

Abstract
Objective
STXBP1‐related disorders are rare genetic epilepsies and neurodevelopmental disorders, but the impact of symptoms across clinical domains is poorly understood. Disease concept models are formal frameworks to assess the lived experience of individuals and their families and provide a basis for generating outcome measures.
Methods
We conducted semistructured, qualitative interviews with 19 caregivers of 16 individuals with STXBP1‐related disorders and 7 healthcare professionals. We systematically coded themes using NVivo software and grouped concepts into the domains of symptoms, symptom impact, and caregiver impact. We quantified the frequency of concepts throughout the lifespan and across clinical subgroups stratified by seizure history and developmental trajectories.
Results
Over 25 hours of interviews, we coded a total of 3626 references to 38 distinct concepts. In addition to well‐recognized clinical features such as developmental delay (n = 240 references), behavior (n = 201), and seizures (n = 147), we identified previously underrepresented symptoms including gastrointestinal (n = 68) and respiratory symptoms (n = 24) and pain (n = 30). The most frequently referenced symptom impacts were autonomy (n = 96), socialization (n = 64), and schooling (n = 61). Emotional impact (n = 354), support (n = 200), and daily life & activities (n = 108) were highly cited caregiver impacts. We found that seizures were more commonly referenced in infancy than in other age groups, while behavior and socialization were more likely to be referred to in childhood. We found that caregivers of individuals with ongoing seizures were less likely to reference developmental delay, possibly due to the relatively high impact of seizures.
Significance
STXBP1‐related disorders are complex conditions affecting a wide range of clinical and social domains. We comprehensively mapped symptoms and their impact on families to generate a comprehensive disease model as a foundation for clinical endpoints in future trials.
Keywords: developmental and epileptic encephalopathy, disease concept model, outcome measures, quality of life, STXBP1‐related disorders
Key points.
Disease concept models (DCMs) are formal frameworks that capture the relationship between symptoms, concerns, and impact on daily life based on qualitative interviews.
Endpoints for future clinic studies involving STXBP1‐related disorder need to be relevant to patients and families.
Interviewing a diverse cohort of caregivers and healthcare professionals allows DCMs to be generalizable, and reveals high‐priority and novel disease concepts for treatment.
Disease concepts can vary depending on an affected individual's age, necessitating a longitudinal record of concepts.
Disease concepts can differ across clinical subgroups stratified by epilepsy and developmental histories making the inclusion of variable disorder presentations important.
1. INTRODUCTION
STXBP1‐related disorders are rare developmental and epileptic encephalopathies (DEEs), a group of early‐onset epilepsies and neurodevelopmental disorders. 1 , 2 , 3 First described in 2008 in a cohort of individuals with Ohtahara syndrome, 4 STXBP1‐related disorders are now known to present with a broader range of clinical features and neurodevelopmental trajectories. 3 , 5 , 6 , 7 While largely unknown only several years ago, remarkable progress has been made towards understanding the clinical spectrum and longitudinal disease trajectory in individuals with STXBP1‐related disorders. We previously identified emerging genotype–phenotype relationships within a larger cohort of 534 individuals, identifying several clinical features associated with specific variant types. For example, protein‐truncating variants including stop codon mutations, splice‐site variants, and frameshift variants were associated with a three‐fold increased risk of infantile spasms, while the recurrent p.R406C/H variant was associated with a two‐fold increased risk of spasticity and suppression‐burst EEG. 3 In addition, the adult phenotype 8 and the interaction between early seizures and development are increasingly being investigated. 9 Furthermore, significant progress has been made towards disease‐specific therapies within the last 2 years, including chemical chaperones 10 and novel avenues for targeted gene therapies and precision medicine approaches.
However, little is known about the full impact of STXBP1‐related disorders on affected individuals and their caregivers, an understanding of which is critical to identify outcomes of importance to patients and inform optimal windows for interventions. Disease concept models (DCMs) are formal frameworks that capture the lived experience of a disorder based on qualitative interviews. While clinical studies are often limited to predefined symptoms such as seizures or achievement of developmental milestones, DCMs use an open‐ended approach to capture the entirety of the features associated with a given condition. Within frameworks for drug development, regulators such as the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) increasingly rely on formal DCMs to prioritize outcomes for clinical trials. 11 , 12 , 13 DCMs have already been developed for Angelman syndrome and Dravet syndrome, leading to significant progression in understanding the overall impact of clinical features beyond epilepsy and neurodevelopmental symptoms in these disorders. 14 , 15
Here, we develop a DCM for STXBP1‐related disorders based on formal qualitative interviews with caregivers and healthcare professionals revealing 38 concepts, spanning symptoms, symptom impacts, and caregiver impact domains. We demonstrate a dynamic landscape of symptoms and their impact on families, capturing subgroups stratified by seizure history and developmental trajectories across the age span, providing insight for future natural history studies and trial design.
2. METHODS
2.1. Study inclusion of caregivers and healthcare providers
Caregivers of individuals with STXBP1‐related disorders were recruited through the Epilepsy Neurogenetics Initiative (ENGIN) and the Epilepsy Genetics Research Project (EGRP) at Children's Hospital of Philadelphia (CHOP) and the STXBP1 Foundation. To ensure a diverse patient population, we included caregivers of individuals with STXBP1‐related disorders of different ages, variable clinical spectrums regarding seizure histories, developmental trajectories, demographic features, and family living situations. Healthcare professionals were recruited through snowball sampling.
2.2. Framework conceptualization and interview process
Semistructured interviews were conducted virtually based on a formally developed framework consisting of open‐ended questions. A genetic counseling student trained in methods of qualitative interviews was the primary interviewer. The interviews comprised discussion about the diagnosis history, symptoms, the impact of STXBP1‐related symptoms on daily life, the care, the participation of the individual in the community, and challenges for caregivers (File S1–S7). No questions, prompting a particular concept (e.g., “Does your child have GI problems?”) were asked to ensure that participants focused on concepts that most affect daily life. An inductive thematic analysis approach was used to identify concepts described by participants rather than defined a priori. 16 Transcripts were analyzed using NVivo, a HIPAA‐approved qualitative data analysis computer software. Coding was performed based on a formally developed codebook consisting of specific concepts, which were categorized into the domains: symptoms, symptom impacts, and caregiver impacts (File S8). For example, concepts, including references to behavior, seizures, and tone were classified as symptoms, while schooling, socialization, and daily life & activities were defined as symptom impacts. Two members of the study team coded in parallel and comparative methods were used to ensure consistency. 17
We assessed the frequency of references to certain concepts across all interviews. To elucidate age‐related impacts of STXBP1‐related disorders, we measured concepts within defined lifespans including infancy (<12 months), toddlerhood (1–4 years), childhood (5–12 years), teenage & adolescence (13–17 years) and adulthood (18+ years). We also compared concepts across clinical subgroups of seizure history and the ability of independent ambulation and verbal communication. One individual, who was under 1 year of age at the time of study inclusion, was excluded from these subgroup analyses.
