

Abstract
Objectives
We describe the Residras registry, dedicated to Dravet syndrome (DS) and to other phenotypes related to SCN1A mutations, as a paradigm of registry for rare and complex epilepsies. Our primary objectives are to present the tools and framework of the integrative platform, the main characteristics emerging from the patient cohort included in the registry, with emphasis on demographic, clinical outcome, and mortality.
Methods
Standardized data of enrolled pediatric and adult patients were collected in 24 Italian expert centers and regularly updated at least on a yearly basis. Patients were prospectively enrolled, at registry starting, but historical retrospective data were also included.
Results
At present, 281 individuals with DS and a confirmed SCN1A mutation are included. Most patients have data available on epilepsy (n = 263) and their overall neurological condition (n = 255), based on at least one follow‐up update. Median age at first clinical assessment was 2 years (IQR 0–9) while at last follow‐up was 11 years (IQR 5–18.5). During the 7‐year activity of the registry, five patients died resulting in a mortality rate of 1.84 per 1000‐person‐years. When analyzing clinical changes over the first 5‐year follow‐up, we observed a significant difference in cognitive function (P < 0.001), an increased prevalence of behavioral disorders including attention deficit (P < 0.001), a significant worsening of language (P = 0.001), and intellectual disability (P < 0.001).
Significance
The Residras registry represents a large collection of standardized national data for the DS population. The registry platform relies on a shareable and interoperable framework, which promotes multicenter high‐quality data collection. In the future, such integrated platform may represent an invaluable asset for easing access to cohorts of patients that may benefit from clinical trials with emerging novel therapies, for drug safety monitoring, and for delineating natural history. Its framework makes it improvable based on growing experience with its use and easily adaptable to other rare and complex epilepsy syndromes.
Keywords: epilepsy syndrome, natural history, rare disease, registry, SCN1A
Key Points.
The Residras registry aims to gather a large collection of standardized national data of patients with Dravet Syndrome.
The registry platform relies on a shareable and interoperable framework, which promotes multicentre high‐quality data collection.
Such integrated platform could be easily adaptable to other rare and complex epilepsy syndromes.
1. INTRODUCTION
Dravet syndrome (DS) is one of the most common developmental and epileptic encephalopathies (DEEs) with an incidence of 6.5 per 100 000 live births (95% CI 3.2–10.00). 1 Its hallmark clinical presentation is with prolonged, febrile and afebrile, generalized clonic or hemiclonic seizures with onset within the first year of life. Other seizure types with later onset include myoclonic, atypical absence and focal seizures. Episodes of seizure clusters and status epilepticus are frequent. In addition to seizures, most children, often during late infancy or early childhood, manifest cognitive, motor, and behavioral impairment. 2 DS is caused by loss‐of‐function pathogenic variants in the gene coding for the α1 subunit of the sodium channel (SCN1A), which is important for action potential initiation in inhibitory GABAergic interneurons. 3 There is a wide spectrum of clinical entities caused by SCN1A mutations ranging from genetic epilepsy with febrile seizure plus (GEFS+) to DS, with variable disease course even within the core DS phenotype. 4
Epilepsy is treatment‐resistant although the seizure burden is highly variable. As in all severe DEEs, the most acceptable clinical poise should be individually established based on a balance between reduction in seizure severity and frequency and minimisation of treatment‐related adverse effects. 5 Controlled trials have demonstrated antiseizure efficacy of stiripentol in association with clobazam and of add‐on cannabidiol and fenfluramine. 6 , 7 , 8 , 9 However, there are no current treatments that address the overall disease, in addition to seizures. Precision medicine approaches such as antisense oligonucleotides (ASO) and adeno‐associated virus (AAV)‐delivered gene modulation are potential treatment options for DS, which are currently being investigated on a research basis, with some now transitioning to clinical trials. 10
Disease course is variable and not fully characterized, and the extent to which prompt treatment with the most effective medications can alter prognosis is unclear. The few available studies of outcomes in adulthood show that epilepsy severity progressively decreases from childhood to adolescence and throughout adulthood, and reduced frequency of convulsive status epilepticus is associated with better seizure outcome. 11 On the other hand, persistence of seizures in adolescents and adults correlates with cognitive and neurologic impairment, and there is ongoing cognitive dysfunction in adulthood, independent of seizure control. Early onset of seizures, especially myoclonic, correlates with the severity of intellectual disability and language impairment. 12 , 13 Most adults with DS require a considerable amount of support and are unable to live independently. 14 , 15 , 16 Also, the incidence of premature mortality, including sudden unexpected death in epilepsy (SUDEP), is elevated in childhood, but data on this ominous outcome are lacking in adulthood. 17 , 18
Although DS is likely the most studied genetic DEE and is the target of regulatory trials for orphan drug designation, there is still limited knowledge on several clinical aspects including natural history, risk of comorbidities, mortality, treatment response, and safety. Such knowledge becomes crucial for developing novel treatment strategies especially in view of identifying the potential for disease modifying therapeutic interventions in the precision medicine framework. In this perspective, disease registries represent an ideal tool to move forward in research and improve knowledge in the field of rare diseases. 19
The Italian Registry of Dravet Syndrome and Other Syndromes correlated with SCN1A and PCDH19 mutation (Residras) was established to provide an integrative infrastructure for collection of standardized molecular and clinical data of patients with DS from national centers 20 where they are diagnosed and regularly followed up.
The primary purposes of this study are to present: (a) the tools and framework of the integrative platform; (b) the main characteristics of the patient cohort ie, demographic, clinical outcome, and mortality; (c) the concept of the Residras registry, which we progressively adapted to the emerging diagnostic and therapeutic challenges of an epilepsy syndrome under intense clinical investigation; and (d) to set the basis for a wider international reach of the registry, which is now being adopted in additional EU countries.
2. METHODS
2.1. History of the registry
The “Residras” project started in 2010 under the auspices of the Scientific Committee of Dravet Italia Onlus, 21 an association of patients and physicians, focusing on research on DS and related syndromes. The Scientific Committee identified a minimum of mandatory items to be included in the dataset. Members of the Scientific Committee participated with their centers in a pilot phase. Meyer Children's Hospital, in Florence, was selected as the coordinating center. Fondazione Monasterio, a public institution for healthcare research 22 with a particular experience in rare diseases registries, was identified as a partner for the platform creation, data storage, confidentiality, and management of privacy. By June 2013, the dataset for the pilot phase was ready, and the first patients were enrolled to test the process of data inclusion in the platform. Once the process was improved in terms of consistency, reliability, and ease, pilot centers started to enroll all their patients, either new or in follow‐up, and in April 2015, Residras was opened to all Italian centers upon their specific request and ethics committee approval.
2.2. Current format of the registry—coverage, interface for clinicians, data use, and research applications
Registry coordination is entrusted by a Scientific Advisory Committee (10 expert clinicians on DS), an external Steering Committee for management (Fondazione Monasterio), an external Advisor on Rare Disease (National Centre on Rare Diseases), a patient association (Dravet Italia Onlus), and three patients' representatives. 20
Twenty‐four centers are currently participating to data input (Figure 1). Written informed consent or assent was provided by adult patients with capacity to consent or by families or legal guardians for children and adults lacking capacity.
FIGURE 1.
