

Abstract
Cancer cachexia is a systemic hypoanabolic and catabolic syndrome that diminishes the quality of life of cancer patients, decreases the efficiency of therapeutic strategies and ultimately contributes to decrease their lifespan. The depletion of skeletal muscle compartment, which represents the primary site of protein loss during cancer cachexia, is of very poor prognostic in cancer patients. In this review, we provide an extensive and comparative analysis of the molecular mechanisms involved in the regulation of skeletal muscle mass in human cachectic cancer patients and in animal models of cancer cachexia. We summarize data from preclinical and clinical studies investigating how the protein turnover is regulated in cachectic skeletal muscle and question to what extent the transcriptional and translational capacities, as well as the proteolytic capacity (ubiquitin–proteasome system, autophagy–lysosome system and calpains) of skeletal muscle are involved in the cachectic syndrome in human and animals. We also wonder how regulatory mechanisms such as insulin/IGF1–AKT–mTOR pathway, endoplasmic reticulum stress and unfolded protein response, oxidative stress, inflammation (cytokines and downstream IL1ß/TNFα–NF‐κB and IL6–JAK–STAT3 pathways), TGF‐ß signalling pathways (myostatin/activin A‐SMAD2/3 and BMP‐SMAD1/5/8 pathways), as well as glucocorticoid signalling, modulate skeletal muscle proteostasis in cachectic cancer patients and animals. Finally, a brief description of the effects of various therapeutic strategies in preclinical models is also provided. Differences in the molecular and biochemical responses of skeletal muscle to cancer cachexia between human and animals (protein turnover rates, regulation of ubiquitin‐proteasome system and myostatin/activin A‐SMAD2/3 signalling pathways) are highlighted and discussed. Identifying the various and intertwined mechanisms that are deregulated during cancer cachexia and understanding why they are decontrolled will provide therapeutic targets for the treatment of skeletal muscle wasting in cancer patients.
Keywords: Autophagy–lysosome, Cancer cachexia, Glucocorticoids, Inflammation, Myostatin, Oxidative stress, Proteostasis, Skeletal muscle, Ubiquitin–proteasome
Introduction
Cancer cachexia is a systemic hypoanabolic and catabolic syndrome characterized by a progressive unintentional loss of body mass that cannot be reversed by nutritional support. 1 The prevalence of cachexia is quite variable, reaching up to 70% in pancreatic cancer patients. 2 Skeletal muscle represents the primary site of protein loss during cancer cachexia. 3 Image analysis indicates that muscle depletion varies from 7 to 30%, 4 , 5 , S1–S6 (references S1 to S215 are listed as a supplementary reference list) the depletion worsening with the severity of the disease. 5 Another noteworthy feature is muscle weakness, which often may precede muscle loss S7,S8 and which aggravates with cachexia. S7 Besides muscle, the syndrome can also affect other tissues including bone, heart, liver and/or adipose tissue. 6 Overall, cancer cachexia increases the risk of surgical complications 7 , ,S9 and chemotherapy toxicity S10–S13 and lead to functional impairments, respiratory complications and fatigue that markedly reduce patients' quality of life and ultimately patients' survival. It has been estimated that in 2013, 15.8 subjects per 10 000 of the total population in European Union suffered from cancer cachexia. 8
Over the past 15 years, considerable progress has been made in elucidating the molecular pathways involved in muscle mass loss, thus providing outstanding information about the pathophysiological mechanisms involved in cancer cachexia. However, our current knowledge comes mainly from animal models. Even though the number of human studies exploring the biological mechanisms of muscle wasting is increasing, human studies remain scarce, thus raising the question of the translatability of animal findings to human clinical research. This is a critical point as therapeutic targets may differ between human and animals. Surprisingly, a detailed comparative analysis between cachectic human patients and cachectic cancer animals is missing.
This review provides a comprehensive analysis of the molecular mechanisms involved in cancer cachexia‐associated muscle mass loss in preclinical and clinical studies. Article database of the US National Library of Medicine (PubMed) were searched using ‘cancer cachexia’ AND (muscle or ‘skeletal muscle’) AND specific terms (corresponding to the studied scientific area of each section). Any additional relevant literature was obtained from the reference lists of the published papers. This methodology has been applied for each section of the current review. Preclinical and clinical studies dealing with cancer, but for which cachexia was not characterized or demonstrated, were not included.
Protein synthesis and degradation rates
Little information is presently available on muscle protein turnover in cachectic cancer patients (Figure 1). A decline in muscle protein synthesis rate has been reported in cachectic cancer patients compared with healthy subjects. 9 , 10 Colorectal cancer patients with lower leg muscle mass than control subjects display unchanged muscle protein synthesis and a trend towards increased muscle protein breakdown, but reduced postprandial muscle protein synthesis, 11 which overall may confer a net catabolic status. Surprisingly, a lower protein degradation rate has been reported in weight‐losing cancer patients than in healthy controls. 12 This was also accompanied by a blunted anabolic response to feeding. 12 Higher protein synthesis rates have even been documented in skeletal muscle of cachectic cancer patients, 13 , 14 suggesting the existence of a compensatory mechanism that may bridle the magnitude of muscle mass loss.
Figure 1.
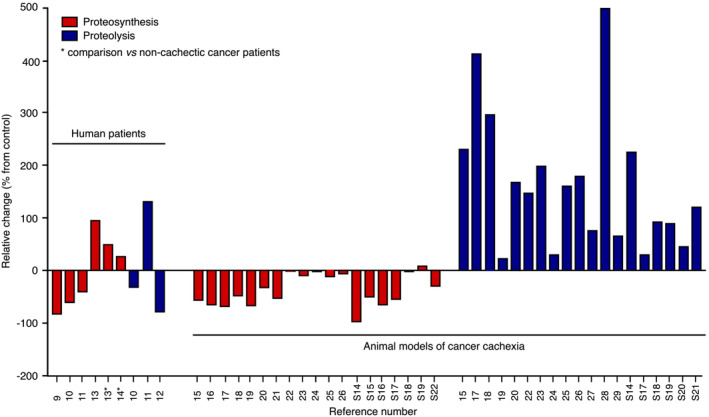
Relative variation in protein synthesis and degradation rates in clinical and preclinical studies. Variations have been calculated from data reported in quoted references. All data are expressed relative to controls or non‐cachectic cancer patients (indicated by an asterisk).
Animal studies clearly show a reduction in muscle protein synthesis 15 , 16 , 17 , 18 , 19 , 20 , 21 , ,S14–S17 and an increase in protein breakdown, 15 , 17 , 18 , 19 , 20 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , ,S14,S17–S21 (Figure 1), the extent of the variation being associated with the severity of cachexia. 17 , 18 , ,S16 Of note, unchanged muscle protein synthesis rates have also been frequently reported in animal experiments. 22 , 23 , 24 , 25 , 26 , ,S18,S19,S22
Therefore, currently available human data on muscle protein turnover in cachectic cancer patients are very limited and sometimes contradictory, even if a reduction in protein synthesis emerges as a mechanism that contributes to a net catabolic status. In animal models, a decrease in protein synthesis and an increase in protein degradation lower muscle mass. It is important to remind that cancer cachexia develops much more slowly in patients compared with animal models of cancer cachexia. Thus, it would be expected that protein degradation rate would be less easily identified as increased in human, compared with animals. More sensitive methods applied to the kinetic analysis of protein turnover during disease progression would be therefore necessary to provide a clear picture of skeletal muscle protein turnover in cachectic cancer patients.
Gene expression capacity during cancer cachexia
Transcriptional and translational capacities are essential for skeletal muscle homoeostasis. The transcriptional capacity is determined by the efficiency of the transcriptional machinery and the number of myonuclei, whereas the translational capacity depends on the ribosomal content and the global cellular RNA pool that is available to sustain the synthesis of myofibrillar protein.
Myonuclear death
Although not systematically observed, S23 hallmarks of nuclear death by apoptosis have been reported in skeletal muscle of cachectic cancer patients (PARP and DNA fragmentation, S24 increased BAX pro‐apoptotic factor mRNA level, S25 increased p53 phosphorylation, 30 activation of caspase‐8 and caspase‐9 30 ). Animal studies also show the presence of TUNEL‐positive nuclei, 27 , 31 , ,S20,S26–S28 an increase in PARP cleavage S29 and DNA fragmentation 32 , ,S30–S32 in cachectic skeletal muscle. Increases in BAX pro‐apoptotic‐to‐BCL2 anti‐apoptotic protein and mRNA ratios S30,S33–S35 and BAX mRNA level S36 as well as increased expression S33,S34 and cleavage S35 of caspase‐3, along with increased caspase‐1, caspase‐3, caspase‐6, caspase‐7, caspase‐8 and caspase‐9 activities, 32 , ,S29,S37 have also been described. Accordingly, transcriptomic studies highlight heightened expression of apoptosis genes in muscle of cachectic cancer mice. 33 , 34
As skeletal muscle is a very heterogeneous tissue, where approximately half of its nuclei reside outside of muscle fibres, S38 it is essential to distinguish myonuclei from those of neighbouring mononuclear cells. Whereas most of studies presented above did not report the localization of nuclei, 27 , 30 , ,S20,S26,S28 some animal studies detected TUNEL‐positive nuclei outside (mononucleated cells) 31 , ,S27 and inside S27 the muscle fibre, suggesting the existence of myonuclear death. The multinucleated nature of the muscle fibre also implies that the death of a single myonucleus does not mean the destruction of the entire fibre. Accordingly, the whole number of fibres is maintained in skeletal muscle of human cachectic patients S3,S39 and cancer mice. 31 Therefore, myonuclear death during cancer cachexia may trigger individual nuclei decay segmentally along the fibre, which may, over an extended period of time, locally weaken the transcriptional capacity of the fibre and contribute to atrophy and muscle dysfunction.
Muscle fibre microenvironment
Pioneering studies at the beginning of the 20th century 35 and later S40 reported an increased number of nuclei in the vicinity of the sarcolemma. More recently, inflammatory cells, 36 macrophages and fibro‐adipogenic progenitors cells 37 have been identified in muscle of cachectic cancer patients. A higher number of activated satellite cells, 38 activated stem cells, 38 undifferentiated cells S41,S42 and inflammatory cells 27 , 39 , 40 , ,S20,S26,S28 have also been detailed in muscle of cachectic cancer mice.
The presence of inflammatory cells and macrophages, together with an augmented cytokine content (see below), suggests the persistence of unresolved inflammation in cachectic skeletal muscle, which may alter the properties of myogenic precursors. S43 It has thus been shown that although progenitor cells do commit to the myogenic programme, they do not completely differentiate. 38 Muscle frailty may also cause localized episodes of muscle regeneration, as exhibited in cachectic cancer patients 37 , ,S44 and in cancer mice, 27 , 39 , ,S20,S28 and also illustrated by the presence of centralized myonuclei. 27 , 37 , 39 , ,S20,S28,S44 Furthermore, skeletal muscle of cachectic cancer mice regenerates less efficiently after freeze clamping‐induced 38 , ,S45 or cardiotoxin‐induced S46 muscle injury. Therefore, cancer cachexia is associated with an altered regenerative capacity of skeletal muscle that could lead to a reduction in the renewal of myonuclei by myogenic precursor cells, whose activity would be further impaired by unresolved inflammation, thus ultimately contributing to decrease the transcriptional capacity of the fibre.
Ribosomal content
Reductions in skeletal muscle rRNA level, 16 , 20 28S rRNA level S47 and ribosomal S6 protein content 41 have been reported in skeletal muscle of tumour‐bearing animals, along with defective transcriptional activity of polymerase I, S48 the enzyme responsible for ribosomal gene transcription and a drop in ribosomal gene transcription. 42 Ribophagy (selective ribosome degradation by the autophagy–lysosome pathway) would be also increased. 42 Consequently, ribosomal gene transcription and protein machineries appear to be compromised in muscle during cancer cachexia. Clearly, the mechanisms regulating skeletal muscle ribosomal content warrants further research to determine the functional relevance of the translational capacity in controlling muscle wasting during cancer cachexia.
