

Abstract
Blockade of CD47, the “do not eat me” signal, has limited effects in solid tumors despite its potent antitumor effects in hematopoietic malignancies. Taking advantage of the high expression of cytotoxic T lymphocyte–associated protein 4 (CTLA-4) on Treg cells and abundant Fc receptor–expressing active phagocytes inside the tumor microenvironment (TME), we designed and tested a heterodimer combining an anti–CTLA-4 antibody, which targets Treg cells, with the CD47 ligand, signal regulatory protein α (SIRPα), to selectively block CD47 on intratumoral Treg cells. We hypothesized that heterodimer treatment would increase antibody-dependent cellular phagocytosis of the targeted Treg cells. We found that anti–CTLA-4×SIRPα preferentially depleted ICOShigh immunosuppressive Treg cells in the TME and enhanced immunity against solid tumors, including MC38 and CT26 murine colon cancers. Mechanistically, we found that CD47 expression on Treg cells limited anti–CTLA-4–mediated depletion and Fc on the heterodimer-enhanced depletion. Furthermore, anti-human CTLA-4×SIRPα depleted tumor Treg cells and exhibits less toxicity than anti-human CTLA-4 in a humanized mouse model. Collectively, these results demonstrate that simultaneously modulating both “eat me” and do not eat me signals induces Treg cell depletion inside the TME and may be an effective strategy for treating solid tumors.
INTRODUCTION
Regulatory T (Treg) cells play a key role in the control of autoimmunity. However, Treg cells orchestrate cellular and molecular networks to create an immunosuppressive environment favoring tumorigenesis (1). Several studies have indicated that the infiltration of Treg cells correlates with poor prognosis in cancer (2–4) and that the balance between effector T cells and Treg cells dictates the outcome of antitumor immune responses (5, 6). Although Treg cell depletion with therapeutic antibodies such as anti-CD25, anti–glucocorticoid-induced tumor necrosis factor (TNF) receptor (GITR), and anti-OX40 shows tumor control in preclinical models (3, 7–9), clinical efficacy is limited. To avoid systemic side effects and to increase efficacy, it would be desirable to selectively deplete Treg cells in the tumor but not in the periphery.
Cytotoxic T lymphocyte antigen 4 (CTLA-4) has been identified as an important immune checkpoint that modulates T cell activation. CTLA-4 blockade with anti–CTLA-4 (ipilimumab) induces durable remission in patients with advanced melanoma (10, 11), suggesting that CTLA-4 is an attractive target for cancer immunotherapy. However, a major hurdle for anti–CTLA-4 therapy in the clinic is immune-related adverse effects (irAEs), with 90% of patients developing irAEs in any grade and 40% of patients in grades 3 to 4. Most irAEs are associated with autoimmune effects in normal tissues caused by the overactivation of systemic immune responses (12, 13). Of importance, these irAEs are likely due to the blocking effect of anti–CTLA-4 on peripheral Treg cells. Compared with effector T cells, Treg cells have much higher CTLA-4 expression especially inside the tumor microenvironment (TME). In mouse tumor models, anti–CTLA-4 antibodies have been shown to deplete tumor Treg cells through engagement with Fc receptors (FcRs). The involvement of phagocytotic cells and activating FcRs in the tumor co-defines antitumor effects of anti–CTLA-4 therapy (14–16). Studies also show that enhancing anti–CTLA-4 binding to FcRs could potentially improve clinical outcomes. Nonetheless, the extent to which Treg cell depletion contributes to clinical benefit is difficult to determine and remains controversial, as is whether depletion occurs in the TME or in the periphery (17, 18). Preferential depletion of Treg cells in the TME might be more desirable clinically than systemic blockade of CTLA-4 function.
It is possible that FcR-based “eat me” signals could be restricted by a “do not eat me” signal. CD47, a broadly expressed molecule, delivers a do not eat me signal that limits phagocytosis upon the engagement with its ligand, signal regulatory protein α (SIRPα) (19). Up-regulation of CD47 has been reported in a wide range of human and murine tumors. CD47 blockade has antitumor effect in various preclinical tumor models, particularly in the context of hematopoietic malignancies (20, 21). This antitumor effect is especially strong when it is combined with a potent therapeutic antibody such as rituximab, providing an FcR-restricted eat me signal (22–24). Both increased phagocytosis and enhanced adaptive immunity contribute to CD47 blockade–mediated antitumor effects (20, 25–27). However, there are two major obstacles that limit translation of CD47 blockade therapy into the clinic. One is the large CD47 “antigen sink” in hematopoietic cell lineages, especially red blood cells (RBCs) and platelets. To saturate this peripheral antigen sink, high-dose and intensive treatment must be conducted, which, in turn, causes severe toxicity such as anemia. The other is the limited efficacy of CD47 blockade in solid tumors, which is likely due to the lack of large amounts of prophagocytic receptors and the reduced access that such antibodies have to solid tumors (28).
We hypothesized that if we could develop a therapy that preferentially blocks CD47 on Treg cells in the TME while delivering an eat me signal, then we could increase Treg depletion efficiency and antitumor efficacy in solid tumors. Therefore, we designed an anti–CTLA-4×SIRPα heterodimer that targets both CTLA-4 and CD47 on the same cell to modulate these two signals using one molecule with lower affinity for each target. The reduced affinity to each molecule provides the anti–CTLA-4×SIRPα with the ability to avoid nonspecific binding to peripheral cells, including RBCs and systemic Treg cells, and facilitates its access to Treg cells specifically in the TME. We further compared its antitumor efficacy in relation to combination therapy with separate molecule delivery and dissected its mechanism of action.
RESULTS
Anti–CTLA-4×SIRPα heterodimer selectively targets intratumoral Treg cells
To test the feasibility of CD47 blockade on Treg cells, we first analyzed CD47 and CTLA4 expression pattern on T cells from human tumor tissues. On the basis of single-cell sequencing data from published datasets (29, 30), we found the universal expression of CD47 and enriched expression of CTLA4 on tumor Treg cells compared with other T cells in samples from patients with colorectal cancer or non–small cell lung cancer (NSCLC) (Fig. 1A and fig. S1, A to C). Treg cells in the tumor had the highest CD47 and CTLA4 double-positive cells among all T cell subsets (fig. S1D). We then evaluated CD47 expression in MC38 tumor–bearing mice. As expected, we found universal and high expression of CD47 on RBCs and on splenic and tumor Treg cells (fig. S1, E and F), illustrating the large peripheral antigen sink problem in CD47 blockade therapy. We also evaluated CTLA-4 expression and observed a higher abundance on Treg cells in the tumor compared with Treg cells in the blood or spleen (Fig. 1, B and C). Consistent with patient data, we observed that the proportion of CTLA-4+CD47+ cells was highest in Treg cells among intratumoral T cell subsets evaluated (fig. S1, G and H). Moreover, CTLA-4 expression density was higher on Treg cells than that on effector T cells (fig. S1I).
Fig. 1. Anti–CTLA-4×SIRPα heterodimer selectively targets intratumoral Treg cells.
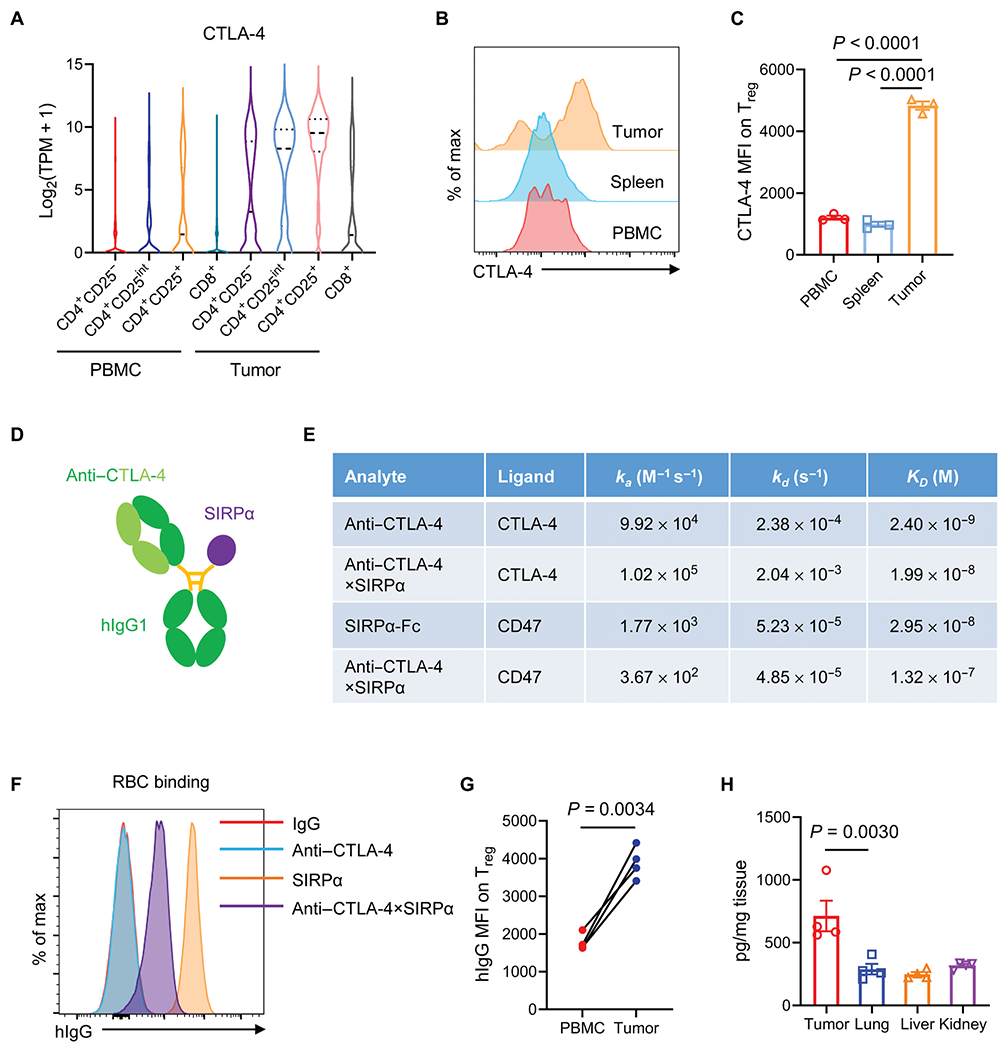
(A) Expression of CTLA-4 transcripts in tumors and PBMCs isolated from patients with colorectal cancer was determined on the basis of single-cell sequencing online database (29). (B and C) CTLA-4 expression on Treg cells from PBMCs, spleens, and tumor cells in MC38 tumor–bearing mice was quantified on day 14. A representative flow cytometry histogram (B) and quantification of CTLA-4 median fluorescence intensity (MFI) (C) are shown. (D) Schematic diagram of anti–CTLA-4×SIRPα heterodimer shows the anti–CTLA-4 region, the SIRPα region, and the hIgG1 Fc region. (E) Kinetic association (ka), dissociation (Kd), and calculated affinity (KD) for binding of the indicated analyte to mouse CTLA-4 or CD47 antigen were measured by surface plasmon resonance. (F) Binding of SIRPα, CTLA-4, and anti–CTLA-4×SIRPα to RBCs from C57BL/6 mice was evaluated by flow cytometry. IgG indicates the isotype control used (n = 4 mice per group). (G) Anti–CTLA-4×SIRPα binding was quantified on Treg cells isolated from PBMCs or tumors from MC38 tumor–bearing mice based on hIgG MFI. (H) C57BL/6 mice were inoculated with 5 × 105 MC38 tumor cells. After 13 days, 20 μg of anti–CTLA-4×SIRPα was intraperitoneally injected. Five days later, mice were perfused with PBS, and tumors and indicated tissues were harvested. The concentration of anti–CTLA-4×SIRPα protein in each tissue was determined by enzyme-linked immunosorbent assay (n = 4 mice per group). Data are shown as median and range (A) and means ± SEM from two to three independent experiments (C, G, and H). P values were determined by one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test (C and H) or two-tailed paired t test and (G). The normality of data was confirmed by Shapiro-Wilk test.
