

Abstract
Familial adult myoclonic epilepsy (FAME) is an adult‐onset neurological disease characterized by cortical tremor, myoclonus, and seizures due to a pentanucleotide repeat expansion: a combination of pathogenic TTTCA expansion associated with a TTTTA repeat in introns of six different genes. Repeat‐primed PCR (RP‐PCR) is an inexpensive test for expansions at known loci. The analysis of the SAMD12 locus revealed that the repeats have different size, configuration, and composition. The TTTCA repeats can be very long (>1000 repeats) but also very short (14 being the shortest identified). Here, we report siblings of European descent with the clinical diagnosis of FAME yet a negative RP‐PCR test. Using short‐read genome sequencing, we identified the pentanucleotide expansion in intron 4 of SAMD12, which was confirmed by CRIPSR‐Cas9‐mediated enrichment and long‐read sequencing to be of (TTTTA)~879(TTTCA)3(TTTTA)7(TTTCA)7 configuration. Our finding is the first to associate the SAMD12 locus in European patients with FAME and currently represents the shortest identified TTTCA expansion. Our results suggest that the SAMD12 locus should be tested in patients with suspected FAME independent of ethnicity. Furthermore, RP‐PCR may miss the underlying mutation, and genome sequencing may be needed to confirm the pathogenic repeat.
Keywords: FAME, long‐read sequencing, optical genome mapping, repeat expansion, SAMD12, short‐read sequencing
Key points.
Familial adult myoclonic epilepsy (FAME) is caused by repeat expansions at different loci.
We report the first FAME family of European descent with a SAMD12 repeat expansion identified using short‐read genome sequencing.
The expansion was not seen on repeat‐primed PCR due to the very short TTTCA expansion (7 repeats) but confirmed on long‐read sequencing.
Our finding suggests that the SAMD12 locus should be tested in patients with suspected FAME independent of ethnicity.
Repeat expansions with short TTTCA repeat may escape detection by repeat‐primed PCR.
1. INTRODUCTION
Familial adult myoclonic epilepsy (FAME) is an autosomal dominant disease, characterized by an adult‐onset of cortical tremor, myoclonus, and epilepsy. 1 , 2 Ishiura and colleagues 3 identified that FAME was caused by a combination of a pathogenic TTTCA repeat associated with a polymorphic TTTTA repeat 3 in introns of either SAMD12, TNRC6A, or RAPGEF2 genes. Three additional loci have been described in introns of MARCH6, 4 YEATS2, 5 and STARD7. 6 The observation of abnormal RNA foci (UUUCA from the TTTCA repeat) in cortical neurons and Purkinje cells in the brain of patients, along with transcriptional dysregulation, suggested that the genes in which the TTTTA/TTTCA repeat expansions are localized had minor involvement in the pathogenicity of FAME, while the TTTCA repeat was central. 3
Mizuguchi and colleagues 7 recently combined CRISPR‐Cas9 mediated enrichment of the SAMD12 locus with Nanopore sequencing to describe repeat sizes, configuration, and composition in 22 families. Their work revealed variability in the repeat size for both the polymorphic TTTTA (28 to >1000 repeats) and pathogenic TTTCA repeats (14 to >1000 repeats). 7 The repeat configuration was also variable; configuration 1 [(TTTTA)exp(TTTCA)exp] was seen most frequently, followed by configuration 2 [(TTTTA)exp(TTTCA)exp(TTTTA)exp], and a new configuration 3 [(TTTTA)exp(TTTGA)exp(TTTCA)exp] was uncovered in one patient. 7 Finally, the repeat composition was highly variable as well and the proportions of the pathogenic TTTCA repeat in the overall repeat sequence ranged from 1% to 90%, 7 with even a repeat size of only 14 TTTCA pentanucleotides resulting in FAME. This suggests a complex relationship between the combination of repeat expansions and disease manifestation; a finding that warrants further investigation.
Common ancestral founder effects seem to play a role as individual loci expansions have been only associated with specific populations. 3 , 8 , 9 For example, SAMD12 expansions have hitherto only been reported in patients of Japanese, Chinese, Thai, Sri Lanka, and Indian descents. 3 , 8 , 9
Genetic testing for FAME is challenging as chromosomal microarray, gene panel sequencing and exome sequencing are likely to miss an intronic repeat expansion. Repeat‐primed PCR (RP‐PCR) is an inexpensive targeted test to confirm a repeat expansion but only at a known locus and for a known motif. However, recently developed and published bioinformatics tools, such as ExpansionHunter Denovo, allow to explore the whole genome for known and unknown repeats regardless of their size using short reads. 10
Here we report a FAME family of European descent with a repeat expansion in SAMD12 that had previously only been described in families with Asian inheritance. The TTTCA repeat in this family is unusually short and could not be identified using RP‐PCR.
