

Abstract
Current therapeutic strategies for treating non-alcoholic steatohepatitis (NASH) have failed to alleviate liver fibrosis, which is a devastating feature leading to hepatic dysfunction. Here, we integrated single-nucleus transcriptomics and epigenomics to characterize all major liver cell types during NASH development in mice and humans. The bifurcation of hepatocyte trajectory with NASH progression was conserved between mouse and human. At the non-alcoholic fatty liver (NAFL) stage, hepatocytes exhibited metabolic adaptation, whereas at the NASH stage, a subset of hepatocytes was enriched for the signatures of cell adhesion and migration, which was mainly demarcated by receptor tyrosine kinase EphB2. EphB2, acting as a downstream effector of Notch signaling in hepatocytes, was sufficient to induce cell-autonomously inflammation. Knockdown of Ephb2 in hepatocytes ameliorated inflammation and fibrosis in mouse models of NASH. Thus, EphB2 expressing hepatocytes contribute to NASH progression and may serve as a potential therapeutic target.
One Sentence Summary:
Single-nucleus multi-omics profiling of mouse and human NASH reveals Ephb2-expressing hepatocytes contribute to NASH progression.
INTRODUCTION
With obesity and diabetes becoming more prevalent across the globe, nonalcoholic fatty liver disease (NAFLD) is now recognized as an increasingly common form of chronic liver disease (1). NAFLD represents a spectrum of diseases that range from non-alcoholic fatty liver (NAFL) with simple lipid accumulation to advanced stage non-alcoholic steatohepatitis (NASH) characterized by progressive inflammation and fibrosis. Current pharmacological targeting strategies in clinical trials for treating NASH often improved metabolic profiles but failed to ameliorate fibrosis (2, 3). Liver fibrosis poses the greatest risk of liver-related morbidity and mortality in patients with NASH (4), but approved therapy for treating fibrosis in NASH is still lacking.
NASH pathophysiology is complicated by the involvement of multiple cell types within the liver, but recent studies that have attempted to take the cellular diversity of the liver into account using single-cell RNA-seq (scRNA-seq) (5–7) have been challenged by limited recovery of hepatocytes, the major tissue-specific cell type of the liver. Moreover, the characterization of NASH livers in these studies has largely been either restricted to the end stage or over a relatively short time window. Thus, the underlying mechanisms of NASH progression are not fully understood.
Here we use single-nucleus RNA-seq (snRNA-seq) and snATAC-seq to efficiently capture the information regarding hepatocytes as well as other liver cell types in the same livers of mice and humans during NASH progression. This approach provided single-cell resolution of the transcriptomes as well as insights into key transcription factors contributing to the transition. We identified one subset of murine hepatocytes unexpectedly expressing receptor tyrosine kinase EphB2 at the NASH stage, which was corroborated in human NASH livers. The activation of EphB2 in hepatocytes was sufficient to generate cell-autonomous inflammation in hepatocytes. Further, inactivating EphB2 in hepatocytes mitigated fibrosis in NASH models. Thus, the hepatocytes demarcated by EphB2 contribute to the development of NASH.
RESULTS
Characterization of mouse models of NASH progression
We evaluated the Gubra Amylin NASH (GAN) diet (8) and the modified American Lifestyle-Induced Obesity Syndrome (ALIOS) diet (9) in C57BL/6J male mice, beginning at 4 weeks of age, both of which led to hepatomegaly after 3 months (mo) of feeding (fig. S1A). The ALIOS diet model resulted in a more rapid increase in inflammatory and fibrogenic gene expression (fig. S1B and data file S1), and we therefore focused on the ALIOS model in our study. The mice fed the ALIOS diet gained weight (fig. S1C and data file S1) and developed modest glucose intolerance (fig. S1D and data file S1), as often seen in human patients with NASH. To understand NASH progression, we extended the feeding period to 9 mo and observed more pronounced up-regulation of inflammatory and fibrogenic gene expression (fig. S1E and data file S1). Liver histopathologic evaluation, which is the gold standard for diagnosis of NASH, demonstrated increased fibrosis (fig. S1F), and blinded analysis revealed increased fibrosis stage (fig. S1G and data file S1) and NAFLD activity score (NAS) (fig. S1H and data file S1) from 3 to 9 mo on the ALIOS diet. Based on the NAS, 6 out of 7 mice were still in the NAFL stage with only 1 diagnosed with NASH at 3 mo, whereas when the feeding time was extended to 9 mo, all the mice fed the ALIOS diet were diagnosed with NASH (fig. S1I). Of note, no malignantly transformed cells were observed in any of the animals. Thus, characterization of livers from mice fed the ALIOS diet for 3 and 9 mo allowed us to capture the natural disease progression from NAFL to NASH over time.
Global transcriptomic feature shifts across all major mouse liver cell types during NASH progression
To profile the transcriptional changes of major liver cell types, we performed snRNA-seq on livers from mice that were given the ALIOS diet for 3 months or 9 months (referred to as 3moALIOS and 9moALIOS). To exclude an effect of aging, age-matched mice on normal chow (referred to as 3moNC and 9moNC) were used as controls (Fig. 1A). A total of 28,308 nuclei from the 4 conditions were analyzed by snRNAseq, leading to the identification of clusters of hepatocytes and non-parenchymal cells (NPCs) including macrophages, endothelial cells, stellate cells, and cholangiocytes (Fig. 1B and data file S1). In agreement with previous scRNA-seq studies on mouse or human NASH livers (5−7, 10), extensive transcriptomic remodeling was observed in ALIOS groups compared to NC groups (fig. S2A left, fig. S2B and data file S1). The samples from the 4 different conditions showed comparable recovery of the number of genes per nucleus, as well as quality as assessed by mitochondrial fraction, both overall (fig. S2C and data file S1) and at the level of cell clusters (fig. S2D and data file S1).
Fig. 1. snRNA-seq reveals hepatocyte-switching from macronutrient processing to cell adhesion and migration during murine NASH.
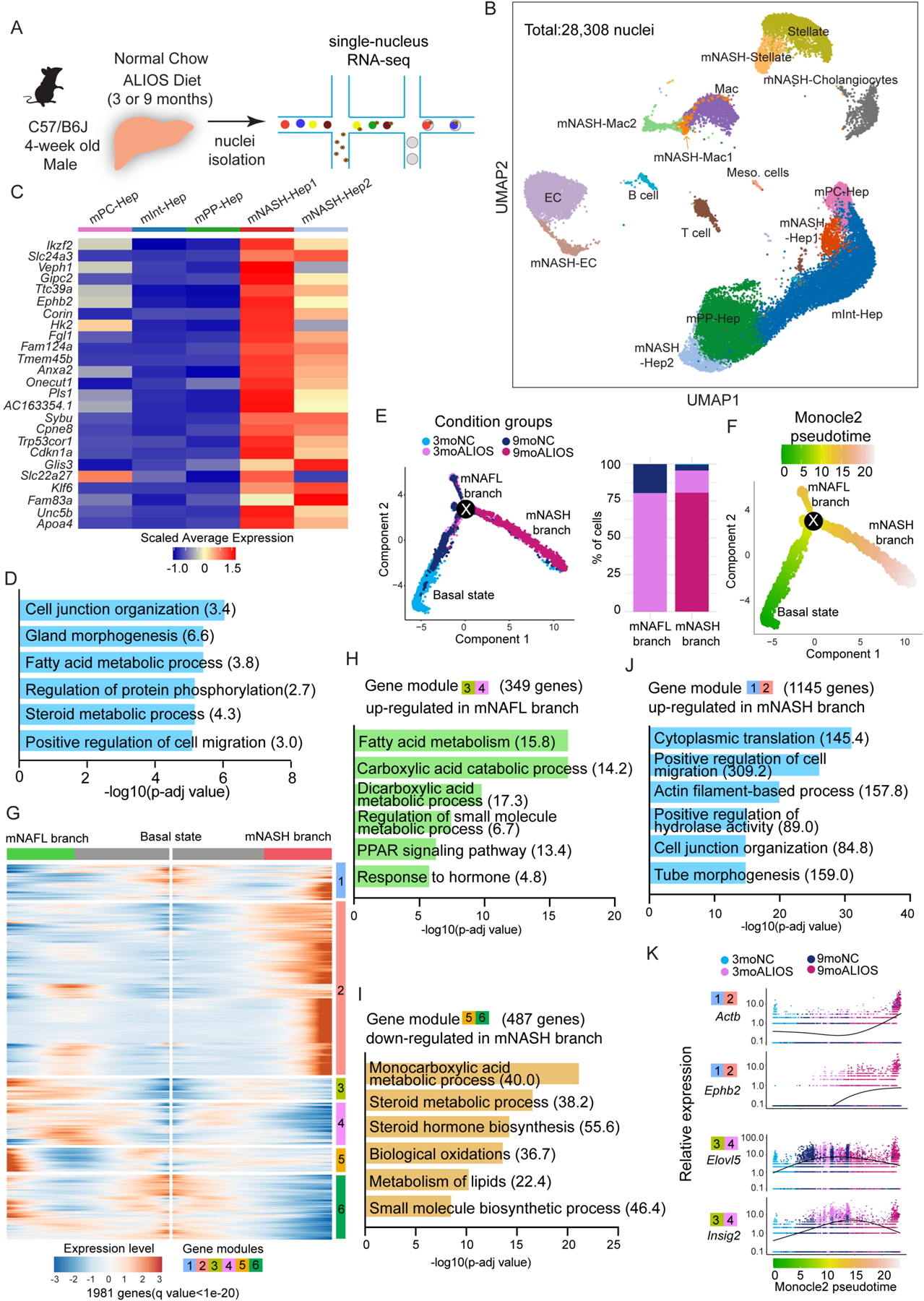
(A) Experimental scheme of snRNA-seq in profiling transcriptomic changes in all major liver cells during NASH progression in mice.
(B) UMAP plot visualization of the unsupervised cell clusters containing 28,308 nuclei from livers of mice with four feeding paradigms including 3moNC,3moALIOS,9moNC, and 9moALIOS. Hep: hepatocytes; Mac: macrophages; EC: endothelial cells; Meso.cells: mesothelial cells.
(C-D) Heatmap (C) and GO analysis (D) of top enriched genes in mNASH-Hep1/2 (Odds ratio indicated after each term).
(E) Pseudotime analysis by Monocle2 revealed hepatocyte trajectory bifurcation during NASH progression, color coded by condition groups (left) and composition of the condition groups of mNAFL and mNASH branches (right).
(F) Pseudotime analysis by Monocle2 revealed hepatocyte trajectory bifurcation during NASH progression color coded by Monocle2 pseudotime value.
(G) Heatmap of the 1981 genes (q value < 1e-20) that determined hepatocyte trajectory bifurcation using branched expression analysis modeling (BEAM) in Monocle2. These genes are clustered into 6 different modules based on their gene expression pattern across Monocle2 pseudotime. Left: gene expression patterns from basal state to NAFL state (upper branch in Fig. 1E); Right: gene expression patterns from basal state to NASH state (lower branch in Fig. 1E). (H-J) GO analysis of gene modules that determined hepatocyte trajectory bifurcation (Odds ratio indicated after each term).
(K) Example genes of gene module 1/2 and 3/4 (the gene module color is consistent with Fig. 1H,J and the Monocle2 pseudotime color is consistent with Fig. 1F).