2.3. Assessment of concept saturation and statistical analysis
Concept saturation is defined as the point in the collection of qualitative data when new data no longer adds to the body of knowledge. To properly reach saturation, sampling must be widely inclusive for all categories, which might influence qualitative responses. Concept saturation was assessed using a data saturation matrix generated in NVivo Qualitative Software. Concept saturation was achieved with 16 interviews, suggesting an adequate sample size. When stratifying by STXBP1‐related disorder symptoms, references were normalized to the total number of children within each subgroup. Quantitative analyses of subgroups were performed using the R statistical framework to determine associations between concepts. However, as our study utilizes a phenomenological approach based on semistructured interviews, statistical testing was only used as a supplementary method to provide insight into the concepts that differed between subgroups based on caregiver interviews. This approach can also serve as a framework that highlights the potential significance of concepts for quantitative follow‐up studies.
3. RESULTS
3.1. Generation of a disease concept model identified 38 concepts
We developed a semistructured interview guide using a collaborating organizations' preliminary STXBP1‐related disorder disease concept model (DCM) and published interview guides from Dravet syndrome. 14 Disease features were reviewed and tailored to individuals with STXBP1‐related disorders and their families based on current literature and the study team's clinical experience in caring for 101 unique individuals with STXBP1‐related disorders. The resulting interview guide consisted of 36 open questions, leading to conversations achieving both breadth and depth (File S1–S7, Table S1). We transcribed a total of 25 hours and 23 minutes of interview time, resulting in 3626 references within 38 concepts and achieved an overall intercoder reliability of 88.2%.
3.2. Interviewed caregivers and healthcare providers represented a wide demographic spectrum
The interview process included 19 caregivers and seven healthcare providers. Caregivers, including eight male participants, had a mean age of 42.5 years and cared for a total of 16 individuals with STXBP1‐related disorders (Table 1). Additional demographic features included participants in same‐sex couples, parental STXBP1 pathogenic variant mosaicism, and households that speak English as a second language. We interviewed seven healthcare providers including two physicians, one genetic counselor, two physical therapists, one occupational therapist, and one educator.
TABLE 1.
Participant demographics and clinical subgroups
| Caregivers (n = 19) | Healthcare providers (n = 7) | ||
|---|---|---|---|
| Age (years) | Age (years) | ||
| Mean | 42.5 | Mean | 47.9 |
| Min, max | 33, 64 | Min, max | 28, 70 |
| Gender, n (%) | Gender, n (%) | ||
| Male | 8 (42%) | Male | 3 (42.9%) |
| Female | 11 (58%) | Female | 4 (57.1%) |
| Race, n (%) | Race, n (%) | ||
| White | 16 (84%) | White | 6 (85.7%) |
| Non‐white | 3 (16%) | Asian | 1 (14.3%) |
| Ethnicity, n (%) | Ethnicity, n | ||
| Hispanic | 4 (21%) | Hispanic | 0 |
| Non‐Hispanic | 15 (79%) | Non‐Hispanic | 7 |
| Clinical specialty, n | |||
| Physician | 2 | ||
| Physical therapist | 2 | ||
| Educator | 1 | ||
| Genetic Counselor | 1 | ||
| Occupational Therapist | 1 |
| Individuals with STXBP1‐related disorders (n = 16) | |||
|---|---|---|---|
| Age at interview (years), n (%) | Race, n (%) | ||
| <1 | 1 (6%) | White | 13 (81%) |
| 1–5 | 6 (38%) | Non‐white | 3 (19%) |
| 6–9 | 3 (19%) | Ethnicity, n (%) | |
| 10–13 | 2 (13%) | Hispanic | 4 (25%) |
| 14–17 | 0 (0%) | Non‐Hispanic | 12 (75%) |
| 18+ | 4 (25%) | Seizures (n = 16), n (%) | |
| Age of diagnosis (years), n (%) | No seizures | 3 (19%) | |
| <1 | 7 (44%) | Seizures | 13 (81%) |
| 1–5 | 4 (25%) | Past seizures | 8 (62%) |
| 6–9 | 2 (13%) | Current seizures | 5 (38%) |
| 10–13 | 0 (0%) | Development (n = 15), n (%) | |
| 14–17 | 1 (6%) | Global DD | 15 (100%) |
| 18+ | 2 (13%) | Ambulation (n = 15), n (%) | |
| Years since diagnosis | Nonambulatory | 5 (33%) | |
| Mean | 4.78 | Ambulatory | 10 (67%) |
| Min, max | 0.31, 11 | Verbal communication (n = 15), n (%) | |
| Gender, n (%) | Verbal/some words | 3 (20%) | |
| Male | 7 (44%) | Nonverbal | 12 (80%) |
| Female | 9 (60%) | ||
3.3. Individuals with STXBP1 ‐related disorders largely represent a pediatric patient population
We included seven male and nine female individuals with STXBP1‐related disorders with a median age of 7.5 years with the youngest individual being 4 months old and the oldest being 26 years old (Table 1). This cohort includes four adult individuals but largely represents a pediatric patient population. The mean years since STXBP1‐related disorders diagnosis was 4.8 with a minimum and maximum of 0.3 and 11 years, respectively. Seven individuals in our cohort (43.75%) were diagnosed with STXBP1‐related disorders within the first 6 months of life after the onset of seizures, which is consistent with the critical periods of seizure onset in STXBP1‐related disorders and the availability of clinical genetic testing. Outside of the first year of life, the average age of diagnosis was 8.8 years, which included individuals with and without seizure histories. Three individuals had a confirmed missense variant including two recurrent variants (p.(Arg292His), p.(Arg406Cys)). Four individuals had a splice‐site variant, four had a stop mutation, one a frameshift mutation, and one a larger deletion. All but three individuals (n = 13/16, 81.3%) had a history of seizures including five individuals with ongoing seizures at the time of the interview. These individuals were considered in the “current seizures” group and individuals with seizure remission in the “past seizures” group. All individuals in this cohort experienced a global developmental delay, 10 out of 15 children (66.6%) achieved some degree of independent ambulation (e.g., crawling or walking), and three out of 15 children (20%) had some verbal expressive language or words.
