

Abstract
Background:
The ichthyoses are rare genetic keratinizing disorders that share the characteristics of an impaired epidermal barrier and increased risk of microbial infections. Although ichthyotic diseases share a T helper (Th) 17 cell immune signature, including increased expression of antimicrobial peptides, the skin microbiota of ichthyoses is virtually unexplored.
Objectives:
To analyse the metagenome profile of skin microbiome for major congenital ichthyosis subtypes.
Methods:
Body site-matched skin surface samples were collected from the scalp, upper arm and upper buttocks of 16 healthy control participants and 22 adult patients with congenital forms of ichthyosis for whole metagenomics sequencing analysis.
Results:
Taxonomic profiling showed significant shifts in bacteria and fungi abundance and sporadic viral increases across ichthyosis subtypes. Cutibacterium acnes and Malassezia were significantly reduced across body sites, consistent with skin barrier disruption and depletion of lipids. Microbial richness was reduced, with specific increases in Staphylococcus and Corynebacterium genera, as well as shifts in fungal species, including Malassezia. Malassezia globosa was reduced at all body sites, whereas M. sympodialis was reduced in the ichthyotic upper arm and upper buttocks. Malassezia slooffiae, by contrast, was strikingly increased at all body sites in participants with congenital ichthyosiform erythroderma (CIE) and lamellar ichthyosis (LI). A previously undescribed Trichophyton species was also detected as sporadically colonizing the skin of patients with CIE, LI and epidermolytic ichthyosis subtypes.
Conclusions:
The ichthyosis skin microbiome is significantly altered from healthy skin with specific changes predominating among ichthyosis subtypes. Skewing towards the Th17 pathway may represent a response to the altered microbial colonization in ichthyosis. What is already known about this topic? The skin microbiome of congenital ichthyoses is largely unexplored. Microbes play an important role in pathogenesis, as infections are common. The relative abundances of staphylococci and corynebacteria is increased in the cutaneous microbiome of patients with Netherton syndrome, but extension of these abundances to all congenital ichthyoses is unexplored. What does this study add? A common skin microbiome signature was observed across congenital ichthyoses. Distinct microbiome features were associated with ichthyosis subtypes. Changes in microbiome may contribute to T helper 17 cell immune polarization. What is the translational message? These data provide the basis for comparison of the microbiome with lipidomic and transcriptomic alterations in these forms of ichthyosis and consideration of correcting the dysbiosis as a therapeutic intervention.
Graphical Abstract
• A common skin microbiome signature was observed across congenital ichthyoses • Distinct microbiome features were associated with ichthyosis subtypes • Changes in microbiome may contribute to Th17 immune polarization
The ichthyoses are a group of lifelong genetic disorders characterized by skin thickening, scaling and underlying cutaneous inflammation.1,2 The most common types are usually not present at birth: ichthyosis vulgaris, affecting 1 in 100 individuals3 and recessive X-linked ichthyosis, which manifests in 1 in 4500 male individuals.4 All congenital subtypes are orphan forms (< 1 in 200 000 individuals). Among these, most prevalent are epidermolytic ichthyosis (EI; most often variants in KRT10), Netherton syndrome (NS; variants in SPINK5, leading to excessive epidermal serine protease activity),5 lamellar ichthyosis (LI; variants in TGM1), and congenital ichthyosiform erythroderma (CIE; variants in at least 10 genes involved in epidermal barrier lipid biosynthesis).2 LI and CIE are phenotypically different subtypes within the classification autosomal recessive congenital ichthyosis (ARCI).
The outer epidermis is a critical physical and innate immune barrier against the ingress of pathogenic organisms and the home for numerous commensal organisms.6,7 Studies of the microbiome in other skin disorders with an altered barrier have shown a correlation between disease activity and reductions in normal flora.8-10 The prototypic example is atopic dermatitis, in which flares have been associated with relative increases in Staphylococcus aureus and loss of commensal organism diversity.11 Although S. aureus has long been recognized to promote T helper (Th)2 cell-skewed immunity and drive cutaneous inflammation in atopic dermatitis, skin commensal organisms have more recently been recognized to modulate innate and adaptive immune responses.12 Furthermore, many commensal organisms have direct anti-S. aureus activity, leading to early clinical trials with S. hominis, which is deficient in the lesional skin of people with atopic dermatitis and who have S. aureus toxicity.13-16
The high levels of expression of the Th17 pathway components across the spectrum of congenital ichthyoses, including of genes encoding antimicrobial peptides β-defensins and cathelicidin, suggest the possibility of alterations in the ichthyosis microbiome.17-19 Furthermore, individuals with ichthyosis are at increased risk of developing S. aureus and fungal (candida and dermatophyte) infections, despite the increased antimicrobial response, suggestive of a permissive microenvironment for colonization of pathobionts.20 However, the microbiome of orphan forms of ichthyosis is poorly understood. To date, investigation has only focused on NS. In one investigation comparing the bacterial and fungal microbiota in patients with mild atopic dermatitis, ichthyosis vulgaris, NS and healthy controls (each n = 3), the greatest increases in Firmicutes (Staphylococcus) and Actinobacteria (Corynebacterium) were seen in people with NS vs. healthy controls, although these organisms were also increased in people with ichthyosis vulgaris and atopic dermatitis vs. controls.21 Participants with NS also showed reduced Clostridia and Lactobacillus compared with controls (whereas those with ichthyosis vulgaris and atopic dermatitis did not) and greater alteration vs. controls in the fungal profile than participants with atopic dermatitis and ichthyosis vulgaris, mostly driven by expansion of Ascomycota (Cladosporium), which was correlated with elevations in serum IgE levels.21 Expansion of staphylococcal species was also described based on shotgun metagenomic analysis in the affected skin of 10 patients with NS.22 These staphylococcal species have been shown to augment the increase in protease activity found in those with NS, further promoting barrier dysfunction.22
To date, no studies have compared the spectrum of congenital ichthyoses, despite their unique barrier alterations, and none has used metagenomics to capture the entire multikingdom fingerprint, including bacteria, fungi and viruses, in the same analysis. We have profiled the whole skin metagenome and analysed microbiome kingdoms in a cohort of 22 patients with rare congenital forms of ichthyosis.
Materials and methods
Study participants
Participants who were 17 years of age or older with a genotype-confirmed diagnosis of an orphan form of ichthyosis (ARCI–CIE, ARCI–LI, EI, NS) and age-matched healthy controls were recruited from Northwestern University Feinberg School of Medicine and its Lurie Children’s Hospital, Chicago and Mount Sinai Icahn School of Medicine, New York (Figure S1; see Supporting Information). All participants provided written informed consent, approved by the Institutional Review Boards. Patients with ichthyosis were genotyped for sequence variants in candidate genes, as well as for FLG, to exclude any additional modifier effect to the skin barrier as previously described23 (see Appendix S1 in the Supporting Information for further details about the Methods).
Study design
Recruited participants provided their personal and family medical history, details of current medication and topical product use, transepidermal water loss (TEWL) measurements and microbiome samples. Two trained physicians concurrently graded severity using the Ichthyosis Area and Severity Index (IASI),19 a nonvalidated tool with components on scaling and erythema, scored 0–4 for intensity and weighted by extent of involvement by body region (all participants had generalized involvement; Figure S2; see Supporting Information).
Microbiome sample collection
Investigators swabbed the skin of a 2–4 cm2 area of the upper outer arm (nondominant hand), upper buttock and scalp 20 times for 15 s using sterile nylon-flocked dry swabs (Copan Diagnostics, Murrieta, CA, USA). Swabs were placed in sterile 2-mL micro tubes and stored frozen in 400-μL Tris-EDTA buffer. Environmental controls were obtained by swab exposure to room air.
Skin physiological measurements
Triplicate TEWL measurements were obtained using AquaFlux AF200 (Biox, London, UK) 2–3 cm away from the sampled areas on the arm and buttock.
DNA extraction for shotgun metagenomics
DNA was extracted from swabs according to standard protocols and quantified prior to storage at –20 °C. Detailed methods are provided in Appendix S1 (see Supporting Information).
Library construction and read preprocessing for metagenomics
Extracted DNA was prepared for sequencing using NEBNext Ultra II FS (New England Biolabs, Ipswich, MA, USA) kit followed by Illumina sequencing. Detailed methods are provided in Appendix S1 (see Supporting Information). Human reads were filtered leaving an average of 31·3% of the reads used for microbiome analysis (Table S1; see Supporting Information).
Taxonomic, diversity and differential abundance analysis
Relative abundance of microbes was performed with MetaPhlAn2 with default parameters.24 Alpha and beta diversity were computed using the R package ‘vegan’ (v2·5–6, http://www.r-project.org/). Bray–Curtis dissimilarity indices were computed from community composition data using ‘vegdist’ function from ‘vegan’. Hierarchical clustering was performed using ‘pvclust’.25 Scripts and pipeline information are available at github (https://github.com/lch14forever/shotgun-metagenomics-pipeline). Further analysis was performed for eukaryote with CLC Genomics Workbench 20·0·4 (QIAGEN, Germantown, MD, USA) and CLC Microbial Genomics Module 20·1·1 (QIAGEN). Details are available in Appendix S1 (see Supporting Information).
Statistical analysis
Differential abundance analyses were undertaken at both species and genus levels for the three body sites separately and for samples from the three sites combined. All analyses, including the differential abundance analysis, other statistical tests, generation of the principal coordinates analysis (PCoA) plots, box plots and stacked bar charts, were produced in R, version 3·6·2 with appropriate statistical tests, described in the results and/or figure legends. Full detailed descriptions are provided in Appendix S1 (see Supporting Information).