Among NPCs, the macrophage cluster mNASH-Mac2, abundantly expressing pro-inflammatory genes such as Ccr2, was comprised of cells predominantly from 9moALIOS (fig. S2A) (7). The endothelial cell cluster with cells mostly derived from NC group (EC) was enriched with liver sinusoid endothelial markers Stab2 whereas mNASH-EC, mainly consisting of cells from ALIOS, exhibited increased abundance of the endothelial cell adhesion molecule PECAM1 (fig. S2A), consistent with the notion that sinusoid endothelial cells lose their fenestration during NASH progression (11). Stellate cell subgroup mNASH-Stellate, predominantly derived from 9moALIOS, showed abundant expression of genes known to contribute to fibrosis and extracellular matrix assembly such as Col1a1 (fig. S2A) (6, 12). Moreover, the pronounced increase of 9moALIOS origin in cholangiocyte cluster mNASH-Cholangiocytes (fig. S2A) reflected a common pathological feature ductular reaction in NASH (13). Thus, the consistencies between our findings using snRNA-seq and previous studies largely on NPCs using scRNA-seq demonstrated the validity of our mouse model and single-nucleus approach in resembling human NASH progression.
The transcriptomic signature of hepatocytes switches from macronutrient processing to cell migration during NASH progression in mice
snRNA-seq has a major advantage over scRNA-seq in the evaluation of NAFLD/NASH livers because snRNA-seq more faithfully reports on larger cells and is thus preferable for studying hepatocytes, especially in fatty liver (14). The unsupervised clustering identified 14,484 hepatocyte nuclei that represented about 60% of all recovered nuclei. Cell clustering visualized by UMAP plot revealed 5 hepatocyte clusters (Fig. 1B and fig.S2A).
We next examined the hepatocytes in greater detail. Three subclusters of the mouse hepatocytes were assigned as the pericentral zone hepatocyte cluster (mPC-Hep), intermediate zone hepatocyte cluster (mInt-Hep), and periportal zone hepatocyte cluster (mPP-Hep) (Fig. 1B), as they showed distinct expression patterns of liver zonation markers (15, 16) including Glul, Aldh3a2, and Gls2, respectively (fig. S2A). There were more shared changes across all 3 zones at the NAFL stage (fig. S2E), while the NASH stage was characterized by a greater degree of zone-specific changes (fig. S2F).
At the NAFL stage, the shared up-regulated genes were involved in metabolic processes and PPAR signaling (fig. S2G), whereas at the NASH stage elevated genes were associated with the electron transport chain and cell-cell adhesion (fig. S2H). Regarding the zone-specific changes, mPC-Hep showed enrichment of bile acid synthesis at the NAFL stage (fig. S2I) and mPP-Hep exhibited an increase of Wnt signaling regulation at the NASH stage (fig. S2J). Among these 3 zonated-hepatocyte clusters, PP-Hep and Int-Hep, but not PC-Hep, showed a reduced percentage of cells from 9moALIOS, suggesting the periportal and intermediate zones were impacted more severely than the pericentral zone during NASH progression (fig. S2A).
In addition to the 3 zonated-hepatocyte groups, another 2 hepatocyte clusters (hereafter referred to as mNASH-Hep1 and mNASH-Hep2) were identified as highly NASH-specific because the majority of cells in these 2 clusters were derived from 9moALIOS (Fig. 1B and fig. S2A). mNASH-Hep1/2 exhibited distinct gene signatures from mPC-Hep, mInt-Hep and mPP-Hep (Fig. 1C). These genes included the hepatokine fibrinogen-like protein 1 (Fgl1), phospholipid-binding protein Annexin A2 protein (Anxa2), as well as the senescence marker Cdkn1a (P21), all of which have been reported to correlate with NAFLD/NASH in mice and are also dysregulated in human NASH (17–19). In addition, multiple we uncovered markers including zinc finger protein Glis3, plasma membrane sodium/calcium exchanger Slc24a3, and Eph-Ephrin family member Ephb2 with as yet unclear roles in NAFLD/NASH (Fig. 1C and data file S1). Gene ontology (GO) analysis revealed enriched biological processes associated with cell junction organization, cell migration, and cell-matrix adhesion besides the fatty acid metabolic process that reflects the classic function of hepatocytes (Fig. 1D).
To gain more insight into the molecular mechanisms of hepatocyte transition during NASH progression, we performed pseudotime analysis using Monocle2 (20). We found that the hepatocyte trajectory bifurcated at a node (labelled as “X” in Fig. 1E and data file S1). The root to the left of the node contained cells mainly from 3moNC and 9moNC and was defined as the starting point of pseudotime (Monocle2 pseudotime 0 in Fig. 1F) and the basal state of hepatocyte trajectory, which divided into two branches after node X. The upper branch representing the cells mostly from 3moALIOS was defined as the mNAFL branch, whereas the lower branch was considered as the trajectory to NASH and consisted largely of 9moALIOS hepatocytes (Fig. 1E and data file S1). Analysis of the genes that were most affected after the hepatocyte trajectory bifurcation revealed 6 modules, largely consisting of genes up-regulated in NAFL as well as both up- and down-regulated genes in the NASH branch (Fig. 1G and data file S1).
Genes up-regulated in the NAFL branch (Modules 3 and 4) were largely involved in lipid metabolism (Fig. 1H and data file S1) including Elovl5 and Insig2 (Fig. 1K), supporting an adaptive enhancement of both de novo lipogenesis and fatty acid oxidation to process excessive nutrients in NAFL (21). Genes down-regulated as hepatocytes progressed from NAFL to NASH (Modules 5 and 6) were largely associated with macronutrient processing (Fig. 1I and data file S1) such as Acly and Cyp1a2 (fig. S2K). By contrast, genes induced in the mNASH branch of hepatocytes (Modules 1 and 2) play roles in cell migration and cytoskeleton organization (Fig. 1J and data file S1) such as Actb, Ephb2 and Grip1 (Fig. 1K and fig. S2K), suggesting that hepatocytes lost their canonical metabolic function and gained a new identity during NASH progression.
Transcriptomic signatures of hepatocytes in human NASH resemble those of mouse
We next characterized the transcriptome of human NASH livers at the single-nucleus level, using “healthy” livers from patients without liver diseases as controls (Fig.2A and data file S2). The samples showed comparable recovery of the number of genes per nucleus, as well as quality as assessed by mitochondrial fraction, both overall (fig. S3A) and at the level of cell clusters (fig. S3B). After removing doublets, a total of 54,847 nuclei was recovered from 3 “healthy” and 3 NASH livers. All major liver cell types were identified by snRNA-seq including hepatocytes, endothelial cells, stellate cells, cholangiocytes, vascular smooth muscle cells, and monocytes/macrophages (Fig. 2A, fig. S3C–D and data file S2). Reflecting their diseases status, relative to the “healthy” samples the NASH livers were characterized by fewer hepatocytes and increased NPCs including stellate cells (hNASH-Stellate1/2), vascular smooth muscle cells (hNASH-VSMC), and cholangiocytes (hNASH-Cholangiocytes1/2) (fig. S3E), consistent with the fibrosis and ductular reactions in the NASH livers.
Fig. 2. snRNA-seq uncovers bifurcation of hepatocyte trajectory in human patients with NASH.
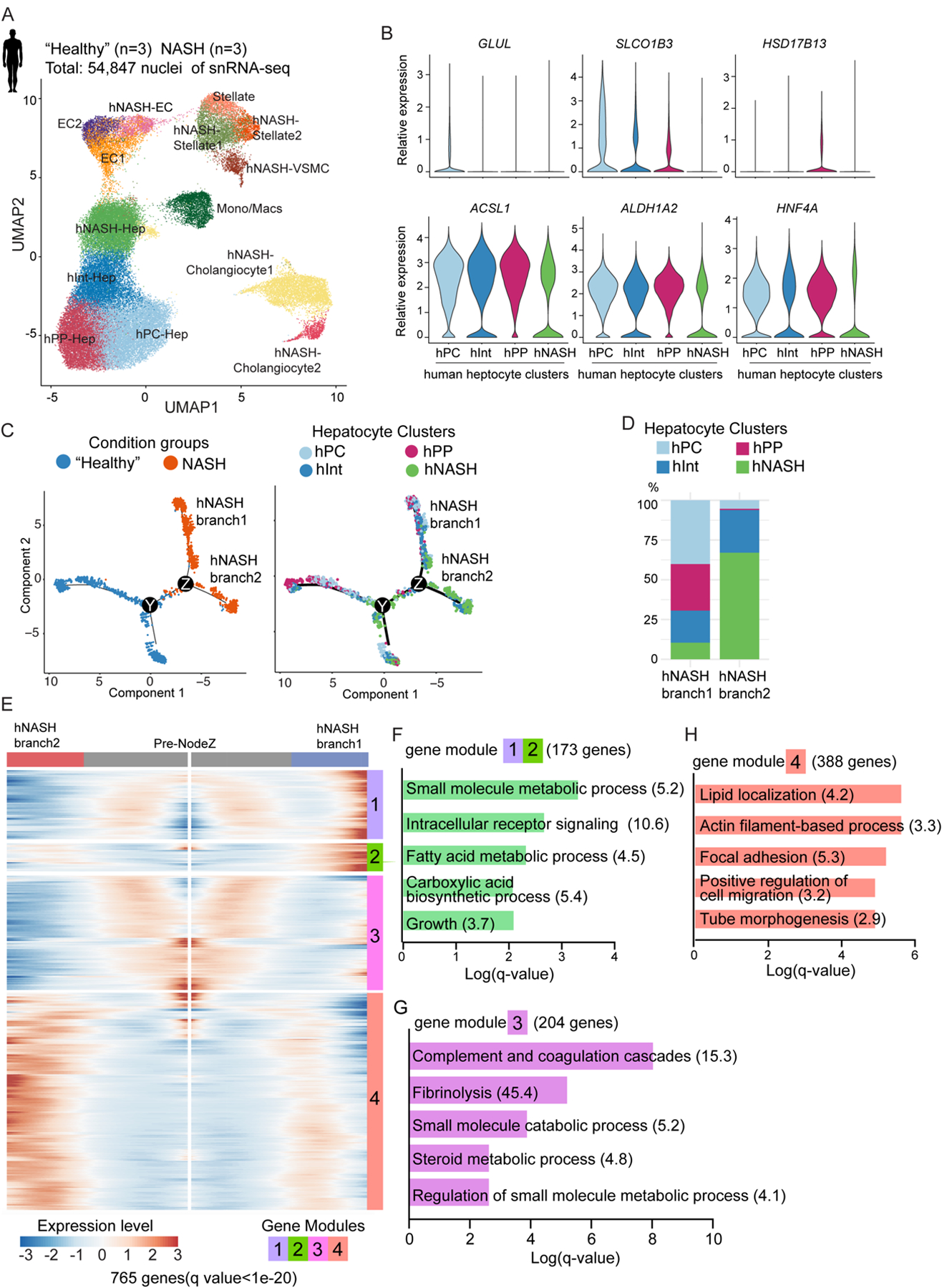
(A) UMAP plot visualization of the unsupervised cell clusters containing 54,847 nuclei from livers of “healthy” and NASH human livers. Hep: hepatocytes; Mono/Mac: monocytes/macrophages; EC: endothelial cells; VSMC: vascular smooth muscle cells.
(B) Violin plot shown differential gene expression across 4 hepatocyte clusters.
(C) Pseudotime analysis by Monocle2 reveals hepatocyte trajectory bifurcation during NASH progression. From left to right, the color code indicates the condition groups and hepatocyte clusters.
(D) The composition of hepatocyte clusters in hNASH branch1 and hNASH branch2.
(E) Heatmap of the 765 genes (q value < 1e-20) that determined hepatocyte trajectory bifurcation at NodeZ in Fig. 2C using branched expression analysis modeling (BEAM) in Monocle2. These genes are clustered into 4 different modules based on their gene expression pattern across Monocle2 pseudotime.
