

Abstract
Heart failure is accompanied by adverse cardiac remodeling involving extracellular matrix (ECM). Cardiac ECM acts as a major reservoir for many proteins including growth factors, cytokines, collagens, and proteoglycans. Activated fibroblasts during cardiac injury can alter the composition and activity of these ECM proteins. Through unbiased analysis of a microarray dataset of human heart tissue comparing normal hearts (n = 135) to hearts with ischemic cardiomyopathy (n = 94), we identified Asporin (ASPN) as the top differentially regulated gene (DEG) in ischemic cardiomyopathy; its gene-ontology terms relate closely to fibrosis and cell death. ASPN is a Class I small leucine repeat protein member implicated in cancer, osteoarthritis, and periodontal ligament mineralization. However, its role in cardiac remodeling is still unknown. Here, we initially confirmed our big dataset analysis through cells, mice, and clinical atrial biopsy samples to demonstrate increased Aspn expression after pressure overload or cardiac ischemia/reperfusion injury. We tested the hypothesis that Aspn, being a TGFβ1 inhibitor, can attenuate fibrosis in mouse models of cardiac injury. We found that Aspn is released by cardiac fibroblasts and attenuates TGFβ signaling. Moreover, Aspn−/− mice displayed increased fibrosis and decreased cardiac function after pressure overload by transverse aortic constriction (TAC) in mice. In addition, Aspn protected cardiomyocytes from hypoxia/reoxygenation-induced cell death and regulated mitochondrial bioenergetics in cardiomyocytes. Increased infarct size after ischemia/reperfusion injury in Aspn−/− mice confirmed Aspn’s contribution to cardiomyocyte viability. Echocardiography revealed greater reduction in left ventricular systolic function post-I/R in the Aspn−/− animals compared to wild type. Furthermore, we developed an ASPN-mimic peptide using molecular modeling and docking which when administered to mice prevented TAC-induced fibrosis and preserved heart function. The peptide also reduced infarct size after I/R in mice, demonstrating the translational potential of ASPN-based therapy. Thus, we establish the role of ASPN as a critical ECM molecule that regulates cardiac remodeling to preserve heart function.
Keywords: Asporin, ECM, Cardiac remodeling, Fibrosis, Ischemia
Introduction
Heart Failure (HF) is the one of the leading causes of morbidity and mortality worldwide, generating significant health and economic burdens. Significant advances to limit ischemic injury by thrombolytic therapy or primary percutaneous coronary intervention (PCI) have decreased mortality from myocardial infarction, but long-term consequences of reperfusion injury and pathological cardiac remodeling contribute significantly to mortality [1]. Dynamic changes in extracellular matrix (ECM) regulate cellular responses mediating cardiac remodeling [2]. Due to increased activity of transforming growth factor (TGF)β1 during pathological remodeling, activated fibroblasts deposit fibrillar collagens and other ECM proteins [3]. Changes in the composition of ECM play crucial roles in providing structural integrity to heart and altering signaling pathways in numerous cell types including cardiomyocytes and fibroblasts [4]. However, adverse cardiac remodeling due to excessive fibrosis increases ventricular stiffness leading to impaired cardiac function and increased risk of mortality.
Despite great advances in the acute management of myocardial I/R injury, strategies to inhibit or reverse adverse cardiac remodeling remains elusive. ECM components are integral to the remodeling process and can play protective and deleterious effects [5]. Among these, small leucine rich proteoglycan (SLRP) decorin, lumican, biglycan, and fibromodulin play crucial roles in heart valve development and cardiac remodeling [6–10]. Waehre et al. [11] showed that decreased presence of SLRPs in heart results in loosely packed ECM, leading to left ventricular dilation and increased mortality after pressure overload. In the current study, we found Asporin (ASPN), an ECM protein, is the top differentially expressed genes (DEGs) in ischemic cardiomyopathy compared to normal controls. ASPN is a member of SLRP class I family, which acts as natural TGFβ inhibitor by regulating the latter’s interaction with its receptor [12]. ASPN, also known as periodontal ligament associated protein-1 (PLAP-1), was first identified in human cartilage where its overexpression was found to be associated with osteoarthritis [13]. ASPN directly binds to type I collagen through its LRR domain [13] and can play a crucial part in collagen fibrillogenesis [13]. Inhibition of ASPN stimulates TGFβ-induced smad2/3 signaling [12]. ASPN contains a pro-peptide sequence, D-repeat region (varies from 9 to 20 aa), 10 tandem leucine-rich repeats (LRR), and cysteine residues on both its N- and C-termini [14]. ASPN has been implicated as an oncoprotein in prostate cancer [15], pancreatic cancer [16] and gastric cancer [17, 18], but as a tumor suppressor in breast cancer [19, 20]. It is also involved in metastatic progression by regulating mesenchymal stromal cell differentiation [21]. However, the role ASPN in cardiac remodeling has not been studied yet.
In the present study, we explored the requirement of Aspn in pathological cardiac remodeling in different preclinical models. TGFβ1 treatment activated fibroblasts and increased the expression and release of Aspn into ECM space, which in turn, can inhibit TGFβ-induced SMAD2/3 signaling. Using paired clinical atrial biopsies obtained before and after cardiopulmonary bypass (CPB), we found increased ASPN expression in human heart tissue as well after surgery; the role of which is further determined using different animal models of cardiac injury. Genetic deficiency of Aspn in mice resulted in exacerbated fibrosis in pressure overload model and cardiomyocyte cell death in ischemia-reperfusion injury model leading to decline in diastolic and systolic function, respectively. Importantly, addition of exogenous ASPN to cultured H9C2 cardiomyocytes reduced cell death induced by hypoxia/reoxygenation. Further, using molecular modeling and docking studies, we designed an ASPN-mimic peptide, which we documented to reduce cardiac fibrosis and cardiomyocyte cell death in vivo. This, in turn, attenuated the decline in heart function after cardiac insults. Thus, our study reveals the important role of ASPN in pathological remodeling and myocardial preservation and may be an attractive candidate for treatment of heart failure.
Results
ASPN is one of the top differentially regulated genes in ischemic cardiomyopathy
In an unbiased approach, we first sought to explore the differential gene expression from ischemic cardiomyopathy clinical samples and compared to control donor hearts. We utilized publicly available microarray dataset of Magnet consortiums obtained from Gene Expression Omnibus (GSE57345). Analysis using R language script pointed to ASPN as one of the top upregulated genes (log2FC=1.77; p = 7.43E-30) in ischemic cardiomyopathy samples (n = 94) compared to non-failing donor heart controls (n = 135) (Supplementary Table 1). Separate analyses of males (Table 1) and females (Table 2) revealed significant upregulation of ASPN in both sexes. Fig. 1A shows the volcano plot for top differentially expressed genes and Supplementary Figure 1A shows the heatmap for top variable genes from microarray dataset analysis. Expression profiles of top 6 differentially expressed genes across all samples are displayed as bar charts (Supplementary Figure 2). ShinyGO analysis showed the involvement of differentially expressed genes in many important processes related to heart including tissue development, ECM organization, hypertrophy and Wnt signaling (Supplementary Figure 1B–C). We performed co-expression analysis and extracted highly co-expressed genes with ASPN visualized with cytoscape and found that many fibrosis-related genes including POSTN, FAP, COL14A1 shows strong co-expression with ASPN (Fig. 1B). ASPN′s co-expression network suggests biological processes in Ischemic cardiomyopathy that can act as a putative therapeutic target. We then extracted the experimentally validated protein-protein interaction network of ASPN from Reactome and String database (Fig. 1C–D) where ASPN is shown to interact with proteins involved in fibrosis mainly TGFβ, Fibromodulin (FMOD) and COL3A1. We further confirmed the microarray analysis findings using publicly available RNA-seq dataset (GSE46224) and found ASPN to be differentially expressed with 2.25 log fold increase (1.653e-7) in ischemic cardiomyopathy samples compared to non-failing donor heart tissue samples. The data has been shown as volcano plots (Fig. 1E) for the top differentially expressed genes and heatmap (Supplementary Figure 1C) for the top variable genes. Analysis with ShinyGO of top differentially regulated genes points to their involvement in development process, cell communication, receptor signaling and response to various stimuli including TGFβ (Supplementary Figure 1E–F). Comparison of the expression of other Class I SLRP genes including decorin (DCN) and biglycan (BGN) pointed to the ASPN as among the top differentially expressed gene in both microarray and RNA-seq dataset analysis (Supplementary Figure 3A–B).
Table 1.
List of top DEGs in ischemia cardiomyopathy samples compared with normal control samples in males.
| Gene.symbol | adj.P.Val | logFC |
|---|---|---|
| SERPINA3 | 1.28E-35 | −2.61 |
| FCN3 | 6.78E-35 | −1.95 |
| PLA2G2A | 8.71E-16 | −1.88 |
| IL1RL1 | 2.31E-21 | −1.76 |
| MYH6 | 1.8E-19 | −1.75 |
| CD163 | 2.34E-18 | −1.47 |
| LYVE1 | 6.2E-17 | −1.46 |
| VSIG4 | 5.62E-20 | −1.39 |
| LUM | 8.84E-29 | 1.35 |
| FRZB | 1.59E-27 | 1.35 |
| MXRA5 | 2.14E-19 | 1.37 |
| COL14A1 | 3.56E-21 | 1.41 |
| OGN | 3.56E-24 | 1.44 |
| HBB | 3.23E-09 | 1.73 |
| NPPA | 0.000000157 | 1.79 |
| ASPN | 2.38E-26 | 1.81 |
| SFRP4 | 1.05E-22 | 1.95 |
Table 2.
