23. Because this cross-correlation score (Xcorr) is influenced by peptide length, expectation values (E-values) are now computed in Kojak 2.0, which are more useful than the cross-correlation score when performing downstream CSM validation. The E-value is computed from a linear least squares regression of the log transform of the cumulative distribution function of the histogram of all cross-correlation scores to that MS2 spectrum. The user can define a minimum size of the histogram. If insufficient cross-correlation scores were recorded, additional cross-correlation scores to random peptide sequences of the same approximate mass are computed and added to the histogram. Additional E-value calculations are also performed for the individual peptides in the crosslinked sequence pair. In these cases, histograms are generated at a user defined size to include randomized peptide sequences of equivalent mass to the observed sequence, with a randomized site of linkage to the complementary peptide. These E-values better represent how good a Xcorr score is given the peptide length and observed spectrum peaks and are preferred to Xcorr scores when evaluating CSMs.
Isotope labeling for identification of protein dimer interactions
Differentiation between self-linked proteins and crosslinks between different subunits in homodimers and homomultimers is performed using 15N-labeled techniques24,25. Briefly, the protein of interest is purified in both normal and 15N-labeled forms. The resulting mass spectra following crosslinking and acquisition of data by mass spectrometry are then analyzed with Kojak 2.0 using the new 15N_filter parameter. This parameter specifies a unique identifier word that is added to the FASTA protein identification line for the protein sequence that is to be analyzed with mass adjustments pertaining to the number of nitrogen atoms in each peptide from that protein sequence. For example, if a mixed normal and 15N-labeled “protein X” were crosslinked, the FASTA file would contain (1) the sequence for protein X, identified as “>protein-X”, and (2) the sequence repeated for protein X, identified as, “>15n_protein-X”. The 15N_filter parameter would be set to “15n_”, indicating that masses from peptides from “>15n_protein-X” are to be adjusted for the heavy nitrogen, while the masses from peptides from “>protein-X” are to be calculated as normal. In this manner, peptides of identical sequence will have different masses depending on the protein sequence of origin. Crosslinks identified as containing a normal and a 15N-labeled peptide are therefore evidence of interaction between two separate protein subunits. For efficiency, it is not necessary for Kojak to search 15N-labeled peptides for every sequence in the database, instead limiting this step to only the proteins that were labeled in the experiment.
Cleavable crosslinker analysis
Mass spectrometry cleavable crosslinkers contain one or more labile bonds that break during collision-induced dissociation. When performing MS/MS analysis of peptides linked with cleavable crosslinkers, bond breakage can occur both along either peptide and at the labile crosslinker bonds. This creates fragment ion series that contain either the intact crosslinker and complement peptide, or simply a small mass addition equal to the remaining crosslinker after breaking at its labile bond. These predictable fragment ion products can be exploited to improve identification of crosslinked peptide sequences.26 When a cleavable crosslinker is specified, Kojak considers the additional product masses that occur from breakage of the labile crosslinker bonds, using these masses as additional evidence when scoring CSMs.
Crosslink peptide spectrum match validation
The use of the Trans-Proteomic Pipeline (TPP)27 software tools of PeptideProphet28 and iProphet29 to validate the spectrum matches produced by Kojak has been previously suggested.30 Here, these software tools were updated for the purposes of reading the results and scores as encoded by Kojak in pepXML format and modeling the new types of results produced within the framework of the TPP. The original PeptideProphet method for validation of Kojak CSM results relied on modeling the second-best expectation score in the crosslinked peptide combination as the PeptideProphet f-value, while the second-best Kojak computed score and the top best expectation score were used as additional discriminant models to improve the sensitivity of the PeptideProphet classifier. This approach was not successful in generating accurate or conservative probabilities when applied to Kojak 2.0 results and the validation for the new version of Kojak required a retooling of the models and software applied. The updates to validation software models in PeptideProphet, used to assign probabilities on the level of individual CSMs, consisted of only modeling the second-best expectation score and disabling the additional two discriminant models. This served to improve the accuracy of the estimated probabilities at the cost of reduced sensitivity in the classification of the CSMs. The additional discriminant models described are now optional in the software and can be enabled when running PeptideProphet by options XLSECOND and XLTOPEXP. Using PeptideProphet with the optional EXPECTSCORE flag enabled causes PeptideProphet to use the combined expectation score for the crosslinks as the f-value. Additionally, the models in iProphet have been extended for Kojak 2.0 with the integration of an optional model called HETEROXL. This boolean model computes the likelihoods of observing crosslinks of two peptides from different proteins (i.e., hetero-protein link), versus crosslinks of two peptides from the same protein (i.e., self-protein link), among correct and among incorrect matches, as determined using the protein identifier assigned to each peptide sequence in a crosslink. Like the other models in iProphet, the HETEROXL model is learned by iProphet using the Expectation Maximization algorithm and applied to the results to adjust the probabilities of the CSMs (supplementary figure 1). Although, making these changes to the PeptideProphet model reduced the sensitivity of the PeptideProphet analysis, while improving the accuracy; iProphet with its models (including HETEROXL) enabled is able to recover the sensitivity of the classification given the accurate starting probabilities resultant from the updates to the models in PeptideProphet.
Bovine Arp2/3 Complex Sample Preparation
Bovine Arp2/3 complex was purified from calf thymus (Pel-Freez) as previously described31 with an additional ion exchange column (MonoQ) step used as a final polishing step. After loading on the MonoQ column, the complex was eluted with a gradient of 25–300 mM NaCl in 10mM Tris pH 8.0, 1 mM DTT. Pure fractions were dialyzed into 20 mM Tris pH 8.0, 50 mM NaCl, concentrated, and flash frozen.