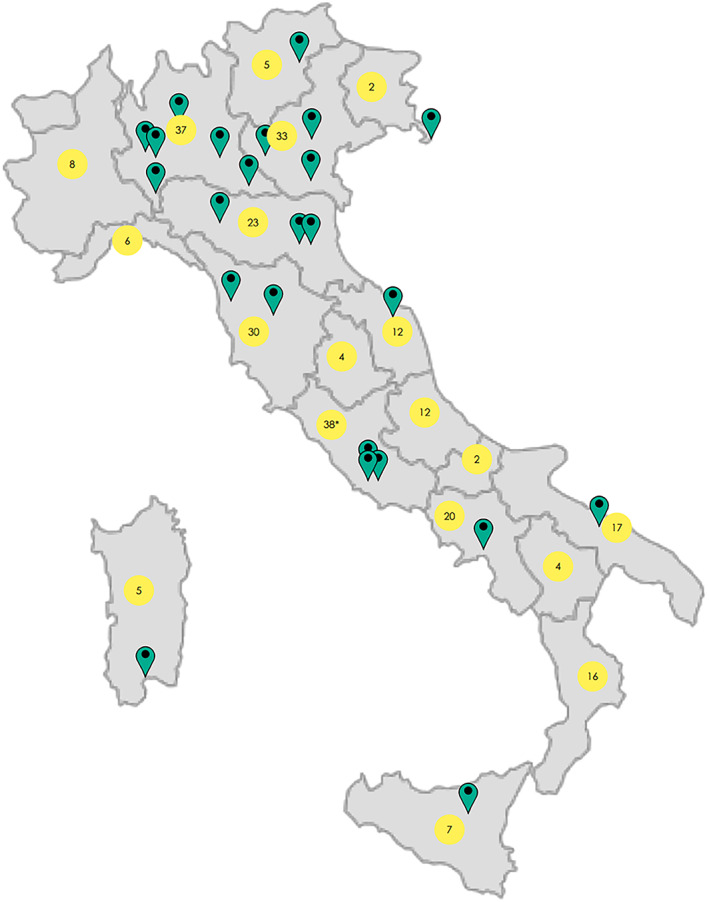
Coverage of Residras with illustration of the participating centre distribution (green pins) and number of enrolled patients who are resident in each italian region (yellow circles). *Patients resident abroad (n = 2).
The Residras registry contains clinical and epidemiological data that are compiled on a standardized online template during regular clinical visits. The information is schematically divided into the following sections: demographic features (date of birth, gender, parental details, place of birth, place of residence, willingness to be contacted on a regular basis to update the registry with longitudinal data and participate in a future clinical study), medical history (including pregnancy, delivery, neonatal period, neurodevelopment, neurological examination), clinical and genetic diagnosis, family history, age at seizure onset, and mortality. Entries for longitudinal follow‐up include seizure assessment (ie, type, frequency, episodes of status epilepticus, hospital admissions), neurological assessment (ie, examination, cognition, behavior, formal neuropsychological testing), treatment (ie, type, max dosage, duration, response, adverse events), EEG (ie, date, background activity, epileptiform abnormalities, photosensitivity, recorded seizures), and other investigations (eg, neuroimaging). Seizure assessment was based on clinical criteria, and seizures were classified as hemiclonic, generalized motor (including tonic‐clonic and clonic), absence, focal‐onset, reflex, massive (ie, generalized) myoclonus, and action myoclonus (ie, triggered by voluntary movement). 23 Status epilepticus was defined as per the latest International League Against Epilepsy (ILAE) definition. 24 A descriptive assessment of cognition and behavior was based on indicators of everyday functioning, informal cognitive tasks (eg, biographical info, remembering objects, making a judgment, playing with toys), contextual information (eg, language and education level), and presenting mental state (behavior, orientation, speech, mood, and perception). On this basis, cognitive function was classified by the clinician, into the following categories: normal, borderline, mildly impaired, moderately impaired, and severely impaired. Schooling progress was established based on the reported educational level and classified as normal, mildly reduced, moderately reduced, severely reduced, or absent, based on the level expected for the patient's age. Assessment of intellectual disability and language function was based on formal cognitive testing and classified as normal cognitive function, borderline, mild, moderate, severe, or profound. Formal cognitive assessment was performed according to age and verbal function, using one or more of the following neuropsychological tests: Brunet‐Lezine, Griffiths mental development scale, Bayley Scales of Infant and Toddler Development (Bayley‐III), Leiter International Performance Scale‐Revised (Leiter‐R), Raven's Coloured Progressive Matrices, Stanford‐Binet test, Wechsler Preschool and Primary Intelligence test (WPPSI), Wechsler Intelligence Scale for Children (WISC), Wechsler Adult Intelligence Scale | Fourth Edition (WAIS‐IV), and Mini‐Mental State Examination (MMSE). At the first visit, a total of 178 patients (70%) were assessed by neuropsychological tests, while the number of patients with available formal testing decreased at subsequent follow‐up, as shown in Table S1. Autism spectrum disorder was formally diagnosed through administration of the Autism Diagnostic Observation Schedule (ADOS); 25 autistic spectrum symptoms were defined when only some of the diagnostic criteria listed in the Diagnostic and Statistical Manual of Mental disorders, 5th edition (DSM‐5), were present; 26 Defiant, disobedient, or disruptive behavior was classified as “behavioral abnormalities.” Diagnoses of attention deficit, with or without hyperactivity, or obsessive‐compulsive disorder, were defined according to the International Classification of Diseases 10th Revision (ICD‐10). Unavoidably, data were not systematically collected at the same time points for all patients, and not using the same psychometric tools and scales in all centers, given the intrinsic design of the Registry, the different ages at first inclusion in the registry, and the inconstant availability of direct neuropsychological expertise in the various sites.
As a quality control step, a set of mandatory fields need to be filled to save information provided in each section. Additional quality control elements include a centralized system to check for duplicate case enrolment. Residras applies the principles of Findability, Accessibility, Interoperability, and Reusability (FAIR) for humans and computers, 27 thus enabling efficient analysis of data across multiple sources and making data “as open as possible and as closed as necessary.” 28 The platform has been customized for data collection specific to DS and includes 14 out of 16 Common Data Elements (CDEs) for Rare Diseases Registration released by the Joint Research Centre of the European Commission. 29 The two elements not included are 6.3 (undiagnosed cases are not included) and 7 (there is no collection of biological samples). The ontology codes used include Unified Medical Language System (UMLS), 30 Human Phenotype Ontology (HPO), 31 and Orphanet Rare Disease Ontology (ORDO). 32
To access the system, each user is assigned a personal username and password. The online input and access to the data are restricted to healthcare practitioners from each center. The access codes are generated by administrators once the user has signed a written agreement. Healthcare practitioners have an online and secure access to patients' data. Only fully anonymized data are available to researchers and for analysis—all enrolled participants have a registry identification code, which is automatically generated.
Investigators from expert centres who wish to access data for clinical research purposes are required to submit a research protocol to the internal Scientific Committee. When unanimous approval is obtained, the proposing team can access a subset of anonymized data depending on the study requirements.
The interface layout has been designed ad hoc to facilitate navigation and allows the use of various tools integrated into the system. The Residras home page describes SCN1A‐related conditions, the Residras platform (including aims, participating centres, statistical data, instruction to accredit a new center), and news in the field of SCN1A‐related disease. 20 Healthcare practitioners can access with their personal login and complete or create a new patient follow‐up. Patient health is summarized in a dashboard that helps physicians to have a detailed overview of the collected longitudinal data and to edit synopsizes or medical reports.