List of top DEGs in ischemia cardiomyopathy samples compared with normal control samples in females.
| Gene.symbol | adj.P.Val | logFC |
|---|---|---|
| SERPINA3 | 2.82E-09 | −2.76 |
| PLA2G2A | 4.28E-06 | −2.18 |
| FCN3 | 7.12E-08 | −2.07 |
| IL1RL1 | 1.82E-05 | −2.05 |
| SERPINE1 | 0.0103 | −1.73 |
| CD163 | 4.31E-07 | −1.63 |
| MGST1 | 5.17E-06 | −1.63 |
| AOX1 | 1.11E-05 | −1.51 |
| CYP4B1 | 0.000314 | −1.5 |
| SLC27A6 | 0.000195 | 1.31 |
| FRZB | 1.27E-05 | 1.39 |
| NEB | 0.00413 | 1.41 |
| OGN | 6.99E-05 | 1.46 |
| DSC1 | 0.000108 | 1.48 |
| NPPA | 0.0563 | 1.52 |
| SFRP4 | 8.17E-06 | 1.53 |
| ASB14 | 0.00475 | 1.53 |
| HBB | 0.0305 | 1.59 |
| FRZB | 1.27E-05 | 1.39 |
| NEB | 0.00413 | 1.41 |
| OGN | 6.99E-05 | 1.46 |
| DSC1 | 0.000108 | 1.48 |
| NPPA | 0.0563 | 1.52 |
| SFRP4 | 8.17E-06 | 1.53 |
| ASB14 | 0.00475 | 1.53 |
| HBB | 0.0305 | 1.59 |
| ASPN | 0.000158 | 1.86 |
Fig. 1. ASPN is one of the top differentially regulated gene in ischemic cardiomyopathy.

(A) Volcano plot of differentially expressed genes from microarray dataset (GSE57345) analysis in Ischemic vs the Nonfailing condition; (B) Gene-gene co-expression network of ASPN showing high co-expression with genes. The color denotes logFC at a gene level from microarray analysis in Ischemia vs Non failing hearts in males; Protein-protein interaction networks extracted from (C) Reactome and (D) String database; (E) Volcano plot of differentially expressed genes in Ischemic vs the Nonfailing condition from RNA-seq analysis of publicly available dataset GSE46224.
ASPN is induced in response to cardiac ischemia-reperfusion injury
To confirm the increase in Aspn expression in vivo, we employed ischemia (30 min) /reperfusion (24 h) mouse model and quantified the Aspn expression in area at risk zone and remote zone. Western blot analysis showed Aspn expression increased only in area at risk (AAR), near the site of injury, compared to remote zone and sham controls where no change in expression was seen (Fig. 2A and B). We didn’t find any significant change at transcript level in any of the groups (Fig. 2C). The findings were confirmed by IHC staining where Aspn protein staining was found to be more pronounced in AAR compared to remote zone (Fig. 2D). Double fluorescent staining of Aspn and vimentin (activated fibroblasts) reveals intensely positive Aspn-expressing cells were also vimentin positive in infarct and border zones. Further, increased expression of extracellular Aspn was found in the vicinity of vimentin-positive cells compared to remote region (Fig. 2E), highlighting the possibility of activated fibroblasts as main source of extracellular Aspn. As Aspn is a secretory protein, we next determined the Aspn levels in blood and found ~3 times (9.63±3.82 ng/ml vs 2.71±.43 ng/ml) increased levels in mice subjected to I/R compared to control mice, suggesting that cardiac cells release Aspn in response to cardiac stress/injury (Fig. 2F). To confirm if the increase in ASPN expression after cardiac stress is a global phenomenon, we used paired clinical atrial biopsies obtained before and after cardiopulmonary bypass (CPB) with cold cardioplegia for coronary arterial bypass graft and/or valve surgery from a cohort of 12 patients and found ASPN to be significantly upregulated after CPB surgery (Fig. 2G–H).
Fig. 2. Increase in Aspn expression in response to cardiac ischemia-reperfusion injury.
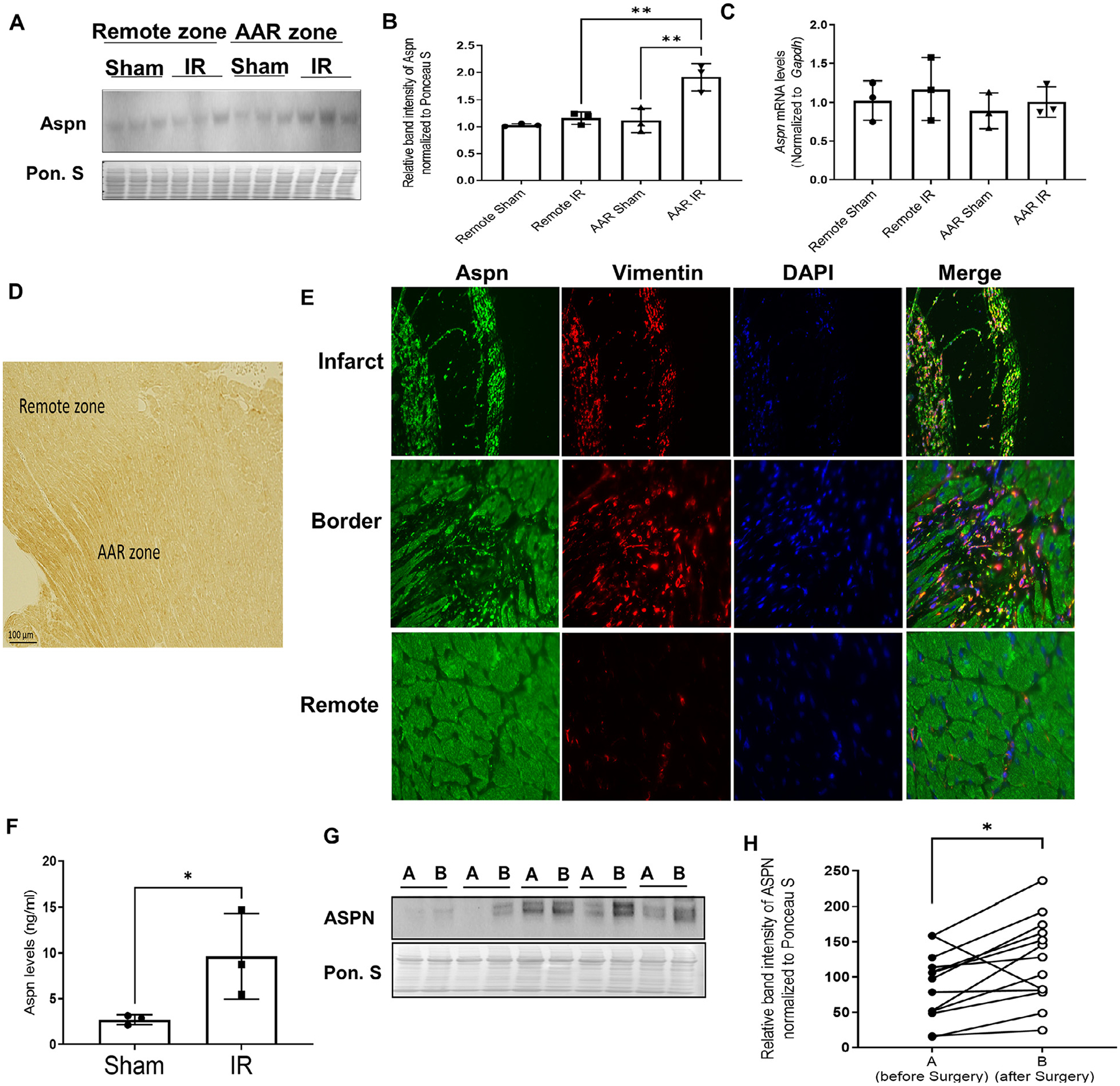
(A) Wild type C57 mice subjected to ischemia (30 mins) – reperfusion (24 hrs) injury and mice were sacrificed to separate remote zone and area at risk (AAR) zone from mice hearts. Western blot of tissue lysates for Aspn. Ponceau S staining was used to normalize the protein expression; (B) Bar graphs representing the expression of Aspn normalized to Ponceau S (n = 3); (C) qPCR analysis done on remote and AAR zone for Aspn expression normalized to GAPDH (n = 3). One-way ANOVA with Tukey multiple comparisons test was employed, **p<0.01; (D) Representative image for IHC staining on the mice heart subjected to ischemia-reperfusion injury; (E) Representative images of infarct, border and remote zone from mice heart subjected to ischemia-reperfusion injury model, co-stained with Aspn (Green), Vimentin (Red) and DAPI (blue). (F) Bar graph showing the Aspn levels from serum of the mice as detected by ELISA (n = 3). Data are expressed as mean with SD, *p ≤ 0.05 by unpaired t-test; (G) Representative western blot for Aspn and corresponding ponceau stain from tissue lysates of human atrial heart biopsies obtained before and after cardiac surgery involving cardiopulmonary bypass (CPB) and cardioplegia; (H) Quantitation of A before (A, solid circles) and after (B, empty circles) CPB (n = 13); *p<0.05 by paired t-test.
ASPN regulates TGFβ1 signaling from fibroblasts
Previous studies showed that Aspn is expressed and released predominantly from fibroblasts in different tissues [15, 18, 20]; thus, we explored the possibility of Aspn release from cardiac fibroblasts during cardiac injury. To investigate, we initially isolated primary fibroblasts from mouse hearts and treated the cells with angiotensin (100 nM and 1 μM) for 24 h. Western blot analysis revealed the increase in Aspn expression in a dose dependent manner (Fig. 3A). We also validated these findings in pressure overload TAC model—which activates fibroblasts—and demonstrated that Aspn expression increased compared to sham controls at both transcript and protein level (Fig. 3B–D). We also determined the Aspn expression in infarct and remote region after 24 h of myocardial infarction model and surprisingly didn’t find any difference (Supplementary Figure 4A–B). To gain further insight on the role of Aspn regulation and its release, 3T3 embryonic fibroblasts were treated with TGFβ1 (5 ng/ml) where increased expression as well as increased release of Aspn (Fig. 3E–H) into the extracellular media was detected. Although differentiated H9c2 cardiomyocytes showed endogenous Aspn expression, no significant induction or any release in extracellular media was observed in cardiomyocytes after TGFβ1 stimulation, suggesting secretion of Aspn predominantly derives from fibroblasts in the injured heart. Next, we determined if Aspn can also regulate TGFβ signaling; to test, we used double nickase plasmid gene editing mediated disruption to knockout Aspn in 3T3 cells. Supplementary Figure 4C–D shows effective transfection of plasmids along with reduction in Aspn expression. When these cells were treated with TGFβ1, Aspn deletion stimulated enhanced pSmad2/3 expression compared to control (Fig. 3I–J). Overall, the findings highlight the interplay between Aspn and TGFβ1 in a negative regulatory feedback loop and implicates Aspn’s critical role in cardiac remodeling.