3.4. Symptoms (domain 1): Most frequently reported symptoms by caregivers included developmental delay, behavior, and seizures
Developmental delay, behavior, and seizures were mentioned across all caregiver interviews (Figures 1 and 5). We found that developmental delay was referenced 240 times throughout the interviews and was frequently double‐coded with other concepts, such as medical interaction (general medical; n = 92), gross motor skills (n = 58), expressive communication (n = 56), and impacts on autonomy for the child with STXBP1‐related disorders (n = 52, Figure 2). Caregivers referenced behavioral symptoms 201 times, categorizing them as negative in 61%, positive in 23%, and neutral in 17% of cases (Figure S1). Behavioral symptoms were frequently referenced with medical interaction (n = 42), caregiver emotional impacts (n = 41), caregiver impacts on daily life & activities (n = 34), and developmental delays (n = 34). Seizures were referenced 147 times and often mentioned in the context of medical interaction (n = 120), emotional caregiver impacts (n = 55), impact on the child's sleep (n = 15), and caregiver impacts on support (n = 12).
FIGURE 1.
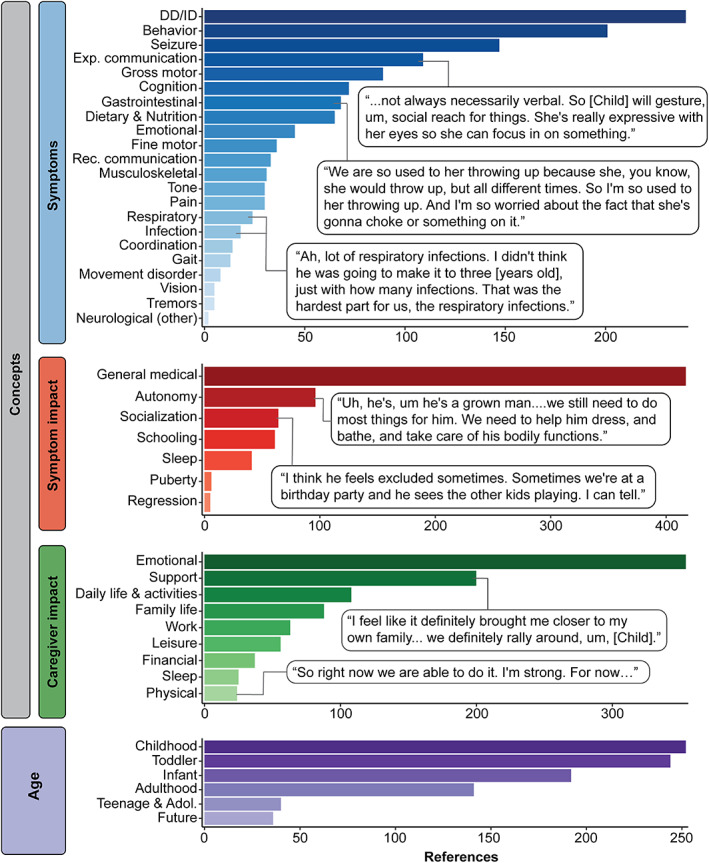
Disease concept model of STXBP1‐related disorders. A total of 3626 references across 19 caregivers were made referring to 38 distinct concepts categorized in the domains: symptoms, symptom impact, caregiver impacts, and across the lifespan (infancy: <12 months, toddlerhood: 1–4 years, childhood: 5–12 years, teenage & adolescence: 13–17 years, and adulthood 18+ years). In the domain of symptoms, expressive communication and receptive communication are notated as “exp. communication” and “rec. communication,” respectively.
FIGURE 5.
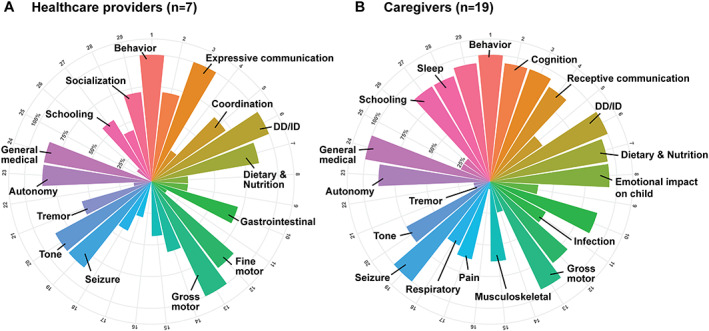
Proportion of interviews in which concepts were referenced at least once by healthcare providers or caregivers. (A) Distribution of concepts in the symptoms and symptom impact domains across healthcare providers. (B) Distribution of concepts in the symptoms and symptom impact domains across caregivers. We found that healthcare providers were significantly less likely to reference behavior and more likely to mention developmental delay and movement disorders compared with caregivers of individuals with STXBP1‐related disorders.
FIGURE 2.
