

Abstract
Background
Skeletal muscle dysfunction is an important co‐morbidity in patients with chronic obstructive pulmonary disease (COPD) and is significantly associated with increased mortality. Oxidative stress has been demonstrated an important trigger for COPD‐related skeletal muscle dysfunction. Glycine‐histidine‐lysine (GHK) is an active tripeptide, which is a normal component of human plasma, saliva, and urine; promotes tissue regeneration; and acts as an anti‐inflammatory and antioxidant properties. The purpose of this study was to determine whether GHK is involved in COPD‐related skeletal muscle dysfunction.
Methods
The plasma GHK level in patients with COPD (n = 9) and age‐paired healthy subjects (n = 11) were detected using reversed‐phase high‐performance liquid chromatography. The complex GHK with Cu (GHK‐Cu) was used in in vitro (C2C12 myotubes) and in vivo experiments (cigarette smoking [CS]‐exposure mouse model) to explore the involvement of GHK in CS‐induced skeletal muscle dysfunction.
Results
Compared with healthy control, plasma GHK levels were decreased in patients with COPD (70.27 ± 38.87 ng/mL vs. 133.0 ± 54.54 ng/mL, P = 0.009). And plasma GHK levels in patients with COPD were associated with pectoralis muscle area (R = 0.684, P = 0.042), inflammatory factor TNF‐α (R = −0.696, P = 0.037), and antioxidative stress factor SOD2 (R = 0.721, P = 0.029). GHK‐Cu was found to rescue CSE‐induced skeletal muscle dysfunction in C2C12 myotubes, as evidenced by increased expression of myosin heavy chain, reduced expression of MuRF1 and atrogin‐1, elevated mitochondrial content, and enhanced resistance to oxidative stress. In CS‐induced muscle dysfunction C57BL/6 mice, GHK‐Cu treatment (0.2 and 2 mg/kg) reduces CS‐induced muscle mass loss (skeletal muscle weight (1.19 ± 0.09% vs. 1.29 ± 0.06%, 1.40 ± 0.05%; P < 0.05) and muscle cross‐sectional area elevated (1055 ± 552.4 μm2 vs. 1797 ± 620.9 μm2, 2252 ± 534.0 μm2; P < 0.001), and also rescues CS‐induced muscle weakness, indicated by improved grip strength (175.5 ± 36.15 g vs. 257.6 ± 37.98 g, 339.1 ± 72.22 g; P < 0.01). Mechanistically, GHK‐Cu directly binds and activates SIRT1(the binding energy was −6.1 kcal/mol). Through activating SIRT1 deacetylation, GHK‐Cu inhibits FoxO3a transcriptional activity to reduce protein degradation, deacetylates Nrf2 and contribute to its action of reducing oxidative stress by generation of anti‐oxidant enzymes, increases PGC‐1α expression to promote mitochondrial function. Finally, GHK‐Cu could protect mice against CS‐induced skeletal muscle dysfunction via SIRT1.
Conclusions
Plasma glycyl‐l‐histidyl‐l‐lysine level in patients with chronic obstructive pulmonary disease was significantly decreased and was significantly associated with skeletal muscle mass. Exogenous administration of glycyl‐l‐histidyl‐l‐lysine‐Cu2+ could protect against cigarette smoking‐induced skeletal muscle dysfunction via sirtuin 1.
Keywords: Chronic obstructive pulmonary disease, Skeletal muscle dysfunction, Glycine‐histidine‐lysine, SIRT1
Introduction
Chronic obstructive pulmonary disease (COPD) is a chronic airway disease characterized by persistent respiratory symptoms and airflow limitation, 1 often accompanied by multiple co‐morbidities involving cardiovascular, gastrointestinal, haematologic, musculoskeletal, and other systems. 2 Among them, reduced exercise tolerance due to skeletal muscle dysfunction was associated with poor prognosis, hospitalization, and increased morbidity and mortality. The literatures reported that approximately 15%–45% of patients with stable COPD had skeletal muscle dysfunction. 3 Patients with stable COPD with combined sarcopenia have increased dyspnoea index scores and decreased exercise tolerance, whereas decreased exercise capacity and reduced activity lead to wasting muscle atrophy, which the vicious cycle of disease progression is further aggravated. 4
Oxidative stress is emerging as an important contributor to both the lung and systemic pathologies of COPD, 5 and has been demonstrated an important trigger for COPD‐related skeletal muscle dysfunction. Oxidative stress refers to the imbalance between oxidative damage (mediated by reactive oxygen species) and antioxidant defence system. Several studies indicated that reactive oxygen species in skeletal muscle cells is inversely correlated with cross‐sectional area of skeletal muscle fibres. 6 In addition, antioxidant capacity of skeletal muscle was correlated with muscle mass and strength in patients with COPD. 7 Mechanistically, oxidative stress could directly or indirectly modulate transcription factors and kinases inducing proteolytic pathways (increasing the expression of muscle atrophy‐related genes, atrogin‐1 and myostatin), and decreasing mitochondrial content and function. 8 Thus, therapy aimed at reducing oxidative stress in muscle could be of benefit for improving the muscle pathology in COPD.
Glycine‐Histidine‐Lysine (GHK) is an active tripeptide that is a normal component of human plasma, saliva, and urine, and its levels decline with age. 9 Initial studies showed that GHK is present in the alpha 2 chain of type I collagen and is released into the site of injury to exert local healing effects when injury activates protein hydrolases, an early signal for skin repair. 10 In recent years, as research has progressed, the ability of GHK to promote tissue regeneration and anti‐inflammatory and antioxidant properties has been demonstrated in tissues such as bone, 11 and liver. 12 In vivo, GHK readily forms the complex glutamate‐histidine‐lysine‐copper with Cu (GHK‐Cu), enhancing the bioavailability of GHK. Studies have shown that exogenous administration of GHK and GHK‐Cu has potential therapeutic value in pulmonary fibrosis, 13 and acute lung injury, 14 by mechanisms closely related to its anti‐inflammatory antioxidant effects.
The purpose of this study was to determine the involvement of GHK in COPD related skeletal muscle dysfunction. We aimed to evaluate the relationship between plasma GHK level and skeletal muscle function in patients with COPD. Meanwhile, we try to explore whether exogenous administration of GHK‐Cu ameliorate cigarette smoke‐induced skeletal muscle dysfunction.
Methods
Human subjects
Patients with COPD (n = 9) and age‐matched healthy controls (n = 11) were recruited in the First Hospital of China Medical University between December 2020 and March 2021. The inclusion criterion was a diagnosis of stable COPD according to the Global Initiative for Chronic Obstructive Lung Disease (GOLD) criteria. The exclusion criteria were as follows: COPD exacerbation within the last 1 month; presence of severe cardiovascular disease or active lung disease; concomitant disease affecting the musculoskeletal system; long‐term systemic steroid therapy; and inability to read or understand the informed consent documents.
Measurement of plasma GHK level
As previous study reported, 15 o‐phenanthroline was added to blood samples at a final concentration of 5 mg/mL immediately after blood collection due to the instability of GHK in human plasma, and then plasma was preserved at −80°C after blood samples centrifugation. Then with specific conditions, a reversed‐phase high‐performance liquid chromatography (RP‐HPLC) equilibrated with a C18 column (4.5 mm × 250 mm, Shimadzu Corporation LC‐20A, Kyoto, Japan). First, two eluents consisting of solvent A (in distilled water with 0.1% (v/v) TFA) and solvent B (acetonitrile with 0.1% (v/v) trifluoroacetate) were eluted linearly at a flow rate of 1.0 mL/min for 30 min. The monitoring wavelengths at this stage were 220 nm and 280 nm, respectively. The GHK level could be determined.
C2C12 cell culture and differentiation
Mouse C2C12 myoblasts were obtained from the China Infrastructure of Cell Line Resource and maintained in DMEM with 10% FBS (fetal bovine serum). To initiate differentiation, the cells were grown to 80% confluence and incubated with DMEM containing 2% horse serum for 4 days. Medium was refreshed every other day. The fully differentiated myotubes used for next experiments.
Preparation of cigarette smoke extract (CSE)
CSE was prepared as described previously. 16 Briefly, one non‐filtered Marlboro cigarette (Philip Morris Companies, 0.8 mg of Nicotine, 10 mg of Tar, and 10 mg of carbon monoxide per cigarette) was burned, and the smoke was passed through 4 ml of PBS. The extract was used when adjusting the pH to between 7.00 and 7.40 after filtering through a filter with 0.22‐μM pores to remove particles and bacteria. The fresh CSE‐PBS solution was prepared for each experiment. In this study, cells were treated with CSE for 48 h.
Cell viability
Cell viability was determined by CCK8 assay as described previously. 17 C2C12 cells were seeded in 96‐well plates at a density of 1 × 104 cells per well. After fully differentiated, the myotubes were treated with different concentrations of GHK‐Cu, with or without CSE. For CCK8 assay, 10 μL CCK8 solution (C0038) were obtained from Beyotime Technology Corp., Ltd. Shanghai, China.
MitoTracker green staining
C2C12 cells were seeded with 1.0 × 105 cells per well in six‐well plates. After fully differentiation, myotubes were treated with CSE, with or without GHK‐Cu. Living cells were directly stained with MitoTracker Green (C1048, Beyotime) following the manufacturer's instructions.
Antioxidant index measurement
Different biochemical assays were performed in homogenates of freshly procured muscle tissues and cell samples. The muscle tissue homogenates were prepared in ice‐cold saline at 4°C, and the supernatants were collected following centrifugation at 2500 rpm and 4°C for 10 min. The supernatant was used to detect antioxidant biomarkers, such as the total antioxidant capacity (T‐AOC) and GSH content and malondialdehyde (MDA) levels as a measure of lipid peroxidation in the lung tissue, according to the manufacturer's instructions (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). The protein concentration was determined using a bicinchoninic acid (BCA) protein assay kit (Beyotime Institute of Biotechnology), with bovine serum albumin as the standard. The T‐AOC contents were calculated, and the results were expressed as activity per milligram of protein.