Results
Distinct skin microbiome communities are associated with congenital ichthyosis
Twenty-two participants with orphan forms of ichthyosis were enrolled (Table 1; Table S2; see Supporting Information). All participants with LI had sequence variants in TGM1 (n = 7), all those with NS had sequence variants in SPINK5 (n = 6), and all those with EI had sequence variants in KRT10 (n = 4). Among the five with CIE, two participants had ALOX12B variants and one each had PNPLA1, NIPAL4 and CYP4F22 variants (Table S2). No ichthyosis or control participants had a loss-of-function mutation in FLG.
Table 1.
Demographic and clinical data for the participants with ichthyosis and the controls
| Control (n = 16) |
Ichthyosis group – all (n = 22) |
ARCI–CIE type (n = 5) |
ARCI–lamellar type (n = 7) |
Epidermolytic ichthyosis (n = 4) |
Netherton syndrome (n = 6) |
|
|---|---|---|---|---|---|---|
| Sex, n (%) | ||||||
| Female | 9 (56) | 14 (64) | 1 (20) | 6 (86) | 2 (50) | 5 (83) |
| Male | 7 (44) | 8 (36) | 4 (80) | 1 (14) | 2 (50) | 1 (17) |
| Age, years | ||||||
| Mean (SD) | 36·0 (10·7) | 34·9 (13·3) | 45·5 (5·1) | 35·7 (13·4) | 37·5 (15·6) | 23·5 (5·2) |
| Median (range) | 34·3 (22·0–56·0) | 32·7 (17·2–59·7) | 42·8 (40·6–54·3) | 32·2 (19·2–59·7) | 37·4 (18·2–57·0) | 23·8 (17·2–33·3) |
| Ethnicity, n (%) | ||||||
| Hispanic | 1 (6) | 2 (9) | 0 (0) | 1 (14) | 1 (25) | 0 (0) |
| Non-Hispanic | 15 (94) | 20 (91) | 5 (100) | 6 (86) | 3 (75) | 6 (100) |
| Race, n (%) | ||||||
| Asian/Pacific Islander | 3 (19) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Black/African American | 2 (13) | 2 (9) | 0 (0) | 1 (14) | 0 (0) | 1 (17) |
| White | 10 (63) | 20 (91) | 5 (100) | 6 (86) | 4 (100) | 5 (83) |
| Other/unknown | 1 (6) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| TEWL, mean (SD) | ||||||
| Upper arm | 17·7 (10·5)** | 30·7 (14·7) | 38·4 (5·6) | 21·5 (8·2) | 24·6 (6·0) | 39·2 (20·5) |
| Buttock | 18·1 (8·5)** | 30·6 (13·1) | 32·9 (7·7) | 20·3 (7·6) | 24·9 (4·9) | 43·5 (12·7) |
| IASI, mean (SD) | ||||||
| Total score | N/A | 34·4 (5·9) | 31·8 (6·0) 7 | 36·9 (3·3) | 38·3 (2·7) | 31·2 (6·9) |
| Erythema subscore | N/A | 17·0 (3·1) | 16·4 (3·2) | 16·1 (2·2) | 19·1 (2·6) | 17·3 (3·5) |
| Scaling subscore | N/A | 17·4 (4·2) | 15·4 (3·2) | 20·8 (2·2) | 19·2 (0·) | 13·8 (4·4) |
ARCI, autosomal recessive congenital ichthyosis; CIE, congenital ichthyosiform erythroderma; IASI, Ichthyosis Area and Severity Index; N/A, not applicable; TEWL, transepidermal water loss.
P < 0·01 for unpaired t-test of all patients with ichthyosis vs. controls.
Across the entire cohort, 14 (64%) participants were women and eight (36%) were men, with a mean age of 34·9 years. The control participants (n = 16) were similarly distributed by sex (56% female), age (mean, 36·0 years of age) and ethnicity (Table 1). TEWL measurements of epidermal barrier integrity were higher for the ichthyosis cohort than for healthy controls, as expected [upper arm: mean 30·7 (SD 14·7) vs. 17·7 (SD 10·5), P < 0·01; upper buttock: 30·6 (SD 13·1) vs. 18·1 (SD 8·5), P < 0·01]. The severity of condition in the cohort was moderate–severe according to IASI scores [mean 34·4 (SD 5·9) out of possible 48] and relatively consistent across subtypes (Table 1). The mean severity for the IASI-Erythema subscore was 17·0 (SD 3·1) out of a possible score of 24 and for the IASI-Scaling subscore was 17·4 (SD 4·2) out of a possible score of 24.
All participants in the healthy control and ichthyosis groups had skin swabbing at the three sites for whole metagenomic sequencing (n = 114) (Table S1 and Appendix S1; see Supporting Information).10 Taxonomic analysis showed distinct skin microbiome community assemblages in ichthyosis skin compared with healthy skin at all sites (scalp permutation test P-value < 0·001; outer arm permutation P-value <0·001; upper buttock permutation P-value <0·001; Figure S3; see Supporting Information). Plotting of PCoA using the Bray–Curtis index with samples from all sites resulted in a cluster enriched for healthy skin microbiomes that segregated from ichthyosis samples. However, in the ichthyosis group the ichthyosis disease subtypes did not separate from each other on the PCoA plot (Figure 1a). Similar clustering patterns were also replicated when each body site was visualized independently (PCoA; Figure S4; see Supporting Information). Additionally, hierarchical clustering with bootstrapped confidence intervals confirmed the presence of a group containing predominantly healthy skin microbiome and groups with predominantly ichthyosis skin microbiomes (Figure S5; see Supporting Information).
Figure 1.

Distinct skin microbiome community structures in congenital ichthyosis. (a) Bray–Curtis dissimilarity species-level analysis from multiple body sites. Principal coordinates analysis (PCoA) plot with contours to indicate sample density. (b) Box plots showing the Bray–Curtis dissimilarity of taxonomic profiles within control participants and within participants with ichthyosis subtypes. Statistical significance was determined using the permutation test on the Wilcoxon rank sum test in 100 000 times of random reshuffling of the participant subgroup labels with Bonferroni multiple testing correction (**P < 0·01; as described in the Method). CIE, congenital ichthyosiform erythroderma; EI, epidermolytic ichthyosis; LI, lamellar ichthyosis; NS, Netherton syndrome.
The ichthyosis disease subtypes, except EI, were each significantly different from the control group when comparing for dissimilarity (permutation test on the Wilcoxon rank sum test in 100 000 times of random reshuffling; Figure 1b). These results indicate that the structure and composition of the skin microbiome of patients with ichthyosis is altered at three distinct body sites and produces compositional changes among ichthyosis subtypes.
Skin microbiome composition of ichthyotic skin has reduced microbial richness and a predominance of Staphylococcus and Corynebacterium species
We observed reduced microbial richness in the skin microbiome of patients with ichthyosis compared with healthy controls (Figure S6a; see Supporting Information). This statistically significant reduction in species richness was observed for NS at each body site and CIE on the scalp and outer arm (Figure 2a). However, Shannon’s and Simpson’s diversity indices, measures of richness and evenness, were not significantly altered in ichthyosis analysis groupings when compared with control skin (Figure S7 and Figure S8 and see detailed description of statistical analysis in Appendix S1; see Supporting Information).
Figure 2.

Major shifts in key community members in control and ichthyosis subtype skin. (a) Species richness is reduced in congenital ichthyosiform erythroderma (CIE) and Netherton syndrome (NS) on the scalp and the outer arm, and NS on the upper buttock. Kruskal–Wallis tests for three body sites are all significant (P-value of 3·22 × 10−4, 8·20 × 10−4 and 8·42 × 10−3 respectively for scalp, outer arm and upper buttock) and Bonferroni adjusted P-values from Dunn’s post hoc tests were used to indicate the statistical significance in comparison of subtypes vs. the control group (*P < 0·05; **P < 0·01). Raw and adjusted P-values can be found in Table S8; see Supporting Information. (b) Major significant changes in key microbial species were observed in all subtypes, including a reduction of Cutibacterium acnes and Malassezia globosa, and an increase in species in the Staphylococcus and Corynebacterium genera. Statistical significance was determined using the permutation test on the Wilcoxon rank sum test statistic with multiple testing correction using familywise error rate as described in Appendix S1 (see Supporting Information) (*P < 0·05; **P < 0·01). EI, epidermolytic ichthyosis; LI, lamellar ichthyosis; ns, not significant.