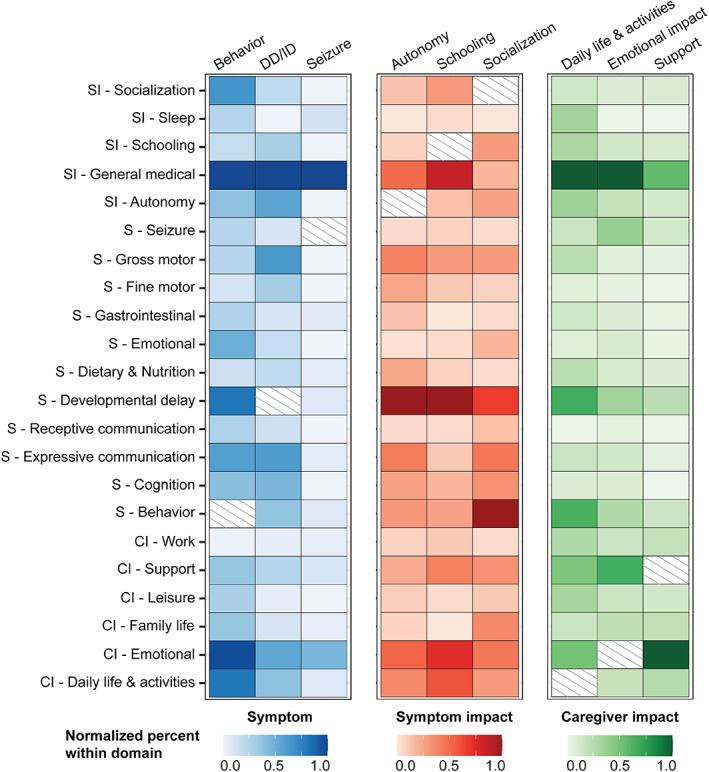
Distribution of co‐codes among the most referenced concepts in each domain. Behavior, developmental delay or intellectual disability (DD/ID), and seizures were the most common symptoms, while autonomy, schooling, and socialization were the most common symptom impacts. The most frequent caregiver impacts were daily life & activities, emotional impact, and support. Most common co‐coded concepts in each domain are shown, normalized in relation to the most frequently co‐coded concept.
We also identified symptoms underrepresented in the literature including gastrointestinal symptoms (68 references, across 87.5% of interviews), pain (24 references, across 56.3% of interviews), and respiratory symptoms (30 references, across 62.5% of interviews) (Figures 1 and 5). In our previously published cohort of 534 individuals across 19 973 harmonized clinical terms, respiratory symptoms were reported only in 33 individuals (6.2%), gastrointestinal symptoms in 31 individuals (5.8%), and pain in nine individuals (1.7%). 3
3.5. Symptom impact (domain 2): Caregivers referenced autonomy, socialization, and schooling as key symptom impacts on individuals with STXBP1 ‐related disorders
We found that symptom impacts play an important role in the daily lives of families with STXBP1‐related disorders, covering almost 66% of the interview time. Ninety‐six references regarding the child's autonomy (87.5% across all interviews), followed by socialization (n = 64, 93.8% across all interviews) and schooling (n = 61, 87.5% across all interviews) were recorded (Figures 1 and 5). Impacts on the child's autonomy were frequently double coded with developmental delay (n = 52), caregivers' emotions (n = 52), and medical interaction (n = 26, Figure 2). Caregivers described autonomy impacts as negative in 61%, positive in 18%, and neutral in 21% of cases (Figure S1). By contrast, socialization impacts were categorized as positive in 68% and negative in 32% of cases and frequently double coded with behavior (n = 27), developmental delay (n = 18), and caregivers' emotions (n = 12). Impacts on schooling were frequently double coded with developmental delay (n = 27), medical interaction (n = 23), caregiver impacts on emotions (n = 20), and impacts on daily life & activities (n = 16).
3.6. Caregiver impact (domain 3): Emotional, support, and daily life & activities concepts reflect major caregiver impacts
Besides the impacts of symptoms on individuals with STXBP1‐related disorders, we found that the impact of STXBP1 on caregivers is also a major component covering almost 45% of the interview time. Caregivers described emotional, support, and changes to daily life & activities as the main impacts (Figure 1). Emotional impact, support, and daily life & activities were mentioned across almost all interviews. Emotional impacts were referenced 354 times and frequently double coded with other concepts, such as medical interaction (n = 144), support (n = 94), seizures (n = 55), and developmental delays (n = 50) (Figure 2). Support, or the participant's support system, was referenced 200 times across all interviews and were frequently double coded with caregiver impacts on emotions (n = 94), medical interaction (n = 53), daily life & activities (n = 25), and developmental delays (n = 22). The category daily life & activities were referenced 108 times across all interviews and was frequently double coded with medical interaction (n = 53), developmental delay (n = 35), behavior (n = 34), and caregiver impacts on emotions (n = 27).
3.7. Lived experience for STXBP1 ‐related disorders varied across clinical subgroups
We stratified STXBP1‐related symptoms and their impacts across clinical subgroups including seizure history, ambulation, and verbal communication (Figure 3 ). Although our qualitative methodology does not allow for precise statistical comparison of concepts between subgroups, the following listed differences would be significant if a quantitative approach was used (Table S2). Comparing individuals with a history of seizures (n = 13), including those with ongoing seizures (n = 5) and those in seizure remission (n = 8), we found that caregivers of individuals with ongoing seizures were more likely to reference seizures. They were less likely to reference tone and developmental delay. By contrast, caregivers of individuals in seizure remission were more likely to reference receptive communication and schooling.
FIGURE 3.
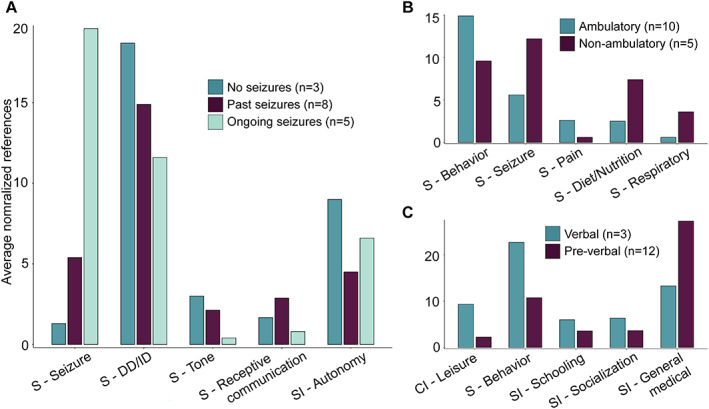
Analysis of subgroups stratified by seizure histories and developmental trajectories. Highlighted are the concepts that were significant across subgroups after correction for multiple testing with a false discovery rate of 5%. Height of each bar indicates the average references of concepts normalized to the number of individuals in each respective subgroup. (A) Differences in concepts across individuals without any history of seizures (n = 3), individuals in seizure remission (n = 8), and individuals with ongoing seizures (n = 5). (B) Differences in concepts across individuals who did not achieve independent ambulation at the time of study inclusion (n = 5) compared to those who achieved independent ambulation (n = 10). (C) Differences in concepts across individuals who did not use words to communicate (nonverbal, n = 12) compared to those who used words to communicate (verbal, n = 3).