A diagnosis of DS is made based on the clinical definition and is distinct by other SCN1A‐related epilepsies. 2 Although inclusion of patients in Residras is based on clinical criteria, for the purpose of this study, we only considered DS individuals with a proven SCN1A mutation, classified as pathogenic or likely pathogenic according to the international guidelines of the American College of Medical Genetics and Genomics (ACMG). 33
2.3. Statistical analysis
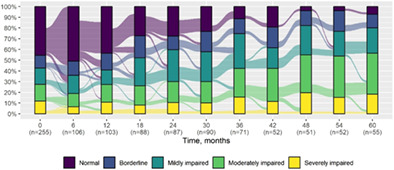
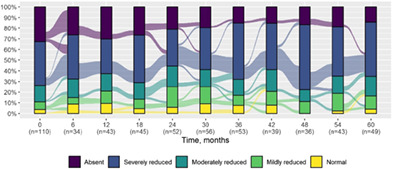
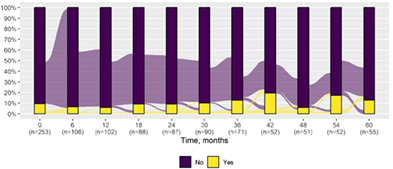
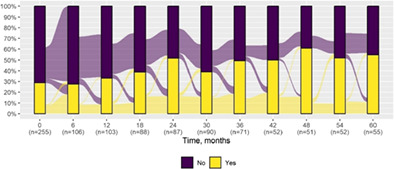
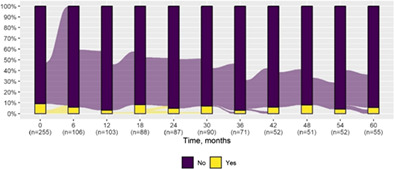
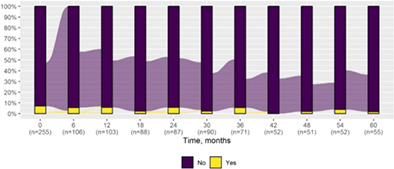
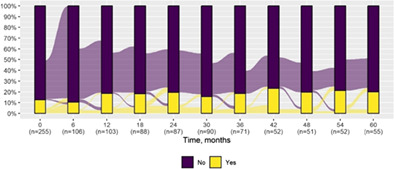
We present data as absolute number and percentage for categorical variable and median and interquartile range for continuous variables. We assessed clinical differences between first visit and after 5‐year follow‐up (Tables 1 and 2), and between first and last visit (Tables S2–S5), using the Chi‐square test for independent data. We analyzed patterns of changes in neurological condition and epilepsy features between first visit and after 5‐year follow‐up, after stratifying patients by age at first visit (Tables 3 and 4). When sensitivity analyses were performed using McNemar test for paired data, for subjects reporting both first and last visit data, results did not change. The mortality rate was calculated as number of observed events (death) divided by the person time at risk. Alluvial diagrams were used to illustrate the flows of variation in clinical conditions, from the first visit (time 0), every 6 months until the fifth year of follow‐up (time 60) (Tables 1 and 2), and after stratification by age at first visit (Figures S1–S22); each bar shows the frequency distribution of the variable over the corresponding time, change in the condition over time is represented by the flow stream direction going through different colors.
TABLE 1.
Neurological condition at first visit and after 5‐year follow‐up with alluvial diagrams showing variation over time.
| First visit | Last visit at 5th year | ||
|---|---|---|---|
| N | 255 | 55 | |
| Gender | |||
| Females | 132 (51.8) | 27 (49.1) | |
| Age | |||
| Median (IQR) | 2.0 (0–9.0) | 6.0 (5.0–9.5) | |
| Cognitive function* | |||
| Normal | 116 (45.5) | 4 (7.3) |
|
| Borderline | 30 (11.8) | 7 (12.7) | |
| Mildly impaired | 39 (15.3) | 13 (23.6) | |
| Moderately impaired | 40 (15.7) | 21 (38.2) | |
| Severely impaired | 30 (11.8) | 10 (18.2) | |
| Schooling progress* |
|
||
| Normal | 4 (3.6) | 2 (4.1) | |
| Mildly reduced | 8 (7.3) | 6 (12.2) | |
| Moderately reduced | 17 (15.5) | 9 (18.4) | |
| Severely reduced | 45 (40.9) | 25 (51.0) | |
| Absent | 36 (32.7) | 7 (14.3) | |
| Not applicable | 145 | 6 | |
| Behavior* | |||
| Oppositional defiant disorder | 24 (9.5) | 7 (12.7) |
|
| Attention deficit* | 45 (17.8) | 26 (47.3) |
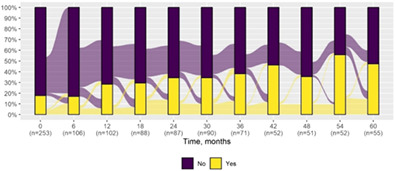
|
| Autism spectrum symptoms* | 14 (5.5) | 8 (14.5) |
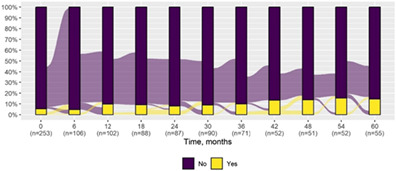
|
| Obsessive compulsive disorder | 6 (2.4) | 2 (3.6) |
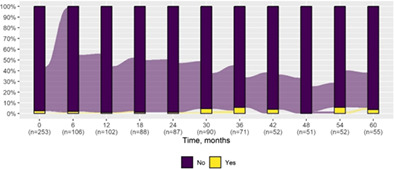
|
| Neurological examination | |||
| Normal* | 129 (50.6) | 11 (20.0) |
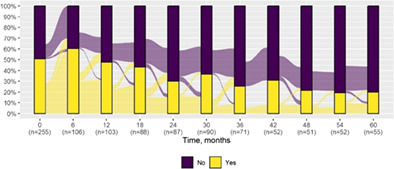
|
| Ataxia* | 73 (28.6) | 30 (54.5) |
|
| Pyramidal signs | 23 (9.0) | 3 (5.5) |
|
| Extrapyramidal signs | 18 (7.1) | 1 (1.8) |
|
| Action myoclonus | 32 (12.5) | 11 (20.0) |
|
| Gait abnormality* | 44 (17.4) | 19 (34.5) |
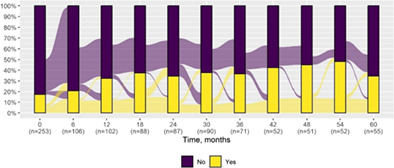
|
| Neuropsychological assessment | |||
| Language* | |||
| Normal | 79 (44.4) | 1 (5.0) |
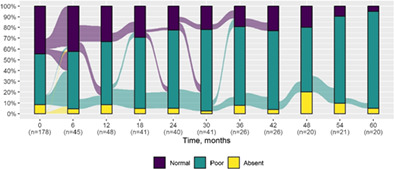
|
| Poor | 84 (47.2) | 18 (90.0) | |
| Absent | 15 (8.4) | 1 (5.0) | |
| Not assessable | 32 | 2 | |
| Intellectual disability* | |||
| Normal cognitive function | 70 (47.3) | — |
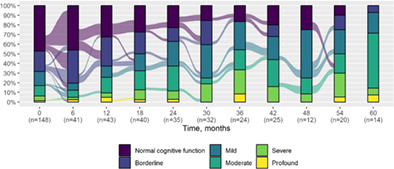
|
| Borderline | 31 (20.9) | 1 (7.1) | |
| Mild | 22 (14.9) | 3 (21.4) | |
| Moderate | 16 (10.8) | 8 (57.1) | |
| Severe | 7 (4.7) | 1 (7.1) | |
| Profound | 2 (1.4) | 1 (7.1) | |
| Not assessable | 32 | 2 | |
Statistically significant difference between first and last visit (p‐value <0.05).