(F-H) GO analysis of gene modules which determined hNASH branch1 and 2 (Odds ratio indicated after each term).
We focused on human hepatocytes, as we identified a group of NASH-specific hepatocytes in mouse NASH models. Among the 4 subclusters of hepatocytes recovered by snRNA-seq on human livers, the identities of pericentral zone hepatocytes (hPC-Hep), intermediate zone hepatocytes(hInt-Hep), and periportal zone hepatocytes (hPP-Hep) were assigned based on the zonation landmarks GLUL, SLCO1B3, and HSD17B13 (Fig. 2B upper panels). The cellular composition of hepatocytes revealed that periportal hepatocytes were impaired most severely in human NASH compared with other zones (fig. S3F), in line with our observation in mice. We identified a fourth cluster of hepatocytes that scarcely expressed specific zonation genes (Fig. 2B upper panels) nor canonical metabolic and lineage markers of hepatocytes (Fig. 2B lower panels and fig. S3D). Because this cluster of hepatocytes was present in higher proportion in NASH livers (fig. S3F), we refer to it as hNASH-Hep.
To capture the molecular changes of hepatocytes with NASH progression, we performed Monocle2 pseudotime analysis. Consistent with the findings in mice, hepatocyte trajectories divided into two branches (Fig. 2C, Node Z and data file S2) during the progression from “healthy” to NASH. The upper branch “hNASH branch1” was mostly derived from hPC-Hep and hPP-Hep, whereas the lower branch “hNASH branch2” was predominantly comprised of hNASH-Hep, suggesting a unique feature of this branch (Fig. 2D and data file S2). Therefore, we further analyzed Node Z on the pseudotime trajectory and identified gene modules that determined the bifurcation. Analysis of the genes that were most affected after the hepatocyte trajectory bifurcation revealed 4 modules (Fig. 2E and data file S2).
Gene modules 1 and 2, containing 173 genes elevated in the hNASH branch1 (Fig. 2F), were enriched for liver metabolic processes such as the master regulator of gluconeogenesis FOXO1 and glucose transporter SLC2A2 (GLUT2), suggesting that hNASH branch1 hepatocytes underwent metabolic adaption. Gene module 3, with 204 genes that were down-regulated in both NASH branches, was involved in coagulation process and steroid metabolic process (Fig. 2G). Gene module 4, with 388 genes determining the hNASH branch2, displayed augmentation in cytoskeleton organization and cell migration (Fig. 2H). The identification of gene signatures associated with cell adhesion and migration in the human as well as the mouse hepatocyte trajectories suggested common molecular mechanisms in the pathogenesis of NASH.
EphB2 demarcates a subset of NASH hepatocytes engaged in intercellular crosstalk and trajectory bifurcation
To explore intercellular crosstalk in the development of NASH, we used CellChat (22) to compute all putative ligand-receptor interactions of each mouse cluster and rank their interaction strength. mNASH-Stellate demonstrated the most intercellular communication based on the sum of outgoing and the incoming signals (fig. S4A and data file S3). Macrophages are well-known to activate stellate cells (23), and indeed this analysis uncovered the incoming signaling from macrophages (mNASH-Mac1/2) including TGFb and PDGF signaling which have been reported to activate stellate cells (23) (fig. S4B–C and data file S3). The mNASH-Hep1/2 cluster communicated with mNASH-Stellate to a greater degree than with macrophages (fig. S4B). Thus, we focused on the interactions between mNASH-Hep and mNASH-Stellate (Fig. 3A). We identified signals such as HGF and PDGF, whose roles in liver fibrosis have been previously identified (24, 25). We also identified Eph-Ephrin signaling pathway as one of the top ranked pathways (Fig. 3A and data file S3). This pathway is well-known to regulate cell adhesion and migration, consistent with the common transcriptomic signatures of the mNASH branches in mouse and human hepatocytes. The Eph-Ephrin signaling pathway consists of 14 transmembrane receptors and 8 membrane-tethered ligands, and signaling can be bidirectionally transduced into such that changes occur in both receptor-expressing cells as well as ligand-expressing cells (26). However, the role of Eph-Ephrin signaling in hepatocytes is unknown.
Fig. 3. A subset of hepatocytes from mouse and human NASH livers exhibit enhanced EphB2 activity.
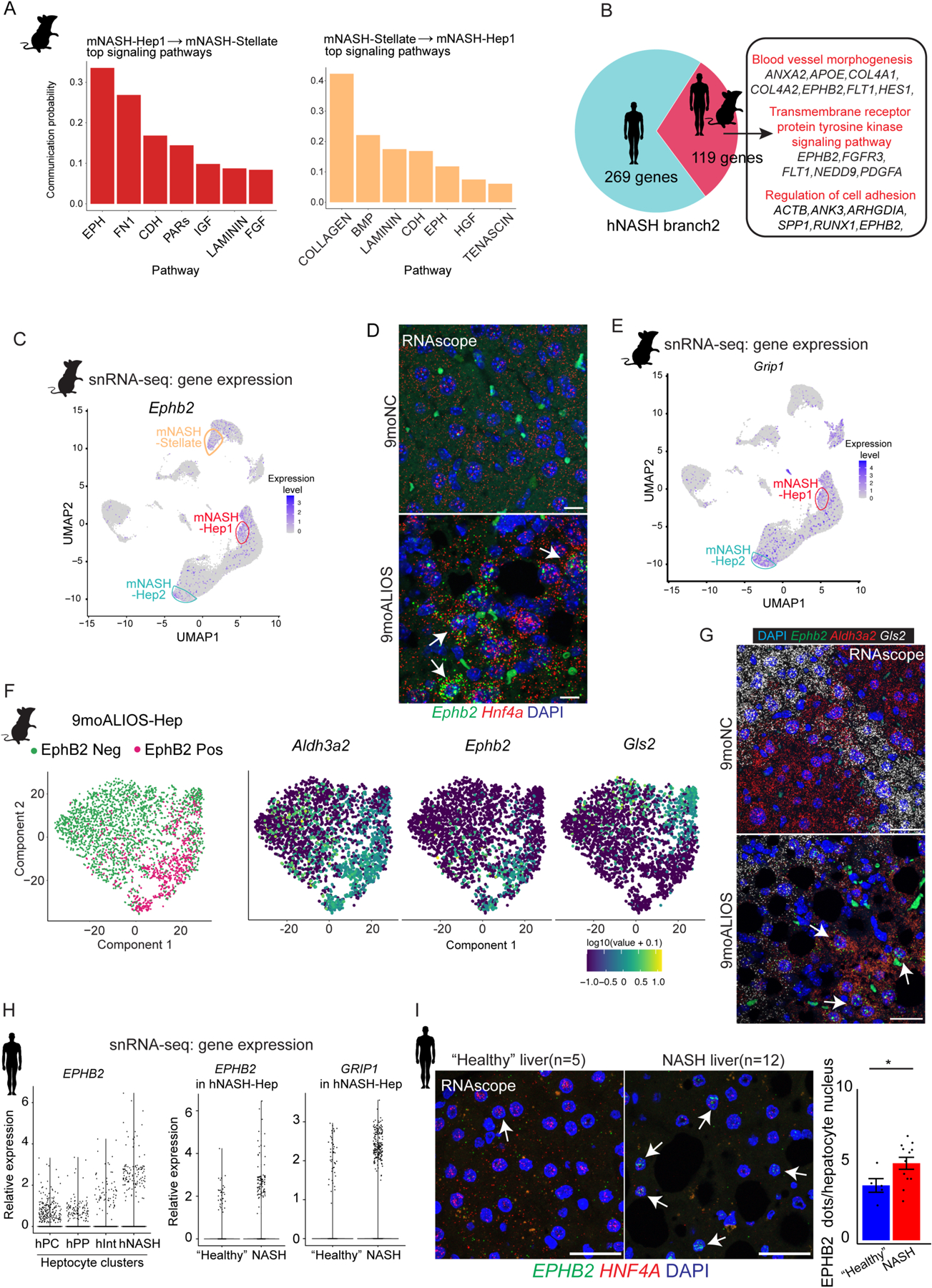
(A) Top signaling pathways mediated intercellular communication between mNASH-Hep1 and mNASH-Stellate.
(B) 119 common genes including EPHB2 between mouse and human were identified as contributing to mNASH branch/hNASH branch2.
(C) UMAP plot of snRNA-seq showing mNASH-Hep1/2 Ephb2 RNA expression.
(D) Elevated transcript abundance of Ephb2 in hepatocytes from 9moALIOS liver was validated by RNAscope (white arrows pointing to the hepatocyte nuclei labelled by Hnf4a, scale bar=20μm, n=3 in each group).
(E) UMAP plot of snRNA-seq shown Grip1 was prominently elevated in mNASH-Hep1/2.
(F) Subclustering of hepatocytes from 9moALIOS liver based on Ephb2 transcript abundance and expression pattern of zonation landmarks Aldh3a2 and Gls2 in EphB2-positive and EphB2-negative clusters.
(G) Co-staining Ephb2 with zonation landmarks using RNAscope validated that Ephb2-expressing hepatocytes mostly localized in pericentral zone (scale bar=25μm, n=3 in each group). (H) EPHB2 expression across different hepatocyte clusters in human snRNA-seq and differential expression of EPHB2 and GRIP1 between “healthy” and NASH conditions within the hNASH-Hep cluster.
(I) RNA in situ hybridization using RNAscope showed increased EPHB2 expression (green, white arrows) in hepatocyte nuclei stained with HNF4A (red) in livers from human patients with NASH (n=12) compared with “healthy” donor livers (n=5) (*p<0.05, Mann-Whitney U test, scale bar=25μm).
Eph ligands and Ephrin receptors were detected across several different cell types. The detectable Ephrin ligands, except Efnb2 was specifically expressed in mNASH-EC, Efna1 and Efna5 were present in hepatocytes or stellate cells regardless of NASH condition. In contrast, the Eph receptors Epha1, Epha2, and Ephb2 displayed restricted expression in mNASH-Hep1/2 (fig. S4D). However, Ephb2 from mNASH-Hep was predicted to interact with the ligand Efna5 secreted from mNASH-Stellate with the highest communication probability (fig. S4E). Furthermore, we overlapped the genes determining the bifurcations of NASH hepatocyte trajectories in both human and mouse and indeed, EPHB2 was among the common genes (Fig. 3B). Although EphB2 has mostly been studied in the nervous system (26), it has been shown to be expressed in a mouse liver fibrosis model induced by chronic carbon tetrachloride (CCl4); in that model EphB2 expression was reported to be increased in activated stellate cells but not in other cell types (27). We therefore focused on the unexpected expression of EphB2 in NASH hepatocytes.
In mouse hepatocytes, Ephb2 was sparsely expressed in NAFL, but prominently elevated in NASH (Fig. 3C). Of note, activation of Ephb2 was also found in the NASH stellate cells (mNASH-Stellate), as was reported in other liver fibrosis models (27, 28). To validate the RNA and protein abundance of EphB2 in hepatocytes, we performed RNA in situ hybridization using RNAscope probe and immunofluorescence (IF) staining on the mouse liver tissue sections. Indeed, Ephb2 RNA was expressed in a subset of hepatocytes labelled with Hnf4a RNAscope probe in 9moALISO livers but barely detected in 9moNC group (arrows in Fig. 3D, dotted line circled regions in fig. S4F). The EphB2 positive cells co-stained with hepatocyte marker HNF4α by IF, confirming that the increased protein abundance was in hepatocytes of 9moALIOS liver (fig. S4G, white arrows). Furthermore, EphB2 was increased in another diet-induced NASH model, the choline-deficient L-amino acid-defined high fat diet (CDAHFD) administered for 2 months (2moCDAHFD) (fig. S4H). We found that Grip1, which encodes glutamate receptor interacting protein and is absent in normal hepatocytes, was elevated in 9moALIOS hepatocytes in the snRNA-seq data (Fig. 3E) and validated by RNA scope (fig. S4I). GRIP1 has been shown to control dendrite morphogenesis by promoting the translocation of EphB2 to the cell surface (29, 30), suggesting a concordant function acquired in mNASH-Hep.