Comparing ambulatory (n = 6) and nonambulatory (n = 9) individuals, caregivers of individuals who were ambulatory were more likely to reference behavior and pain. On the other hand, seizures, visual impairment, respiratory symptoms, and dietary & nutrition were mentioned less in this subgroup.
Caregivers of individuals with verbal communication (n = 3) were more likely to reference behavior, schooling, socialization, and leisure, and less likely to reference general medical issues than caregivers of individuals who were nonverbal.
3.8. Most referenced STXBP1 ‐related concepts evolved across the lifespan
To assess the impact of STXBP1‐related disorders across the lifespan, we associated referenced concepts with predefined age groups (Figure 4). Sixteen caregivers made references to infancy (<12 months, 542 references), 15 to toddlerhood (1–4 years, 893 references), 19 to childhood (5–12 years, 798 references), four to teenage years (13–17 years, 133 references), four to adulthood (18+ years, 444 references) and 13 to the future (114 references). We found seizures, general medical issues, and caregivers' emotions to be referenced more likely related to infancy than to any other age category. In toddlerhood, tone, developmental delay, and fine motor skills were referenced more often by caregivers than in any other age group. By contrast, seizures were less likely to be mentioned with regard to toddlerhood. In the context of childhood, behavior, schooling, socialization, and leisure were more likely to be referenced. With respect to teenage years, musculoskeletal symptoms and pain were more likely to be referenced. Related to adulthood, infection, seizures, sleep, and daily life & activities were overrepresented references compared with other age groups. When referring to considerations for the future, autonomy was sevenfold more likely to be mentioned.
FIGURE 4.
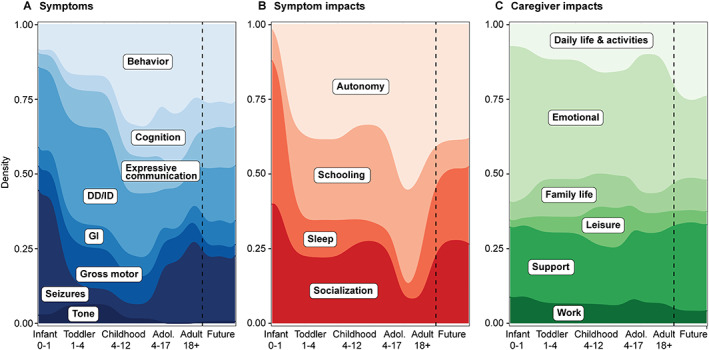
Distribution of symptoms, symptom impacts, and caregiver impacts across the lifespan in STXBP1‐related disorders. Concepts were referenced to age groups, including infancy (<12 months), toddlerhood (1–4 years), childhood (5–12 years), teenage & adolescence (13–17 years), adulthood (18+ years), and future considerations. (A) Relative density of the most frequently referenced symptoms. (B) Relative density of the most frequently referenced symptom impacts. (C) Relative density of the most frequently referenced caregiver impacts.
3.9. Caregivers and healthcare providers differ with respect to symptom references
To assess the impact of STXBP1‐related symptoms, we also interviewed seven healthcare providers involved in the care of individuals with STXBP1‐related disorders, complementing the caregivers' perspectives. While the average interview concept frequency was higher among caregivers (73%) than among healthcare professionals (51%), the concepts elicited by healthcare professionals largely reflect concepts provided by caregivers. However, some concepts were more likely to be referenced by either caregivers or healthcare providers. Healthcare providers were less likely to reference behavior and dietary & nutrition and more likely to mention developmental delay and movement disorders, including tremors (Figure 5).
4. DISCUSSION
Characterization of rare diseases has historically been based on collections of physician observations, perspectives, and case reports. However, in 1995, Wilson and Cleary modeled a conceptual framework that comprehensively surveyed the entirety of an affected individual or caregiver's lived experience with complex or rare disease, thereby centering the patients' experience within the resulting disease definition. 18 Using this model, several studies have created disease concept models (DCMs) of rare diseases, also referred to as conceptual models, conceptual disease models, and patient‐centered models, to ensure that patient and caregiver voices contribute to the description of the disorder. 14 , 15 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 In addition, regulating entities have endorsed these patient‐centered conceptual frameworks and increasingly encouraged drug development to involve patients and caregivers so that their perspectives are prioritized in disease treatment and understanding. 27 , 28 , 29 , 30
Here, we report the first DCM for STXBP1‐related disorders, a group of genetic epilepsies that is increasingly receiving attention due to the potential availability of precision medicine therapies. Building on previously generated DCMs for other neurodevelopmental and epilepsy disorders, 14 , 15 , 25 , 26 our study reinforces the need to incorporate diverse caregiver and clinician perspectives to fully describe a rare disease. We enrolled participants who varied in ages, sex, race, ethnicities, and diagnostic odysseys so that our DCM could be generalizable to many individuals with STXBP1‐related disorders. Whereas previously published models have heavily relied on physician opinions to generate the final DCM model, we sought out other expert multidisciplinary providers who could comment on the diagnosis, medical care, therapy needs, and education of individuals with STXBP1‐related disorders.
Standard within qualitative research, semistructured caregiver and family interviews are assessed for recurrent themes that are formally evaluated using a thematic analysis framework. 16 We find that the lived experience of families with STXBP1‐related disorders ranges across three major domains: symptoms of the condition itself, the impact of these symptoms on the individual with STXBP1‐related disorders, and their impact on caregivers.
We find that developmental delay, behavior, and seizures are the predominant symptoms in STXBP1‐related disorders. However, the entire range of reported symptoms spans across a total of 22 distinct concepts, including features that have previously been underrepresented in the literature such as gastrointestinal and respiratory symptoms.