Fig. 3. TGFβ1 induces Aspn expression while Aspn suppresses TGFβ signaling.

(A) Western blot for Aspn protein expression in isolated primary adult fibroblasts stimulated with Ang II or Ctrl for 24 hrs. (B-C) Western blot and quantification of Aspn in the heart homogenates from wild type mice following 4 weeks of TAC (n = 6). Ponceau staining was used as loading control. (D) The Aspn mRNA levels from heart tissue after sham controls (n = 5) and TAC surgery (n = 6). (E-F) 3T3 embryonic fibroblasts were treated with vehicle or TGFβ1 (5 ng/ml) for 24 h and expression of Aspn was determined. Representative western blot and quantification of intracellular ASPN (n = 4). (G-H) Extracellular media was collected from the same set of experiments. Representative western blot and quantification of extracellular Aspn (n = 4). (I) Aspn knockdown was done by employing Double Nickase Plasmids and cells were treated with TGFβ1 (5 ng/ml) for 24 h to assess the expression of pSMAD2/3 and Smad2/3. Representative western blot shows the expression of these proteins along with loading control, Ponceau S. (J) Quantification of the protein expression normalized to Ponceau S (n = 3). Data are expressed as mean with SD and unpaired t-test was employed to test for significance *p<0.05, **p<0.01.
Exogenous aspn is sufficient to ameliorate cardiomyocyte cell death while loss of ASPN impairs cardiac function
Next, we investigated the effects of extracellular ASPN on cardiomyocytes and we hypothesized that providing exogenous ASPN could protect cardiomyocyte cell death induced by I/R injury. To test, we used differentiated H9c2 cardiomyocytes and exposed them to hypoxia/reoxygenation and determined lactate dehydrogenase (LDH) release as a marker of cell death. Hypoxia/reoxygenation significantly increased LDH release compared to normoxia. However, cardiomyocytes treated with rASPN showed reduced cell death following H/R (Fig. 4A). The findings were further validated by TUNEL staining whereby rASPN treatment reduced percent TUNEL positive cells induced by hypoxia-reoxygenation (Fig. 4B). Using Aspn KO mice, we found that the presence of Aspn significantly reduced infarct size induced by ischemia (30 mins) /reperfusion (24 h) injury (Fig. 4C–E), suggesting the importance of Aspn in supporting cardiomyocyte survival. We also extended our study to 4 weeks of reperfusion to determine cardiac function (Supplementary Table 2 and Supplementary figure 5) in these mice. Heart weight expressed as a ratio to body weight 28 d after IR was significantly higher in Aspn KO mice compared to wild type mice (Fig. 4F). Further, ejection fraction (EF) and fractional shortening (FS) consistently declined in Aspn KO mice as opposed to wild type controls with time where it tended to stabilize (Fig. 4G–H). Overall, these data support the involvement of Aspn as a protective mechanism in cardiac remodeling.
Fig. 4. Aspn protects against cardiomyocytes cell death induced by ischemia-reperfusion injury and improves cardiac function.

(A) Differentiated H9c2 cardiomyocytes were treated with vehicle or rASPN protein (1 nM) for 24 hrs prior to exposing the cells to hypoxia (2 hrs) and reoxygenation (2 hrs) (HR). The media was collected and LDH levels were determined by ELISA. Bar graph representing the levels of LDH in the media from different groups normalized to control normoxia group (n = 4). Data are expressed as mean with SD and one-way ANNOVA with Turkey’s multiple comparison test, ***p<0.001, *p<0.01; (B) TUNEL positive nuclei were counted from many fields, each containing 10–15 cells and expressed as a ratio to DAPI stained cells in the field. Data are expressed as mean with SD. (C) Wild type and Aspn KO mice were subjected to ischemia (30 mins) – reperfusion (24 hrs) injury and left ventricular tissue were stained with TTC. Representative infarct size images, showing viable tissue (in blue), area at risk (in red) and infarct (in white). (D) Infarct size as a ratio to area at risk was calculated and represented as bar graph, with individual points presented (n = 6 for wild type; n = 7 for Aspn KO). (E) Bar graph representing area at risk in 2 groups. (F) Heart weight is represented as ratio to body weight as assessed at Day 28 after I/R (n = 7 for wild type; n = 5 for Aspn KO). Data are expressed as mean with SD and unpaired t-test was employed to test for significance *p<0.05, ****p<0.0001. (G) Line graph represents ejection fraction and (H) fractional shortening as determined by echocardiography at Day 0 (before surgery), Day 3, 7, 14 and 28 of I/R injury model.
ASPN treatment increases mitochondrial respiratory capacity
Mitochondria play an integral role in maintaining ATP levels in cardiomyocytes and can act as a mediator of cardiomyocyte viability [22]. To gain insights into the role of mitochondrial function in ASPN’s protective effects, we performed real-time measurements of the mitochondrial respiration on cardiomyocytes treated with rASPN or vehicle, using Seahorse XFe analyzer. Treatment with rASPN increased basal OCR and ATP production while reducing non- mitochondrial OCR, suggesting effective mitochondrial electron transport (Fig. 5A–E). When FCCP as uncoupler was administered, ASPN-treated cardiomyocytes showed significant increase in maximal respiration and spare respiratory capacity (Fig. 5F–G). Hypoxia-reoxygenation stimulated ROS production which was significantly reduced by rASPN treatment as determined by MitoSOX staining (Fig. 5H–I), further reinforcing the notion that mitochondria from these cells can better cope with stress conditions compared to untreated cells.
Fig. 5. Exogenous ASPN improves mitochondrial respiration and ATP production.

Equal number of H9c2 cells were plated in 24-well seahorse culture plate, followed by differentiation to H9c2 cardiomyocytes. After 5 days of differentiation, cells were treated with vehicle or rASPN (1 nM) for 24 hrs followed by seahorse respirometry analysis. OCR was measured at several time points. (A) OCR under basal conditions and after addition of oligomycin, FCCP and antimycin A/Rotenone as indicated. Calculations were done from the traces and compared between two groups and illustrated in bar graph representing (B) basal respiration, (C) Proton leak, (D) Non-mitochondrial respiration, (E) ATP production, (F) Spare respiratory capacity, and (G) Maximal respiration. Data are expressed as mean with SD (n = 3). *p<0.05 and **p<0.01 by unpaired t-test. (H) Representative images of differentiated H9c2 cardiomyocytes treated with either vehicle or rASPN subjected to simulated hypoxia-reoxygenation model and stained with MitoSOX red dye. Psedocolor green was selected during imaging using Keyonce microscope. Control cells were kept under normal culture conditions. (I) Bar graph representing the relative MitoSOX fluorescence intensity compared to control group. Data are expressed as mean with SD and one-way ANNOVA with Turkey’s multiple comparison test, ****p<0.0001, **p<0.001.
ASPN-mimic peptide reduces fibrosis and infarct size
Study [23] by Kou et al. identified the ASPN protein region between amino acid 159–205 to be critical for TGFβ1 interaction, and Maris et al. [20] showed that the peptide fragment mimicking the site can inhibit TGFβ1-induced pSmad2/3 phosphorylation in human breast cancer cells. To translate our findings, we similarly designed a peptide mimic based on homologous sequence in mouse and used molecular modeling and docking studies to predict the key amino acids involved in the interaction with TGFβ. The three-dimensional structure of the peptide sequence was modeled using comparative modeling based on a template structure of Chain A, Crystal Structure of the Biglycan Dimer Core Protein (PDB-ID 3FTA). Ramachandran plot and Prosa Z-score of −1.29 (Supplementary Figure 6A–D) showed favorable energy conformations and sequence similarity score of 52.00%. The molecular docking of ASPN peptide with the target TGFβ1 active chain (PDB ID: 4KV5; residues 279–390) were performed using HADDOCK tool [24, 25]. HADDOCK clustered 146 water refined structures in 13 clusters. Models in cluster-2 were the most reliable based on Haddock score and RMSD from the overall lowest-energy structure of 109.9 +/− 3.4 and 1.3 +/− 1.6 respectively. The best model was selected on Haddock score and lowest Z score of −2.3 showing stable protein peptide complex with 9 polar contacts stabilizing interaction (Fig. 6A–C). The amino acids involved in the interaction are shown in Fig. 6B and Supplementary Table 3. Further, MST was performed to determine the binding affinity using FITC-labeled ASPN peptide. The analysis revealed that the rTGFβ–1 protein interacts with the asporin peptide with a binding constant (KD) of 15 nM (Fig. 6D). Additionally, in vitro studies showed that peptide preincubation with TGFβ1 reduces the collagen expression induced by latter in 3t3 cells, documenting the efficiency of peptide to bind to and inhibit TGFβ1 (Fig. 6E–F).
Fig. 6. ASPN-mimic peptide reduces fibrosis and improves cardiac function.


(A) Docked complex of TGFβ (PDBID-4KV5: Chain A (residues 279–390)) with molecular modeled peptide ASPN (in green). Amino acids (TGFβ) (Dark blue) involved in interaction with ASPN peptide are highlighted in golden color. Inset shows zoomed-in view showing amino acids from peptides forming polar bonds with TGFβ residues. (B) Amino acid sequence of ASPN peptide and predicted amino acids involved in interaction are highlighted in bold red (H-bond acceptor) and bold black (H-bond donor) color. (C) 3-dimensional structure of ASPN peptide in cartoons and translucent surface colored based on hydrophobicity. (D) Graph shows the MST dose response curve obtained by titrating TGFβ–1 protein (4 μM to 122 pM) against 25 Nm FITC-labeled ASPN peptide. A binding constant (KD) of 15 nM was obtained for this interaction. (E) ASPN Peptide and TGFβ1 were incubated together or alone at 37 °C for 30 min. The complexes were added on to 3t3 cells for 24 h and cells were lysed. Representative western blot for Col1a1 protein expression and (F) quantification from different treatment groups. Ponceau staining was used as loading control. (G) Mice were subjected to pressure overload TAC surgery (4 weeks) and hearts sections were stained by mason-trichrome to determine fibrosis. Representative images show the stained heart sections from saline or peptide-treated mice (3x/week). (H) Fibrosis was quantified by reviewer blinded to the study groups and scores are represented in the form of a violon plot-graph, with individual points highlighted. Mann-Whitney test was employed for non-parametric comparison. (I) Various parameters were assessed using tissue Doppler imaging at day 28. Violon-plot graph represents ratio of E’/A’ in saline or peptide treated mice at day 28 (n = 7). (J) Myocardial performance index (MPI) is calculated at Day 28 and represented as violon-plots, with individual points highlighted. Unpaired t-test was employed for comparison, *p<0.05.
We next tested the peptide in vivo in pressure-overload TAC model for its protective effects on fibrosis and cardiac function. 2 h after TAC surgery, mice started receiving either vehicle (saline) or ASPN peptide (1 mg/kg: in 100 μl saline via i.p. injection) 3 times/week. Peptide therapy significantly reduced cardiac fibrosis (Fig. 6G–H). Tissue Doppler echocardiography showed a lower E’/A’ (early to late ventricular filling velocities ratio) at day 28, indicating diastolic dysfunction in control animals, which was prevented with ASPN peptide therapy (Fig. 6I). Also, myocardial performance index (MPI: (IVCT + IVRT)/ET; IVCT: isovolumic contraction time; IVRT: isovolumic relaxation time, ET: ejection time), which is inverse to the cardiac function, was significantly reduced with peptide therapy (Fig. 6J). Supplementary Table 4 details the parameters obtained from tissue Doppler echocardiography.
Next, we tested if the peptide, like ASPN protein, could attenuate cardiomyocyte cell death. In mice subjected to I/R, peptide (1 mg/kg; i.v) was administered 5 min before reperfusion and mice were sacrificed 24 h after reperfusion (Fig. 7A). TTC staining showed reduced infarct size in ASPN peptide-injected mice compared to saline-injected mice (Fig. 7B–D). However, when peptide was tested for its efficacy in altering mitochondrial respiration in cardiomyocytes using differentiated H9c2 cells, we found no statistical difference in either mitochondrial or non-mitochondrial OCR (Fig. 7F–K), suggesting the peptide effects are specific to extracellular TGFβ binding. Overall, the findings validate the novel role of ASPN and ASPN-based therapies in mediating protective effects to heart.
Fig. 7. ASPN mimic peptide reduces infarct size in ischemia-reperfusion injury mice model.
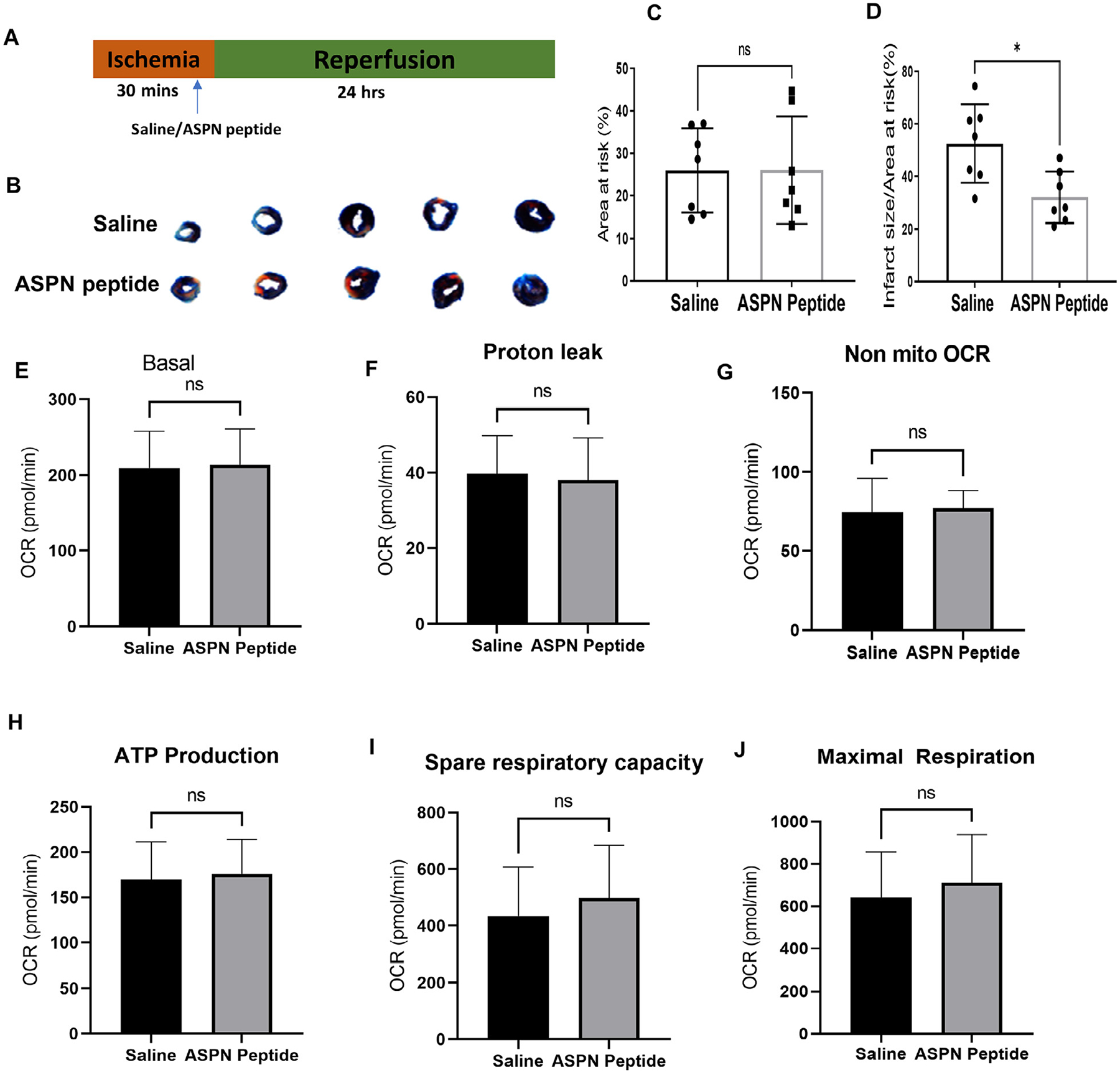
Mice were subjected to ischemia (30 mins) followed by reperfusion (24 h). Vehicle or peptide (1 mg/kg in saline) was injected (intravenous) to the mice, 5 min before reperfusion. Harvested left ventricular tissue were stained for TTC. (A) Schematic representation of saline/peptide treatment in ischemia-reperfusion model. (B) Representative images of heart after TTC staining. (C) Bar graph represents area at risk between two groups. (D) Infarct size as a ratio to area at risk was calculated and represented as bar graph. Data are shown as mean with SD; Unpaired t-test was used for the comparison, *p<0.05. (E) Equal number of H9c2 cells were plated in 24-well seahorse culture plate, followed by differentiation to H9c2 cardiomyocytes. After 5 days of differentiation, cells were treated with vehicle or ASPN peptide (50 μg/ml) for 24 hrs followed by seahorse respirometry analysis. OCR was measured at several time points. Calculations were done from the traces and compared between two groups and illustrated in bar graph representing (E) basal respiration, (F) Proton leak, (G) Non-mitochondrial respiration, (H) ATP production, (I) Spare respiratory capacity, and (J) Maximal respiration. Data are expressed as mean with SD (n = 9).
Discussion
In the last 2 decades or so, many advances have been made to better understand the regulation of adverse cardiac remodeling in HF. Controlled scarring is a normal repair process of cardiac remodeling; however, excessive fibrosis is deleterious [26]. Stress signals can also induce compensatory mechanisms to inhibit adverse cardiac signaling [27]. Here, we report the comprehensive role of an ECM protein, Aspn, in cardiac remodeling for the first time as an important compensatory signal. We integrated the big dataset analysis of microarray and RNA-seq to discover that ASPN was found to be among the top differentially regulated genes in ischemic cardiomyopathy. We further confirmed these findings, both in vitro and in vivo, using different TAC and IR models and showed that Aspn is upregulated and secreted specifically by fibroblasts. These findings are complemented by previous findings by Wang et al. [28] where they also documented increase in Aspn expression by cardiac fibroblasts, but not by cardiomyocytes, during cardiac remodeling. Interestingly, our data using I/R model showed upregulation of Aspn near the site of injury while sparing the remote region. Further, Aspn was also found to colocalize with vimentin positive cells and extracellular Aspn was found more prevalent in vicinity of activated fibroblasts. In addition, Aspn is further upregulated in mouse models in which cardiac fibrosis was blocked by interfering with distal pathways [29, 30] suggesting that Aspn might function as a critical proximal regulator moderating profibrotic signals to fibroblasts.
Although Aspn induction by TGFβ1 is well known [20, 31], its involvement in adverse cardiac remodeling has not been studied yet. Cardiac remodeling after myocardial infarction, I/R, or TAC induces fibroblast activation which upregulates active TGFβ1 expression and release to stimulate canonical and non-canonical signaling pathways [3, 32]. Aspn is known to bind to extracellular TGFβ1 [20], thus can restrict these signaling cascades to alleviate fibrotic changes. Our in vitro data and double fluorescent staining highlight the possibility that fibroblasts, but not cardiomyocytes, are the major source of Aspn. However, it’s possible that cardiomyocytes or other cell types apart from fibroblasts also release Aspn in mouse hearts and will need to be explored in future studies using conditional knockout mice. Previous studies support the role of ASPN-mediated inhibition of TGFβ1 signaling in various other cell types including, breast cancer cells, intervertebral annulus cells and chondrogenic cells [20, 33, 34]. Consistently, we also documented that knockdown of Aspn from cardiac fibroblasts upregulated TGFβ1-mediated canonical Smad signaling.