We further characterized the EphB2 positive cell population by clustering 9moALIOS hepatocytes based on Ephb2 mRNA abundance. The clusters with and without detectable Ephb2 were defined as EphB2-positive (EphB2 Pos) and EphB2-negative (EphB2 Neg), respectively (Fig. 3F,left panel). Ephb2-positive cells abundantly expressed pericentral marker Aldh3a2 but lacked periportal marker Gls2 (Fig. 3F, right panel). RNAscope validated that Ephb2-positive hepatocytes mainly localized in the pericentral zone stained by Aldh3a2 but rarely in the periportal zone labelled by Gls2 (Fig. 3G).
In humans, hNASH-Hep expressed the greatest amount of EPHB2 compared to hPC-Hep, hPP-Hep and hInt-Hep (Fig. 3H, left panel). Within hNASH-Hep, cells from NASH groups also showed higher expression of EPHB2 as well as GRIP1 (Fig. 3H, right 2 panels). We further examined liver tissue sections from 5 “healthy” donors and 12 patients with NASH. Co-staining EPHB2 with hepatocyte marker HNF4A using RNAscope demonstrated increased EPHB2-positive hepatocytes in human NASH livers (Fig. 3I and data file S3). Of note, unlike in mice where EphB2-positive hepatocytes localized at the pericentral zone, human NASH-Hep enriched for EPHB2 expression were not zone-specific.
Notch induces EphB2 expression in hepatocytes
To determine the epigenomic landscape of liver cell types and define the factors contributing to the transcriptomic alterations in NASH, we performed snATAC-seq on mouse livers with the same diet-feeding paradigm as described for snRNA-seq (Fig. 4A). All samples were recovered with comparable quality (fig. S5A and data file S4). We next integrated snATAC-seq with snRNA-seq and transferred cell clustering labels from snRNA-seq to snATAC-seq. The clustering of snATAC-seq exhibited a similar cluster identity with snRNA-seq (Fig. 4A and data file S4). Each cluster exhibited cluster-specific open chromatin regions (fig. S5B). Among NPCs, macrophage subcluster mNASH-Mac1/2, endothelial cell subcluster mNASH-EC, stellate cell subcluster mNASH-Stellate, and mNASH-Cholangiocyte were proportionally more prevalent in 9moALIOS liver (fig. S5C and data file S4).
Fig. 4. Notch induces EphB2 expression in hepatocytes.
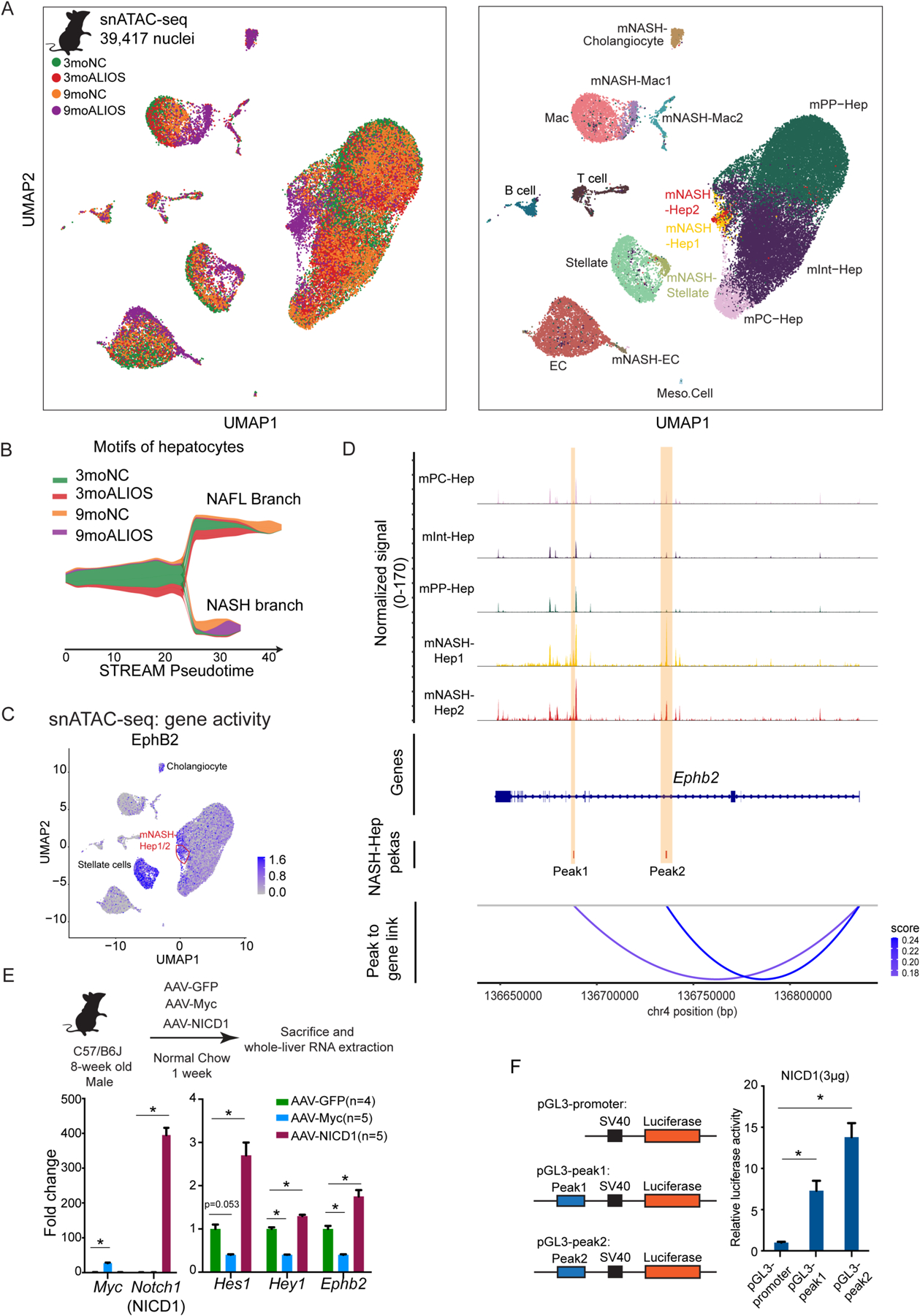
(A) Left: UMAP plot of snATAC-seq of mouse livers colored by sample identity. Right: UMAP plot of snATAC-seq colored by clusters (Hep: hepatocytes; EC: endothelial cells; MAC: macrophages).
(B) Using ChromVAR motif-cell z-score matrix as input, STREAM pesudotime density plot uncovered a bifurcation of hepatocyte TF motif trajectory during NASH progression. NAFL branch and NASH branch were assigned based on the experimental group composition in the corresponding branch.
(C) UMAP plot of snATAC-seq demonstrated that mNASH Hep1/2 acquired elevated gene activity of EphB2 (orange circle).
(D) snATAC-seq peak tracks across different hepatocyte clusters at Ephb2 locus. Peak to gene links were calculated based on the correlations between peak accessibility in snATAC-seq and gene expression in snRNA-seq. Two cis-elements (regions highlighted in yellow as Peak1 and Peak2) were predicted to positively control Ephb2 expression in hepatocytes.
(E) Top: experimental scheme of forced activation of NICD and Myc in hepatocytes. Bottom: NICD but not Myc in hepatocytes up-regulated Ephb2 expression. Data are expressed as mean ± SEM, *p<0.05, Mann-Whitney U test.
(F) NICD promoted luciferase activity through the two cis-regulatory elements at Ephb2 locus identified by the integration of snRNA-seq and snATAC-seq. Data are expressed as mean ± SD, n=3 in each group, * p<0.05 Mann-Whitney U test.
We next focused on the hepatocyte clusters. In agreement with snRNA-seq, mInt-Hep and mPP-Hep had less cells from 9moALIOS whereas mPC-Hep contained similar a proportion from each condition, suggesting that periportal hepatocytes were more profoundly disrupted by NASH compared to pericentral hepatocytes (fig. S5C). Consistent with the transcriptomic bifurcation, enriched transcription factor (TF) motifs also displayed distinct trajectories between NAFL and NASH, both in the density of the cell distribution (Fig. 4B and data file S4) and at the single-cell level (fig. S5D and data file S4), demonstrating clear stage-specific epigenomic signatures. The intersection between TFs that positively contributed to the bifurcation with a published dataset that provided their transcriptional abundances in bulk (GSE162876) suggested that TFs such as CUX2 and PPARg contributed to NAFL (fig. S5E–F and data file S4). By contrast, the top TFs in the branch leading to NASH consisted of three major groups: GLIS2/3, the FOS/JUN family, and the TEAD family (fig. S5G–H). Indeed, the function of GLIS2 in NASH progression was highlighted in a recent study where knockdown of Glis2 in hepatocytes attenuated inflammation and fibrosis in mouse NASH liver (31).
In accordance with the elevated transcriptional expression in mNASH-Hep1/2, the gene activity of EphB2, reflected by ATAC signals on the gene body, was also enhanced compared with other hepatocyte clusters (Fig. 4C). To interrogate upstream signals that drive EphB2 expression in NASH hepatocytes, predicted functioning regulatory regions were assessed based on the correlation between peak accessibility (snATAC-seq) and gene expression (transferred snRNA-seq information). Compared with other hepatocyte clusters, two mNASH-Hep-specific peaks were found at the Ephb2 locus (Fig. 4D, highlighted in yellow; Peak1 shown in higher resolution in fig. S6A) that exhibited correlation with Ephb2 RNA abundance (Fig. 4D, Peak to Gene links track). The binding motifs of 7 TFs, including HES5, HEY1, and MYCN, were found in both peaks, and another 62 TFs, including TCF7L1, MYC, and RBPJ, were detected in 1 of the identified peaks (fig. S6B and data file S4). To prioritize these 69 TFs, we performed enrichment analysis of signaling transduction pathways that revealed enrichment for Notch signaling, Hippo signaling, and Wnt signaling (fig. S6C), which are major pathways driving NASH progression and transition to HCC (32, 33).
We focused on the Notch pathway, which has been previously implicated in NAFLD/NASH (32, 34), as well as MYC, a critical node at which multiple signaling pathways converge including Hippo and Wnt signaling (35, 36). The Notch pathway and Myc were induced by transducing hepatocytes with constitutively activated Notch1 intracellular domain (NICD1) or Myc for 1 week in mice fed NC diet (Fig. 4E, top and bottom left). NICD1 was sufficient to drive Ephb2 transcription to a degree comparable to Notch canonical downstream targets Hes1 and Hey1 whereas Myc repressed Ephb2 expression (Fig. 4E, bottom right and data file S4). To exclude the possibility that Notch activated Ephb2 expression in NPCs, especially stellate cells, we performed IF staining. Enhanced Ephb2 was detected in the hepatocytes with forced expression of Flag epitope-tagged NICD across different zones of livers (fig. S6D), supporting the conclusion that NICD induced Ephb2 transcription in a cell-autonomous manner. The enhancer activities of the 2 identified ATAC-seq peaks at the Ephb2 locus were tested by cloning them upstream of the SV40 promoter in luciferase reporter vectors (Fig. 4F, left). Both sites conferred NICD-responsiveness in AML12 hepatocyte cells (Fig. 4F, right and data file S4), suggesting that Notch signaling activates Ephb2 transcription in hepatocytes through these 2 identified regulatory regions during NASH progression.