Our study has three main findings: first, known clinical features in STXBP1‐related disorders only represent a subset of the lived experience of families and caregivers. In addition to a wide range of reported clinical characteristics, the full impact of the STXBP1 mutation on individuals with STXBP1‐related disorders and their families had not been assessed to date. We find that the impact of symptoms and the impact on caregivers play an important role in the daily lives of families of individuals with STXBP1‐related disorders, as 42% of all concepts are related to these two domains. For future clinical trials, identifying appropriate outcome measures across these domains will be critical. Validated measurement models have been developed to capture the impacts of various diseases on the daily life of affected families. 31 , 32 Furthermore, ongoing work examines the nuances in developmental and epileptic encephalopathies, such as the association of quality of life (QoL) with the proportion of days that are minimally disrupted by seizures. 33
Our second main finding is the prominent difference with regard to concepts across the age range. Caregivers of individuals with STXBP1‐related disorders frequently cite seizures and general medical issues when referring to infancy compared with other age groups. In childhood, social aspects such as behavior and leisure become more prominent. When referring to adulthood, symptoms such as seizures and infections were more frequently mentioned. These findings reflect the dynamic nature of seizure onset, offset, and recurrence and the impact of social aspects of STXBP1‐related disorders that parallels other neurodevelopmental and epilepsy DCMs 14 , 15 , 25 , 26 , 34 where there is a higher impact of developmental delay and behavioral disorders with a decrease in seizure frequency as the affected individual gets older. 35 This variability across the age span highlights a challenge for DCMs: when lived experience changes across age groups, establishing general instruments to assess symptom and caregiver impacts may be difficult. Further research is needed to determine how this difficulty can be addressed to assure that valid frameworks to capture symptom impact and caregiver impact can be reliably assessed across the lifespan.
The third main finding emerging from our study is the difference between clinical subgroups. We included this analysis in our study as STXBP1‐related disorders are traditionally considered to have a very broad range of clinical presentation, a feature that sets STXBP1‐related disorders apart from other conditions where DCMs have been developed. Families of individuals with seizures made more references to seizures than those without epilepsy, which is not surprising given the disruptive role of seizures in the families' lives and made fewer references to developmental and behavioral symptoms. By contrast, families of individuals who walked independently or communicated verbally made more references to behavior and fewer references to general medical concerns. These findings suggest that the impact of behavior in individuals with STXBP1‐related disorders may at least partially be dependent on comorbidities and the overall disease burden on an individual. As our study is one of the first DCMs attempting to compare lived experience across disease subgroups, it is currently unclear whether the inverse relationship of overall disease burden with developmental and behavioral concerns is specific to STXBP1‐related disorders or a more general feature in neurodevelopmental disorders. There is some literature to suggest that caregivers of children with developmental differences who have both greater caregiver needs and difficult behaviors during caregiving activities experience higher levels of stress. 36 Additionally, within pediatric populations with cerebral palsy (CP), research has shown that families report greater levels of self‐efficacy when caregiving to children with higher gross motor function. 37
Furthermore, we captured a difference between caregivers and healthcare providers. These groups differed with regard to relative references to symptom and symptom impacts. We found that healthcare providers made fewer references to behavior and dietary & nutrition domains to caregivers. Other DCMs have demonstrated differences between caregiver and healthcare provider references, which identifies potential discrepancies between these cohorts for treatment prioritization and impact importance. Additionally, when comparing symptoms referenced in our cohort to the disease spectrum captured in a previously published cohort of 534 individuals, we found that gastrointestinal symptoms, respiratory symptoms, and pain are underrepresented in clinical documentation despite being consistently mentioned across our interviews. 3 This highlights a potential gap in clinical care that could be addressed by providers who follow individuals with STXBP1‐related disorders.
Limitations of our study include the limited number of healthcare providers interviewed per subspecialty, number of adults with STXBP1‐related disorder included, and power to compare between subgroups. While few healthcare providers were interviewed per clinical specialty, this multidisciplinary panel had over 140 years of professional experience and followed approximately 110 individuals with STXBP1‐related disorders ranging from infancy to adulthood. Our study greatly benefitted from the diverse perspectives of multidisciplinary experts reflective of a clinical team assembled to manage individuals with STXBP1‐related disorder. We acknowledge that our study needed to be based on caregiver interviews and not the affected person themselves; additionally, while our study population was diverse, it may not represent the experience of all caregivers and healthcare providers of individuals with STXBP1‐related disorders. Nevertheless, due to the small number of adult individuals diagnosed with STXBP1‐related disorders, our cohort is limited to four adult individuals and may not represent all adult histories. We expect that the conceptual model of STXBP1‐related disorders will change as current patients age, and more patients from a wider spectrum of severity are diagnosed in the coming decades. Furthermore, this study reflects the experience of caregivers and providers of individuals with STXBP1‐related disorders receiving care from a selection of institutions in the United States. Nevertheless, DCMs of Dravet Syndrome, developed with families living in four different countries, showed that the main disease concepts are similar despite different healthcare systems. 14 For STXBP1‐related disorders, we also expect similar disease concepts to be referenced by families living outside of the US. This study uses a qualitative method to measure the impact of each concept on the daily life of families. Therefore, we cannot exclude that an individual with STXBP1‐related disorders experienced more symptoms than reported by caregivers and providers. Also, due to the small sample size, we are not able to stratify genetic subgroups in this study. However, it has been shown in our previous work that phenotypic variability in STXBP1 is not fully explained by an individual's genotype, as STXBP1‐related disorders present with marked genetic and phenotypic variability and heterogeneity. 3 Therefore, we do not expect genotype to significantly influence concepts that are most important to STXBP1 families as a group. 3
In summary, we developed a disease concept model for STXBP1‐related disorders containing 38 distinct concepts to present the lived experiences of families with STXBP1‐related disorders. We captured symptoms and their impact on individuals and their caregivers across different clinical subgroups and age ranges. We found clinical features previously underrepresented in literature and less frequently referenced by healthcare providers compared with caregivers. This first comprehensive disease model of STXBP1‐related disorders is essential as a foundation for developing standard‐of‐care protocols to identify endpoints in future clinical trials, focusing on the concepts that are important to affected individuals and their caregivers.