Proteolytic capacity during cancer cachexia
Ubiquitin–proteasome system
Skeletal muscle proteins targeted for degradation are marked by the 26S proteasome complex through an ATP‐dependent ubiquitination process (Figure 2). The covalent attachment of a chain of ubiquitin molecules to the substrate protein involves a three‐step enzymatic cascade driven by E1 (ubiquitin‐activating enzymes), E2 (ubiquitin‐conjugating enzymes) and E3 (ubiquitin‐ligase enzymes). Ubiquitinated proteins are then docked to the proteasome for degradation. S49 The activation of the ubiquitin–proteasome system has largely been inferred from increased expression of the E3 ligases, MuRF1 (TRIM63) and MAFbx/Atrogin1 (FBXO32) in multiple atrophy conditions. S50 MuRF1 targets sarcomeric proteins (actin, myosin heavy chain, troponin) for degradation, S51–S53 whereas MAFbx targets MyoD and the translational enhancer EIF3F for degradation. S54,S55
Figure 2.
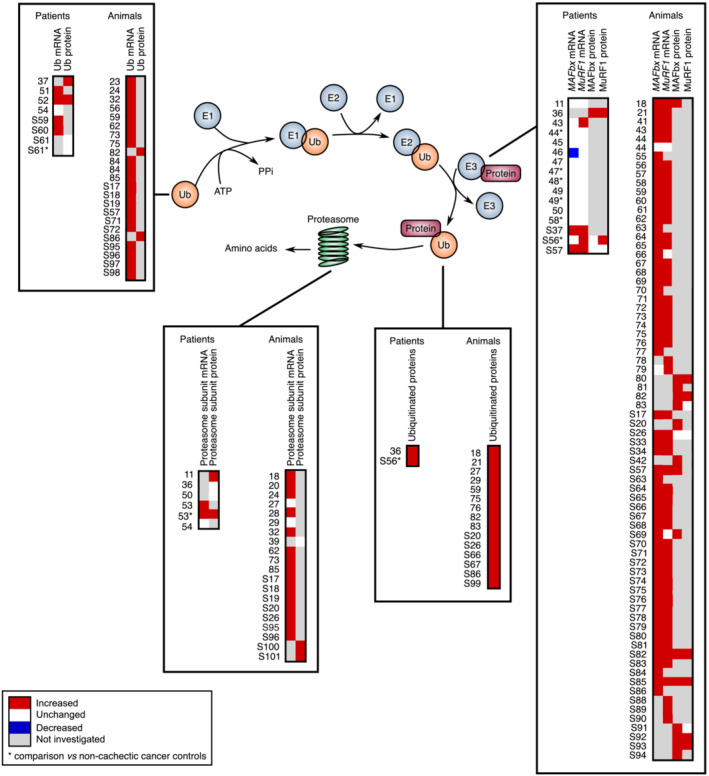
Comparative analysis of the transcript and protein levels of components of the ubiquitin–proteasome system in cachectic skeletal muscle of cancer patients and animals. Proteins are targeted for degradation by the 26S proteasome through covalent attachment of a chain of ubiquitin molecules. The E1 ubiquitin‐activating enzyme hydrolyses ATP to bind ubiquitin. E2 ubiquitin‐conjugating enzymes receive ubiquitin from E1 and brings it to the E3 ubiquitin‐ligase enzymes, which catalyse the transfer of the ubiquitin from E2 to the substrate. This reaction is the rate‐limiting step of the ubiquitination process. The ubiquitinated protein is then docked to the proteasome for degradation. One gene encodes the E1 enzyme, whereas one hundred genes encode the E2 enzymes and almost one thousand genes the E3 enzymes. Significant variations are reported in red (increase) or blue (decrease). Unchanged levels are reported in white.
Although some studies show increased transcript and protein levels of MuRF1, 36 , 43 , ,S37,S56,S57 and MAFbx, 36 , ,S37,S57 in skeletal muscle of cachectic cancer patients, a majority of investigations report unchanged expression of MuRF1, 11 , 44 , 45 , 46 , 47 , 48 , 49 , 50 , ,S37,S58 and MAFbx, 11 , 43 , 44 , 45 , 47 , 48 , 49 , 50 , ,S37,S58 in muscle of cachectic cancer patients compared with non‐cachectic patients or healthy subjects (Figure 2). A decrease in MAFbx expression has even been documented. 46 By contrast, increased ubiquitin mRNA 51 , 52 , ,S59,S60 and protein 37 , 52 levels, ubiquitinated proteins 36 , ,S56 and mRNA and protein levels of proteasome subunits 11 , 53 have been frequently reported in skeletal muscle of cancer patients, even if some discrepancies still persist. 36 , 50 , 54 , ,S61 The activity of the ubiquitin–proteasome system also correlates with the severity of body mass loss. S59 Finally, transcriptomic analyses provide divergent results with either increased, S62 unchanged S58 or decreased 45 expression of genes related to the ubiquitin–proteasome system in muscle of cachectic cancer patients.
Animal models consistently highlight an augmentation in the mRNA level of MAFbx, 18 , 21 , 41 , 43 , 44 , 55 , 56 , 57 , 58 , 59 , 60 , 61 , 62 , 63 , 64 , 65 , 66 , 67 , 68 , 69 , 70 , 71 , 72 , 73 , 74 , 75 , 76 , 77 , ,S17,S26,S33,S34,S57,S63–S87 and MuRF1, 18 , 21 , 41 , 43 , 44 , 56 , 57 , 58 , 59 , 60 , 61 , 62 , 64 , 65 , 67 , 68 , 69 , 71 , 72 , 73 , 74 , 75 , 76 , 78 , 79 , ,S17,S26,S33,S34,S57,S64–S68,S70–S83,S85,S87–S90 in skeletal muscle of cachectic cancer animals (Figure 2). This has also been confirmed at the protein level. 18 , 80 , 81 , 82 , 83 , ,S20,S42,S57,S69,S82,S85,S91–S94 Ubiquitin mRNA 23 , 24 , 32 , 56 , 59 , 62 , 73 , 75 , 84 , 85 ,S17–S19,S57,S71,S72,S95–S98 and protein 82 , ,S86 levels, the content of ubiquitinated proteins 18 , 21 , 27 , 29 , 59 , 75 , 76 , 82 , 83 , ,S20,S26,S66,S67,S86,S99 and the transcript 18 , 20 , 24 , 28 , 32 , 62 , 73 , 85 , ,S17–S20,S26,S95,S96 and protein S100,S101 levels of several proteasome subunits are also increased in muscle of cachectic cancer mice. Transcriptomic studies also feature an up‐regulation of genes related to the ubiquitin–proteasome system in muscle of cachectic cancer animals. 33 , 34 , 55 , 73 , 78 , 86 , 87 , 88 , ,S70,S72,S92,S102,S103 Consistent with these observations, proteasome enzyme activity is elevated in muscle of cachectic cancer mice. 28 , 89 , ,S75,S100,S104
Regardless of a lesser number of investigations in human cancer patients, the analysis of MuRF1 and MAFbx expression presented above suggests that the ubiquitin–proteasome system does not contribute to muscle mass loss in cancer patients, which is in line with the observation that there is currently no clear evidence of an increase in muscle proteolysis rate in cachectic cancer patients. This would thus give prominence to a major interspecies difference. However, a careful examination of the data reveals that whereas the picture is effectively contrasted when looking at MuRF1 and MAFbx, a much more nuanced picture appears when looking at ubiquitin mRNA level, proteasome subunit mRNA level, ubiquitinated proteins and ubiquitin–proteasome activity (Figure 2). Therefore, when considering all markers of the ubiquitin‐proteasome system, and not solely MuRF1 and MAFbx, currently available data rather indicate that the ubiquitin–proteasome system would be activated in human cachectic cancer patients. One may also consider the function of other E3 ligases such as MUSA1/Fbxo30 S105 and SMART/Fbxo21, S106 all required for skeletal muscle atrophy. Although one study reports a decrease in MUSA1 transcript level in muscle of tumour‐bearing mice, S66 other studies show that MUSA1 21 , 43 , 71 , 73 and SMART 43 transcript levels are increased. Expression of MUSA1 is also heightened in skeletal muscle of cachectic cancer patients. 43 Expression of Fbxo31, a novel E3 ligase that is induced during denervation and fasting, S106 is also elevated in muscle of cachectic cancer mice. 71 , 73 It now remains to determine which substrates are targeted for degradation by these E3 ligases and to what extent they are associated with the regulation of the ubiquitin–proteasome system. Finally, the kinetic of cancer cachexia is very different between patients and animals. In tumour‐bearing mice, the subcutaneous injection of cancer cells induces a rapid and violent tumour burden in a couple of days/weeks and a fast and important depletion of muscle compartment. This allows for a well‐controlled and accurate kinetic analysis of the ubiquitin–proteasome system. This is also true for genetic models of cancer cachexia, even if they display a greater heterogeneity in disease progression and appearance of cachexia‐associated symptoms. By contrast, the complexity of the clinical context in cancer patients, the difficulty of determining the onset of the tumour surge and the onset of cachexia and the lower rate of skeletal muscle depletion render the kinetic analysis of the ubiquitin–proteasome system during the cachectic syndrome particularly challenging.
Autophagy–lysosome system
The autophagy–lysosome system is responsible for eliminating long‐lived proteins and large supramolecular structures, including dysfunctional mitochondria. S49 Proteins and organelles to be degraded are engulfed during the formation of a double‐membrane structure called the autophagosome, which then fuse with lysosomes allowing acidic proteolytic degradation of their contents by cathepsins. Because autophagy is constantly active to remove damaged proteins and organelles, a defect in autophagy will result in muscle functional impairment, S107 but excessive autophagy will also contribute to muscle mass loss. S108,S109 Therefore, a tight regulation of the autophagy–lysosome system is necessary for skeletal muscle homoeostasis.
The protein level of autophagy‐related genes such as ATG5, ATG7, Beclin1 and LC3B is increased in skeletal muscle of cachectic cancer patients, 30 , 47 , 68 , 90 , ,S39,S56 together with the number of autophagosomes, S56 suggesting an increase in autophagosome formation. The number of autophagosomes results from a dynamic equilibrium between autophagosome formation and autophagosome docking and fusion with lysosomes. Therefore, an accumulation of autophagosomes can be also interpreted as a default in autophagosome clearance. The multidomain protein p62/SQSTM1 is a cargo adaptor protein involved in selectively targeting protein aggregates to autophagosomes. S110 Because p62 is constantly removed by autophagy, a rise in p62 protein content is a good marker of disturbances in autophagosome turnover. p62 protein content 68 , 90 , ,S56 and p62 aggregates 37 are increased in skeletal muscle of cachectic cancer patients, an observation that is consistent with a disrupted clearance of autophagosomes and suggests that defects in lysosomal rejuvenation/biogenesis and proteolytic capacity could be involved in impaired autophagosome turnover. Nevertheless, the mRNA levels of TFEB, 90 a master regulator of lysosome biogenesis, S111 and cathepsin D 54 are unchanged in muscle of cachectic cancer patients, whereas the mRNA level of cathepsin B 54 is even increased. Although very limited, these data suggest that disturbances in autophagosome turnover may primarily be due to induction of pathways that promote autophagosome formation that are not properly matched to those promoting autophagosome clearance. Of note, some studies do not report any difference in the mRNA 44 , 45 , 49 and protein 36 levels of autophagy markers nor in autophagosome number, 36 suggesting that autophagy can be also properly balanced in muscle of cachectic cancer patients.
Expression of autophagy‐related genes is increased in skeletal muscle of cachectic cancer animals. 27 , 41 , 44 , 67 , 68 , 72 , 73 , 77 , 79 , 80 , 81 , 91 , ,S26,S28,S66,S78,S88,S112 This has also been confirmed by transcriptomic analysis. 73 , 86 , 88 , ,S70,S72 Cathepsin expression 20 , 32 , 62 , 69 , 77 , 79 , 92 , ,S17,S81,S95 and activity 25 , 26 , 29 , ,S95 as well as lysosomal proteolysis, 18 , 29 , 77 , ,S95 are also increased. Together, these findings indicate that autophagy and the clearance of autophagosomes are activated in skeletal muscle of cachectic cancer mice. However, one should note that some studies mention either unchanged 24 or even decreased 77 , 84 cathepsin activity and unchanged lysosomal proteolysis 24 , 89 in muscle of cachectic cancer animals. Furthermore, an accumulation of p62 in muscle of cachectic cancer animals, 68 , 77 , 80 , 91 , ,S26,S112 with an increase in autophagosome number, 68 suggests a disequilibrium between autophagosome formation and clearance.