EphB2 is sufficient to induce cell-autonomous inflammation in mouse and human hepatocytes
To test the function of EphB2-expressing hepatocytes, we first performed a gain-of-function study. Analysis of published bulk mouse NASH liver RNA-seq data (GSE119340) revealed the most abundant EphB2 isoform (ENSMUST00000059287.13) (fig. S7A and data file S5). This EphB2 isoform was tagged at its C-terminus with the Flag epitope and expressed in the hepatocytes of 8-week-old male C57/B6J mice (Hep-EphB2) using the hepatocyte-specific AAV8-TBG expression system (37) that were studied one week later to capture the direct effects of EphB2 (Fig. 5A). Mice injected with AAV-TBG-GFP were used as controls (Hep-GFP) (Fig. 5A). Hepatic expression of EphB2 was confirmed by immunoblotting (fig. S7B). IF staining using the anti-Flag antibody confirmed that the EphB2 expression was restricted to hepatocytes and not found in stellate cells (fig. S7C–D). Moreover, ALT and AST concentrations were normal (fig. S7E and data file S5) without evidence of steatosis (fig. S7F and data file S5) or fibrosis (fig. S7G).
Fig. 5. Hepatocyte-specific EphB2 activation in mice fed NC diet induces cell-autonomous inflammation.
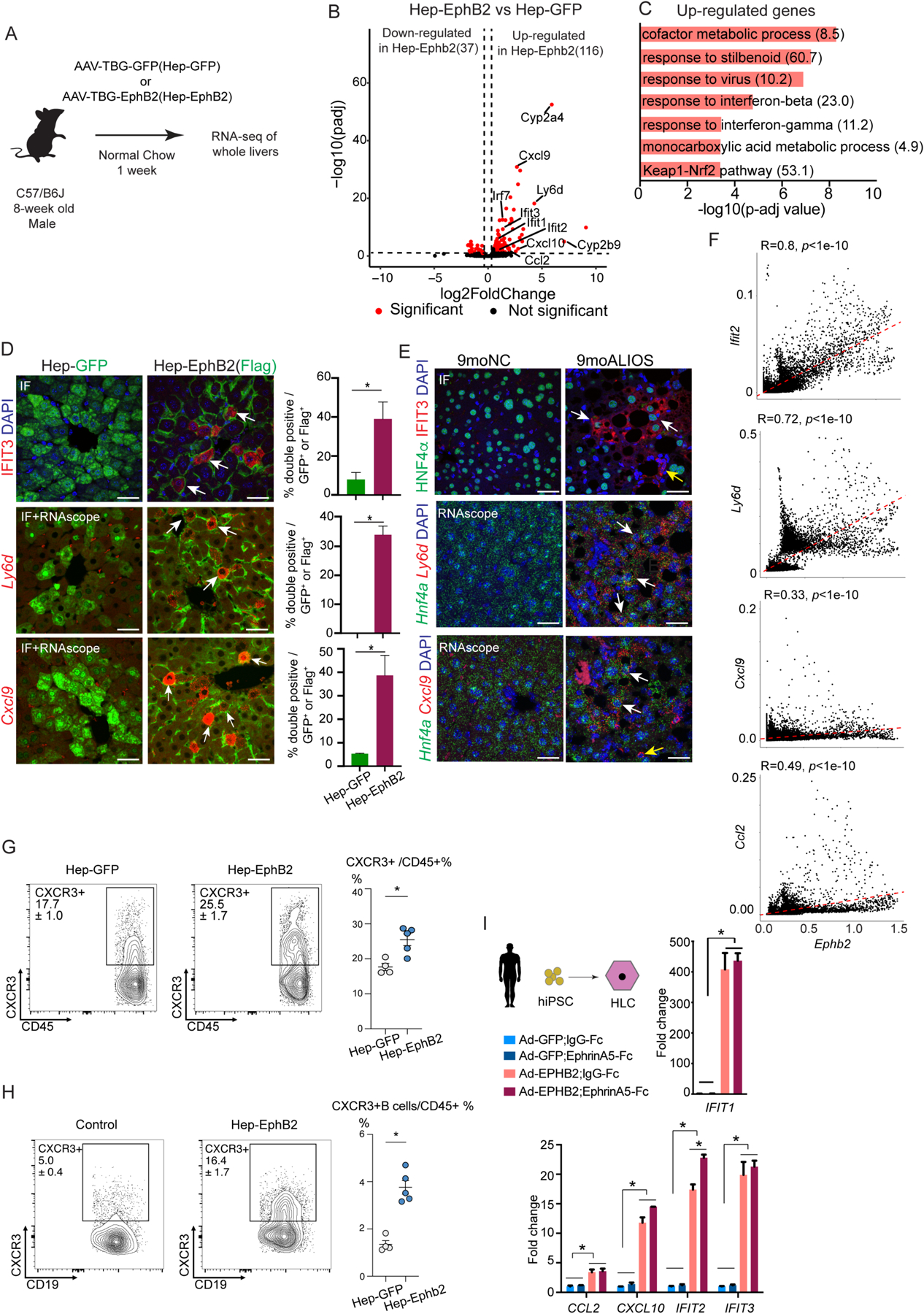
(A) Experimental scheme of RNA-seq of whole livers from the mice with forced expression of EphB2 (Hep-EphB2, n=3) or GFP (Hep-GFP, n=3).
(B) Volcano plot of the differentially expressed genes (shown as red dots with cutoff padj<0.1 and abs(log2FC) > 0.5) between Hep-EphB2 and Hep-GFP (Ephb2 is not shown on this plot).
(C) GO analysis demonstrated enrichment of metabolic processes, interferon response pathways, oxidative stress pathways, and Keap1-Nrf2 in up-regulated genes (Odds ratio indicated after each term).
(D) IF staining (IFIT3) and RNA-scope (Ly6d and Cxcl9) showing induced inflammatory response in hepatocytes with ectopic expression of EphB2 (white arrows). GFP and Flag antibodies stained the transduced hepatocytes from AAV-GFP and AAV-EphB2-Flag tail-vein injected mouse livers respectively. Data are expressed as mean ± SD, n=3 in each group, *p<0.05, Mann-Whitney U test, scale bar=25μm.
(E) IF staining (IFIT3) and RNA-scope (Ly6d and Cxcl9) in hepatocytes from mice treated with ALIOS diet for 9 months (white arrows). Antibody against Hnf4α or RNAscope probe detecting Hnf4a was used to label hepatocytes. Ifit3 and Cxcl9 were also detected in the non-hepatocytes in 9moALIOS livers (yellow arrows, scale bar=25μm).
(F) Correlation analysis of snRNA-seq demonstrated Ifit2, Ly6d, Cxcl9, and Ccl2 positively correlated with Ephb2 expression in hepatocytes.
(G-H) Flow cytometry showing an increased percentage of CXCR3 and CXCR3+ B cells in Hep-Ephb2 livers. Data are expressed as mean ± SEM, n=4/5 in each group, *p<0.05.
(I) Activation of EPHB2 in hiPSC-HLC induced inflammatory response and the effect was largely EphrinA5-independent. Data are expressed as mean ± SD, n=3 in each group, * p<0.05, Mann–Whitney U test.
RNA-seq of whole livers identified 116 up-regulated genes and 37 down-regulated genes (Fig. 5B and data file S5). The up-regulated genes in Hep-EphB2 livers were associated with interferon (IFN) pathways and inflammatory responses, including Ifit1/2/3, Irf7, Cxcl9/10, and Ccl2, as well as oxidative stress and metabolic genes such as Gsta1/2, Gstm1/2/3/4/6, Cyp2a4, and Cyp2b9 in Hep-EphB2 livers (Fig. 5B), and indeed these pathways were enriched in GO analysis (Fig. 5C). Many of these genes have been shown to be elevated in mouse and human NASH livers, including IFN-induced Ifit1/2/3 and Irf7 (38) and inflammatory genes Ccl2 and Cxcl9/10 (38–40). Moreover, the down-regulated genes in Hep-EphB2 were enriched for retinol metabolism (fig. S7H), which is disrupted in patients with NAFLD (41). Of note, forced expression of EphB2 in hepatocytes did not cause apparent cell apoptosis (fig. S7I).
To determine which cell type was responsible for the enhanced inflammatory response, we performed IF and RNA in situ hybridization on liver tissue sections. Consistent with the gene expression data, Flag-positive cells, representing hepatocytes with forced expression of EphB2, displayed marked up-regulation of the selected inflammatory markers including IFIT3, Ly6d, and Cxcl9 (Fig. 5D). Increased expression of IFIT3, Ly6d, and Cxcl9 was also observed in the hepatocytes from 9moALIOS livers compared to 9moNC (Fig. 5E). Moreover, snRNA-seq showed a positive correlation between hepatocyte expression of these inflammatory genes and Ephb2 (Fig. 5F and data file S5). These data suggested that the expression of EphB2 in hepatocytes induced a cell-autonomous inflammatory response that resembled the changes that occur in NASH.
Because hepatic expression of EphB2 induced chemokines CXCL9/CXCL10 and CCL2, we performed flow cytometry to examine immune cells with CXCR3 and CCR2 expression which receptors mediate the inflammatory effect of these chemokines. Although the percentage of CCR2+ cells among all immune cells (CCR2+/CD45+) was not different between Hep-Ephb2 and controls (fig. S8A), a higher percentage of CXCR3 (CXCR3+/CD45+) was found in Hep-EphB2 livers (Fig. 5G and data file S5), which were further confirmed by IF (fig. S8B). This change was mainly ascribed to the increased infiltration of CXCR3+ B cells (Fig. 5H and data file S5). Increased abundance of CXCR3 and accumulation of B cells are characteristic of both mouse and human NASH (42, 43).
To determine whether EphB2 functions similarly in human hepatocytes, we derived hepatocyte-like cells (HLCs) from the human induced pluripotent stem cells (hiPSC) (44). After 21 days of differentiation, HLCs stained positive for hepatocyte markers E-Cadherin and HNF4α, confirming their hepatocyte-like identity (fig. S8C). To express EPHB2 in HLCs, we analyzed a bulk RNA-seq dataset from human NASH livers and identified ENST00000400191.7 as the most abundant transcript that also most strongly correlated with fibrosis stage (fig. S8D and data file S5). We then used an adenoviral delivery system to express this EPHB2 isoform (Ad-EPHB2) in HLCs, and the cells were treated with the putative EPHB2 ligand EphrinA5-Fc or IgG-Fc control. The expression of EPHB2 was efficiently induced in HLCs (fig. S8E and data file S5), and none of the treatments affected cell morphology after 24hr (fig. S8F). Of note, and consistent with the findings in mouse hepatocytes, Ad-EPHB2 robustly activated inflammatory response genes IFIT1/2/3, CCL2, and CXCL10 in the human HLCs (Fig. 5I). The addition of putative EPHB2 ligand had minimal effect in this in vitro HLC system (Fig. 5I), suggesting that the ligand is already present endogenously or that EPHB2 acts ligand-independently in these cells.