Cigarette smoking exposure mice model
All animal experiments were approved by the Animal Ethics Committee of China Medical University (No. KT2018061). Male C57BL/6J mice 8–10 weeks of age were purchased from Liaoning Changsheng Biotechnology Company (Benxi, China). Mice were exposed to CS using a systemic exposure system (8050II, Hepu, Tianjin, China) within a barrier facility. Mice were exposed to CS (20 cigarettes per session, twice/day, 6 days/week) using Marlboro cigarettes (Philip Morris Companies, USA) for 3 months.
Molecular docking
The SIRT1 protein crystal structure was obtained from the RCSB database (PDB ID: 4ZZH); the 3D structure of GHK‐Cu was downloaded from the PubChem database (PubChem CID: 342538) and energy minimized under the MMFF94 force field using AVOGADR 1.2.0. In this study, AutoDock Vina 1.1.2 software was used for molecular docking work. All receptor proteins were first hydrogenated using PyMol Academic Open Source Edition. Then, all processed small molecules and receptor proteins were converted to PDBQT format using ADFRsuite 1.0. Before docking, the centre of mass of the protein was used as the centre of the box (see Table 1 for the centre coordinates of the protein), and the appropriate X, Y, and Z edge lengths were adjusted to construct the box and to fully wrap the entire protein (see Table 1 for the docking Box X, Y, and Z edge length parameters). For docking, the grid boxes and the PDBQT files of the processed proteins and small molecules were used as input files and docked using Vina, with the exhaustive degree of docking global search set to 32, and the remaining parameters were kept at default settings. Finally, the output docking conformation with the highest score was considered as the binding conformation, and the docking results were visualized and analysed using PyMol Academic Open Source Edition.
Table 1.
The centre coordinates of the protein and the parameters of box.
| Protein | The centre coordinates of the protein (X, Y, and Z) | Length parameters of the docking box (X, Y, and Z) |
|---|---|---|
| SIRT1 | 18.298, −29.719, −11.525 | 64.0, 86.0, 80.0 |
Sirtuin1 deacetylase activity
SIRT1 deacetylase activity was quantified using the SIRT1 Activity Fluorometric Assay Kit (ab156065, Abcam) according to the manufacturer's instructions. Fluorescence intensity was monitored at an excitation wavelength of 355 nm and an emission wavelength of 450 nm, using a SpectraMax M5 microplate reader, and normalized by the amount of protein. The SIRT1 deacetylase activity of GHK‐Cu‐treated cells was represented as fold of the vehicle control group.
Statistical analysis
All data were expressed as mean ± SD and analysed by SPSS 13.0 software (IBM). Histograms, Q‐Q (quantile–quantile), and Shapiro–Wilk's test were used to check if the data obeyed a normal distribution. Differences between two groups were assessed using a t‐test (normally distributed data) or the Mann–Whitney test (non‐normal distribution). For comparisons between multiple groups, data with a normal distribution analysed using an ANOVA test and with subsequent multiple comparisons using Tukey–Kramer correction; data that do not show a normal distribution were analysed using Kruskal–Wallis test with Dunn's multiple‐comparison test. P‐values less than 0.05 were considered statistically significant.
Additional materials and methods can be found in the supporting information.
Results
The plasma level and clinical relevance of GHK in patients with COPD
A total of nine patients with COPD and 11 age‐matched healthy controls were enrolled in the final analysis. The baseline characteristics of the patients are listed in Table S1. There were no statistically significant differences in age, BMI, or sex between COPD patients and healthy controls. Pulmonary function (FEV1, FEV1%predicted, FVC, and FEV1/FVC), exercise tolerance, muscle function was significantly decreased in patients with COPD compared with that in healthy controls. To analysed the expression level and clinical relevance of GHK in patients with COPD and healthy subjects, the plasma levels of GHK, inflammatory factor, and antioxidative stress factor were determined. Our data showed that patients with COPD exhibited lower levels of GHK (70.27 ± 38.87 ng/mL vs. 133.0 ± 54.54 ng/mL, P = 0.009) (Figure 1A), higher level of the pro‐inflammatory cytokines TNF‐α (Figure 1B), and lower level of the antioxidative stress factor SOD2 (Figure 1C) in plasma compared with those of healthy controls. Next, we analysis the relationship between plasma GHK level and skeletal muscle mass in patients with COPD. The pectoralis muscle area (PMA) was quantifiable by single chest CT image superior to the aortic arch. As shown in Figure 1D, the plasma GHK level was positive correlated (R = 0.684, P = 0.042) with PMI (PMA divided by height2) in patients with COPD. In additional, the plasma GHK level was positive correlated with 6MWD (R = 0.671, P = 0.048) and qMVC(R = 0.683, P = 0.043) in patients with COPD (Figure 1E). Next, the correlation between GHK, and inflammation, oxidative stress was applied. As shown in Figure 1F, decreased GHK plasma levels were negatively correlated with the levels of the inflammatory factor TNF‐α (R = −0.696, P = 0.037). Similarly, GHK plasma levels were positively correlated with the levels of the antioxidative stress factor SOD2 (R = 0.721, P = 0.029). Our findings suggest that GHK may play an important role in skeletal muscle dysfunction in patients with COPD.
Figure 1.
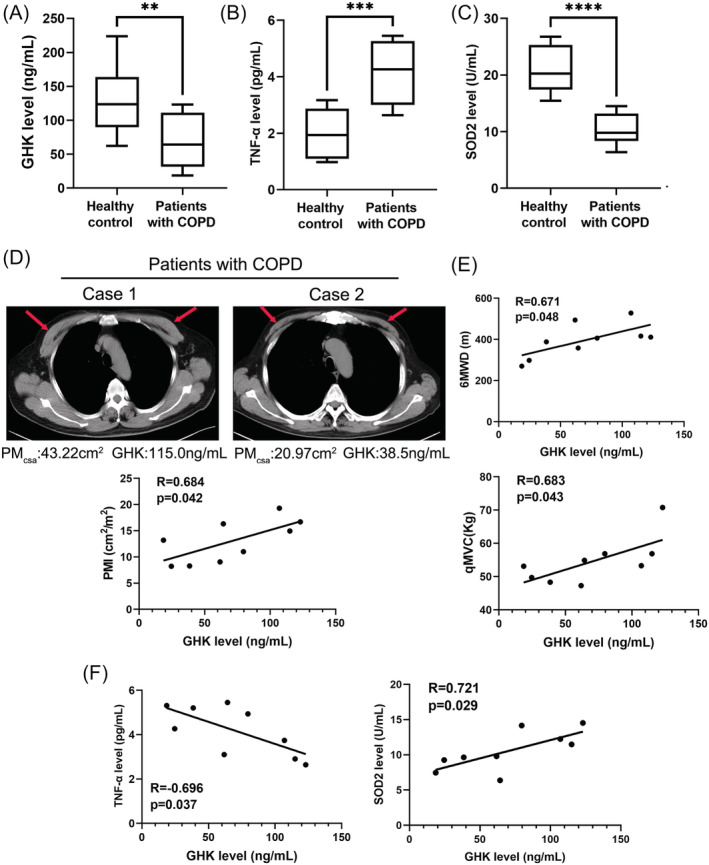
The plasma level and clinical relevance of GHK in patients with COPD. (A) The plasma levels of GHK in healthy control (n = 11) and patients with COPD (n = 9). (B) The plasma levels of TNF‐α in healthy control and patients with COPD. (C) The plasma levels of SOD2 in healthy control and patients with COPD. (D) Representative images of the pectoralis muscle area (PMA) in chest CT image of patients with COPD; and the relationship between the plasma GHK level and pectoralis muscle index (PMI) in patients with COPD. (E) The relationship between the plasma GHK level and 6MWD (6‐minute walk distance), qMVC (quadriceps maximum voluntary contraction) in patients with COPD. (F) The relationship between the plasma GHK level and the plasma levels of TNF‐α (left), and SOD2 (right) in patients with COPD. **P < 0.01; ***P < 0.001.
GHK‐Cu suppressed CSE‐induced C2C12 myotube alteration
The complex GHK with Cu (GHK‐Cu) was used in in vitro and in vivo experiments due to it enhanced the bioavailability of GHK. To assess the protective effect of GHK‐Cu against CS‐induced skeletal muscle dysfunction, we first assessed the effect of GHK‐Cu on C2C12 myotubes. The structure of GHK‐Cu was shown in Figure 2A. CCK8 cell viability assay (Figure 2B) shows that GHK‐Cu treatment alone did not show apparent cytotoxicity on C2C12 myotubes, and GHK‐Cu could prevent CSE‐induced cell death concentration‐dependently. As previous study, 18 CSE treatment robustly increased the expression of muscle specific ubiquitin E3 ligases (atrogin‐1 and MuRF1). GHK‐Cu treatment completely reversed CSE‐induced increase of atrogin‐1 and MuRF1 expression (Figure 2C). AKT/FoxO3a pathway is a critical pathway that regulates muscle atrophy and protein degradation. 19 We also found GHK‐Cu could significantly up‐regulated CSE induced decrease of AKT/FoxO3a pathway. CSE significantly decreased p‐Akt and p‐FoxO3a, which was totally reversed by GHK‐Cu treatment (Figure 2C). Immunofluorescence staining showed that CSE‐treated myotubes had significantly smaller diameters compared with untreated myotubes, indicating that CSE is capable of inducing myotube atrophy representing muscle atrophy. Meanwhile, GHK‐Cu treatment elevated diameters of myotubes compared with CSE‐treated myotubes in a concentration dependent manner (Figure 2D). Based on above results, GHK‐Cu protects C2C12 myotubes from CSE‐induced atrophy and regulates AKT‐FoxO3a pathway.