TABLE 2.
Epilepsy features at first visit and after 5‐year follow‐up with alluvial diagrams showing variation over time.
| First visit | Last visit at 5th year | ||
|---|---|---|---|
| N | 263 | 83 | |
| Age, years | |||
| Median (IQR) | 2.0 (0–7.8) | 11.0 (5.0–18.0) | |
| Generalized seizures* | 195 (74.1) | 72 (86.7) |
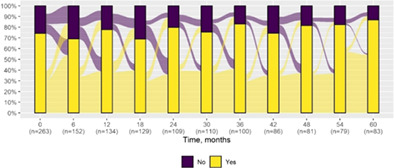
|
| Hemiclonic seizures* | 71 (27.0) | 7 (8.4) |
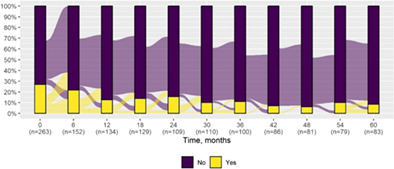
|
| Focal‐onset seizures | 75 (28.5) | 30 (36.1) |
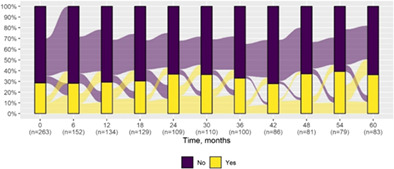
|
| Status epilepticus | 46 (17.5) | 7 (8.4) |
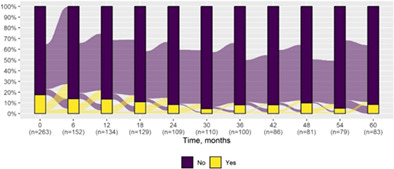
|
| Massive myoclonus | 50 (19.0) | 8 (9.6) |
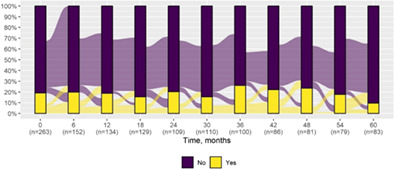
|
| Absence seizures | 42 (16.0) | 20 (24.1) |
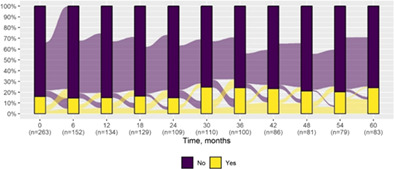
|
| Reflex seizures | 20 (7.6) | 5 (6.0) | |
| Seizure clusters | 30 (11.4) | 16 (19.3) |
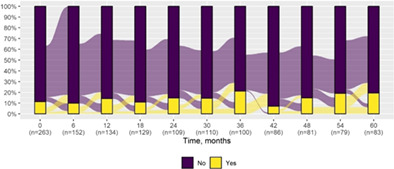
|
| Febrile seizures | 151 (71.9) | 6 (75.0) |
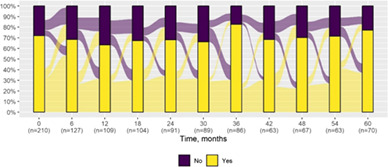
|
Statistically significant difference between first and last visit (p‐value <0.05).
TABLE 3.
Pattern of changes in neurological condition at 5th year, stratified by age at first visit (0–35 months vs over 35 months).
| Overall | 0–35 months | >35 months | |
|---|---|---|---|
| N (%) | 55 | 37 | 18 |
| Deterioration by | |||
| One class | 11 (20.0) | 8 (21.6) | 3 (16.7) |
| Two classes | 14 (25.5) | 11 (29.7) | 3 (16.7) |
| Three classes | 13 (23.6) | 11 (29.7) | 2 (11.1) |
| Four classes | 1 (1.8) | 1 (2.7) | — |
| Improvement | |||
| One class | 1 (1.8) | 1 (2.7) | — |
| Stability | 15 (27.3) | 5 (13.5) | 10 (55.6) |
| Autism spectrum disorder | |||
| Deterioration | 1 (1.8) | 1 (2.7) | — |
| Improvement | — | — | — |
| Stability | 54 (98.2) | 36 (97.3) | 18 (100.0) |
| Behavior | |||
| Oppositional defiant disorder | |||
| Deterioration | 7 (12.7) | 4 (10.8) | 3 (16.7) |
| Improvement | 2 (3.6) | — | 2 (11.1) |
| Stability | 46 (83.6) | 33 (89.2) | 13 (72.2) |
| Attention deficit | |||
| Deterioration | 21 (38.2) | 16 (43.2) | 5 (27.8) |
| Improvement | 2 (3.6) | 2 (5.4) | — |
| Stability | 32 (58.2) | 19 (51.4) | 13 (72.2) |
| Autism spectrum symptoms | |||
| Deterioration | 7 (12.7) | 5 (13.5) | 2 (11.1) |
| Improvement | — | — | — |
| Stability | 48 (87.3) | 32 (86.5) | 16 (88.9) |
| Obsessive compulsive disorder | |||
| Deterioration | 1 (1.8) | — | 1 (5.6) |
| Improvement | 1 (1.8) | — | 1 (5.6) |
| Stability | 53 (96.4) | 37 (100.0) | 16 (88.9) |
| Neurological examination | |||
| Normal | |||
| Deterioration | 26 (47.3) | 21 (56.8) | 5 (27.8) |
| Improvement | 2 (3.6) | 2 (5.4) | ‐ |
| Stability | 27 (49.1) | 14 (37.8) | 13 (72.2) |
| Ataxia | |||
| Deterioration | 22 (40.0) | 20 (54.1) | 2 (11.1) |
| Improvement | 2 (3.6) | 2 (5.4) | — |
| Stability | 31 (56.4) | 15 (40.5) | 16 (88.9) |
| Pyramidal signs | |||
| Deterioration | 2 (3.6) | 2 (5.4) | — |
| Improvement | 1 (1.8) | 1 (2.7) | — |
| Stability | 52 (94.5) | 34 (91.9) | 18 (100.0) |
| Extrapyramidal signs | |||
| Deterioration | 1 (1.8) | — | 1 (5.6) |
| Improvement | 1 (1.8) | 1 (2.7) | — |
| Stability | 53 (96.4) | 36 (97.3) | 17 (94.4) |
| Action myoclonus | |||
| Deterioration | 7 (12.7) | 6 (16.2) | 1 (5.6) |
| Improvement | 2 (3.6) | 1 (2.7) | 1 (5.6) |
| Stability | 46 (83.6) | 30 (81.1) | 16 (88.9) |
| Gait abnormality | |||
| Deterioration | 14 (25.5) | 13 (35.1) | 1 (5.6) |
| Improvement | 3 (5.5) | 2 (5.4) | 1 (5.6) |
| Stability | 38 (69.1) | 22 (59.5) | 16 (88.9) |
| Neuropsychological assessment | |||
| N (%) | 22 | 15 | 7 |
| Language | |||
| Deterioration | 7 (31.8) | 5 (33.3) | 2 (28.6) |
| Improvement | 2 (9.1) | 1 (6.7) | 1 (14.3) |
| Stability | 10 (45.5) | 7 (46.7) | 3 (42.9) |
| Not assessable | 3 (13.6) | 2 (13.3) | 1 (14.3) |
| Intellectual disability | |||
| N (%) | 15 | 11 | 4 |
| Deterioration by | |||
| One class | 1 (6.7) | 1 (9.1) | — |
| Two classes | 4 (26.7) | 3 (27.3) | 1 (25.0) |
| Three classes | 2 (13.3) | 2 (18.2) | — |
| Four classes | 1 (6.7) | 1 (9.1) | — |
| Five classes | 1 (6.7) | 1 (9.1) | — |
| Improvement | |||
| One class | 1 (6.7) | 1 (9.1) | — |
| Stability | 2 (16.7) | — | 2 (50.0) |
| Not assessable | 3 (20.0) | 2 (18.2) | 1 (25.0) |
TABLE 4.