Disease-activated EphB2 in hepatocytes contributes to NASH progression
We next performed a loss of function study using an AAV-based CasRx-mediated RNA targeting system (45) to interrogate the role of EphB2 in hepatocytes during NASH progression. Mice were fed CDAHFD which causes NASH more rapidly than the ALIOS diet (46) and, as noted above, is a potent inducer of EphB2 in hepatocytes (fig. S4H). After two months on the diet, blood was drawn for ALT measurement, and mice were randomly assigned to injection of tandem guide RNA targeting Ephb2 (sgEphB2) or control guide RNA(sgNC), then fed CDAHFD for an additional month while continuing on CDAHFD (Fig. 6A). The analysis focused on mice with comparable CDAHFD-induced liver injury reflected by ALT concentrations (500–600 U/L) at 2 months. At the end of the experiment, a modest reduction in hepatic Ephb2 expression was noted in the livers of mice treated with sgEphB2 (fig. S9A and data file S6). Histology of the sgEphB2 livers showed reduced Sirius Red staining area (Fig. 6B and data file S6), as well as advanced liver fibrosis stage as blindly judged by a pathologist not otherwise involved in the study (Fig. 6C and data file S6). Expression of inflammatory infiltration marker CD11b was also reduced in the sgEphB2 livers (Fig. 6D and data file S6). These data suggest that EphB2-expressing hepatocytes contribute to NASH progression. However, steatosis reflected by hepatic triglycerides (fig. S9B and data file S6) and general liver injury assessed by ALT concentrations (Fig. S9C) were not significantly changed by knocking down EphB2, potentially indicative of a selective effect of Ephb2 on inflammation and fibrosis.
Fig. 6. Hepatocyte-specific EphB2 knockdown ameliorates NASH progression.
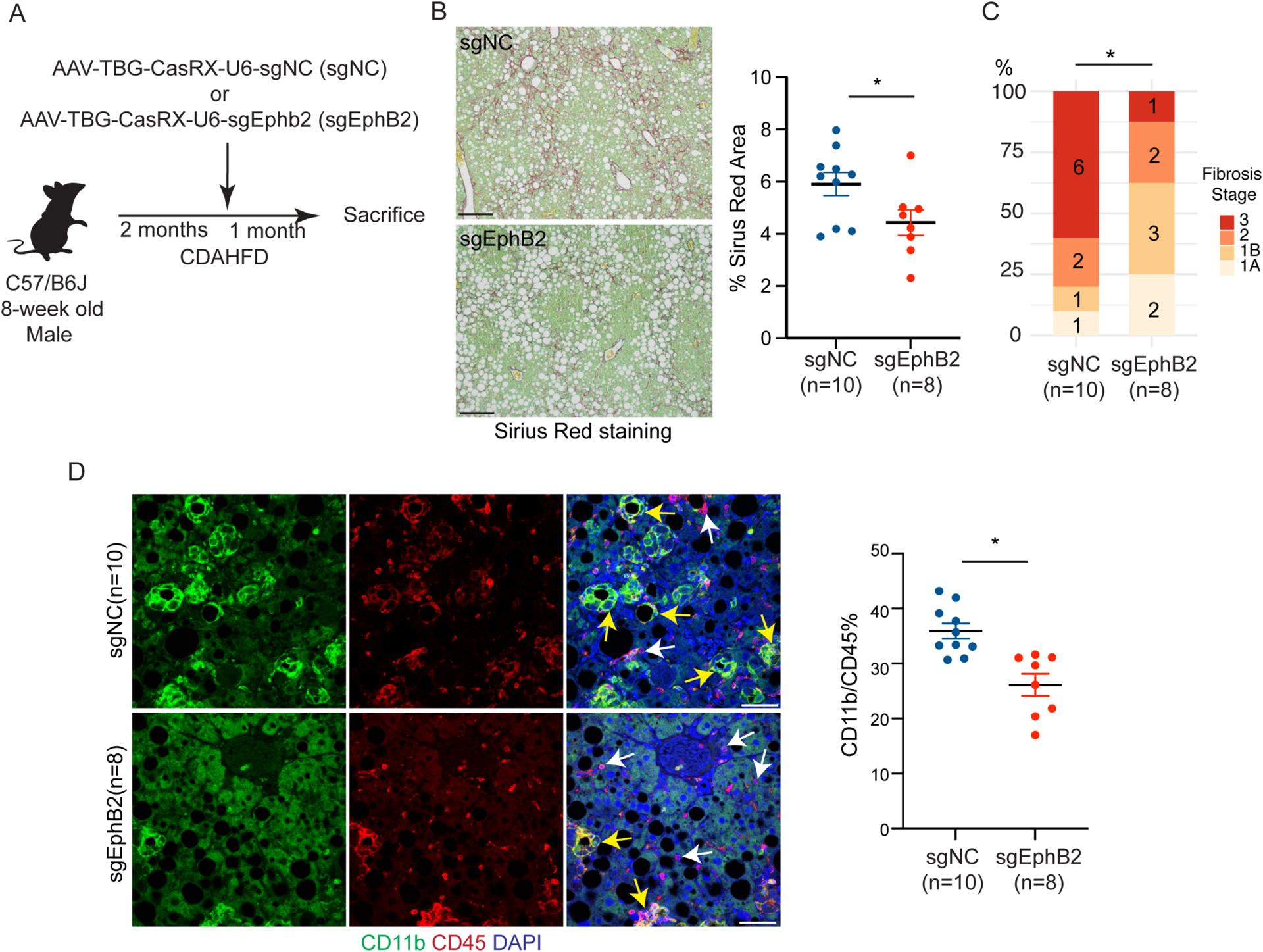
(A) Experimental scheme of inactivating EphB2 in hepatocytes in mice fed CDAHFD.
(B) Representative images and quantification of Sirius red stained area in sgEphB2 and sgNC livers. Data are expressed as mean ± SEM, *p<0.05, Mann–Whitney U test, scale bar=100μm.
(C) NASH fibrosis stage in sgEphB2 mice and sgNC control mice by blinded pathology evaluation (number labelled on the bar graph represented animal numbers in each category, *p<0.05, Mann–Whitney U test).
(D) Reduced CD11b inflammatory cell infiltration in sgEphB2 visualized by IF. Yellow arrows pointed to CD11b+ CD45+ cells and white arrows pointed to Cd45+ cells. Data are expressed as mean ± SEM, *p<0.05, Mann–Whitney U test, scale bar=25μm.
DISCUSSION
Our comprehensive characterization of transcriptomic and epigenomic changes in all major liver cell types during NASH progression revealed two distinct hepatocyte trajectory branches and the implication of TFs potentially responsible for the development of NAFL as well as the transition to NASH in mouse models and human patients. At the NASH stage, a subset of hepatocytes was demarcated by EphB2, whose expression was activated by Notch signaling. EphB2 sufficiently induced a cell-autonomous inflammatory response in hepatocytes and possibly engaged in intercellular communication with NPC including immune cells and stellate cells to contribute to NASH progression (fig. S10A).
Several single-cell transcriptomic studies of NASH development have been reported, but mainly focused on the NPC fraction due to limited information recovered from hepatocytes (5, 7, 38). Here, utilizing single-nucleus transcriptomics and epigenomics, we captured abundant information about hepatocytes as well as NPCs from the same livers of mouse and human, which provided a comprehensive molecular picture of all major liver cell types during NASH progression. During NAFL transits to NASH in mice, the transcriptomic signatures switched from macronutrient processing to cell adhesion and migration. The dichotomous features of hepatocytes were also found in human NASH livers, highlighting the bifurcation is conserved between mouse and human. Importantly, EphB2 is one of the shared genes between mouse and human that determines the branch enriched for cell migration signature in NASH.
In line with the transcriptomic changes in mice and human, the TFs responsible for maintaining hepatocyte lineage and metabolic adaptation were dwindled and replaced with stress-induced TFs during the transition. The distinct signatures of hepatocytes during NASH development provide transcriptomic and epigenomic evidence for the concept that NAFLD does not progress in a linear course (47).
Our findings corroborate the role of stellate cells as a signaling hub in NASH liver (6), but additionally demonstrate that hepatocytes also participate in inter-cellular crosstalk to a great degree. The prominent signaling feature of hepatocytes at the NASH stage, relative to NAFL, prioritizes hepatocytes as a major cell type that might be targeted by NASH treatments. One recent study revealed that Eph-Ephrin signaling positively correlates with the fibrosis stage but not the NASH activity score in the livers of patients with NASH (48), indicating a possible function of Eph-Ephrin in promoting fibrosis. Compared with other Eph-Ephrin family members, EphB2 exhibited a unique NASH-specific expression pattern in hepatocytes. A prior study of primary hepatocytes from Ephb2 −/− mice showed defective inflammatory potential when challenged with red blood cells from malaria parasite-infected mice (28), but the implications for NASH have not been previously explored. Our work provides direct evidence that EphB2 expression, which is absent in normal hepatocytes, is acquired in the degenerating hepatocytes at the NASH stage. However, how Ephb2-expressing hepatocytes communicate with stellate cells or other NPC requires further investigation.
Of note, it is well known that Notch signaling is activated in hepatocytes in response to chronic liver injury and is required for hepatocyte transdifferentiation into biliary epithelial cells (49). Notch signaling is both necessary and sufficient for NASH progression (32, 33). However, the downstream targets of Notch signaling are not completely understood in this context. By integrating snRNA-seq and snATAC-seq of mouse livers, we identified EphB2 as one of downstream effectors of Notch in hepatocytes to relay the effect of Notch signaling activation by demarcating degenerating hepatocytes and mediating their inter-cellular communication.
Interferon (IFN) signaling is best known for modulating immune responses to viral infections (50). Previous work suggested that the type-I IFN pathway is activated in metabolic diseases associated with chronic inflammation (38, 51). The induction of the type-I IFN response in NAFLD livers has been attributed to excessive lipid species (52) or stimulation of STING which senses aberrant double stranded DNA in the cytosol (53), but whether the induction of type-I IFN plays a deleterious or beneficial role during NAFLD progression is controversial (52, 54, 55). Our studies demonstrate that the expression of EphB2 in hepatocytes cell-autonomously elicited prominent type-I and type-III IFN responses both in mice fed normal chow as well as in human iPSC-derived hepatocytes, adding another key player in hepatic IFN activation.
Multiple clinical trials for NASH have targeted nuclear receptors (NRs), for example using agonists of FXR and PPARs, but no approved drug has emerged (3). Our study did reveal signatures of NRs such as PPARα, PPARγ, and FXR at open chromatin in the NAFL state, which is consistent with NAFL being largely a reversible metabolic process and the role of these NRs to sense nutrients and regulate energy balance (49, 50). However, our results show that as NAFL transitions to NASH, NRs are no longer the main contributors to open chromatin in hepatocytes. By contrast, EphB2 is uniquely expressed in hepatocytes at the NASH stage and contributes to the severity of fibrosis and inflammation. Because EphB2 is a transmembrane receptor, it is accessible to therapeutic antibodies and small molecules and may be a more promising target for NASH therapeutics. EphB2 is rarely expressed in liver cells at non-diseased state but abundantly expressed in the nervous system. Therefore, liver-specific targeting would be preferred to reduce the adverse effect of inhibiting EphbB2 activity.
There are limitations to this study. Although our study focused on the cell-autonomous role of EphB2 in hepatocytes after single-nucleus transcriptomic and epigenomic characterization, the communication between EphB2-expressing hepatocytes and other hepatocytes or NPCs during NASH progression remains to be investigated. Furthermore, although we demonstrated that Notch signaling activated the transcription of Ephb2 in hepatocytes, the identification of specific TFs that directly induce EphB2-expressing hepatocytes would be instrumental for targeting the NASH-specific hepatocyte subset. Last, EphB2-specific targeting strategies, including neutralizing antibodies (56) and antagonist peptides (57, 58), have been used to combat other diseases and should be explored in addition to metabolic targets for an effective treatment of NASH.