This suggested the possibility that one could take advantage of the high expression of CTLA-4 on tumor Treg cells to selectively deliver a CD47 blocker to tumor Treg cells. We reasoned that the dual targeting of both CD47 and CTLA-4 on Treg cells that concurrently blocks the do not eat me signal while enhancing the eat me signal should efficiently deplete tumor Treg cells. Coupled with this, we also reasoned that targeting CD47 with lower affinity may aid in bypassing the antigen sink on RBCs and peripheral immune cells. We decided to use SIRPα, the ligand of CD47, to provide lower binding affinity to CD47 as compared with the much higher affinity of anti-CD47 antibodies (22). Thus, we designed an anti–CTLA-4×SIRPα heterodimer to simultaneously target both molecules. The SIRPα arm would provide CD47 binding but with lower affinity, and the antiαCTLA-4 portion provides CTLA-4 targeting but also with reduced affinity because it is a single arm construct (Fig. 1D). This heterodimer was engineered on the basis of “knobs-into-holes” technology (31, 32) and produced with high purity that was confirmed by gel electrophoresis under reducing and nonreducing conditions (fig. S1J). Surface plasmon resonance (SPR) showed that anti–CTLA-4×SIRPα had more than a 10-fold reduction in affinity for CTLA-4 compared to that of the intact anti–CTLA-4 antibody and a 5-fold reduction in affinity for CD47 compared to SIRPα-Fc. Anti–CTLA-4×SIRPα also exhibited a roughly 10-fold lower binding affinity to CD47 than to CTLA-4 (Fig. 1E). These affinity differences should provide an advantage for anti–CTLA-4×SIRPα to direct SIRPα to tumor Treg cells. We also tested the binding of anti–CTLA-4×SIRPα versus SIRPα to RBCs and found a greater than 20-fold reduction in binding to RBCs (Fig. 1F), which should result in reduced toxicity and consumption in peripheral blood.
To further demonstrate that anti–CTLA-4×SIRPα has superior affinity to tumor Treg cells than peripheral Treg cells, we assayed the binding of anti–CTLA-4×SIRPα to MC38 tumorαinfiltrating Treg cells versus peripheral blood mononuclear cell (PBMC) Treg cells and found a higher affinity in tumor Treg cell binding, implying a preferable tumor-targeting effect of anti–CTLA-4×SIRPα in vivo (Fig. 1G). We tested the in vivo half-life of anti–CTLA-4×SIRPα with a single low-dose systemic injection and found that anti–CTLA-4×SIRPα had a half-life of 21.4 hours (fig. S1K). To investigate the tumor-targeting propensity of anti–CTLA-4×SIRPα, we analyzed the tissue distribution 5 days after systemic injection of anti–CTLA-4×SIRPα and found more abundant heterodimer proteins in the tumor compared to that in normal tissues such as lung, liver, and kidney (Fig. 1H), suggesting a preferential enrichment and retention of anti–CTLA-4×SIRPα in the tumor.
Anti–CTLA-4×SIRPα heterodimer preferentially depletes ICOShigh Treg cells in the tumor
To test whether the preferential binding of anti–CTLA-4×SIRPα to tumor Treg cells and the enrichment of anti–CTLA-4×SIRPα in the tumor lead to Treg cell depletion in vivo, we used MC38, a mouse colon cancer model that has high immune cell infiltration including Treg cells and has been extensively used for immunotherapeutic intervention studies (33). We treated mice bearing subcutaneous MC38 tumors at day 13 after tumor inoculation with low-dose (20 μg) anti–CTLA-4×SIRPα, molar equivalents of the combination of anti–CTLA-4 plus SIRPα (Combo), or control human immunoglobulin G (hIgG) (Ctrl) and euthanized mice 5 days after treatment. Analysis of Treg cell abundance in tumor versus nontumor lymphoid tissues showed that anti–CTLA-4×SIRPα specifically depleted Treg cells in the tumor but not in the spleen or in the draining lymph node (dLN). By contrast, Combo treatment at equivalent low doses did not deplete tumor Treg cells (Fig. 2, A and B). To confirm that the Treg depletion by anti–CTLA-4×SIRPα treatment occurred as a direct and not secondary effect, we analyzed tumor-infiltrating Treg cells at earlier time points. Anti–CTLA-4×SIRPα reduced Treg cells in the tumor at 36 and 96 hours after treatment, suggesting a direct depletion effect (fig. S2, A to C). Only a portion of tumor Treg cells were transiently depleted by the Combo treatment, and the Treg cell frequency rebounded later (fig. S2, A to C), showing the limited effect of Combo treatment. Anti–CTLA-4×SIRPα treatment increased the CD8:Treg cell ratio as compared to Combo treatment, suggesting a more active TME (Fig. 2C). To further confirm our results, we took advantage of Foxp3EGFP-cre mice that express enhanced green fluorescent protein (EGFP) driven by endogenous Foxp3 gene promoter (34). Consistent with our results in wild-type (WT) mice, the frequency of intratumoral EGFP+ cells was reduced after anti–CTLA-4×SIRPα treatment, and CD8:Foxp3-EGFP ratio was increased (Fig. 2, D to F). No difference in EGFP+ cell frequency was observed in the spleen or dLN (Fig. 2E). Collectively, these results demonstrated that a low dose of anti–CTLA-4×SIRPα selectively and effectively depletes intratumoral Treg cells.
Fig. 2. Anti–CTLA-4×SIRPα heterodimer preferentially depletes ICOShigh Treg cells in tumors.
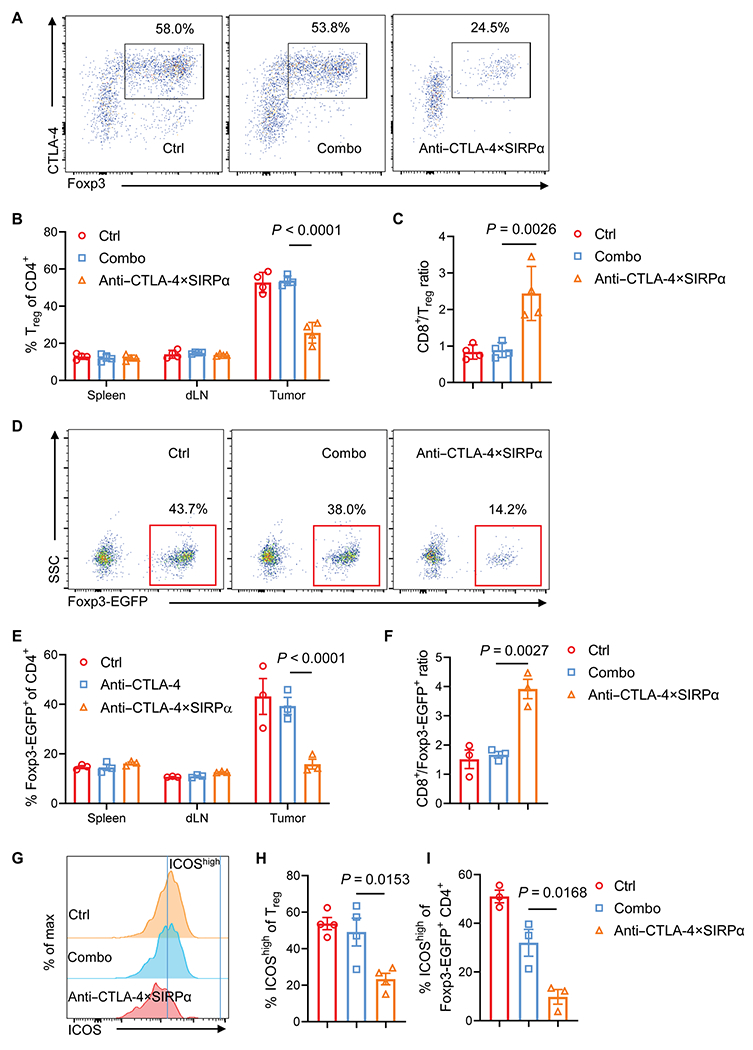
(A to C) C57BL/6 mice were inoculated with 5 × 105 MC38 tumor cells and treated with Combo (anti–CTLA-4 and SIRPα) or anti–CTLA-4×SIRPα heterodimer on day 13. Control (Ctrl) mice were treated with hIgG. Five days later, Treg cells from tumor, spleen, and dLN were analyzed by flow cytometry (A). Treg cell frequency from different treatment groups (B) and the ratio of CD8+ T cells to Treg (C) were quantified (n = 4 mice per group). (D to F) Foxp3EGFP-cre mice were inoculated with 5 × 105 MC38 tumor cells and treated with Combo or anti–CTLA-4×SIRPα on day 13. Ctrl mice were treated with hIgG. Five days later, Foxp3-EGFP+ cells isolated from tumors, spleens, and dLN were analyzed (D). Foxp3-EGFP+ cell frequency from indicated groups (E) and the ratio of CD8+ T cells to Foxp3-EGFP+ cells (F) were quantified (n = 3 mice per group). SSC, side scatter. (G to I) C57BL/6 mice or Foxp3EGFP-Cre mice were treated as above, and samples were stained for expression of ICOS (G). ICOShigh frequency among Treg cells isolated from C57BL/6 mice (H) and ICOShigh frequency among EGFP+ cells from Foxp3EGFP-cre mice (I) are shown (n = 3 to 4 per group). Data are shown as means ± SD from two to three independent experiments. P values were determined by two-way ANOVA with Tukey’s multiple comparisons test (B and E) or oneway ANOVA with Tukey’s multiple comparisons test (C, F, H, and I). The normality of data was confirmed by Shapiro-Wilk test.
Fig. 3. Dual targeting of CTLA-4 and CD47 reduces tumor burden in a T cell–dependent manner.
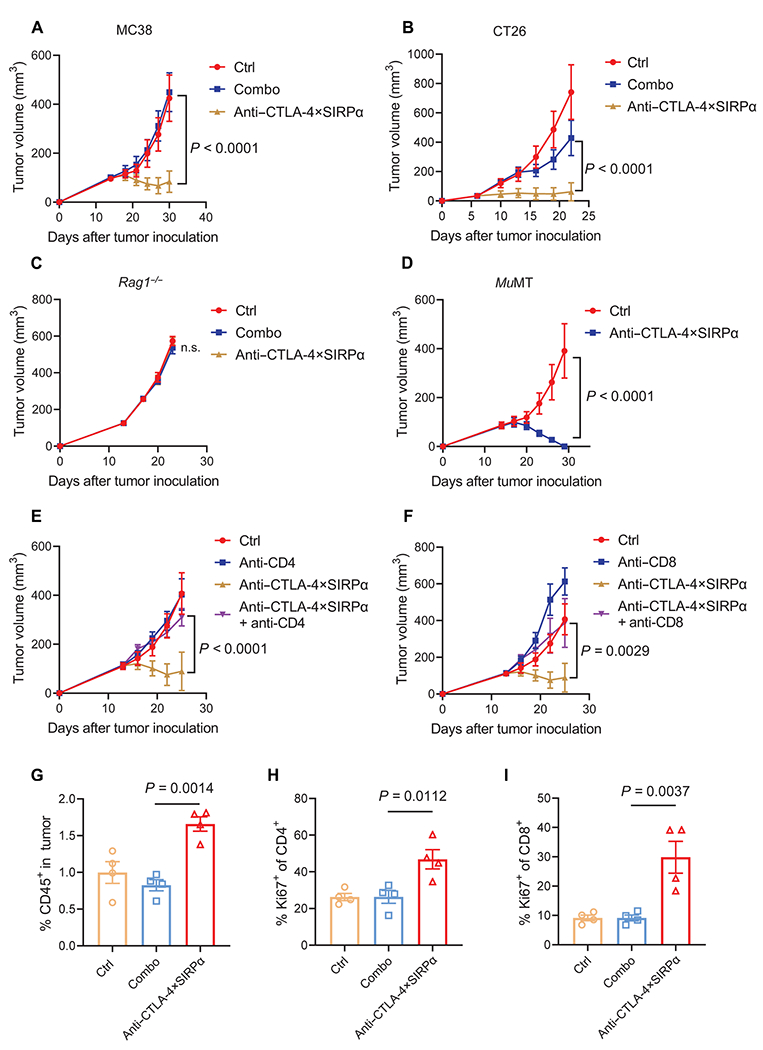
(A) C57BL/6 mice were inoculated with 5 × 105 MC38 tumor cells and treated with molar equivalents of anti–CTLA-4 plus SIRPα (Combo, 12 + 8 μg) or anti–CTLA-4×SIRPα (20 μg) on day 14. Ctrl mice were treated with hIgG. Tumor growth was measured every 3 days (n = 5 mice per group). (B) BALB/c mice were inoculated with 5 × 105 CT26 tumor cells and treated with Combo (30 + 20 μg) or anti–CTLA-4×SIRPα (50 μg) on day 6. Ctrl mice were treated with hIgG. Tumor growth was measured every 3 days (n = 5 mice per group). (C) Rag1−/− mice were inoculated with 1 × 105 MC38 tumor cells and treated with Combo (12 + 8 μg) or anti–CTLA-4×SIRPα (20 μg) on day 13. Ctrl mice were treated with hIgG. Tumor growth was measured every 3 days (n = 5 mice per group). (D) MuMT mice were inoculated with 1.5 × 106 MC38 tumor cells and treated with anti–CTLA-4×SIRPα (20 μg) on day 14. Ctrl mice were treated with hIgG. Tumor growth was measured every 3 days (n = 5 mice per group). (E and F) C57BL/6 mice were inoculated with 5 × 105 MC38 tumor cells and treated with anti–CTLA-4×SIRPα on day 13. Two hundred micrograms of anti-CD4 (E) or anti-CD8 (F) was administrated 1 day before treatment initiation and then twice a week for 2 weeks. Ctrl mice were treated with hIgG. Tumor growth was measured every 3 days (n = 5 mice per group). (G to I) C57BL/6 mice were inoculated with 5 × 105 MC38 tumor cells and treated with Combo or anti–CTLA-4×SIRPα on day 14. Ctrl mice were treated with hIgG. Five days later, TILs were isolated and analyzed for frequency of total CD45+ cells in tumors (G) and Ki67 expression on CD4+ (H) and CD8+ (I) T cells from indicated groups (n = 4 mice per group). Data are shown as means ± SEM from two to three independent experiments. P values were determined by a two-way ANOVA with Geisser-Greenhouse’s correction (A to F) or one-way ANOVA with Tukey’s multiple comparisons test (G to I). The normality of data was confirmed by Shapiro-Wilk test. Not significant (n.s.), P > 0.05.