The E3 ligase TRAF6 mediates the conjugation of Lys63‐linked polyubiquitin chains to ULK1 and BECLIN1, S113 a post‐translational modification that regulates their activity. TRAF6 expression is up‐regulated in skeletal muscle of cancer patients 52 and in tumour‐bearing mice. S66,S88 Muscle‐restricted ablation of TRAF6 also preserves muscle mass in cancer mice S88 and restores LC3B and BECLIN1 expression to control levels, S88 suggesting an important role for TRAF6 during cancer cachexia. To what extent TRAF6 may impact the proteolytic capacity of the autophagy–lysosome system is currently unknown.
Animal studies showed marked alterations in mitochondrial function and network in cachectic muscle. 93 Therefore, the role of mitophagy in controlling mitochondriostasis needs to be questioned. The core mitophagy process is led by the kinase PINK1, which phosphorylates the E3 ligase PARKIN, S113 whereas other proteins (BNIP3, NIX) are involved in ubiquitin‐independent mitophagy. S113 The protein level of PINK1 and PARKIN is either unchanged 90 or decreased S114 in skeletal muscle of cachectic cancer patients, whereas the mRNA and protein levels of BNIP3 and NIX are unchanged. 90 In muscle of cachectic cancer animals, PINK1 protein level is either unchanged 94 or increased, 91 whereas that of PARKIN remains unchanged. 94 Consequently, human and animal data may suggest that mitophagy would be either unchanged or decreased. Mitochondrial fusion and fission proteins are essential in regulating organelle dynamic. The expression of fusion proteins is diminished (MFN2) S114 or unchanged (OPA1), 30 , S114 in muscle of cachectic cancer patients, whereas the mRNA level encoding the fission protein FIS1 is augmented, 30 , S114 Expression of mitochondrial fusion proteins (MFN1, MFN2, OPA1) is unchanged 79 , 94 or reduced 95 , ,S36 in skeletal muscle of cachectic cancer mice, whereas expression of FIS1 is unchanged 94 , ,S78 or elevated. 94 , 95 , ,S36 Therefore, the fission of mitochondria would be favoured in cachectic muscle, which, together with unchanged or decreased mitophagy, would lead to an accumulation of dysfunctional organelles. Clearly, the data currently available suggest that our view of the mechanisms involved in mitophagy during cancer cachexia is still fragmented and needs further exploration.
Calpains
The calpain family consists of a group of 15 calcium‐activated cysteine proteases. S115 The ubiquitously expressed calpain‐1 and calpain‐2 as well as the muscle‐specific isoform calpain‐3 are expressed in skeletal muscle. Calpains have originally been described as facilitators of protein turnover by releasing myofilaments from myofibrills. S115,S116
In human, one study demonstrates increased calpain activity in skeletal muscle of non‐weight‐losing cancer patients, S117 whereas another one does not show any difference. S118 Although former experiments indicated that calcium‐dependent proteolysis would not be involved in muscle proteolysis in cachectic cancer animals, 20 , 24 a more recent examination shows that calcium‐dependent proteolysis is activated in tumour‐bearing mice. S21 Regarding calpain activity per se, investigations show unchanged 23 , ,S75,S119 or increased activity S33,S100,S104 in muscle of cachectic cancer mice. This last result agrees with reports showing heightened expression of calpain isoforms 24 , 27 , 32 , 62 , ,S17,S21,S71,S72,S74 and decreased expression S21,S33 and activity S119 of calpastatin, the endogenous specific inhibitor of calpain. Therefore, considering the major impact of cancer cachexia on skeletal muscle structure and the remodelling function attributed to calpains, S115 a deeper understanding of calpain function during cancer cachexia would be beneficial.
Insulin/IGF1–AKT–mTOR pathway
Insulin and IGF1
Insulin and IGF1 activate a cascade of phosphorylation that coordinately regulate protein synthesis and degradation (Figure 3). IGF1 is mainly produced by the liver under the control of pituitary‐secreted growth hormone and accounts for 70–80% of serum IGF1. S120 However, liver IGF1 only contributes approximately to 30% of adult body size, S121 indicating that IGF1 originating from other tissues, including skeletal muscle, also contributes to the regulation of tissue growth. Among the different IGF1 isoforms expressed in skeletal muscle, IGF1‐Ea, and to a lesser extent IGF1‐Eb (IGF1‐Ec in human), are the most powerful in increasing muscle mass and force in mice. S122
Figure 3.
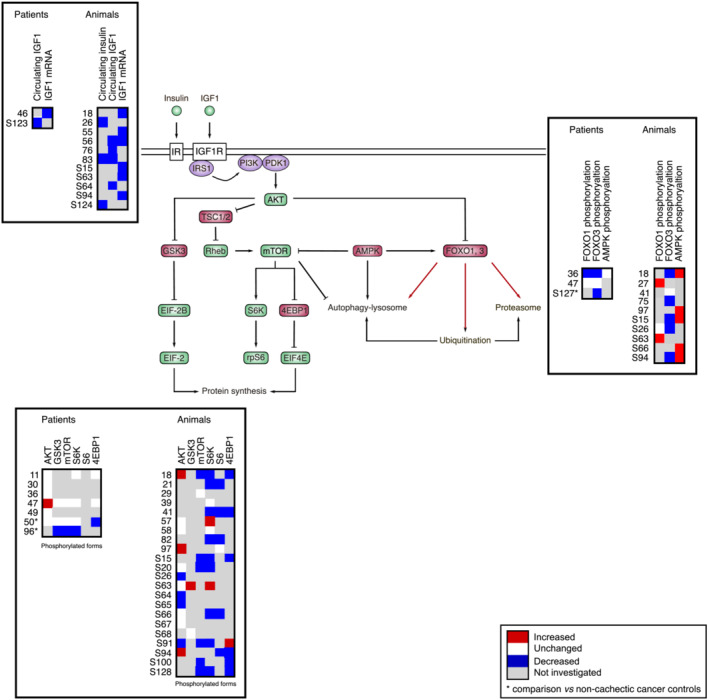
Comparative analysis of the regulation of the insulin/IGF1–AKT–mTOR pathway in clinical and preclinical studies. Upon receptor activation, IRS1 promotes the phosphorylation of phosphatidylinositol 4,5‐bisphosphate into phosphatidylinositol 3,4,5‐triphosphate at the plasma membrane by recruiting the kinase PI3K. Phospholipid phosphorylation promotes AKT recruitment and activation by PDK1. AKT positively or negatively regulates multiple targets including mTOR from the mTORC1 complex (not represented) and FOXO transcription factors. AMPK, whose activity is increased by energy stress, is another important modulator of the pathway. Black arrows indicate post‐translational regulation. Red arrows indicate transcriptional regulation. Significant variations are reported in red (increase) or blue (decrease). Unchanged levels are reported in white.
Data show that the driving force of the pathway is reduced in cachectic muscle. Circulating IGF1 S123 and muscle IGF1‐Ec transcript level 46 are decreased in cachectic cancer patients. The circulating level of insulin, 26 , 83 , ,S124 and IGF1, 56 , 76 , 83 , ,S64 as well as muscle IGF1‐Ea 56 , ,S63 and IGF1‐Eb, 18 , 55 , ,S15,S94 transcript levels, and muscle IGF1 protein content S64 are also decreased in cachectic cancer animals.
The pathway
Insulin/IGF1 signalling impinges a crucial regulatory step in the pathway, that is, the reaction catalysed by the serine/threonine protein kinase AKT. Under its phosphorylated active form, AKT indirectly activates the kinase mTOR. mTOR and then signals to the translation machinery via the activation of S6K, which controls the ribosomal protein S6, and the inhibition of 4EBP1, which hinders translation initiation by sequestering EIF4E. S125 AKT also phosphorylates and inhibits GSK3, thus relieving GSK3‐dependent inhibition of EIF2B. S126
AKT protein level 50 , 96 and the phosphorylation of GSK3, 96 mTOR 96 and S6K 50 , 96 are decreased in skeletal muscle of cachectic cancer patients compared with non‐cachectic cancer patients, along with the phosphorylated inactive form of the translational repressor 4EBP1 50 (Figure 3). Transcriptomic analyses also show that the expression of genes involved in protein anabolism is down‐regulated 45 , ,S127 or altered S62 in skeletal muscle of cachectic cancer patients. Therefore, both transcriptional and post‐translational events regulate the pathway. In animal models, a large majority of studies reports an inhibition of the pathway, as shown by the decrease in the phosphorylation of AKT, S26,S64,S65,S91 mTOR, 18 , ,S15,S20,S91,S100,S128 S6K, 18 , 21 , 41 , 82 , ,S15,S20,S66,S91,S128 S6, 21 , 41 , 82 , ,S66,S94 and 4EBP1. 18 , 41 , ,S15,S94,S100,S128 These events are also associated with weight loss, 18 , ,S15,S128 attesting that the greater is the inhibition of the pathway, the greater is the extent of cachexia. Microarray analyses also highlight a transcriptional regulation of the pathway in skeletal muscle. 33 Consequently, evidence based on global transcript and biochemical analyses indicate that the activity of the insulin/IGF1–AKT–mTOR pathway is reduced in cachectic muscle of cancer patients and cancer animals.
However, some discrepancies exist between studies both in human and animals. Some human investigations reveal unchanged 11 , 30 , 36 , 49 , 50 or even increased AKT phosphorylation 47 in skeletal muscle of cachectic cancer patients, together with unchanged phosphorylation of GSK3, 47 , 50 mTOR, 47 , 50 S6K 11 , 47 , 50 and 4EBP1 11 , 47 (Figure 3). Similarly, AKT phosphorylation remains unchanged, 57 , 58 , ,S20,S63,S66,S67 , or even increased, 18 , 97 , ,S94 in muscle of cachectic cancer animals. A few works also describe unchanged phosphorylation of S6K, 39 , 58 S6 97 and GSK3, S68 and sometimes even increased phosphorylation of S6K, 57 , ,S63 4EBP1 S91 and GSK3. S63 Divergences between studies are important to consider. First, an activation of the pathway, which is at first counter‐intuitive, could be interpreted as a compensatory response aimed at limiting the magnitude of muscle mass loss. Second, these results also strongly suggest that the pathway can be turned on or off depending on the clinical/experimental context (cancer type, severity of the disease and progression through the disease) and that a temporal regulation of the pathway may occur during the time course of the disease. Is the insulin/IGF1–AKT–mTOR pathway constantly repressed to maintain the driving force of cancer cachexia, or are there episodes of reactivation of the pathway that succeed/alternate to episodes of repression? The data presented above hint at the second possibility would be an interesting alternative to consider. Finally, these data also suggest that regulatory influences coming not only from insulin/IGF1 but from other sources may converge to the pathway to modulate its activity. S125 For instance, the accumulation of amino acids released from catabolized proteins may mitigate the down‐regulation of mTOR. S129 Conversely, AMPK, a metabolic sensor that down‐regulates mTOR signaling, S130 is activated in muscle of cachectic cancer mice 18 , 97 , ,S15,S66,S94 and may thus further exacerbate mTOR inhibition.
FOXO transcription factors
AKT inhibits the FOXO family of transcription factors by phosphorylation.S131 FOXO regulate the expression of MuRF1 and MAFbx, as well as that of genes of the autophagy‐lysosome system. S106,S108,S109,S132,S133 AKT thus allows a coordinated regulation of protein synthesis and degradation.
FOXO‐related genes are up‐regulated in skeletal muscle of cachectic cancer patients, 37 and the transcript levels of FOXO1 and FOXO3 negatively correlate with body mass and muscle mass. 98 The ratio of the phosphorylated‐to‐total forms of FOXO1 36 and FOXO3 36 , 96 is diminished in skeletal muscle of cachectic cancer patients (Figure 3), suggesting heightened FOXO nuclear translocation and transcriptional activity. Importantly, the reduction in FOXO1 and FOXO3 phosphorylation reported in Puig‐Vilanova et al. 36 was accompanied by an increase in MuRF1 and MAFbx protein content, which is consistent with a FOXO‐dependent regulation of these E3 ligase. Similarly, unchanged transcript level of MuRF1 and MAFbx was associated with unchanged phosphorylated‐to‐total forms of FOXO1 and FOXO3 in muscle of cachectic cancer patients. 47
In animal models of cancer cachexia, FOXO3, 18 , 75 , ,S15,S26,S94 phosphorylation is lowered, which agrees with increased FOXO3 nuclear localization 83 and transcriptional activity 69 reported by other studies (Figure 3). Furthermore, microarray analysis of FOXO‐regulated transcripts 34 and motif analysis of promoter sequences S70 identify FOXO as a transcription factor involved in muscle atrophy during cancer cachexia. This concurs with the observation showing that blocking FOXO prevents muscle fibre atrophy and spares force deficits in tumour‐bearing mice. 34 Nevertheless, a promoter analysis applied to skeletal muscle of cancer patients shows that weight‐loss associated genes have only fewer FOXO binding sites, S58 highlighting that the molecular characteristics of skeletal muscle from cachectic cancer patients may be different from those of preclinical models.