MATERIALS AND METHODS
Study design
This study aimed to use unbiased methods to identify cellular and molecular contributions to NASH progression that can be used for potential therapeutic targeting. In vivo and in vitro studies were applied to interrogate the role of EphB2-expressing hepatocytes which was a NASH-specific hepatocyte population revealed by single-nucleus transcriptomic and epigenomic characterization of mouse and human NAFL/NASH livers. In general, for in vivo and in vitro studies, at least two independent experiments were conducted or at least two complementary experimental methods were applied to draw conclusions. For each experiment, the sample size was indicated in figures or figure legends or corresponding method section. Liver pathologies were evaluated blindly. All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Pennsylvania. All studies using human samples were approved or exempted by the University of Pennsylvania Institutional Review Board.
Mice
C57BL/6J male mice were obtained from the Jackson laboratory (Cat#000664) and used in all animal experiments in this study. Mice were housed in the animal facility with 12h dark/light cycle (7:00am to 7:00pm) and ad libitum access to food and water. Two types of NASH-promoting diet, GAN diet (Research diet, D09100310) (8) and modified ALIOS diet (Envigo, TD.170428) (9), were evaluated for long-term NASH development. For snRNA-seq and snATAC-seq, 2–3 mice fed either standard mouse chow diet or modified ALIOS diet at 4-week-old of age for 3 months or 9 months were used. In the forced expression of MYC, NICD1 and EphB2 studies, 8-week-old mice were tail-vein injected with AAV-TBG-GFP as control and AAV-TBG-EphB2 or AAV-TBG-MYC or AAV-TBG-NICD1 as experimental groups for 1 week under standard mouse chow diet. In the EphB2 loss of function study, mice at 8-week-old fed L-amino-defined high (60 kcal %) fat diet with 0.1% methionine and no added choline (CDAHFD, Research Diet A06071302) for 8 weeks were injected with AAV-TBG-CasRX-U6-sgNC as controls or AAV-TBG-CasRX-U6-sgEphb2 as experimental groups through tail-vein. To match CDAHFD-induced liver injury severity, mice with similar ALT concentrations (400–500U/L; normal range: 28–132U/L) after 2-month CDAHFD and before AAV injection were selected for NASH severity evaluation.
Human liver samples
Normal human livers and NASH human livers used for snRNA-seq were obtained through the Liver Tissue Cell Distribution System, Minneapolis, Minnesota, which was funded by NIH Contract #HSN276201200017C. Paraffin tissues sections of human livers used for RNA in situ hybridization were acquired from SEKISUI XENOTECH (Tissue Microarray – Non-alcohol steatohepatitis, Lot#2110289).
Quantification and statistical analysis
Contrast and image size of IF images were adjusted with Adobe photoshop. Graphs were produced in R, Python, and GraphPad Prism. All figures were made in Adobe Illustrator. Sample size was indicated on the corresponding graph or figure legend, otherwise n=3, which represents the number of biological replicates that were analyzed in each experimental group. Error bars represent the SEM or SD as indicated in the figure legends. For assays with n<30, non-parametric Mann-Whitney U tests were used. For assays with n>30, non-parametric Mann-Whitney U test was used when data were not normally distributed and Student’s t-test was used when data were normally distributed. For cell origin composition of clusters, Chi-squared test was applied. P values were indicated in the corresponding figures or figure legends. All statistical analyses were performed using SPSS 21.0 or R 3.5.1.
Supplementary Material
Acknowledgements
We thank Yuxia Guan and Isaac J. Celwyn for assistance with animal husbandry. We thank Michael C. Tackenberg for discussions and advice on the manuscript and thank Shian-Huey Chiang, Mohammed Alsuraih and Rong Xin for discussions. We thank Katalin Susztak for access to the Olympus BX43 microscope and Chromium Controller, and Yachen Shen and Hongjun Song for providing AAV packaging plasmid and cells. We thank Kahealani Uehara for help with hepatocyte isolation, ALT/AST measurement, and the Functional Genomics Core, Viral Vector Core and Rodent Metabolic Phenotyping Core of the Penn Diabetes Research Center (P30 DK19525) for next-generation sequencing and providing AAV plasmids and AST/ASL measurements, respectively. We thank Cell & Developmental Biology Microscopy Core for use of the Leica TCS SP8 Confocal microscope. We also thank Kazuhiro Oka and Corinne Sonnet from the Gene Vector Core at Baylor College of Medicine for AAV and adenovirus production. We thank Dr. Rotonya M. Carr and Dr. Rebecca G. Wells for generously providing us human samples.
Funding:
This work was supported by National Institutes of Health grants (R01-DK125573 to M.A.L.; K08-DK116668 to D.A.H.; K01-DK125602 to D.G.) as well as the Cox Medical Research Institute and the JPB Foundation. Y.X. was supported by American Heart Association Training grant #827529 and W.H. was supported by American Diabetes Association Training grant #1-18-PDF-132.
Footnotes
Competing interests:
M.A.L. is an advisory board member and has received research support from Pfizer Inc. He is also a scientific co-founder and advisory board member of Flare Therapeutics, and consultant to Madrigal Pharmaceuticals. The remaining authors declare that they have no competing interests.
List of Supplementary Materials
Data files S1 to S6
Data and materials availability:
All data associated with this study can be found in the paper or supplementary materials. The snRNA-seq, snATAC-seq, and RNA-seq datasets have been deposited in Gene Expression Omnibus (GEO) with accession number GSE189600.
REFERENCES
- 1.Golabi P, Paik JM, AlQahtani S, Younossi Y, Tuncer G, Younossi ZM, Burden of non-alcoholic fatty liver disease in Asia, the Middle East and North Africa: Data from Global Burden of Disease 2009–2019. J Hepatol 75, 795–809 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Albhaisi SAM, Sanyal AJ, New drugs for NASH. Liver Int 41 Suppl 1, 112–118 (2021). [DOI] [PubMed] [Google Scholar]
- 3.Vuppalanchi R, Noureddin M, Alkhouri N, Sanyal AJ, Therapeutic pipeline in non-alcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol 18, 373–392 (2021). [DOI] [PubMed] [Google Scholar]
- 4.Sanyal AJ, Anstee QM, Trauner M, Lawitz EJ, Abdelmalek MF, Ding D, Han L, Jia C, Huss RS, Chung C, Wong VW, Okanoue T, Romero-Gomez M, Muir AJ, Afdhal NH, Bosch J, Goodman Z, Harrison SA, Younossi ZM, Myers RP, Cirrhosis Regression is Associated with Improved Clinical Outcomes in Patients with Nonalcoholic Steatohepatitis. Hepatology, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deczkowska A, David E, Ramadori P, Pfister D, Safran M, At The B, Giladi A, Jaitin DA, Barboy O, Cohen M, Yofe I, Gur C, Shlomi-Loubaton S, Henri S, Suhail Y, Qiu M, Kam S, Hermon H, Lahat E, Ben Yakov G, Cohen-Ezra O, Davidov Y, Likhter M, Goitein D, Roth S, Weber A, Malissen B, Weiner A, Ben-Ari Z, Heikenwälder M, Elinav E, Amit I, XCR1(+) type 1 conventional dendritic cells drive liver pathology in non-alcoholic steatohepatitis. Nat Med 27, 1043–1054 (2021). [DOI] [PubMed] [Google Scholar]
- 6.Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, Zhao XY, Ji Y, Li C, Guo L, Zhou L, Chen Z, Leon-Mimila P, Chung MT, Kurabayashi K, Opp J, Campos-Pérez F, Villamil-Ramírez H, Canizales-Quinteros S, Lyons R, Lumeng CN, Zhou B, Qi L, Huertas-Vazquez A, Lusis AJ, Xu XZS, Li S, Yu Y, Li JZ, Lin JD, Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Molecular cell 75, 644–660.e645 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seidman JS, Troutman TD, Sakai M, Gola A, Spann NJ, Bennett H, Bruni CM, Ouyang Z, Li RZ, Sun X, Vu BT, Pasillas MP, Ego KM, Gosselin D, Link VM, Chong LW, Evans RM, Thompson BM, McDonald JG, Hosseini M, Witztum JL, Germain RN, Glass CK, Niche-Specific Reprogramming of Epigenetic Landscapes Drives Myeloid Cell Diversity in Nonalcoholic Steatohepatitis. Immunity 52, 1057–1074.e1057 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hansen HH, AEgidius HM, Oró D, Evers SS, Heebøll S, Eriksen PL, Thomsen KL, Bengtsson A, Veidal SS, Feigh M, Suppli MP, Knop FK, Grønbæk H, Miranda D, Trevaskis JL, Vrang N, Jelsing J, Rigbolt KTG, Human translatability of the GAN diet-induced obese mouse model of non-alcoholic steatohepatitis. BMC gastroenterology 20, 210 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mells JE, Fu PP, Kumar P, Smith T, Karpen SJ, Anania FA, Saturated fat and cholesterol are critical to inducing murine metabolic syndrome with robust nonalcoholic steatohepatitis. The Journal of nutritional biochemistry 26, 285–292 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, Portman JR, Matchett KP, Brice M, Marwick JA, Taylor RS, Efremova M, Vento-Tormo R, Carragher NO, Kendall TJ, Fallowfield JA, Harrison EM, Mole DJ, Wigmore SJ, Newsome PN, Weston CJ, Iredale JP, Tacke F, Pollard JW, Ponting CP, Marioni JC, Teichmann SA, Henderson NC, Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 575, 512–518 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hammoutene A, Rautou PE, Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J Hepatol 70, 1278–1291 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Dobie R, Wilson-Kanamori JR, Henderson BEP, Smith JR, Matchett KP, Portman JR, Wallenborg K, Picelli S, Zagorska A, Pendem SV, Hudson TE, Wu MM, Budas GR, Breckenridge DG, Harrison EM, Mole DJ, Wigmore SJ, Ramachandran P, Ponting CP, Teichmann SA, Marioni JC, Henderson NC, Single-Cell Transcriptomics Uncovers Zonation of Function in the Mesenchyme during Liver Fibrosis. Cell reports 29, 1832–1847.e1838 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sato K, Marzioni M, Meng F, Francis H, Glaser S, Alpini G, Ductular Reaction in Liver Diseases: Pathological Mechanisms and Translational Significances. Hepatology 69, 420–430 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andrews TS, Atif J, Liu JC, Perciani CT, Ma XZ, Thoeni C, Slyper M, Eraslan G, Segerstolpe A, Manuel J, Chung S, Winter E, Cirlan I, Khuu N, Fischer S, Rozenblatt-Rosen O, Regev A, McGilvray ID, Bader GD, MacParland SA, Single-Cell, Single-Nucleus, and Spatial RNA Sequencing of the Human Liver Identifies Cholangiocyte and Mesenchymal Heterogeneity. Hepatol Commun 6, 821–840 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Halpern KB, Shenhav R, Matcovitch-Natan O, Toth B, Lemze D, Golan M, Massasa EE, Baydatch S, Landen S, Moor AE, Brandis A, Giladi A, Avihail AS, David E, Amit I, Itzkovitz S, Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 542, 352–356 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MacParland SA, Liu JC, Ma XZ, Innes BT, Bartczak AM, Gage BK, Manuel J, Khuu N, Echeverri J, Linares I, Gupta R, Cheng ML, Liu LY, Camat D, Chung SW, Seliga RK, Shao Z, Lee E, Ogawa S, Ogawa M, Wilson MD, Fish JE, Selzner M, Ghanekar A, Grant D, Greig P, Sapisochin G, Selzner N, Winegarden N, Adeyi O, Keller G, Bader GD, McGilvray ID, Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nature communications 9, 4383 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grohmann M, Wiede F, Dodd GT, Gurzov EN, Ooi GJ, Butt T, Rasmiena AA, Kaur S, Gulati T, Goh PK, Treloar AE, Archer S, Brown WA, Muller M, Watt MJ, Ohara O, McLean CA, Tiganis T, Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 175, 1289–1306.e1220 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang G, Duan J, Pu G, Ye C, Li Y, Xiu W, Xu J, Liu B, Zhu Y, Wang C, The Annexin A2-Notch regulatory loop in hepatocytes promotes liver fibrosis in NAFLD by increasing osteopontin expression. Biochimica et biophysica acta. Molecular basis of disease 1868, 166413 (2022). [DOI] [PubMed] [Google Scholar]