Figure 2.
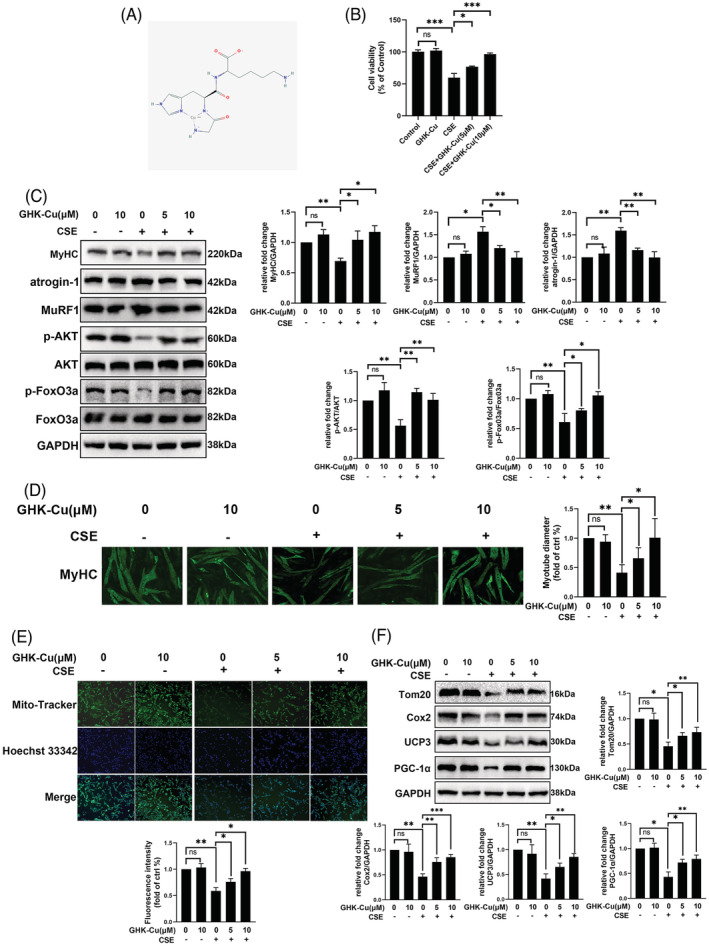
GHK‐Cu suppressed CSE‐induced C2C12 myotube injury alteration. (A) The structure of GHK‐Cu. (B) Cell viability of C2C12 myotubes treated with CSE and different concentrations of GHK‐Cu. (C) Western blot analysis of MyHC, atrogin‐1, MuRF1, AKT/FoxO3a levels in C2C12 myotubes, and relative intensity normalized to the expression of GAPDH. (D) Immunofluorescence staining showed that CSE‐treated myotubes had significantly smaller diameters compared with untreated myotubes. Meanwhile, GHK‐Cu treatment elevated diameters of myotubes compared with CSE‐treated myotubes in a concentration dependent manner (E)MitoTracker Green staining to assess the mitochondrial content of C2C12 myotubes. (F) Western blot analysis of Tom20, Cox2, ucp3, and PGC‐1α levels in C2C12 myotubes, and relative intensity normalized to the expression of GAPDH.
Smoking‐induced skeletal muscle dysfunction is accompanied by mitochondrial loss and dysfunction. 20 Therefore, we try to determine the role of GHK‐Cu in the regulation of mitochondrial function. As shown in Figure 2E, CSE obviously decreased the content of mitochondrial in C2C12 myotubes; and GHK‐Cu treatment elevated mitochondrial content in a concentration dependent manner. In additional, MitoSOX staining experiment to explore the effect of GHK‐Cu on the mitochondrial oxidative stress of CSE‐treated C2C12. As shown in Figure S1, CSE injury drove C2C12 cells to produce excessive mitochondrial oxidative stress as demonstrated via MitoSOX staining, and GHK‐Cu treatment could reduce mitochondrial oxidative stress level in a concentration‐dependent manner. Finally, GHK‐Cu could rescue CSE induced down‐regulation of mitochondrial markers (Tom20, Cox2, and UCP3) and upstream regulatory molecules PCG‐1α (Figure 2F). These results suggested that GHK‐Cu could enhanced mitochondrial content and function in CSE‐treated C2C12 myotubes.
GHK‐Cu attenuates oxidative stress in CSE‐stimulated C2C12 myoblast
Oxidative stress plays an important role in the pathophysiology of COPD and sarcopenia. 21 Studies have shown the association between oxidative stress and severity of COPD leading to medical co‐morbidities and skeletal muscle dysfunction. 22 , 23 Firstly, the capacity to resist oxidative stress, including GSH, SOD2, MDA, and T‐AOC, were evaluated. When CSE was applied to C2C12 cells, the MDA level was increased, and the levels of T‐AOC and GSH were decreased, indicating that CSE stimulates an increase in oxidative stress in C2C12 cells (Figure 3A–D). And GHK‐Cu concentration‐dependently inhibited oxidative stress in CSE‐treated C2C12 myotubes. Finally, GHK‐Cu could rescue CSE‐induced down‐regulation of antioxidative stress markers (Nrf2, SOD2, and HO‐1) (Figure 3E).
Figure 3.
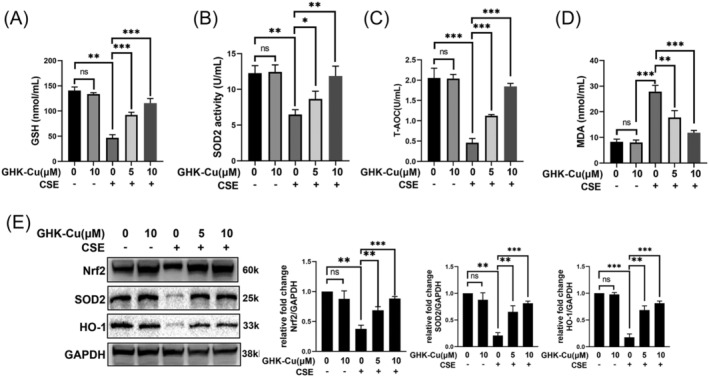
GHK‐Cu attenuates oxidative stress in CSE‐stimulated C2C12 myoblast. The levels of GSH (A), SOD2 (B), T‐AOC (C), and MDA (D) in C2C12 myotubes were detected. (E) Expression levels of Nrf2, SOD2, and HO‐1 in C2C12 myotubes were detected using western blot analysis, and relative intensity normalized to the expression of GAPDH. ns, no significance; *P < 0.05; **P < 0.01; ***P < 0.001.
These results suggested that GHK‐Cu could alleviated the oxidative stress level in CSE‐treated C2C12 myotubes.
GHK‐Cu protects mice against cigarette smoking‐induced skeletal muscle dysfunction
To further validate the therapeutic effect of GHK‐Cu on skeletal muscle dysfunction, we constructed a cigarette smoking exposed mouse model (Figure 4A). After completing 12 weeks of smoke exposure, we first compared the body weights of the mice. As shown in Figure 4B, mice in the smoke exposure group had significantly lower body weight, whereas both high‐dose and low‐dose GHK‐Cu treatments significantly attenuated CS‐induced body weight loss. Further grip strength tests showed that CS significantly reduced muscle strength in mice, which was reversed by high‐dose GHK‐Cu treatment (Figure 4B). Next, we dissected the muscles of the hind legs and weighed them. GHK‐Cu treatment significantly reduced CS‐induced muscle mass loss in mice including Quad (quadriceps), Gast (gastrocnemius), soleus, TA (tibialis anterior), sol (soleus), and EDL (extensor digitorum longus) (Figures 4C and S2A–C). Haematoxylin and eosin (HE) staining of the muscles was performed to assess the effect of GHK‐Cu treatment on muscle morphology. The muscle fibre cross‐sectional area (CSA) of Gast (Figure 4D) and Quad (Figure S2D) were significantly lower in CS‐exposed mice than in controls. GHK‐Cu treatment significantly prevented the CS‐induced reduction in muscle cross‐sectional area. In addition, western blot analysis showed that GHK‐Cu could reverse the CS‐induced elevation of atrogin‐1 and MuRF1 expression. In addition, GHK‐Cu also increased PGC‐1α expression and increased Nrf2 expression in mouse muscle (Figure 4E). Finally, the activities of T‐AOC and antioxidant enzymes (GSH, SOD2, and T‐AOC) in the muscles in the CS group were decreased but partially increased after GHK‐Cu treatment. Correspondingly, the elevated MDA content in the CS group in muscles followed a downward trend after administration of GHK‐Cu (Figure 4F). Taken together, GHK‐Cu would be a potential therapy CS‐induced skeletal muscle dysfunction.
Figure 4.