Finally, some animal studies report unchanged FOXO1 S26 and FOXO3 41 phosphorylation, whereas others even report increased FOXO1 27 , ,S63 phosphorylation and decreased FOXO1 DNA binding activity. S63 This may be interpreted as a compensatory mechanism to limit the extent of muscle mass loss. These data also remind us that additional FOXO‐regulatory mechanisms exist, including phosphorylation by FOXO‐activating kinases, such as AMPK, acetylation, ubiquitination and methylation. S131 This adds another level of complexity to the FOXO code and may dictate the transcription of different FOXO target genes.
Endoplasmic reticulum stress and unfolded protein response
The endoplasmic reticulum (ER) is an organelle involved in the folding, maturation and trafficking of newly synthesized proteins. S134,S135 ER stress, which leads to an abnormal accumulation of unfolded or misfolded proteins, is perceived by three key transducers of the unfolded protein response (UPR). ATF6, IRE1α and PERK act as a surveillance system to relieve ER stress and regulate proteostasis. S136 Although adaptive UPR contributes to skeletal muscle homoeostasis, its prolonged or exacerbated activation leads to muscle atrophy. S137
The expression of several ER stress markers (GRP78, HSP60, HSP70, calnexin and calreticulin mRNA; PDIA3 and PI3K mRNA and protein) and the UPR (ATF6, XBP1 and CHOP mRNA and protein) is up‐regulated in skeletal muscle of cachectic cancer patients, S37 as well as the ratio of the phosphorylated‐to‐total forms of PERK and eIF2α. S37 Together, these data indicate the existence of an ER stress and the activation of the UPR in muscle of cachectic cancer patients. Nevertheless, supplementary investigations are needed to obtain reliable clinical data regarding the implication of ER stress and the UPR in cancer cachexia. In animal models of cancer cachexia, the induction of ER stress and all three arms of the UPR has been demonstrated. S66 These findings were recently corroborated by several animal studies, S138–S141 thus providing robust evidence of ER stress and UPR activation during cancer cachexia. One should note, however, that a study mentions unchanged expression of multiple markers of ER stress and the UPR. S112 This also agrees with repressed phosphorylation of eIF2α reported in skeletal muscle of cachectic cancer rodents. S63
A relevant question raised by these data is the function of ER stress and UPR activation during cancer cachexia. Under experimental ER stress, PERK‐mediated eIF2α phosphorylation contributes to stoppage of translation. S142 The UPR represses the insulin/IGF1–AKT–mTOR pathway and activates autophagy. S143 Furthermore, ATF4 transcription factor (PERK arm), whose expression is increased in cachectic skeletal muscle, 78 promotes muscle atrophy by modulating a subset of atrogenes like Gadd45α, which induces the remodelling of chromatin to repress genes involved in anabolic signalling and to activate pro‐atrophy genes. S144 During cancer cachexia, the activation of ER stress and the UPR is associated with a noticeable inhibition of the insulin/IGF1–AKT–mTOR pathway and increased AMPK activity. S66 Therefore, an interplay between ER stress, the UPR, the insulin/IGF1–AKT–mTOR pathway, AMPK and the expression of atrogenes may contribute to down‐regulate protein synthesis and activate proteolysis. Intriguingly, the use of 4‐phenylbutyrate (ER stress pan‐inhibitor) or the targeted ablation of PERK in skeletal muscle aggravates the deleterious effects of cancer cachexia in tumour‐bearing mice, S66,S138 suggesting that ER stress and the UPR (PERK arm) could be also viewed as a response originally aimed at limiting the effects of cancer cachexia.
Oxidative stress
Oxidative stress (OxS) is due to an excessive production of reactive oxygen species (ROS) and reactive nitrogen species, together with impaired antioxidant defence, which results in an accumulation of oxidized and damaged proteins, organelles, membranes and DNA. S145 In skeletal muscle, OxS reduces muscle strength and triggers atrophy. S146
OxS is increased in skeletal muscle of cachectic cancer patients, as shown by the elevation in total protein carbonylation 36 and malondialdehyde‐protein adducts, 36 , ,S147 and the reduced expression of genes encoding antioxidant proteins, S25 such as superoxide dismutase 2, glutamate–cysteine ligase (involved in glutathione synthesis) and Nrf2 (a transcription factor that regulates the expression of multiple cytoprotective genes). However, an increase in the protein level and activity of the antioxidant enzymes, superoxide dismutase 1 and 2, has also been reported in skeletal muscle of cachectic cancer patients, 36 which may illustrate the induction of a compensatory mechanism to alleviate increased ROS production.
OxS is also increased in skeletal muscle of cachectic cancer mice, as indicated by higher levels of ROS, 91 , 94 , ,S75,S148 lipid peroxidation 40 , 63 , 99 , 100 , ,S75,S149 and protein carbonylation, 27 , 40 , 63 , 99 , 100 , ,S20,S67,S82,S91,S150 as well as by the increase in oxidized‐to‐reduced glutathione ratio, 63 , 91 , ,S74,S75,S112,S149,S151 the decrease in glutathione peroxidase activity S148 and the depletion of reduced glutathione and antioxidant peptides. S152 The expression of antioxidant enzymes has been documented to be either decreased 27 , 55 , ,S20,S28,S75,S148 unchanged 27 , 91 , 94 , 95 , 99 , 100 , ,S28,S67,S148 or even increased, 40 , 55 , 91 , 100 , ,S28,S67,S74 suggesting that the main factor responsible for augmenting OxS in muscle of cachectic cancer rodents is an increased ROS production. Transcriptomic analyses in mice also highlight increased transcriptional response of OxS‐related genes. 33 , 55 , 78 Importantly, OxS would be more significant in Type II myofibres 100 and would also precede muscle mass loss, 94 suggesting that OxS could be a precocious event. Altogether, these data indicate that cachectic skeletal muscle is subjected to a chronic OxS that mainly results from increased ROS production associated or not with lowered antioxidant defence.
This obviously raises the question of the origin of ROS and the mechanisms linking OxS to muscle wasting during cancer cachexia. The main sources of ROS in skeletal muscle are complexes I and III of the mitochondrial respiratory chain and NADH oxidases, but also ER and peroxisomes where enzymatic complexes generate ROS. S145 Sources of ROS are probably multiple during cancer cachexia, but previously reported alterations in muscle mitochondrial metabolism of cachectic cancer mice, 79 , 93 , 101 , ,S153 should significantly contribute to ROS production by increasing leakage of electrons and the subsequent formation of superoxide anion. OxS integrates signal transduction pathways by regulating post‐translational modifications of proteins through the redox regulation of the thiol side chain of cysteine amino acids. S154 Redox proteomic studies have revealed that several hundred proteins contain reactive and potentially modulatory cysteine residues, S155 including transcription factors (FOXO and NF‐κB) and kinases (AMPK and mTOR), which functional implication in the loss of muscle mass during cancer cachexia we have demonstrated. Furthermore, the wide spectrum of action of OxS on other biomolecules (lipid and DNA) also suggests that OxS may target other pathways that contribute to muscle wasting during cancer cachexia.
Inflammatory cytokines and downstream pathways
Circulating and skeletal muscle inflammatory cytokines
Systemic inflammation is a well‐described feature of cancer cachexia. Increased circulating level of C‐reactive protein (CRP) is associated with weight loss in cancer patients. 4 , 47 , 51 , 102 , 103 , ,S4,S156,S157 The circulating levels of pro‐inflammatory cytokines such as IL1ß,S158 IL4, 104 IL6, 47 , 50 , 51 , 104 , 105 , ,S159–S163 IL8, 47 , 104 , 105 , ,S160,S162,S163 IL10, 105 IFNγ 104 and TNFα 104 , ,S158,S162,S164 are also elevated in cachectic cancer patients. Similar observations have been done in mice models of cancer cachexia for the circulating levels of CRP, S91 IL1ß, 75 , ,S82,S91 IL6, 18 , 39 , 43 , 55 , 72 , 74 , 75 , 80 , 87 , 95 , 105 , 106 , ,S8,S15,S42,S73,S82,S85,S165–S167 IL10, 55 IL11, 87 IFNγ, 55 , 80 , ,S168 TNFα 22 , 55 , 75 , 80 , 87 , ,S19,S82,S85,S91,S124,S167 and Tweak. S91
The shock wave of this systemic inflammatory response is largely perceived by skeletal muscle. A large majority of human studies shows increased protein contents of IL1ß, 36 IL6, 50 , ,S169 IFNγ, 36 TNFα S169 and Tweak S91 in skeletal muscle of cachectic cancer patients. Only one work reports unchanged IL6 and TNFα protein levels. 36 IL1ß, S92 IL6, S64,S85 IFNγ 27 and TNFα S28,S64,S85 protein levels are also augmented in muscle of cachectic cancer mice, whereas one report shows unchanged IL1ß, IL6 and TNFα protein levels. 27 One may also consider that skeletal muscle itself can produce cytokines. However, a majority of investigations reports unchanged transcript levels (IL4, S170 IL6 S25 and TNFα 50 , S25 ) in muscle of cachectic cancer patients, whereas one study reports an increase in IL6 mRNA level. 50 The picture is more contrasted in animal models of cancer cachexia with a fairly equivalent number of experiments describing increased (IL1ß, S8,S92 IL6, 21 , 43 , 58 , 106 , ,S8,S74,S77,S92,S94 and TNFα S92 ) or unchanged (IL6, 95 , ,S67,S69 TNFα 58 , ,S67,S74 and Tweak 41 ) mRNA levels in muscle of cachectic cancer mice. Therefore, these data suggest that although non‐muscle tissues (host immune system, tumour cells) seem to mainly contribute to skeletal muscle inflammation, it could also be partly supported by an increase in cytokine production by skeletal muscle. Corollary, the contribution of skeletal muscle fibre, resident and/or recruited mononucleated cells in producing cytokines needs to be evaluated.
IL1ß/TNFα‐NF‐κB pathway
Omic studies in human S62,S127 and animals 33 , 34 , 55 , 87 , 88 , ,S92,S103,S171 indicate that cachectic skeletal muscle is subjected to a persistent activation of a pro‐inflammatory cytokine signalling. NF‐κB transcription factor relays information from various cytokines (mainly IL1ß and TNFα). Five NF‐κB transcription factors [p65 (Rel A), Rel B, c‐Rel, p52, p50] are expressed in skeletal muscle. S172 Activation of cytosolic NF‐κB occurs when the IκB kinase phosphorylates IκB, resulting in its ubiquitination and proteasomal degradation. This allows NF‐κB to translocate into the nucleus and regulate the expression of target genes. NF‐κB activation in mouse skeletal muscle overexpressing IκB kinase leads to severe muscle wasting associated with increased MuRF1 expression and ubiquitin–proteasome proteolysis. S173 Here, we will mainly discuss about the canonical p65 pathway, whose activation is associated with pathological conditions.