Pattern of changes in epilepsy features at 5th year, stratified by age at first visit (0–35 months vs over 35 months).
| Overall | 0–35 months | >35 months | |
|---|---|---|---|
| N (%) | 83 | 66 | 17 |
| Generalized seizures | |||
| Deterioration | 14 (16.9) | 13 (19.7) | 1 (5.9) |
| Improvement | 8 (9.6) | 4 (6.1) | 4 (23.5) |
| Stability | 61 (73.5) | 49 (74.2) | 12 (70.6) |
| Hemiclonic seizures | |||
| Deterioration | 6 (7.2) | 4 (6.1) | 2 (11.8) |
| Improvement | 21 (25.3) | 18 (27.3) | 3 (17.6) |
| Stability | 56 (67.5) | 44 (66.7) | 12 (70.6) |
| Focal onset seizures | |||
| Deterioration | 16 (19.3) | 14 (21.2) | 2 (11.8) |
| Improvement | 11 (13.3) | 10 (15.2) | 1 (5.9) |
| Stability | 56 (67.5) | 42 (63.6) | 14 (82.4) |
| Status epilepticus | |||
| Deterioration | 6 (7.2) | 6 (9.1) | — |
| Improvement | 18 (21.7) | 17 (25.8) | 1 (5.9) |
| Stability | 59 (71.1) | 43 (65.2) | 16 (94.1) |
| Massive myoclonus | |||
| Deterioration | 5 (6.0) | 3 (4.5) | 2 (11.8) |
| Improvement | 18 (21.7) | 15 (22.7) | 3 (17.6) |
| Stability | 60 (72.3) | 48 (72.7) | 12 (70.6) |
| Absence seizures | |||
| Deterioration | 12 (14.5) | 11 (16.7) | 1 (5.9) |
| Improvement | 6 (7.2) | 3 (4.5) | 3 (17.6) |
| Stability | 65 (78.3) | 52 (78.8) | 13 (76.5) |
| Seizure clusters | |||
| Deterioration | 14 (16.9) | 13 (19.7) | 1 (5.9) |
| Improvement | 10 (12.0) | 8 (12.1) | 2 (11.8) |
| Stability | 59 (71.1) | 45 (68.2) | 14 (82.4) |
| Febrile seizures | |||
| Deterioration | 6 (7.2) | 6 (9.1) | — |
| Improvement | 10 (12.0) | 10 (15.2) | — |
| Stability | 45 (54.2) | 32 (48.5) | 13 (76.5) |
| Missing | 22 (26.5) | 18 (27.3) | 4 (23.5) |
3. RESULTS
Although Residras has a nationwide coverage (Figure 1), patients' geographic distribution remained non‐homogeneous with a lower proportional representation from the South of Italy. This is mainly related to the habit of families of seriously ill patients residing in the South to reach hospitals of the northern regions, seeking for a second opinion, and does not have any geographic epidemiological implication. To date, 281 individuals with DS with a confirmed SCN1A mutation have been enrolled. There are available data on epilepsy (n = 263) and on the overall neurological status (n = 255), with at least one follow‐up for most patients (Figure S23). Historical retrospective data were also included, when enrolment date was during a follow‐up visit and the patient had not been previously included in the registry or when a diagnosis of DS was made at a later time after the first clinical evaluation. The time at the first visit was defined as the time at the inclusion in the registry for all patients, including the ones with enrolment date after first clinical assessment.
Median follow‐up from inclusion in the Registry was 5.5. years (2.8–11.3). Median age at the inclusion in the Registry was 2 years (IQR 0–9) while the median age at last follow‐up was 11 years (IQR 5.5–17). During the 7‐year activity of the registry, five patients with a diagnosis of DS had died, with a mortality rate of 1.84 (95% CI 0.77–4.42) per 1000‐person‐years (median age at death 6.3 years, range 2.5–23.4). The causes of death were diverse and included status epilepticus, cerebral hemorrhage, SUDEP, acute encephalopathy of unknown cause (with onset 40 days before death), and brain tumor.
There was a significant difference in cognitive function over the first 5 years of follow‐up (P < 0.001) with evidence of a lower cognitive level after 5 years in the majority (median time to first deterioration in cognitive level 1.5 years, IQR 0.9–2.4). There was a significant change in the schooling progress over time; in particular, we observed increased prevalence of poor schooling progress (from 17.6% at first visit to 45.5% after 5 years) (P < 0.001). There was an increased prevalence of behavioral disorders over time including attention deficit (P < 0.001) and autism spectrum symptoms (P = 0.04). Prevalence of neurological examination abnormalities increased at last visit (P < 0.001) in most patients (median time to first deterioration 1.6 years, IQR 0.9–2.4), including ataxia (P < 0.001) and gait abnormality (P = 0.01).There was also a significant worsening of language (P = 0.001) (median time to first decreased level 1.4 years, IQR 0.8–2.3) and intellectual disability (P < 0.001) (median time to first decreased level 1.5 years, IQR 1.0–2.3), where these were formally assessable.
Significant differences related to the epilepsy features over the first 5‐year follow‐up included an increased prevalence of generalized seizures (P = 0.03) and reduction of hemiclonic seizures (P = 0.001). The main clinical variables included in the analysis and their variation over the first 5‐year follow‐up are illustrated in Tables 1 and 2. The pattern of improvement, stability, or deterioration related to each clinical variable is summarized in Tables 3 and 4, and Tables S4 and S5. The main clinical variables stratified by age group and clinical differences between first and last follow‐up are illustrated in the Figures S1–S22. Different antiseizure treatments were used over time, with the longest treatment duration observed for valproate, clobazam, stiripentol, and topiramate (Table 5). Precise information on treatment duration is negatively affected by the repeated alternations of drugs many patients experienced during seizure exacerbation periods, especially when they were followed at more than one site.
TABLE 5.
Use and duration of antiseizure treatments.
| N (%) | Cumulative duration, years | Median individual duration (IQR), years | |
|---|---|---|---|
| N | 251 | ||
| Drug | |||
| Sodium valproate | 237 (94.4) | 3164.6 | 4.5 (1.0–11.3) |
| Clobazam | 184 (73.3) | 1501.6 | 3.9 (1.0–8.0) |
| Stiripentol | 134 (53.4) | 906.3 | 4.2 (1.5–7.6) |
| Topiramate | 119 (47.4) | 1098.0 | 3.4 (1.0–8.5) |
| Levetiracetam | 92 (36.7) | 505.2 | 1.7 (0.5–5.7) |
| Phenobarbital | 73 (29.1) | 632.2 | 1.3 (0.4–6.9) |
| Clonazepam | 73 (29.1) | 594.3 | 3.2 (1.0–8.0) |
| Fenfluramine | 52 (20.7) | 85.3 | 1.2 (0.2–1.8) |
| Carbamazepine | 48 (19.1) | 130.8 | 0.3 (0.1–2.9) |
| Lamotrigine | 40 (15.9) | 200.5 | 1.1 (0.2–5.7) |
| Ethosuximide | 37 (14.7) | 228.5 | 1.6 (0.5–5.2) |
| ACTH | 21 (8.4) | 16.2 | 0.1 (0.0–0.3) |
| Nitrazepam | 16 (6.4) | 182.2 | 4.7 (1.2–14.6) |
| Magnesium valproate | 16 (6.4) | 209.8 | 12.2 (3.7–22.3) |
| Zonisamide | 16 (6.4) | 69.0 | 1.9 (0.5–3.6) |
| Acetazolamide | 13 (5.2) | 52.5 | 2.3 (0.6–5.4) |
| Ketogenic diet | 13 (5.2) | 36.3 | 0.9 (0.5–3.5) |
| Nervus Vagus Stimolation | 13 (5.2) | 138.8 | 8.7 (5.4–11.3) |
| Vigabatrin | 12 (4.8) | 26.5 | 0.8 (0.3–3.7) |
| Cannabidiol | 10 (4.0) | 24.2 | 2.0 (0.3–2.3) |
| Phenitoin | 10 (4.0) | 32.6 | 1.1 (0.5–5.8) |
| Clinical Trial ZX008 | 9 (3.6) | 15.3 | 0.7 (0.4–3.1) |
| Felbamate | 8 (3.2) | 15.4 | 1.2 (0.5–1.8) |
| Primidone | 6 (2.4) | 80.4 | 13.9 (5.5–19.3) |
| Lacosamide | 5 (2.0) | 5.6 | 0.5 (0.3–1.0) |
| Benzodiazepine | 4 (1.6) | 30.0 | 2.6 (0.8–9.3) |
| Diazepam | 4 (1.6) | 4.2 | 0.5 (0.3–1.2) |
| Oxcarbazepine | 4 (1.6) | 7.7 | 0.7 (0.5–0.8) |
| Perampanel | 4 (1.6) | 5.3 | 1.2 (1.0–1.5) |
| Gabapentin | 3 (1.2) | 2.4 | 0.1 (0.1–1.2) |
| Midazolam | 2 (0.8) | 7.8 | 3.9 (2.0–5.8) |
| Rufinamide | 2 (0.8) | 1.4 | 0.5 (0.4–0.5) |
| Tiagabine | 2 (0.8) | 1.0 | 0.5 (0.3–0.7) |
4. DISCUSSION
Residras is based on a user‐friendly platform that facilitates data collection and analysis of patients with SCN1A‐related epilepsies, and on a network of 24 expert centers across Italy with specific expertise in rare epilepsy syndromes, with a scope to include further national and international centers and promote a longitudinal standardized data collection. The Residras initiative might represent a paradigmatic example to homogenize data collection and improve research in rare and complex epilepsy syndromes. During the registry setup, several control steps were put in place to ensure high data quality such as centralized control for duplicates, set of mandatory fields, use of standardized ontology codes, and a robust security infrastructure. Although Residras has ample potential for expansion at both national and international level so to constitute a unified source of longitudinal phenotypic data for DS and other SCN1A‐related epilepsies, feeding the registry for a complex epilepsy is time demanding, which may in part explain why there are no similar registries in place.
We adopted two different approaches to analyze clinical variation over time. We focussed on the first 5‐year follow‐up to increase specificity of the disease evolution from onset, although clinical assessment at the 5‐year time point was not available for most patients (Tables 1 and 2); we then analyzed variation from first to last visit but noting that follow‐up length was variable, and therefore, results are less specific to interpret disease course (Tables S2 and S3).
Our data confirm worsening of cognitive ability over time in DS, as already reported in cross‐sectional and retrospective studies. 12 , 34 A minority of patients exhibited normal cognitive skills at last follow‐up. Although we excluded febrile convulsions and GEFS+ phenotypes from the analysis, borderline DS phenotypes may explain this finding. Since the longer‐term cognitive outcome is often unpredictable early after onset, excluding patients with less severe outcomes in the aftermath as they do not fit the core clinical definition would be artifactual and not reflect the whole spectrum of the syndrome. The items evaluating cognitive, language, and behavioral skills were designed to capture the granularity of the complex neurodevelopmental phenotype, hence to provide in the longer term a robust basis to assess the impact of existing and novel treatment strategies. Although DS diagnosis does not require the mandatory use of specific neuropsychological and behavioral testing, we are now implementing a more systematic and uniform use of a standardized assessment as the data until now collected in the registry derived from a sum of local clinical practices and do not inform with sufficient detail the cognitive outcome and how this may be affected by novel treatments. For example, we point out that the number of individuals with autistic symptoms is low, but an increased prevalence is observed during the first 5‐year follow‐up. A formal diagnosis through ADOS assessment was obtained only in a minority of patients, and this might be related to an underreporting of comorbid autism spectrum symptoms and the limited access to this diagnostic test, only possible in centers with specifically trained staff. The increased prevalence of autism over time might be in part consequent to difficulties in diagnosing autistic features under the age of 3 years. However, since a formal diagnostic assessment for autism spectrum disorder was not regularly available, we cannot draw any definite conclusion on this comorbidity.
Premature mortality is a recognized unfavorable outcome in DS although most available data are from children, 17 , 18 and there are no survival analyses in the long term. In a cohort of 100 consecutively recruited DS individuals, mortality rate was 15.84 (98% CI 9.01–27.85) per 1000‐person‐years while the rate of SUDEP was 9.32/1000‐person‐years. Living individuals had a median follow‐up of 17 years, while the median age at death was 7 years. 18 We observed a mortality rate of 1.84 (95% CI 0.77–4.42) per 1000‐person‐years, with a median age at death of 6 years. Of the five deaths reported, one was due to SUDEP, and one was epilepsy‐related (ie, status). Multiple factors may concur in generating the far lower mortality rates we observed with respect to abovementioned report. 18 Firstly, mortality data emerging from a registry need to be interpreted with caution as death may occur before a diagnosis is made or before a patient is included or after the latest follow‐up data entry. Secondly, our results might be affected by immortal bias 35 as our cohort was much younger than Cooper et al.'s cohort. 18 In addition, differences in rates of premature mortality may be partly explained by diverse healthcare provision and treatment strategies. The Cooper et al. 18 DS cohort was more heterogeneous in terms of geographic origin and was established almost 20 years earlier, which implies a longer follow‐up and little or no access to more recently introduced drugs with a proven efficacy in DS, particularly stiripentol, cannabidiol, and fenfluramine. 6 , 7 , 8 , 9 Finally, improved management strategies have been applied over the years, including earlier diagnosis in larger number of patients and avoidance of inappropriate drugs, such as sodium channel blockers, or exceedingly sedative drug regimens. 36 , 37 All these factors may contribute to reduced mortality rates in subsequent generations of patients.
Additional highly relevant assets provided by the registry include its reliability in delineating the natural history of DS, the longitudinal comparison of medical practice between DS expert centres, the availability of a wide clinical and genetic spectrum of patients with SCN1A‐related epilepsies whose characteristics, homogeneously recorded, are available for genotype‐phenotype correlations, and comprehensive characterization of comorbidities occurring at different ages.
Accuracy in managing a registry for DS has limitations, as it may be expected for a severe disease starting early in life, associating chronic disability with periods of acute exacerbations and comorbidities that require multiple medical interventions and treatment adjustments. Unavoidably, some data are missing, inclusion of follow‐up information might not respect the set deadlines, the various centers may apply different levels of completeness in reporting relevant information, and application of clinical diagnostic criteria for milder DS forms or other SCN1A‐related epilepsies is not necessarily uniform. Information on the use of rescue medications might be limited if data are not timely included in the registry. Additional limitations include the lack of systematic data on seizure frequency at each follow‐up, as seizure diaries were not regularly used by families or data were not regularly entered by clinicians. Given the likely underreporting of seizure frequency, we omitted them from the analysis. The data on cognitive, language, and behavioral skills gathered in the registry do not yet provide the granularity of the complex neurodevelopmental trajectories. Data on treatment are also limited with lack of systematic information on treatment response but only providing a snapshot on clinicians' prescription habits over follow‐up. However, the aim of this study was not to add novel findings to the existing literature on DS but to provide a registry model for a rare epilepsy syndrome highlighting its strengths and limitations and discussing how this can be further implemented to serve as a basis to collect data on natural history, novel disease‐modifying treatments, and genotype‐phenotype correlation.
A further complication in registry curation for a chronic disease that certainly exists across all health systems is related to transition to adult care. Unless there is continuity of care, for example in a specialized institution, patients may transition to adult neurology centers with limited expertise in DS and propensity to adhere to a registry conceived for an infantile onset disorder. Despite 7 years of Registry activity, we could not obtain regular systematic follow‐up information for the majority of patients, with increasing patients' loss to follow‐up over time, eg, longitudinal data on cognitive function are currently available only for a minority of patients. This is due to a combination of factors including limited personnel resources and geographical bias due to patients residing in the South often traveling or moving to other regions for medical care.
Registry curation is time‐consuming and might be particularly challenging for those centers with high number of patients if no specifically dedicated personnel is available. Multiple associations for specific genetic disorders are being founded with often the aim to establish dedicated registries, but it may become challenging for the treating physicians to fill different registries with variable formats and become familiar with them. The registry inception and initial activities were supported by limited funding raised by a no‐profit patient association (Dravet Italia Onlus). These multiple challenges and initial pitfalls we illustrated should not discourage from establishing registries for specific rare and complex epilepsies as they may represent a basis for funding support within the framework of rare diseases initiatives and private funding. Residras has now been funded by the Italian Ministry of Health (project code PNRR‐MR1‐2022‐12 376 642, https://www.pnrr.salute.gov.it/portale/pnrrsalute/dettaglioBandiPNRRSalute.jsp?lingua=italiano&id=295), based on a project to promote the registry activities, increase its coverage and curation, and limit the number who may be lost to follow‐up.
There are also initiatives by clinicians and scientists to collect information on rare epilepsies such as the Network for Therapy in Rare Epilepsies (NETRE), 38 and there are examples of registries for rare diseases which include epilepsy among their clinical manifestations such as Tuberous Sclerosis Complex. 39 There is a recently started project of a registry for rare and complex epilepsy syndromes, the EpiCARE Registry Project. 40
Applying the Residras model to other rare and complex genetic epilepsies would help gathering up for each of them a critical mass of homogeneously stored information on epidemiology, disease course, attract dedicated funding and easing access to cohorts of patients that may benefit from clinical trials and drug safety monitoring. Therapy development for rare diseases faces several specific challenges, including small populations for clinical studies, difficulty in determining relevant outcome measures and endpoints, and poorly understood natural history.
CONFLICT OF INTEREST STATEMENT
None of the authors has any conflict of interest to disclose.
ETHICS STATEMENT
We confirm that we have read the journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Appendix S1
ACKNOWLEDGMENTS
The Residras Collaboration Group includes: Francesca Bisulli1, Maria Paola Canevini2, Marco Carrozzi3, Paola Costa3, Carlo Fusco4, Laura Licchetta1, Lorenzo Muccioli1, Francesca Felicia Operto5, Elisa Osanni6, Lucio Parmeggiani7, Antonella Pini8, Carlotta Spagnoli4, Claudio Zucca9, Caterina Zanus3.
1Department of Biomedical and Neuromotor Sciences, University of Bologna, Full Member of European Reference Network EpiCARE, Bologna, Italy;
2Epilepsy Center, ASST Santi Paolo Carlo, Health Sciences Department University of Milan, Italy;
3Institute for Maternal and Child Health, IRCCS “Burlo Garofolo”, Trieste, Italy;
4Department of Pediatrics, Child Neurology Unit, AUSL‐ IRCCS of Reggio Emilia, Reggio Emilia, Italy;
5Child Neuropsychiatry Unit, Department of Medicine, Surgery, and Dentistry, University of Salerno, Salerno, Italy;
6Child Neuropsychiatry, Epilepsy and Clinical Neurophysiology Unit, IRCCS “E. Medea”, Conegliano, Treviso, Italy;
7Department of Pediatric Neurology, Bolzano Hospital, Bolzano, Italy;
8IRCCS Istituto delle Scienze Neurologiche di Bologna, Child Neurology and Psychiatry Department, Bologna, Italy;
9Clinical Neurophysiology Unit, Scientific Institute IRCCS E. Medea, 23842 Bosisio Parini, Lecco, Italy.
The project has been supported by the Italian Ministry of Health (project code PNRR‐MR1‐2022‐12376642). Neuroscience Department, Meyer Children's Hospital IRCSS; Neuroscience Department, Bambino Gesù Children's Hospital IRCSS; Pediatric Neuroscience Department, Fondazione IRCSS Istituto Neurologico Carlo Besta; Department of Biomedical and Neuromotor Sciences, University of Bologna, are full members of ERN Epicare.
Balestrini S, Doccini V, Giometto S, Lucenteforte E, De Masi S, Giarola E, et al. Residras Collaboration Group. A registry for Dravet syndrome: The Italian experience. Epilepsia Open. 2023;8:517–534. 10.1002/epi4.12730
REFERENCES
- 1. Symonds JD, Elliott KS, Shetty J, Armstrong M, Brunklaus A, Cutcutache I, et al. Early childhood epilepsies: epidemiology, classification, aetiology, and socio‐economic determinants. Brain. 2021;144(9):2879–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mei D, Cetica V, Marini C, Guerrini R. Dravet syndrome as part of the clinical and genetic spectrum of sodium channel epilepsies and encephalopathies. Epilepsia. 2019;60(Suppl 3):S2–7. [DOI] [PubMed] [Google Scholar]
- 3. Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9(9):1142–9. [DOI] [PubMed] [Google Scholar]
- 4. Li W, Schneider AL, Scheffer IE. Defining Dravet syndrome: an essential pre‐requisite for precision medicine trials. Epilepsia. 2021;62(9):2205–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wirrell EC, Laux L, Donner E, Jette N, Knupp K, Meskis MA, et al. Optimizing the diagnosis and Management of Dravet Syndrome: recommendations from a north American consensus panel. Pediatr Neurol. 2017;68:18–34.e3. [DOI] [PubMed] [Google Scholar]
- 6. Chiron C, Marchand MC, Tran A, Rey E, d'Athis P, Vincent J, et al. Stiripentol in severe myoclonic epilepsy in infancy: a randomised placebo‐controlled syndrome‐dedicated trial. STICLO Study Group. Lancet. 2000;356(9242):1638–42. [DOI] [PubMed] [Google Scholar]
- 7. Devinsky O, Cross JH, Laux L, Marsh E, Miller I, Nabbout R, et al. Trial of cannabidiol for drug‐resistant seizures in the Dravet syndrome. N Engl J Med. 2017;376(21):2011–20. [DOI] [PubMed] [Google Scholar]
- 8. Lagae L, Sullivan J, Knupp K, Laux L, Polster T, Nikanorova M, et al. Fenfluramine hydrochloride for the treatment of seizures in Dravet syndrome: a randomised, double‐blind, placebo‐controlled trial. Lancet. 2019;394(10216):2243–54. [DOI] [PubMed] [Google Scholar]
- 9. Nabbout R, Mistry A, Zuberi S, Villeneuve N, Gil‐Nagel A, Sanchez‐Carpintero R, et al. Fenfluramine for treatment‐resistant seizures in patients with Dravet syndrome receiving Stiripentol‐inclusive regimens: a randomized clinical trial. JAMA Neurol. 2020;77(3):300–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Isom LL, Knupp KG. Dravet syndrome: novel approaches for the Most common genetic epilepsy. Neurotherapeutics. 2021;18(3):1524–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Akiyama M, Kobayashi K, Yoshinaga H, Ohtsuka Y. A long‐term follow‐up study of Dravet syndrome up to adulthood. Epilepsia. 2010;51(6):1043–52. [DOI] [PubMed] [Google Scholar]
- 12. Ragona F, Granata T, Dalla Bernardina B, Offredi F, Darra F, Battaglia D, et al. Cognitive development in Dravet syndrome: a retrospective, multicenter study of 26 patients. Epilepsia. 2011;52(2):386–92. [DOI] [PubMed] [Google Scholar]
- 13. Darra F, Battaglia D, Dravet C, Patrini M, Offredi F, Chieffo D, et al. Dravet syndrome: early electroclinical findings and long‐term outcome in adolescents and adults. Epilepsia. 2019;60(Suppl 3):S49–58. [DOI] [PubMed] [Google Scholar]
- 14. Catarino CB, Liu JYW, Liagkouras I, Gibbons VS, Labrum RW, Ellis R, et al. Dravet syndrome as epileptic encephalopathy: evidence from long‐term course and neuropathology. Brain. 2011;134(Pt 10):2982–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Genton P, Velizarova R, Dravet C. Dravet syndrome: the long‐term outcome. Epilepsia. 2011;52(Suppl 2):44–9. [DOI] [PubMed] [Google Scholar]
- 16. Takayama R, Fujiwara T, Shigematsu H, Imai K, Takahashi Y, Yamakawa K, et al. Long‐term course of Dravet syndrome: a study from an epilepsy center in Japan. Epilepsia. 2014;55(4):528–38. [DOI] [PubMed] [Google Scholar]
- 17. Sakauchi M, Oguni H, Kato I, Osawa M, Hirose S, Kaneko S, et al. Mortality in Dravet syndrome: search for risk factors in Japanese patients. Epilepsia. 2011;52(Suppl 2):50–4. [DOI] [PubMed] [Google Scholar]
- 18. Cooper MS, Mcintosh A, Crompton DE, McMahon JM, Schneider A, Farrell K, et al. Mortality in Dravet syndrome. Epilepsy Res. 2016;128:43–7. [DOI] [PubMed] [Google Scholar]
- 19. Dawkins HJS, Draghia‐Akli R, Lasko P, Lau LPL, Jonker AH, Cutillo CM, et al. Progress in rare diseases research 2010–2016: an IRDiRC perspective. Clin Transl Sci. 2018;11(1):11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Residras . Dravet registry [Internet]. [cited 2022 Jan 3]. Available from https://www.dravet‐registry.com/registry
- 21. Dravet Italia Onlus [Internet]. [cited 2022 Jan 3]. Available from https://www.dravet.it
- 22. Fondazione Monasterio [Internet]. [cited 2022 Jan 3]. Available from https://www.monasterio.it/
- 23. Dravet C, Guerrini R. Dravet syndrome. Cobham, Surrey, England: John Libbey Eurotext; 2023. [Google Scholar]
- 24. Trinka E, Cock H, Hesdorffer D, Rossetti AO, Scheffer IE, Shinnar S, Shorvon S, Lowenstein DH. A definition and classification of status epilepticus–Report of the ILAE Task Force on Classification of Status Epilepticus. Epilepsia. 2015;56(10):1515–23. 10.1111/epi.13121. [DOI] [PubMed] [Google Scholar]
- 25. Lord C, Rutter M, DiLavore P, Risi S, Gotham K, Bishop S. Autism diagnostic observation schedule–2nd edition (ADOS‐2). Los Angeles, CA: Western Psychological Services; 2012. [Google Scholar]
- 26. American Psychiatric Association , DSM‐5 Task Force . (2013). Diagnostic and statistical manual of mental disorders: DSM‐5™ (5th ed.). American Psychiatric Publishing, Inc. 10.1176/appi.books.9780890425596 [DOI] [Google Scholar]
- 27. Wilkinson MD, Dumontier M, Aalbersberg IJJ, Appleton G, Axton M, Baak A, et al. The FAIR guiding principles for scientific data management and stewardship. Sci Data. 2016;15(3):160018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mons B, Neylon C, Velterop J, Dumontier M, da Silva Santos LOB, Wilkinson M. Cloudy, increasingly FAIR; revisiting the FAIR data guiding principles for the European Open Science cloud. Inf Serv Use. 2017;37(1):49–56. [Google Scholar]
- 29. European Commission . Set of Common Data Elements [Internet]. [cited 2022 Jan 3]. Available from https://eu‐rd‐platform.jrc.ec.europa.eu/set‐of‐common‐data‐elements_en
- 30. Bodenreider O. The unified medical language system (UMLS): integrating biomedical terminology. Nucleic Acids Res. 2004;32:D267–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Human Phenotype Ontology (HPO) [Internet]. [cited 2022 Jan 3]. Available from https://hpo.jax.org/app/
- 32. Orphanet Rare Disease Ontology (ORDO) [Internet]. [cited 2022 Jan 3]. Available from https://bioportal.bioontology.org/ontologies/ORDO
- 33. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wolff M, Cassé‐Perrot C, Dravet C. Severe myoclonic epilepsy of infants (Dravet syndrome): natural history and neuropsychological findings. Epilepsia. 2006;47(Suppl 2):45–8. [DOI] [PubMed] [Google Scholar]
- 35. Yadav K, Lewis RJ. Immortal time bias in observational studies. JAMA. 2021;325(7):686–7. [DOI] [PubMed] [Google Scholar]
- 36. Rosati A, De Masi S, Guerrini R. Antiepileptic drug treatment in children with epilepsy. CNS Drugs. 2015;29(10):847–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rintahaka PJ, Nakagawa JA, Shewmon DA, Kyyronen P, Shields WD. Incidence of death in patients with intractable epilepsy during nitrazepam treatment. Epilepsia. 1999;40(4):492–6. [DOI] [PubMed] [Google Scholar]
- 38. Netre [Internet]. Available from http://netre.de/
- 39. Auvin S, Bissler JJ, Cottin V, Fujimoto A, Hofbauer GFL, Jansen AC, et al. A step‐wise approach for establishing a multidisciplinary team for the management of tuberous sclerosis complex: a Delphi consensus report. Orphanet J Rare Dis. 2019;14(1):91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. EpiCare Registry [Internet]. Available from https://epi‐care.eu/epicares‐activities/registry‐project/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1