

Abstract
Objective
The phenotypic and genotypic spectrum of adult patients with epilepsy and intellectual disability (ID) is less clear than in children. We investigated an adult patient cohort to further elucidate this and inform the genetic testing approach.
Methods
Fifty‐two adult patients (30 male, 22 female) with epilepsy, at least mild ID and no known genetic or acquired cause were included and phenotyped. Variants identified through exome sequencing were evaluated using ACMG criteria. Identified variants were compared with commercially available gene panels. Cluster analysis of two features, age at seizure onset and age at ascertainment of cognitive deficits, was performed.
Results
Median age was 27 years (range 20‐57 years) with median seizure onset at 3 years and median ascertainment of cognitive deficits at 1 year. Likely pathogenic/pathogenic variants were identified in 16/52 patients (31%) including 14 (27%) single nucleotide variants and 2 (4%) copy number variants. Simulated yield of commercial gene panels varied between 13% in small (≤144 genes) and 27% in large panels (≥1478 genes).
Cluster analysis (optimal number 3 clusters) identified a cluster with early seizure onset and early developmental delay (developmental and epileptic encephalopathy, n = 26), a cluster with early developmental delay but late seizure onset (ID with epilepsy, n = 16) and a third cluster with late ascertainment of cognitive deficits and variable seizure onset (n = 7). The smaller gene panels particularly missed the genes identified in the cluster with early ascertainment of cognitive deficits and later onset of epilepsy (0/4) as opposed to the cluster with developmental and epileptic encephalopathy (7/10).
Significance
Our data indicates that adult patients with epilepsy and ID represent a heterogeneous cohort that includes grown‐up patients with DEE but also patients with primary ID and later onset of epilepsy. To maximize diagnostic yield in this cohort either large gene panels or exome sequencing should be used.
Keywords: adults, developmental and epileptic encephalopathy, epilepsy, genetic testing, genetics, intellectual disability, phenotype
Key points.
The phenotypic and genotypic spectrum of adult patients with epileptic seizures and intellectual disability is not well described
Our study shows that cohorts of adult patients with intellectual disability and epilepsy are heterogeneous
There are patients with developmental and epileptic encephalopathy and others with primary intellectual disability and later seizure onset
Gene panels with ≤511 genes miss a significant number of causative genes in adult patients with epilepsy and intellectual disability
To increase the diagnostic yield either large gene panels or exome sequencing should be used in this cohort
1. INTRODUCTION
Epilepsy is one of the most frequent chronic neurological disorders and affects more than 70 million people of all age groups worldwide. 1 Epilepsy can have different etiologies, but genetic factors play a role in a significant proportion of patients. Clinical genetic testing is widely available and primarily focuses on patients with suspected monogenic etiology. This includes patients with developmental and epileptic encephalopathy (DEE), which typically arises due to de novo mutations. 2 Next‐generation sequencing, particularly gene panel sequencing approaches, are still commonly used to screen for single nucleotide variants (SNV), but the gene content of the panels varies between different providers. Exome sequencing using a trio approach is thus more powerful as it is not biased by a preselected list of genes and can identify de novo pathogenic variants in the affected child by comparing the genetic information from the patient and parents. However, due to significantly higher costs and the requirement of DNA of both biological parents, it is less accessible.
Most studies looking at diagnostic yield and genes implicated in epilepsy have been done in the pediatric population. Historically, children have more readily accessed genetic services, which are often still under the purview of pediatrics in most jurisdictions. In children with early onset of refractory seizures associated with developmental delay, gene panel testing has become clinical standard with diagnostic yield of around 30%. 3 Chromosomal microarray is used to identify structural chromosomal variants (copy number variants, CNVs) with the highest diagnostic yield of 10.9% in patients with epilepsy and additional comorbid features such as intellectual disability or other conditions. 4 Exome sequencing, ideally in a trio‐based approach, allows an unbiased evaluation of all coding regions and is also able to identify CNVs.
Less is known about the phenotypic and genotypic spectrum of adult patients with epileptic seizures and intellectual disability (ID). These may include grown‐up patients with DEE but also patients with ID (developmental encephalopathy) and later onset of epilepsy. Particularly, the relative contribution of the latter group and its implications for genetic testing has not been clearly delineated yet. These groups of adult patients can be differentiated based on the relationship between age of onset of epilepsy and age of ascertainment of cognitive deficits.
Previous studies investigating adult patients with epilepsy and ID or DEE using gene panel approaches including between 45‐580 genes reported diagnostic rates of 21.8%‐27%. 5 , 6 Studies using exome sequencing or a gene panel approach and/or chromosomal microarray followed by exome sequencing in unsolved cases reported diagnostically relevant results in 25.3%‐47.4%. 7 , 8 , 9 , 10 The yield was significantly lower in cohorts of adult epilepsy patients with lower proportion of reported ID (10.9%‐11%) 11 , 12 , 13 but the use of small panels and the omission of CNV analysis may have confounded the results.
The aim of this study was to clarify the phenotypic and genotypic spectrum of adult patients with epilepsy and ID presenting at two level 4 epilepsy centers using an exome sequencing approach to inform the best diagnostic approach in similar cohorts.
2. MATERIALS AND METHODS
2.1. Probands
Adult patients with a diagnosis of epilepsy were recruited for genetic research from the epilepsy clinics and seizure monitoring units at the two participating sites in Germany, the Epilepsy Center Frankfurt Rhine‐Main and the Epilepsy Center Hessen‐Marburg between January 2014 and September 2019. We also aimed to recruit affected and unaffected first‐degree relatives, particularly parents. DNA was extracted from peripheral blood or saliva samples.