The phosphorylation level of NF‐κBp65 is similar in skeletal muscle of cachectic cancer patients compared with non‐cachectic patients. S39 However, mRNA level of NF‐κB1 (precursor of p50) 50 and NF‐κBp65 protein content 36 , ,S169 are increased compared with healthy controls, suggesting a precocious activation of NF‐κB pathway before the development of cancer cachexia. A majority of animal experiments reports an increase in the phosphorylated active form of NF‐κB, 27 , 57 , 74 , 82 , 97 , ,S8,S20,S26,S67,S94 the phosphorylated inactive form of IκB, 41 the nuclear localization of NF‐κB, S92 NF‐κB DNA binding S88,S89,S92,S101,S173 and transcriptional activity 27 in cachectic muscle. The phosphorylated active form of NF‐κBp65 also negatively correlates with body mass loss and muscle force in cachectic cancer mice. S8 Although still debated, 88 motif analysis of promoter sequences also identified NF‐κB as a transcription factor involved in muscle atrophy during cancer cachexia. S70 Of note, studies report unchanged NF‐κBp65 phosphorylation 41 , 88 , ,S75 and NF‐κB DNA binding activity. 32 , 88 , ,S98,S174
IL6–JAK–STAT3 pathway
The JAK‐STAT pathway is activated by IFNα, IFNβ, IFNγ, IL2 and IL6. S175 Upon cytokine binding, activated JAK tyrosine kinase phosphorylates STAT proteins, which translocate into the nucleus to regulate the expression of target genes. S176 STAT transcriptional activation contributes to muscle wasting by indirect activation of myostatin expression and expression of MAFbx and MuRF1. S177,S178
Limited information in patients indicate that the phosphorylation level of STAT3 is similar in cachectic compared with non‐cachectic muscle. S39 By contrast, numerous evidence of IL6–JAK–STAT3 pathway activation have been reported in preclinical models. Circulating IL6 level correlates with the development of cachexia in tumour‐bearing mice. 95,S166 STAT3 activation 74 , 82 , 95 , 97 , 106 , 107 , 108 , ,S8,S15,S69,S82,S94 and the expression of STAT3 target genes 87 are increased in skeletal muscle of cachectic cancer mice. The active form of STAT3 negatively correlates with body mass and muscle force,S8 and media conditioned with serum of tumour‐bearing mice activate STAT3 in C2C12 myotubes. S178 In silico analysis of transcriptome data also reveals STAT3 as a transcription factor involved in muscle atrophy during cancer cachexia. S70 Finally, hyperactivation of STAT3 in cancer mice exacerbates weight and muscle mass losses compared with control cancer mice. 109 Interestingly, recent results show that the IL6–JAK–STAT3 pathway contributes to the regulation of Noggin, an inhibitor of the anti‐catabolic BMP‐SMAD1/5/8 signalling pathway 43 (see below).
TGF‐ß signalling pathways
Myostatin–SMAD2/3 signalling pathway
Myostatin is a TGF‐ß superfamily member acting as a master negative regulator of skeletal muscle growth during embryonic and postnatal development in animals S179,S180 and human S181 and also in adulthood. 110 Myostatin binds to activin type IIB receptors (ActRIIB and ActRIIA) leading to the recruitment of TGF‐ß Type I receptors (ALK4 and ALK5) S182 and activation of SMAD2 and SMAD3 proteins (Figure 4). Once activated by phosphorylation, SMAD2/3 recruit SMAD4 to regulate the transcription of target genes. S183 Myostatin also inhibits the insulin/IGF1–AKT–mTOR pathway. 111
Figure 4.
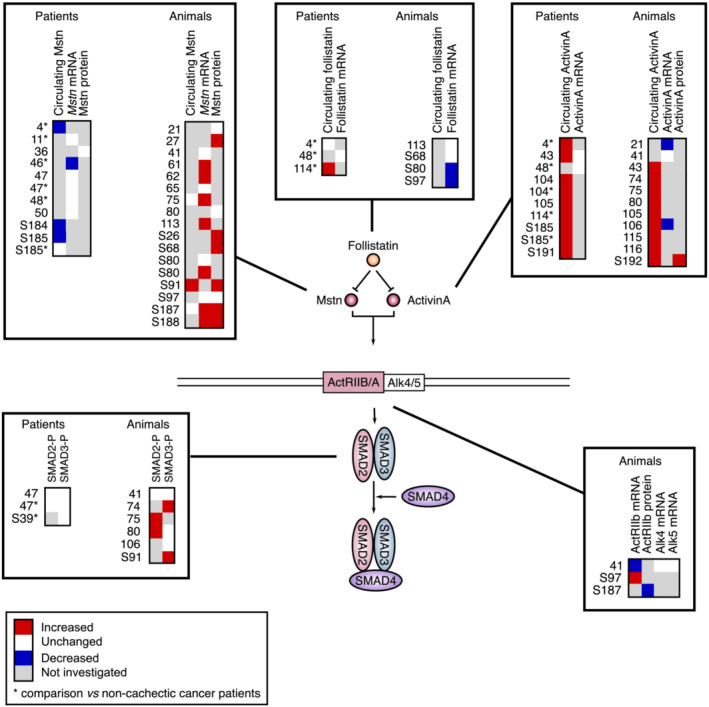
Comparative analysis of the regulation of myostatin/activin A signalling in cachectic skeletal muscle of cancer patients and in animal models of cancer cachexia. Myostatin and activin A bind to activin Type II receptors (ActRIIB/IIA) that activate Type I receptors (ALK4/5/7), which phosphorylate and induce SMAD2/3 to form a complex with SMAD4 and translocate into the nucleus. Myostatin and activin A binding is modulated by the inhibitory action of follistatin. Significant variations are reported in red (increase) or blue (decrease). Unchanged levels are reported in white.
Surprisingly, myostatin circulating level is reduced in cachectic cancer patients compared with healthy controls S184,S185 and decreased 4 or unchanged S185 when compared with non‐cachectic cancer patients (Figure 4). Myostatin mRNA level is also unchanged 11 , 47 , 48 , 50 or diminished 46 in skeletal muscle of cachectic cancer patients. Accordingly, myostatin protein level, 36 as well as the phosphorylation of SMAD2 and SMAD3, 47 , ,S39 is unchanged in muscle of cachectic cancer patients. Circulating 4 and muscle transcript 48 levels of follistatin, an inhibitor of myostatin and activin signalling, are also unchanged. Therefore, these studies indicate that myostatin signalling is not activated in skeletal muscle of cachectic cancer patients, even if this conclusion must be nuanced in the light of investigations showing that genes related to TGF‐ß signalling 37 are up‐regulated in skeletal muscle of cachectic cancer patients and that the transcript level of ActRIIB negatively correlates with muscle mass in cancer patients. 98
How to reconcile the fact that myostatin is a master negative regulator of muscle mass in human S181 and the observation that myostatin expression and signalling are not activated in muscle of cachectic cancer patients? One may first consider that as myostatin is produced by skeletal muscle, myostatin circulating level can thus simply be lowered as a consequence of the reduction in muscle mass during cancer cachexia. Furthermore, the data described above do not exclude the possibility that myostatin expression had increased earlier during the disease when muscle mass had not started to decrease yet. In support of this hypothesis, a strict temporal regulation of myostatin expression has been demonstrated in mice models of atrophy. 112 , ,S186 Kinetic analysis of its expression during the time course of cachexia would allow to answer this question.
Animal studies show that the circulating level of myostatin is either elevated S91 or unchanged 75 , ,S187 (Figure 4). Although some studies report unchanged myostatin expression in skeletal muscle of cachectic cancer rodents, 21 , 41 , 65 , 80 , ,S80,S97 a majority of works report an increase in both myostatin mRNA 61 , 62 , 75 , 113 , ,S80,S187,S188 and protein 27 , ,S26,S68,S91,S187,S188 levels. Downstream, the mRNA level of ActRIIB S97 and the phosphorylation of SMAD2 75 , 80 and SMAD3 74 , ,S91 are also increased in muscle of cachectic cancer mice. Muscle follistatin mRNA level is either unchanged 113 , ,S68 or decreased, S80,S97 which is also consistent with increased myostatin activity. However, a reduction in ActRIIB mRNA level, 41 unchanged ALK4/5 mRNA level 41 and SMAD2/3 phosphorylation 41 , 80 , 106 have also been reported in tumour‐bearing mice. Consequently, and even if this cannot be generalized to all studies, myostatin signalling is commonly activated during cancer cachexia in rodents.
Activin A–SMAD2/3 signalling pathway
Activin A is another TGF‐ß family member that binds to ActRIIA and ActRIIB and activate SMAD2/3 signalling with comparable potencies and efficacy as myostatin does. S189 Increasing circulating activin A levels in mice increases E3 ligase expression, inhibits the insulin/IGF1–AKT–mTOR‐pathway and reduces muscle mass and function. S190
Activin A circulating level is consistently elevated in cachectic cancer patients, 4 , 43 , 104 , 105 , 114 , ,S185,S191 as well as in cachectic cancer mice 43 , 74 , 75 , 80 , 105 , 106 , 115 , 116 , ,S192 (Figure 4). By contrast, studies indicate that skeletal muscle activin A mRNA level is either decreased 21 , 106 , ,S192 or unchanged, 41 , 43 whereas activin A protein level is increased. S192 Therefore, circulating activin A may thus come from another source than skeletal muscle, in particular tumour cells. 21 , 75 , 116 , ,S76,S192–S194 Importantly, while myostatin circulating level is higher in mouse than in human S189 and is the main negative regulator of muscle mass in mouse, S87 activin A circulating level is higher in human than in mouse. S189 Therefore, activin A could play a more prominent role in cachectic cancer patients than myostatin.
BMP–SMAD1/5/8 signalling pathway
BMP (BMP7, BMP13 and BMP14) bind to BMP Type II receptor (BMPRII), ActRIIB or ActRIIA and promote the recruitment and activation of Type I receptors BMPRIA (ALK3), BMPRIB (ALK6) or ActRIA (ALK2). This triggers the phosphorylation of SMAD1, SMAD5 and SMAD8, which, together with SMAD4, regulate the expression of target genes. As SMAD4 is shared by SMAD1/5/8 and SMAD2/3, BMP–SMAD1/5/8 and myostatin–SMAD2/3 pathways operate in parallel, and in opposition. Increased BMP‐SMAD1/5/8 pathway activity in muscle induces hypertrophy, S105,S195 whereas its inhibition causes muscle atrophy S105,S195 and abolishes the hypertrophic phenotype of myostatin‐deficient mice, S105 indicating that this pathway is dominant over myostatin signalling.
Diminished BMP signalling and augmented expression of the BMP inhibitor Noggin are observed in skeletal muscle of cancer patients and mildly cachectic mice. 43 Importantly, both IL6 and activin A trigger the expression of Noggin, 43 a BMP–SMAD1/5/8 pathway inhibitor. BMP signalling inhibition is also associated with neuromuscular junction impairment. 43 Therefore, perturbed BMP signalling appears to be a critical pathogenic mechanism regulating muscle mass and function in cancer patients and animals.
Glucocorticoid signalling
Glucocorticoids are steroid hormones produced by the adrenal glands under the control of the hypothalamic–pituitary axis. The hypothalamus secretes CRH, which stimulates the secretion of ACTH by the anterior pituitary gland. ACTH then binds its receptor on the adrenal cortex to activate the biosynthesis and release of glucocorticoids. Glucocorticoids bind the nuclear receptor NR3C1 of target tissues to activate or inhibit the transcription of multiple target genes. S196 Glucocorticoids are well known to exert strong catabolic effects on skeletal muscle by activating the expression of multiple genes involved in proteolysis while inhibiting those involved in proteosynthesis. S197,S198
Circulating glucocorticoid level is increased in cachectic cancer patients 117 , ,S199,S200 and in cachectic cancer mice. 22 , 23 , 26 , 85 , 86 , 118 , ,S124,S201–S203 Hypothalamic CRH mRNA level 67 and pituitary ACTH secretion S204 are also increased in animals, as well as the adrenal gland mass in cachectic cancer patients S205 and animals. 25 , ,S204,S206 More recently, our group established that the hypothalamic–pituitary–adrenal axis was activated in cachectic cancer mice, along with increased corticosterone level in serum and muscle, and increased skeletal muscle expression of glucocorticoid‐responsive genes. 119 Interestingly, the analysis of transcriptomic data also reveals an increase in the expression of multiple glucocorticoid‐responsive genes in muscle of cachectic cancer mice. 33 , 34 , 44 , 55 , 73 , 78 , 86 , 87 , 88 , ,S70,S72,S83,S92,S102,S103 Therefore, a neuroendocrine mechanism that involves the hypothalamic–pituitary–adrenal axis may also contribute to the transcriptional regulation of skeletal muscle catabolism.
Therapeutic perspectives
A brief description of the effects of therapeutic strategies in preclinical models is presented below.
IGF1
IGF1 treatment reduces weight loss and improves outcome in a rat model of cancer cachexia. S207 Anamorelin is a ghrelin agonist that increases the production of growth hormone from the pituitary gland and stimulates the liver to secrete IGF1. S208,S209 Anamorelin has proven efficacy to limit the extent of cancer cachexia in human, S210,S211 suggesting potential positive effects on the regulation of insulin/IGF1–AKT–mTOR pathway in skeletal muscle. However, care should be taken as IGF1 may promote tumour growth, even if there was no clear trend of increased tumour progression due to anamorelin. S210
ER stress and the UPR
Targeted ablation of XBP1 (IRE1α arm of the UPR) in skeletal muscle reduces muscle mass loss in tumour‐bearing mice, S139 whereas targeted ablation of PERK worsened muscle wasting. S138 Different arms of the UPR may thus be implicated differently and provide different signalling outcomes.