Prior to crosslinking, the Arp2/3 complex was exchanged into HB100D (40 mM HEPES, 100 mM NaCl, 1mM DTT, pH 7) using Pierce Polyacrylamide Spin Desalting Columns (Catalog number: 89849, Thermo Fisher, Scientific). Crosslinking reactions were 100 μL and consisted of 25.32 uL Arp2/3 (60 μg total protein) plus 270 μL HB100D plus 2.34 μL 25 mM CK-666 Arp2/3 complex inhibitor (Millipore-Sigma) plus 9.96 μL 14.5 mM BS2 (BS2G-d0, Thermo Fisher Scientific) in HB100D or 9.96 μL of DSS (Thermo Fisher Scientific) in DMSO. Reactions were mixed and incubated for 10 mins in an Eppendorf Thermomixer at 21°C shaking at 1,000 rpm after which 100 μL was quenched by transfer to fresh 1.5 mL Eppendorf tube containing 10 μL 1M ammonium bicarbonate plus 1 μL 2M β-Mercaptoethanol. Reactions were reduced for 30 mins at 37°C with 10 mM dithiothreitol (DTT) and alkylated for 30 mins at room temperature with 15 mM iodoacetamide. Trypsin digestion was performed at 37°C for 4 hours with shaking at a substrate to enzyme ratio of 15:1 prior to acidification by addition of 250 mM HCl. Mass spectrometry was performed as previously described13 by injection of 3 μL peptide digest onto a fused-silica capillary tip column (75-μm i.d.) packed with 30 cm of Reprosil-Pur C18-AQ (3-μm bead diameter, Dr. Maisch). Peptides were eluted from the column at 0.25 μL/min using an acetonitrile gradient. Mass spectrometry was performed on a QExactive-HF (Thermo Fisher Scientific) in data dependent mode.
Purification of 14N/15N Spc110 covalent heterodimers
Saccharomyces cerevisiae Spc1101–276-SpyCatcher and -SpyTag were transformed into BL21(DE3) CodonPlus RIL (Agilent). Both constructs bore the Spc110-C225S mutation to prevent disulfide-mediated oligomerization. To generate 14N-Spc1101–276-Spc110-SpyTag, cultures were grown in Terrific Broth (Research Products International). For 15N-Spc1101–276-SpyCatcher, cultures were grown in in the following medium32: 50 mM Na2HPO4, 25 mM KH2PO4, 10 mM NaCl, 5 mM MgSO4, 0.2 mM CaCl2, 1% (w/v) glucose, 0.1% (w/v) 15NH4Cl, 0.25x BME vitamins mix (homemade based on the formula in Sigma-Aldrich item B6891), and 0.25x trace metals mixture.33 Cultures were grown at 30 °C until reaching OD600 0.3–0.4. The temperature was then decreased to 18 °C. Once the culture had reached OD600 0.6–0.8, expression was induced with 0.6 mM IPTG for 16–18 h. Cells were harvested by centrifugation then resuspended in lysis buffer (50 mM potassium phosphate pH 8, 300 mM NaCl, 5 mM EDTA, 1 mM DTT, 0.3% Tween-20, 1x cOmplete protease inhibitor, EDTA-free (Millipore-Sigma)). Cells were lysed by Emulsiflex C3 (Avestin). Lysate was cleared by ultracentrifugation at 40,000 rpm for 30 min in a Type 45Ti rotor (Beckman-Coulter). Cleared lysate was applied to cOmplete His-Tag purification resin (Millipore-Sigma) and incubated for 1 h at 4 °C with gentle agitation. The column was then washed with 10 CV lysis buffer followed by 10 CV lysis buffer without Tween-20. Spc110 was then eluted with 4 CV of elution buffer (25 mM Tris pH 8.3, 75 mM NaCl, 5 mM EDTA, 1 mM DTT, 1x cOmplete protease inhibitor, EDTA-free (Millipore-Sigma), and 250 mM imidazole). Eluates were then diluted to < 5 mS/cm conductivity with MonoQ buffer A (25 mM Tris pH 8.3, 1 mM DTT). The diluted eluates were then applied separately to a MonoQ 10/100 GL column (GE) pre-equilibrated in 2.5% MonoQ buffer B (25 mM Tris pH 8.3, 1 M NaCl, 1 mM DTT) in MonoQ buffer A. The column was then washed with 2 CV of 2.5% MonoQ buffer B, then eluted with a linear gradient from 2.5–50% MonoQ buffer B. Spc1101–276 SpyCatcher and -SpyTag typically elute at approximately 17 mS/cm and 9 mS/cm conductivity, respectively. The concentration of the pooled fractions containing Spc1101–276-SpyCatcher or -SpyTag were measured using Bradford protein assay reagent (Bio-Rad) using a BSA standard curve, then combined in a 1:1 molar ratio with the addition of TEV protease to cleave the His-tags. After 1 h, the Spc110 covalent adduct was further purified by size exclusion chromatography on S200 HiLoad 16/600 Superdex 200 pg (Cytiva) equilibrated in HB150 + 10% glycerol. Fractions containing undegraded Spc110 covalent adducts were then pooled, centrifugally concentrated, flash frozen in liquid nitrogen, and stored at −80 °C.
Prior to crosslinking, the protein was first buffer exchanged into HB100 buffer (40 mM HEPES, 100 mM NaCl, pH 7) using Pierce Polyacrylamide Spin Desalting Columns (Catalog number: 89849, Thermofisher, Scientific). A 200 μL crosslinking reaction was made by mixing 23.6 uL desalted Spc110 (42 μg total protein) with 169.6 μL HB100 and adding 6.8 μL 14.5 mM BS3 (in HB100). The reaction was allowed to proceed for 2.5 minutes in an Eppendorf thermomixer at 21°C shaking at 1,000 rpm after which 50 uL was quenched by transfer to fresh 1.5 mL Eppendorf tube containing 5 μL 1M ammonium bicarbonate plus 1 μL 2M β-Mercaptoethanol. 18 μL of crosslinked protein was loaded onto an SDS-PAGE gel (Biorad, Any kD Mini-PROTEAN TGX Precast Protein Gel; catalogue number 4569033) and run according to the manufacturer’s instructions. A single band corresponding to the crosslinked Spc110 dimer was excised from the gel and subjected to in gel digestion using the following procedure. The gel band was cut into small pieces and washed with 200 μL water followed by 200 μL 50% acetonitrile 25 mM ammonium bicarbonate for five min followed by 200 μL acetonitrile for 1 min. Solvent was removed and the sample dried on a speed vac and reconstituted in 50 μL of 25 mM ammonium bicarbonate containing 10 mM TCEP and incubated at 60oC for 1 hour. Excess liquid was removed and 50 μL of 25 mM ammonium bicarbonate containing 10 mM iodoacetamide was added and the sample incubated in the dark for 20 minutes. The sample was washed with 400 μL followed by 200 μL 50% acetonitrile 25 mM ammonium bicarbonate for five min followed by 200 μL acetonitrile for 1 min. Solvent was removed and the sample dried on a speed vac. The sample was reconstituted in 20 μl of 0.01 μg/ul promega trypsin in 25 mM ammonium bicarbonate. Additional 25 mM ammonium bicarbonate was added sufficient to cover the gel slice and the sample was digest overnight at room temperature. After digestion excess solution was removed and transferred to a new 1.5 mL Eppendorf tube. 50 μl acetonitrile was added to the gel slice, vortexed and removed and combined with the solution in the 1.5 mL tube. 50 μl 60% acetonitrile 0.1% formic acid was added, vortexed, removed and combined. The solution in the 1.5 mL tube was then dried in a speed vac and reconstituted in 20 μl 0.1% formic acid. 3 μl of this solution was injected onto on a QExactive HF (Thermo Fisher Scientific) mass spectrometer run in data dependent mode as described above.