The cohort for this study was selected from the recruited participants based on the following inclusion criteria: (1) Diagnosis of epilepsy, (2) age > 18 years at the time of last phenotypic information, (3) at least mild ID, (4) no known genetic or acquired cause explaining the epilepsy and ID, and (5) not included in prior genetic research projects. The severity of intellectual disability was classified according to ICD‐10. Patients with a documented IQ between 50 and 69 or who had difficulties in school but were able to work and had social contacts were classified as mild ID. Patients with a documented IQ of 35‐49 or who were able to communicate but required support for work and during daily life were classified as having moderate ID. Patients with a documented IQ between 20 and 34 or who were unable to read and write, attended a special school and required continuous support were classified as severe ID. Patients with a documented IQ <20 who had no language and were significantly limited in personal care, continence, and movement were classified as having profound ID. Patients with clear history of an acquired cause were excluded. However, a lesional MRI was not an exclusion criterion.
Phenotyping was performed during outpatient visits to the epilepsy clinics and/or inpatient admissions to the seizure monitoring units of the participating sites. Additional data was acquired via telephone interviews. MRI results were classified into epileptogenic lesion (hippocampal sclerosis, cortical lesions including dysplasia and long‐term epilepsy‐associated tumor, bilateral periventricular nodular heterotopia) and normal/nonepileptic abnormalities (atrophy, nonspecific white matter lesions, cerebellar or thalamic lesions). Traumatic structural lesions acquired after epilepsy onset were also included in the latter group. Response to antiseizure medication was classified according to ILAE criteria. 14
2.2. Genotyping
Genotyping was performed at the Cologne Center for Genomics, Germany using the Agilent Sureselect Human All Exon V6 r2 enrichment kit. Sequencing was performed in two batches using an Illumina HiSeq 4000 (2 × 76bp runs) for batch 1 and an Illumina NovaSeq 6000 (2 × 101bp runs, S2 and S4 flowcells) for batch 2. Batch 1 consisted of 66 samples including 15 trio exomes and 21 singleton exomes. Batch 2 consisted of 37 samples including 7 trios (one of these with additional two siblings), 1 proband with one parent and one sibling, 3 probands with one parent, and 5 singleton exomes.
2.3. Genetic analysis
Next‐generation sequencing data was processed using the Varbank pipeline (https://varbank.ccg.uni‐koeln.de) including alignment to the hg19 (batch 1) and hg38 reference genome (batch 2 and CNV analysis) followed by variant calling with GATK. 15 The samples were analyzed using multiple strategies. De novo variants in trio exomes were identified via analysis of the BAM files using denovogear. 16 Homozygous variants in trio exome data were identified using the online platform Varbank (https://varbank.ccg.uni‐koeln.de).
All samples were additionally analyzed using a comprehensive virtual gene panel using a custom script for heterozygous dominant, homozygous, and compound heterozygous recessive as well as hemizygous variants. The panel was constructed using gene associations from the Human Phenotype Ontology 17 for the HPO terms “Seizure” (1501 associated genes, accessed May 5, 2020, https://hpo.jax.org/app/browse/term/HP:0001250) and “Neurodevelopmental abnormality” (2171 genes, accessed May 5, 2020, https://hpo.jax.org/app/browse/term/HP:0012759, includes child terms “Neurodevelopmental delay”, “Intellectual disability”, “Specific learning disability”, “Developmental stagnation” and “Developmental regression”). The gene lists were then combined resulting in a total of 2364 genes. Variants were annotated using Annovar 18 followed by filtering for exonic or splice‐site location and frequency in control cohorts (gnomAD 19 ). Deleterious copy number variants >50 kb were identified using the Varbank pipeline with filtering for overlapping variations including XHMM 20 (ZRD < ‐3.3), CoNIFER 21 (ZPRKM<‐1.5) and ExomeDepth 22 (RT <0.2).
The identified variants were manually reviewed regarding alignment plots (Varbank and Varbank 2), gene‐disease validity, mode of inheritance (autosomal or X‐chromosomal dominant or recessive, compound heterozygous), and functional effect. Possible pathogenic SNVs were validated using Sanger sequencing in patients and all available relatives. In some cases, additional relatives had been recruited since performing the exome sequencing and were included into the Sanger sequencing validation. Variants were manually classified according to ACMG criteria 23 incorporating the interpretation of Franklin by Genoox (https://franklin.genoox.com).
2.4. Clustering
Clustering analysis was performed on two continuous features, age of onset of epilepsy and age of ascertainment of cognitive deficits. These features were chosen as their relationship reflects if patients have DEE, where the onset of developmental delay closely follows seizure onset, or if they have primary developmental delay with later onset of seizures. As we included two continuous variables without high dimensionality, k‐means clustering was used. The analysis was performed in R with multiple random seeds using the stats package. Forty‐nine probands where information on timing of developmental delay and epilepsy was available were included. The optimal number of clusters between 2 and 10 was chosen using the Silhouette method (cluster package). The identified clusters and the cohorts of patients with and without an identified genetic cause were compared using the tbl_summary function (gtsummary package).
2.5. Simulated gene panels
The genes with identified variants were compared with commercially available gene panels to simulate the diagnostic yield of these panels. The content of the commercial gene panels was obtained from the respective provider's website. The websites were accessed on April 23, 2022 (Ambry Genetics EpilepsyNext panel, Ambry Genetics NeurodevelopmentNext‐Expanded panel, Blueprint Comprehensive Epilepsy Gene panel, Invitae Neurodevelopmental Disorders panel, Invitae Epilepsy panel, Prevention Comprehensive Epilepsy, and Seizure panel) and on May 1, 2022 (GeneDx Comprehensive Epilepsy Panel).
2.6. Ethics declaration
This study was performed in line with the principles of the Declaration of Helsinki and was approved by the ethics committees of the Philipps‐University Marburg (86/13, 163/14) and Goethe University Frankfurt (132/15). All participants or their legal guardians provided written informed consent prior to inclusion in the study.