Oxidative stress
Administration of different antioxidants (α‐tocopherol, dehydroepiandrosterone, cocktail of catechins, quercetin and vitamin C) have provided contrasted results. Some studies show a sparing effect on muscle mass loss, S75 reduced expression of E3 ligases 63 , S75 and a restoration of MyoD and myogenin expression, S75 , whereas another investigation reports an acceleration of cachexia by increasing muscle atrophy and promoting tumour growth. S76
TNFα–NF‐κB
Therapeutic strategies invalidating TNFα receptor, S18,S19,S31 as well as the injection of anti‐TNFα antibody, 22 failed to provide beneficial results. However, the pharmacological inhibition of NF‐κB has proven its efficiency in cancer cachectic animals. 27 Besides, selective pharmacological inhibition of iNOS, which is a downstream effector of NF‐κB pathway and is highly expressed in cachectic muscle, 120 ameliorates cancer cachexia in mouse. 120
IL6–JAK–STAT3
Cancer mice lacking IL6 107 , 109 , ,S69 do not develop cachexia. Injection of an anti‐IL6 antibody S166 or an anti‐IL6 receptor antibody 18 prevents cachexia progression in tumour‐bearing animals. STAT3 inhibition also reduces muscle wasting in tumour‐bearing mice. S178,S212
Myostatin/activin A‐SMAD1/3
Cancer cachexia is blocked by myostatin gene invalidation, 41 the administration of a myostatin antisense RNA, S188 a myostatin antibody, 31 a soluble form of ActRIIB, 21 , 62 , 75 , 113 , ,S17,S192 an ALK4/5 receptor antagonist, 66 and by the administration of IMB0901 (myostatin signalling inhibitor). S93 AAV‐targeted inhibition of myostatin and activin A also prevents muscle wasting in tumour‐bearing mice. S87
BMP–SMAD1/5/8
Increasing BMP signalling in skeletal muscle of tumour‐bearing mice by gene delivery or pharmacological means can prevent muscle wasting. 43
Glucocorticoids
Ablation of adrenal glands does not attenuate cachexia 26 , ,S213 in tumour‐bearing animals. However, adrenalectomy itself induces weight loss. S214 The steroid inhibitor RU486 shows contrasted results, with some positive effects on the attenuation of body weight loss S202 or not. 118 ,S201 However, RU486 also exerts anti‐progestogenic and anti‐androgenic effects, which may mitigate the potential anti‐catabolic effects of glucocorticoid inhibition. Muscle wasting is abrogated in muscle‐specific glucocorticoid receptor knockout mice inoculated with LLC cells. 61 This spatially targeted tissue approach suggests that targeting glucocorticoids through the hypothalamic–pituitary–adrenal axis by specific molecular tools may be promising in preclinical models of cancer cachexia.
Conclusion and future directions
This analysis of the literature highlights several points and redraw the contours of some accepted ideas.
Our analysis shows the existence of species‐dependent molecular and biochemical responses. This is an essential factor to consider when evaluating the relevance of preclinical data to the clinical field. This also raises the question of the preclinical models used for research that should reproduce as closely as possible the complexity of the clinical context. Important factors are the model used (syngeneic ectopic/orthotopic graft, human tumour xenograft, genetic engineered mouse), the rate of disease progression and tumour growth, the age of the animals, the presence of metastasis or not and the use of additional chemotherapy or not. S215
There are still uncertainties whether protein degradation rate and MuRF1 and MAFbx expression are increased in skeletal muscle of cachectic cancer patients. This remains a key issue, even though data suggest that the ubiquitin–proteasome system would be activated in muscle of human cancer patients.
Activin A may play a more pre‐eminent role in cachectic cancer patients than myostatin, the reverse being true in animals.
The regulation of the molecular mechanisms involved in cancer cachexia is not a linear process during the natural history of the disease. A temporal analysis of the mechanisms involved in muscle proteostasis should be performed in preclinical models. In cancer patients, due to the complexity of the clinical context and the impossibility to determine the onset of tumour growth, a time‐course analysis of muscle atrophy and muscle function will help to more accurately determine the cachectic state of the patients throughout the course of the disease.
Emerging evidence indicate that muscle fibre microenvironment contributes to cancer cachexia. Whether or not this is quantitatively important in determining muscle wasting remains to investigate.
One major unsolved question is the influence of gender on the molecular and biological expression of cancer cachexia. This is a key issue that needs to be addressed.
Finally, although substantial progress has been made in developing therapeutic strategies in preclinical models, treatments of cancer cachexia are not available in the clinic. Considering the numerous molecular mechanisms that are coordinately regulated within a specific time frame during the disease, multi‐targeted strategies could be more effective in addressing the diversity and complexity of cancer cachexia.
Funding
A.M. was financially supported by the Ministère de l'Enseignement Supérieur de la Recherche et de l'Innovation. This work was supported by La Ligue contre le cancer (Comité de la Loire) et la Fondation ARC pour la Recherche sur le Cancer.
Conflict of interest
A.M., Y.S.G. and D.F. declare that they have no conflict of interest.
Supporting information
Supplementary reference list.
Acknowledgements
The authors certify that they comply with the ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle. S215
Martin A., Gallot Y. S., and Freyssenet D. (2023) Molecular mechanisms of cancer cachexia‐related loss of skeletal muscle mass: data analysis from preclinical and clinical studies, Journal of Cachexia, Sarcopenia and Muscle, 14, 1150–1167, 10.1002/jcsm.13073
Agnès Martin and Yann S. Gallot share first authorship.
References
- 1. Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 2011;12:489–495. [DOI] [PubMed] [Google Scholar]
- 2. Baracos VE, Martin L, Korc M, Guttridge DC, Fearon KCH. Cancer‐associated cachexia. Nat Rev Dis Primers 2018;4:17105. [DOI] [PubMed] [Google Scholar]
- 3. Cohn SH, Gartenhaus W, Sawitsky A, Rai K, Zanzi I, Vaswani A, et al. Compartmental body composition of cancer patients by measurement of total body nitrogen, potassium, and water. Metabolism 1981;30:222–229. [DOI] [PubMed] [Google Scholar]
- 4. Loumaye A, de Barsy M, Nachit M, Lause P, Frateur L, van Maanen A, et al. Role of activin A and myostatin in human cancer cachexia. J Clin Endocrinol Metab 2015;100:2030–2038. [DOI] [PubMed] [Google Scholar]
- 5. Prado CM, Sawyer MB, Ghosh S, Lieffers JR, Esfandiari N, Antoun S, et al. Central tenet of cancer cachexia therapy: do patients with advanced cancer have exploitable anabolic potential? Am J Clin Nutr 2013;98:1012–1019. [DOI] [PubMed] [Google Scholar]
- 6. Argiles JM, Stemmler B, Lopez‐Soriano FJ, Busquets S. Inter‐tissue communication in cancer cachexia. Nat Rev Endocrinol 2018;15:9–20. [DOI] [PubMed] [Google Scholar]
- 7. Weerink LBM, van der Hoorn A, van Leeuwen BL, de Bock GH. Low skeletal muscle mass and postoperative morbidity in surgical oncology: a systematic review and meta‐analysis. J Cachexia Sarcopenia Muscle 2020;11:636–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Anker MS, Holcomb R, Muscaritoli M, von Haehling S, Haverkamp W, Jatoi A, et al. Orphan disease status of cancer cachexia in the USA and in the European Union: a systematic review. J Cachexia Sarcopenia Muscle 2019;10:22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Emery PW, Edwards RH, Rennie MJ, Souhami RL, Halliday D. Protein synthesis in muscle measured in vivo in cachectic patients with cancer. Br Med J (Clin Res Ed) 1984;289:584–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dworzak F, Ferrari P, Gavazzi C, Maiorana C, Bozzetti F. Effects of cachexia due to cancer on whole body and skeletal muscle protein turnover. Cancer 1998;82:42–48. [PubMed] [Google Scholar]
- 11. Williams JP, Phillips BE, Smith K, Atherton PJ, Rankin D, Selby AL, et al. Effect of tumor burden and subsequent surgical resection on skeletal muscle mass and protein turnover in colorectal cancer patients. Am J Clin Nutr 2012;96:1064–1070. [DOI] [PubMed] [Google Scholar]
- 12. Lundholm K, Bennegard K, Eden E, Svaninger G, Emery PW, Rennie MJ. Efflux of 3‐methylhistidine from the leg in cancer patients who experience weight loss. Cancer Res 1982;42:4807–4811. [PubMed] [Google Scholar]
- 13. Shaw JH, Humberstone DA, Douglas RG, Koea J. Leucine kinetics in patients with benign disease, non‐weight‐losing cancer, and cancer cachexia: studies at the whole‐body and tissue level and the response to nutritional support. Surgery 1991;109:37–50. [PubMed] [Google Scholar]
- 14. MacDonald AJ, Johns N, Stephens N, Greig C, Ross JA, Small AC, et al. Habitual myofibrillar protein synthesis is normal in patients with upper GI cancer cachexia. Clin Cancer Res 2015;21:1734–1740. [DOI] [PubMed] [Google Scholar]
- 15. Beck SA, Smith KL, Tisdale MJ. Anticachectic and antitumor effect of eicosapentaenoic acid and its effect on protein turnover. Cancer Res 1991;51:6089–6093. [PubMed] [Google Scholar]
- 16. Emery PW, Lovell L, Rennie MJ. Protein synthesis measured in vivo in muscle and liver of cachectic tumor‐bearing mice. Cancer Res 1984;44:2779–2784. [PubMed] [Google Scholar]
- 17. Smith KL, Tisdale MJ. Increased protein degradation and decreased protein synthesis in skeletal muscle during cancer cachexia. Br J Cancer 1993;67:680–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. White JP, Baynes JW, Welle SL, Kostek MC, Matesic LE, Sato S, et al. The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the Apc (Min/+) mouse. PLoS ONE 2011;6:e24650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Samuels SE, Knowles AL, Tilignac T, Debiton E, Madelmont JC, Attaix D. Higher skeletal muscle protein synthesis and lower breakdown after chemotherapy in cachectic mice. Am J Physiol Regul Integr Comp Physiol 2001;281:R133–R139. [DOI] [PubMed] [Google Scholar]
- 20. Baracos VE, DeVivo C, Hoyle DH, Goldberg AL. Activation of the ATP‐ubiquitin‐proteasome pathway in skeletal muscle of cachectic rats bearing a hepatoma. Am J Physiol Endocrinol Metab 1995;268:E996–E1006. [DOI] [PubMed] [Google Scholar]
- 21. Nissinen TA, Hentila J, Penna F, Lampinen A, Lautaoja JH, Fachada V, et al. Treating cachexia using soluble ACVR2B improves survival, alters mTOR localization, and attenuates liver and spleen responses. J Cachexia Sarcopenia Muscle 2018;9:514–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Costelli P, Carbo N, Tessitore L, Bagby GJ, Lopez‐Soriano FJ, Argiles JM, et al. Tumor necrosis factor‐alpha mediates changes in tissue protein turnover in a rat cancer cachexia model. J Clin Invest 1993;92:2783–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Costelli P, Garcia‐Martinez C, Llovera M, Carbo N, Lopez‐Soriano FJ, Agell N, et al. Muscle protein waste in tumor‐bearing rats is effectively antagonized by a beta 2‐adrenergic agonist (clenbuterol). Role of the ATP‐ubiquitin‐dependent proteolytic pathway. J Clin Invest 1995;95:2367–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Temparis S, Asensi M, Taillandier D, Aurousseau E, Larbaud D, Obled A, et al. Increased ATP‐ubiquitin‐dependent proteolysis in skeletal muscles of tumor‐bearing rats. Cancer Res 1994;54:5568–5573. [PubMed] [Google Scholar]