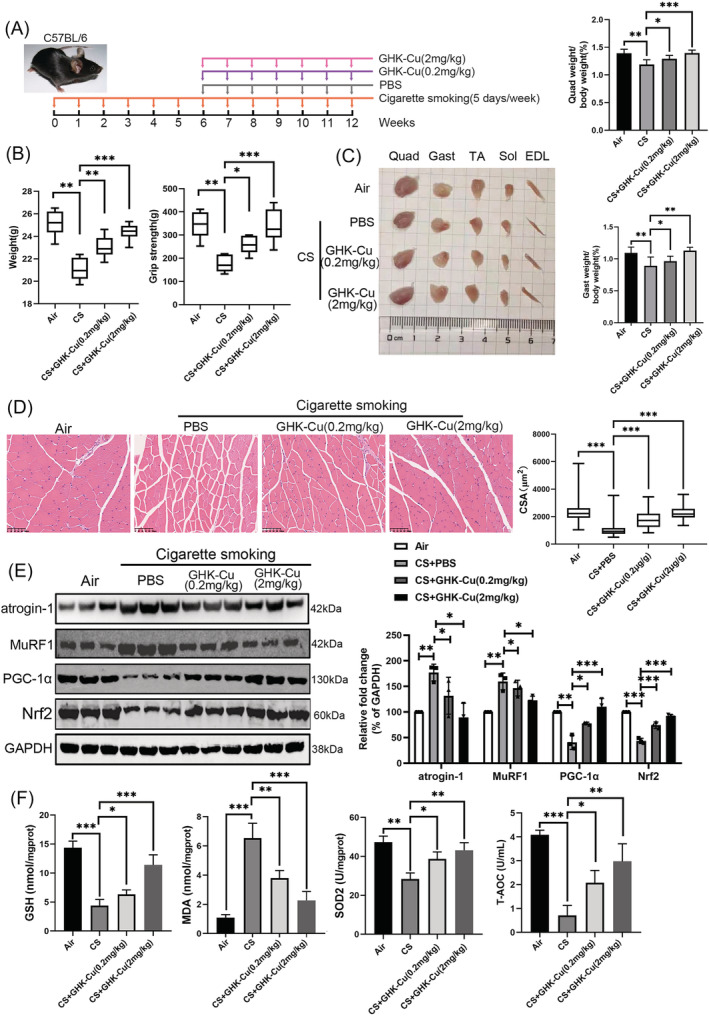
GHK‐Cu protects against CS‐induced muscle dysfunction in mice. (A) Schematic diagram of the intraperitoneal administration of GHK‐Cu (GHK‐Cu; 0.2 mg/kg or 2 mg/kg body weight every week, 7 weeks; n = 8), or vehicle (PBS every week, 7 weeks; n = 8) in CS‐exposed mice model. (B) Body weight (left) and grip strength (right) in each group. (C) Comparison of representative samples of dissected skeletal muscle (left), including Quad (quadriceps), Gast (gastrocnemius), soleus, TA (tibialis anterior), sol (soleus), and EDL (extensor digitorum longus); the ratios of Gast and Quad muscle weight to body weight (right). (D) Representative H&E staining of myofibre cross‐section of Gast (left), and the muscle cross‐sectional area of Gast muscle fibre (right). (E) Western blot analysis of atrogin‐1, MuRF1, PGC‐1α, and Nrf2 levels in mouse Gast muscle. (F) The levels of GSH, MDA, SOD2, and T‐AOC in mouse Gast muscle; *P < 0.05; **P < 0.01; ***P < 0.001.
GHK‐Cu could activate SIRT1 and potential bound it
We next used network pharmacology and molecular docking to explore the potential mechanism of action of GHK‐Cu (Figure 5A). Fifty‐one potential targets of GHK‐Cu were obtained from SwissTargetPrediction and BATMAN website. PPI analysis was carried out for selecting hub protein among all target proteins (Figure 5B). Among them, SIRT1 caught our attention due to its plays a critical role in muscle function. 24 To evaluate if GHK‐Cu could bind to SIRT1, virtual docking analysis was conducted (Figure 5C). A total of nine different conformations of the complex were generated from the software. After ranking the conformations by Gibbs free energy, the first rank was regarded as the optimal docking result and was selected for downstream analysis. The binding energy of the complex was −6.1 kcal/mol. GHK‐Cu binds to the pocket of the SIRT1 protein surrounded by amino acids PRO419, VAL412, GLY364, ALA367, ILE360, GLN421, GLU420, GLU410, THR368, and SER365, where it forms hydrogen bonds with GLN421, GLU420, GLU410, THR368, and SER365. Furthermore, western blot and the fluorometric SIRT1 assay was performed on GHK‐Cu‐treated cells to evaluate whether GHK‐Cu affected SIRT1 expression and deacetylase activity. As results in Figure 5D, E, CSE suppressed SIRT1 expression and deacetylase activity; GHK‐Cu dose‐dependently increased SIRT1 expression and deacetylase activity in CSE‐treated myotubes. Thus, GHK‐Cu could activate SIRT1 and potential bound it in CSE‐treated myotubes.
Figure 5.
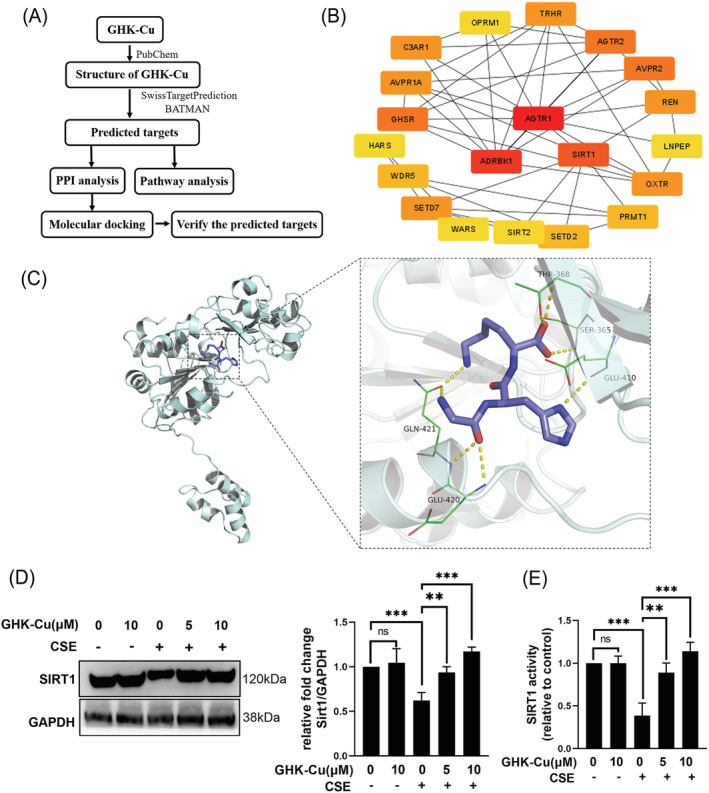
GHK‐Cu directly bound and activated SIRT1. (A) Process of target screening. (B) Interaction of potential targets. (C) Interactive sites between GHK‐Cu and SIRT1 by docking analysis. (D) GHK‐Cu increased SIRT1 expression dose‐dependently in CSE‐treated C2C12 myotubes. (E) GHK‐Cu enhanced SIRT1 deacetylase activity in C2C12 myotubes. ns, no significance; *P < 0.05; **P < 0.01; ***P < 0.001.
GHK‐Cu protects C2C12 myotubes against CSE‐induced damage via activating SIRT1
To verify whether the protective effect of GHK‐Cu against CSE‐induced myotube atrophy is mediated through activating SIRT1, C2C12 myotubes were treated with a SIRT1 specific inhibitor, EX527, together with GHK‐Cu. CCK8 cell viability assay (Figure 6A) show that co‐treated with EX527, the protective effect in GHK‐Cu‐treated myotubes were almost abolished. Immunofluorescence staining showed that co‐treated with EX527, the protective effect of myotubes diameters in GHK‐Cu‐treated myotubes were almost abolished (Figure S3). As we know SIRT1 rescued muscle atrophy by blocking the activation of FoxO3a. 25 Our further experiments likewise formalized this: the addition of EX527 caused GHK‐Cu to lose its positive regulatory effect on the AKT/FoxO3a pathway modulation (Figure 6B). FoxO3a involved in the transcriptional regulation of atrogin‐1, MuRF1. And SIRT1 could directly deacetylates FoxO3a. 25 As shown in Figure 6C, GHK‐Cu completely reversed the CSE‐induced increase in total and acetylated FoxO3a levels; whereas co‐treatment with EX‐527 attenuated the effect of GHK‐Cu. Therefore, GHK‐Cu mitigated CSE‐induced muscle atrophy in C2C12 myotubes via the SIRT1‐Akt‐FoxO3a axis.
Figure 6.
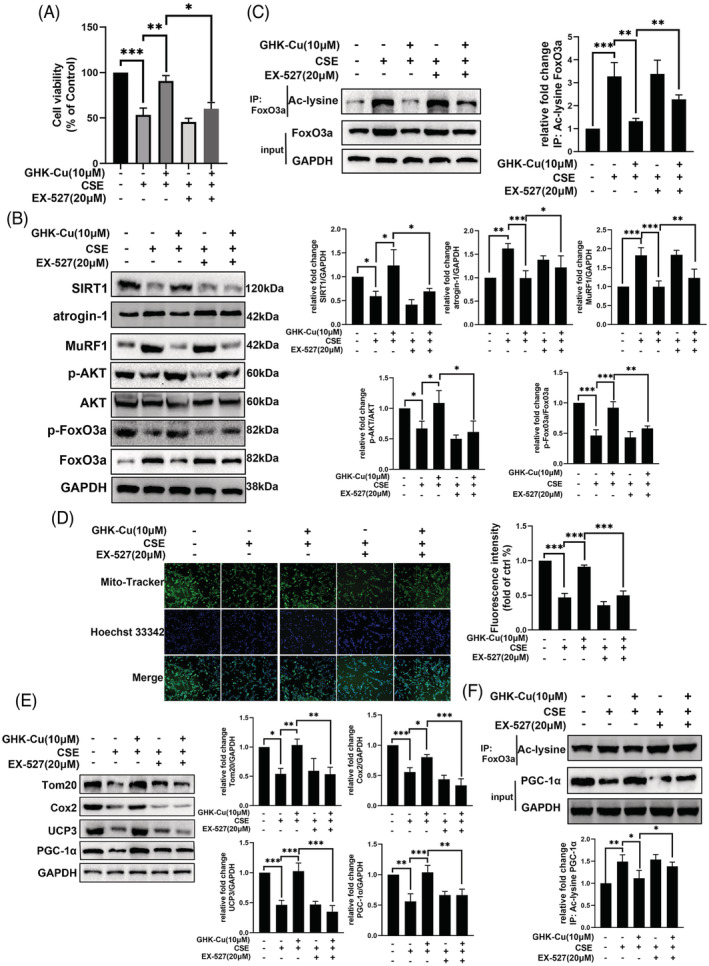
GHK‐Cu protects C2C12 myotubes against CSE‐induced damage via activating SIRT1. (A) Cell viability of C2C12 myotubes treated with CSE, GHK‐Cu or/and SIRT1 specific inhibitor EX527 show that co‐treated with EX527, the protective effect in GHK‐Cu‐treated myotubes were almost abolished. (B) Combination of GHK‐Cu with EX‐527 almost completely blocked the regulatory effect of CSE on AKT/FoxO3a pathway, atrogin‐1 and MuRF1. (C) GHK‐Cu reversed CSE‐induced increases of total and acetylated FoxO3a levels, which were abolished by co‐treatment of EX‐527. (D) MitoTracker Green staining to assess the mitochondrial content of C2C12 myotubes show that the protective effect of GHK‐Cu on mitochondrial function was reversed after application of EX527 in CSE‐treated C2C12 myotubes. (E)co‐treatment of EX‐527 abolished the effect of GHK‐Cu on expression of mitochondrial‐associated proteins (Tom20, UCP3, and PCG‐1α). (F) GHK‐Cu reversed CSE‐induced decrease of PGC‐1α expression and increase of acetylated PGC‐1α level, which were abolished by co‐treatment of EX‐527.