3. RESULTS
3.1. Cohort description
Out of 1413 patients with epilepsy recruited for genetic research, 52 unrelated probands (30 male, 22 female) who fulfilled the inclusion criteria were included (Figure 1). These 52 patients underwent exome sequencing together with 51 relatives. Phenotypic details of the included probands are provided in Table 1. Median age was 27 years (range 20‐57 years). Median age of seizure onset was 3.0 years with a range from 0‐26 years. Fourteen probands had seizure onset within the first year of life and 19 after age 6 years. Median age of ascertainment of cognitive deficits was 1.0 years with a range from 0‐12 years. Twelve patients had focal epilepsy, 8 multifocal epilepsy, 18 generalized epilepsy, and in 14 patients the epilepsy classification was unknown. Ten had an epileptogenic lesion on MRI. Thirty‐four were drug resistant.
FIGURE 1.
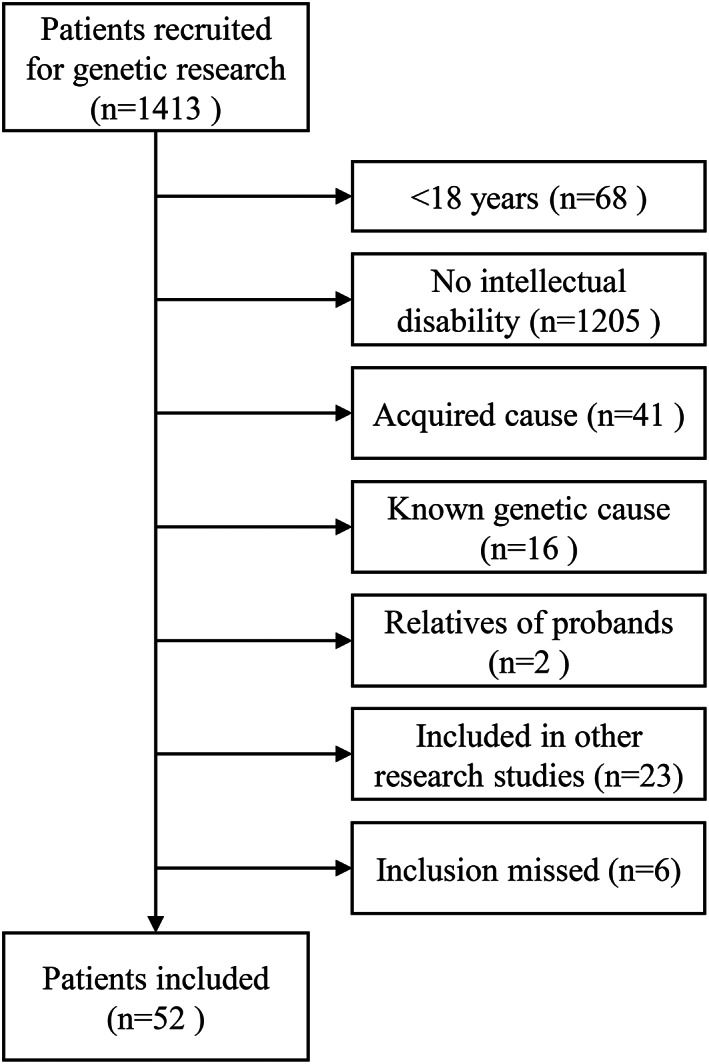
CONSORT diagram for study recruitment. Known and excluded genetic etiologies were: Angelman syndrome, CDKL5 encephalopathy, Dravet syndrome (SCN1A, 3x), familial focal epilepsy with variable foci with focal cortical dysplasia (DEPDC5), genetic epilepsy with febrile seizures plus (SCN1A), Kabuki syndrome, Klinefelter syndrome, linear sebaceous nevus syndrome, microcephaly (CENPJ), progressive myoclonus epilepsy, trisomy 21, tuberous sclerosis (2x).
TABLE 1.
Phenotypic data of the whole study cohort and comparison of patients with and without identified genetic cause.
| Characteristic | Whole cohort | Patients without identified genetic cause | Patients with identified genetic cause | Comparison identified vs. no identified genetic cause |
|---|---|---|---|---|
| n = 52 a | n = 36 a | n = 16 a | P‐value b | |
| Age | 27 (range 20‐57, IQR 23‐33) | 29 (range 20‐57, IQR 26‐34) | 24 (range 21‐46, IQR 23‐30) | 0.13 |
| Sex | 0.049 | |||
| Male | 30 (58%) | 24 (67%) | 6 (38%) | |
| Female | 22 (42%) | 12 (33%) | 10 (62%) | |
| Epilepsy type | 0.2 | |||
| Focal | 12 (32%) | 10 (42%) | 2 (14%) | |
| Multifocal | 8 (21%) | 5 (21%) | 3 (21%) | |
| Generalized | 18 (47%) | 9 (38%) | 9(64%) | |
| Unknown | 14 | 12 | 2 | |
| Epileptogenic lesion on MRI | 10 (24%) | 9 (33%) | 1 (7.1%) | 0.12 |
| Unknown | 11 | 9 | 2 | |
| Febrile seizures | 8 (17%) | 4 (12%) | 4 (25%) | 0.4 |
| Unknown | 4 | 4 | 0 | |
| Age of onset – Epilepsy | 3.0 (range 0‐26, IQR 0.6‐10.0) | 4.5 (range 0.1‐26, IQR 0.6‐10.0) | 2.8 (range 0‐18, IQR 1.6‐10.2) | 0.8 |
| Unknown | 2 | 2 | 0 | |
| Age of ascertainment – Cognitive deficits | 1.00 (range 0‐12, IQR 0.25‐3.00) | 1.50 (range 0‐12, IQR 0.40‐3.00) | 0.88 (range 0‐8, IQR 0.08‐2.62) | 0.4 |
| Unknown | 3 | 3 | 0 | |
| Seizure frequency at last follow‐up (over last 3 months) | 0.4 | |||
| No seizures | 21 (40%) | 13 (36%) | 8 (50%) | |
| <1/month | 1 (1.9%) | 0 (0%) | 1 (6.2%) | |
| 1/week – 1/month | 8 (15%) | 6 (17%) | 2 (12%) | |
| 1/day – 1/week | 6 (12%) | 4 (11%) | 2 (12%) | |
| Daily | 16 (31%) | 13 (36%) | 3 (19%) | |
| Seizure free for 1 year c | 12 (23%) | 7 (19%) | 5 (31%) | 0.5 |
| Drug resistant | 34 (65%) | 24 (67%) | 10 (62%) | 0.8 |
| Degree of intellectual disability | 0.8 | |||
| Mild | 14 (27%) | 11 (31%) | 3 (19%) | |
| Moderate | 18 (35%) | 11 (31%) | 7 (44%) | |
| Severe | 10 (19%) | 7 (19%) | 3 (19%) | |
| Profound | 10 (19%) | 7 (19%) | 3 (19%) | |
| Family history of epilepsy | 17 (33%) | 13 (36%) | 4 (25%) | 0.4 |
| Genetic cause identified | 16 (31%) | 0 (0%) | 16 (100%) |
Median (range, IQR: interquartile range); n (%).