- 25. Tessitore L, Costelli P, Bonetti G, Baccino FM. Cancer cachexia, malnutrition, and tissue protein turnover in experimental animals. Arch Biochem Biophys 1993;306:52–58. [DOI] [PubMed] [Google Scholar]
- 26. Tessitore L, Costelli P, Baccino FM. Pharmacological interference with tissue hypercatabolism in tumour‐bearing rats. Biochem J 1994;299:71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chacon‐Cabrera A, Fermoselle C, Urtreger AJ, Mateu‐Jimenez M, Diament MJ, de Kier Joffe ED, et al. Pharmacological strategies in lung cancer‐induced cachexia: effects on muscle proteolysis, autophagy, structure, and weakness. J Cell Physiol 2014;229:1660–1672. [DOI] [PubMed] [Google Scholar]
- 28. Khal J, Wyke SM, Russell ST, Hine AV, Tisdale MJ. Expression of the ubiquitin‐proteasome pathway and muscle loss in experimental cancer cachexia. Br J Cancer 2005;93:774–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lorite MJ, Thompson MG, Drake JL, Carling G, Tisdale MJ. Mechanism of muscle protein degradation induced by a cancer cachectic factor. Br J Cancer 1998;78:850–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. de Castro GS, Simoes E, Lima J, Ortiz‐Silva M, Festuccia WT, Tokeshi F, et al. Human cachexia induces changes in mitochondria, autophagy and apoptosis in the skeletal muscle. Cancers (Basel) 2019;11:1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Murphy KT, Chee A, Gleeson BG, Naim T, Swiderski K, Koopman R, et al. Antibody‐directed myostatin inhibition enhances muscle mass and function in tumor‐bearing mice. Am J Physiol Regul Integr Comp Physiol 2011;301:R716–R726. [DOI] [PubMed] [Google Scholar]
- 32. Busquets S, Figueras MT, Fuster G, Almendro V, Moore‐Carrasco R, Ametller E, et al. Anticachectic effects of formoterol: a drug for potential treatment of muscle wasting. Cancer Res 2004;64:6725–6731. [DOI] [PubMed] [Google Scholar]
- 33. Nosacka RL, Delitto AE, Delitto D, Patel R, Judge SM, Trevino JG, et al. Distinct cachexia profiles in response to human pancreatic tumours in mouse limb and respiratory muscle. J Cachexia Sarcopenia Muscle 2020;11:820–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Judge SM, Wu CL, Beharry AW, Roberts BM, Ferreira LF, Kandarian SC, et al. Genome‐wide identification of FoxO‐dependent gene networks in skeletal muscle during C26 cancer cachexia. BMC Cancer 2014;14:997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jewesbury RC, Topley WW. Pathological changes in voluntary muscles in general diseases. Proc R Soc Med 1912;5:161–178. [PMC free article] [PubMed] [Google Scholar]
- 36. Puig‐Vilanova E, Rodriguez DA, Lloreta J, Ausin P, Pascual‐Guardia S, Broquetas J, et al. Oxidative stress, redox signaling pathways, and autophagy in cachectic muscles of male patients with advanced COPD and lung cancer. Free Radic Biol Med 2015;79:91–108. [DOI] [PubMed] [Google Scholar]
- 37. Judge SM, Nosacka RL, Delitto D, Gerber MH, Cameron ME, Trevino JG, et al. Skeletal muscle fibrosis in pancreatic cancer patients with respect to survival. JNCI Cancer Spectr 2018;2:pky043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. He WA, Berardi E, Cardillo VM, Acharyya S, Aulino P, Thomas‐Ahner J, et al. NF‐kappaB‐mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J Clin Invest 2013;123:4821–4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mehl KA, Davis JM, Berger FG, Carson JA. Myofiber degeneration/regeneration is induced in the cachectic ApcMin/+ mouse. J Appl Physiol 2005;99:2379–2387. [DOI] [PubMed] [Google Scholar]
- 40. Salazar‐Degracia A, Busquets S, Argiles JM, Lopez‐Soriano FJ, Barreiro E. Formoterol attenuates increased oxidative stress and myosin protein loss in respiratory and limb muscles of cancer cachectic rats. PeerJ 2017;5:e4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gallot YS, Durieux AC, Castells J, Desgeorges MM, Vernus B, Plantureux L, et al. Myostatin gene inactivation prevents skeletal muscle wasting in cancer. Cancer Res 2014;74:7344–7356. [DOI] [PubMed] [Google Scholar]
- 42. Kim HG, Huot JR, Pin F, Guo B, Bonetto A, Nader GA. Reduced rDNA transcription diminishes skeletal muscle ribosomal capacity and protein synthesis in cancer cachexia. FASEB J 2021;35:e21335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sartori R, Hagg A, Zampieri S, Armani A, Winbanks CE, Viana LR, et al. Perturbed BMP signaling and denervation promote muscle wasting in cancer cachexia. Sci Transl Med 2021;13. [DOI] [PubMed] [Google Scholar]
- 44. Talbert EE, Cuitino MC, Ladner KJ, Rajasekerea PV, Siebert M, Shakya R, et al. Modeling human cancer‐induced cachexia. Cell Rep 2019;28:1612–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gallagher IJ, Stephens NA, MacDonald AJ, Skipworth RJ, Husi H, Greig CA, et al. Suppression of skeletal muscle turnover in cancer cachexia: evidence from the transcriptome in sequential human muscle biopsies. Clin Cancer Res 2012;18:2817–2827. [DOI] [PubMed] [Google Scholar]
- 46. Bonetto A, Penna F, Aversa Z, Mercantini P, Baccino FM, Costelli P, et al. Early changes of muscle insulin‐like growth factor‐1 and myostatin gene expression in gastric cancer patients. Muscle Nerve 2013;48:387–392. [DOI] [PubMed] [Google Scholar]
- 47. Op den Kamp CM, Langen RC, Snepvangers FJ, de Theije CC, Schellekens JM, Laugs F, et al. Nuclear transcription factor kappa B activation and protein turnover adaptations in skeletal muscle of patients with progressive stages of lung cancer cachexia. Am J Clin Nutr 2013;98:738–748. [DOI] [PubMed] [Google Scholar]
- 48. D'Orlando C, Marzetti E, Francois S, Lorenzi M, Conti V, di Stasio E, et al. Gastric cancer does not affect the expression of atrophy‐related genes in human skeletal muscle. Muscle Nerve 2014;49:528–533. [DOI] [PubMed] [Google Scholar]
- 49. Stephens NA, Skipworth RJ, Gallagher IJ, Greig CA, Guttridge DC, Ross JA, et al. Evaluating potential biomarkers of cachexia and survival in skeletal muscle of upper gastrointestinal cancer patients. J Cachexia Sarcopenia Muscle 2015;6:53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Murton AJ, Maddocks M, Stephens FB, Marimuthu K, England R, Wilcock A. Consequences of late‐stage non‐small‐cell lung cancer cachexia on muscle metabolic processes. Clin Lung Cancer 2017;18:e1–e11. [DOI] [PubMed] [Google Scholar]
- 51. DeJong CH, Busquets S, Moses AG, Schrauwen P, Ross JA, Argiles JM, et al. Systemic inflammation correlates with increased expression of skeletal muscle ubiquitin but not uncoupling proteins in cancer cachexia. Oncol Rep 2005;14:257–263. [PubMed] [Google Scholar]
- 52. Sun YS, Ye ZY, Qian ZY, Xu XD, Hu JF. Expression of TRAF6 and ubiquitin mRNA in skeletal muscle of gastric cancer patients. J Exp Clin Cancer Res 2012;31:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Khal J, Hine AV, Fearon KC, Dejong CH, Tisdale MJ. Increased expression of proteasome subunits in skeletal muscle of cancer patients with weight loss. Int J Biochem Cell Biol 2005;37:2196–2206. [DOI] [PubMed] [Google Scholar]
- 54. Jagoe RT, Redfern CP, Roberts RG, Gibson GJ, Goodship TH. Skeletal muscle mRNA levels for cathepsin B, but not components of the ubiquitin‐proteasome pathway, are increased in patients with lung cancer referred for thoracotomy. Clin Sci (Lond) 2002;102:353–361. [PubMed] [Google Scholar]
- 55. Constantinou C, Fontes de Oliveira CC, Mintzopoulos D, Busquets S, He J, Kesarwani M, et al. Nuclear magnetic resonance in conjunction with functional genomics suggests mitochondrial dysfunction in a murine model of cancer cachexia. Int J Mol Med 2011;27:15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Costelli P, Muscaritoli M, Bossola M, Penna F, Reffo P, Bonetto A, et al. IGF‐1 is downregulated in experimental cancer cachexia. Am J Physiol Regul Integr Comp Physiol 2006;291:R674–R683. [DOI] [PubMed] [Google Scholar]
- 57. Acharyya S, Butchbach ME, Sahenk Z, Wang H, Saji M, Carathers M, et al. Dystrophin glycoprotein complex dysfunction: a regulatory link between muscular dystrophy and cancer cachexia. Cancer Cell 2005;8:421–432. [DOI] [PubMed] [Google Scholar]
- 58. Murphy KT, Struk A, Malcontenti‐Wilson C, Christophi C, Lynch GS. Physiological characterization of a mouse model of cachexia in colorectal liver metastases. Am J Physiol Regul Integr Comp Physiol 2013;304:R854–R864. [DOI] [PubMed] [Google Scholar]
- 59. Acharyya S, Ladner KJ, Nelsen LL, Damrauer J, Reiser PJ, Swoap S, et al. Cancer cachexia is regulated by selective targeting of skeletal muscle gene products. J Clin Invest 2004;114:370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Braun TP, Zhu X, Szumowski M, Scott GD, Grossberg AJ, Levasseur PR, et al. Central nervous system inflammation induces muscle atrophy via activation of the hypothalamic‐pituitary‐adrenal axis. J Exp Med 2011;208:2449–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Braun TP, Grossberg AJ, Krasnow SM, Levasseur PR, Szumowski M, Zhu XX, et al. Cancer‐ and endotoxin‐induced cachexia require intact glucocorticoid signaling in skeletal muscle. FASEB J 2013;27:3572–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Busquets S, Toledo M, Orpi M, Massa D, Porta M, Capdevila E, et al. Myostatin blockage using actRIIB antagonism in mice bearing the Lewis lung carcinoma results in the improvement of muscle wasting and physical performance. J Cachexia Sarcopenia Muscle 2012;3:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Guarnier FA, Cecchini AL, Suzukawa AA, Maragno AL, Simao AN, Gomes MD, et al. Time course of skeletal muscle loss and oxidative stress in rats with Walker 256 solid tumor. Muscle Nerve 2010;42:950–958. [DOI] [PubMed] [Google Scholar]
- 64. Julienne CM, Dumas JF, Goupille C, Pinault M, Berri C, Collin A, et al. Cancer cachexia is associated with a decrease in skeletal muscle mitochondrial oxidative capacities without alteration of ATP production efficiency. J Cachexia Sarcopenia Muscle 2012;3:265–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kir S, Komaba H, Garcia AP, Economopoulos KP, Liu W, Lanske B, et al. PTH/PTHrP receptor mediates cachexia in models of kidney failure and cancer. Cell Metab 2016;23:315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Levolger S, Wiemer EAC, van Vugt JLA, Huisman SA, van Vledder MG, van Damme‐van Engel S, et al. Inhibition of activin‐like kinase 4/5 attenuates cancer cachexia associated muscle wasting. Sci Rep 2019;9:9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Michaelis KA, Zhu X, Burfeind KG, Krasnow SM, Levasseur PR, Morgan TK, et al. Establishment and characterization of a novel murine model of pancreatic cancer cachexia. J Cachexia Sarcopenia Muscle 2017;8:824–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pigna E, Berardi E, Aulino P, Rizzuto E, Zampieri S, Carraro U, et al. Aerobic exercise and pharmacological treatments counteract cachexia by modulating autophagy in colon cancer. Sci Rep 2016;6:26991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Reed SA, Sandesara PB, Senf SM, Judge AR. Inhibition of FoxO transcriptional activity prevents muscle fiber atrophy during cachexia and induces hypertrophy. FASEB J 2011;26:987–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Schwarzkopf M, Coletti D, Sassoon D, Marazzi G. Muscle cachexia is regulated by a p53‐PW1/Peg3‐dependent pathway. Genes Dev 2006;20:3440–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Shakri AR, Zhong TJ, Ma W, Coker C, Kim S, Calluori S, et al. Upregulation of ZIP14 and altered zinc homeostasis in muscles in pancreatic cancer cachexia. Cancers (Basel) 2019;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Talbert EE, Metzger GA, He WA, Guttridge DC. Modeling human cancer cachexia in colon 26 tumor‐bearing adult mice. J Cachexia Sarcopenia Muscle 2014;5:321–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wang G, Biswas AK, Ma W, Kandpal M, Coker C, Grandgenett PM, et al. Metastatic cancers promote cachexia through ZIP14 upregulation in skeletal muscle. Nat Med 2018;24:770–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Winbanks CE, Murphy KT, Bernardo BC, Qian H, Liu Y, Sepulveda PV, et al. Smad7 gene delivery prevents muscle wasting associated with cancer cachexia in mice. Sci Transl Med 2016;8:348ra398. [DOI] [PubMed] [Google Scholar]
- 75. Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010;142:531–543. [DOI] [PubMed] [Google Scholar]
- 76. Huot JR, Pin F, Narasimhan A, Novinger LJ, Keith AS, Zimmers TA, et al. ACVR2B antagonism as a countermeasure to multi‐organ perturbations in metastatic colorectal cancer cachexia. J Cachexia Sarcopenia Muscle 2020;11:1779–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Penna F, Costamagna D, Pin F, Camperi A, Fanzani A, Chiarpotto EM, et al. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am J Pathol 2013;182:1367–1378. [DOI] [PubMed] [Google Scholar]
- 78. Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, et al. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J 2004;18:39–51. [DOI] [PubMed] [Google Scholar]
- 79. Penna F, Ballaro R, Martinez‐Cristobal P, Sala D, Sebastian D, Busquets S, et al. Autophagy exacerbates muscle wasting in cancer cachexia and impairs mitochondrial function. J Mol Biol 2019;431:2674–2686. [DOI] [PubMed] [Google Scholar]
- 80. Marino FE, Risbridger G, Gold E. Activin‐beta modulates cachexia by repressing the ubiquitin‐proteasome and autophagic degradation pathways. J Cachexia Sarcopenia Muscle 2015;6:365–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhang G, Liu Z, Ding H, Zhou Y, Doan HA, Sin KWT, et al. Tumor induces muscle wasting in mice through releasing extracellular Hsp70 and Hsp90. Nat Commun 2017;8:589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hardee JP, Counts BR, Gao S, VanderVeen BN, Fix DK, Koh HJ, et al. Inflammatory signalling regulates eccentric contraction‐induced protein synthesis in cachectic skeletal muscle. J Cachexia Sarcopenia Muscle 2018;9:369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Koutnik AP, Poff AM, Ward NP, DeBlasi JM, Soliven MA, Romero MA, et al. Ketone bodies attenuate wasting in models of atrophy. J Cachexia Sarcopenia Muscle 2020;11:973–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Llovera M, Garcia‐Martinez C, Agell N, Marzabal M, Lopez‐Soriano FJ, Argiles JM. Ubiquitin gene expression is increased in skeletal muscle of tumour‐bearing rats. FEBS Lett 1994;338:311–318. [DOI] [PubMed] [Google Scholar]
- 85. Carbo N, Lopez‐Soriano J, Costelli P, Busquets S, Alvarez B, Baccino FM, et al. Interleukin‐15 antagonizes muscle protein waste in tumour‐bearing rats. Br J Cancer 2000;83:526–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Goncalves MD, Hwang SK, Pauli C, Murphy CJ, Cheng Z, Hopkins BD, et al. Fenofibrate prevents skeletal muscle loss in mice with lung cancer. Proc Natl Acad Sci U S A 2018;115:E743–E752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Bonetto A, Aydogdu T, Kunzevitzky N, Guttridge DC, Khuri S, Koniaris LG, et al. STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS One 2011;6:e22538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Cornwell EW, Mirbod A, Wu CL, Kandarian SC, Jackman RW. C26 cancer‐induced muscle wasting is IKKbeta‐dependent and NF‐kappaB‐independent. PLoS One 2014;9:e87776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Llovera M, Garcia‐Martinez C, Agell N, Lopez‐Soriano FJ, Argiles JM. Muscle wasting associated with cancer cachexia is linked to an important activation of the ATP‐dependent ubiquitin‐mediated proteolysis. Int J Cancer 1995;61:138–141. [DOI] [PubMed] [Google Scholar]
- 90. Aversa Z, Pin F, Lucia S, Penna F, Verzaro R, Fazi M, et al. Autophagy is induced in the skeletal muscle of cachectic cancer patients. Sci Rep 2016;6:30340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ballaro R, Penna F, Pin F, Gomez‐Cabrera MC, Vina J, Costelli P. Moderate exercise improves experimental cancer cachexia by modulating the redox homeostasis. Cancers (Basel) 2019;11:285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Deval C, Mordier S, Obled C, Bechet D, Combaret L, Attaix D, et al. Identification of cathepsin L as a differentially expressed message associated with skeletal muscle wasting. Biochem J 2001;360:143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Martin A, Freyssenet D. Phenotypic features of cancer cachexia‐related loss of skeletal muscle mass and function: lessons from human and animal studies. J Cachexia Sarcopenia Muscle 2021;12:252–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Brown JL, Rosa‐Caldwell ME, Lee DE, Blackwell TA, Brown LA, Perry RA, et al. Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour‐bearing mice. J Cachexia Sarcopenia Muscle 2017;8:926–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. White JP, Baltgalvis KA, Puppa MJ, Sato S, Baynes JW, Carson JA. Muscle oxidative capacity during IL‐6‐dependent cancer cachexia. Am J Physiol Regul Integr Comp Physiol 2011;300:R201–R211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Schmitt TL, Martignoni ME, Bachmann J, Fechtner K, Friess H, Kinscherf R, et al. Activity of the Akt‐dependent anabolic and catabolic pathways in muscle and liver samples in cancer‐related cachexia. J Mol Med (Berl) 2007;85:647–654. [DOI] [PubMed] [Google Scholar]
- 97. Puppa MJ, Murphy EA, Fayad R, Hand GA, Carson JA. Cachectic skeletal muscle response to a novel bout of low‐frequency stimulation. J Appl Physiol 1985;2014:1078–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Johns N, Stretch C, Tan BH, Solheim TS, Sorhaug S, Stephens NA, et al. New genetic signatures associated with cancer cachexia as defined by low skeletal muscle index and weight loss. J Cachexia Sarcopenia Muscle 2017;8:122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Barreiro E, de la Puente B, Busquets S, Lopez‐Soriano FJ, Gea J, Argiles JM. Both oxidative and nitrosative stress are associated with muscle wasting in tumour‐bearing rats. FEBS Lett 2005;579:1646–1652. [DOI] [PubMed] [Google Scholar]
- 100. Marin‐Corral J, Fontes CC, Pascual‐Guardia S, Sanchez F, Olivan M, Argiles JM, et al. Redox balance and carbonylated proteins in limb and heart muscles of cachectic rats. Antioxid Redox Signal 2010;12:365–380. [DOI] [PubMed] [Google Scholar]
- 101. Neyroud D, Nosacka RL, Judge AR, Hepple RT. Colon 26 adenocarcinoma (C26)‐induced cancer cachexia impairs skeletal muscle mitochondrial function and content. J Muscle Res Cell Motil 2019;40:59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. O'Gorman P, McMillan DC, McArdle CS. Longitudinal study of weight, appetite, performance status, and inflammation in advanced gastrointestinal cancer. Nutr Cancer 1999;35:127–129. [DOI] [PubMed] [Google Scholar]
- 103. Scott HR, McMillan DC, Forrest LM, Brown DJ, McArdle CS, Milroy R. The systemic inflammatory response, weight loss, performance status and survival in patients with inoperable non‐small cell lung cancer. Br J Cancer 2002;87:264–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Lerner L, Hayes TG, Tao N, Krieger B, Feng B, Wu Z, et al. Plasma growth differentiation factor 15 is associated with weight loss and mortality in cancer patients. J Cachexia Sarcopenia Muscle 2015;6:317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lerner L, Tao J, Liu Q, Nicoletti R, Feng B, Krieger B, et al. MAP 3K11/GDF15 axis is a critical driver of cancer cachexia. J Cachexia Sarcopenia Muscle 2016;7:467–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Chen JL, Walton KL, Qian H, Colgan TD, Hagg A, Watt MJ, et al. Differential effects of IL6 and activin A in the development of cancer‐associated cachexia. Cancer Res 2016;76:5372–5382. [DOI] [PubMed] [Google Scholar]
- 107. Baltgalvis KA, Berger FG, Pena MM, Davis JM, Muga SJ, Carson JA. Interleukin‐6 and cachexia in ApcMin/+ mice. Am J Physiol Regul Integr Comp Physiol 2008;294:R393–R401. [DOI] [PubMed] [Google Scholar]
- 108. Segatto M, Fittipaldi R, Pin F, Sartori R, Dae Ko K, Zare H, et al. Epigenetic targeting of bromodomain protein BRD4 counteracts cancer cachexia and prolongs survival. Nat Commun 2017;8:1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Miller A, McLeod L, Alhayyani S, Szczepny A, Watkins DN, Chen W, et al. Blockade of the IL‐6 trans‐signalling/STAT3 axis suppresses cachexia in Kras‐induced lung adenocarcinoma. Oncogene 2017;36:3059–3066. [DOI] [PubMed] [Google Scholar]
- 110. Durieux AC, Amirouche A, Banzet S, Koulmann N, Bonnefoy R, Pasdeloup M, et al. Ectopic expression of myostatin induces atrophy of adult skeletal muscle by decreasing muscle gene expression. Endocrinology 2007;148:3140–3147. [DOI] [PubMed] [Google Scholar]
- 111. Amirouche A, Durieux AC, Banzet S, Koulmann N, Bonnefoy R, Mouret C, et al. Down‐regulation of akt/mammalian target of rapamycin signaling pathway in response to myostatin overexpression in skeletal muscle. Endocrinology 2009;150:286–294. [DOI] [PubMed] [Google Scholar]
- 112. Morel J, Palao JC, Castells J, Desgeorges M, Busso T, Molliex S, et al. Regulation of Akt‐mTOR, ubiquitin‐proteasome and autophagy‐lysosome pathways in locomotor and respiratory muscles during experimental sepsis in mice. Sci Rep 2017;7:10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Benny Klimek ME, Aydogdu T, Link MJ, Pons M, Koniaris LG, Zimmers TA. Acute inhibition of myostatin‐family proteins preserves skeletal muscle in mouse models of cancer cachexia. Biochem Biophys Res Commun 2010;391:1548–1554. [DOI] [PubMed] [Google Scholar]
- 114. Paajanen J, Ilonen I, Lauri H, Jarvinen T, Sutinen E, Ollila H, et al. Elevated circulating activin A levels in patients with malignant pleural mesothelioma are related to cancer cachexia and reduced response to platinum‐based chemotherapy. Clin Lung Cancer 2020;21:e142–e150. [DOI] [PubMed] [Google Scholar]
- 115. Matzuk MM, Finegold MJ, Mather JP, Krummen L, Lu H, Bradley A. Development of cancer cachexia‐like syndrome and adrenal tumors in inhibin‐deficient mice. Proc Natl Acad Sci U S A 1994;91:8817–8821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Parajuli P, Kumar S, Loumaye A, Singh P, Eragamreddy S, Nguyen TL, et al. Twist1 activation in muscle progenitor cells causes muscle loss akin to cancer cachexia. Dev Cell 2018;45:e716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Flint TR, Janowitz T, Connell CM, Roberts EW, Denton AE, Coll AP, et al. Tumor‐induced IL‐6 reprograms host metabolism to suppress anti‐tumor immunity. Cell Metab 2016;24:672–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Llovera M, Garcia‐Martinez C, Costelli P, Agell N, Carbo N, Lopez‐Soriano FJ, et al. Muscle hypercatabolism during cancer cachexia is not reversed by the glucocorticoid receptor antagonist RU38486. Cancer Lett 1996;99:7–14. [DOI] [PubMed] [Google Scholar]
- 119. Martin A, Castells J, Allibert V, Emerit A, Zolotoff C, Cardot‐Ruffino V, et al. Hypothalamic‐pituitary‐adrenal axis activation and glucocorticoid‐responsive gene expression in skeletal muscle and liver of Apc mice. J Cachexia Sarcopenia Muscle 2022;16:1686–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Sadek J, Hall DT, Colalillo B, Omer A, Tremblay AK, Sanguin‐Gendreau V, et al. Pharmacological or genetic inhibition of iNOS prevents cachexia‐mediated muscle wasting and its associated metabolism defects. EMBO Mol Med 2021;13:e13591. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary reference list.