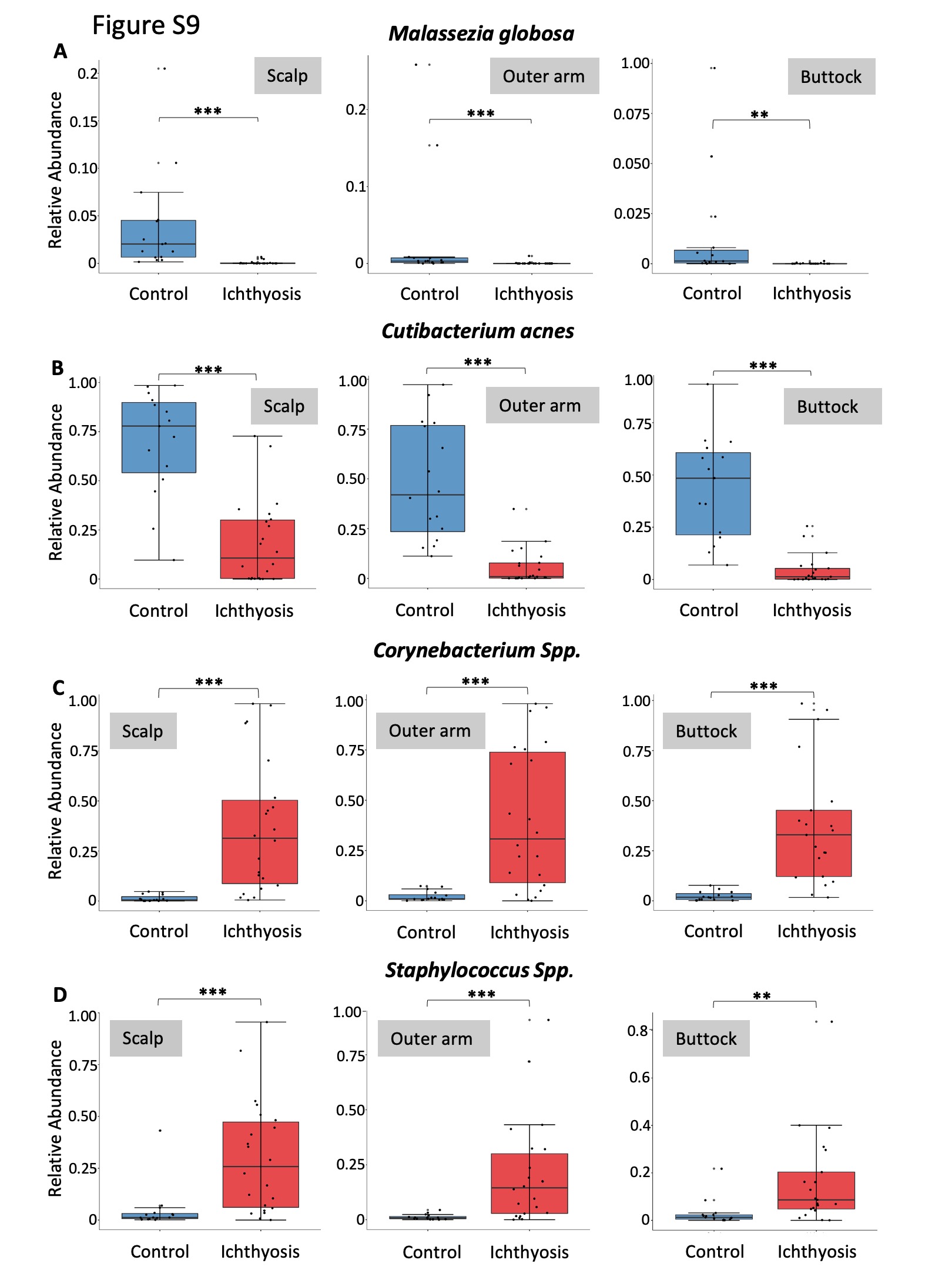
A shared feature of ichthyotic skin was the altered skin microbiome community structure, with significant loss of key skin microbiome commensals, such as lipophilic microbes Cutibacterium acnes and Malassezia globosa, as well as an increase in microbes from the Staphylococcus and Corynebacterium genera that have previously been associated with dry skin conditions (Table 2; Figure 2b and Figure S6b; see Supporting Information).21 This observation was most pronounced on the scalp, generally the most sebaceous gland-enriched body site analysed, but was also observed at other body sites (Figure S9; see Supporting Information).26
Table 2.
Significantly altered microbial species in the skin microbiome of participants with ichthyosis subtypes compared with the control group, based on the MetaPhlAn2 databasea
| FDR |
||||
|---|---|---|---|---|
| Location and species | CIE vs. control group | LI vs. control group |
EI vs. control group |
NS vs. control group |
| Scalp | ||||
| Corynebacterium bovis | 0·3664 | 0·0229 | 0·0039 | 0·0488 |
| Corynebacterium massiliense | 0·3664 | 0·2154 | 0·0039 | 0·6275 |
| Corynebacterium resistens | 0·6633 | 0·0229 | 0·5044 | NA |
| Corynebacterium striatum | 0·6986 | 0·2154 | 0·7049 | 0·0639 |
| Corynebacterium tuberculostearicum | 0·2362 | 0·7084 | 0·0285 | 0·6275 |
| Enhydrobacter aerosaccus | 0·5352 | 0·7608 | 0·1767 | 0·0488 |
| Malassezia globosa | 0·2362 | 0·0245 | 0·0285 | 0·0488 |
| Propionibacterium acnes | 0·4218 | 0·1517 | 0·1330 | 0·0488 |
| Propionibacterium granulosum | 0·6986 | 0·7084 | 0·7049 | 0·0488 |
| Pseudomonas unclassified | 0·6986 | 0·7608 | 0·5666 | 0·0488 |
| Staphylococcus aureus | 0·6633 | 0·0665 | 0·7049 | 0·1029 |
| Staphylococcus caprae/capitis | 0·2362 | 0·0028 | 0·1355 | 0·6275 |
| Staphylococcus hominis | 0·1240 | 0·0952 | 0·7049 | 0·0488 |
| Staphylococcus lugdunensis | 0·0048 | 0·0229 | 0·1355 | NA |
| Stenotrophomonas maltophilia | 0·6623 | 0·7084 | 0·1355 | 0·0488 |
| Streptococcus mitis/oralis/pneumoniae | 0·6986 | 0·7084 | 0·4154 | 0·0488 |
| Veillonella unclassified | 0·6986 | 0·9847 | 0·2519 | 0·0778 |
| Upper outer arm | ||||
| Corynebacterium bovis | 0·3605 | 0·0534 | 0·0029 | 0·2616 |
| Corynebacterium massiliense | 0·3605 | 0·6989 | 0·0029 | 0·2616 |
| Corynebacterium resistens | 0·6841 | 0·0703 | 0·1013 | 0·2616 |
| Corynebacterium striatum | 0·6841 | 0·5797 | 0·7255 | 0·0568 |
| Corynebacterium tuberculostearicum | 0·6841 | 0·6989 | 0·0932 | 0·0535 |
| Halomonas stevensii | 0·6841 | 0·6989 | 0·0932 | 0·6320 |
| Malassezia globosa | 0·2309 | 0·0618 | 0·0932 | 0·2382 |
| Neisseria unclassified | 0·–2309 | 0·6989 | 0·1339 | 0·0981 |
| Propionibacterium acnes | 0·2309 | 0·0534 | 0·0392 | 0·0568 |
| Propionibacterium granulosum | 0·3605 | 0·0703 | 0·1060 | 0·2791 |
| Pseudomonas unclassified | 0·8643 | 0·7791 | 0·0932 | 0·6520 |
| Staphylococcus caprae/capitis | 0·4751 | 0·0618 | 0·1013 | 0·6320 |
| Staphylococcus pettenkoferi | 0·6841 | 0·6989 | 0·0932 | 0·6320 |
| Stenotrophomonas maltophilia | 0·6841 | 0·7379 | 0·0932 | 0·5759 |
| Stenotrophomonas unclassified | 0·4143 | 0·6989 | 0·1013 | 0·0611 |
| Upper buttock | ||||
| Arthroderma benhamiae | 0·5872 | NA | 0·0345 | NA |
| Corynebacterium bovis | 0·4023 | 0·0107 | 0·0046 | 0·1081 |
| Corynebacterium massiliense | 0·4023 | 0·0447 | 0·0046 | 0·2019 |
| Corynebacterium resistens | 0·5872 | 0·0107 | 0·1538 | 0·2019 |
| Corynebacterium striatum | 0·5872 | 0·1031 | 0·7325 | 0·0324 |
| Corynebacterium tuberculostearicum | 0·0343 | 0·7997 | 0·1657 | 0·0049 |
| Propionibacterium acnes | 0·4023 | 0·0107 | 0·0345 | 0·0049 |
| Staphylococcus caprae/capitis | 0·5563 | 0·0352 | 0·1538 | 0·1493 |
ARCI, autosomal recessive congenital ichthyosis; CIE, congenital ichthyosiform erythroderma; IASI, Ichthyosis Area and Severity Index; N/A, not applicable; TEWL, transepidermal water loss.
P < 0·01 for unpaired t-test of all patients with ichthyosis vs. controls.
The importance of Th17 pathways in patients with ichthyosis has been previously highlighted.17-19 Th17 responses can be stimulated in mouse models by microbial infections, such as from S. aureus27 and Malassezia.28 In this study we see a reduction in abundance of M. globosa on the body sites investigated, but an increased abundance in various staphylococcal species and other Malassezia spp. (Figure S9; see Supporting Information). Staphylococcus aureus has been associated with the development of skin inflammation in atopic dermatitis and observed on the skin of people with NS.22 However, we observed a mixture of staphylococcal species at various abundances and not a predominance of S. aureus, similar to a recent study focusing on NS (Figure 3a).21 We also observed an increase in a consortium of Corynebacterium spp., but not a predominance of one particular species in ichthyosis (Figure 3b).
Figure 3.