Wilcoxon rank sum test; Pearson's Chi‐squared test; Fisher's exact test.
Patients with ongoing continuous spike‐and‐wave during sleep were not considered seizure free.
DNA for segregation analysis was available from both parents in 26 of 52 probands (50%) and from one parent in 9 probands (17%). The other parent had passed away in two cases, could not be reached in two cases and decided not to participate in three cases. No parental DNA was available in 17 probands (32%). Parents could not be reached in one case and decided not to participate in 4 cases. In the remaining cases no further recruitment efforts were undertaken as no candidate variants had been identified.
At the time of recruitment, prior clinical genetic test results for epilepsy were available in 19 patients (37%) including 13 epilepsy gene panels, 9 chromosomal microarrays, and 4 karyotypes. Two patients had sequencing for SCN1A or SLC2A1 only. These tests did not reveal a cause for the epilepsy or ID. No genetic test results for epilepsy were available in the other 33 patients (63%).
3.2. Genetic analysis
Exome sequencing was done with a mean coverage of 66‐108x (mean 82x) in batch 1 and 56‐119x (mean 87x) in batch 2. Pathogenic or likely pathogenic SNVs were identified and confirmed by Sanger sequencing in 14 probands (Table S1). This included 9 individuals with heterozygous autosomal variants (2xSCN1A, CACNA1A, CHD2, CTCF, KDM6B, NUS1, PACS2, SYNGAP1) of which 3 were proven to be de novo (KDM6B, SCN1A, SYNGAP1), 1 individual with a de novo heterozygous X‐chromosomal variant (NEXMIF; based on the diagnosis in this study this patient was also included into a collaborative study 24 ) and 4 individuals with homozygous variants (ALDH7A1, NSUN2, TANGO2, TDP2). The patient with the ALDH7A1 variant was on pyridoxine supplementation when he presented to the adult service. His mother reported that the supplementation was started after a status epilepticus episode at age 46 days with some benefit. However, as a formal diagnosis or reports from that time were unavailable, he was included into the study. One variant (NSUN2) was identified in a patient who had negative epilepsy gene panel sequencing previously. In addition, two CNVs were identified in the cohort (del 6q21‐22.31, de novo del 9q34.11, Figure S1).
The total diagnostic rate in our cohort using exome sequencing was 31% (16/52). The diagnostic yield for patients with mild ID was 21% (3/14), for moderate ID 39% (7/18), for severe ID 30% (3/10), and profound ID 30% (3/10). Thirteen of the identified 14 SNVs were included in the list of 1501 genes created using the HPO term “Seizure.” Only CTCF was not included in this list but was included in the 2171 genes associated with the HPO term “Neurodevelopmental abnormality.”
The availability of trio exome sequencing data reduced the number of variants that needed to be segregated by Sanger sequencing but did not result in a higher diagnostic rate. In total, pathogenic or likely pathogenic SNVs were identified in 5/22 (23%) of trio exomes, 7/26 (27%) single exomes, and 2/4 exomes (50%) with 1 parent. However, in several of these parental DNA became available later for Sanger segregation analysis. Overall, a pathogenic or likely pathogenic SNV was identified in 3/17 patients (18%) where no parental DNA was eventually available, in 4/9 (44%) where only one parent was available, and in 7/26 (27%) where both parents were available. All SNVs were also found on the virtual panel analysis including the 2364 genes associated with the HPO terms “Seizure” or “Neurodevelopmental abnormality”.
3.3. Comparison of phenotype between patients with and without identified genetic cause
There were no significant differences in the examined phenotypic features between these cohorts except for a higher frequency of genetic diagnosis in females (Table 1).
3.4. Phenotypic clusters
Clustering analysis of the two features, age of onset of epilepsy and age of ascertainment of cognitive deficits, resulted in an ideal cluster number of 3 (Silhouette score of 0.525, 1: highly dense and separated clusters, 0: overlapping clusters, −1: incorrect clusters, Figure 2, Table 2). Cluster A included patients with early onset of epilepsy (median 1.5 years, range 0‐5 years) and developmental delay (median 1.25 years, range 0‐5 years), i.e., DEE (n = 26). Cluster B (ID with epilepsy, n = 16) included patients with early ascertainment of cognitive deficits (median 0.38 years, range 0‐3 years) but later onset of epilepsy (median 11.5 years, range 8‐26 years) and cluster C (n = 7) included patients with late ascertainment of cognitive deficits (median 8 years, range 5.5‐12 years) and variable onset of epilepsy (median 6 years, range 0.5‐12 years). Comparison of several phenotypic features between cluster A and B revealed no other phenotypic differences except for age of onset of the epilepsy (Table 2). Most pathogenic variants in this study were identified in cluster A and B with no significant difference in diagnostic rates between the clusters (cluster A: 38% vs. cluster B: 31%, P = 0.6). One pathogenic CNV (14%) was identified in cluster C.