AUTHOR CONTRIBUTIONS
Katie R Sullivan involved in conceptualization, data curation, formal analysis, investigation, methodology, software, validation, visualization, writing—original draft preparation, and writing—review and editing. Sarah M Ruggiero involved in conceptualization, data Curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, software, supervision, validation, visualization, writing—original draft preparation, and writing—review and editing. Julie Xian involved in conceptualization, data curation, formal analysis, investigation, methodology, software, validation, visualization, writing—original draft preparation, and writing—review and editing. Kim M Thalwitzer involved in conceptualization, data curation, formal analysis, investigation, methodology, software, validation, visualization, writing—original draft preparation, and writing—review and editing. Rahma Ali involved in project administration and writing—review and editing. Sydni Stewart involved in data curation and methodology. Mahgenn Cosico involved in data curation and methodology. Jackie Steinburg involved in conceptualization, funding acquisition, investigation, validation, methodology, and writing—review and editing. James Goss involved in conceptualization, data curation, formal analysis, funding acquisition, investigation, validation, methodology, and writing—review and editing. Anna Pfalzer involved in conceptualization, methodology, and writing—review and editing. Kyle J Horning involved in conceptualization, methodology, and writing—review and editing. Nicole Weitzel involved in conceptualization, methodology, and writing—review and editing. Sydney Corey involved in conceptualization, methodology, and writing—review and editing. Laura Conway involved in supervision, project administration, and writing—review and editing. Charlene Son Rigby involved in conceptualization, data curation, formal analysis, funding acquisition, investigation, validation, methodology, and writing—review and editing. Terry Jo Bichell involved in conceptualization, data curation, formal analysis, methodology, supervision, investigation, validation, and writing—review and editing. Ingo Helbig involved in conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, software, supervision, validation, visualization, writing—original draft preparation, and writing—review and editing.
FUNDING INFORMATION
I.H. was supported by The Hartwell Foundation through an Individual Biomedical Research Award. This work was also supported by the National Institute for Neurological Disorders and Stroke (K02 NS112600), the Eunice Kennedy Shriver National Institute of Child Health and Human Development through the Intellectual and Developmental Disabilities Research Center (IDDRC) at Children's Hospital of Philadelphia and the University of Pennsylvania (U54 HD086984), and by intramural funds of the Children's Hospital of Philadelphia through the Epilepsy NeuroGenetics Initiative (ENGIN). Research reported in this publication was also supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number UL1TR001878. This project was also supported in part by the Institute for Translational Medicine and Therapeutics' (ITMAT) Transdisciplinary Program in Translational Medicine and Therapeutics at the Perelman School of Medicine of the University of Pennsylvania. The study also received support through the EuroEPINOMICS‐Rare Epilepsy Syndrome (RES) Consortium, which provided the capacity for exome sequencing, by the German Research Foundation (HE5415/3‐1 to I.H.) within the EuroEPINOMICS framework of the European Science Foundation, by the German Research Foundation (DFG; HE5415/5‐1, HE5415/6‐1 to I.H.) by the DFG/FNR INTER Research Unit FOR2715 (We4896/4‐570 1, and He5415/7‐1 to I.H.), and by the Genomics Research and Innovation Network (GRIN, grinnetwork.org).
CONFLICT OF INTEREST
The authors declare no conflicts of interest. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Appendix S1:
ACKNOWLEDGMENTS
We thank the families and individuals with STXBP1‐related disorders for making this study possible. This study was funded in part by the STXBP1 Foundation. Individuals from COMBINEDBrain provided essential input guiding study design and data collection efforts.
Sullivan KR, Ruggiero SM, Xian J, Thalwitzer KM, Ali R, Stewart S, et al. A disease concept model for STXBP1‐related disorders. Epilepsia Open. 2023;8:320–333. 10.1002/epi4.12688
REFERENCES
- 1. Stamberger H, Nikanorova M, Willemsen MH, Accorsi P, Angriman M, Baier H, et al. STXBP1 encephalopathy: a neurodevelopmental disorder including epilepsy. Neurology. 2016;86(10):954–62. [DOI] [PubMed] [Google Scholar]
- 2. Symonds JD, McTague A. Epilepsy and developmental disorders: next generation sequencing in the clinic. Eur J Paediatr Neurol. 2020;24:15–23. [DOI] [PubMed] [Google Scholar]
- 3. Xian J, Parthasarathy S, Ruggiero SM, Balagura G, Fitch E, Helbig K, et al. Assessing the landscape of STXBP1‐related disorders in 534 individuals. Brain. 2022;145(5):1668–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Saitsu H, Kato M, Mizuguchi T, Hamada K, Osaka H, Tohyama J, et al. De novo mutations in the gene encoding STXBP1 (MUNC18‐1) cause early infantile epileptic encephalopathy. Nat Genet. 2008;40(6):782–8. [DOI] [PubMed] [Google Scholar]
- 5. Loussouarn A, Doummar D, Beaugendre Y, Bienvenu T, Charles P, Depienne C, et al. Tremor‐like subcortical myoclonus in STXBP1 encephalopathy. Eur J Paediatr Neurol. 2021;34:62–6. [DOI] [PubMed] [Google Scholar]
- 6. O'Brien S, Ng‐Cordell E, The DDD Study , Astle DE, Scerif G, Baker K. STXBP1‐associated neurodevelopmental disorder: a comparative study of behavioural characteristics. J Neurodev Disord. 2019;11(1):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Suri M, Evers JMG, Laskowski RA, O'Brien S, Baker K, Clayton‐Smith J, et al. Protein structure and phenotypic analysis of pathogenic and population missense variants in STXBP1. Mol Genet Genomic Med. 2017;5(5):495–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stamberger H, Crosiers D, Balagura G, Bonardi CM, Basu A, Cantalupo G, et al. Natural history study of STXBP1‐developmental and epileptic encephalopathy into adulthood. Neurology. 2022;99:e233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Balagura G, Xian J, Riva A, Marchese F, Ben Zeev B, Rios L, et al. Epilepsy course and developmental trajectories in STXBP1‐DEE. Neurol Genet. 2022;8(3):e676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guiberson NGL, Pineda A, Abramov D, Kharel P, Carnazza KE, Wragg RT, et al. Mechanism‐based rescue of Munc18‐1 dysfunction in varied encephalopathies by chemical chaperones. Nat Commun. 2018;9(1):3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Food and Drug Administration . Guidance For Industry: Patient‐Reported Outcome Measures: Use In Medical Product Development To Support Labelling Claims. Rockville, MD: FDA; 2009. [Google Scholar]