The bidirectional cross talk between myocytes and non-myocytes determines the extent of adverse or beneficial cardiac remodeling. ECM produced by fibroblasts presents signals to cardiomyocytes and other cell types present in heart [35, 36]. Studies have documented the importance of proteoglycans in heart development and remodeling after injury [9, 37–40]. Among them, SLRPs have received significant attention in the last decade for their role in numerous functions of the cell, especially in regulating collagen deposition and fibrosis [41]. Decorin is increased during heart failure, and overexpression of decorin in cardiac tissue attenuates fibrosis and adverse remodeling [42]. Biglycan deletion in mice improves cardiac function by attenuating hypertrophy and fibrosis in pressure overload model, suggesting its important role in pro-fibrotic signaling [43]. However, these mice showed increased mortality in myocardial infarction model due to cardiac rupture [44]. These studies led us to hypothesize that, like decorin and biglycan, Aspn can also impact cardiomyocytes in the heart. Using Aspn null mice, we documented that Aspn can protect cardiomyocytes from I/R-induced cell death in vivo. Also, the cardiac function in these mice declined with time. Further, validation of the Aspn protective effects in vitro suggested the effects might be independent of TGFβ1-mediated signaling in ECM. Mitochondria serve as the primary source of energy production in heart and their dysfunction has been implicated in adverse cardiac remodeling [45, 46]. Disturbance in homeostasis between ATP demand and supply during heart failure can lead to decreased cardiomyocyte viability [47]. Exogenous ASPN increased the respiratory capacity of mitochondria and the stress-resistant mitochondrial population increased ATP production, implicating ASPN’s crucial effect on bioenergetics. However, it still needs to be determined whether ASPN directly affects mitochondria or acts indirectly. It will also be critical to delineate the intracellular effects of ASPN in future studies.
Based on the known interaction site of ASPN and TGFβ1 and molecular modeling/docking analysis, we designed an ASPN mimic peptide which we validated for its binding to and inhibiting TGFβ1 signaling in vitro. The peptide was also shown to alleviate fibrotic responses in pressure overload and to preserve cardiac function in vivo. The peptide was further found to reduce infarct size in I/R injury, suggesting that mimic peptide can also preserve cardiomyocyte viability in vivo. Further, mitochondria respiration analysis on cardiomyocytes in vitro suggests the possibility of the peptide protective effects can be due to extracellular TGFβ1-specific inhibitory signaling.
In summary, we demonstrate a novel role of Aspn in cardiac remodeling whereby Aspn protects against excessive fibrosis and cardiomyocyte cell death. This, in turn, can limit the decline of cardiac function during pressure overload or I/R injury. Further, targeted ASPN-mimic therapy can prevent these deleterious effects, suggesting the usefulness of ASPN-based therapy in heart failure. Thus, our study reveals the necessity of ASPN to prevent adverse cardiac remodeling.
Experimental procedures
Cell culture
H9c2 and 3T3 cells were obtained from ATCC and maintained in growth media (DMEM: 10 mM glucose, 10% FBS, antibiotic and antimycotic, pH 7.4). For H9c2 cells, differentiation was initiated by switching to differentiation media (DMEM: 10 mM glucose, 1% FBS, antibiotic and antimycotic, 1 nM retinoic acid, pH 7.4) in the manner described previously [48]. Differentiation was sustained for 5 days before starting experiments.
Animal ethics
All animal procedures followed the National Institutes of Health standards and were approved by the Institutional Animal Care and Use Committee of Cedars-Sinai Medical Center. Aspn −/− mice were procured from MMRRC (B6;129S5-Aspntm1Lex/Mmucd). Exon 1 of Aspn was targeted using homologous recombination to create null mice. Mice exhibiting discomfort and distress were treated with buprenorphine (1 mg/kg, i.p.) or euthanized when appropriate. At the end of experimental procedures, mice were anesthetized via isoflurane and euthanized by cervical dislocation. Depth of anesthesia was confirmed by loss of flexor muscle response from paw pinch.
Differential expression analysis
Microarray dataset was obtained from publicly available Gene Expression Omnibus (GSE57345) to compare the gene expression between ischemic cardiomyopathy and normal hearts [49]. The dataset contained raw data from microarrays performed on left ventricle free wall tissue. The tissue was obtained from heart failure samples at the time of cardiac surgery undergoing transplantation. Empirical Bayes method from Bioconductor package “LIMMA” and “GEO2R” [50, 51] was used to compute differential expression of genes in Microarray data analysis. The p-values determined using T test were then subjected to multiple test correction using false discovery rate method to correct false positive occurrences and for obtaining statistically significant genes. Minimum absolute value of log (FC) 1.5 was determined as threshold cutoff value for DEGs. The volcano-plot were constructed using Enhanced volcano plot(1R) on top differentially expressed genes and heatmaps are generated using pheatmap package r(1R) on normalized expression profiles of top variable genes with significant adjusted p Values. The clustering is performed on rows, that is, genes are based on Euclidean distance.
RNA-seq analysis
We analyzed RNA-seq data GSE46224 for identifying DEGs in Ischemic cardiomyopathy vs Normal donor controls. The RNA Sequencing data was trimmed using Trimgalore and aligned using Star aligner on reference GRch38 [52]. The mapped reads were sorted by coordinates and read counts for the conditions were computed using HTseq-count [53]. Differential expression was computed using DeSeq2 [54]. Minimum absolute value of log (FC) 1 was determined as threshold cutoff value for DEGs.
Gene enrichment analysis
Gene Ontology (GO) classification for biological processes were performed on unique genes and DEGs using ShinyGO v0.61 [55]. Top DEGs from microarray and RNA-seq dataset analysis were uploaded on to ShinyGo platform.
Gene gene co-expression networks
The gene co-expression network was constructed using The Weighted Gene Co-expression Network Analysis package (WGCNA) [56, 57]. The networks were then preserved using optimal β parameter for reconstruction of networks following scale free property. Topologies overlap measure is used for computation of edge weights of on any two connected genes. The edge weight scaled between 0 and 1 is a measure for strength of genes co-expressing (expression correlation) with threshold edge weight of 0.05. Cytoscape was utilized for visualization of the large network and to extract the subnetworks of interest [58]. Several centrality measures for networks were computed using Netanalyzer and Centiscape [59, 60].
String and reactome network analysis
We retrieved interactions of ASPN protein from STRING [61] with confidence score of greater than 0.7 and experimentally validated protein-protein interactions from Reactome version 7.0 for humans to construct protein-protein interaction networks.
Mouse model of ischemia/reperfusion injury
Mice were subjected to ischemia-reperfusion injury as previously described [62]. Briefly, 8–12 week-old mice were anesthetized with an i.p. injection of ketamine (50 mg/kg) and xylazine (10 mg/kg). Isoflurane anesthesia was used throughout the procedure with 1% isoflurane mixed with 1.0 L/min 100% O2. Volume-controlled ventilation (Harvard Apparatus 683) was maintained at a rate of about 140 bpm and a pressure of 2 cm H2O. A PE-10 tube was placed on the surface of the left anterior descending artery (LAD) which was ligated together with an 6–0 silk suture for 30 min. The suture was then released but left in position, and after verifying adequate reperfusion, the chest was closed, and the mice were allowed to recover for 24 hr. To determine infarct size and risk area, the suture was re-tied, and heart was perfused with 1% Evans Blue and then stained with triphenyl tetrazolium chloride (TTC). The area at risk (AAR) represented the myocardial perfusion region distal to an occluded coronary artery. Wavefront phenomenon is followed to detect AAR in the sections [63].
Mouse model of pressure overload
8–12-week-old mice were anesthetized with ketamine(50 mg/kg)/xylazine(10 mg/kg) and anesthesia was maintained using 0.5% isoflurane mixed with 1.0 L/min 100% O2 by mechanical ventilation. Mice were then placed in a supine position on the heating pad at 37 °C ± 1 °C. Partial thoracotomy to the second rib was performed under a surgical microscope and the sternum retracted. Following identification of the transverse aorta, a small piece of a 7.0 silk suture was placed between the innominate and left carotid arteries. A small piece of a 27-gage blunt needle was placed parallel to the transverse aorta and two loose knots were tied around the transverse aorta to yield a constriction of ~0.4 mm in diameter. The rib cage and the skin were closed using a 6.0 monofilament suture and mice were allowed to recover.
Mouse model of myocardial infarction
Mice were anesthetized with ketamine(50 mg/kg)/xylazine(10 mg/kg) and anesthesia was maintained using 0.5% isoflurane mixed with 1.0 L/min 100% O2 by mechanical ventilation. A left thoracotomy was performed to expose the heart, followed by opening of pericardium. Then, a 7–0 silk suture was placed around the proximal left coronary artery 2 mm below the left atrial appendage using a curved 27.5 G needle. The vessel was ligated, chest closed, and animals allowed to recover [64].
Echocardiography assessments
Transthoracic echocardiography was performed using a high-resolution Vevo 3100 equipped with a linear transducer (Fujifilm VisualSonics, Inc.). Briefly, during ultrasound scanning, the animal was anesthetized with an inhaled mixture of 2% isoflurane gas at 1 L/min O2 flow rate delivered by nose cone. Short-axis two-dimensional view of the heart was obtained at the level of papillary muscles and corresponding M-mode imaging was recorded. The heart rate was determined from cardiac cycles recorded on M-mode. Ultrasound gel was warmed to body temperature. To measure tissue Doppler and transmitral pulse wave Doppler of E, E′ and E/E, a consistent sampling position was identified as described previously [65]. Left ventricle (LV) dimensions were presented as the average of measurements of 3 consecutive sinus beats using the leading-edge technique. EF% was then calculated from M-mode-derived LV dimensions using the formula; LV Volume = [7/ (2.4 + LVID)] * LVID3; EF= (LV vol,d-LV vol,s)/LV vol,d *100; MPI= (IVRT+IVCT)/ET.
Histology
Hearts were harvested, perfused with PBS and stored in 4% paraformaldehyde (PFA) for 24 hours, followed by 70% ethanol storage for subsequent histopathology staining and microscopic analysis (Keyence Biorevo BZ-9000). Sections were stained with Masson’s trichrome (Sigma) following manufacturer’s protocol for visualization of fibrosis. Briefly, slide sections were deparaffinized using xylene and an ethanol gradient (100–70%), followed by hydration in deionized water. Sections were placed in a container with Bouin solution overnight, washed and stained in Weigert’s iron hematoxylin and Biebrich scarlet-acid fuchsin. Further steps included decolorizing with phosphomolybdic/phosphotungstic acid solution, aniline blue staining and clarification in 1% acetic acid solution. Sections were dehydrated in ethanol 95–100%, cleared in xylene and mounted [64]. The staining was visualized under the microscope and fibrosis scoring was done by an independent reviewer blinded to the study groups.
Mason-Trichrome staining
Briefly, all images were taken with EVOS7000 microscope at same exposure time and then analyzed by ImageJ. Color channels were split to different color channel, then images were adjusted for the threshold to label blue or red. Area was calculated through ImageJ.
Primary fibroblast isolation
3 mouse hearts were used per isolation. Briefly, tissue was collected from freshly euthanized mice and placed them on a petri dish containing ice cold PBS under sterile conditions. Tissue was minced using scissor and blade into small pieces. Minced tissues were transferred to digestion buffer (containing collagenase II; 100 U/ml and trypsin; 0.1%) under constant stirring at 37 °C for 5 mins. Tissue was allowed to settle down and first supernatant containing debris and blood cells was discarded. Digestion buffer was added, followed by constant stirring at 37 °C for 10 min. Supernatant was collected and fibroblast growth medium was added. The above steps were repeated 5–8 times until most of the tissue is dissolved. Cells were spun for 5 min at 300 g on 4 °C. Cells were resuspended in fibroblast growth medium and plated in 100 mm culture dishes. After 2-hour incubation, cells were washed with PBS 3 times to remove dead cells and debris. The cells are expanded and split when these were about to reach 90% confluence. All experiments are done within 20 days of fibroblast isolation [66].
Immunofluorescent staining
Slide sections were deparaffinized with xylene treatment twice and ethanol gradient for 100% to 70%, followed by, hydration with deionized water. Permeabilization and blocking was done using PBS containing 5% horse serum, 0.1% BSA and 0.3% Triton X-100 followed by washing with PBS containing 0.1% BSA. Samples were then incubated with primary antibodies (Abcam ab31303) and Vimentin (Cell signaling 5741S) overnight at 4 °C, washed with TBS 3 times and incubated with secondary antibodies for 2 h at room temperature in the dark. Further cells were washed with PBS twice and visualized for imaging using a Keyence BZ-9000 microscope (Keyence; Japan).
Western blot analysis
Protein concentration was measured using the Sigma Bradford reagent protein assay kit. Equal amount of protein was resolved on Bolt™ 4–12% Bis-Tris Plus gels (Invitrogen by Thermo Fisher Scientific) and transferred to nitrocellulose membranes. The membranes were blocked with 5% nonfat dry milk for 45 min, then incubated with 1:1000 diluted primary antibodies against: ASPN (Abcam ab31303), Smad2 (Cell Signaling 5339S), pSmad2 (Cell Signaling 3108S) at 4 °C overnight. Membranes were washed with Tris-buffered saline Ph 7.6 containing Tris–HCl (20 mM) and NaCl (150 mM) with 0.1% Tween-20 (Sigma-Aldrich) (TBS-T) at room temperature and incubated with KPL peroxidase Labeled secondary antibodies for 2 h at room temperature. Membranes were washed 3 times in TBS-T. Immunoreactive bands were developed with Clarity™ Western ECL Substrate (Bio-Rad Laboratories Inc.) and imaged using a ChemiDoc XRS system (Bio-Rad Laboratories Inc.). Densitometry was performed using NIH Image J software.
Real time PCR analysis
RNA was isolated using the standard Trizol protocol, with a few changes. Briefly, frozen heart sections were lysed in Trizol using the TissueLyzer LT (Qiagen) followed by chloroform addition. The samples were centrifuged for 15 min at 12,000 × g at 4 °C. The upper aqueous phase was transferred to a new tube and 500 μL of isopropanol was added to each sample, followed by another incubation for 2 h at −20 °C. Samples were centrifuged for 10 min at 12,000 × g at 4 °C. The supernatants were discarded, and the pellets were resuspended in 1 mL of 75% ethanol. The tubes were vortexed and centrifuged for 5 min at 7500 × g at 4 °C, and the supernatants were discarded. The pellets were air-dried for 10 min and resuspended in 50 μL of RNAse-free water and stored at −80 °C until further analysis. For cDNA synthesis, 1 μg of RNA was used for each 20 μL reaction following the iScript Reverse Transcription Supermix protocol (Bio-Rad). ASPN (5′-TCCAGCAAAGTTGGTGGTAG-3′, 5′-CCTCTTGAGAACAACGGGATAG-3′) expression analysis by quantitative real-time PCR reaction (qPCR) was performed in the CFX96™ Real-Time System (Bio-Rad) using 50 ng of cDNA and following the iTaq Universal Sybr Green Supermix (Bio-Rad). Gapdh was used as endogenous control. Expression results were analyzed using the 2-del-taCt formula and represented in graphs as fold change.
TUNEL staining
Cells were fixed using 2% paraformaldehyde for 15 mins at room temperature. Cells were then washed three times with PBS and permeabilized with 0.1% Triton X-100. Apoptosis in the permeabilized cells was detected by In Situ Cell Death Detection Kit, Fluorescein (11,684,795,910 Roche) following manufacturer’s protocol. DapI was used to stain the nucleus and cells were imaged using Keyence BZ-9000 microscope (Keyence, Japan).
Aspn ELISA analysis
Quantification of Aspn in serum samples was done following manufacturer’s protocol (My Bio-Source; MBS451291).
Aspn knockdown
3T3 embryonic fibroblast cells were plated and allowed to grow for 60–70% confluency. Aspn gene was silenced by Aspn Double Nickase Plasmids (1ug) (Santa Cruz) using Lipofectamine LTX and PlusTM reagent (Invitrogen) following recommended protocol for 48 hrs. The transfected cells were further selected using puromycin (1ug/ml) in growth medium and used for further experiments.
Seahorse respirometry analysis
H9c2 cells were seeded in 24-well Seahorse culture plate at a density of 15,000 cells/well and differentiated using the protocol described above. After 5 days of differentiation, the media was then changed to XF base medium supplemented with glucose, sodium pyruvate and sodium L-glutamine. Cells were allowed to equilibrate in a non-CO2 incubator at 37 °C for 45 mins before starting the assay. In the meantime, Seahorse sensor cartridge was prepared, and drugs were added in A (oligomycin), B (FCCP) and C (antimycin A) ports and loaded into the Seahorse analyzer. After the drugs were loaded into the injection ports, cartridge was replaced with Seahorse culture plate and oxygen consumption rate (OCR) was monitored.
Molecular modeling and docking
The three-dimensional structure of the peptide sequence (ASPN peptide) were modeled using comparative modeling on a template structure of Chain A, Crystal Structure of the Biglycan Dimer Core Protein (PDB-ID 3FTA) [67] identified with well-defined protocol used in previous studies [68, 69]. Comparative modeling consisted of four main steps: i) Performed search with BLAST(Basic Local Alignment Tool) tool for query peptide sequence (ASPN peptide) against PDB database using BlastP (Basic Local Alignment Tool-protein) of NCBI to obtain one or more than one best matches [70]. BLAST search for query peptide sequence against PDB database obtained 3FTA_A as a best hit with 52.17% percent identity, evaluate of 2E-11 and query coverage of 97%. ii) alignment between the query sequence and selected PDB template. Protein sequences were aligned in .pir format with CLUS-TALW [71], iii) model constructed on the query sequence aligned with PDB template using MODELER and modweb toolkit, and iv) Evaluation of stereo chemical quality was done by analyzing the allowed conformations of amino acids using hard sphere model on the basis of the Ramachandran plot [72, 73] and Z score was computed using Prosa-web tool to check the fitness of the sequences relative to the obtained structures and to assign a scoring function [74, 75]. The molecular docking of ASPN peptide with the target human pro-TGFβ1 (PDB ID- 4KV5 Chain A 5VQP) was performed using HADDOCK tool with the constrained docking to the interface residues. Best docked complexes was then selected based on Z-score computed by HADDOCK and the model with most negative score was further evaluated for polar bonds and visualized using Chimera [76].
Protein-peptide binding analysis
The binding of ASPN peptide to the TGFβ–1 protein was measured by Microscale Thermophoresis (MST), using the Monolith NT.115 instrument (Nanotemper Technologies, München, Germany). The fluorescein isothiocyanate (FITC)-labeled asporin peptide (FITC-ε-Ahx-Asporin; custom generated from Genscript, purity >92.5%) and the recombinant mouse TGFβ–1 protein were procured commercially from BioLegend (San Diego, CA). The assay was performed in phosphate buffered saline (PBS) supplied with 0.05% Tween-20. A serial dilution of the TGFβ–1 protein was titrated against the FITC labeled asporin peptide. For the titration, 10 μL of each concentration of TGFβ–1 was mixed with 10 μL of FITC labeled asporin. The final concentration of peptide in the assay was 25 nM, while the highest and lowest concentrations of the TGFβ–1 protein were 4 μM and 122 pM, respectively. The MST measurement settings include medium MST power, 100% excitation power, Nano-BLUE excitation type and 25 °C thermostat temperature. Affinity of the asporin peptide binding to TGFβ–1 was determined by the MO.Affinity Analysis Software (Nanotemper Technologies, München, Germany).
Statistical analysis
Standard Student’s t-test was used to compare data with 2 groups only. Data with 3 or more groups was analyzed using Analysis of Variance (ANOVA) with Tukey posthoc test. Mann-whitney test was employed for non-parametric comparison analysis. p < 0.05 was considered as statistically significant.
Supplementary Material
Acknowledgments
We would like to acknowledge the Cedars-Sinai Biobank and Translational research core for histology services. We thank Ian Williamson for help in getting Aspn knock out mice and excellent lab management. We also would like to acknowledge Dr. Tanuj Sharma (Yonsei University, South Korea) for in-depth discussion of molecular docking models.
Funding
This work was supported by the NHLBI HL144509 (RAG), NHLBI HL155553 (HP) and institutional funding (RAG).
Abbreviations:
- ECM
Extracellular Matrix
- TGF
Transforming growth factor
- I/R
Ischemia-reperfusion
- SLRP
Small leucine rich proteoglycan
- ASPN
Asporin
- PLAP
Periodontal ligament associated protein
- LRR
Leucine rich repeat
- CPB
Cardiopulmonary bypass
- i.p.
Intraperitoneal
- DEG
Differentially expressed gene
- FC
Fold change
- GO
Gene ontology
- WGCNA
Weighted gene co-expression network analysis package
- EF
Ejection fraction
- FS
Fractional shortening
- LV
Left ventricle
- MPI
Myocardial performance index
- PDB
Protein databank
- TAC
Transverse aortic constriction
- AAR
Area at risk
- LDH
Lactate dehydrogenase
- rASPN
Recombinant asporin protein
- OCR
Oxygen consumption rate
- ATP
Adenosine 5′-triphosphate
- FCCP
Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone
- TTC
Triphenyl tetrazolium chloride
- Col
Collagen
Footnotes
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.matbio.2022.04.005.
References
- [1].Hausenloy DJ, Yellon DM, Myocardial ischemia-reperfusion injury: a neglected therapeutic target, J. Clin. Invest 123 (1) (2013) 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Frangogiannis NG, The extracellular matrix in myocardial injury, repair, and remodeling, J. Clin. Invest 127 (5) (2017) 1600–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Humeres C, Frangogiannis NG, Fibroblasts in the Infarcted, Remodeling, and Failing Heart, JACC Basic Transl. Sci 4 (3) (2019) 449–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bowers SL, Banerjee I, Baudino TA, The extracellular matrix: at the center of it all, J. Mol. Cell Cardiol 48 (3) (2010) 474–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Fan D, Creemers EE, Kassiri Z, Matrix as an interstitial transport system, Circ. Res 114 (5) (2014) 889–902. [DOI] [PubMed] [Google Scholar]
- [6].Dupuis LE, Kern CB, Small leucine-rich proteoglycans exhibit unique spatiotemporal expression profiles during cardiac valve development, Dev. Dyn 243 (4) (2014) 601–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dupuis LE, Berger MG, Feldman S, Doucette L, Fowlkes V, Chakravarti S, Thibaudeau S, Alcala NE, Bradshaw AD, Kern CB, Lumican deficiency results in cardiomyocyte hypertrophy with altered collagen assembly, J. Mol. Cell Cardiol 84 (2015) 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Andenaes K, Lunde IG, Mohammadzadeh N, Dahl CP, Aronsen JM, Strand ME, Palmero S, Sjaastad I, Christensen G, Engebretsen KVT, Tonnessen T, The extracellular matrix proteoglycan fibromodulin is upregulated in clinical and experimental heart failure and affects cardiac remodeling, PLoS ONE 13 (7) (2018) e0201422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Westermann D, Mersmann J, Melchior A, Freudenberger T, Petrik C, Schaefer L, Lullmann-Rauch R, Lettau O, Jacoby C, Schrader J, Brand-Herrmann SM, Young MF, Schultheiss HP, Levkau B, Baba HA, Unger T, Zacharowski K, Tschope C, Fischer JW, Biglycan is required for adaptive remodeling after myocardial infarction, Circulation 117 (10) (2008) 1269–1276. [DOI] [PubMed] [Google Scholar]
- [10].Jahanyar J, Joyce DL, Southard RE, Loebe M, Noon GP, Koerner MM, Torre-Amione G, Youker KA, Decorin-mediated transforming growth factor-beta inhibition ameliorates adverse cardiac remodeling, J. Heart Lung Transplant 26 (1) (2007) 34–40. [DOI] [PubMed] [Google Scholar]
- [11].Waehre A, Halvorsen B, Yndestad A, Husberg C, Sjaastad I, Nygard S, Dahl CP, Ahmed MS, Finsen AV, Reims H, Louch WE, Hilfiker-Kleiner D, Vinge LE, Roald B, Attramadal H, Lipp M, Gullestad L, Aukrust P, Christensen G, Lack of chemokine signaling through CXCR5 causes increased mortality, ventricular dilatation and deranged matrix during cardiac pressure overload, PLoS ONE 6 (4) (2011) e18668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nakajima M, Kizawa H, Saitoh M, Kou I, Miyazono K, Ikegawa S, Mechanisms for asporin function and regulation in articular cartilage, J. Biol. Chem 282 (44) (2007) 32185–32192. [DOI] [PubMed] [Google Scholar]
- [13].Lorenzo P, Aspberg A, Onnerfjord P, Bayliss MT, Neame PJ, Heinegard D, Identification and characterization of asporin. a novel member of the leucine-rich repeat protein family closely related to decorin and biglycan, J. Biol. Chem 276 (15) (2001) 12201–12211. [DOI] [PubMed] [Google Scholar]
- [14].Xu L, Li Z, Liu SY, Xu SY, Ni GX, Asporin and osteoarthritis, Osteoarthritis Cartilage 23 (6) (2015) 933–939. [DOI] [PubMed] [Google Scholar]
- [15].Rochette A, Boufaied N, Scarlata E, Hamel L, Brimo F, Whitaker HC, Ramos-Montoya A, Neal DE, Dragomir A, Aprikian A, Chevalier S, Thomson AA, Asporin is a stromally expressed marker associated with prostate cancer progression, Br. J. Cancer 116 (6) (2017) 775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang L, Wu H, Wang L, Zhang H, Lu J, Liang Z, Liu T, Asporin promotes pancreatic cancer cell invasion and migration by regulating the epithelial-to-mesenchymal transition (EMT) through both autocrine and paracrine mechanisms, Cancer Lett 398 (2017) 24–36. [DOI] [PubMed] [Google Scholar]
- [17].Ding Q, Zhang M, Liu C, Asporin participates in gastric cancer cell growth and migration by influencing EGF receptor signaling, Oncol. Rep 33 (4) (2015) 1783–1790. [DOI] [PubMed] [Google Scholar]
- [18].Satoyoshi R, Kuriyama S, Aiba N, Yashiro M, Tanaka M, Asporin activates coordinated invasion of scirrhous gastric cancer and cancer-associated fibroblasts, Oncogene 34 (5) (2015) 650–660. [DOI] [PubMed] [Google Scholar]
- [19].Castellana B, Escuin D, Peiro G, Garcia-Valdecasas B, Vazquez T, Pons C, Perez-Olabarria M, Barnadas A, Lerma E, ASPN and GJB2 Are Implicated in the Mechanisms of Invasion of Ductal Breast Carcinomas, J. Cancer 3 (2012) 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Maris P, Blomme A, Palacios AP, Costanza B, Bellahcene A, Bianchi E, Gofflot S, Drion P, Trombino GE, Di Valentin E, Cusumano PG, Maweja S, Jerusalem G, Delvenne P, Lifrange E, Castronovo V, Turtoi A, Asporin Is a Fibroblast-Derived TGF-beta1 Inhibitor and a Tumor Suppressor Associated with Good Prognosis in Breast Cancer, PLoS Med 12 (9) (2015) e1001871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hughes RM, Simons BW, Khan H, Miller R, Kugler V, Torquato S, Theodros D, Haffner MC, Lotan T, Huang J, Davicioni E, An SS, Riddle RC, Thorek DLJ, Garraway IP, Fertig EJ, Isaacs JT, Brennen WN, Park BH, Hurley PJ, Asporin Restricts Mesenchymal Stromal Cell Differentiation, Alters the Tumor Microenvironment, and Drives Metastatic Progression, Cancer Res 79 (14) (2019) 3636–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kuznetsov AV, Javadov S, Margreiter R, Grimm M, Hagenbuchner J, Ausserlechner MJ, The Role of Mitochondria in the Mechanisms of Cardiac Ischemia-Reperfusion Injury, Antioxidants (Basel) 8 (10) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kou I, Nakajima M, Ikegawa S, Binding characteristics of the osteoarthritis-associated protein asporin, J. Bone Miner. Metab 28 (4) (2010) 395–402. [DOI] [PubMed] [Google Scholar]
- [24].Dominguez C, Boelens R, Bonvin AM, HADDOCK: a protein-protein docking approach based on biochemical or biophysical information, J. Am. Chem. Soc 125 (7) (2003) 1731–1737. [DOI] [PubMed] [Google Scholar]
- [25].van Zundert GCP, Rodrigues J, Trellet M, Schmitz C, Kastritis PL, Karaca E, Melquiond ASJ, van Dijk M, de Vries SJ, Bonvin A, The HADDOCK2.2 Web Server: user-Friendly Integrative Modeling of Biomolecular Complexes, J. Mol. Biol 428 (4) (2016) 720–725. [DOI] [PubMed] [Google Scholar]
- [26].Rog-Zielinska EA, Norris RA, Kohl P, Markwald R, The Living Scar–Cardiac Fibroblasts and the Injured Heart, Trends Mol. Med 22 (2) (2016) 99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Liu Y, Chen X, Zhang HG, Editorial: cardiac Hypertrophy: from Compensation to Decompensation and Pharmacological Interventions, Front. Pharmacol 12 (2021) 665936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wang HB, Huang R, Yang K, Xu M, Fan D, Liu MX, Huang SH, Liu LB, Wu HM, Tang QZ, Identification of differentially expressed genes and preliminary validations in cardiac pathological remodeling induced by transverse aortic constriction, Int. J. Mol. Med 44 (4) (2019) 1447–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Khalil H, Kanisicak O, Vagnozzi RJ, Johansen AK, Maliken BD, Prasad V, Boyer JG, Brody MJ, Schips T, Kilian KK, Correll RN, Kawasaki K, Nagata K, Molkentin JD, Cell-specific ablation of Hsp47 defines the collagen-producing cells in the injured heart, JCI Insight 4 (15) (2019) e128722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sawaki D, Hou L, Tomida S, Sun J, Zhan H, Aizawa K, Son BK, Kariya T, Takimoto E, Otsu K, Conway SJ, Manabe I, Komuro I, Friedman SL, Nagai R, Suzuki T, Modulation of cardiac fibrosis by Kruppel-like factor 6 through transcriptional control of thrombospondin 4 in cardiomyocytes, Cardiovasc. Res 107 (4) (2015) 420–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jazayeri R, Qoreishi M, Hoseinzadeh HR, Babanejad M, Bakhshi E, Najmabadi H, Jazayeri SM, Investigation of the asporin gene polymorphism as a risk factor for knee osteoarthritis in Iran, Am. J. Orthop. (Belle Mead NJ) 42 (7) (2013) 313–316. [PubMed] [Google Scholar]
- [32].Shinde AV, Frangogiannis NG, Mechanisms of Fibroblast Activation in the Remodeling Myocardium, Curr. Pathobiol. Rep 5 (2) (2017) 145–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Jiang X, Wu CA, Wang Y, Shi KJ, Jiang XZ, Zheng S, Tian W, Knockdown of asporin affects transforming growth factor-beta1-induced matrix synthesis in human intervertebral annulus cells, J. Orthop. Translat 7 (2016) 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kizawa H, Kou I, Iida A, Sudo A, Miyamoto Y, Fukuda A, Mabuchi A, Kotani A, Kawakami A, Yamamoto S, Uchida A, Nakamura K, Notoya K, Nakamura Y, Ikegawa S, An aspartic acid repeat polymorphism in asporin inhibits chondrogenesis and increases susceptibility to osteoarthritis, Nat. Genet 37 (2) (2005) 138–144. [DOI] [PubMed] [Google Scholar]