FIGURE 2.
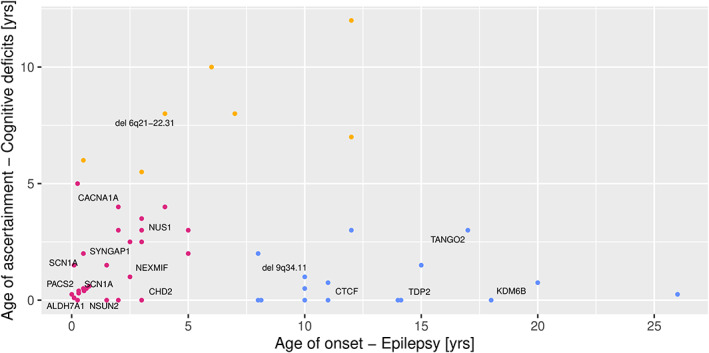
Cluster analysis of the study cohort. The analysis identified 3 clusters. Cluster A (shown in red) represents developmental and epileptic encephalopathy. Cluster B (shown in blue) represents intellectual disability with later onset of epilepsy. Cluster C (yellow) consists of patients with late ascertainment of cognitive deficits and variable onset of epilepsy.
TABLE 2.
Phenotypic data of cluster A (Developmental and epileptic encephalopathy), cluster B (Intellectual disability with epilepsy) and cluster C (Late ascertainment of cognitive deficits with variable onset of epilepsy).
| Cluster A Developmental and epileptic encephalopathy | Cluster B Intellectual disability with epilepsy | Comparison of cluster A and B | Cluster C Late ascertainment of cognitive deficits | |
|---|---|---|---|---|
| n = 26 a | n = 16 a | P‐value b | N = 7 a | |
| Age | 27 (range 21‐57, IQR 24‐33) | 22 (range 21‐36, IQR 22‐32) | 0.084 | 37 (range 20‐46, IQR 28‐43) |
| Sex | 0.3 | |||
| Male | 12 (46%) | 10 (62%) | 6 (86%) | |
| Female | 14 (54%) | 6 (38%) | 1 (14%) | |
| Epilepsy type | 0.4 | |||
| Focal | 5 (25%) | 5 (50%) | 1 (20%) | |
| Multifocal | 5 (25%) | 2 (20%) | 1 (20%) | |
| Generalized | 10 (50%) | 3 (30%) | 3 (60%) | |
| Unknown | 6 | 6 | 2 | |
| Epileptogenic lesion | 6 (27%) | 2 (15%) | 0.7 | 1 (25%) |
| Unknown | 4 | 3 | 3 | |
| Febrile seizures | 3 (12%) | 1 (6.2%) | >0.9 | 3 (60%) |
| Unknown | 1 | 0 | 2 | |
| Age of onset ‐ Epilepsy | 1.5 (range 0‐5, IQR 0.3‐2.9) | 11.5 (range 8‐26, IQR 10.0‐15.5) | <0.001 | 6.0 (range 0.5‐12, IQR 3.5‐9.5) |
| Age of ascertainment – Cognitive deficits | 1.25 (range 0‐5, IQR 0.32‐2.88) | 0.38 (range 0‐3, IQR 0.00‐1.12) | 0.065 | 8.00 (range 5.5‐12, IQR 6.50‐9.00) |
| Seizure frequency at last follow‐up (over last 3 months) | 0.5 | |||
| No seizures | 9 (35%) | 9 (56%) | 2 (29%) | |
| <1/month | 1 (3.8%) | 0 (0%) | 0 (0%) | |
| 1/week – 1/month | 4 (15%) | 1 (6.2%) | 1 (14%) | |
| 1/day – 1/week | 3 (12%) | 3 (19%) | 0 (0%) | |
| Daily | 9 (35%) | 3 (19%) | 4 (57%) | |
| Seizure free for 1 year c | 5 (19%) | 5 (31%) | 0.5 | 1 (14%) |
| Drug resistant | 19 (73%) | 9 (56%) | 0.3 | 5 (71%) |
| Degree of intellectual disability | >0.9 | |||
| Mild | 5 (19%) | 4 (25%) | 3 (43%) | |
| Moderate | 8 (31%) | 6 (38%) | 3 (43%) | |
| Severe | 6 (23%) | 3 (19%) | 1 (14%) | |
| Profound | 7 (27%) | 3 (19%) | 0 (0%) | |
| Family history of epilepsy | 11 (42%) | 3 (19%) | 0.12 | 2 (29%) |
| Genetic cause identified | 10 (38%) | 5 (31%) | 0.6 | 1 (14%) |
Median (range, IQR: interquartile range); n (%).
Wilcoxon rank sum test; Pearson's Chi‐squared test; Fisher's exact test.
Patients with ongoing continuous spike‐and‐wave during sleep were not considered seizure free.
3.5. Simulated gene panels
We simulated the diagnostic rates for SNVs that would have resulted from using commercial epilepsy gene panels (Table S1). Both the Ambry Genetics EpilepsyNext panel (124 genes) and the GeneDx Comprehensive Epilepsy Panel (144 genes) would have identified the same 7 variants (diagnostic yield: 13%). The Invitae Neurodevelopmental disorders Panel (241 genes) would have identified 9 variants (diagnostic yield: 17%). The Invitae Epilepsy panel (320 genes) and the Blueprint Comprehensive Epilepsy Gene Panel (511 genes) would have identified the same 10 variants (diagnostic yield: 19%). Both the Prevention Comprehensive Epilepsy and Seizure panel (1478 genes) and the Ambry Genetics NeurodevelopmentNext‐Expanded panel (1527 genes) would have identified all 14 SNVs (diagnostic yield: 27%).