We then tested whether anti–CTLA-4×SIRPα treatment depletes Treg cells in normal tissues, which might cause potential autoimmune problems. We did not observe differences in Treg subsets in the liver, lung, and kidney after anti–CTLA-4×SIRPα treatment (fig. S2D), suggesting that anti–CTLA-4×SIRPα has limited effect on normal tissues. We then quantified the absolute CTLA-4 and CD47 receptor numbers on Treg cells from different tissues. We found that intratumoral Treg cells expressed the highest numbers of CTLA-4 molecules (average of 14,373 per cell) and moderate numbers of CD47 molecules (average of 1935 per cell) (fig. S2E). These data suggest that CTLA-4 expression dictates the depletion effect of anti–CTLA-4×SIRPα and further support our design rationale for this heterodimer.
Previous studies showed that the inducible T cell costimulator (ICOS) expression on Treg cells defines an activated subset with more immunosuppressive features, and ICOShigh Treg cells are adversely correlated with survival in patients with cancer (35–37). We found that ICOShigh Treg cells expressed CTLA-4 and CD47 at higher densities as compared with ICOSlow Treg cells (fig. S2, F to H). After anti–CTLA-4×SIRPα treatment, ICOShigh Treg cells were preferentially depleted among all Treg cells (Fig. 2, G to I, and fig. S2I). To evaluate whether these ICOShigh Treg cells are more immunosuppressive in our MC38 model, we sorted EGFP+ICOS+ and EGFP+ICOS− Treg cells from Foxp3EGFP-cre mice and cocultured them with anti-CD3–stimulated EGFP− splenocytes. We found that ICOS+ Treg cells exhibited stronger inhibitory effects on CD8+ T cell proliferation, as measured by carboxyfluorescein succinimidyl ester (CFSE) dilution (fig. S2J). This suggested that anti–CTLA-4×SIRPα treatment results in the preferential depletion of the more immunosuppressive Treg cell population. Thus, although anti–CTLA-4×SIRPα did not eliminate tumor Treg cells, the selective depletion of ICOShigh subset may be sufficient to convert an immunosuppressive TME to an immune-active one.
Dual targeting of CTLA-4 and CD47 reduces tumor burden in a T cell–dependent manner
We then sought to study whether the depletion of Treg cells by anti–CTLA-4×SIRPα has a beneficial outcome for tumor control. We found that a single low dose of 20 μg of anti–CTLA-4×SIRPα administered intraperitoneally on day 14 after tumor implantation inhibited tumor growth in well-established MC38 tumors, whereas an equivalent dose of Combo treatment had no effect (Fig. 3A). The antitumor effect of anti–CTLA-4×SIRPα was not restricted to the MC38 tumor model; a similar tumor burden reduction was observed in CT26, a mouse colon cancer on BALB/c background (Fig. 3B). We observed partial tumor growth inhibition when we raised the anti–CTLA-4 dose to 100 μg (fig. S3A), suggesting that anti–CTLA-4×SIRPα is much more potent than Combo treatment. To further show that anti–CTLA-4×SIRPα is preferential to other treatment designs, we made a control heterodimer with the anti–CTLA-4 arm only but no SIRPα expression on the other arm and found that the control heterodimer could not exert antitumor effects (fig. S3B). This indicates an essential role of SIRPα for tumor control after anti–CTLA-4×SIRPα treatment. In addition, a heterodimer with a single-chain version of anti–CTLA-4 paired with SIRPα was not as efficacious as anti–CTLA-4×SIRPα with the Fab version of anti–CTLA-4, likely due to the low affinity of the single-chain antibody (fig. S3C). To demonstrate that the antitumor effect of anti–CTLA-4×SIRPα is not restricted to one clone of anti–CTLA-4 (4F10), we compared another clone of anti–CTLA-4 (9D9) paired with SIRPα (9D9×SIRPα) with 4F10×SIRPα and observed similar antitumor effects between these two clones when administered at a dose of 40 μg, with 4F10×SIRPα slightly better at a dose of 20 μg (fig. S3, D and E).
Because anti–CTLA-4×SIRPα potentially activates both the innate arm and the adaptive arm of immune system, we used Rag1−/− mice that lack recombination activating protein 1 (RAG1), which knocks out the adaptive arm of the immune response. We found that the antitumor effect of anti–CTLA-4×SIRPα was abolished in Rag1−/− mice, indicating a requirement for adaptive immune responses (Fig. 3C). We then inoculated MC38 tumor into muMt mice that lack mature B cells and found that anti–CTLA-4×SIRPα inhibited tumor growth effectively (Fig. 3D), suggesting a dispensable role of B cells. We further validated the role of T cells in anti–CTLA-4×SIRPα–mediated antitumor effect. Depleting either CD4+ or CD8+ T cells abolished the antitumor effect of anti–CTLA-4×SIRPα (Fig. 3, E and F), further demonstrating an essential role for T cells. We then explored how anti–CTLA-4×SIRPα changes immune cell components in the TME. We analyzed the tumor-infiltrating lymphocytes (TILs) 5 days after anti–CTLA-4×SIRPα treatment in MC38 tumor–bearing mice and found an increase in the frequency of CD45+ TILs (Fig. 3G). Further analysis showed increases of both CD4+ and CD8+ T cell proliferation marked by elevated Ki67 expression relative to Combo treatment (Fig. 3, H and I, and fig. S3F). These data indicate an active immune microenvironment after anti–CTLA-4×SIRPα treatment.
Dual targeting of CTLA-4 and CD47 enhances tumor-specific T cell responses in an interferon-γ–dependent manner
To evaluate whether anti–CTLA-4×SIRPα increases antigen-specific T cells, we used a tetramer recognizing the MC38-specific antigen epitope KSPWFTTL(KSP) presented in the H-2Kb major histocompatibility class I molecule to identify tumor-specific T cells. We observed an increase of KSP-specific CD8+ T cells after anti–CTLA-4×SIRPα treatment relative to Combo treatment (Fig. 4A and fig. S4A). To explore whether anti–CTLA-4×SIRPα treatment changes the cytokine milieu in the TME to favor antigen-specific T cell responses, we tested the expression of a broad spectrum of cytokines and chemokines in tumors 48 hours after Combo or anti–CTLA-4×SIRPα treatment. We observed an increase in the concentration of multiple cytokines including interleukin-1α/β (IL-1α/β), C-C motif chemokine ligand 5 (CCL5), CCL17, and interferon-γ (IFN-γ) in both treatment groups compared to control group (fig. S4B). IFN-γ was increased in the anti–CTLA-4×SIRPα–treated group but not in the Combo group (fig. S4B), suggesting an enhanced T cell activation unique to the anti–CTLA-4×SIRPα group. Consistent with this, we observed an increase of IFN-γ+TNF+ T cells after anti–CTLA-4×SIRPα treatment (fig. S4, C and D). We reasoned that the increased T cell activation is tumor-specific instead of a bystander effect. To test our hypothesis, we assessed IFN-γ production by coculturing TILs isolated from Combo- or anti–CTLA-4×SIRPα–treated mice with irradiated MC38 tumor cells (tumor-specific) or Lewis lung cancer (LLC) tumor cells (controls). Irradiated tumor cells were used to avoid space occupation and metabolic competition by overgrowing tumor cells. We observed a marked increase of IFN-γ production stimulated by MC38 tumor cells, but not LLC cells, in the anti–CTLA-4×SIRPα–treated group, whereas Combo treatment showed no difference with the control group (Fig. 4B). Similar results were observed in the dLN (Fig. 4C). These results further showed that anti–CTLA-4×SIRPα treatment, but not Combo treatment, increased tumor-specific immune responses. To investigate whether IFN-γ is essential in anti–CTLA-4×SIRPα treatment–induced antitumor immunity, we inoculated MC38 tumor into Ifng−/− mice and observed no tumor control benefit after anti–CTLA-4×SIRPα treatment (Fig. 4D). To exclude the potential bias for genetically deficient mice, we confirmed the results with the administration of IFN-γ–neutralizing antibodies before and during the treatment with anti–CTLA-4×SIRPα in MC38 tumor–bearing WT mice. Consistent with our findings in the Ifng−/− mice, IFN-γ neutralization abolished anti–CTLA-4×SIRPα treatment–mediated tumor inhibition (Fig. 4E), demonstrating an essential role of IFN-γ in anti–CTLA-4×SIRPα treatment.
Fig. 4. Dual targeting of CTLA-4 and CD47 enhances tumor-specific T cell responses in an IFN-γ-dependent manner.
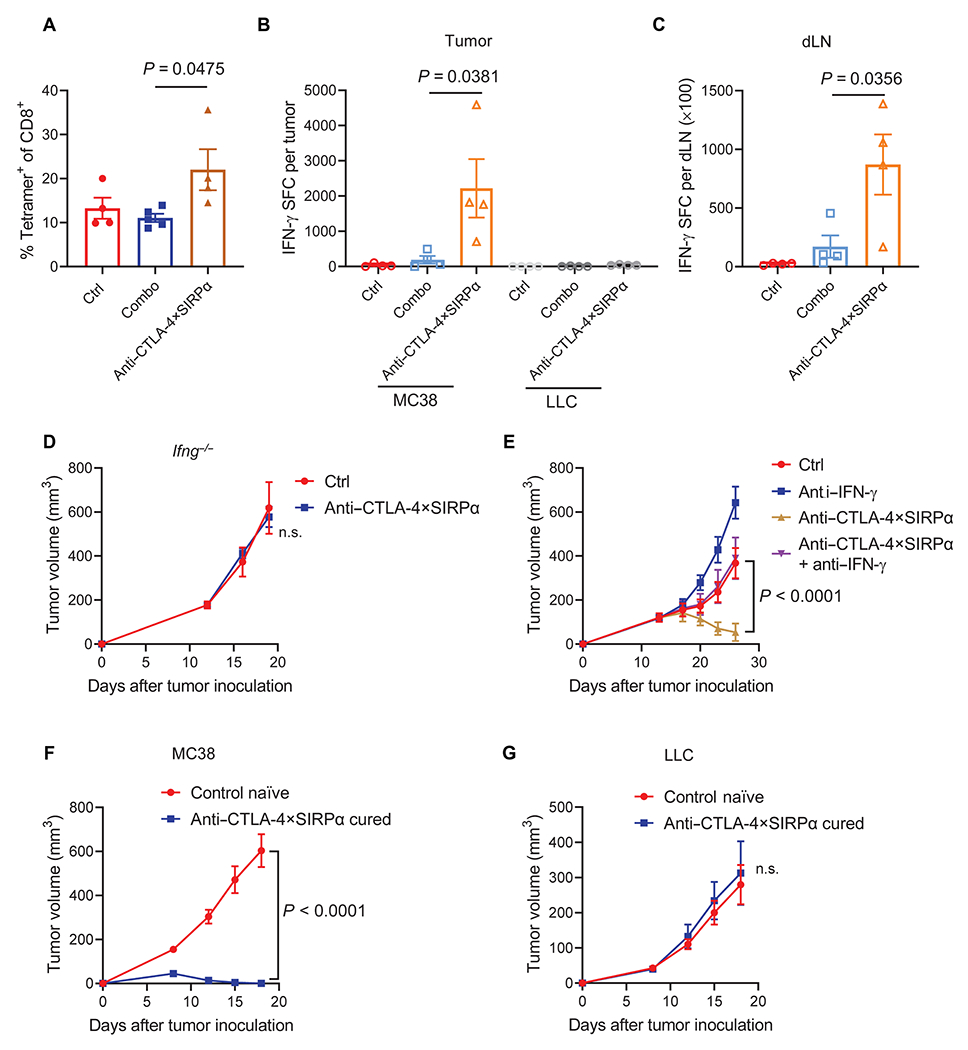
(A) C57BL/6 mice were inoculated with 5 × 105 MC38 tumor cells and treated with Combo or anti–CTLA-4×SIRPα on day 13. Ctrl mice were treated with hIgG. Five days later, TILs were analyzed for tumor-specific T cells using an H-2Kb KSP(KSPWFTTL) tetramer. The frequency of tetramer+ cells of total CD8+ T cells was evaluated by flow cytometry (n = 4 to 5 mice per group). (B and C) C57BL/6 mice were inoculated with 5 × 105 MC38 tumor cells and treated with Combo or anti–CTLA-4×SIRPα on day 13. Ctrl mice were treated with hIgG. Five days later, tumors and dLNs were isolated. TILs were purified with CD45-positive selection magnetic beads. TILs were restimulated with irradiated MC38 tumor cells or irrelevant control Lewis lung cancer (LLC) cells for 48 hours (B), and dLN cells were stimulated with irradiated MC38 tumor cells for 48 hours (C). IFN-γ-producing cell counts were determined by ELISPOT assay (n = 4 mice per group). SFC, spot-forming cells. (D) WT or Ifng−/− C57BL/6 mice were inoculated with 5 × 105 MC38 tumor cells and treated with anti–CTLA-4×SIRPα on day 13. Ctrl mice were treated with hIgG. Tumor growth was measured every 3 days (n = 4 mice per group). (E) C57BL/6 mice were inoculated with 5 × 105 MC38 tumor cells and treated with anti–CTLA-4×SIRPα on day 13. One hundred fifty micrograms of anti–IFN-γ was administrated 1 day before treatment initiation and then twice a week for 2 weeks. Ctrl mice were treated with hIgG. Tumor growth was measured every 3 days (n = 5 mice per group). (F and G) C57BL/6 mice were inoculated with 5 × 105 MC38 tumor cells and treated with anti–CTLA-4×SIRPα on day 14. Six weeks later, anti–CTLA-4×SIRPα-cured mice and control naive mice were rechallenged with 5 × 106 MC38 tumor cells on the left flank (opposite to the original injection flank) (F), and 5 × 105 LLC tumor cells were injected on the right flank (G). Tumor growth was measured every 3 days (n = 5 mice per group). Data are shown as means ± SEM from two to three independent experiments. P values were determined by one-way ANOVA with Tukey’s multiple comparisons test (A to C) or two-way ANOVA with Geisser-Greenhouse’s correction (D to G). The normality of data was confirmed by Shapiro-Wilk test. n.s., P > 0.05.
To determine whether anti–CTLA-4×SIRPα treatment induces a protective memory response, which is a unique feature for adaptive immunity, we rechallenged anti–CTLA-4×SIRPα–treated mice with complete tumor resolution after 5 weeks with 10 times more MC38 tumor cells on the opposite flank to the original tumor inoculation site. The flank bearing the initial MC38 tumor was inoculated with control LLC tumor cells. All anti–CTLA-4×SIRPα–cured mice spontaneously rejected MC38, but not LLC, tumors. By contrast, both MC38 and LLC tumors grew aggressively on naïve control mice with the same tumor inoculation scheme (Fig. 4, F and G). Together, these data suggested that anti–CTLA-4×SIRPα treatment generates tumor-specific immune memory.
CD47 expression on Treg cells is essential for anti–CTLA-4×SIRPα–mediated antitumor effect
Our earlier data showed that a heterodimer without the SIRPα arm cannot control tumor growth (fig. S3B), suggesting a critical role of CD47 blockade in anti–CTLA-4×SIRPα treatment. Because CD47 is ubiquitously expressed on both tumor and host cells, we decided to explore which cell population expressing CD47 is essential in anti–CTLA-4×SIRPα-mediated antitumor effect. We first tested Cd47−/− MC38 tumor cells (fig. S5A) (38). Cd47−/− tumor cells did not grow as aggressively as the parental MC38 tumor cells. Nonetheless, Combo treatment did not slow down tumor growth, whereas anti–CTLA-4×SIRPα treatment reduced mouse tumor burden (Fig. 5A), indicating that expression of CD47 on tumor cells is dispensable for antitumor responses to anti–CTLA-4×SIRPα treatment. We then compared antitumor effects of anti–CTLA-4×SIRPα in WT versus Cd47−/− mice and found that anti–CTLA-4×SIRPα treatment retained antitumor effects in WT mice but lost its efficacy in Cd47−/− mice (Fig. 5B). These data suggest that host cell expression of CD47 is required in anti–CTLA-4×SIRPα treatment–induced antitumor effects. To further dissect whether T cell–intrinsic or T cell–extrinsic expression of CD47 is essential, we transferred WT or Cd47−/− T cells into recipient Rag1−/− mice, inoculated MC38 tumor cells 1 day later, and treated the mice with anti–CTLA-4×SIRPα after tumor establishment. Tumors grew in a similar rate between WT and Cd47−/− T cell recipients. Anti–CTLA-4×SIRPα treatment reduced tumor burden in WT T cell recipients. In contrast, no tumor control benefit was observed in Cd47−/− T cell recipients (Fig. 5C). To further demonstrate that CD47 expression on Treg cells is indispensable for efficacy of anti–CTLA-4×SIRPα therapy, we transferred a combination of WT Treg or Cd47−/− Treg cells plus Treg-depleted WT CD4+ and CD8+ T cells to Rag−/− mice. We then inoculated mice with MC38 tumors and treated the mice with anti–CTLA-4×SIRPα (fig. S5B). We found that anti–CTLA-4×SIRPα treatment slowed tumor growth in WT Treg cell recipient group but had no impact in Cd47−/− Treg cell recipient group (fig. S5, C and D). These results demonstrated that Treg cell–intrinsic expression of CD47 is essential for anti–CTLA-4×SIRPα treatment–induced antitumor responses. These data further implied that the absence of do not eat me signal is not sufficient on its own, and an additional eat me signal is required. To make a side-by-side comparison of tumor-CD47 targeting versus intratumoral Treg cell–CD47 targeting approaches, we generated anti–epidermal growth factor receptor (EGFR)×SIRPα that targets tumor cell–expressed CD47 with a similar heterodimer structure. Using EGFR-overexpressing MC38 tumors (39), we showed that anti-EGFR×SIRPα could not control tumor growth as compared to anti–CTLA-4×SIRPα (Fig. 5D). These findings supported our hypothesis and suggested that the modulation of eat me and do not eat me signals on Treg cells is more effective than that on tumor cells.
Fig. 5. CD47 expression on Treg cells is essential for anti–CTLA-4×SIRPα-mediated control of tumor burden.
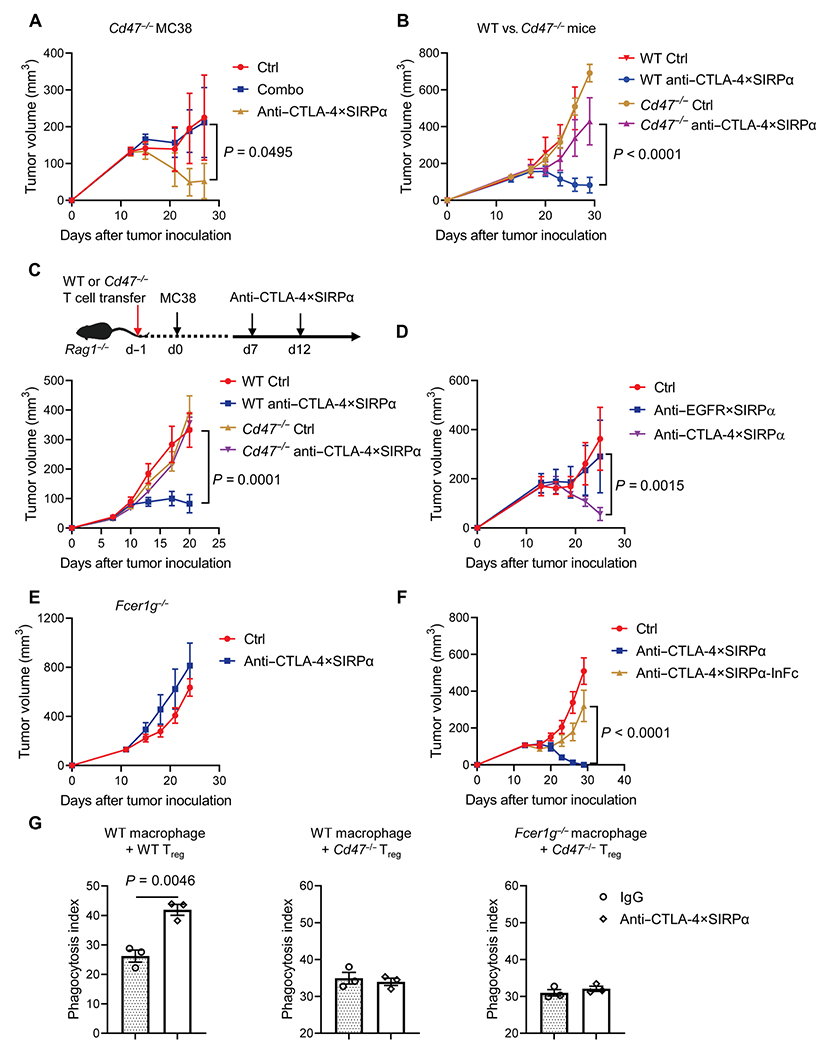
(A) Cd47−/− tumor-bearing C57BL/6 mice were treated with Combo or anti–CTLA-4×SIRPα on day 13. Ctrl mice were treated with hIgG. Tumor growth was measured every 3 days (n = 5 mice per group). (B) WT or Cd47−/− C57BL/6 mice were inoculated with 5 × 105 MC38 tumor cells and treated with anti–CTLA-4×SIRPα on day 14. Ctrl mice were treated with hIgG. Tumor growth was measured every 3 days (n = 5 mice per group). (C) WT or Cd47−/− T cells were purified and intravenously transferred to Rag1−/− mice. A total of 2 × 105 MC38 tumor cells were inoculated into the recipient mice the next day. Mice were treated with 40 μg of anti–CTLA-4×SIRPα on days 7 and 12. Ctrl mice were treated with hIgG. Experiment scheme is shown in the top, and tumor growth is shown in the bottom (n = 5 mice per group). (D) C57BL/6 mice were inoculated with 5 × 105 MC38-cEGFR tumor cells and treated with 30 μg of anti-EGFR×SIRPα or anti–CTLA-4×SIRPα on day 14. Ctrl mice were treated with hIgG. Tumor growth was measured every 3 days (n = 3 to 4 mice per group). (E) Fcer1g−/− C57BL/6 mice were inoculated with 1 × 105 MC38 tumor cells and treated with anti–CTLA-4×SIRPα on day 13. Ctrl mice were treated with hIgG. Tumor growth was measured every 3 days (n = 5 mice per group). (F) WT C57BL/6 mice were inoculated with 5 × 105 MC38 tumor cells and treated with anti–CTLA-4×SIRPα or anti–CTLA-4×SIRPα with mutant Fc (anti–CTLA-4×SIRPα-InFc) on day 14. Ctrl mice were treated with hIgG. Tumor growth was measured every 3 days (n = 5 mice per group). (G) BM–derived macrophages from WT or Fcer1g−/− mice were mixed with CFSE-labeled in vitro differentiated Treg cells from WT or Cd47−/− mice for 3 hours. Phagocytosis was determined by flow cytometry. Phagocytosis index was defined as the percentage of CFSE+ macrophages among total macrophages (n = 3 replicates per group). Data are shown as means ± SEM from two to three independent experiments. P values were determined by two-way ANOVA with Geisser-Greenhouse’s correction (A to D and F) or unpaired t test (G). The normality of data was confirmed by Shapiro-Wilk test.
We further investigated whether an Fc-dependent eat me signal is involved in anti–CTLA-4×SIRPα treatment. We found that anti–CTLA-4×SIRPα treatment was not able to control tumor growth in Fcer1g−/− mice, which lack the common γ chain of FcRs (40) and thus are void of antibody-dependent cellular phagocytosis (ADCP) function (Fig. 5E). To exclude the potential bias of genetic deficiency mice, we made an “LALA-PG” mutant of the Fc region of anti–CTLA-4×SIRPα (anti–CTLA-4×SIRPα-InFc) to silence the heterodimer’s ADCP function (41). anti–CTLA-4×SIRPα-InFc did not exhibit an antitumor effect as compared to treatment with the original anti–CTLA-4×SIRPα (Fig. 5F). These results illustrated that an Fc-dependent effect is essential in anti–CTLA-4×SIRPα treatment. To directly evaluate the contribution of eat me and do not eat me signals, we performed phagocytosis assay with in vitro differentiated Treg cells and bone marrow (BM)–derived macrophages. anti–CTLA-4×SIRPα increased phagocytosis in WT macrophages cocultured with WT Treg cells but not with Cd47−/− Treg cells (Fig. 5G and fig. S5E). Together, both CD47 expression on Treg cells and host FcR engagement are required to modulate the eat me and do not eat me signals that coordinately mediate the effects of anti–CTLA-4×SIRPα treatment.
A humanized version of anti–CTLA-4×SIRPα heterodimer depletes Treg cells in a humanized mouse tumor model
To study whether our findings in mouse tumors reflect a similar scenario in human tumor settings, we confirmed the CD47 and CTLA-4 expressions on Treg cells of tumor tissues versus PBMCs in patients with NSCLC. Consistent with single-cell sequencing data (Fig. 1A and fig. S1, A to C), the CTLA-4 expression abundance was higher on tumor Treg cells compared with PBMC Treg cells isolated from patients. CD47 expression was slightly reduced on tumor Treg cells, which might make Treg cells more susceptible to CD47 blockade if properly targeted (Fig. 6, A and B). A similar coexpression of CD47 and CTLA-4 was observed in tumor tissues from a patient with NSCLC by immunohistochemical staining (fig. S6A). We then made a humanized version of anti–CTLA-4×SIRPα heterodimer (anti–hCTLA-4×SIRPα) and examined its binding to the Jurkat leukemia cell line, which expresses CD47 constituently, and to engineered CTLA-4–expressing Jurkat cells, which express both CD47 and CTLA-4. We observed that SIRPα bound to both Jurkat variants similarly. In comparison, anti–hCTLA-4×SIRPα bound to both cell lines, but binding was associated with a much higher median fluorescence intensity for CTLA-4–expressing Jurkat cells (Fig. 6, C and D), suggesting a potential targeting role of anti–hCTLA-4×SIRPα for CTLA-4–expressing cells in vivo.
Fig. 6. A humanized version of anti–CTLA-4×SIRPα heterodimer depletes Treg cells in a humanized mouse tumor model.
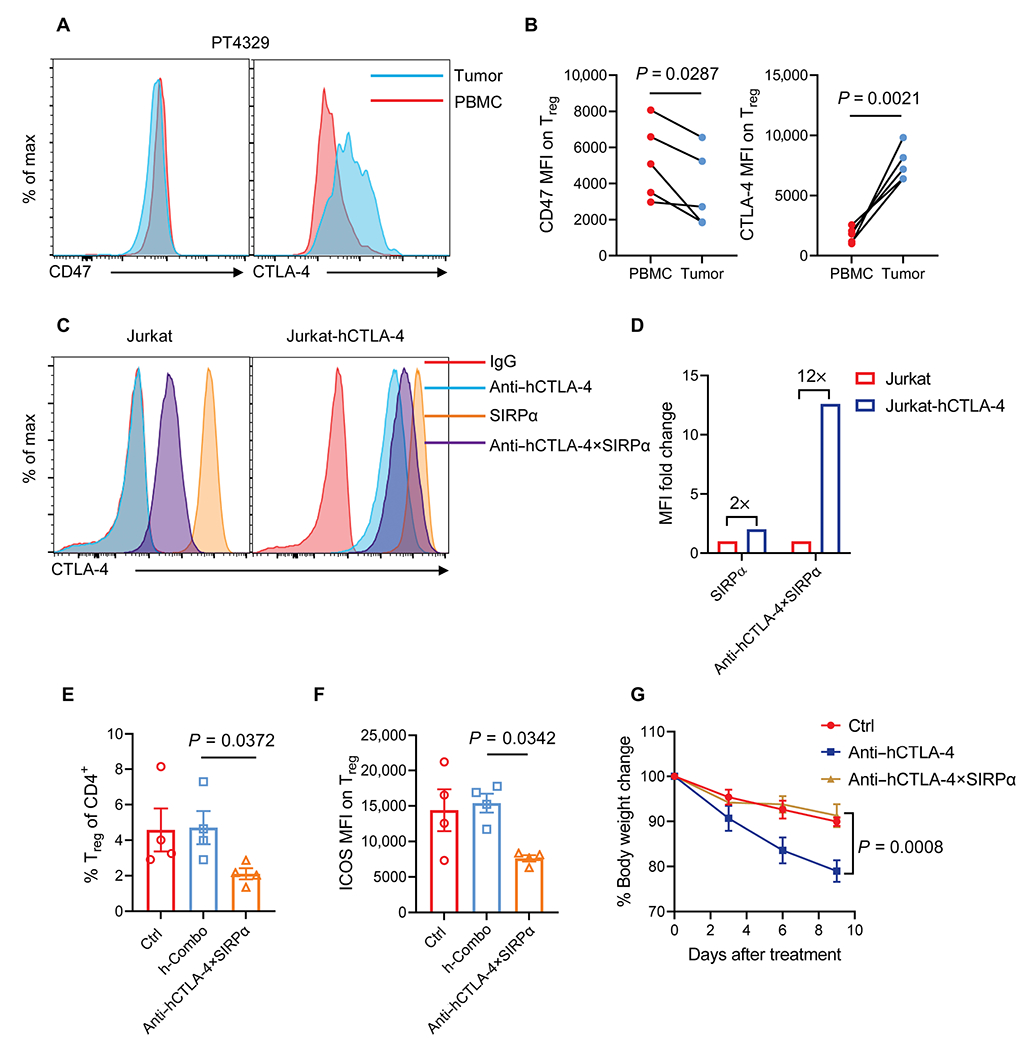
(A and B) Representative flow cytometry histograms (A) and quantification pooled from different patients (B) show CD47 and CTLA-4 expression on Treg cells of PBMCs and tumor tissues from patients with NSCLC. (C and D) Representative flow cytometry histogram (C) shows anti–hCTLA-4, SIRPα, and anti–hCTLA-4×SIRPα binding on Jurkat and Jurkat-hCTLA-4–expressing cells. Anti–hCTLA-4×SIRPα binding on Jurkat and Jurkat-hCTLA-4 cells was quantified on the basis of MFI (D). (E and F) PBMC-humanized NSG mice were inoculated with 2.5 × 106 A549 tumor cells and treated with anti–hCTLA-4 plus SIRPα (h-Combo, 18 + 12 μg) or anti–hCTLA-4×SIRPα (30 μg) on day 12. Ctrl mice were treated with hIgG. Two days later, the frequency of tumor-infiltrating Treg cells of total CD4 T cells (E) and ICOS expression on Treg cells (F) were analyzed (n = 4 mice per group). (G) PBMC-humanized NSG mice were treated with 200 μg of anti–hCTLA-4 or anti–hCTLA-4×SIRPα twice a week for four times. Ctrl mice were treated with hIgG. Mouse body weight was monitored every 3 days to measure evidence of systemic toxicity (n = 5 mice per group). Data are shown as means ± SEM from two independent experiments. P values were determined by paired t test (B), Kruskal-Wallis test with Dunn’s comparison test (E), one-way ANOVA with Tukey’s multiple comparisons test (F), or two-way ANOVA with Geisser-Greenhouse’s correction (G). The normality of data was evaluated by Shapiro-Wilk test.
To demonstrate that anti–hCTLA-4×SIRPα efficiently depletes Treg cells with dual targeting of CD47 and CTLA-4 in human tumor setting in the presence of T cells, we used the human PBMC reconstituted nonobese diabetic (NOD) scid gamma (NSG) mouse model that allowed the rapid expansion of human T cells including Treg cells. We inoculated NSG mice with A549 lung cancer cells subcutaneously and treated with anti–hCTLA-4×SIRPα or Combo on day 12, after tumors were well established. An analysis of the TME showed a reduction of Treg cells inside tumors by anti–hCTLA-4×SIRPα compared to Combo, although Treg cells were less prevalent in human PBMC–repopulated mice than in syngeneic mouse tumors (Fig. 6E). We also observed a reduction of ICOS expression density on Treg cells after anti–hCTLA-4×SIRPα treatment, which recapitulated our findings in syngeneic mouse tumors (Fig. 6F). Last, we evaluated the toxicity of anti–hCTLA-4×SIRPα—the key issue for anti–CTLA-4 treatment in the clinic (12). Although we markedly reduced the dose with enhanced tumor Treg cell targeting, high-dose treatment might still be toxic. To address this, we compared the potential side effects of high-dose and prolonged treatments in the human PBMC–repopulated mouse model. With control treatment, we observed a gradual body weight loss likely due to graft-versus-host disease (GVHD). As expected, anti–CTLA-4 treatment increased mouse body weight loss, whereas the anti–hCTLA-4×SIRPα-treated group showed no difference compared to the control-treated group (Fig. 6G). Similarly, we observed no toxicity toward RBCs after high-dose anti–CTLA-4×SIRPα treatment, whereas high-dose anti-CD47 antibodies depleted RBCs and reduced hemoglobin concentration and hematocrit ratios (fig. S6, B to D). Therefore, anti–hCTLA-4×SIRPα treatment had efficient Treg cell depletion but no obvious toxicity in humanized mouse models in vivo, suggesting that it may be a promising approach for tumor Treg cell depletion in humans.
Together, these results show that humanized anti–CTLA-4×SIRPα heterodimer preferentially blocks CD47 on CTLA-4high tumor-infiltrating Treg cells and spares peripheral cells like RBCs and Treg cells that have low CTLA-4 expression. The concurrent blocking of the do not eat me signal and enhancing of the eat me signal efficiently deplete Treg cells in an Fc-dependent manner (fig. S6E). This suggests that dual targeting of CD47 and CTLA-4 on Treg cells can be a safe and effective approach in cancer immunotherapy.
DISCUSSION
CD47 therapeutic targeting has been recently tested in preclinical models and clinical trials with some promising results for B cell lymphoma when combined with tumor-targeting antibodies (42). Nonetheless, it has been challenging to solve the problems related to CD47 antigen sink and undetectable therapeutic activity in solid tumors. Here, we report a rational design of anti–CTLA-4×SIRPα heterodimer that takes advantage of the high expression of CTLA-4 on tumor Treg cells to selectively deliver SIRPα to block CD47 on tumor Treg cells. This design allows concurrent blockade of the do not eat me signal by SIRPα and enhancement of the eat me signal by anti–CTLA-4 on Treg cells. This heterodimer treatment results in efficient tumor Treg cell depletion, particularly depletion of the more immunosuppressive ICOShigh Treg cells, using a relatively low-dose anti–CTLA-4×SIRPα. We also showed that both CD47 expression on Treg cells and host FcR engagement are required for anti–CTLA-4×SIRPα-mediated antitumor effects, substantiating our initial hypothesis for the antitumor mechanism of action of anti–CTLA-4×SIRPα. These antitumor effects of anti–CTLA-4×SIRPα show that leveraging innate checkpoints on adaptive immune regulatory cells may be effective approaches for enhancing adaptive antitumor immunity.
Tremendous efforts have been made to overcome the antigen sink problem for CD47 blockade, including selecting non-RBC binding anti-CD47 antibody or using a low-affinity ligand (43–45). Nonetheless, the ubiquitous expression pattern of CD47 on somatic cells is still problematic, so targeted delivery of a CD47 blocker to the tumor and bypassing the normal tissue is a preferred yet challenging approach. Our data showed that tumor-infiltrating Treg cell–targeted delivery with anti–CTLA-4 in the heterodimer is effective. One reason is the high expression of CTLA-4 on tumor Treg cells, but not on other cell populations, making it an ideal candidate for target delivery. The other reason is that CTLA-4 blockade can induce a strong eat me signal, making Treg cells more susceptible to Fc-dependent depletion when combined with blocking do not eat me signal. Ongoing clinical trials also show that when combined with additional eat me signals such as the anti-CD20 antibody, rituximab, treatment with anti-CD47 antibody can have clinical benefits (42). In our study, anti–CTLA-4×SIRPα is more potent than the simple combination therapy with the two proteins, implying that there are still improvement possibilities for clinical translation. Last, compared with tumor cell–targeted strategies that tend to use high doses (46, 47), the delivery of SIRPα to Treg cells instead of tumor cells reduces the effective dose needed for an antitumor response. These data also suggest a central role for Treg cell depletion in reversing inhibition of adaptive antitumor immune responses.
In a previous study, Ingram et al. (48) tried to block CD47 on tumor Treg cells to make them more susceptible to antibody-mediated cellular phagocytosis. They combined a high-affinity CD47 nanobody (A4) with anti–CTLA-4 antibodies but did not observe synergy in the B16F10 melanoma model. Systemic delivery of A4 could not overcome the CD47 antigen sink even with high doses (48). The anti–CTLA-4×SIRPα heterodimer presented here has different features when compared with the A4 nanobody. One feature of anti–CTLA-4×SIRPα heterodimer is the reduction of affinity of each arm for their targets but not overall avidity. This is critical to reduce the peripheral binding to CD47 in nondesirable targets, such as RBCs and platelets, that do not express CTLA-4 and to enhance the stable binding to the desirable targets, tumor Treg cells. The relatively higher affinity for CTLA-4 versus CD47 is also crucial, because this directs anti–CTLA-4×SIRPα to target to CTLA-4 high-expressing cells, which are predominantly Treg cells in the TME. Another feature is the preservation of Fc activity. FcR-positive cells such as myeloid cells and natural killer cells are abundant inside the TME. Fc-FcR engagement–mediated eat me signaling is strongly induced by therapeutic antibodies but is restrained by the presence of the do not eat me signal mediated by CD47. Our employment of SIRPα to block the do not eat me signal instead of adding a second eat me signal (a current popular strategy for bispecific antibodies) turns out to be an effective strategy. Anti–CTLA-4×SIRPα heterodimer sensitizes Fc-dependent eat me signal and leads to efficient Treg cell depletion at low treatment doses. In particular, the synergy of eat me signal and do not eat me signals requires a close physical interaction of the two molecules, because the combination of anti–CTLA-4 and SIRPα delivered separately at equivalent doses fails to do so. As a result, FcR-expressing phagocytic cells are primarily responsible for depleting intratumoral Treg cells. The deletion of Treg cells diminished immune suppression and expanded tumor-specific conventional T cells, which are responsible for tumor control.
Many studies have linked Treg cell abundance in tumors with poor prognosis in clinical outcome (2, 49). Treg cells inhibit effector T cell functions through multiple mechanisms. Recently, an interesting study shows that even apoptotic intratumoral Treg cells dampen checkpoint blockade immunotherapy through the production of immunosuppressive adenosine (50). Therefore, efficient Treg cell depletion is critical for unleashing antitumor immunity. However, therapeutic manipulation of overall Treg cells must be approached cautiously, given the potential for systemic toxicity. How to precisely target tumor Treg cells and bypass depletion in peripheral tissues is of high priority in Treg cell–targeted therapy. Our anti–CTLA-4×SIRPα addresses this key issue by (i) using single-arm anti–CTLA-4 that reduces antibody affinity to Treg cells in the periphery but retains the ability to target to tumor Treg cells due to the high CTLA-4 density expressed on the latter and (ii) reducing the dose for anti–CTLA-4 through pairing with SIRPα, which retains the tumor Treg cell depletion capacity by the additional blockade of this do not eat me signal. We have shown a safer profile for anti–CTLA-4×SIRPα when compared with anti–CTLA-4 antibodies. This suggests a wide therapeutic window for clinical translation.
We also observed similar expression patterns of CTLA-4 and CD47 on Treg cells in patients with cancer. We also demonstrated using a human version of anti–CTLA-4×SIRPα heterodimer that a relatively low-dose treatment could efficiently deplete human Treg cells in a humanized mouse tumor model. Looking forward, it is intriguing to consider using anti–CTLA-4×SIRPα as a tumor Treg cell depletion candidate that may be combined with programmed cell death protein 1(PD-1)/PD-ligand 1 (PD-L1) checkpoint blockade, because current ipilimumab (which targets CTLA-4) plus nivolumab (which targets PD-1) combination therapy clearly benefits patients with cancer but is limited by more aggravated toxicity (51–53).
Although our study shows that anti–CTLA-4×SIRPα can be a promising approach for immunotherapy, there are several limitations in the current study. First, in cold tumor where immune cell infiltration is limited, targeting intratumoral Treg depletion may not be an effective approach. A proper combination therapy is yet to be explored. Second, we are not able to evaluate the antitumor effects because of the short experimental window before GVHD develops in the PBMC-humanized mouse model. The translation of our findings in mouse model into the clinic remains to be further explored. Whether the anti–CTLA-4×SIRPα heterodimer can be produced in large-scale manufacture remains to be determined.
Overall, our findings demonstrate that dual targeting of CTLA-4 and CD47 on tumor Treg cells with the anti–CTLA-4×SIRPα heterodimer is a potent strategy to selectively deplete intratumoral Treg cells. This is accomplished by simultaneously enhancing an eat me signal and blocking a do not eat me signal. These results highlight the idea that leveraging innate checkpoints on adaptive immune regulatory cells may be a strategy for developing effective and safe immunotherapeutic approaches against solid tumors.
MATERIALS AND METHODS
Study design
The aim of this study was to test the antitumor effect and dissect the mechanism of action of an anti–CTLA-4×SIRPα heterodimer that selectively blocks CD47 on intratumoral Treg cells. We produced different bispecific proteins with a FreeStyle 293-F expression system as comparison. We used different tumor models and genetically modified mouse strains to test the antitumor effect of anti–CTLA-4×SIRPα. We also validated preclinical findings with samples from patients with NSCLC and publicly available datasets. Sample size was chosen empirically on the basis of results of previous studies. In general, experiments aimed to include three to five mice per group and were repeated two to three times with precise numbers for each individual experiment provided in the figure legends. Mice were randomly assigned to different treatment groups. Investigators were not blinded. No data outliers were excluded. Human NSCLC samples and PBMCs were obtained from patients at the University of Texas (UT) Southwestern Medical Center with informed consent, according to an Institutional Review Boards (IRB)–approved protocol (IRB no. STU 102010-51).
Mice
Female C57BL/6J, BALB/cJ, Ifng−/−, Cd47−/−, Fcer1g−/−, muMT and Foxp3EGFP-cre transgenic mice, and NOD scid gamma mice were purchased from the Jackson laboratory. Rag1−/− mice on C57BL/6 background were purchased from the UT Southwestern mice breeding core. All mice were maintained under specific pathogen–free conditions. Animal care and experiments were carried out under institutional and National Institutes of Health protocol and guidelines. This study has been approved by the Institutional Animal Care and Use Committee of the UT Southwestern Medical Center.
Cell lines and reagents
MC38, CT26, LLC, Jurkat, and A549 cells were purchased from the American Type Culture Collection. Jurkat-CTLA-4 cells were purchased from Promega. Cd47−/− MC38 and MC38-cEGFR tumor cells were generated in our laboratory (38, 39). All cell lines were routinely tested using a mycoplasma contamination kit (R&D Systems). Jurkat cell was cultured in RPMI 1640 medium, and other cell lines were cultured in Dulbecco’s modified Eagle’s medium. All media were supplemented with 10% heat-inactivated fetal bovine serum (FBS), penicillin (100 U/ml), and streptomycin (100 U/ml). β-Mercaptoethanol (55 μM) was added to Jurkat cell culture medium. All cells were cultivated at 37°C in a humidified atmosphere containing 5% CO2.
Anti-CD4 (GK1.5), anti-CD8 (53-5.8), and anti-IFN-γ (XMG1.2) monoclonal antibodies were purchased from Bio X Cell. Anti–CTLA-4 (ipilimumab) was provided by the UT Southwestern Simmons Cancer Center Pharmacy.
Production of anti–CTLA-4×SIRPα heterodimer
Light chain or heavy chain with hIgG1-Fc genes of anti–CTLA-4 antibody (clone 4F10) was separately cloned into pEE6.4 (Lonza). The extracellular domain of SIRPα gene was fused with hIgG1-Fc with 3×GGGGS linker and cloned into pEE6.4. Heterodimerization of anti–CTLA-4 and SIRPα-Fc was achieved by the knobs-into-holes approach (31, 32). Anti–CTLA-4×SIRPα-InFc was generated on the basis of the LALA-PG mutation in Fc region as previously described (41). Plasmids encoding anti–CTLA-4 light chain, heavy chain, and SIRPα-Fc were cotransfected into FreeStyle 293-F cells (Thermo Fisher Scientific) at a ratio of 1:2:1. The supernatant containing fusion proteins was purified with Protein A affinity chromatography (Repligen) according to the manufacturer’s instructions. Anti–CTLA-4 antibody, SIRPα-Fc, anti–CTLA-4×SIRPα-InFc, anti–CTLA-4(9D9)×SIRPα, and anti-EGFR×SIRPα were purified by the same procedure. Anti-human CTLA-4 (ipilimumab)×SIRPα heterodimer was generated with a similar strategy.
Tumor growth and treatment
A total of 5 × 105 MC38 (or Cd47−/− MC38) or 5 × 105 CT26 cells were subcutaneously inoculated into the right dorsal flanks of mice in 100 μl of phosphate-buffered saline (PBS). Tumor-bearing mice were randomly grouped into different treatment groups when tumors grew to 80 to 150 mm3. For MC38 tumor, 20 μg of anti–CTLA-4×SIRPα or molar equivalents of anti–CTLA-4 plus SIRPα were intraperitoneally administrated once on about day 13 (days 12 to 14). For CT26 tumor, 50 μg of anti–CTLA-4×SIRPα or molar equivalents of anti–CTLA-4 plus SIRPα were intraperitoneally administrated once on day 6. For depletion or neutralization experiments, 200 μg of CD4+ and CD8+ T cell depletion antibodies or 150 μg of IFN-γ blockade antibodies was intraperitoneally injected in 200 μl of PBS 1 day before treatment initiation and then twice a week for 2 weeks. For the rechallenge study, 5 × 106 MC38 or 5 × 105 LLC tumors were subcutaneously injected into the left (MC38) or right (LLC) dorsal flanks. Tumor volumes were measured by the length (a), width (b), and height (h) and calculated as tumor volume = abh/2.
PBMC-humanized mouse tumor model
A total of 1 × 107 PBMCs (Ancell) were intravenously injected into recipient NSG mouse. One week later, mice were bled to confirm the human cell reconstitution, and then 2.5 × 106 A549 tumor cells were subcutaneously inoculated onto the right flank. Thirty micrograms of anti–CTLA-4×SIRPα or molar equivalents of anti–CTLA-4 plus SIRPα were intraperitoneally administrated on day 12. Mice were sacrificed for Treg cell depletion analysis 48 hours later. Alternatively, 200 μg of anti–CTLA-4×SIRPα or anti–CTLA-4 was intraperitoneally administrated twice a week for a total of four times. Mouse body weights were monitored every 3 days until severe GVHD developed.
Adoptive transfer of T cells
T cells from WT or Cd47−/− C57BL/6 mice were purified with negative selection beads (STEMCELL), and then 2.5 × 106 T cells in 100 μl of PBS were intravenously transferred into recipient Rag1−/− mice by retro-orbital injection. A total of 2 × 105 MC38 tumor cells were inoculated into mice 1 day later. Mice were randomly grouped and treated with 30 μg of anti–CTLA-4×SIRPα on day 7.
Tissue homogenate preparation
For preparing tumor single-cell suspension, tumor tissues were homogenized with tissue dissociator (Miltenyi) and digested with collagenase I (1 mg/ml; Sigma-Aldrich) and deoxyribonuclease I (0.5 mg/ml; Roche) and gently incubated in a shaker for 30 min at 37°C. Tumor was then passed through a 70-μm cell strainer to remove large pieces of undigested tumor. Tumor-infiltrating cells were washed twice with flow cytometry staining [fluorescence-activated cell sorting (FACS)] buffer (PBS containing 2% FBS) and resuspended for further use.
For preparing tissue for lysis, anti–CTLA-4×SIRPα–treated mice were perfused with PBS, and then hearts, kidneys, livers, and tumors were harvested and homogenized in cell lysis buffer containing protease and phosphatase inhibitor cocktails (Thermo Fisher Scientific) with the FastPrep-24 5G homogenizer (MP Biomedicals). Homogenized tissues were centrifuged for 20 min at 12,000 rpm, and supernatant was collected and stored at −80°C for further use.
Flow cytometry analysis
Single-cell suspensions of cells were incubated with 1:100 dilution of anti-FcγIII/I receptor (clone 2.4G2, BD Biosciences) in FACS buffer for 15 min to block the nonspecific binding before staining with the conjugated antibodies and then incubated with indicated antibodies at 1:200 dilution in FACS buffer for 30 min at 4°C in the dark. Fixable viability dye eFlour780 (eBioscience) was used at a 1:1000 dilution in PBS to exclude the dead cells. Detailed flow cytometry antibodies are listed in table S1. H-2Kb MuLV p15E Tetramer-KSPWFTTL-PE was purchased from MBL. Foxp3 and Ki67 were intracellularly stained with True-Nuclear transcription factor buffer set (BioLegend) following the manufacturer’s instructions. For anti–CTLA-4×SIRPα or anti–CTLA-4 or SIRPα binding in vitro, 0.5 μg of proteins was first stained and washed, and then PE anti-human IgG (Jackson ImmunoResearch) was used as a secondary antibody and diluted at 1:200 in FACS buffer. Data were collected on a CytoFLEX flow cytometer (Beckman Coulter) and analyzed using FlowJo (Tree Star) software.
IFN-γ enzyme-linked immunosorbent spot assay
MC38 tumors were subcutaneously injected on the right flank of C57BL/6 mice, and 20 μg of anti–CTLA-4×SIRPα or molar equivalents of anti–CTLA-4 plus SIRPα were intraperitoneally administrated on day 13. Five days after treatment, tumors and dLN were collected. Tumors were processed as described in the “Tissue homogenate preparation” section. Tumor-infiltrating immune cells were isolated by biotinylated anti-CD45 antibody and EasySep Biotin Positive Selection Kit II (STEMCELL) according to the manufacturer’s instructions. Irradiated MC38 tumor cells and control LLC tumor cells were used to restimulate tumor-specific T cells. A total of 1.5 × 105 dLN cells or 2 × 104 TILs and 7.5 × 104 irradiated tumor cells (40 grays) were cocultured for 48 hours, and enzyme-linked immunosorbent spot assay (ELISPOT) was performed using the IFN-γ ELISPOT kit (BD Biosciences) according to the manufacturer’s instructions. IFN-γ spots were enumerated with the CTL-ImmunoSpot S6 Analyzer (Cellular Technology Limited).
Enzyme-linked immunosorbent assay
Microtiter plates (Corning Costar) were coated with capture antibody (2 μg/ml; AffiniPure goat anti-human IgG, Jackson ImmunoResearch) overnight at 4°C. After washing and blocking, diluted tissue lysates from anti–CTLA-4×SIRPα-treated mice were added and incubated at 37°C for 1 hour. After washing, alkaline phosphatase–conjugated goat anti-human IgG (H + L) (Jackson ImmunoResearch) was added at 1:2000 and incubated at 37°C for 30 min. Last, the plates were visualized using phosphatase substrate system (SeraCare) following the manufacturer’s instructions and read at 405 nm using the SPECTROstar Nano (BMG LABTECH).
In vitro phagocytosis assay
To obtain BM-derived macrophages, single-cell suspensions of BM cells were collected from tibias and femurs of C57BL/6 mice. The BM cells were placed in a 10-cm dish and cultured with complete RPMI 1640 medium containing 30% supernatant collected from macrophage colony-stimulating factor–secreting L929 cells. Fresh medium was changed on days 3 and 6. Macrophages were harvested on day 7.
To obtain Treg cells, CD4+ T cells were isolated from lymph nodes and spleens of C57BL/6 mice with a negative CD4+ T cell isolation kit (STEMCELL) and cultured in complete RPMI 1640 medium supplemented with transforming growth factor-β (20 ng/ml; BioLegend) and IL-2 (20 ng/ml; Prometheus) in a 24-well plate coated with anti-CD3/anti-CD28 antibodies (5 μg/ml). After 3 days, cells were moved to another well and changed to medium containing IL-2 (20 ng/ml). After 6 days, cells were collected and labeled with CFSE (Sigma-Aldrich). A total of 10 μg/ml of anti–CTLA-4×SIRPα or control hIgG was added to CFSE-labeled Treg cells that were seeded in a 96-well round-bottom plate and kept on ice for 30 min. Macrophages were then added to Treg cells at a 1:1 ratio and incubated at 37°C for 3 hours. Cells were washed and analyzed by flow cytometry.
Surface plasmon resonance
The kinetics of anti–CTLA-4×SIRPα binding to recombinant CTLA-4 and CD47 antigen were determined by SPR-based measurements with the Open SPR instrument (Nicoya). The ligand (recombinant CTLA-4 or CD47, Sino Biological) was immobilized to a carboxyl sensor chip (Nicoya). The test proteins were diluted in PBS and slowly flowed over the sensor chip for a 5-min interaction time. SPR data were analyzed using TraceDrawer software (Nicoya) assuming a one-to-one binding mode.
Quantification and statistical analysis
All the data analyses were performed with GraphPad Prism statistical software and shown as means ± SEM. P value was determined by two-way analysis of variance (ANOVA) with Geisser-Greenhouse’s correction for tumor growth curve and mouse body weight change with time. One-way ANOVA with Tukey’s multiple comparisons test or Kruskal-Wallis test with Dunn’s comparison test or paired or unpaired two-tailed t tests were used for other analysis. The normality of data was evaluated by Shapiro-Wilk test with a P value of >0.05. A value of P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments:
We acknowledge the support of the UT Southwestern Medical Center Animal Research Center. We thank the UT Southwestern Simmons Cancer Center Pharmacy for providing clinical reagents. We thank J. Xie for providing technical help. We also thank all the members of Fu laboratory for sharing experiment materials and helpful discussions.
Funding:
This work was, in part, supported by Texas CPRIT grants RR150072 (to Y.-X.F.) and RP180725 (to Y.-X.F.) and NCI SPORE P50CA070907 (to J.D.M.). Y.-X.F. holds the Mary Nell and Ralph B. Rogers Professorship in Immunology. Patient tissues were provided by Tissue Management Shared Resource, which was supported, in part, by a National Cancer Institute Cancer Center Support grant (P30CA14142543).
Footnotes
Competing interests: Y.-X.F. is a consulting adviser for Aetio Biotherapy, and K.-F.T. is an employee of Aetio Biotherapy. K.-F.T. has a pending patent related to this work (Bi-Specific Fusion Proteins for Depletion of Regulatory T Cells; file no. AEBI:2003WO). All other authors declare that they have no competing interests.
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. Reagents derived from this work can be requested from Y.-X.F. (Yang-Xin.Fu@UTSouthwestern.edu) and will be shared with the scientific community via a material transfer agreement.
REFERENCES AND NOTES
- 1.Plitas G, Rudensky AY, Regulatory T cells in cancer. Annu. Rev. Cancer Biol 4, 459–477 (2020). [Google Scholar]
- 2.Fu J, Xu D, Liu Z, Shi M, Zhao P, Fu B, Zhang Z, Yang H, Zhang H, Zhou C, Yao J, Jin L, Wang H, Yang Y, Fu YX, Wang FS, Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology 132, 2328–2339 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E, Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor α) monoclonal antibody. Cancer Res. 59, 3128–3133 (1999). [PubMed] [Google Scholar]
- 4.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W, Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med 10, 942–949 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Shen Z, Zhou S, Wang Y, Li RL, Zhong C, Liang C, Sun Y, Higher intratumoral infiltrated Foxp3+ Treg numbers and Foxp3+/CD8+ ratio are associated with adverse prognosis in resectable gastric cancer. J. Cancer Res. Clin. Oncol 136, 1585–1595 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quezada SA, Peggs KS, Curran MA, Allison JP, CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J. Clin. Invest 116, 1935–1945 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mahne AE, Mauze S, Joyce-Shaikh B, Xia J, Bowman EP, Beebe AM, Cua DJ, Jain R, Dual roles for regulatory T-cell depletion and costimulatory signaling in agonistic GITR targeting for tumor immunotherapy. Cancer Res. 77, 1108–1118 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Piconese S, Valzasina B, Colombo MP, OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J. Exp. Med 205, 825–839 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vargas FA, Furness AJS, Solomon I, Joshi K, Mekkaoui L, Lesko MH, Miranda Rota E, Dahan R, Georgiou A, Sledzinska A, Ben Aissa A, Franz D, Werner Sunderland M, Wong YNS, Henry JY, O’Brien T, Nicol D, Challacombe B, Beers SA; Melanoma TRACERx Consortium; Renal TRACERx Consortium; Lung TRACERx Consortium, Turajlic S, Gore M, Larkin J, Swanton C, Chester KA, Pule M, Ravetch JV, Marafioti T, Peggs KS, Quezada SA, Fc-optimized anti-CD25 depletes tumor-infiltrating regulatory T cells and synergizes with PD-1 blockade to eradicate established tumors. Immunity 46, 577–586 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ, Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med 363, 711–723 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Patt D, Chen TT, Berman DM, Wolchok JD, Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J. Clin. Oncol 33, 1889–1894 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weber JS, Dummer R, de Pril V, Lebbé C, Hodi FS; MDX010-20 Investigators, Patterns of onset and resolution of immune-related adverse events of special interest with ipilimumab: Detailed safety analysis from a phase 3 trial in patients with advanced melanoma. Cancer 119, 1675–1682 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Sosa A, Lopez Cadena E, Simon Olive C, Karachaliou N, Rosell R, Clinical assessment of immune-related adverse events. Ther. Adv. Med. Oncol 10, 1758835918764628 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA, Wilson NS, Dranoff G, Brogdon JL, Activating Fc γ receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J. Exp. Med 210, 1685–1693 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, Korman AJ, Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol. Res 1,32–42 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, Peggs KS, Ravetch JV, Allison JP, Quezada SA, Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med 210, 1695–1710 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, Solomon I, Lesko MH, Ruef N, Roddie C, Henry JY, Spain L, Aissa AB, Georgiou A, Wong YNS, Smith M, Strauss D, Hayes A, Nicol D, O’Brien T, Martensson L, Ljungars A, Teige I, Frendeus B; TRACERx Melanoma; TRACERx Renal; TRACERx Lung consortia, Pule M, Marafioti T, Gore M, Larkin J, Turajlic S, Swanton C, Peggs KS, Quezada SA, Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell 33, 649–663.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharma A, Subudhi SK, Blando J, Scutti J, Vence L, Wargo J, Allison JP, Ribas A, Sharma P, Anti-CTLA-4 immunotherapy does not deplete FOXP3+ regulatory T cells (Tregs) in human cancers. Clin. Cancer Res 25, 1233–1238 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barclay AN, Van den Berg TK, The interaction between signal regulatory protein alpha (SIRPα) and CD47: Structure, function, and therapeutic target. Annu. Rev. Immunol 32, 25–50 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD Jr., van Rooijen N, Weissman IL, CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 138, 286–299 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Willingham SB, Volkmer J-P, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, Wang J, Contreras-Trujillo H, Martin R, Cohen JD, Lovelace P, Scheeren FA, Chao MP, Weiskopf K, Tang C, Volkmer AK, Naik TJ, Storm TA, Mosley AR, Edris B, Schmid SM, Sun CK, Chua M-S, Murillo O, Rajendran P, Cha AC, Chin RK, Kim D, Adorno M, Raveh T, Tseng D, Jaiswal S, Enger PO, Steinberg GK, Li G, So SK, Majeti R, Harsh GR, van de Rijn M, Teng NN, Sunwoo JB, Alizadeh AA, Clarke MF, Weissman IL, The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U.S.A. 109, 6662–6667 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weiskopf K, Ring AM, Ho CC, Volkmer JP, Levin AM, Volkmer AK, Ozkan E, Fernhoff NB, van de Rijn M, Weissman IL, Garcia KC, Engineered SIRPα variants as immunotherapeutic adjuvants to anticancer antibodies. Science 341, 88–91 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chao MP, Alizadeh AA, Tang C, Myklebust JH, Varghese B, Gill S, Jan M, Cha AC, Chan CK, Tan BT, Park CY, Zhao F, Kohrt HE, Malumbres R, Briones J, Gascoyne RD, Lossos IS, Levy R, Weissman IL, Majeti R, Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell 142, 699–713 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao XW, van Beek EM, Schornagel K, Van der Maaden H, Van Houdt M, Otten MA, Finetti P, Van Egmond M, Matozaki T, Kraal G, Birnbaum D, van Elsas A, Kuijpers TW, Bertucci F, van den Berg TK, CD47–signal regulatory protein-α (SIRPα) interactions form a barrier for antibody-mediated tumor cell destruction. Proc. Natl. Acad. Sci. U.S.A 108, 18342–18347 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu X, Pu Y, Cron K, Deng L, Kline J, Frazier WA, Xu H, Peng H, Fu YX, Xu MM, CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat. Med 21, 1209–1215 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tseng D, Volkmer JP, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB, Seita J, Inlay MA, Weiskopf K, Miyanishi M, Weissman IL, Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc. Natl. Acad. Sci. U.S.A. 110, 11103–11108 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weiskopf K, Jahchan NS, Schnorr PJ, Cristea S, Ring AM, Maute RL, Volkmer AK, Volkmer JP, Liu J, Lim JS, Yang D, Seitz G, Nguyen T, Wu D, Jude K, Guerston H, Barkal A, Trapani F, George J, Poirier JT, Gardner EE, Miles LA, de Stanchina E, Lofgren SM, Vogel H, Winslow MM, Dive C, Thomas RK, Rudin CM, van de Rijn M, Majeti R, Garcia KC, Weissman IL, Sage J, CD47-blocking immunotherapies stimulate macrophage-mediated destruction of small-cell lung cancer. J. Clin. Invest 126, 2610–2620 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen J, Zhong MC, Guo H, Davidson D, Mishel S, Lu Y, Rhee I, Perez-Quintero LA, Zhang S, Cruz-Munoz ME, Wu N, Vinh DC, Sinha M, Calderon V, Lowell CA, Danska JS, Veillette A, SLAMF7 is critical for phagocytosis of haematopoietic tumour cells via Mac-1 integrin. Nature 544, 493–497 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, Gao R, Kang B, Zhang Q, Huang JY, Konno H, Guo X, Ye Y, Gao S, Wang S, Hu X, Ren X, Shen Z, Ouyang W, Zhang Z, Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 564, 268–272 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, Kang B, Liu Z, Jin L, Xing R, Gao R, Zhang L, Dong M, Hu X, Ren X, Kirchhoff D, Roider HG, Yan T, Zhang Z, Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med 24, 978–985 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Ridgway JB, Presta LG, Carter P, ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 9, 617–621 (1996). [DOI] [PubMed] [Google Scholar]
- 32.Wei H, Cai H, Jin Y, Wang P, Zhang Q, Lin Y, Wang W, Cheng J, Zeng N, Xu T, Zhou A, Structural basis of a novel heterodimeric Fc for bispecific antibody production. Oncotarget 8, 51037–51049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhong W, Myers JS, Wang F, Wang K, Lucas J, Rosfjord E, Lucas J, Hooper AT, Yang S, Lemon LA, Guffroy M, May C, Bienkowska JR, Rejto PA, Comparison of the molecular and cellular phenotypes of common mouse syngeneic models with human tumors. BMC Genomics 21, 2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haribhai D, Lin W, Relland LM, Truong N, Williams CB, Chatila TA, Regulatory T cells dynamically control the primary immune response to foreign antigen. J. Immunol 178, 2961–2972 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Sim GC, Martin-Orozco N, Jin L, Yang Y, Wu S, Washington E, Sanders D, Lacey C, Wang Y, Vence L, Hwu P, Radvanyi L, IL-2 therapy promotes suppressive ICOS+ Treg expansion in melanoma patients. J. Clin. Invest 124, 99–110 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Y, Shen S, Gorentla BK, Gao J, Zhong XP, Murine regulatory T cells contain hyperproliferative and death-prone subsets with differential ICOS expression. J. Immunol 188, 1698–1707 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nagase H, Takeoka T, Urakawa S, Morimoto-Okazawa A, Kawashima A, Iwahori K, Takiguchi S, Nishikawa H, Sato E, Sakaguchi S, Mori M, Doki Y, Wada H, ICOS+Foxp3+TILs in gastric cancer are prognostic markers and effector regulatory T cells associated with Helicobacter pylori. Int. J. Cancer 140, 686–695 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Liu X, Liu L, Ren Z, Yang K, Xu H, Luan Y, Fu K, Guo J, Peng H, Zhu M, Fu YX, Dual targeting of innate and adaptive checkpoints on tumor cells limits immune evasion. Cell Rep. 24, 2101–2111 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Qiao J, Liu Z, Dong C, Luan Y, Zhang A, Moore C, Fu K, Peng J, Wang Y, Ren Z, Han C, Xu T, Fu YX, Targeting tumors with IL-10 prevents dendritic cell-mediated CD8+ T cell apoptosis. Cancer Cell 35, 901–915.e4 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV, FcR gamma chain deletion results in pleiotrophic effector cell defects. Cell 76, 519–529 (1994). [DOI] [PubMed] [Google Scholar]
- 41.Schlothauer T, Herter S, Koller CF, Grau-Richards S, Steinhart V, Spick C, Kubbies M, Klein C, Umana P, Mossner E, Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Eng. Des. Sel. 29, 457–466 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Advani R, Flinn I, Popplewell L, Forero A, Bartlett NL, Ghosh N, Kline J, Roschewski M, LaCasce A, Collins GP, Tran T, Lynn J, Chen JY, Volkmer JP, Agoram B, Huang J, Majeti R, Weissman IL, Takimoto CH, Chao MP, Smith SM, CD47 blockade by Hu5F9-G4 and rituximab in non-Hodgkin’s lymphoma. N. Engl. J. Med 379, 1711–1721 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Puro RJ, Bouchlaka MN, Hiebsch RR, Capoccia BJ, Donio MJ, Manning PT, Frazier WA, Karr RW, Pereira DS, Development of AO-176, a next-generation humanized anti-CD47 antibody with novel anticancer properties and negligible red blood cell binding. Mol. Cancer Ther 19, 835–846 (2020). [DOI] [PubMed] [Google Scholar]
- 44.Ma L, Zhu M, Gai J, Li G, Chang Q, Qiao P, Cao L, Chen W, Zhang S, Wan Y, Preclinical development of a novel CD47 nanobody with less toxicity and enhanced anti-cancer therapeutic potential. J Nanobiotechnology 18, 12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Petrova PS, Viller NN, Wong M, Pang X, Lin GH, Dodge K, Chai V, Chen H, Lee V, House V, Vigo NT, Jin D, Mutukura T, Charbonneau M, Truong T, Viau S, Johnson LD, Linderoth E, Sievers EL, Vareki SM, Figueredo R, Pampillo M, Koropatnick J, Trudel S, Mbong N, Jin L, Wang JC, Uger RA, TTI-621 (SIRPαFc): A CD47-blocking innate immune checkpoint inhibitor with broad antitumor activity and minimal erythrocyte binding. Clin Cancer Res. 23, 1068–1079 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Piccione EC, Juarez S, Tseng S, Liu J, Stafford M, Narayanan C, Wang L, Weiskopf K, Majeti R, SIRPα-antibody fusion proteins selectively bind and eliminate dual antigen-expressing tumor cells. Clin. Cancer Res 22, 5109–5119 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Yang Y, Guo R, Chen Q, Liu Y, Zhang P, Zhang Z, Chen X, Wang T, A novel bispecific antibody fusion protein co-targeting EGFR and CD47 with enhanced therapeutic index. Biotechnol. Lett 40, 789–795 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Ingram JR, Blomberg OS, Sockolosky JT, Ali L, Schmidt FI, Pishesha N, Espinosa C, Dougan SK, Garcia KC, Ploegh HL, Dougan M, Localized CD47 blockade enhances immunotherapy for murine melanoma. Proc. Natl. Acad. Sci. U.S.A 114, 10184–10189 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Petersen RP, Campa MJ, Sperlazza J, Conlon D, Joshi M-B, Harpole DH Jr., Patz EF Jr., Tumor infiltrating Foxp3+ regulatory T-cells are associated with recurrence in pathologic stage I NSCLC patients. Cancer 107, 2866–2872 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, Zhao L, Vatan L, Shao I, Szeliga W, Lyssiotis C, Liu JR, Kryczek I, Zou W, Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol 18, 1332–1341 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, Burke MM, Caldwell A, Kronenberg SA, Agunwamba BU, Zhang X, Lowy I, Inzunza HD, Feely W, Horak CE, Hong Q, Korman AJ, Wigginton JM, Gupta A, Sznol M, Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med 369, 122–133 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, Morse MA, Van Cutsem E, McDermott R, Hill A, Sawyer MB, Hendlisz A, Neyns B, Svrcek M, Moss RA, Ledeine JM, Cao ZA, Kamble S, Kopetz S, Andre T, Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J. Clin. Oncol 36, 773–779 (2018). [DOI] [PubMed] [Google Scholar]
- 53.Motzer RJ, Tannir NM, McDermott DF, Aren Frontera O, Melichar B, Choueiri TK, Plimack ER, Barthelemy P, Porta C, George S, Powles T, Donskov F, Neiman V, Kollmannsberger CK, Salman P, Gurney H, Hawkins R, Ravaud A, Grimm MO, Bracarda S, Barrios CH, Tomita Y, Castellano D, Rini BI, Chen AC, Mekan S, McHenry MB, Wind-Rotolo M, Doan J, Sharma P, Hammers HJ, Escudier B; CheckMate 214 Investigators, Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med 378, 1277–1290 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. Reagents derived from this work can be requested from Y.-X.F. (Yang-Xin.Fu@UTSouthwestern.edu) and will be shared with the scientific community via a material transfer agreement.