Next, we try to explore whether the regulation of GHK‐Cu to mitochondrial function is mediated through activating SIRT1. The protective effect of GHK‐Cu on mitochondrial function was reversed after application of EX527 in CSE‐treated C2C12 myotubes (Figure 6D). Western blot results similarly confirmed this effect: the up‐regulation of mitochondrial markers (Tom20 and UCP3) and PCG‐1α was dismissed in C2C12 myotubes exposed to CSE after the combination of GHK‐Cu and EX527 (Figure 6E). As we know, 26 PGC‐1α is one of the deacetylated substrates of SIRT1 and the deacetylated PGC‐1α is the bioactivated form. The results of immunoprecipitation showed that GHK‐Cu treatment reversed the CSE‐induced elevation of acetylated PGC‐1α levels, and the treatment of GHK‐Cu and EX‐527 almost blocked the effect of GHK‐Cu on total and acetylated PGC‐1α expression (Figure 6F). Overall, these results suggest that GHK‐Cu regulates mitochondrial function via SIRT1 pathway.
GHK‐Cu attenuates oxidative stress in CSE‐stimulated C2C12 myoblast via activating SIRT1
We tried to clarify whether the regulatory effect of GHK‐Cu on oxidative stress in C2C12 cells is dependent on SIRT‐1. The alleviative effect of GHK‐Cu on oxidative stress was reversed after application of EX527 in CSE‐treated C2C12 myotubes (Figure 7A–D). Western blot results similarly confirmed this effect: the up‐regulation of antioxidative stress markers (Nrf2, SOD2, and HO‐1) was dismissed in C2C12 myotubes exposed to CSE after the combination of GHK‐Cu and EX527 (Figure 7E). Sirtuins are known to deacetylate Nrf2 and contribute to its action of reducing ROS by generation of anti‐oxidant enzymes. 27 In Figure 7F, GHK‐Cu treatment reversed the CSE‐induced elevation of acetylated Nrf2 levels, and the treatment of GHK‐Cu and EX‐527 almost blocked the effect of GHK‐Cu on total and acetylated Nrf2 expression.
Figure 7.
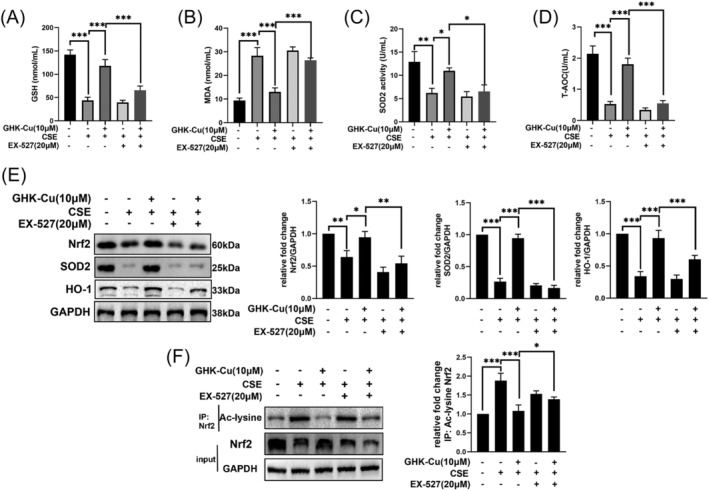
GHK‐Cu attenuates oxidative stress in CSE‐stimulated C2C12 myoblast via activating SIRT1. The alleviative effect of GHK‐Cu on oxidative stress (contained GSH (A), SOD2 (B), T‐AOC (C), and MDA (D) levels) was reversed after application of EX527 in CSE‐treated C2C12 myotubes (upper). (E) Co‐treatment of EX‐527 abolished the effect of GHK‐Cu on expression of oxidative stress‐related proteins (lower). (F) GHK‐Cu reversed CSE‐induced decrease of total and increase of acetylated Nrf2 levels, which were abolished by co‐treatment of EX‐527. ns, no significance; *P < 0.05; **P < 0.01; ***P < 0.001.
These results suggest that GHK‐Cu could alleviated the oxidative stress level in CSE‐treated C2C12 myotubes is dependent on SIRT‐1.
GHK‐Cu protects mice against CS‐induced skeletal muscle dysfunction via SIRT1
To further verify the regulatory role of GHK‐Cu and SIRT1, we co‐administered GHK‐Cu and SIRT1 inhibitor (EX527) in mouse model (Figure 8A). In Figure 8B, co‐treatment of EX527 abolished the protective effects of GHK‐Cu on body weight, and muscle strength in CS‐exposed mice. In Figures 8C and S4A–C, co‐treatment of EX527 abolished the protective effects of GHK‐Cu on muscle (including Quad, Gast, TA, Sol, and EDL.) mass loss in CS‐exposed mice. Next, we dissected the muscles of the hind legs and performed HE staining. By comparing muscle mass and muscle fibre cross‐sectional area of Quad and Gast (Figures 8D and S4D), we found that EX527 treatment blocked the protective effect of GHK‐Cu against muscle atrophy (muscle mass and muscle fibre cross‐sectional area). Co‐treatment of EX527 with GHK‐Cu almost hindered the changes of atrogin‐1, MuRF1, PGC‐1α, and Nrf2 in CS‐exposed mouse skeletal muscle tissues by GHK‐Cu (Figure 8E). Finally, EX527 treatment blocked the protective effect of GHK‐Cu against oxidative stress via decreased T‐AOC, GSH, SOD2 level and increased MDA content (Figure 8F). Overall, these results suggest that GHK‐Cu could protects mice against CS‐induced skeletal muscle dysfunction via SIRT1.
Figure 8.
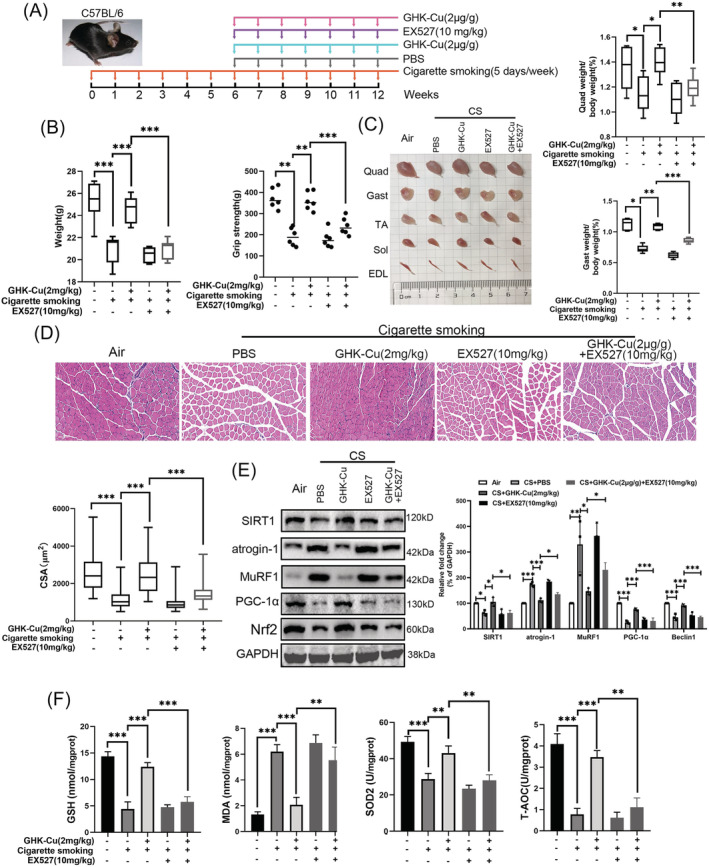
GHK‐Cu protects mice against CS‐induced skeletal muscle dysfunction via SIRT1. (A) Schematic diagram of the control group (exposed to room air), and other four groups of mice exposure to cigarette smoking (5 days/week, 12 weeks) with PBS (every week, 7 weeks; n = 8), high dosage of GHK‐Cu (2 mg/kg body weight every week, 7 weeks; n = 8), EX527 solution (10 mg/kg body weight every week, 7 weeks; n = 8), and high dosage of GHK‐Cu with EX527 solution. (B) The difference of body weight (left) and grip strength (right) between each group. (C) The representative samples of dissected skeletal muscle (left), including Quad (quadriceps), Gast (gastrocnemius), soleus, TA (tibialis anterior), sol (soleus), and EDL (extensor digitorum longus), and the difference of the ratios of Gast and Quad muscle weight to body weight (right). (D) The representative H&E staining of myofibre cross‐section of Gast (upper), and the difference of muscle cross‐sectional area of Gast muscle fibre between each group (lower). (E) Western blot analysis showed the expression of atrogin‐1, MuRF1, PGC‐1α, and Nrf2 levels in mouse Gast muscle; (F) The levels of GSH, MDA, SOD2, and T‐AOC in mouse Gast muscle between each group; *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
Skeletal muscle dysfunction is present in approximately one‐third of patients with COPD and is associated with poor clinical outcomes. 3 Although a growing body of evidence reveals the mechanisms underlying co‐morbid skeletal muscle dysfunction in patients with COPD, only a small number of therapeutic agents are available, such as supplementation with androgens that promote muscle protein synthesis and antioxidants that improve oxidative damage. 28 There are more preclinical studies currently underway. However, there is still a lack of well‐documented effective drugs for clinical treatment. In this study, we firstly reported the potential role of plasma GHK in skeletal muscle dysfunction in patients with COPD. Moreover, exogenous administration of GHK‐Cu could alleviate CS‐induced skeletal muscle dysfunction.
Our result showed that plasma GHK level in patients with COPD was lower than those in age paired healthy control. Importantly, we also found plasma GHK level was significantly associated with skeletal muscle mass, inflammatory factor TNF‐α, and antioxidative stress factor SOD2. TNF‐α level has been reported was significantly associated with grip strength and skeletal muscle mass, which are important determinants of sarcopenia in patients with stable COPD. 3 And the increase in oxidative stress‐related factors (including SOD2) are associated with COPD‐related sarcopenia. 29 Overall, we found that plasma GHK levels were significantly reduced in patients with COPD. And GHK level was negatively correlated with skeletal muscle mass, which possibly related to inflammatory factor and oxidative stress component. Previous study 30 has identified GHK as a compound that can reverse the gene‐expression signature associated with emphysematous destruction and induce expression patterns consistent with TGFβ pathway activation. In this study, our results add new clinical evidence for the protective effects of GHK to skeletal muscle in patients with COPD.
The maintenance of skeletal muscle function is based on the dynamic balance of muscle protein synthesis and degradation. Excessive protein degradation is a basic feature of skeletal muscle dysfunction, and the ubiquitin‐proteasome system (UPS) is the main mediating pathway. 31 FoxO transcription factors could be dephosphorylated and/or acetylated to induce nuclear translocation, which enhances the expression of E3 ubiquitin ligases, MuRF1 and atrogin‐1, and leads to muscle atrophy. In this study, we found that GHK‐Cu could against cigarette smoking‐induced muscle protein degradation via AKT/FoxO3a pathway. Previous study 32 indicated that the disease severity was significantly related to the loss of mitochondrial content based on the results of quadriceps muscle biopsies from COPD patients. In this study, we found CSE obviously decreased the content of mitochondrial in CSE‐treated C2C12 myotubes and GHK‐Cu treatment elevated mitochondrial content. Meanwhile, GHK‐Cu could upregulate PCG‐1α. PGC‐1α could directly interacts with the nuclear respiratory factor 1 to translocate to the mitochondria and activate mitochondrial DNA replication, resulting in mitochondrial biogenesis. Therefore, GHK‐Cu could enhance mitochondrial content and function. Oxidative stress is an important contributor to both the lung and systemic pathologies of COPD. The elevated levels of oxidative damage observed in the muscle of COPD patients and the link between oxidative stress and muscle strength. 33 Our results indicated that GHK‐Cu could alleviating the oxidative stress level via Nrf2/HO‐1 pathway. Overall, these results suggest that GHK‐Cu would may be a candidate therapy for skeletal muscle dysfunction of patients with COPD.
Another finding of our study was that GHK could directly bound and activated SIRT1 to protect mice against cigarette smoking‐induced skeletal muscle dysfunction. SIRT1 is highly involved in regulating skeletal muscle remodelling, which directly deacetylates and activates PGC‐1α to regulate mitochondrial biogenesis, 34 modulates FoxOs transcriptional activity to control loss of muscle mass, 25 and affects NRF2 to inhibit oxidative damage. 27 Therefore, activation of SIRT1 is a potential therapeutic approach to prevent and/or rescue muscle dysfunctional wasting. In this study, we demonstrated that GHK‐Cu could directly bound and activated SIRT1 in C2C12 cells based on the results of molecular docking, and SIRT1 activity assay. Moreover, the protective effect of GHK‐Cu to against CS‐induced skeletal muscle dysfunction was dependent on the expression of SIRT1.
Previous study 35 showed that GHK could alleviates astrocytes injury of intracerebral haemorrhage by regulating Akt/miR‐146a‐3p/AQP4 pathway. Another study 36 from our team indicated that GHK‐Cu presented a protective effect by suppressing TGFβ1/Smad2/3 pathway in pulmonary fibrosis. The direct target of GHK remains unclear. Only one study 37 demonstrated GHK could interacts with AT1 receptors (angiotensin type 1 receptor) in rat hepatocytes. We demonstrated for the first time that GHK‐Cu functions by directly regulating the expression and activity of SIRT1. Thus, GHK‐Cu is identified as a novel SIRT1 activator.
In this study, we found GHK could modulate the expression and activity of SIRT1, we speculated that GHK may regulate the expression of SIRT1 through protein interaction. Peptide‐mediated interactions are gaining increased attention due to their predominant roles in the many regulatory processes that involve dynamic interactions between proteins. 38 , 39 Therefore, as an active tripeptide, GHK may regulated the expression of SIRT1 via peptide‐mediated interactions. In additional, the main limitation of this study was that the regulation mechanism of GHK and SIRT1 expression and activity was not deeply explored. In the future, we will apply more novel technologies (e.g., constructing SIRT1 mutant plasmid) to explore the mechanism of GHK and SIRT1.
There are some limitations to this study that need to be considered. Firstly, due to the limited sample size, we were not able to analyse the difference of plasma GHK level between patients with or without sarcopenia. Therefore, the clinical value of GHK still requires further comprehensive and in‐depth analyses. In additional, we used the SIRT1‐specific inhibitor EX527 in this study rather than muscle‐specific SIRT1 knockout mice to interfere with SIRT1 expression and activity is another important limitation.
Conclusions
Plasma GHK level in patients with COPD was significantly decreased and was significantly associated with skeletal muscle mass. Moreover, exogenous administration of GHK‐Cu could execute the preventive function against CS‐induced skeletal muscle dysfunction by reducing muscle protein degradation, promoting mitochondrial biogenesis, and alleviating the oxidative stress level via directly bound and activated SIRT1.
Funding
This research was supported by National High Level Hospital Clinical Research Funding (2022‐NHLHCRF‐LX‐01), the Elite Medical Professionals Project of China‐Japan Friendship Hospital (No. ZRJY2021‐BJ08), the Nonprofit Central Research Institute Fund of Chinese Academy of Medical Sciences (No. 2020‐PT320‐001), the National Natural Science Foundation of China (No. 81900040), the Liaoning Education Ministry Supporting Foundation (No. QN2019014), and the Liaoning Science and Technology Ministry Supporting Foundation (No. 2019‐ZD‐0766).
Conflict of interest
All authors declare that they have no conflict of interest.
Supporting information
Figure S1. CSE injury drove C2C12 cells to produce excessive mitochondrial oxidative stress as demonstrated via MitoSOX staining, and GHK‐Cu treatment could reduce mitochondrial oxidative stress level in a concentration‐dependent manner.
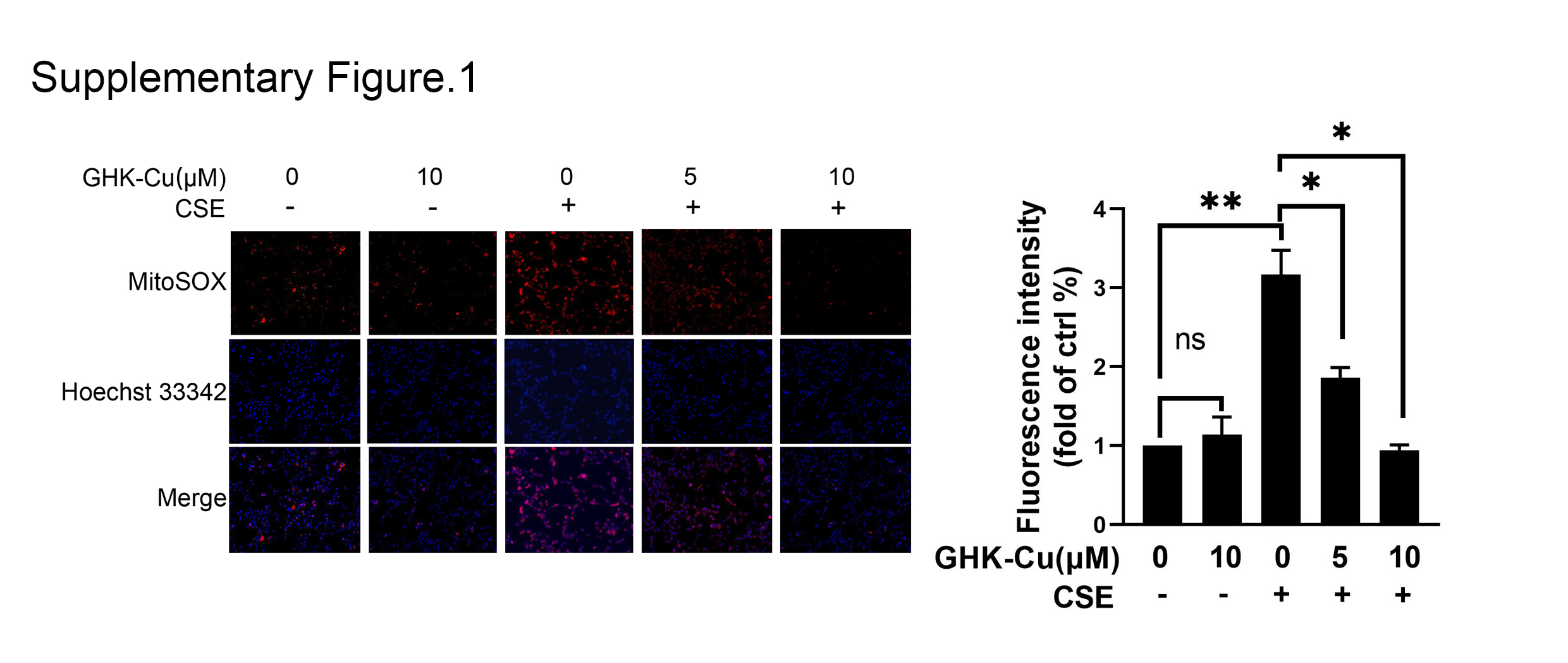{kind=link}
Figure S2. The ratios of TA (A), soleus (B) and EDL (C) muscle to body weight. The difference of muscle cross sectional area of Quad muscle fibre between each group(D). *p < 0.05; **p < 0.01; ***p < 0.001.
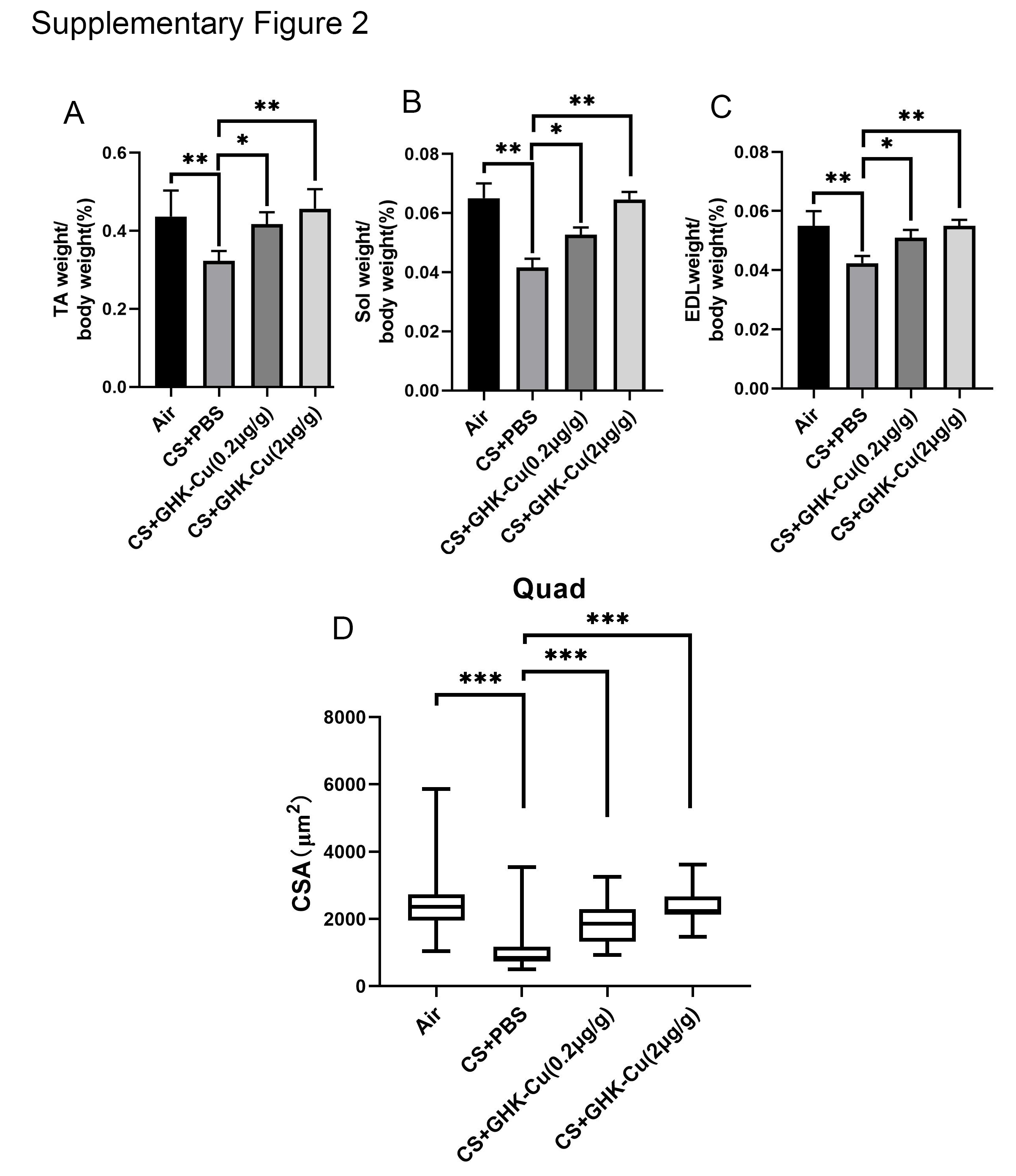{kind=link}
Figure S3. Images of immunofluorescent staining of MyHC in eacg groups. *p < 0.05; **p < 0.01; ***p < 0.001.
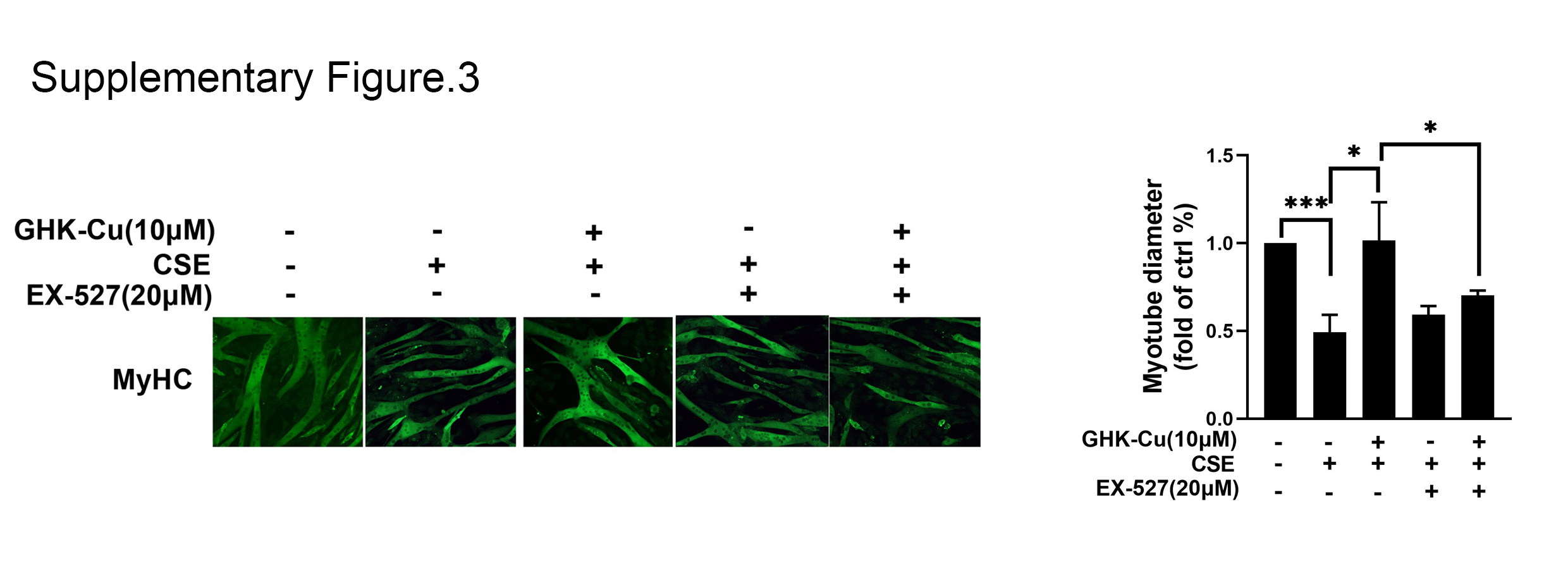{kind=link}
Figure S4. The ratios of soleus (A), TA (B) and EDL (C) muscle to body weight. The difference of muscle cross sectional area of Quad muscle fibre between each group(D). *p < 0.05; **p < 0.01; ***p < 0.001.
{kind=link}
Table S1. Basic characteristics of the study population
Acknowledgements
The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. 40 We would like to thank Jia Liu (Shanghai Institute of Materia Medica, Chinese Academy of Sciences) for helpful assistance with the measurement of plasma GHK levels.
Deng M., Zhang Q., Yan L., Bian Y., Li R., Gao J., et al (2023) Glycyl‐l‐histidyl‐l‐lysine‐Cu2+ rescues cigarette smoking‐induced skeletal muscle dysfunction via a sirtuin 1‐dependent pathway, Journal of Cachexia, Sarcopenia and Muscle, 14, 1365–1380, 10.1002/jcsm.13213
References
- 1. Rabe KF, Watz H. Chronic obstructive pulmonary disease. Lancet (London, England) 2017;389:1931–1940. [DOI] [PubMed] [Google Scholar]
- 2. Vanfleteren LEGW, Spruit MA, Wouters EFM, Franssen FME. Management of chronic obstructive pulmonary disease beyond the lungs. Lancet Respir Med 2016;4:911–924. [DOI] [PubMed] [Google Scholar]
- 3. Byun MK, Cho EN, Chang J, Ahn CM, Kim HJ. Sarcopenia correlates with systemic inflammation in COPD. Int J Chron Obstruct Pulmon Dis 2017;12:669–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Limpawattana P, Inthasuwan P, Putraveephong S, Boonsawat W, Theerakulpisut D, Sawanyawisuth K. Sarcopenia in chronic obstructive pulmonary disease: A study of prevalence and associated factors in the Southeast Asian population. Chron Respir Dis 2018;15:250–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barnes PJ. Oxidative stress‐based therapeutics in COPD. Redox Biol 2020;33:101544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mathur S, Brooks D, Carvalho CRF. Structural alterations of skeletal muscle in copd. Front Physiol 2014;5:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sepúlveda‐Loyola W, de Castro LA, Matsumoto AK, Camillo CA, Barbosa DS, Galvan CCR, et al. NOVEL antioxidant and oxidant biomarkers related to sarcopenia in COPD. Heart Lung 2021;50:184–191. [DOI] [PubMed] [Google Scholar]
- 8. Abrigo J, Simon F, Cabrera D, Vilos C, Cabello‐Verrugio C. Mitochondrial dysfunction in skeletal muscle pathologies. Curr Protein Pept Sci 2019;20:536–546. [DOI] [PubMed] [Google Scholar]
- 9. Pani A, Porta C, Cosmai L, Melis P, Floris M, Piras D, et al. Glomerular diseases and cancer: evaluation of underlying malignancy. J Nephrol 2016;29:143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maquart FX, Pickart L, Laurent M, Gillery P, Monboisse JC, Borel JP. Stimulation of collagen synthesis in fibroblast cultures by the tripeptide‐copper complex glycyl‐L‐histidyl‐L‐lysine‐Cu2+. FEBS Lett 1988;238:343–346. [DOI] [PubMed] [Google Scholar]
- 11. Pohunková H, Stehlík J, Váchal J, Cech O, Adam M. Morphological features of bone healing under the effect of collagen‐graft‐glycosaminoglycan copolymer supplemented with the tripeptide Gly‐His‐Lys. Biomaterials 1996;17:1567–1574. [DOI] [PubMed] [Google Scholar]
- 12. Pickart L, Vasquez‐Soltero JM, Margolina A. The human tripeptide GHK‐Cu in prevention of oxidative stress and degenerative conditions of aging: implications for cognitive health. Oxid Med Cell Longev 2012;2012:324832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhou X‐M, Wang G‐L, Wang X‐B, Liu L, Zhang Q, Yin Y, et al. GHK peptide inhibits bleomycin‐induced pulmonary fibrosis in mice by suppressing TGFβ1/Smad‐mediated epithelial‐to‐mesenchymal transition. Front Pharmacol 2017;8:904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Park J‐R, Lee H, Kim S‐I, Yang S‐R. The tri‐peptide GHK‐Cu complex ameliorates lipopolysaccharide‐induced acute lung injury in mice. Oncotarget 2016;7:58405–58417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Endo T, Miyagi M, Ujiie A. Simultaneous determination of glycyl‐L‐histidyl‐L‐lysine and its metabolite, L‐histidyl‐L‐lysine, in rat plasma by high‐performance liquid chromatography with post‐column derivatization. J Chromatogr B Biomed Sci Appl 1997;692:37–42. [DOI] [PubMed] [Google Scholar]
- 16. Chen L, Luo L, Kang N, He X, Li T, Chen Y. The Protective effect of HBO1 on cigarette smoke extract‐induced apoptosis in airway epithelial cells. Int J Chron Obstruct Pulmon Dis 2020;15:15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Deng M, Liu B, Zhang Z, Chen Y, Wang Y, Wang X, et al. Loss of G‐protein‐signaling modulator 2 accelerates proliferation of lung adenocarcinoma via EGFR signaling pathway. Int J Biochem Cell Biol 2020;122:105716. [DOI] [PubMed] [Google Scholar]
- 18. Xiong J, Le Y, Rao Y, Zhou L, Hu Y, Guo S, et al. RANKL mediates muscle atrophy and dysfunction in a cigarette smoke‐induced model of chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 2021;64:617–628. [DOI] [PubMed] [Google Scholar]
- 19. Schiaffino S, Dyar KA, Ciciliot S, Blaauw B, Sandri M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J 2013;280:4294–4314. [DOI] [PubMed] [Google Scholar]
- 20. Puente‐Maestu L, Pérez‐Parra J, Godoy R, Moreno N, Tejedor A, González‐Aragoneses F, et al. Abnormal mitochondrial function in locomotor and respiratory muscles of COPD patients. Eur Respir J 2009;33:1045–1052. [DOI] [PubMed] [Google Scholar]
- 21. Singh S, Verma SK, Kumar S, Ahmad MK, Nischal A, Singh SK, et al. Evaluation of oxidative stress and antioxidant status in chronic obstructive pulmonary disease. Scand J Immunol 2017;85:130–137. [DOI] [PubMed] [Google Scholar]
- 22. Ben Anes A, Fetoui H, Bchir S, Ben Nasr H, Chahdoura H, Chabchoub E, et al. Increased oxidative stress and altered levels of nitric oxide and peroxynitrite in Tunisian patients with chronic obstructive pulmonary disease: correlation with disease severity and airflow obstruction. Biol Trace Elem Res 2014;161:20–31. [DOI] [PubMed] [Google Scholar]
- 23. Sepúlveda Loyola WA, Vilaça Cavallari Machado F, Araújo de Castro L, Hissnauer Leal Baltus T, Rampazzo Morelli N, Landucci Bonifácio K, et al. Is oxidative stress associated with disease severity, pulmonary function and metabolic syndrome in chronic obstructive pulmonary disease? Rev Clin Esp (Barc) 2019;219:477–484. [DOI] [PubMed] [Google Scholar]
- 24. Kuno A, Horio Y. SIRT1: a novel target for the treatment of muscular dystrophies. Oxid Med Cell Longev 2016;2016:6714686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee D, Goldberg AL. SIRT1 protein, by blocking the activities of transcription factors FoxO1 and FoxO3, inhibits muscle atrophy and promotes muscle growth. J Biol Chem 2013;288:30515–30526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cantó C, Gerhart‐Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009;458:1056–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Singh V, Ubaid S. Role of silent information regulator 1 (SIRT1) in regulating oxidative stress and inflammation. Inflammation 2020;43:1589–1598. [DOI] [PubMed] [Google Scholar]
- 28. Simoes DCM, Vogiatzis I. Can muscle protein metabolism be specifically targeted by exercise training in COPD? J Thorac Dis 2018;10:S1367–S1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lage VKS, de Paula FA, Dos Santos JM, Costa HS, da Silva GP, Lima LP, et al. Are oxidative stress biomarkers and respiratory muscles strength associated with COPD‐related sarcopenia in older adults? Exp Gerontol 2022;157:111630. [DOI] [PubMed] [Google Scholar]
- 30. Campbell JD, McDonough JE, Zeskind JE, Hackett TL, Pechkovsky DV, Brandsma C‐A, et al. A gene expression signature of emphysema‐related lung destruction and its reversal by the tripeptide GHK. Genome Med 2012;4:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sartori R, Romanello V, Sandri M. Mechanisms of muscle atrophy and hypertrophy: implications in health and disease. Nat Commun 2021;12:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Leermakers PA, Schols AMWJ, Kneppers AEM, Kelders MCJM, de Theije CC, Lainscak M, et al. Molecular signalling towards mitochondrial breakdown is enhanced in skeletal muscle of patients with chronic obstructive pulmonary disease (COPD). Sci Rep 2018;8:15007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Passey SL, Hansen MJ, Bozinovski S, McDonald CF, Holland AE, Vlahos R. Emerging therapies for the treatment of skeletal muscle wasting in chronic obstructive pulmonary disease. Pharmacol Ther 2016;166:56–70. [DOI] [PubMed] [Google Scholar]
- 34. Gurd BJ. Deacetylation of PGC‐1α by SIRT1: importance for skeletal muscle function and exercise‐induced mitochondrial biogenesis. Appl Physiol Nutr Metab 2011;36:589–597. [DOI] [PubMed] [Google Scholar]
- 35. Zhang H, Wang Y, Lian L, Zhang C, He Z. Glycine‐histidine‐lysine (GHK) alleviates astrocytes injury of intracerebral hemorrhage via the Akt/miR‐146a‐3p/AQP4 Pathway. Front Neurosci 2020;14:576389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ma W‐H, Li M, Ma H‐F, Li W, Liu L, Yin Y, et al. Protective effects of GHK‐Cu in bleomycin‐induced pulmonary fibrosis via anti‐oxidative stress and anti‐inflammation pathways. Life Sci 2020;241:117139. [DOI] [PubMed] [Google Scholar]
- 37. García‐Sáinz JA, Olivares‐Reyes JA. Glycyl‐histidyl‐lysine interacts with the angiotensin II AT1 receptor. Peptides 1995;16:1203–1207. [DOI] [PubMed] [Google Scholar]
- 38. Lin J, Wang S, Wen L, Ye H, Shang S, Li J, et al. Targeting peptide‐mediated interactions in omics. Proteomics 2022;e2200175. [DOI] [PubMed] [Google Scholar]
- 39. London N, Raveh B, Schueler‐Furman O. Peptide docking and structure‐based characterization of peptide binding: from knowledge to know‐how. Curr Opin Struct Biol 2013;23:894–902. [DOI] [PubMed] [Google Scholar]
- 40. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2021. J Cachexia Sarcopenia Muscle 2021;12:2259–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. CSE injury drove C2C12 cells to produce excessive mitochondrial oxidative stress as demonstrated via MitoSOX staining, and GHK‐Cu treatment could reduce mitochondrial oxidative stress level in a concentration‐dependent manner.
Figure S2. The ratios of TA (A), soleus (B) and EDL (C) muscle to body weight. The difference of muscle cross sectional area of Quad muscle fibre between each group(D). *p < 0.05; **p < 0.01; ***p < 0.001.
Figure S3. Images of immunofluorescent staining of MyHC in eacg groups. *p < 0.05; **p < 0.01; ***p < 0.001.
Figure S4. The ratios of soleus (A), TA (B) and EDL (C) muscle to body weight. The difference of muscle cross sectional area of Quad muscle fibre between each group(D). *p < 0.05; **p < 0.01; ***p < 0.001.
Table S1. Basic characteristics of the study population