The smaller gene panels particularly missed genes identified in cluster B with early ascertainment of cognitive deficits and later onset of epilepsy. Panels including ≤241 genes would have identified none of the variants in this cluster (compared to 7/10 in the DEE cluster), panels between 320‐511 genes 1/4 variants (25%, compared to 8‐9/10 in the DEE cluster), and panels ≥1478 genes would have identified all variants.
4. DISCUSSION
We performed exome sequencing in a cohort of 52 adult probands with epilepsy and ID that were selected from a large cohort of patients recruited for genetic research (n = 1413). We identified pathogenic or likely pathogenic variants in 31% (27% SNVs, 4% CNVs). Our diagnostic rate lies within the range reported by previous studies using exome sequencing (25.3%‐47.3%). 7 , 9 , 10 The cluster analysis identified a cohort with early onset of epilepsy and developmental delay which corresponds well to DEE (53% of the cohort). However, a second cluster included patients who had early ascertainment of cognitive deficits and later onset of epilepsy (≥8 years) which is inconsistent with the concept of DEE (33% of the cohort) as seizures starting at this age are less likely to significantly affect development. Pathogenic or likely pathogenic variants were identified in both of these clusters indicating that testing strategies in older patients need to address both phenotypes. A third cluster included patients with late ascertainment of cognitive deficits and variable onset of epilepsy (14% of the cohort) in which one CNV was identified. It should be noted that all patients with an identified genetic cause had onset of developmental delay before age 8 years but epilepsy could start as late as age 18 years. This suggests that genetic testing should be done in all patients presenting with intellectual disability and epilepsy independent of age of onset.
Our results also provide insight into the most appropriate type of next‐generation sequencing in adults with epilepsy and ID. Exome sequencing was able to identify substantially more variants than smaller gene panels with only a few hundred genes, which only identified 50%‐71% of the variants. The smaller gene panels particularly missed the genes identified in the cluster with early ascertainment of cognitive deficits and later onset of epilepsy. However, large panels of around 1500 genes would have identified the same number of variants. Most of the smaller gene panels are focused on established epilepsy genes whereas the larger panels include more broadly neurodevelopmental disorder genes. These may or may not have been associated with epilepsy previously but nevertheless played a role in our adult cohort, at least explaining the ID phenotype. This suggests that in adults with epilepsy and ID, either exome sequencing or large gene panels focusing broadly on neurodevelopmental disorders should be used to maximize diagnostic yield. One caveat is that smaller gene panels are typically focusing on well‐established genes and do not include new genes for which only limited evidence exists. Two of the variants identified in our cohort were located in such genes (TDP2, KDM6B) which may explain why these were not included in the smaller panels.
A prior study has concluded that exome sequencing and small to medium gene panels (38‐455 genes) are most cost‐effective in children. 25 Our study suggests that in adult patients with epilepsy and ID, exome sequencing or large gene panels should be used due to their considerably higher diagnostic yield compared to smaller gene panels. Exome sequencing also allows exome‐wide identification of CNVs which further increases yield. Future studies will need to clarify if this diagnostic strategy results in higher cost‐effectiveness.
Three of the 14 identified SNVs were located in genes for which seizures had been described in <20% of patients (CTCF, KDM6B, NSUN2), accounting for 1/10 variants (10%) identified in cluster A and 2/4 identified in cluster B (50%). These genes are clearly responsible for the ID but their effect on the occurrence of epileptic seizures may be less pronounced. This again supports the conclusion that genetic testing in adults with epilepsy and ID needs to be broader including genes primarily associated with ID.
This study has some limitations. At variance to the approach of some previous studies, 6 , 7 , 8 , 9 we aimed to recruit an unbiased cohort independent of the clinical decision to go ahead with genetic testing. However, our cohort still represents a biased sample as participants with ID can only be recruited if informed consent can be obtained from a legal guardian. These do not always accompany the patient during the clinic visit requiring additional steps to obtain consent with lower chance of recruitment. In addition, we excluded patients who had previously been included in other research studies or where a genetic diagnosis was already known. This may have shifted the phenotypic spectrum in our cohort. On the other hand, this cohort resembles clinical practice in an adult epilepsy center where obvious and clinically recognizable early childhood phenotypes are typically already diagnosed and therefore not presenting for genetic workup in adulthood, or where severe onset early presentations may have already been accessed by previous genetic testing. The missing distinctive pediatric phenotypes likely explain why comparison of the phenotype of patients with and without an identified genetic cause did not show significant differences in our cohort. This suggests that genetic testing is warranted in adult patients with epilepsy and intellectual disability independent of epilepsy type, seizure severity, and presence of an epileptogenic lesion. However, the limited number of patients included in the study may have precluded the recognition of minor phenotypic differences. Furthermore, we used available bioinformatic tools to call CNVs from exome sequencing data which are less reliable than chromosomal microarray. 26 To avoid false‐positive results, we restricted the CNV analysis to CNVs >50 kb. Hence, smaller CNVs may have been missed by our analysis. We also noted that recruitment of the parents was difficult in our adult cohort as some parents had passed away, could not be reached or decided not to participate. Both parents could only be recruited for 26 patients which limited the ability to test for de novo variants. These issues provide significant challenges both for clinical genetic testing and research studies in adult patients with epilepsy and ID. In this respect, our cohort is representative for a typical clinical cohort and our results may reflect what can be expected in a typical clinical setting. Even in participants where both parents were not available, a diagnosis could be made in 18%. These were either known pathogenic variants, null variants, or homozygous variants where segregation information was not required to be classified as likely pathogenic according to ACMG criteria. 23
In conclusion, our results indicate that genetic testing is worthwhile in adult patients with epilepsy and ID, even if parents are unavailable for segregation analysis. Identification of a genetic cause has implications for genetic counseling and patient management including possible precision medicine approaches that can still be effective in adult patients. 6 , 27 Furthermore, our results indicate that both grown‐up patients with DEE and patients with primary ID and later onset of epilepsy are encountered in adult epilepsy clinics. Unfortunately, obtaining a detailed history of early life development in an adult patient with ID and seizures can be challenging as parents are often not available or may not remember details from the past. Therefore, exome sequencing or large gene panels (~1500 genes) are required for maximum diagnostic yield in this heterogenous cohort.
AUTHOR CONTRIBUTIONS
Sophie von Brauchitsch: Major role in the acquisition of data; analyzed the data; drafted the manuscript for intellectual content. Denise Haslinger: Major role in the acquisition of data; analyzed the data, revised the manuscript for intellectual content. Silvia Lindlar: Major role in the acquisition of data; analyzed the data. Holger Thiele: Major role in the acquisition of data; analyzed the data, revised the manuscript for intellectual content. Natalie Bernsen: Major role in the acquisition of data. Felix Zahnert: Major role in the acquisition of data, revised the manuscript for intellectual content. Philipp S. Reif: Major role in the acquisition of data. Yunus Balcik: Major role in the acquisition of data. Ping Yee Billie Au: Analyzed the data, revised the manuscript for intellectual content. Colin B. Josephson: Analyzed the data, revised the manuscript for intellectual content. Janine Altmüller: Major role in the acquisition of data; analyzed the data. Adam Strzelczyk: Major role in the acquisition of data; revised the manuscript for intellectual content. Susanne Knake: Major role in the acquisition of data; revised the manuscript for intellectual content. Felix Rosenow: Major role in the acquisition of data; revised the manuscript for intellectual content. Andreas Chiocchetti: Major role in the acquisition of data; analyzed the data; revised the manuscript for intellectual content. Karl Martin Klein: Designed and conceptualized study; analyzed the data; drafted the manuscript for intellectual content.
FUNDING INFORMATION
The study was supported by the Federal State Hessen, Germany, through the LOEWE program.
CONFLICT OF INTEREST STATEMENT
S. v. B. reports no disclosures relevant to the manuscript. D. H. reports no disclosures relevant to the manuscript. S. L. reports no disclosures relevant to the manuscript. H. T. reports no disclosures relevant to the manuscript. N. B. reports no disclosures relevant to the manuscript. F. Z. reports no disclosures relevant to the manuscript. P. S. R. reports grants from the Federal State Hessen, Germany, through the LOEWE program, Y. B. reports no disclosures relevant to the manuscript. P. Y. B. A. reports no disclosures relevant to the manuscript. C. B. J. reports unrestricted educational grants from UCB Inc. and Eisai Inc., as well as New Investigator Grant from Epilepsy Canada for work unrelated to this project. J. A. reports no disclosures relevant to the manuscript. A. S. reports personal fees and grants from Angelini Pharma, Desitin Arzneimittel, Eisai, Jazz (GW) Pharmaceuticals, Marinus Pharmaceuticals, Precisis, Takeda, UCB Pharma, UNEEG medical, and Zogenix. S. K. reports personal fees from Angelini Pharma, Bial, Desitin, Eisai, UCB Pharma and Zoogenix. F. R. reports personal fees from Angelini Pharma/Arvelle Therapeutics, Eisai GmbH, GSK, GW Pharmaceuticals companies/Jazz Pharma, Roche Pharma, UCB Pharma, and grants from Eisai, from the Federal State Hessen, Germany, through the LOEWE program, Grants from the German Research Foundation (DGF), the European Union (FP7), the Detlev‐Wrobel Fonds for Epilepsy Research, Frankfurt, The HAJA Foundation Frankfurt, and the German Minister for Education and Research (BMBF). A. G. C. reports no disclosures relevant to the manuscript. K. M. K. reports personal fees from UCB Pharma, Novartis Pharma AG, Eisai, and GW Pharmaceuticals, grants from the Federal State Hessen, Germany, through the LOEWE program and from the Canadian Institutes of Health Research.
ETHICAL APPROVAL
The study was approved by the ethics committees of the Philipps‐University Marburg (86/13, 163/14) and Goethe University Frankfurt (132/15).
PATIENT CONSENT STATEMENT
All participants or their legal guardians provided written informed consent.
Supporting information
Figure S1.
Table S1.
ACKNOWLEDGMENTS
We thank the patients and their families and carers for participation in our study. The study was supported by the Federal State Hessen, Germany, through the LOEWE program. Open Access funding enabled and organized by Projekt DEAL.
von Brauchitsch S, Haslinger D, Lindlar S, Thiele H, Bernsen N, Zahnert F, et al. The phenotypic and genotypic spectrum of epilepsy and intellectual disability in adults: Implications for genetic testing. Epilepsia Open. 2023;8:497–508. 10.1002/epi4.12719
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
DATA AVAILABILITY STATEMENT
The de‐identified phenotypic data will be made available upon reasonable request from any qualified investigator. The exome sequencing data can be made available if the respective proband or their legal guardian consented to sharing of the de‐identified genetic data.
REFERENCES
- 1. Singh A, Trevick S. The epidemiology of global epilepsy. Neurol Clin. 2016;34:837–47. 10.1016/j.ncl.2016.06.015 [DOI] [PubMed] [Google Scholar]
- 2. Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–21. 10.1038/nature12439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mercimek‐Mahmutoglu S, Patel J, Cordeiro D, Hewson S, Callen D, Donner EJ, et al. Diagnostic yield of genetic testing in epileptic encephalopathy in childhood. Epilepsia. 2015;56:707–16. 10.1111/epi.12954 [DOI] [PubMed] [Google Scholar]
- 4. Coppola A, Cellini E, Stamberger H, Saarentaus E, Cetica V, Lal D, et al. Diagnostic implications of genetic copy number variation in epilepsy plus. Epilepsia. 2019;60:689–706. 10.1111/epi.14683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Borlot F, Almeida BI, Combe SL, Andrade DM, Filloux FM, Myers KA. Clinical utility of multigene panel testing in adults with epilepsy and intellectual disability. Epilepsia. 2019;60:1661–9. 10.1111/epi.16273 [DOI] [PubMed] [Google Scholar]
- 6. Johannesen KM, Nikanorova N, Marjanovic D, Pavbro A, Larsen LHG, Rubboli G, et al. Utility of genetic testing for therapeutic decision‐making in adults with epilepsy. Epilepsia. 2020;61:1234–9. 10.1111/epi.16533 [DOI] [PubMed] [Google Scholar]
- 7. Snoeijen‐Schouwenaars FM, van Ool JS, Verhoeven JS, van Mierlo P, Braakman HMH, Smeets EE, et al. Diagnostic exome sequencing in 100 consecutive patients with both epilepsy and intellectual disability. Epilepsia. 2019;60:155–64. 10.1111/epi.14618 [DOI] [PubMed] [Google Scholar]
- 8. Benson KA, White M, Allen NM, Byrne S, Carton R, Comerford E, et al. A comparison of genomic diagnostics in adults and children with epilepsy and comorbid intellectual disability. Eur J Hum Genet. 2020;28:1066–77. 10.1038/s41431-020-0610-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zacher P, Mayer T, Brandhoff F, Bartolomaeus T, le Duc D, Finzel M, et al. The genetic landscape of intellectual disability and epilepsy in adults and the elderly: a systematic genetic work‐up of 150 individuals. Genet Med. 2021;23:1492–7. 10.1038/s41436-021-01153-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Minardi R, Licchetta L, Baroni MC, Pippucci T, Stipa C, Mostacci B, et al. Whole‐exome sequencing in adult patients with developmental and epileptic encephalopathy: it is never too late. Clin Genet. 2020;98:477–85. 10.1111/cge.13823 [DOI] [PubMed] [Google Scholar]
- 11. McKnight D, Bristow SL, Truty RM, Morales A, Stetler M, Westbrook MJ, et al. Multigene panel testing in a large cohort of adults with epilepsy. Neurol Genet. 2021;8:e650. 10.1212/NXG.0000000000000650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li J, Toffa DH, Lefèbvre M, Tétreault M, Cossette P, Samarut É, et al. Usage of genetic panels in an adult epilepsy clinic. Can J Neurol Sci. 2022;1–21:1–7. 10.1017/cjn.2022.49 [DOI] [PubMed] [Google Scholar]
- 13. Jiang Y, Song C, Wang Y, Zhao J, Yang F, Gao Q, et al. Clinical utility of exome sequencing and reinterpreting genetic test results in children and adults with epilepsy. Front Genet. 2020;11:591434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Allen Hauser W, Mathern G, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc task force of the ILAE commission on therapeutic strategies: definition of drug resistant epilepsy. Epilepsia. 2009;51:1069–77. 10.1111/j.1528-1167.2009.02397.x [DOI] [PubMed] [Google Scholar]
- 15. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome res. 2010;20:1297–303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ramu A, Noordam MJ, Schwartz RS, Wuster A, Hurles ME, Cartwright RA, et al. DeNovoGear: de novo indel and point mutation discovery and phasing. Nat Methods. 2013;10:985–7. 10.1038/nmeth.2611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Köhler S, Carmody L, Vasilevsky N, Jacobsen JOB, Danis D, Gourdine JP, et al. Expansion of the human phenotype ontology (HPO) knowledge base and resources. Nucleic Acids res. 2019;47:D1018–27. 10.1093/nar/gky1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc. 2015;10:1556–66. 10.1038/nprot.2015.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Genome Aggregation Database Consortium , Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–43. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fromer M, Moran JL, Chambert K, Banks E, Bergen SE, Ruderfer DM, et al. Discovery and statistical genotyping of copy‐number variation from whole‐exome sequencing depth. Am J Hum Genet. 2012;91:597–607. 10.1016/j.ajhg.2012.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Krumm N, Sudmant PH, Ko A, O’Roak BJ, Malig M, Coe BP, et al. Copy number variation detection and genotyping from exome sequence data. Genome res. 2012;22:1525–32. 10.1101/gr.138115.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. 2012;28:2747–54. 10.1093/bioinformatics/bts526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stamberger H, Hammer TB, Gardella E, Vlaskamp DRM, Bertelsen B, Mandelstam S, et al. NEXMIF encephalopathy: an X‐linked disorder with male and female phenotypic patterns. Genet Med. 2021;23:363–73. 10.1038/s41436-020-00988-9 [DOI] [PubMed] [Google Scholar]
- 25. Fernández IS, Loddenkemper T, Gaínza‐Lein M, Sheidley BR, Poduri A. Diagnostic yield of genetic tests in epilepsy: a meta‐analysis and cost‐effectiveness study. Neurology. 2019;92:e418–28. 10.1212/WNL.0000000000006850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gordeeva V, Sharova E, Babalyan K, Sultanov R, Govorun VM, Arapidi G. Benchmarking germline CNV calling tools from exome sequencing data. Sci Rep. 2021;11:14416. 10.1038/s41598-021-93878-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Balestrini S, Chiarello D, Gogou M, Silvennoinen K, Puvirajasinghe C, Jones WD, et al. Real‐life survey of pitfalls and successes of precision medicine in genetic epilepsies. J Neurol Neurosurg Psychiatry. 2021;92:1044–52. 10.1136/jnnp-2020-325932 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Table S1.
Data Availability Statement
The de‐identified phenotypic data will be made available upon reasonable request from any qualified investigator. The exome sequencing data can be made available if the respective proband or their legal guardian consented to sharing of the de‐identified genetic data.