- 19.Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, Day CP, Burt A, Palmer A, Anstee QM, Grellscheid SN, Hoeijmakers JHJ, Barnhoorn S, Mann DA, Bird TG, Vermeij WP, Kirkland JL, Passos JF, von Zglinicki T, Jurk D, Cellular senescence drives age-dependent hepatic steatosis. Nature communications 8, 15691 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qiu X, Hill A, Packer J, Lin D, Ma YA, Trapnell C, Single-cell mRNA quantification and differential analysis with Census. Nature methods 14, 309–315 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guan D, Xiong Y, Borck PC, Jang C, Doulias PT, Papazyan R, Fang B, Jiang C, Zhang Y, Briggs ER, Hu W, Steger D, Ischiropoulos H, Rabinowitz JD, Lazar MA, Diet-Induced Circadian Enhancer Remodeling Synchronizes Opposing Hepatic Lipid Metabolic Processes. Cell 174, 831–842.e812 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan CH, Myung P, Plikus MV, Nie Q, Inference and analysis of cell-cell communication using CellChat. Nature communications 12, 1088 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tacke F, Targeting hepatic macrophages to treat liver diseases. J Hepatol 66, 1300–1312 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Nakamura T, Sakai K, Nakamura T, Matsumoto K, Hepatocyte growth factor twenty years on: Much more than a growth factor. Journal of gastroenterology and hepatology 26 Suppl 1, 188–202 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Kikuchi A, Singh S, Poddar M, Nakao T, Schmidt HM, Gayden JD, Sato T, Arteel GE, Monga SP, Hepatic Stellate Cell-Specific Platelet-Derived Growth Factor Receptor-α Loss Reduces Fibrosis and Promotes Repair after Hepatocellular Injury. The American journal of pathology 190, 2080–2094 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kania A, Klein R, Mechanisms of ephrin-Eph signalling in development, physiology and disease. Nature reviews. Molecular cell biology 17, 240–256 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Mimche PN, Lee CM, Mimche SM, Thapa M, Grakoui A, Henkemeyer M, Lamb TJ, EphB2 receptor tyrosine kinase promotes hepatic fibrogenesis in mice via activation of hepatic stellate cells. Scientific reports 8, 2532 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mimche PN, Brady LM, Bray CF, Lee CM, Thapa M, King TP, Quicke K, McDermott CD, Mimche SM, Grakoui A, Morgan ET, Lamb TJ, The receptor tyrosine kinase EphB2 promotes hepatic fibrosis in mice. Hepatology 62, 900–914 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoogenraad CC, Milstein AD, Ethell IM, Henkemeyer M, Sheng M, GRIP1 controls dendrite morphogenesis by regulating EphB receptor trafficking. Nature neuroscience 8, 906–915 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Misra C, Ziff EB, EphB2 gets a GRIP on the dendritic arbor. Nature neuroscience 8, 848–850 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Loft A, Alfaro AJ, Schmidt SF, Pedersen FB, Terkelsen MK, Puglia M, Chow KK, Feuchtinger A, Troullinaki M, Maida A, Wolff G, Sakurai M, Berutti R, Ekim Üstünel B, Nawroth P, Ravnskjaer K, Diaz MB, Blagoev B, Herzig S, Liver-fibrosis-activated transcriptional networks govern hepatocyte reprogramming and intra-hepatic communication. Cell metabolism 33, 1685–1700.e1689 (2021). [DOI] [PubMed] [Google Scholar]
- 32.Zhu C, Kim K, Wang X, Bartolome A, Salomao M, Dongiovanni P, Meroni M, Graham MJ, Yates KP, Diehl AM, Schwabe RF, Tabas I, Valenti L, Lavine JE, Pajvani UB, Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Science translational medicine 10, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu C, Tabas I, Schwabe RF, Pajvani UB, Maladaptive regeneration - the reawakening of developmental pathways in NASH and fibrosis. Nat Rev Gastroenterol Hepatol 18, 131–142 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu J, Zhu C, Wang X, Kim K, Bartolome A, Dongiovanni P, Yates KP, Valenti L, Carrer M, Sadowski T, Qiang L, Tabas I, Lavine JE, Pajvani UB, Hepatocyte TLR4 triggers inter-hepatocyte Jagged1/Notch signaling to determine NASH-induced fibrosis. Science translational medicine 13, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiao W, Wang J, Ou C, Zhang Y, Ma L, Weng W, Pan Q, Sun F, Mutual interaction between YAP and c-Myc is critical for carcinogenesis in liver cancer. Biochemical and biophysical research communications 439, 167–172 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Dang CV, MYC on the path to cancer. Cell 149, 22–35 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guan D, Xiong Y, Trinh TM, Xiao Y, Hu W, Jiang C, Dierickx P, Jang C, Rabinowitz JD, Lazar MA, The hepatocyte clock and feeding control chronophysiology of multiple liver cell types. Science (New York, N.Y.) 369, 1388–1394 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiong X, Wang Q, Wang S, Zhang J, Liu T, Guo L, Yu Y, Lin JD, Mapping the molecular signatures of diet-induced NASH and its regulation by the hepatokine Tsukushi. Molecular metabolism 20, 128–137 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang X, Shen J, Man K, Chu ES, Yau TO, Sung JC, Go MY, Deng J, Lu L, Wong VW, Sung JJ, Farrell G, Yu J, CXCL10 plays a key role as an inflammatory mediator and a non-invasive biomarker of non-alcoholic steatohepatitis. J Hepatol 61, 1365–1375 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Wasmuth HE, Lammert F, Zaldivar MM, Weiskirchen R, Hellerbrand C, Scholten D, Berres ML, Zimmermann H, Streetz KL, Tacke F, Hillebrandt S, Schmitz P, Keppeler H, Berg T, Dahl E, Gassler N, Friedman SL, Trautwein C, Antifibrotic effects of CXCL9 and its receptor CXCR3 in livers of mice and humans. Gastroenterology 137, 309–319, 319.e301–303 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhong G, Kirkwood J, Won KJ, Tjota N, Jeong H, Isoherranen N, Characterization of Vitamin A Metabolome in Human Livers With and Without Nonalcoholic Fatty Liver Disease. The Journal of pharmacology and experimental therapeutics 370, 92–103 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang X, Han J, Man K, Li X, Du J, Chu ES, Go MY, Sung JJ, Yu J, CXC chemokine receptor 3 promotes steatohepatitis in mice through mediating inflammatory cytokines, macrophages and autophagy. J Hepatol 64, 160–170 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Huby T, Gautier EL, Immune cell-mediated features of non-alcoholic steatohepatitis. Nature reviews. Immunology 22, 429–443 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu W, Jiang C, Kim M, Yang W, Zhu K, Guan D, Lv W, Xiao Y, Wilson JR, Rader DJ, Pui CH, Relling MV, Lazar MA, Individual-specific functional epigenomics reveals genetic determinants of adverse metabolic effects of glucocorticoids. Cell metabolism 33, 1592–1609.e1597 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Konermann S, Lotfy P, Brideau NJ, Oki J, Shokhirev MN, Hsu PD, Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 173, 665–676.e614 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matsumoto M, Hada N, Sakamaki Y, Uno A, Shiga T, Tanaka C, Ito T, Katsume A, Sudoh M, An improved mouse model that rapidly develops fibrosis in non-alcoholic steatohepatitis. International journal of experimental pathology 94, 93–103 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Semova I, Biddinger SB, Triglycerides in Nonalcoholic Fatty Liver Disease: Guilty Until Proven Innocent. Trends in pharmacological sciences 42, 183–190 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hoang SA, Oseini A, Feaver RE, Cole BK, Asgharpour A, Vincent R, Siddiqui M, Lawson MJ, Day NC, Taylor JM, Wamhoff BR, Mirshahi F, Contos MJ, Idowu M, Sanyal AJ, Gene Expression Predicts Histological Severity and Reveals Distinct Molecular Profiles of Nonalcoholic Fatty Liver Disease. Scientific reports 9, 12541 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yanger K, Zong Y, Maggs LR, Shapira SN, Maddipati R, Aiello NM, Thung SN, Wells RG, Greenbaum LE, Stanger BZ, Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes & development 27, 719–724 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Katze MG, He Y, Gale M Jr., Viruses and interferon: a fight for supremacy. Nature reviews. Immunology 2, 675–687 (2002). [DOI] [PubMed] [Google Scholar]
- 51.Gregor MF, Hotamisligil GS, Inflammatory mechanisms in obesity. Annual review of immunology 29, 415–445 (2011). [DOI] [PubMed] [Google Scholar]
- 52.Wieser V, Adolph TE, Grander C, Grabherr F, Enrich B, Moser P, Moschen AR, Kaser S, Tilg H, Adipose type I interferon signalling protects against metabolic dysfunction. Gut 67, 157–165 (2018). [DOI] [PubMed] [Google Scholar]
- 53.Luo X, Li H, Ma L, Zhou J, Guo X, Woo SL, Pei Y, Knight LR, Deveau M, Chen Y, Qian X, Xiao X, Li Q, Chen X, Huo Y, McDaniel K, Francis H, Glaser S, Meng F, Alpini G, Wu C, Expression of STING Is Increased in Liver Tissues From Patients With NAFLD and Promotes Macrophage-Mediated Hepatic Inflammation and Fibrosis in Mice. Gastroenterology 155, 1971–1984.e1974 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ghazarian M, Revelo XS, Nøhr MK, Luck H, Zeng K, Lei H, Tsai S, Schroer SA, Park YJ, Chng MHY, Shen L, D’Angelo JA, Horton P, Chapman WC, Brockmeier D, Woo M, Engleman EG, Adeyi O, Hirano N, Jin T, Gehring AJ, Winer S, Winer DA, Type I Interferon Responses Drive Intrahepatic T cells to Promote Metabolic Syndrome. Science immunology 2, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Møhlenberg M, Terczynska-Dyla E, Thomsen KL, George J, Eslam M, Grønbæk H, Hartmann R, The role of IFN in the development of NAFLD and NASH. Cytokine 124, 154519 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Mao W, Luis E, Ross S, Silva J, Tan C, Crowley C, Chui C, Franz G, Senter P, Koeppen H, Polakis P, EphB2 as a therapeutic antibody drug target for the treatment of colorectal cancer. Cancer research 64, 781–788 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Ma B, Kolb S, Diprima M, Karna M, Tosato G, Yang Q, Huang Q, Nussinov R, Investigation of the interactions between the EphB2 receptor and SNEW peptide variants. Growth factors (Chur, Switzerland: ) 32, 236–246 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saha N, Robev D, Mason EO, Himanen JP, Nikolov DB, Therapeutic potential of targeting the Eph/ephrin signaling complex. The international journal of biochemistry & cell biology 105, 123–133 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study can be found in the paper or supplementary materials. The snRNA-seq, snATAC-seq, and RNA-seq datasets have been deposited in Gene Expression Omnibus (GEO) with accession number GSE189600.