Stacked bar plots showing relative abundance of (a) Staphylococcus and (B) Corynebacterium spp. (P < 0·05) present in the healthy control and the congenital ichthyosis skin microbiome and grouped by body site. Healthy controls are labelled on the x-axis as control in the blue box and participants with ichthyosis are grouped according to disease subtype [congenital ichthyosiform erythroderma (CIE); lamellar ichthyosis (LI); epidermolytic ichthyosis (EI) and Netherton syndrome (NS)]. Box plots showing significant changes in relative abundance of (c) Staphylococcus and (d) Corynebacterium spp. In the ichthyosis subtypes in comparison with the healthy control group. Statistical significance was determined using the permutation test on the Wilcoxon rank sum test statistic with multiple testing correction using familywise error rate as described in Appendix S1 (see Supporting Information) (*P < 0·05; **P < 0·01) was computed for (c) and (d). ns, not significant.
Among the staphylococcal and corynebacterial species, S. hominis was significantly reduced in CIE, LI and NS subtypes (Figure 3c), whereas S. caprae/capitis, and C. bovis, appeared as the predominant species from each genera in ichthyotic skin (Figure 3c, d). Although no one species reached false discovery rate (FDR) significance, S. caprae/capitis was positively correlated with IASI-E score and TEWL in participants with LI for outer arm and upper buttock samples (ρ > 0·75; P ≤ 0·05) (Table S3; see Supporting Information). Interestingly, S. caprae/capitis predominance was similarly found in affected skin of people with psoriasis,29 who share an interleukin (IL)-17-dominant immune profile with patients with congenital ichthyosis.18,19
The reduced and altered organization of epidermal lipids and proteins in ichthyotic skin impairs skin barrier function and disrupts the skin surface microenvironment. Microenvironment alterations likely result in the observed reduction in microbial richness, increase in staphylococcal and corynebacterial species, and loss of key lipophilic microbes, Cutibacterium and Malassezia, which can both negatively regulate staphylococcal colonization.30,31
Participants with congenital ichthyosis show shifts in kingdom contributions to the skin microbiome
Multikingdom analysis plots were generated from the relative abundance of each microbial kingdom using MetaPhlAn2 analysis (Table S4 and Appendix S1; see Supporting Information). Relative abundance of bacterial kingdom within the whole skin metagenome significantly increased in participants with LI, EI and NS (Figure 4a). The contribution of eukaryote (fungal species) was reduced in people with ichthyosis, particularly on the scalp with its high fungal load in healthy controls (Figure 4b).
Figure 4.

Multikingdom analysis of skin microbiome. Multikingdom analysis plot from MetaPhlAn2 analysis shows relative abundance of (a) bacteria, increased in the participants with ichthyosis subtypes lamellar ichthyosis (LI); epidermolytic ichthyosis (EI) and Netherton syndrome (NS). (b) Eukaryota are significantly reduced in participants with ichthyosis subtypes compared with the healthy control group. (c) The viral kingdom analysis plot shows that the level of viruses on the skin of participants with ichthyosis is low, except for patients with congenital ichthyosiform erythroderma (CIE), who have a trend towards increased relative abundance at the three body sites that did not reach significance. The false discovery rate (FDR)-corrected Wilcoxon rank sum test P-value (*P < 0·05; **P < 0·01), undertaken separately for three body sites was used to determine the statistical significance. FDR multiple testing correction were undertaken over all the 12 tests of the three kingdoms and for the three body sites separately. Raw and adjusted P-values can be found in Table S8 (see Supporting Information). ns, not significant.
The viral kingdom analysis showed an increased level of viruses in patients with CIE compared with control participants, primarily on the arm and upper buttock and from sporadic individual dominance of Merkel cell polyomavirus (Table S4; see Supporting Information); however, given the limited numbers of patients, this increase did not reach significance (Figure 4c). Viral relative abundance was also increased in participants with EI on the arm site, driven by sampling in one participant with particularly high viral reads mapping (37·6% relative abundance), but this failed to reach significance (Table S4).
Major alterations in fungal species colonization
Malassezia and Trichophyton spp. are known to activate the IL-17 pathway,28,32 which is polarized in patients with ichthyosis.19 However, the MetaPhlAn2 reference database covers only one species from both Malassezia (M. globosa) and Trichophyton (T. verrucosum) genera. To interrogate the eukaryota kingdom further we used the CLC Genomics Workbench and Microbial Genomics Module (QIAGEN) for taxonomic profiling against a fungal reference database (Table S5; see Supporting Information) that covers 15 Malassezia spp. and 11 Trichophyton spp. (Supplemental Appendix S1 and Table S6; see Supporting Information).
We confirmed a reduction in the Malassezia genus in the ichthyosis skin microbiome at the three sites (Figure S10a; see Supporting Information), most pronounced in patients with NS (Figure 5a). The stacked bar plot of relative abundance showed that Malassezia spp. were altered in the ichthyosis group with significant reductions of M. globosa, M. sympodialis and M. restricta and increases of M. slooffiae (Figure 5b; Table 3). Interestingly, M. globosa was primarily detected on healthy scalps, whereas M. sympodialis was detected mainly on healthy outer arms. Both species were significantly reduced in all ichthyosis subtypes; however, M. restricta was only reduced in participants with LI and NS (Figure 5c). In contrast to M. globosa and M. sympodialis, M. slooffiae, rarely reported in the healthy skin microbiome, was markedly increased on all subtypes of ichthyotic skin except NS (Figure 5c; Figure S10b; see Supporting Information).
Figure 5.

Mycobiome analysis for the control and ichthyosis groups. (a) Malassezia genus level alterations separated for ichthyosis subtypes. (b) Stacked bar charts of mycobiome highlighting only Malassezia spp. (P < 0·05) across the spectrum of ichthyosis subtypes. (c) Malassezia globosa and M. sympodialis were significantly reduced in all ichthyosis subtypes, whereas M. slooffiae was significantly increased in ichthyosis skin [except Netherton syndrome (NS)]. Malassezia restricta was significantly reduced in lamellar ichthyosis (LI) and NS subtypes. (d) Trichophyton at the genus level was augmented in all subtypes (except NS). (e) Stacked bar charts of mycobiome highlighting only Trichophyton spp. (P < 0·05) across the spectrum of ichthyosis subtypes. (f) Trichophyton unclassified species was significantly increased in all subtypes (except NS). Statistical significance was determined using permutation test on the Wilcoxon rank sum test statistic with multiple testing correction using familywise error rate as described in Appendix S1 (see Supporting Information) (*P < 0·05; **P < 0·01) was computed for (a), (c), (d) and (f). CIE, congenital ichthyosiform erythroderma; epidermolytic ichthyosis (EI); ns, not significant.
Table 3.
Significantly altered Malassezia and Trichophyton spp. in the skin microbiome of participants with ichthyosis subtypes compared with the control group, based on the CLC fungal databasea
| Location and species | FDR | |||
|---|---|---|---|---|
| CIE vs. control group | LI vs. control group | EI vs. control group | NS vs. control group | |
| Scalp | ||||
| (Unclassified species) Malassezia | 0·6919 | 0·0212 | 0·7136 | 0·5041 |
| Malassezia globosa | 0·0968 | 0·0034 | 0·0980 | 0·0785 |
| Malassezia restricta | 0·6919 | 0·0108 | 0·7136 | 0·6996 |
| Malassezia slooffiae | 0·0968 | 0·0108 | 0·4934 | 0·1530 |
| Malassezia sympodialis | 0·4391 | 0·0620 | 0·4934 | 0·2343 |
| Upper outer arm | ||||
| (Unclassified species) Trichophyton | 0·1129 | 0·2103 | 0·0365 | NA |
| Malassezia globosa | 0·0759 | 0·0331 | 0·0847 | 0·0969 |
| Malassezia restricta | 1·0000 | 0·0576 | 0·7165 | 0·1097 |
| Malassezia slooffiae | 0·1129 | 0·0143 | 0·2505 | 0·6944 |
| Malassezia sympodialis | 0·2174 | 0·0576 | 0·1244 | 0·0330 |
| Trichophyton rubrum | 0·1129 | 0·3153 | 0·0847 | NA |
| Trichophyton violaceum | 0·2174 | 0·3153 | 0·0847 | NA |
| Upper buttock | ||||
| (Unclassified species) Trichophyton | 0·2631 | 0·0742 | 0·0157 | NA |
| Malassezia globosa | 0·2831 | 0·0175 | 0·0615 | 0·0049 |
| Malassezia restricta | 0·7174 | 0·0175 | 0·4586 | 0·1931 |
| Malassezia slooffiae | 0·2852 | 0·0381 | 0·3691 | 0·6419 |
| Malassezia sympodialis | 0·2831 | 0·0742 | 0·1569 | 0·0528 |
| Trichophyton rubrum | 0·3085 | 0·3462 | 0·0157 | NA |
| Trichophyton tonsurans | 0·3085 | NA | 0·0771 | NA |
| Trichophyton violaceum | 0·3085 | NA | 0·0157 | NA |
| Trichophyton yaoundei | 0·3085 | NA | 0·0771 | NA |
Separated by body sites; false discovery rate (FDR)-corrected Wilcoxon rank sum test P-values (< 0·1). Green shading indicates increased and blue shading indicates reduced relative abundance. CIE, congenital ichthyosiform erythroderma; EI, epidermolytic ichthyosis; LI, lamellar ichthyosis; NA, not available; NS, Netherton syndrome.
In addition, we observed an increase in the Trichophyton genus in the ichthyosis group skin microbiome compared with the healthy control group (Figure 5d). Sporadic significant increases in Trichophyton unclassified species in participants with ichthyosis were observed at the three body sites (Figure 5e, f). To further characterize Trichophyton spp. in people with ichthyosis, we extracted the Trichophyton unclassified species sequencing reads for additional taxonomic profiling against all available complete genomes of Trichophyton spp. deposited in NCBI (Table S7 and Appendix S1; see Supporting Information). However, Trichophyton unclassified species remained unidentified, suggesting that a novel Trichophyton genome, not previously described in the NCBI genomes, is a frequent colonizer of ichthyotic skin.
Discussion
In this study, we analysed the skin microbiome of participants with ichthyosis who had monogenic loss-of-function mutations in ALOX12B (CIE), CYP4F22 (CIE), NIPAL4 (CIE), PNPLA1 (CIE), TGM1 (LI), KRT10 (EI) and SPINK5 (NS). Disruption in any of these genes was sufficient to cause cutaneous dysbiosis of the skin microbiome, whereby total bacteria were increased in the ichthyotic skin microbiome, with higher abundance of staphylococci and corynebacteria, but lower abundance of C. acnes. This finding is consistent with recent reports that the relative abundance of staphylococci and corynebacteria are augmented in patients with NS,21,22 and extends the microbiome findings across the spectrum of congenital ichthyoses. Increases of staphylococcal and corynebacterial abundances have also been reported in other related dry skin conditions, ichthyosis vulgaris and atopic dermatitis.21 In addition, staphylococcal abundance was found to be higher in affected lesions in a cohort with psoriasis29 and a cohort with atopic dermatitis.10 Interestingly, S. capitis/caprae were a predominant species in our ichthyosis group and in the affected lesions of participants with psoriasis,29 and S. capitis also had a strong presence in atopic dermatitis.10 The lipophilic microbes, C. acnes and M. globosa, were reduced in our ichthyosis group, and this is also similar to our previous report in atopic dermatitis,33 and in people with psoriasis.29
The mechanism by which members from staphylococcal and corynebacterial genera flourish in ichthyotic skin remains elusive. Dramatic change in the skin microenvironment owing to gene variants leading to various lipid- and protein-related barrier defects produced similar levels and types of microbiome dysbiosis and similar alterations in Cutibacterium, staphylococci and corynebacteria genera. However, we also observed species differences in staphylococci and corynebacteria in the specific subtypes, with C. bovis significantly increased across body sites in the ichthyosis group in all the ichthyosis subtypes except for CIE. We also observed genus alterations in the fungal contributions, with a more pronounced increase in Trichophyton in the participants with EI and, interestingly, no significant reduction of the lipophilic Malassezia genus in participants with CIE, which features defects in the barrier related to lipid and fatty acid synthesis and metabolism. We did not observe any reduction in Clostridia and Lactobacillus or an expansion of Cladosporium as was previously reported for patients with NS;21 however, Trichophyton, which is also an Ascomycota, was increased.
We also performed Spearman rank correlation between the microbes and ichthyosis severity, as measured by IASI, or the skin barrier defect, as measured by TEWL, for each ichthyosis subtype and the entire group with ichthyosis at the three body sites. Numerous correlations were observed at P ≤ 0·05 significance (summarized in Table S3; see Supporting Information); however, no significant FDR ≥ 0·3 were observed, suggesting a prominent role for alternative host defence properties in modulating the ichthyosis skin microenvironment. Atopic dermatitis scanning electron microscopy images have shown S. aureus colonizing dry skin crevices, associated with a biofilm-like extracellular matrix;34 similar localization in the dry skin of people with ichthyosis may promote staphylococcal and corynebacterial, rather than C. acnes, growth and survival. Cutibacterium acnes can also be selectively killed by antimicrobial peptides produced by S. capitis,35 the most abundant staphylococcal species identified in ichthyotic skin. Reduction in short-chain fatty acids produced by C. acnes that inhibit staphylococcal biofilm formation could also promote staphylococcal overgrowth in ichthyosis.31
Coagulase-negative staphylococci (CoNS) and corynebacteria are considered skin commensal organisms, capable of competing with S. aureus using antimicrobial peptides, lantibiotics and noncognate autoinducing peptides.36 However, S. aureus, S. epidermis and corynebacteria are able to activate IL-23/IL-17-mediated inflammation in mouse skin27,37,38 and may, in the setting of ichthyosis, in which CoNS and corynebacteria predominate, have proinflammatory roles via the staphylococcal agr quorum-sensing system39 and the corynebacterial cell wall mycolic acids.38 Malassezia and Trichophyton spp. may also contribute to the Th17 skewing of ichthyosis.40
Mycobiome analysis showed increased abundance of M. slooffiae and Trichophyton spp. in ichthyosis samples, implicating fungal antigens in driving ichthyosis Th17 polarization, consistent with activation of the murine IL-17 immunity axis mediated by Malassezia and Trichophyton spp.28,32,41 Conversely, M. globosa and M. sympodialis, which predominated in healthy scalp and the outer arm, respectively, were reduced significantly in ichthyosis samples. Malassezia globosa, but not M. sympodialis preference for a sebum-rich microenvironment, such as the scalp, suggests that the former may have greater potency in hydrolysing sebum as a lipid source and is consistent with M. globosa having the highest activity of secreted lipases among several Malassezia spp.42 Malassezia globosa also secretes proteases with antibiofilm properties against S. aureus,30 which may prevent staphylococcal overgrowth in healthy skin. Taken together, staphylococci, corynebacteria, M. slooffiae and Trichophyton spp. may promote the Th17 skewing, whereas C. acnes, M. globosa and M. sympodialis may foster a homeostatic host–microbe interaction. Concurrent immunological, skin microbiome, and host metabolomic assessments using longitudinally collected specimens would be required to address whether staphylococcal and corynebacterial virulence factors or host-derived endogenous factors serve to trigger Th17 polarization in ichthyotic skin.19
There are potential limitations to sequencing-based studies for skin metagenomes. Also, dry swabbing may result in low biomass and potential bias at extremely low abundance, leading us to cut-off relative abundance at 0·1% to reduce this possibility. Because metagenomic shotgun sequencing captures all DNA, including human, we sequenced with sufficient depth to accurately capture microbes (average 31·3% of microbial reads; Table S1, see Supporting Information). Microbial databases can limit the output microbiome profiles and we used MetaPhlAn2, which is well established; however, species from Malassezia genus are not well covered. We therefore employed a second database (QIAGEN CLC Microbial Genomics Module) to map additional reads and capture a more representative profile for Malassezia and Trichophyton (Table S6; see Supporting Information).
Congenital forms of the ichthyoses are known to be associated with an increased risk of developing S. aureus and fungal infections, but little is known about the microbial community as a whole and the role of alterations, including in commensal organisms, in promoting these infections in people with ichthyosis. During upcoming years, new therapeutic approaches will be tested for orphan forms of ichthyosis, among them repurposed biologics, topical gene therapy and transplanted gene-altered skin, and biochemical approaches, such as to suppress kallikrein activity in NS. Our study provides the framework for analysis of the impact of new therapeutics on microbial balance in understanding mechanism of action. Finally, topically applied commensal organisms are now being tested as a treatment for atopic dermatitis13-15 based on the demonstrated ability of specific commensals to control pathogens such as S. aureus. With deeper knowledge of the shifts in the skin microbiome of patients with ichthyosis, a similar possibility of biotherapeutics could be part of the treatment strategy for ichthyosis.
Supplementary Material
Figure S1 Study design. Schematic of study design indicating the criteria for participant recruitment and the data and sample collection process.
{kind=link}
Figure S2 Description of the Ichthyosis Area and Severity Index tool that was used to categorize the severity of condition of the participants in the study.