2. MATERIALS AND METHODS
Two out of four affected members of a Canadian family of self‐described European descent were recruited and phenotyped by direct interview and review of existing medical records.
RP‐PCR in proband II‐3 was performed for FAME2, FAME3, and subsequently FAME1. The genomes of both siblings were sequenced with short‐read whole genome sequencing (srWGS) at The Hospital for Sick Children (SICKKIDS) using the Illumina TruSeq PCR‐free DNA library and the Illumina NovaSeq 6000 with a targeted coverage of 40×. The average depth of coverage achieved was 36 for individual II‐2 and 41 for II‐3. The data were analyzed to detect potentially causative single nucleotide variants (SNVs; as in Maroilley et al., 2022 11 ) and structural variants (SVs; based on a semi‐automated pipeline first developed using model organism genomes, and later adapted to human genomes 12 ). The ancestry of both individuals was assessed using the R package EthSeq. 13 Repeat expansions were identified using ExpansionHunter De Novo v0.9.0 10 (Outlier and Control/Case analysis modules with 50 control samples) followed by genotyping using Expansion Hunter v3.2.2 14 and visualization using GraphAlignmentViewer (https://github.com/Illumina/GraphAlignmentViewer).
Optical Genome Mapping (OGM) is a complementary approach to genome sequencing that enables the detection of structural variants and complex genome rearrangements, albeit not at base‐pair resolution. 15 We used OGM, Bionano's Saphyr genome mapping for individual II‐2.
CRISPR‐Cas9‐targeted enriched long‐range sequencing (LRS) was recently shown as an effective way to apply long‐read sequencing to a specific locus of the human genome without having to sequence an entire genome. 7 , 16 We designed guide RNAs for intron 4 of SAMD12 (chr8:118364471–118 368 363, 3893 bp, hg38) followed by the CRISPR‐Cas9‐based amplification‐free targeted enrichment for individual II‐2 and sequenced with Oxford Nanopore long‐read technology (MinION flow cell R9.4.1 on ONT GridION device).
Due to insufficient DNA quality, OGM and LRS could not be performed for individual II‐3.
The study was approved by the Conjoint Health Research Ethics Board (CHREB) at the University of Calgary (REB18‐2099). The patients provided written informed consent. The sequencing data from this study are not publicly available due to privacy.
3. RESULTS
We present a European family with four family members consistent with FAME of which two were available for the study (Figure 1A). The proband II‐3 is a 58‐year‐old female with onset of cortical tremor (postural and action, Figure 1B) in her mid‐30s. Seizure onset was at age 48 years with frequent myoclonic seizures (multiple/day) and rare focal impaired awareness seizures (FIAS, few/year), as well as sleep‐related bilateral tonic–clonic seizures (BTCS, 6‐8/y). The electroencephalogram (EEG) showed a posterior dominant generalized spike wave (Figure 1D) and a photoparoxysmal reaction grade 4 (Figure 1E). 17 Her 64‐year‐old brother II‐2 had an onset of cortical tremor around age 30 years (Figure 1C) followed by the onset of myoclonic seizures (1‐2/mo), and infrequent FIAS (0‐2/y), as well as BTCS (<1/y). His EEG showed a photoparoxysmal reaction grade 3 (Figure 1F). 17 Magnetic resonance imaging of both siblings (II‐2, II‐3) was nonlesional. Comorbidities were consistent with FAME in European ancestry families including migraine with visual aura in II‐2 and II‐3 18 and major depressive disorder, which had resolved on medication in II‐3. 19 The deceased mother I‐2 had an onset of cortical tremor and BTCS around 40–50 years of age. The father I‐1 and the maternal half‐brother II‐5 were unaffected. The son of the proband III‐3 was 33 years old and reported to have recent onset of cortical tremor and seizures but was unavailable for the study.
FIGURE 1.
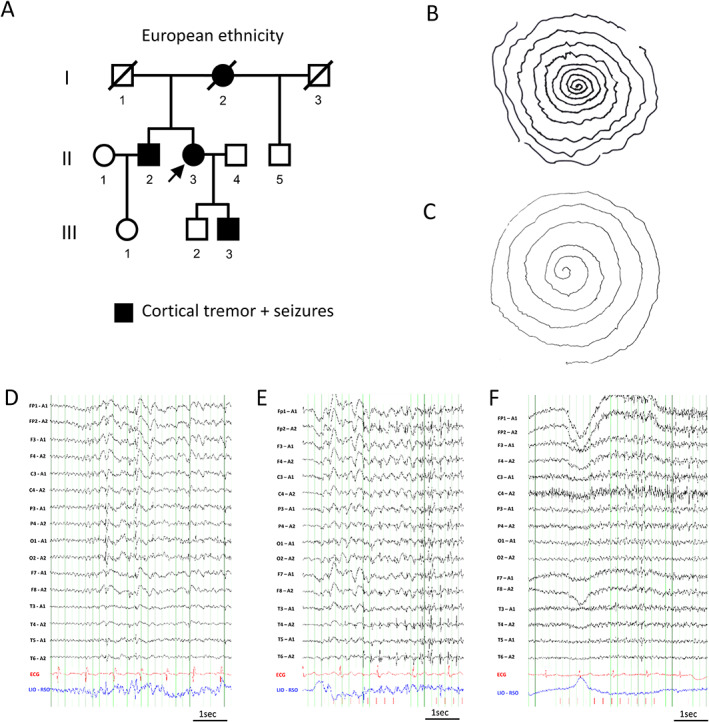
Clinical diagnosis of FAME in family of European descent. A, Family pedigree depicting affected family members. The arrow indicates the proband. B, Archimedes spiral drawn by the proband II‐3 and C, her brother II‐2 showing cortical tremor with variable frequency and amplitude. D–F, Electroencephalograms (low‐frequency filter 0.3 Hz, high‐frequency filter 70 Hz) D, Proband II‐3 at age 51 years after sleep deprivation (sensitivity 15 μV/mm). E, Proband II‐3 at age 55 years during 4 Hz photic stimulation (sensitivity 20 μV/mm). F, Brother II‐2 at age 63 years during 4 Hz photic stimulation (sensitivity 10 μV/mm).
First, the family was tested for repeat expansions using RP‐PCR targeting all known loci for European‐based ancestry (FAME2, FAME3), with negative results. We then hypothesized that siblings may either have a repeat expansion in a new locus or an unusual FAME variant. 7 Hence, the genomes of both siblings were sequenced with srWGS. We first confirmed the European ancestry of both siblings (EUR:75%/AMR:25%). Then, we analyzed the short‐read genome sequencing data for SNVs and SVs that could explain the phenotype (see Section 2), but none were found. However, our analyses of repeat expansions revealed the TTTTA/TTTCA repeat expansion on chromosome 8 in intron 4 of SAMD12 in both siblings (Figure 2A,B).
FIGURE 2.
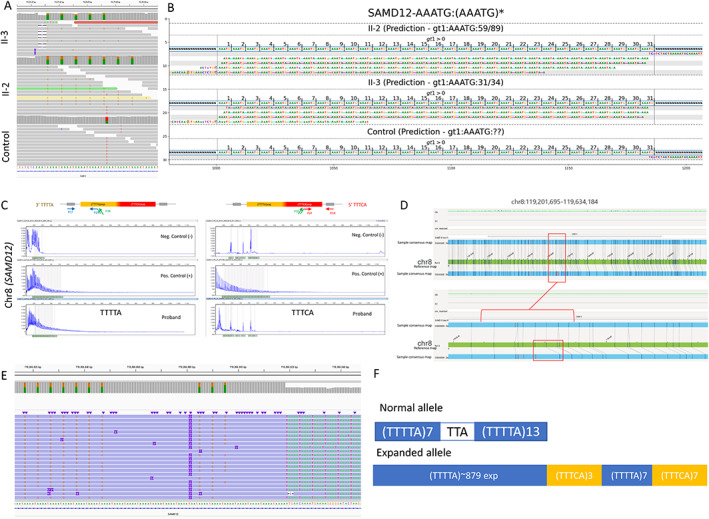
Combined genomics to uncover repeat expansion in FAME siblings. A, Short‐read whole genome sequencing depicted in the Integrative Genomic Viewer (IGV) image from II‐2 and the control clearly showing the heterozygous (TTTCA)7 expansion in the II‐2 and II‐3 but not in unrelated control genomes. B, TTTCA repeats visualized with GraphAlignmentViewer based on ExpansionHunter predictions showing the presence of the TTTCA motif, expanded in II‐2 and II‐3, but absent in unrelated control. C, Repeat‐primed PCR depicting TTTTA repeat expansion in II‐3 but no clear evidence for TTTCA repeat expansion. For TTTTA, positive control and proband show a tail of peaks extending up to more than 600 bp length representing the expansion, which is not present in the negative control individual. However, for TTTCA, the abnormal tail in the positive control is not present in the proband. D, Optical Genome Mapping Bionano's Saphyr depicting heterozygous 4.1 kb repeat expansion in the intron 4 of SAMD12 in II‐2; the region depicted is chr8:119201695‐119634184, hg19. The bottom allele shows a greater size between labels in the consensus map compared with the labels in the reference map. This might denote a small insertion between these two labels, corroborated by the misalignment of subsequent labels in that region. E, CRISPR/Cas9‐targeted enriched LRS showing the heterozygous TTTTA and TTTCA repeat expansion in II‐2. F, Configuration of the repeat expansion in II‐2 based on (D).
To further validate the findings and to better understand the size, configuration, and composition of the repeat, we combined several genomics approaches (Figure 2). RP‐PCR was ordered next with a specific focus on chromosome 8 and SAMD12. The RP‐PCR was able to successfully detect the presence of a TTTTA repeat expansion on 5′ and 3′ ends but failed to detect the TTTCA repeat (Figure 2C). Optical Genome Mapping (OGM) confirmed that the II‐2 genome contains a single repeat expansion on chromosome 8 in intron 4 of SAMD12 (Figure 2D) and estimated the size of the repeat expansion to be 4.1 kb. CRISPR‐Cas9‐targeted enriched LRS (Figure 2E) was able to detect a large repeat expansion at the SAMD12 locus of similar size as predicted by OGM (~4 kb). Importantly, as predicted by the srWGS (Figure 2A,B), the Nanopore sequencing determined that most of the repeat expansion is due to the TTTTA repeat, while the TTTCA repeat is much smaller. We next phased the flanking reads into two groups based on the read length, and we were able to determine that the configuration of the repeat in our family is (TTTTA)~879(TTTCA)3(TTTTA)7(TTTCA)7 (Figure 2F). The haplotype analyses of individuals II‐2 and II‐3 (Figure 1A) in our study revealed no evidence that the haplotype is different from the common founder haplotype reported in patients of Asian descent (Table S1).
4. DISCUSSION
The SAMD12 repeat expansion has only been reported in patients of Japanese, Chinese, Thai, Sri Lanka, and Indian descent 3 , 8 , 9 and is considered to have arisen from a common founder. 8 Here, we report two European kindreds with FAME caused by TTTTA/TTTCA repeat expansions in intron 4 of SAMD12, both presenting a haplotype similar to the common founder haplotype reported in Asian patients. Thus, our findings show that the TTTTA/TTTCA SAMD12 repeat expansion can affect individuals with non‐Asian ethnicity. Therefore, the SAMD12 locus should be tested regardless of the patient's origin. In fact, all six known loci should be tested in all individuals with a clinical suspicion of FAME regardless of ancestry.
The reported family also shows a novel and unusual repeat configuration 3 , 7 that could not be detected as pathogenic using RP‐PCR. It has been reported before that RP‐PCR may miss the pathogenic TTTCA repeat in patients who either have the TTTCA embedded deep into the TTTTA repeat (>150–200 bp from the extremities) or where the TTTCA repeat is short (e.g., 14 repeats). 7 Based on our srWGS and CRISPR‐CAS9 LRS data, our FAME patients had a very short TTTCA expansion with only 7 repeats (Figure 2B) which is the shortest reported so far. This might result in the inability of the RP‐PCR to create a product of sufficient length that could be detected (Figure 2C) leading to potential misdiagnoses and diagnostic odyssey.
Albeit the TTTCA repeats in the configuration reported here are the shortest known to date, the proportions of the TTTCA repeats in the overall repeat sequence length (10/896 = 1.1%) and the overall size of the repeat configuration (4.1 kb) are within the range reported recently. 7 Therefore, as suggested by Mizuguchi and colleagues, 7 our finding supports the possibility that the TTTCA insertion itself plays an important role in the pathogenicity of the disease regardless of the size.
We demonstrate that srWGS has the potential for uncovering new repeat configurations. Moreover, we were the first to report the application of the new OGM technology on FAME patients, establishing OGM as a new cost‐effective method to uncover new loci for repeat expansions in patients with the clinical diagnosis of FAME. However, CRISPR/Cas9 target enriched LRS was decisive in characterizing the configuration of the repeat as its length was superior to the short‐read length. A combination of approaches might be our best chance to discover new FAME loci and/or new repeat configurations, and therefore help gain new knowledge about disease mechanisms.
5. CONCLUSION
Our findings indicate that repeat expansions in SAMD12 may also occur in patients with non‐Asian ethnicity suggesting that all known FAME loci should be tested in patients with suspected FAME independent of ethnicity. We also show that RP‐PCR may not be able to reliably detect pathogenic repeat expansions suggesting that further workup is required in patients where a TTTTA, but no TTTCA repeat is identified.
AUTHOR CONTRIBUTIONS
M.T.G. and K.M.K. conceptualized and supervised the study. T.M., M.H.T., R.M., S.K., Ch.D., and M.T.G. analyzed the data. C.D. and M.K. prepared samples. K.M.K. phenotyped the patients. M.T.G., T.M., M.H.T., and K.M.K. wrote the manuscript. M.T.G., K.M.K., and M.H.T. obtained funding. All authors read and approved the final version of the manuscript.
FUNDING INFORMATION
This work was funded by the Alberta Children's Hospital Foundation and the Clinical Research Fund award to M.T.G. Department of Clinical Neurosciences, Hotchkiss Brain Institute and Alberta Children's Hospital Research Institute, University of Calgary, to K.M.K. Chang Gung Medical Foundation (CMRPG8L1331 and CMRPG8M1091), National Health Research Institute (EX111‐11022NI), and Ministry of Science & Technology (110‐2314‐B‐182A‐076‐MY3) to M.H.T. This research was enabled in part by support provided by the Research Computing Services group at the University of Calgary.
CONFLICT OF INTEREST STATEMENT
K.M.K. reports personal fees from UCB Pharma, Novartis Pharma AG, Eisai, and GW Pharmaceuticals, grants from the federal state Hessen, Germany, through the LOEWE program and from the Canadian Institutes of Health Research. The other authors declare that they have no conflict of interests.
ETHICAL APPROVAL
The study was approved by the CHREB at the University of Calgary (REB18‐2099).
PATIENT CONSENT STATEMENT
To search for the genetic cause in this family, written informed consent was obtained and both siblings were enrolled in the Genetics of paroxysmal neurological disorder research study. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Table S1
ACKNOWLEDGMENTS
The authors thank the family for their participation in the research study and acknowledge the Bionano Genomics Services Lab and Health GeneTech, Taiwan, for valuable technical assistance. This research was funded by Alberta Children's Hospital Foundation and the Clinical Research Fund award to M.T.G., Chang Gung Memorial Hospital, National Health Research Institute and Ministry of Science and Technology, Taiwan, to M.H.T. and the Department of Clinical Neurosciences, Hotchkiss Brain Institute and Alberta Children's Hospital Research Institute, University of Calgary, to K.M.K. This research was enabled in part by support provided by the Research Computing Services group at the University of Calgary.
Maroilley T, Tsai M‐H, Mascarenhas R, Diao C, Khanbabaei M, Kaya S, et al. A novel FAME1 repeat configuration in a European family identified using a combined genomics approach. Epilepsia Open. 2023;8:659–665. 10.1002/epi4.12702
Tatiana Maroilley and Meng‐Han Tsai equal first authors.
Maja Tarailo‐Graovac and Karl Martin Klein equal senior and co‐corresponding authors.
Contributor Information
Maja Tarailo‐Graovac, Email: maja.tarailograovac@ucalgary.ca.
Karl Martin Klein, Email: karl.klein@ucalgary.ca.
DATA AVAILABILITY STATEMENT
The sequencing data from this study are not publicly available due to privacy.
REFERENCES
- 1. Ikeda A, Kakigi R, Funai N, Neshige R, Kuroda Y, Shibasaki H. Cortical tremor: a variant of cortical reflex myoclonus. Neurology. 1990;40(10):1561–5. [DOI] [PubMed] [Google Scholar]
- 2. Peters L, Depienne C, Klebe S. Familial adult myoclonic epilepsy (FAME): clinical features, molecular characteristics, pathophysiological aspects and diagnostic work‐up. Medizinische Genetik. 2021;33(4):311–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ishiura H, Doi K, Mitsui J, Yoshimura J, Matsukawa MK, Fujiyama A, et al. Expansions of intronic TTTCA and TTTTA repeats in benign adult familial myoclonic epilepsy. Nat Genet. 2018;50(4):581–90. [DOI] [PubMed] [Google Scholar]
- 4. Florian RT, Kraft F, Leitão E, Kaya S, Klebe S, Magnin E, et al. Unstable TTTTA/TTTCA expansions in MARCH6 are associated with familial adult myoclonic epilepsy type 3. Nat Commun. 2019;10(1):4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yeetong P, Pongpanich M, Srichomthong C, Assawapitaksakul A, Shotelersuk V, Tantirukdham N, et al. TTTCA repeat insertions in an intron of YEATS2 in benign adult familial myoclonic epilepsy type 4. Brain. 2019;142(11):3360–6. [DOI] [PubMed] [Google Scholar]
- 6. Corbett MA, Kroes T, Veneziano L, Bennett MF, Florian R, Schneider AL, et al. Intronic ATTTC repeat expansions in STARD7 in familial adult myoclonic epilepsy linked to chromosome 2. Nat Commun. 2019;10(1):4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mizuguchi T, Toyota T, Miyatake S, Mitsuhashi S, Doi H, Kudo Y, et al. Complete sequencing of expanded SAMD12 repeats by long‐read sequencing and Cas9‐mediated enrichment. Brain. 2021;144(4):1103–17. [DOI] [PubMed] [Google Scholar]
- 8. Bennett MF, Oliver KL, Regan BM, Bellows ST, Schneider AL, Rafehi H, et al. Familial adult myoclonic epilepsy type 1 SAMD12 TTTCA repeat expansion arose 17,000 years ago and is present in Sri Lankan and Indian families. Eur J Hum Genet. 2020;28(7):973–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yeetong P, Chunharas C, Pongpanich M, Bennett MF, Srichomthong C, Pasutharnchat N, et al. Founder effect of the TTTCA repeat insertions in SAMD12 causing BAFME1. Eur J Hum Genet. 2021;29(2):343–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dolzhenko E, Bennett MF, Richmond PA, Trost B, Chen S, van Vugt JJFA, et al. ExpansionHunter Denovo: a computational method for locating known and novel repeat expansions in short‐read sequencing data. Genome Biol. 2020;21(1):102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maroilley T, Wright NAM, Diao C, MacLaren L, Pfeffer G, Sarna JR, et al. Case report: biallelic loss of function ATM due to pathogenic synonymous and novel deep intronic variant c.1803‐270T>G identified by genome sequencing in a child with ataxia‐telangiectasia. Front Genet. 2022;13:815210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maroilley T, Li X, Oldach M, Jean F, Stasiuk SJ, Tarailo‐Graovac M. Deciphering complex genome rearrangements in C. elegans using short‐read whole genome sequencing. Sci Rep. 2021;11(1):18258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Romanel A, Zhang T, Elemento O, Demichelis F. EthSEQ: ethnicity annotation from whole exome sequencing data. Bioinformatics. 2017;33(15):2402–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dolzhenko E, van Vugt JJFA, Shaw RJ, Bekritsky MA, van Blitterswijk M, Narzisi G, et al. Detection of long repeat expansions from PCR‐free whole‐genome sequence data. Genome Res. 2017;27(11):1895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barseghyan H, Tang W, Wang RT, Almalvez M, Segura E, Bramble MS, et al. Next‐generation mapping: a novel approach for detection of pathogenic structural variants with a potential utility in clinical diagnosis. Genome Med. 2017;9(1):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Miller DE, Sulovari A, Wang T, Loucks H, Hoekzema K, Munson KM, et al. Targeted long‐read sequencing identifies missing disease‐causing variation. Am J Hum Genet. 2021;108(8):1436–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Waltz S, Christen HJ, Doose H. The different patterns of the photoparoxysmal response—a genetic study. Electroencephalogr Clin Neurophysiol. 1992;83(2):138–45. [DOI] [PubMed] [Google Scholar]
- 18. Crompton DE, Sadleir LG, Bromhead CJ, Bahlo M, Bellows ST, Arsov T, et al. Familial adult myoclonic epilepsy: recognition of mild phenotypes and refinement of the 2q locus. Arch Neurol. 2012;69(4):474–81. [DOI] [PubMed] [Google Scholar]
- 19. Coppola A, Caccavale C, Santulli L, Balestrini S, Cagnetti C, Licchetta L, et al. Psychiatric comorbidities in patients from seven families with autosomal dominant cortical tremor, myoclonus, and epilepsy. Epilepsy Behav. 2016;56:38–43. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Data Availability Statement
The sequencing data from this study are not publicly available due to privacy.