Crosslinking Benchmark Standard
Additional analyses were performed using a previously published ground truth crosslinking dataset.34 These data are available via ProteomeXchange with identifier PXD014337.
Computational Data Analyses
All raw mass spectrometer data files were converted to mzML using msconvert (--mzML –zlib –filter “peakPicking true 1-“ –filter “zeroSamples removeExtra”) from ProteoWizard35 prior to analysis. All computational analyses were performed using Kojak version 2.0 within the Trans-Proteomic Pipeline using the automated decoy sequence generation described above. Parameters for each tool for each analysis are provided in Supplementary Table 1. Kojak is available in standalone format at http://kojak-ms.org and bundled with the TPP at http://www.tppms.org. Novel data acquired for this study have been deposited to the ProteomeXchange Consortium via the PRIDE36 partner repository with identifier PXD037492.
Results
The newest features of Kojak 2.0 were explored using a ground truth dataset.34 In this dataset, crosslinking was performed between twelve sets of synthesized peptides belonging to S. pyogenes Cas9. Crosslinking only occurred between peptides within a set, and sets consisted of seven to nine peptides. By design, all possible crosslinks are known beforehand, and incorrect results are CSMs that contain a non-Cas9 peptide, or are Cas9 peptides from two different groups. Peptides were crosslinked with either DSS or DSSO.
Data were searched using Kojak 2.0 for CSM identification and validated using PeptideProphet and iProphet. A CSM probability cutoff of 0.9 was set, which gave an estimated <1% FDR. For the DSS-linked results, the searches of the data from three replicate injections were combined prior to validation, then CSMs were tallied from each replicate following validation. The number of correct and incorrect CSMs found in each replicate are reported in Table 1. These numbers met or exceeded the published results of this dataset at this error threshold, with an observed error rate that remained below the estimate. That same study also identified 157–265 CSMs per replicate with StavroX and 312–438 CSMs per replicate with Xi at a 1% FDR threshold.34 pLink identified 585–644 CSMs at a 1% FDR threshold, but with error rates in excess of the threshold in all replicates, including higher than 4% in one replicate. The CSMs were then grouped by unique peptide combinations, as correct CSMs are likely to be redundantly observed while incorrect CSMs tend to be random pairings. Here the number of unique crosslinks (156–166 per replicate) were again similar or better to previously published results, with notably better error rates. Across all three replicates, the observed error rate was 1.62%, which, though higher than the CSM-level error threshold, is expected and remained low. For comparison, StavroX identified 90–124 unique CSMs per replicate at error rates of 0–3.1% and Xi identified 141–163 unique CSMs at error rates of 1.4–3.0%. pLink identified 189–218 unique CSMs per replicate, but with error rates of 4.0–11.6%, well in excess of the desired 1% threshold. This same study also reported unique CSM results for an earlier version of Kojak, but using a 5% FDR threshold, and found 120–128 CSMs per replicate at error rates of 1.4–3.2%.34 Overall, Kojak 2.0 with PeptideProphet and iProphet analysis showed high sensitivity and accuracy in its results.
Table 1:
Kojak identified DSS-crosslinked CSMs at PeptideProphet/iProphet estimated <1% error rate (CSM-level).
| 1% Estimated Error Rate (CSM-level) | ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Correct (CSMa) | Incorrect (CSM) | Error Rate (CSM) | Correct (XLb) | Incorrect (XL) | Error Rate (XL) | |
| R1 | 405 | 2 | 0.49% | 156 | 2 | 1.27% |
| R2 | 539 | 3 | 0.55% | 166 | 2 | 1.19% |
| R3 | 490 | 1 | 0.20% | 165 | 1 | 0.60% |
| Total | 1434 | 6 | 0.42% | 182 | 3 | 1.62% |
Results are evaluated for each CSM above the 0.9 probability threshold.
Multiple CSMs to the same crosslinked pair of peptides are combined and results are evaluated for each unique crosslinked peptide pair.
Next, the search was performed on the DSSO-linked data from the same benchmarking standard to showcase the new cleavable crosslinking analysis features of Kojak 2.0. Here two sets of parameters were compared, either using the new cleavable cross-linking settings or not. All other parameters remained identical during the analysis. To replicate the stringency level of the analysis from the original publication, a larger, more challenging sequence database was used that contained the singular S. pyogenes Cas9 sequence, plus more than 100 additional sequences from the CRAPome.37 A CSM probability cutoff of 0.9 was set, which gave an estimated <1% FDR. Using cleavable crosslinker optimizations showed an increase in the number of correct CSMs detected in the analysis, though an increase in the error rate was observed (Table 2). When looking at the unique crosslinked peptide pairs, the cleavable crosslinker optimizations more than doubled the number of correctly identified crosslinks. The observed error rate among unique crosslinked peptides was higher than the desired 1% threshold set at the CSM-level, as expected, though the number of correct crosslinks identified (216) was greater than the previously published results, and often at lower error rates. For example, XlinkX found 128 unique crosslinked peptides at a 29% error rate, and MeroX-RiseUP mode found 149 unique crosslinked peptides at 11% error rate. MeroX-Rise mode found 124 unique crosslinked peptides, but at a notably low 0.8% error rate.34
Table 2:
Additional Kojak CSMs identified from DSSO cleavable crosslinker analysis at PeptideProphet/iProphet estimated <1% error rate (CSM-level).
| 1% Estimated Error Rate (CSM-level) | ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Correct (CSMa) | Incorrect (CSM) | Error Rate (CSM) | Correct (XLb) | Incorrect (XL) | Error Rate (XL) | |
| DSSO | 760 | 3 | 0.39% | 88 | 3 | 3.30% |
| DSSO, Cleavable | 2261 | 37 | 1.61% | 216 | 13 | 5.68% |
Results are evaluated for each CSM above the 0.9 probability threshold.
Multiple CSMs to the same crosslinked pair of peptides are combined and results are evaluated for each unique crosslinked peptide pair.
To illustrate Kojak 2.0 and PeptideProphet/iProphet validation with protein complex analysis, mass spectrometry data from bovine Arp2/3 complex crosslinked with BS2 or DSS were analyzed. A larger protein sequence database consisting of seven Arp2/3 complex subunit sequences and sixty bovine and human contaminant sequences was used in the analysis. Decoy sequences were produced in Kojak using the new decoy database generation feature. Following Kojak 2.0 and PeptideProphet/iProphet analysis, CSMs were uploaded to ProXL38 and visualized by mapping to PDB structure 3UKU (Figure 1). When viewing the DSS results, at a 1% FDR estimated from the iProphet probability scores on the CSMs, 57 unique crosslinked residue pairs were mapped to six of the seven protein subunits (Figure 1A). All but one were within the expected crosslink distance restraints of 35 Å, with 51 of them falling within 25 Å. A distance density plot (Figure 1B) compares the observed CSM distance restraints compared to the set of all possible distance restraints obtainable from the structure, and shows the validated CSMs to belong solely to the small fraction of all possible CSMs that are within the expected distance restraints for DSS. The same analysis was repeated with the shorter crosslinker, BS2, and 35 unique crosslinks were identified (Figure 1C), and all but three were within a 30 Å distance restraint threshold across all seven ARP2/3 subunits. The three outliers were only slightly beyond the threshold (all less than 36 Å), and the distribution of CSMs shows a bias towards short distance restraints when compared to the distribution expected from randomly assigning linked residues (Figure 1D). Together, these results show high conformity between prophet validated CSMs identified with Kojak and crystal structures.
Figure 1.
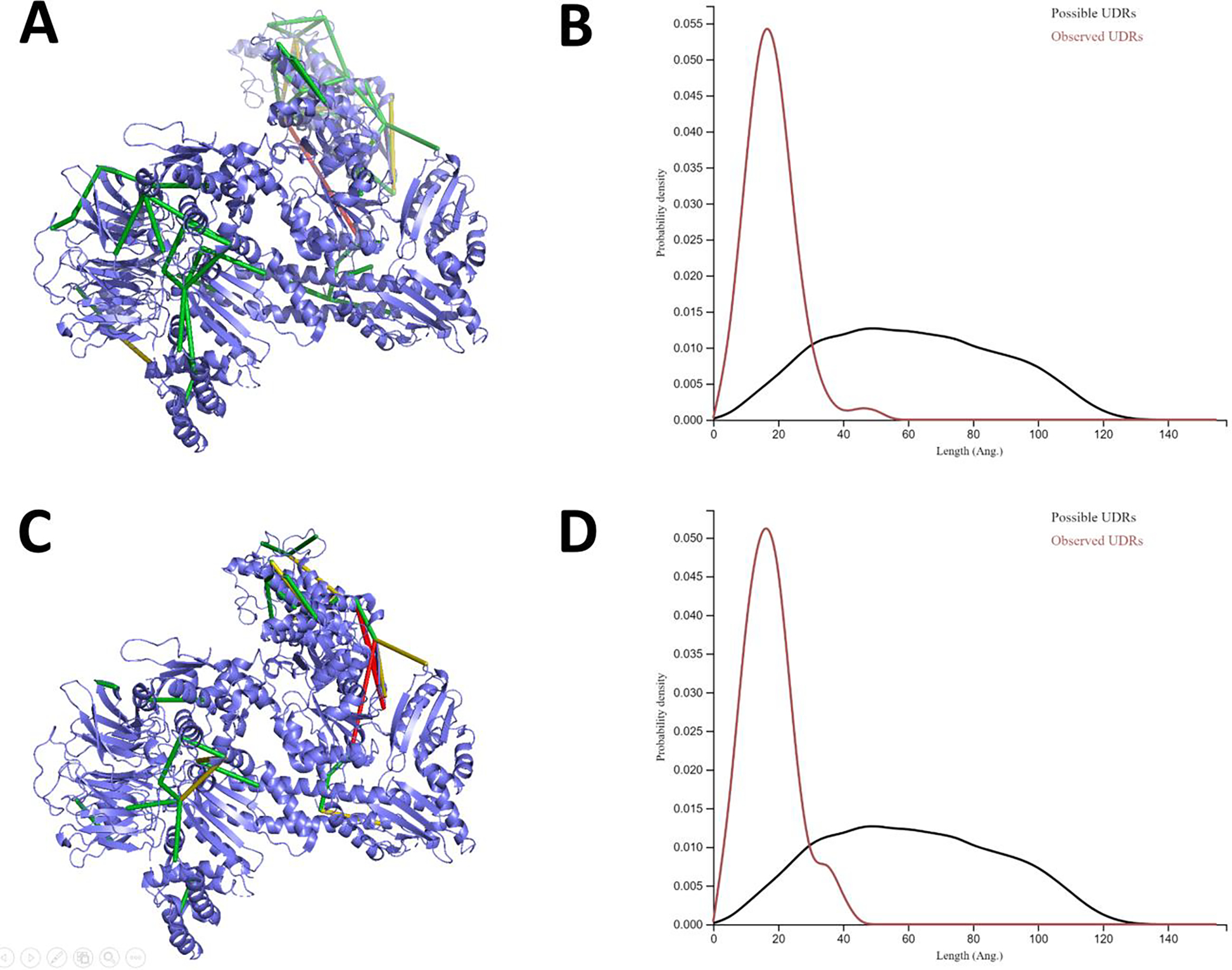
Structural views and crosslink distance restraint distributions for bovine Arp2/3 complex crosslinked with DSS (A and B) and BS2 (C and D). Crosslink distances are colored to represent Cα Lys-Lys distance constraints within 25 Å (green), 35 Å (yellow), and >35 Å (red) for DSS, and 20 Å (green), 30 Å (yellow), and >30 Å (red) for BS2. The distribution of observed unique distance restraints (UDRs) versus all possible UDRs are shown in panel B for DSS and panel D for BS2.
Homodimers, and by extension homomultimers, are often difficult to study by typical XL-MS because it is unclear whether or not the two interacting peptides originate from the same or different subunits. A solution is to mix 15N-labeled and unlabeled forms of the identical subunits and identify XL interactions between subunits as a mix of labeled and unlabeled peptide sequences. The observation of both 15N-labeled and unlabeled fragment ions for two peptides crosslinked together provides spectral evidence that the crosslinked peptides originated from different protein subunits. The S. cerevisiae gamma tubulin small complex binds to the nuclear face of the spindle pole body via interaction with the dimeric coiled-coil protein Spc110. Identification of interacting domains between the two Spc110 subunits was performed by mixing unlabeled and 15N-labeled Spc1101–276 protein. To maximize the likelihood of forming a heavy-light dimer, the SpyCatcher-SpyTag system39 was used. Unlabeled Spc1101–276-SpyTag was produced and purified. 15N-Spc1101–276-SpyCatcher was produced and purified separately. Spc1101–276-SpyTag and 15N-Spc1101–276-SpyCatcher were then mixed. Dimers subsequently formed between 15N-Spc1101–276-SpyCatcher and unlabeled Spc1101–276-SpyTag are permanently trapped by formation of a covalent bond between the SpyCatcher and SpyTag. In this way samples were selectively enriched for Spc1101–276 dimers between 15N and unlabeled Spc110 subunits. Crosslinked spectra database searching with Kojak 2.0 was performed using a FASTA sequence database with two instances of the Spc110 sequence; however, one was annotated with a unique identifier to indicate all its peptides have additional mass due to the excess of 15N. CSMs were validated with the prophets and uploaded to ProXL for visualization (Figure 2). Crosslinks were plotted following a CSM probability threshold of 0.9 (<1% FDR estimation) and high frequency in the coiled-coil region of Spc1101–276, which includes residues 164–276 of each subunit (Figure 2A). Self-links were found as well (i.e. unlabeled-to-unlabeled and 15N-to-15N peptides, Figure 2B), as expected when the crosslinker binds to two locations on the same subunit. These types of crosslinks were distributed across the entirety of the dimer, in contrast to the mixed-label crosslinks that indicate interaction between two subunits.
Figure 2.
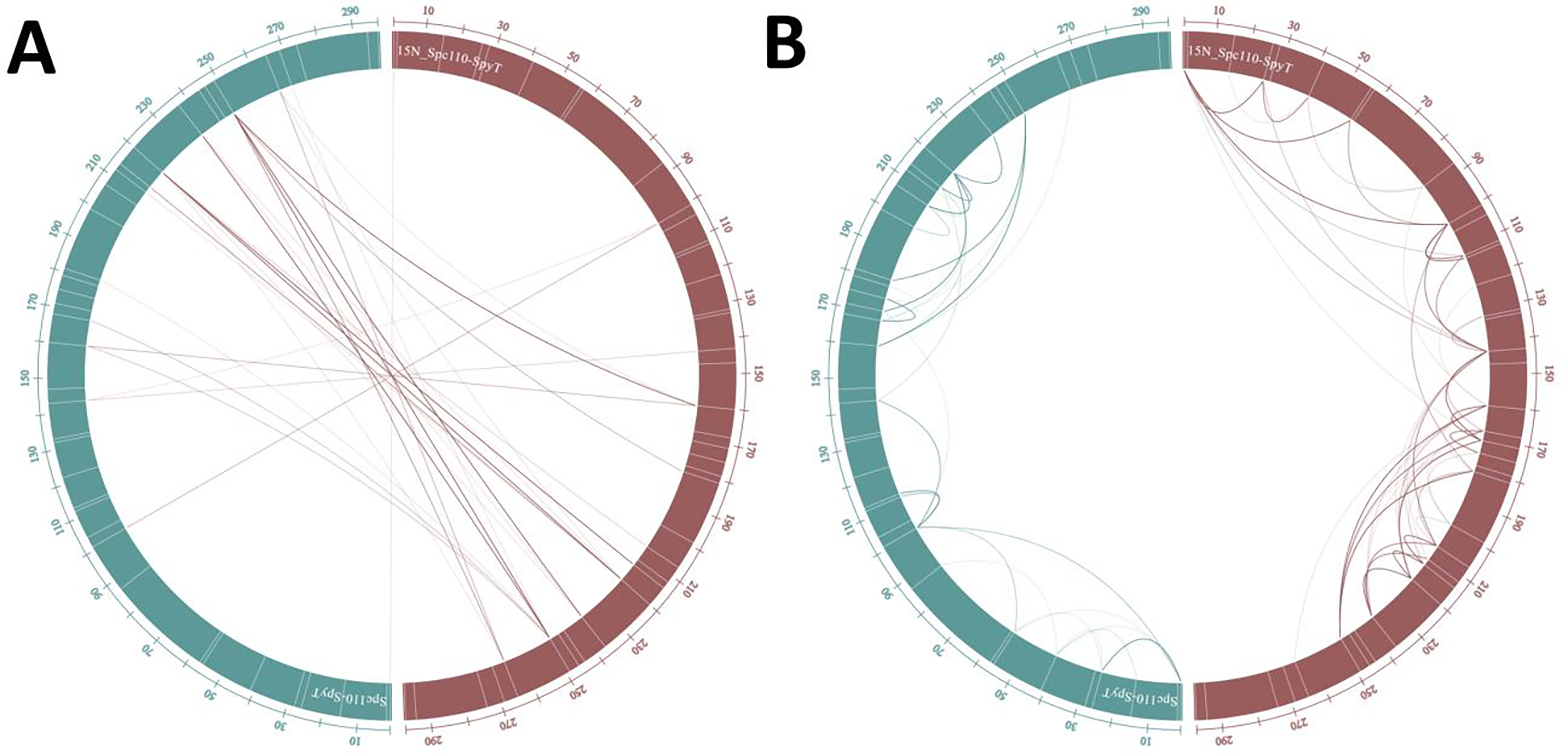
Crosslink distribution between unlabeled Spc-1101–276 (teal) and 15N-Spc-1101–276 (red). Potential sites of crosslinker binding are marked at each position and observed crosslinks as the arcs connecting two positions. Crosslink line weight indicates frequency of observing the interaction. (A) Inter-protein crosslinks most heavily connect the coiled-coil regions of the homodimer (residues 164–276). (B) Self-crosslinks within each subunit are distributed across the entire sequence.
Discussion
Kojak 2.0 has been improved through code optimization and new feature implementation. Combined within a large data processing suite, it reflects the diversity of new technology and analyses available for XL-MS, particularly cleavable crosslinker and 15N-labeled homomultimer analyses. However, regardless of the nature of the crosslinking study, accurately estimating the error rate of the results is critical to the success of any XL-MS study. Kojak does not report error rates in its results by design, instead requiring use of the existing tools for such tasks. Kojak 2.0 now includes the ability to generate decoy sequences on-the-fly, to facilitate target-decoy validation strategies employed by many tools for error estimation. Previously, we have shown how to use Percolator40 for CSM validation.16 Here, we have integrated Kojak 2.0 into the Trans-Proteomic Pipeline to make use of PeptideProphet and iProphet for CSM validation. This versatility is meant to make Kojak extensible to still other validation tools (e.g., XiFDR19), and pipelines. These tools have different capabilities that can be tailored to the needs of the study. For example, Percolator is, as of the time of this writing, best for use only at the CSM-level, while iProphet extends error estimation to the peptide level. These levels might be sufficient for small studies, but large-scale analyses such as whole cell linking require even stronger thresholds and tools appropriate for them.18,21 The plug-and-play nature of Kojak 2.0 makes it easy to adapt to the different tools required for XL-MS analysis at any scale.
It is important to note that beyond the scope of the latest features presented here, Kojak 2.0 remains open source and supporting open formats. Upon initial release, Kojak supported mzXML and mzML for input, and provided tab-delimited output. Because many existing tools for both preprocessing and post-processing of searched spectral data require specific formats, Kojak 2.0 was extended to support MGF for input and pepXML30 and mzIdentML41 for output. These additional formats allow for the integration of Kojak into existing pipelines,30,38,42,43 as well as facilitate integration to future pipelines. Such adaptations are particularly beneficial over all-in-one software suites, particularly if tools exist elsewhere that offer tangential features, such as spectral preprocessing, CSM validation, and results visualization that exceed the capabilities contained within any single software suite. We expect this versatility empower users in the analysis of particularly difficult datasets, while still providing a fast and simple interface accessible to anyone for use in most crosslinking analyses.
Conclusions
Kojak 2.0 offers many new features and capabilities for XL-MS data analysis. Because it is open source and adheres to open data formats and standards, it is easily incorporated into computational pipelines. We have demonstrated several of these new features and shown how, through use of the TPP, Kojak can be integrated into a robust pipeline for crosslinked peptide identification and validation.
Supplementary Material
Supplementary Figure 1: HETEROXL model of iProphet.
Supplementary Table 1: Software analysis parameters.
Supplementary File 1: Configuration files and FASTA search databases for Kojak.
Acknowledgements
This work was funded in part by the National Institutes of Health grants from the National institute General Medical Sciences R01GM087221, the National Heart, Lung, and Blood Institute R01HL133135, the Office of the Director S10OD026936, the National Science Foundation awards 1920268, Howard Hughes Medical Institute (DAA), National Institute of General Medical Sciences R01 GM031627, R35GM118099, and P01 GM105537 (DAA), National Science Foundation Graduate Research Fellowship Grant No. 1144247 (ASL), UCSF Discovery Fellowship (ASL), and National institute General Medical Sciences P41GM103533 (MJM).
Footnotes
SUPPORTING INFORMATION:
The following supporting information is available free of charge at ACS website http://pubs.acs.org
References
- (1).Leitner A; Bonvin AMJJ; Borchers CH; Chalkley RJ; Chamot-Rooke J; Combe CW; Cox J; Dong M-Q; Fischer L; Götze M; Gozzo FC; Heck AJR; Hoopmann MR; Huang L; Ishihama Y; Jones AR; Kalisman N; Kohlbacher O; Mechtler K; Moritz RL; Netz E; Novak P; Petrotchenko E; Sali A; Scheltema RA; Schmidt C; Schriemer D; Sinz A; Sobott F; Stengel F; Thalassinos K; Urlaub H; Viner R; Vizcaíno JA; Wilkins MR; Rappsilber J Toward Increased Reliability, Transparency, and Accessibility in Cross-Linking Mass Spectrometry. Structure 2020, 28 (11), 1259–1268. 10.1016/j.str.2020.09.011. [DOI] [PubMed] [Google Scholar]
- (2).O’Reilly FJ; Rappsilber J Cross-Linking Mass Spectrometry: Methods and Applications in Structural, Molecular and Systems Biology. Nat Struct Mol Biol 2018, 25 (11), 1000–1008. 10.1038/s41594-018-0147-0. [DOI] [PubMed] [Google Scholar]
- (3).Iacobucci C; Piotrowski C; Aebersold R; Amaral BC; Andrews P; Bernfur K; Borchers C; Brodie NI; Bruce JE; Cao Y; Chaignepain S; Chavez JD; Claverol S; Cox J; Davis T; Degliesposti G; Dong M-Q; Edinger N; Emanuelsson C; Gay M; Götze M; Gomes-Neto F; Gozzo FC; Gutierrez C; Haupt C; Heck AJR; Herzog F; Huang L; Hoopmann MR; Kalisman N; Klykov O; Kukačka Z; Liu F; MacCoss MJ; Mechtler K; Mesika R; Moritz RL; Nagaraj N; Nesati V; Neves-Ferreira AGC; Ninnis R; Novák P; O’Reilly FJ; Pelzing M; Petrotchenko E; Piersimoni L; Plasencia M; Pukala T; Rand KD; Rappsilber J; Reichmann D; Sailer C; Sarnowski CP; Scheltema RA; Schmidt C; Schriemer DC; Shi Y; Skehel JM; Slavin M; Sobott F; Solis-Mezarino V; Stephanowitz H; Stengel F; Stieger CE; Trabjerg E; Trnka M; Vilaseca M; Viner R; Xiang Y; Yilmaz S; Zelter A; Ziemianowicz D; Leitner A; Sinz A First Community-Wide, Comparative Cross-Linking Mass Spectrometry Study. Anal Chem 2019, 91 (11), 6953–6961. 10.1021/acs.analchem.9b00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Chu F; Baker PR; Burlingame AL; Chalkley RJ Finding Chimeras: A Bioinformatics Strategy for Identification of Cross-Linked Peptides. Mol Cell Proteomics 2010, 9 (1), 25–31. 10.1074/mcp.M800555-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Götze M; Pettelkau J; Schaks S; Bosse K; Ihling CH; Krauth F; Fritzsche R; Kühn U; Sinz A StavroX--a Software for Analyzing Crosslinked Products in Protein Interaction Studies. J Am Soc Mass Spectrom 2012, 23 (1), 76–87. 10.1007/s13361-011-0261-2. [DOI] [PubMed] [Google Scholar]
- (6).Rinner O; Seebacher J; Walzthoeni T; Mueller LN; Beck M; Schmidt A; Mueller M; Aebersold R Identification of Cross-Linked Peptides from Large Sequence Databases. Nat Methods 2008, 5 (4), 315–318. 10.1038/nmeth.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Yang B; Wu Y-J; Zhu M; Fan S-B; Lin J; Zhang K; Li S; Chi H; Li Y-X; Chen H-F; Luo S-K; Ding Y-H; Wang L-H; Hao Z; Xiu L-Y; Chen S; Ye K; He S-M; Dong M-Q Identification of Cross-Linked Peptides from Complex Samples. Nat Methods 2012, 9 (9), 904–906. 10.1038/nmeth.2099. [DOI] [PubMed] [Google Scholar]
- (8).Mohr JP; Perumalla P; Chavez JD; Eng JK; Bruce JE Mango: A General Tool for Collision Induced Dissociation-Cleavable Cross-Linked Peptide Identification. Anal Chem 2018, 90 (10), 6028–6034. 10.1021/acs.analchem.7b04991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Götze M; Pettelkau J; Fritzsche R; Ihling CH; Schäfer M; Sinz A Automated Assignment of MS/MS Cleavable Cross-Links in Protein 3D-Structure Analysis. J Am Soc Mass Spectrom 2015, 26 (1), 83–97. 10.1007/s13361-014-1001-1. [DOI] [PubMed] [Google Scholar]
- (10).Leitner A; Walzthoeni T; Aebersold R Lysine-Specific Chemical Cross-Linking of Protein Complexes and Identification of Cross-Linking Sites Using LC-MS/MS and the XQuest/XProphet Software Pipeline. Nat Protoc 2014, 9 (1), 120–137. 10.1038/nprot.2013.168. [DOI] [PubMed] [Google Scholar]
- (11).Jaiswal M; Crabtree N; Bauer MA; Hall R; Raney KD; Zybailov BL XLPM: Efficient Algorithm for the Analysis of Protein-Protein Contacts Using Chemical Cross-Linking Mass Spectrometry. BMC Bioinformatics 2014, 15 Suppl 11, S16. 10.1186/1471-2105-15-S11-S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Leitner A; Joachimiak LA; Bracher A; Mönkemeyer L; Walzthoeni T; Chen B; Pechmann S; Holmes S; Cong Y; Ma B; Ludtke S; Chiu W; Hartl FU; Aebersold R; Frydman J The Molecular Architecture of the Eukaryotic Chaperonin TRiC/CCT. Structure 2012, 20 (5), 814–825. 10.1016/j.str.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Zelter A; Bonomi M; Kim JO; Umbreit NT; Hoopmann MR; Johnson R; Riffle M; Jaschob D; MacCoss MJ; Moritz RL; Davis TN The Molecular Architecture of the Dam1 Kinetochore Complex Is Defined by Cross-Linking Based Structural Modelling. Nat Commun 2015, 6, 8673. 10.1038/ncomms9673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Schweppe DK; Chavez JD; Bruce JE XLmap: An R Package to Visualize and Score Protein Structure Models Based on Sites of Protein Cross-Linking. Bioinformatics 2016, 32 (2), 306–308. 10.1093/bioinformatics/btv519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Schweppe DK; Zheng C; Chavez JD; Navare AT; Wu X; Eng JK; Bruce JE XLinkDB 2.0: Integrated, Large-Scale Structural Analysis of Protein Crosslinking Data. Bioinformatics 2016, 32 (17), 2716–2718. 10.1093/bioinformatics/btw232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hoopmann MR; Zelter A; Johnson RS; Riffle M; MacCoss MJ; Davis TN; Moritz RL Kojak: Efficient Analysis of Chemically Cross-Linked Protein Complexes. J Proteome Res 2015, 14 (5), 2190–2198. 10.1021/pr501321h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Eng JK; Jahan TA; Hoopmann MR Comet: An Open-Source MS/MS Sequence Database Search Tool. Proteomics 2013, 13 (1), 22–24. 10.1002/pmic.201200439. [DOI] [PubMed] [Google Scholar]
- (18).Lenz S; Sinn LR; O’Reilly FJ; Fischer L; Wegner F; Rappsilber J Reliable Identification of Protein-Protein Interactions by Crosslinking Mass Spectrometry. Nat Commun 2021, 12 (1), 3564. 10.1038/s41467-021-23666-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Fischer L; Rappsilber J Quirks of Error Estimation in Cross-Linking/Mass Spectrometry. Anal Chem 2017, 89 (7), 3829–3833. 10.1021/acs.analchem.6b03745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Trnka MJ; Baker PR; Robinson PJJ; Burlingame AL; Chalkley RJ Matching Cross-Linked Peptide Spectra: Only as Good as the Worse Identification. Mol Cell Proteomics 2014, 13 (2), 420–434. 10.1074/mcp.M113.034009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).de Jong L; Roseboom W; Kramer G Towards Low False Discovery Rate Estimation for Protein-Protein Interactions Detected by Chemical Cross-Linking. Biochim Biophys Acta Proteins Proteom 2021, 1869 (7), 140655. 10.1016/j.bbapap.2021.140655. [DOI] [PubMed] [Google Scholar]
- (22).Chen Z-L; Meng J-M; Cao Y; Yin J-L; Fang R-Q; Fan S-B; Liu C; Zeng W-F; Ding Y-H; Tan D; Wu L; Zhou W-J; Chi H; Sun R-X; Dong M-Q; He S-M A High-Speed Search Engine PLink 2 with Systematic Evaluation for Proteome-Scale Identification of Cross-Linked Peptides. Nat Commun 2019, 10 (1), 3404. 10.1038/s41467-019-11337-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Eng JK; Fischer B; Grossmann J; Maccoss MJ A Fast SEQUEST Cross Correlation Algorithm. J Proteome Res 2008, 7 (10), 4598–4602. 10.1021/pr800420s. [DOI] [PubMed] [Google Scholar]
- (24).Taverner T; Hall NE; O’Hair RAJ; Simpson RJ Characterization of an Antagonist Interleukin-6 Dimer by Stable Isotope Labeling, Cross-Linking, and Mass Spectrometry. J Biol Chem 2002, 277 (48), 46487–46492. 10.1074/jbc.M207370200. [DOI] [PubMed] [Google Scholar]
- (25).Lima DB; Melchior JT; Morris J; Barbosa VC; Chamot-Rooke J; Fioramonte M; Souza TACB; Fischer JSG; Gozzo FC; Carvalho PC; Davidson WS Characterization of Homodimer Interfaces with Cross-Linking Mass Spectrometry and Isotopically Labeled Proteins. Nat Protoc 2018, 13 (3), 431–458. 10.1038/nprot.2017.113. [DOI] [PubMed] [Google Scholar]
- (26).Kolbowski L; Lenz S; Fischer L; Sinn LR; O’Reilly FJ; Rappsilber J Improved Peptide Backbone Fragmentation Is the Primary Advantage of MS-Cleavable Crosslinkers. Anal Chem 2022, 94 (22), 7779–7786. 10.1021/acs.analchem.1c05266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Deutsch E; Mendoza L; Shteynberg D; Hoopmann M; Sun Z; Eng J; Moritz R The Trans-Proteomic Pipeline: Robust Mass Spectrometry-Based Proteomics Data Analysis Suite; preprint; Chemistry, 2022. 10.26434/chemrxiv-2022-3c75n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Keller A; Nesvizhskii AI; Kolker E; Aebersold R Empirical Statistical Model to Estimate the Accuracy of Peptide Identifications Made by MS/MS and Database Search. Anal Chem 2002, 74 (20), 5383–5392. 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- (29).Shteynberg D; Deutsch EW; Lam H; Eng JK; Sun Z; Tasman N; Mendoza L; Moritz RL; Aebersold R; Nesvizhskii AI IProphet: Multi-Level Integrative Analysis of Shotgun Proteomic Data Improves Peptide and Protein Identification Rates and Error Estimates. Mol Cell Proteomics 2011, 10 (12), M111.007690. 10.1074/mcp.M111.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Hoopmann MR; Mendoza L; Deutsch EW; Shteynberg D; Moritz RL An Open Data Format for Visualization and Analysis of Cross-Linked Mass Spectrometry Results. J Am Soc Mass Spectrom 2016, 27 (11), 1728–1734. 10.1007/s13361-016-1435-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Doolittle LK; Rosen MK; Padrick SB Purification of Native Arp2/3 Complex from Bovine Thymus. Methods Mol Biol 2013, 1046, 231–250. 10.1007/978-1-62703-538-5_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Sivashanmugam A; Murray V; Cui C; Zhang Y; Wang J; Li Q Practical Protocols for Production of Very High Yields of Recombinant Proteins Using Escherichia Coli. Protein Sci 2009, 18 (5), 936–948. 10.1002/pro.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Studier FW Protein Production by Auto-Induction in High Density Shaking Cultures. Protein Expr Purif 2005, 41 (1), 207–234. 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- (34).Beveridge R; Stadlmann J; Penninger JM; Mechtler K A Synthetic Peptide Library for Benchmarking Crosslinking-Mass Spectrometry Search Engines for Proteins and Protein Complexes. Nat Commun 2020, 11 (1), 742. 10.1038/s41467-020-14608-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Chambers MC; Maclean B; Burke R; Amodei D; Ruderman DL; Neumann S; Gatto L; Fischer B; Pratt B; Egertson J; Hoff K; Kessner D; Tasman N; Shulman N; Frewen B; Baker TA; Brusniak M-Y; Paulse C; Creasy D; Flashner L; Kani K; Moulding C; Seymour SL; Nuwaysir LM; Lefebvre B; Kuhlmann F; Roark J; Rainer P; Detlev S; Hemenway T; Huhmer A; Langridge J; Connolly B; Chadick T; Holly K; Eckels J; Deutsch EW; Moritz RL; Katz JE; Agus DB; MacCoss M; Tabb DL; Mallick P A Cross-Platform Toolkit for Mass Spectrometry and Proteomics. Nat Biotechnol 2012, 30 (10), 918–920. 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Perez-Riverol Y; Bai J; Bandla C; García-Seisdedos D; Hewapathirana S; Kamatchinathan S; Kundu DJ; Prakash A; Frericks-Zipper A; Eisenacher M; Walzer M; Wang S; Brazma A; Vizcaíno JA The PRIDE Database Resources in 2022: A Hub for Mass Spectrometry-Based Proteomics Evidences. Nucleic Acids Res 2022, 50 (D1), D543–D552. 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Mellacheruvu D; Wright Z; Couzens AL; Lambert J-P; St-Denis NA; Li T; Miteva YV; Hauri S; Sardiu ME; Low TY; Halim VA; Bagshaw RD; Hubner NC; Al-Hakim A; Bouchard A; Faubert D; Fermin D; Dunham WH; Goudreault M; Lin Z-Y; Badillo BG; Pawson T; Durocher D; Coulombe B; Aebersold R; Superti-Furga G; Colinge J; Heck AJR; Choi H; Gstaiger M; Mohammed S; Cristea IM; Bennett KL; Washburn MP; Raught B; Ewing RM; Gingras A-C; Nesvizhskii AI The CRAPome: A Contaminant Repository for Affinity Purification-Mass Spectrometry Data. Nat Methods 2013, 10 (8), 730–736. 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Riffle M; Jaschob D; Zelter A; Davis TN ProXL (Protein Cross-Linking Database): A Platform for Analysis, Visualization, and Sharing of Protein Cross-Linking Mass Spectrometry Data. J Proteome Res 2016, 15 (8), 2863–2870. 10.1021/acs.jproteome.6b00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Li L; Fierer JO; Rapoport TA; Howarth M Structural Analysis and Optimization of the Covalent Association between SpyCatcher and a Peptide Tag. J Mol Biol 2014, 426 (2), 309–317. 10.1016/j.jmb.2013.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).The M; MacCoss MJ; Noble WS; Käll L Fast and Accurate Protein False Discovery Rates on Large-Scale Proteomics Data Sets with Percolator 3.0. J Am Soc Mass Spectrom 2016, 27 (11), 1719–1727. 10.1007/s13361-016-1460-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Vizcaíno JA; Mayer G; Perkins S; Barsnes H; Vaudel M; Perez-Riverol Y; Ternent T; Uszkoreit J; Eisenacher M; Fischer L; Rappsilber J; Netz E; Walzer M; Kohlbacher O; Leitner A; Chalkley RJ; Ghali F; Martínez-Bartolomé S; Deutsch EW; Jones AR The MzIdentML Data Standard Version 1.2, Supporting Advances in Proteome Informatics. Mol Cell Proteomics 2017, 16 (7), 1275–1285. 10.1074/mcp.M117.068429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Riffle M; Jaschob D; Zelter A; Davis TN Proxl (Protein Cross-Linking Database): A Public Server, QC Tools, and Other Major Updates. J Proteome Res 2019, 18 (2), 759–764. 10.1021/acs.jproteome.8b00726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Kertesz-Farkas A; Adoquaye Acquaye FLN; Bhimani K; Eng JK; Fondrie WE; Grant C; Hoopmann MR; Lin A; Lu YY; Moritz RL; MacCoss MJ; Noble WS The Crux Toolkit for Analysis of Bottom-up Tandem Mass Spectrometry Proteomics Data; preprint; Bioinformatics, 2022. 10.1101/2022.10.02.510538. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: HETEROXL model of iProphet.
Supplementary Table 1: Software analysis parameters.
Supplementary File 1: Configuration files and FASTA search databases for Kojak.