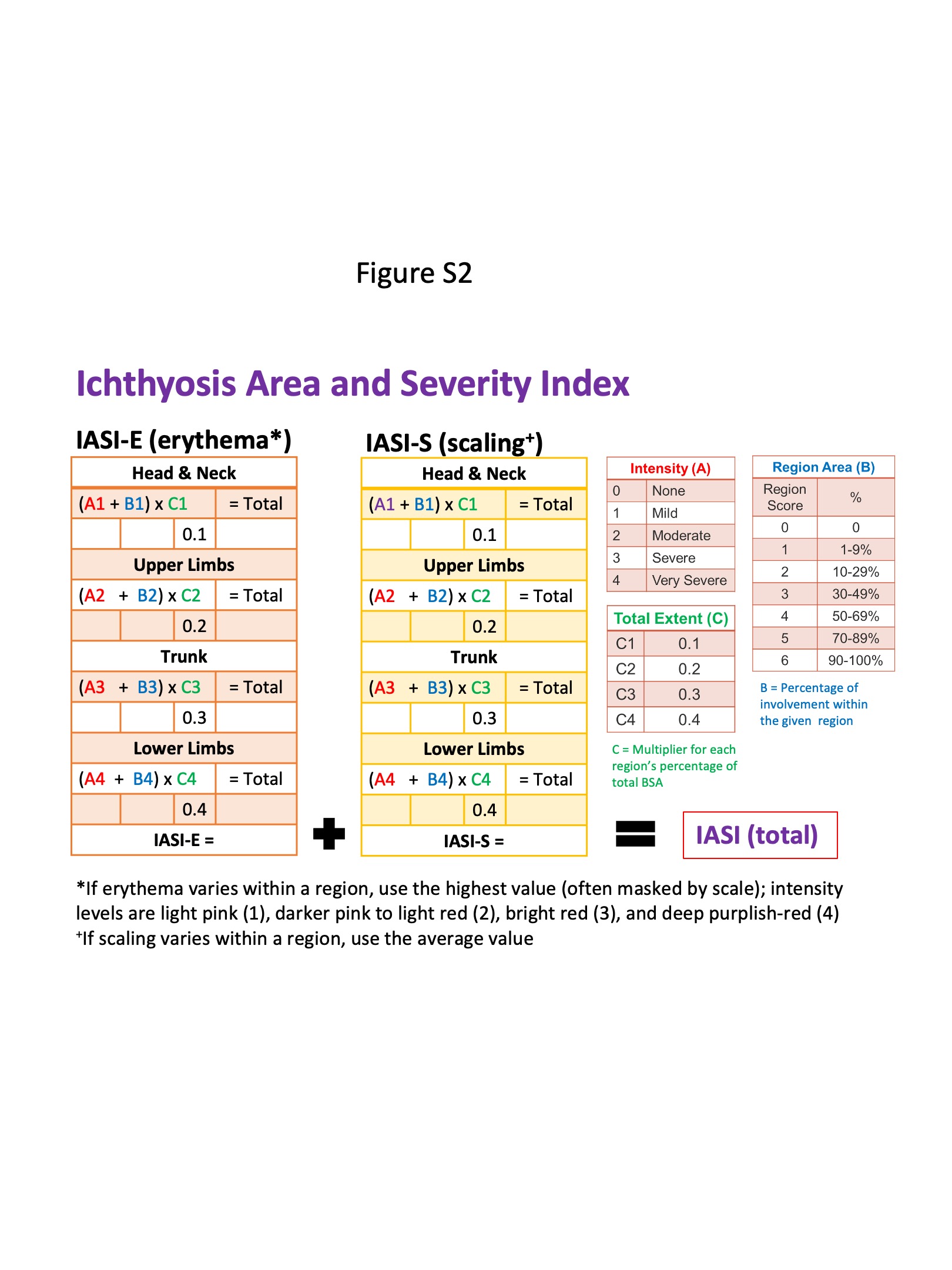{kind=link}
Figure S3 Box plots showing the Bray–Curtis dissimilarity of taxonomic profiles from all body sites (a) and separated body sites (b, c, d) within control participants and within participants with ichthyosis.
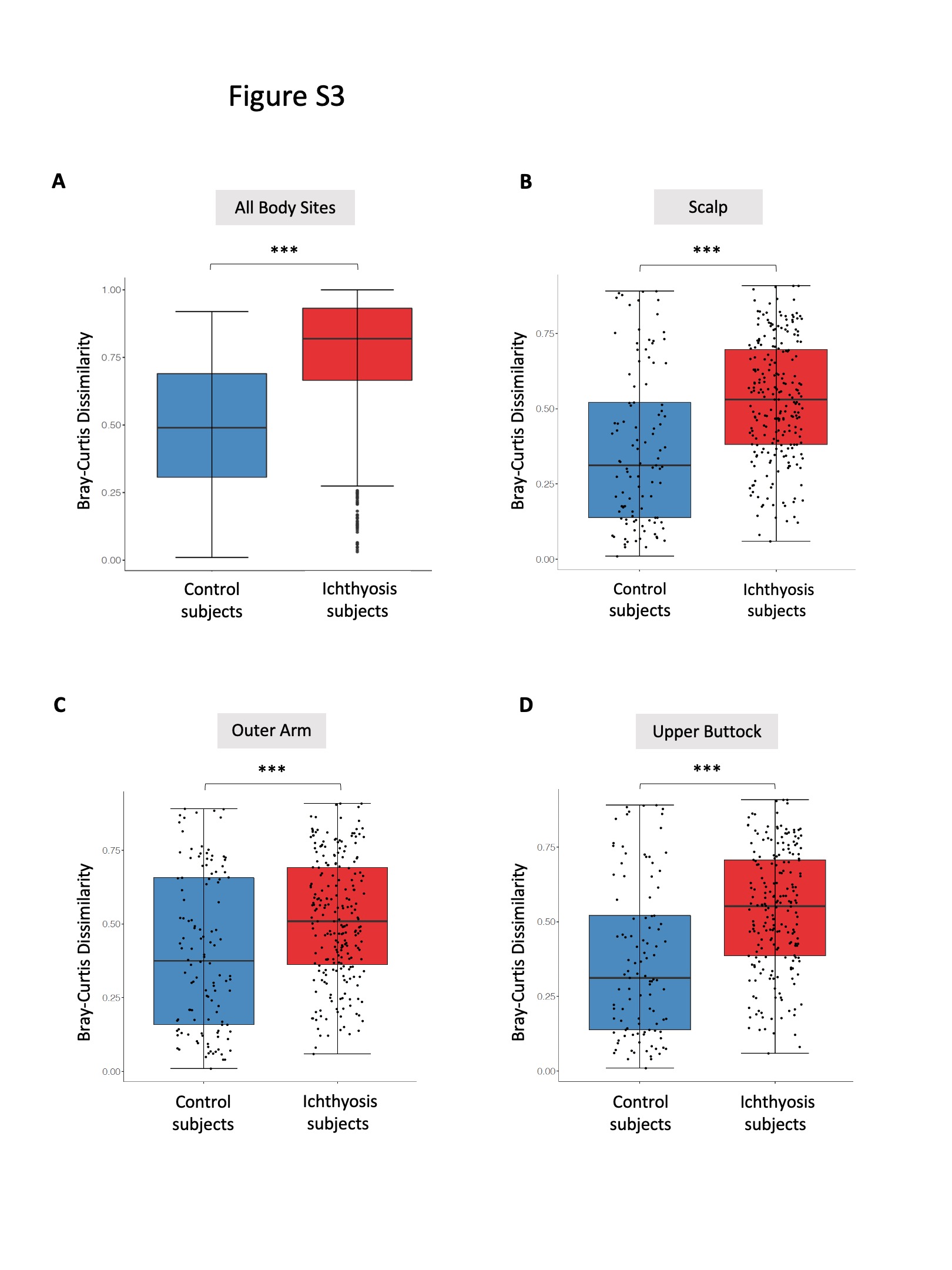{kind=link}
Figure S4 Plotting of principal coordinates analysis using Bray–Curtis dissimilarity index species level with samples from the (a) scalp, (b) outer arm and (c) upper buttock (C).
{kind=link}
Figure S5 Hierarchical clustering of ichthyosis and control skin microbiome profiles species level of samples from the (a) scalp, (b) outer arm and (c) upper buttock.
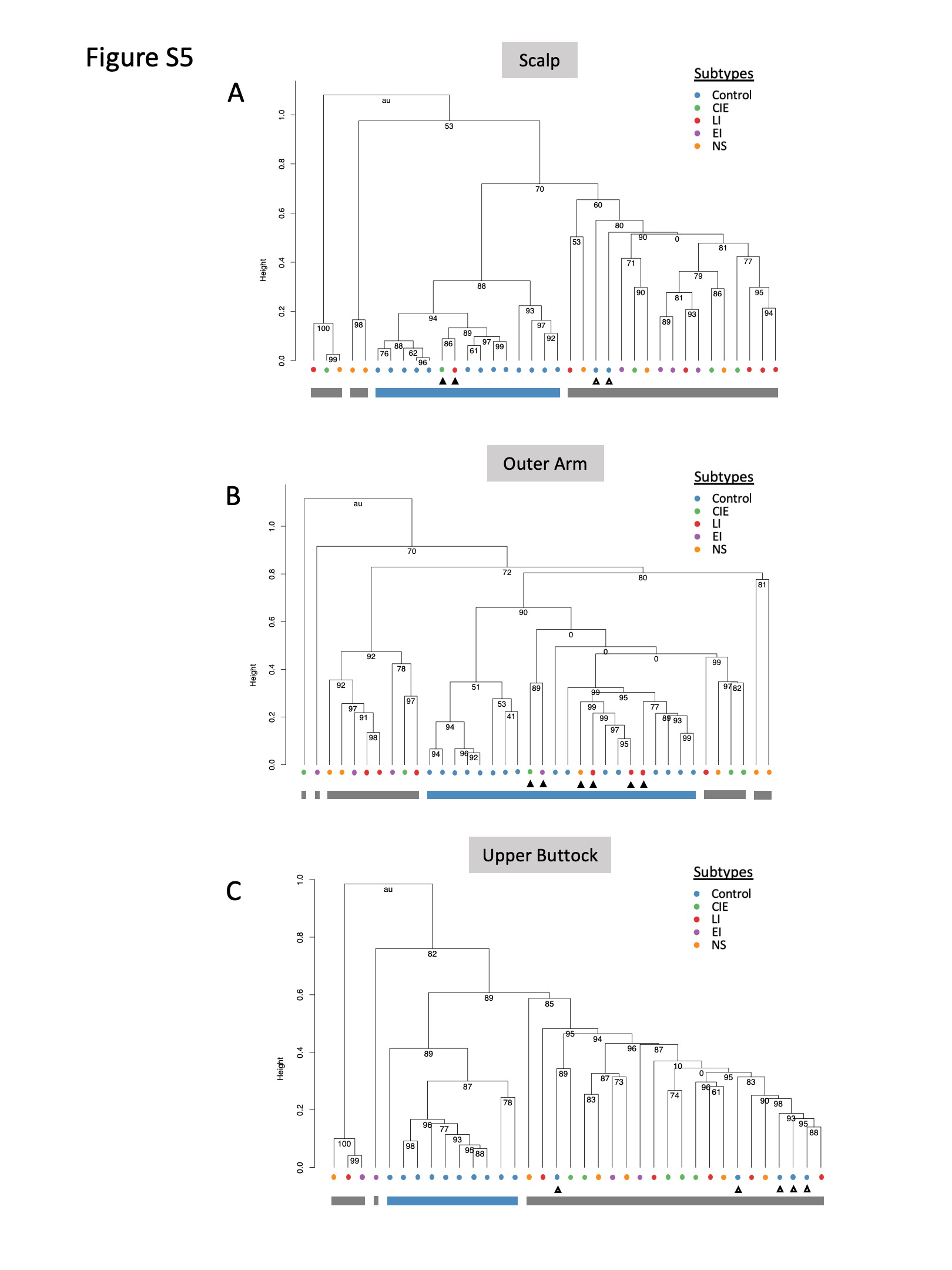{kind=link}
Figure S6 Major shifts in key community members in the skin of participants in the control and ichthyosis groups.
{kind=link}
Figure S7 Shannon’s diversity of control group vs. all the ichthyosis group (a) and for specific ichthyosis subtype (b) separated by body site.
{kind=link}
Figure S8 Simpson’s diversity of control group vs. all the ichthyosis group (a) and for specific ichthyosis subtype (b) separated by body site.
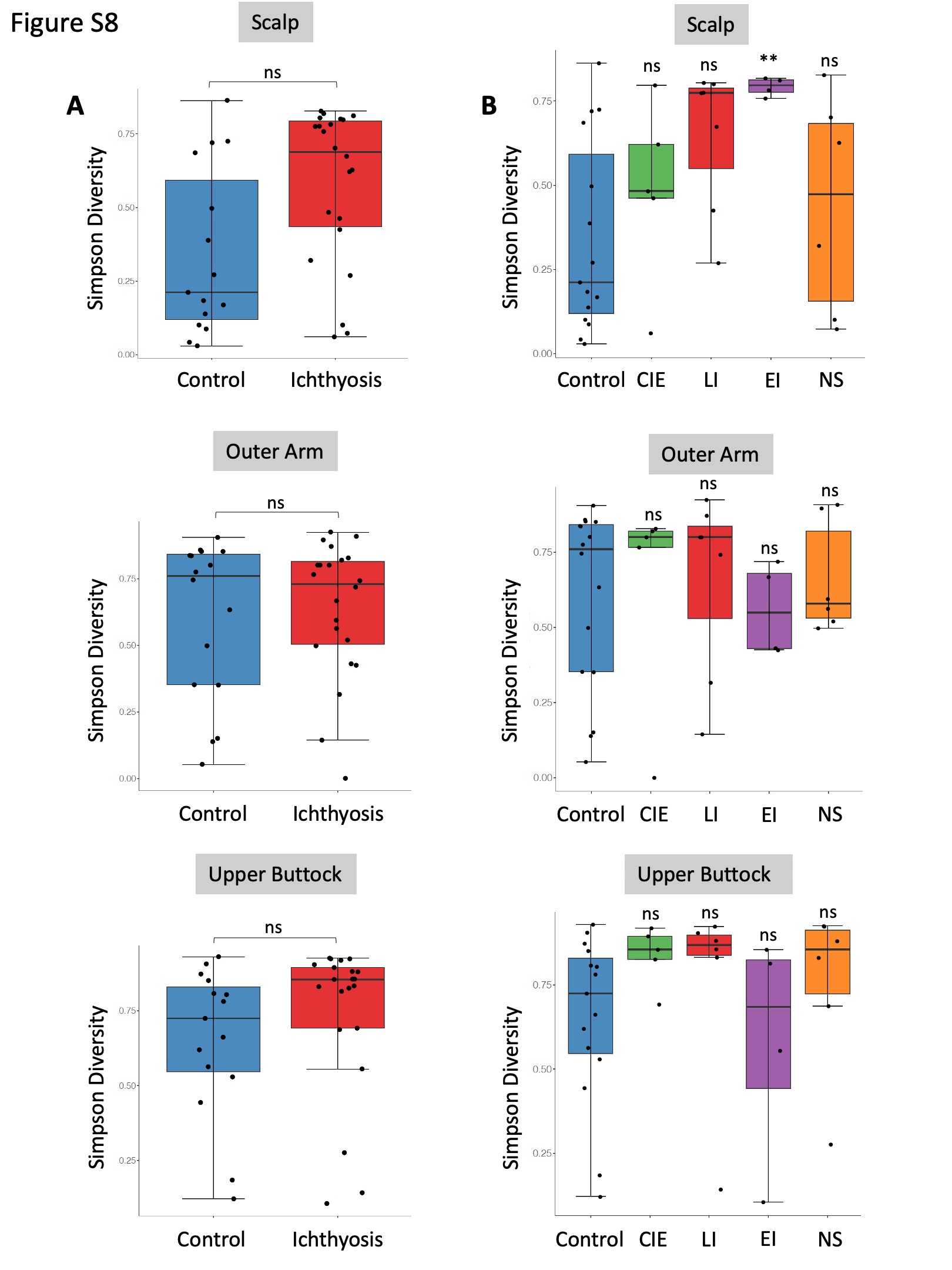{kind=link}
Figure S9 Major shifts in key community members, Malassezia globosa (a); Cutibacterium acnes (b), Staphylococcus spp. (c) and Corynebacterium spp. (d) in healthy control and ichthyotic skin displayed for each body site.
{kind=link}
Figure S10 Malassezia analysis for the control and ichthyosis groups.
{kind=link}
Table S1 Sequencing statistics from metagenomics.
Table S2 Individual demographic and clinical data.
Table S3 Spearman rank correlation analysis between microbes and participants’ disease scores.
Table S4 Relative abundance from MetaPhlAn2 to represent the contribution of specific microbes to the skin microbiome.
Table S5 CLC fungal genome reference database.
Table S6 (a) Relative abundance from CLC to represent the contribution of fungal microbes to the skin microbiome. (b) Sequencing statistics from the CLC database mapping.
Table S7 Trichophyton genomes from NCBI used to further analyse unclassified Trichophyton species.
Table S8 Raw and adjusted P-values for (a) species richness, (b) kingdom analysis of scalp, (c) outer arm and (d) upper buttock body sites.
Table S9 Multi-fastqc table of sequencing reads and statistics associated with each participant.
What is already known about this topic?
The skin microbiome of congenital ichthyoses is largely unexplored.
Microbes play an important role in pathogenesis, as infections are common.
The relative abundances of staphylococci and corynebacteria is increased in the cutaneous microbiome of patients with Netherton syndrome, but extension of these abundances to all congenital ichthyoses is unexplored.
What does this study add?
A common skin microbiome signature was observed across congenital ichthyoses.
Distinct microbiome features were associated with ichthyosis subtypes.
Changes in microbiome may contribute to T helper 17 cell immune polarization.
What is the translational message?
These data provide the basis for comparison of the microbiome with lipidomic and transcriptomic alterations in these forms of ichthyosis and consideration of correcting the dysbiosis as a therapeutic intervention.
Acknowledgments
We thank the participants for taking part in this study, Dr Stephen Wearne for project management support and Dr Keith Choate for genotyping some of the patients with ichthyosis.
Funding sources
This work was supported by funding from Agency for Science, Technology and Research (A*STAR) and A*STAR BMRC EDB IAF-PP grants – H17/01/a0/004 ‘Skin Research Institute of Singapore’ and H18/01a0/016 ‘Asian Skin Microbiome Program’ (K.-C.T., S.S.L., X.F.C.C.W., J.E.A.C.). Although not part of the trial, some of the patients were swabbed at baseline of a clinical trial (NCT03041038), funded as an investigator-initiated grant from Novartis CAIN457AUS05T (A.S.P., E.G.-Y.). Biosample management of samples in this study was through NIH P30AR075049 (Northwestern’s Skin Biology and Disease Resource-based Center/SBDRC) (A.S.P.). R.L. received partial salary funding from a National Psoriasis Foundation fellowship grant. B.L. and K.D. are part of the SIgN Immunomonitoring platform (supported by BMRC IAF 311006 grant, BMRC transition funds H16/99/b0/011, BMRC IAF-PP H19/01/a0/024 SIGNAL grant and NRF SIS NRF2017_SISFP09 grant).
Footnotes
Conflicts of interest
The authors declare they have no conflicts of interest.
Ethics statement
All participants provided written informed consent, approved by the Institutional Review Boards of Mount Saini Medical School and Northwestern University.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher’s website:
Data availability
All sequencing reads are available from the NCBI sequence read archive under accession number PRJNA814343.
References
- 1.Oji V, Tadini G, Akiyama M et al. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Soreze 2009. J Am Acad Dermatol 2010; 63:607–41. [DOI] [PubMed] [Google Scholar]
- 2.Vahlquist A, Fischer J, Torma H. Inherited nonsyndromic ichthyoses: an update on pathophysiology, diagnosis and treatment. Am J Clin Dermatol 2018; 19:51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown SJ, Relton CL, Liao H et al. Filaggrin haploinsufficiency is highly penetrant and is associated with increased severity of eczema: further delineation of the skin phenotype in a prospective epidemiological study of 792 school children. Br J Dermatol 2009; 161:884–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Craig WY, Roberson M, Palomaki GE et al. Prevalence of steroid sulfatase deficiency in California according to race and ethnicity. Prenat Diagn 2010; 30:893–8. [DOI] [PubMed] [Google Scholar]
- 5.Chavanas S, Bodemer C, Rochat A et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat Genet 2000; 25:141–2. [DOI] [PubMed] [Google Scholar]
- 6.Byrd AL, Belkaid Y, Segre JA. The human skin microbiome. Nat Rev Microbiol 2018; 16:143–55. [DOI] [PubMed] [Google Scholar]
- 7.Egawa G, Kabashima K. Multifactorial skin barrier deficiency and atopic dermatitis: Essential topics to prevent the atopic march. J Allergy Clin Immunol 2016; 138:350–8. [DOI] [PubMed] [Google Scholar]
- 8.Fyhrquist N, Muirhead G, Prast-Nielsen S et al. Microbe-host interplay in atopic dermatitis and psoriasis. Nat Commun 2019; 10:4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kong HH, Oh J, Deming C et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res 2012; 22:850–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tay ASL, Li C, Nandi T et al. Atopic dermatitis microbiomes stratify into ecologic dermotypes enabling microbial virulence and disease severity. J Allergy Clin Immunol 2021; 147:1329–40. [DOI] [PubMed] [Google Scholar]
- 11.Paller AS, Kong HH, Seed P et al. The microbiome in patients with atopic dermatitis. J Allergy Clin Immunol 2019; 143:26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geoghegan JA, Irvine AD, Foster TJ. Staphylococcus aureus and atopic dermatitis: a complex and evolving relationship. Trends Microbiol 2018; 26:484–97. [DOI] [PubMed] [Google Scholar]
- 13.Nakatsuji T, Chen TH, Narala S et al. Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci Transl Med 2017; 9:4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakatsuji T, Gallo RL, Shafiq F et al. Use of autologous bacteriotherapy to treat staphylococcus aureus in patients with atopic dermatitis: a randomized double-blind clinical trial. JAMA Dermatol 2021; 157:978–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakatsuji T, Hata TR, Tong Y et al. Development of a human skin commensal microbe for bacteriotherapy of atopic dermatitis and use in a phase 1 randomized clinical trial. Nat Med 2021; 27:700–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams MR, Costa SK, Zaramela LS et al. Quorum sensing between bacterial species on the skin protects against epidermal injury in atopic dermatitis. Sci Transl Med 2019; 11:8329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Czarnowicki T, He H, Leonard A et al. The major orphan forms of ichthyosis are characterized by systemic T-cell activation and Th-17/Tc-17/Th-22/Tc-22 polarization in blood. J Invest Dermatol 2018; 138:2157–67. [DOI] [PubMed] [Google Scholar]
- 18.Malik K, He H, Huynh TN et al. Ichthyosis molecular fingerprinting shows profound TH17 skewing and a unique barrier genomic signature. J Allergy Clin Immunol 2019; 143:604–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paller AS, Renert-Yuval Y, Suprun M et al. An IL-17-dominant immune profile is shared across the major orphan forms of ichthyosis. J Allergy Clin Immunol 2017; 139:152–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miao H, Dong R, Zhang S et al. Inherited ichthyosis and fungal infection: an update on pathogenesis and treatment strategies. J Dtsch Dermatol Ges 2021; 19:341–50. [DOI] [PubMed] [Google Scholar]
- 21.Moosbrugger-Martinz V, Hackl H, Gruber R et al. Initial evidence of distinguishable bacterial and fungal dysbiosis in the skin of patients with atopic dermatitis or Netherton syndrome. J Invest Dermatol 2021; 141:114–23. [DOI] [PubMed] [Google Scholar]
- 22.Williams MR, Cau L, Wang Y et al. Interplay of staphylococcal and host proteases promotes skin barrier disruption in Netherton syndrome. Cell Rep 2020; 30:2923–33e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wong X, Denil S, Foo JN et al. Array-based sequencing of filaggrin gene for comprehensive detection of disease-associated variants. J Allergy Clin Immunol 2018; 141:814–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Segata N, Waldron L, Ballarini A et al. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat Methods 2012; 9:811–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzuki R, Shimodaira H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 2006; 22:1540–2. [DOI] [PubMed] [Google Scholar]
- 26.Grice EA, Kong HH, Conlan S et al. Topographical and temporal diversity of the human skin microbiome. Science 2009; 324:1190–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakagawa S, Matsumoto M, Katayama Y et al. Staphylococcus aureus virulent PSMalpha peptides induce keratinocyte alarmin release to orchestrate IL-17-dependent skin inflammation. Cell Host Microbe 2017; 22:667–77e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sparber F, De Gregorio C, Steckholzer S et al. The skin commensal yeast Malassezia triggers a Type 17 response that coordinates antifungal immunity and exacerbates skin inflammation. Cell Host Microbe 2019; 25:389–403e6. [DOI] [PubMed] [Google Scholar]
- 29.Tett A, Pasolli E, Farina S et al. Unexplored diversity and strain-level structure of the skin microbiome associated with psoriasis. NPJ Biofilms Microbiomes 2017; 3:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Goh BN, Teh WK et al. Skin commensal malassezia globosa secreted protease attenuates Staphylococcus aureus biofilm formation. J Invest Dermatol 2018; 138:1137–45. [DOI] [PubMed] [Google Scholar]
- 31.Nakamura K, O’Neill AM, Williams MR et al. Short chain fatty acids produced by Cutibacterium acnes inhibit biofilm formation by Staphylococcus epidermidis. Sci Rep 2020; 10:21237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heinen MP, Cambier L, Antoine N et al. Th1 and Th17 immune responses act complementarily to optimally control superficial dermatophytosis. J Invest Dermatol 2019; 139:626–37. [DOI] [PubMed] [Google Scholar]
- 33.Chng KR, Tay AS, Li C et al. Whole metagenome profiling reveals skin microbiome-dependent susceptibility to atopic dermatitis flare. Nat Microbiol 2016; 1:16106. [DOI] [PubMed] [Google Scholar]
- 34.Sonesson A, Przybyszewska K, Eriksson S et al. Identification of bacterial biofilm and the Staphylococcus aureus derived protease, staphopain, on the skin surface of patients with atopic dermatitis. Sci Rep 2017; 7:8689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Neill AM, Nakatsuji T, Hayachi A et al. Identification of a human skin commensal bacterium that selectively kills Cutibacterium acnes. J Invest Dermatol 2020; 140:1619–28e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parlet CP, Brown MM, Horswill AR. Commensal staphylococci influence Staphylococcus aureus skin colonization and disease. Trends Microbiol 2019; 27:497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Naik S, Bouladoux N, Linehan JL et al. Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature 2015; 520:104–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ridaura VK, Bouladoux N, Claesen J et al. Contextual control of skin immunity and inflammation by Corynebacterium. J Exp Med 2018; 215:785–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dufour P, Jarraud S, Vandenesch F et al. High genetic variability of the agr locus in Staphylococcus species. J Bacteriol 2002; 184:1180–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sparber F, LeibundGut-Landmann S. Interleukin-17 in antifungal immunity. Pathogens 2019; 8:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakamura T, Nishibu A, Yasoshima M et al. Analysis of Trichophyton antigen-induced contact hypersensitivity in mouse. J Dermatol Sci 2012; 66:144–53. [DOI] [PubMed] [Google Scholar]
- 42.Juntachai W, Oura T, Murayama SY et al. The lipolytic enzymes activities of Malassezia species. Med Mycol 2009; 47:477–84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Study design. Schematic of study design indicating the criteria for participant recruitment and the data and sample collection process.
Figure S2 Description of the Ichthyosis Area and Severity Index tool that was used to categorize the severity of condition of the participants in the study.
Figure S3 Box plots showing the Bray–Curtis dissimilarity of taxonomic profiles from all body sites (a) and separated body sites (b, c, d) within control participants and within participants with ichthyosis.
Figure S4 Plotting of principal coordinates analysis using Bray–Curtis dissimilarity index species level with samples from the (a) scalp, (b) outer arm and (c) upper buttock (C).
Figure S5 Hierarchical clustering of ichthyosis and control skin microbiome profiles species level of samples from the (a) scalp, (b) outer arm and (c) upper buttock.
Figure S6 Major shifts in key community members in the skin of participants in the control and ichthyosis groups.
Figure S7 Shannon’s diversity of control group vs. all the ichthyosis group (a) and for specific ichthyosis subtype (b) separated by body site.
Figure S8 Simpson’s diversity of control group vs. all the ichthyosis group (a) and for specific ichthyosis subtype (b) separated by body site.
Figure S9 Major shifts in key community members, Malassezia globosa (a); Cutibacterium acnes (b), Staphylococcus spp. (c) and Corynebacterium spp. (d) in healthy control and ichthyotic skin displayed for each body site.
Figure S10 Malassezia analysis for the control and ichthyosis groups.
Table S1 Sequencing statistics from metagenomics.
Table S2 Individual demographic and clinical data.
Table S3 Spearman rank correlation analysis between microbes and participants’ disease scores.
Table S4 Relative abundance from MetaPhlAn2 to represent the contribution of specific microbes to the skin microbiome.
Table S5 CLC fungal genome reference database.
Table S6 (a) Relative abundance from CLC to represent the contribution of fungal microbes to the skin microbiome. (b) Sequencing statistics from the CLC database mapping.
Table S7 Trichophyton genomes from NCBI used to further analyse unclassified Trichophyton species.
Table S8 Raw and adjusted P-values for (a) species richness, (b) kingdom analysis of scalp, (c) outer arm and (d) upper buttock body sites.
Table S9 Multi-fastqc table of sequencing reads and statistics associated with each participant.
Data Availability Statement
All sequencing reads are available from the NCBI sequence read archive under accession number PRJNA814343.