- [35].Hall C, Gehmlich K, Denning C, Pavlovic D, Complex Relationship Between Cardiac Fibroblasts and Cardiomyocytes in Health and Disease, J. Am. Heart Assoc 10 (5) (2021) e019338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Manabe I, Shindo T, Nagai R, Gene expression in fibroblasts and fibrosis: involvement in cardiac hypertrophy, Circ. Res 91 (12) (2002) 1103–1113. [DOI] [PubMed] [Google Scholar]
- [37].Rienks M, Papageorgiou AP, Frangogiannis NG, Heymans S, Myocardial extracellular matrix: an ever-changing and diverse entity, Circ. Res 114 (5) (2014) 872–888. [DOI] [PubMed] [Google Scholar]
- [38].Weis SM, Zimmerman SD, Shah M, Covell JW, Omens JH, Ross J Jr., Dalton N, Jones Y, Reed CC, Iozzo RV, McCulloch AD, A role for decorin in the remodeling of myocardial infarction, Matrix Biol 24 (4) (2005) 313–324. [DOI] [PubMed] [Google Scholar]
- [39].Van Aelst LN, Voss S, Carai P, Van Leeuwen R, Vanhoutte D, Sanders-van Wijk S, Eurlings L, Swinnen M, Verheyen FK, Verbeken E, Nef H, Troidl C, Cook SA, Brunner-La Rocca HP, Mollmann H, Papageorgiou AP, Heymans S, Osteoglycin prevents cardiac dilatation and dysfunction after myocardial infarction through infarct collagen strengthening, Circ. Res 116 (3) (2015) 425–436. [DOI] [PubMed] [Google Scholar]
- [40].Deckx S, Heggermont W, Carai P, Rienks M, Dresselaers T, Himmelreich U, van Leeuwen R, Lommen W, van der Velden J, Gonzalez A, Diez J, Papageorgiou AP, Heymans S, Osteoglycin prevents the development of age-related diastolic dysfunction during pressure overload by reducing cardiac fibrosis and inflammation, Matrix Biol 66 (2018) 110–124. [DOI] [PubMed] [Google Scholar]
- [41].Iozzo RV, Karamanos N, Proteoglycans in health and disease: emerging concepts and future directions, FEBS J 277 (19) (2010) 3863. [DOI] [PubMed] [Google Scholar]
- [42].Faust SM, Lu G, Wood SC, Bishop DK, TGF beta Neutralization within Cardiac Allografts by Decorin Gene Transfer Attenuates Chronic Rejection, J. Immunol 183 (11) (2009) 7307–7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Beetz N, Rommel C, Schnick T, Neumann E, Lother A, Monroy-Ordonez EB, Zeeb M, Preissl S, Gilsbach R, Melchior-Becker A, Rylski B, Stoll M, Schaefer L, Beyersdorf F, Stiller B, Hein L, Ablation of biglycan attenuates cardiac hypertrophy and fibrosis after left ventricular pressure overload, J. Mol. Cell. Cardiol 101 (2016) 145–155. [DOI] [PubMed] [Google Scholar]
- [44].Westermann D, Mersmann J, Melchior A, Freudenberger T, Petrik C, Schaefer L, Lullmann-Rauch R, Lettau O, Jacoby C, Schrader J, Brand-Herrmann SM, Young MF, Schultheiss HP, Levkau B, Baba HA, Unger T, Zacharowski K, Tschope C, Fischer JW, Biglycan is required for adaptive remodeling after myocardial infarction, Circulation 117 (10) (2008) 1269–1276. [DOI] [PubMed] [Google Scholar]
- [45].Tsutsui H, Kinugawa S, Matsushima S, Mitochondrial oxidative stress and dysfunction in myocardial remodelling, Cardiovasc. Res 81 (3) (2009) 449–456. [DOI] [PubMed] [Google Scholar]
- [46].Zhou B, Tian R, Mitochondrial dysfunction in pathophysiology of heart failure, J. Clin. Invest 128 (9) (2018) 3716–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Chiong M, Wang ZV, Pedrozo Z, Cao DJ, Troncoso R, Ibacache M, Criollo A, Nemchenko A, Hill JA, Lavandero S, Cardiomyocyte death: mechanisms and translational implications, Cell Death. Dis 2 (2011) e244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Jaishy B, Zhang Q, Chung HS, Riehle C, Soto J, Jenkins S, Abel P, Cowart LA, Van Eyk JE, Abel ED, Lipid-induced NOX2 activation inhibits autophagic flux by impairing lysosomal enzyme activity, J. Lipid Res 56 (3) (2015) 546–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Liu Y, Morley M, Brandimarto J, Hannenhalli S, Hu Y, Ashley EA, Tang WH, Moravec CS, Margulies KB, Cappola TP, Li M, consortium MA, RNA-Seq identifies novel myocardial gene expression signatures of heart failure, Genomics 105 (2) (2015) 83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Davis S, Meltzer PS, GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor, Bioinformatics 23 (14) (2007) 1846–1847. [DOI] [PubMed] [Google Scholar]
- [51].Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK, limma powers differential expression analyses for RNA-sequencing and microarray studies, Nucleic. Acids. Res 43 (7) (2015) e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: ultrafast universal RNA-seq aligner, Bioinformatics 29 (1) (2013) 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Anders S, Pyl PT, Huber W, HTSeq–a Python framework to work with high-throughput sequencing data, Bioinformatics 31 (2) (2015) 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Anders S, Huber W, Differential expression analysis for sequence count data, Genome Biol 11 (10) (2010) R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ge SX, Jung D, Yao R, ShinyGO: a graphical gene-set enrichment tool for animals and plants, Bioinformatics 36 (8) (2020) 2628–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Langfelder P, Horvath S, WGCNA: an R package for weighted correlation network analysis, BMC Bioinformatics 9 (2008) 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zhang B, Horvath S, A general framework for weighted gene co-expression network analysis, Stat. Appl. Genet. Mol. Biol 4 (2005) Article17. [DOI] [PubMed] [Google Scholar]
- [58].Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T, Cytoscape 2.8: new features for data integration and network visualization, Bioinformatics 27 (3) (2011) 431–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Assenov Y, Ramirez F, Schelhorn SE, Lengauer T, Albrecht M, Computing topological parameters of biological networks, Bioinformatics 24 (2) (2008) 282–284. [DOI] [PubMed] [Google Scholar]
- [60].Scardoni G, Petterlini M, Laudanna C, Analyzing biological network parameters with CentiScaPe, Bioinformatics 25 (21) (2009) 2857–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Snel B, Lehmann G, Bork P, Huynen MA, STRING: a web-server to retrieve and display the repeatedly occurring neighbourhood of a gene, Nucleic. Acids. Res 28 (18) (2000) 3442–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Huang C, Andres AM, Ratliff EP, Hernandez G, Lee P, Gottlieb RA, Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1, PLoS ONE 6 (6) (2011) e20975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Redfors B, Shao Y, Omerovic E, Myocardial infarct size and area at risk assessment in mice, Exp. Clin. Cardiol 17 (4) (2012) 268–272. [PMC free article] [PubMed] [Google Scholar]
- [64].Germano JF, Huang C, Sin J, Song Y, Tucker KC, Taylor DJR, Saadaeijahromi H, Stotland A, Piplani H, Gottlieb RA, Mentzer RM Jr., Andres AM, Intermittent Use of a Short-Course Glucagon-like Peptide-1 Receptor Agonist Therapy Limits Adverse Cardiac Remodeling via Parkin-dependent Mitochondrial Turnover, Sci. Rep 10 (1) (2020) 8284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Dhutia NM, Cole GD, Willson K, Rueckert D, Parker KH, Hughes AD, Francis DP, A new automated system to identify a consistent sampling position to make tissue Doppler and transmitral Doppler measurements of E, E’ and E/E’, Int. J. Cardiol 155 (3) (2012) 394–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Zafiriou MP, Noack C, Unsold B, Didie M, Pavlova E, Fischer HJ, Reichardt HM, Bergmann MW, El-Armouche A, Zimmermann WH, Zelarayan LC, Erythropoietin responsive cardiomyogenic cells contribute to heart repair post myocardial infarction, Stem Cells 32 (9) (2014) 2480–2491. [DOI] [PubMed] [Google Scholar]
- [67].Scott PG, Dodd CM, Bergmann EM, Sheehan JK, Bishop PN, Crystal structure of the biglycan dimer and evidence that dimerization is essential for folding and stability of class I small leucine-rich repeat proteoglycans, J. Biol. Chem 281 (19) (2006) 13324–13332. [DOI] [PubMed] [Google Scholar]
- [68].Autiero I, Costantini S, Colonna G, Human sirt-1: molecular modeling and structure-function relationships of an unordered protein, PLoS ONE 4 (10) (2008) e7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Sharma A, Gautam V, Costantini S, Paladino A, Colonna G, Interactomic and pharmacological insights on human sirt-1, Front. Pharmacol 3 (2012) 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ, Basic local alignment search tool, J. Mol. Biol 215 (3) (1990) 403–410. [DOI] [PubMed] [Google Scholar]
- [71].Thompson JD, Higgins DG, Gibson TJ, CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice, Nucleic. Acids. Res 22 (22) (1994) 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Ramakrishnan C, Ramachandran GN, Stereochemical criteria for polypeptide and protein chain conformations. II. Allowed conformations for a pair of peptide units, Biophys. J 5 (6) (1965) 909–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Gopalakrishnan K, Sowmiya G, Sheik SS, Sekar K, Ramachandran plot on the web (2.0), Protein Pept. Lett 14 (7) (2007) 669–671. [DOI] [PubMed] [Google Scholar]
- [74].Sippl MJ, Recognition of errors in three-dimensional structures of proteins, Proteins 17 (4) (1993) 355–362. [DOI] [PubMed] [Google Scholar]
- [75].Wiederstein M, Sippl MJ, ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins, Nucleic. Acids. Res 35 (Web Server issue) (2007) W407–W410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE, UCSF Chimera–a visualization system for exploratory research and analysis, J. Comput. Chem 25 (13) (2004) 1605–1612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.