- 12. E.M. Agency . Reflection Paper on the Regulatory Guidance for the use of Health‐Related Quality of Life (HRQL) Measures in the Evaluation of Medicinal Products. London: European Medicines Agency; 2005. [Google Scholar]
- 13. Food and Drug Administration . Patient‐Focused Drug Development: Selecting, Developing, or Modifying Fit‐for‐Purpose Clinical Outcome Assessments. Rockville, MD: FDA; 2022. [Google Scholar]
- 14. Nabbout R, Auvin S, Chiron C, Thiele E, Cross H, Scheffer IE, et al. Perception of impact of Dravet syndrome on children and caregivers in multiple countries: looking beyond seizures. Dev Med Child Neurol. 2019;61(10):1229–36. [DOI] [PubMed] [Google Scholar]
- 15. Willgoss T, Cassater D, Connor S, Krishnan ML, Miller MT, Dias‐Barbosa C, et al. Measuring what matters to individuals with Angelman syndrome and their families: development of a patient‐centered disease concept model. Child Psychiatry Hum Dev. 2021;52(4):654–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Joffe HYL. Content and thematic analysis. Research Methods for Clinical and Health Psychology. London: Sage Publications; 2004. p. 56–68. [Google Scholar]
- 17. Glaser BG. The constant comparative method of qualitative analysis*. Soc Probl. 2014;12(4):436–45. [Google Scholar]
- 18. Wilson IB, Cleary PD. Linking clinical variables with health‐related quality of life. A conceptual model of patient outcomes. JAMA. 1995;273(1):59–65. [PubMed] [Google Scholar]
- 19. Kanters TA, Redekop WK, Rutten‐van Mölken MPMH, Kruijshaar ME, Güngör D, van der Ploeg AT, et al. A conceptual disease model for adult Pompe disease. Orphanet J Rare Dis. 2015;10:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Karantzoulis S, Heuer K, Sparling N, Teynor M. The patient experience of Wilson disease: a conceptual model based on qualitative research. Orphanet J Rare Dis. 2021;16(1):437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Williams K, Davidson I, Rance M, Boehnke A, Buesch K, Acaster S. Symptoms and impacts of ambulatory nonsense mutation Duchenne muscular dystrophy: a qualitative study and the development of a patient‐centred conceptual model. J Patient Rep Outcomes. 2021;5(1):75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hamed A, An Haack K, Gwaltney C, Baranowski E, Stewart A, Krupnick R, et al. Qualitative interviews to improve patient‐reported outcome measures in late‐onset Pompe disease: the patient perspective. Orphanet J Rare Dis. 2021;16(1):428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shields AL, Lamoureux RE, Taylor F, Barth JA, Mulberg AE, Kessler V, et al. FABry Disease Patient‐Reported Outcome‐GastroIntestinal (FABPRO‐GI): a new Fabry disease‐specific gastrointestinal outcomes instrument. Qual Life Res. 2021;30(10):2983–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gallop K, Lloyd AJ, Olt J, Marshall J. Impact of developmental and epileptic encephalopathies on caregivers: a literature review. Epilepsy Behav. 2021;124:108324. [DOI] [PubMed] [Google Scholar]
- 25. Grieco JC, Romero B, Flood E, Cabo R, Visootsak J. A conceptual model of Angelman syndrome and review of relevant clinical outcomes assessments (COAs). Patient. 2019;12(1):97–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nabbout R, Auvin S, Chiron C, Irwin J, Mistry A, Bonner N, et al. Development and content validation of a preliminary core set of patient‐ and caregiver‐relevant outcomes for inclusion in a potential composite endpoint for Dravet syndrome. Epilepsy Behav. 2018;78:232–42. [DOI] [PubMed] [Google Scholar]
- 27. Walton MK, Powers JH 3rd, Hobart J, Patrick D, Marquis P, Vamvakas S, et al. Clinical outcome assessments: conceptual foundation‐report of the ISPOR clinical outcomes assessment – emerging good practices for outcomes research task force. Value Health. 2015;18(6):741–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Benjamin K, Vernon MK, Patrick DL, Perfetto E, Nestler‐Parr S, Burke L. Patient‐reported outcome and observer‐reported outcome assessment in rare disease clinical trials: an ISPOR COA emerging good practices task force report. Value Health. 2017;20(7):838–55. [DOI] [PubMed] [Google Scholar]
- 29. Patrick DL, Burke LB, Powers JH, Scott JA, Rock EP, Dawisha S, et al. Patient‐reported outcomes to support medical product labeling claims: FDA perspective. Value Health. 2007;10(Suppl 2):S125–37. [DOI] [PubMed] [Google Scholar]
- 30. Morel T, Cano SJ. Measuring what matters to rare disease patients – reflections on the work by the IRDiRC taskforce on patient‐centered outcome measures. Orphanet J Rare Dis. 2017;12(1):171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Varni JW, Seid M, Rode CA. The PedsQL: measurement model for the pediatric quality of life inventory. Med Care. 1999;37(2):126–39. [DOI] [PubMed] [Google Scholar]
- 32. Downs J, Jacoby P, Leonard H, Epstein A, Murphy N, Davis E, et al. Psychometric properties of the quality of life inventory‐disability (QI‐disability) measure. Qual Life Res. 2019;28(3):783–94. [DOI] [PubMed] [Google Scholar]
- 33. Cohen SR, Helbig I, Kaufman MC, Schust Myers L, Conway L, Helbig KL. Caregiver assessment of quality of life in individuals with genetic developmental and epileptic encephalopathies. Dev Med Child Neurol. 2022;64(8):957–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Helbig I, Tayoun AA. Understanding genotypes and phenotypes in epileptic encephalopathies. Mol Syndromol. 2016;7(4):172–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chemaly N, Kuchenbuch M, Teng T, Marie E, D'Onofrio G, Lo Barco T, et al. A European pilot study in Dravet syndrome to delineate what really matters for the patients and families. Epilepsia Open. 2021. 10.1002/epi4.12557. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Plant KM, Sanders MR. Predictors of care‐giver stress in families of preschool‐aged children with developmental disabilities. J Intellect Disabil Res. 2007;51(Pt 2):109–24. [DOI] [PubMed] [Google Scholar]
- 37. Pierce SR, Skorup J, Paremski AC, Prosser LA. The relationship between the family empowerment scale and gross motor function Measure‐66 in young children with cerebral palsy. Child Care Health Dev. 2021;47(1):112–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: