

Significance
Here, we devised a practical and efficient method, termed LOCK, which utilizes a donor with hybrid “3′-overhang dsDNA” (odsDNA) structure for efficient and accurate knock-in of large DNA fragment. This donor could be readily synthesized with low cost. In addition, the odsDNA donor could be directly or indirectly attached to Cas9 fusion protein and extended sgRNA and achieve efficient knock-in of 2,500 bp genes in situ.
Keywords: CRISPR knock-in, double-stranded break, phosphorothioate modification, 3′-overhangs dsDNA, off-target effect
Abstract
CRISPR/Cas9 genome-editing tools have tremendously boosted our capability of manipulating the eukaryotic genomes in biomedical research and innovative biotechnologies. However, the current approaches that allow precise integration of gene-sized large DNA fragments generally suffer from low efficiency and high cost. Herein, we developed a versatile and efficient approach, termed LOCK (Long dsDNA with 3′-Overhangs mediated CRISPR Knock-in), by utilizing specially designed 3′-overhang double-stranded DNA (odsDNA) donors harboring 50-nt homology arm. The length of the 3′-overhangs of odsDNA is specified by the five consecutive phosphorothioate modifications. Compared with existing methods, LOCK allows highly efficient targeted insertion of kilobase-sized DNA fragments into the mammalian genomes with low cost and low off-target effects, yielding >fivefold higher knock-in frequencies than conventional homologous recombination-based approaches. This newly designed LOCK approach based on homology-directed repair is a powerful tool suitable for gene-sized fragment integration that is urgently needed for genetic engineering, gene therapies, and synthetic biology.
CRISPR technologies have transformed our ability to manipulate genomes for research and gene therapy over the past decade (1, 2). The canonical nonhomologous end joining (c-NHEJ) pathway usually dictates direct ligation (for blunt ends), or minimal processing (for 1 to 2-nt overhangs) of the double-stranded break (DSB) ends, often thereby causing imprecise DSB repair. By comparison, the homology-directed repair (HDR) at the DSB site allows for precise knock-in (KI) of genes using exogenous donors with homology arms (HAs) as templates (3, 4). However, the HDR pathway is inherently inefficient, especially for large DNA donor templates. One of the reasons could be ascribed to the low concentration of exogenous DNA donors locally available in the DSB editing sites. Therefore, enormous efforts have recently focused on promoting the nuclear entry or increasing the stability, of the double-stranded DNA (dsDNA) donor templates, such as using adeno-associated viral (AAV) donors, chromatin-packaged donors, Strep-biotin tethering or chemical modification of ends of dsDNA (5–12). By comparison, short single-stranded DNA (ssDNA) donors, which are readily synthesized with a length <200-nt, showed a higher KI rate, as well as lower cytotoxicity or off-target changes when utilized as donor templates, compared with dsDNA donors (13, 14).
On the other hand, long ssDNA (lssDNA) donors are valuable for in vivo genome-editing in mouse embryos, induced pluripotent stem cells (iPS), and CAR-T cells owing to their high efficiency and low cellular toxicity (8, 15, 16). Nonetheless, the major caveat with lssDNA lies in the difficulties in its production with sufficient amounts, which clearly impedes the wide application of lssDNA-mediated HDR for genome editing. Compared with the restriction of synthesis length by chemical methods, enzymatic synthesis of lssDNA has been gradually developed and utilized, such as reverse transcription (RT) (16) and exonuclease digestion (17, 18), but it also has some disadvantages, such as time- or cost-prohibitive (15, 19, 20). Moreover, the multiple-step purifications during the lssDNA production often generate impure lssDNA pools with residual or truncated dsDNAs, or inaccurate lssDNA sequences owing to the weak proofreading activity of the RT enzyme. Strikingly, recent long-read amplicon sequencing analyses showed that, in contrast to previous results, ssDNA-mediated KI is not superior to dsDNA with respect to the KI efficiency and off-target events when utilized as the HDR donors (21). These data suggest that further optimization of the HDR donors is required to improve the CRISPR/Cas9-mediated gene-sized KI applications in biomedical sciences and therapeutic applications.
To leverage the distinct advantages of both dsDNA and ssDNA in a single donor. Herein, we report an efficient and versatile method to carry out CRISPR-Cas9-guided large-fragment KI by designing a hybrid “ssDNA-dsDNA” donor that harbors an optimal 10-nt 3′-overhang at both ends, referred to as overhang dsDNA (odsDNA). We first provide an affordable approach for the preparation of odsDNA donors with any length of 3′-overhang by designing PCR primers with PT modification at specified nucleotides. By comparing KI frequencies of gene-sized (1.1 kb and 2.5 kb) exogenous DNA donors, we demonstrated that odsDNA significantly improved the targeted precise KI rates by up to 4.3-fold as compared to conventional dsDNA donors, while maintaining low off-target events across multiple genomic loci in a host of mammalian cells. When applied in conjugation with tethering technology by harnessing the 3′ single-stranded overhang, the KI efficiency using odsDNA donors could be improved by up to 5.2-fold compared with dsDNA donors. Together, we developed an efficient large-fragment integration method via Long dsDNA with 3′-Overhangs-mediated CRISPR Knock-in (LOCK). We envision that the LOCK strategy harbors advantageous features that make it suitable for time-saving and cost-effective KI in place of dsDNA or lssDNA donors in the future.
Results
Evidence for Rational Design of odsDNA Donor Templates.
The repair pathway choice for CRISPR-induced DSBs is largely dependent on the processing of DSB ends (22). The c-NHEJ is the predominant pathway of DSB repair that requires no or minimal end processing and is thus innately error-prone. The precise integration of exogenous large-fragment donor templates is achieved via HDR, using ssDNA donors or dsDNA donors, which requires the long 3′-overhang produced by strand resection (Fig. 1 A, i and Fig. 1 B, iv) (23). In general, HDR is inefficient in mammalian cells. However, recent advances showed that the microhomology revealed by the short-range strand resection directs the microhomology-mediated end-joining (MMEJ) pathway, which dominates the end-joining events over c-NHEJ in CRISPR-induced DSB repair, suggesting that microhomology-directed base-pairing is more efficient in DSB repair than c-NHEJ or HDR (4, 24, 25). On the other hand, short ssDNAs have been widely employed as donors for the highly efficient HDR repair through the noncanonical HDR-mediated single-strand annealing (SSA) pathway (Fig. 1 A, ii and iii), which is defined by longer microhomology length (26, 27). The ssDNA donors exhibit a higher KI rate and lower cell cytotoxicity than dsDNA donors, presumably due to the distinct nature of the strands and repair mechanisms (Fig. 1 B, v and vi). However, for gene-sized KI, the preparation of lssDNA is laborious and remarkably costly (15, 19, 20). As such, we sought to devise a chimeric “ssDNA-dsDNA” donor, i.e., 3′-overhang dsDNA (odsDNA), which we anticipated harbors both advantages of the dsDNA and ssDNA donors (28). The 3′-overhang in the non-target strand tends to be released earlier from the Cas9 complex (Fig. 1 D, vii), and thus anneals to one end of the odsDNA or ssDNA (29–32). In comparison, the other 3′-end of the complementary strand of the odsDNA, but not the ssDNA donor (3, 33), could have the complementary homology to the 3′-overhang in the target strand in host genome. In this scenario, both ends of the odsDNA donor harbor the complementary homology sequences to the resected DSB overhangs, resulting in the highly efficient repair through the SSA pathway (Fig. 1 D, viii–x).
Fig. 1.

Evidence for rational design of odsDNA donor for gene-sized KI. (A) A known putative model for precise Cas9-induced DSB repair using ssDNA donor. The 3′-overhangs flanking the DSB breakage site in the genome are exposed following bidirectional end resection (i). The 3′ ssDNA end anneals to one DSB end via microhomology-mediated SSA and is thus efficiently repaired through the non-canonical HDR-mediated SSA pathway (ii). At the 5′ ssDNA donor end, owing to the lack of complementarity between 5′ ssDNA end and the DSB overhang, it is precisely repaired through relatively low-efficient HDR dependent on synthesis-dependent strand annealing (SDSA) (iii). (B) HDR-mediated repair for Cas9-generated DSBs using dsDNA donor. Both the newly produced DSB overhangs independently invade the dsDNA donor template and synthesize new strand with the dsDNA template. Both DSB ends are precisely repaired through HR-mediated HDR. (C) A rational design for a hybrid ‘ssDNA-dsDNA’ donor model, referred to as 3′-overhang dsDNA (odsDNA). (D) In this hypothesized odsDNA model, both 3′- overhangs of odsDNA would bias the DSB repair toward more efficient SSA-mediated non-canonical HDR repair, similar to the 3′ ssDNA end repair in A. Overhang binding site, OBS. The structure of Cas9 (no. 4CMP) from PDB database.
Therefore, in such a further optimized design in this study, we envisioned that the optimized odsDNA donors (Fig. 1C) possess the following advantages: i) easy preparation with low cost; ii) improved KI efficiency and low cellular toxicity; iii) enhanced KI rate when conjugated with facile tethering of odsDNA by promoting nuclear entry.
Validation of Phosphorothioate Bonds to Prevent dsDNA Digestion by Lambda Exonuclease for the Preparation of odsDNA Donors.
We sought out to devise a method that robustly produces 3′-overhangs in any PCR-amplified dsDNA products. Taking the time and labor cost into consideration, we focused on an approach with lambda exonuclease digestion of the dsDNA in a 5′- to 3′- direction under the protection by phosphorothioate (PT) bonds among selected deoxynucleotides adjacent to the 5′-end, leading to the production of odsDNA (Fig. 2A). The PT bond is a covalent phosphodiester linkage where one of the two nonbridging oxygens is replaced by a sulfur (the dashed box in Fig. 2A). Owing to its ease and low cost through commercial synthesis, PT modification has been widely adopted for either 5′- or 3′-end DNA-strand decoration for the sake of protecting of DNA ends from degradation by exonucleases in vitro or in vivo (9–11, 34).
Fig. 2.
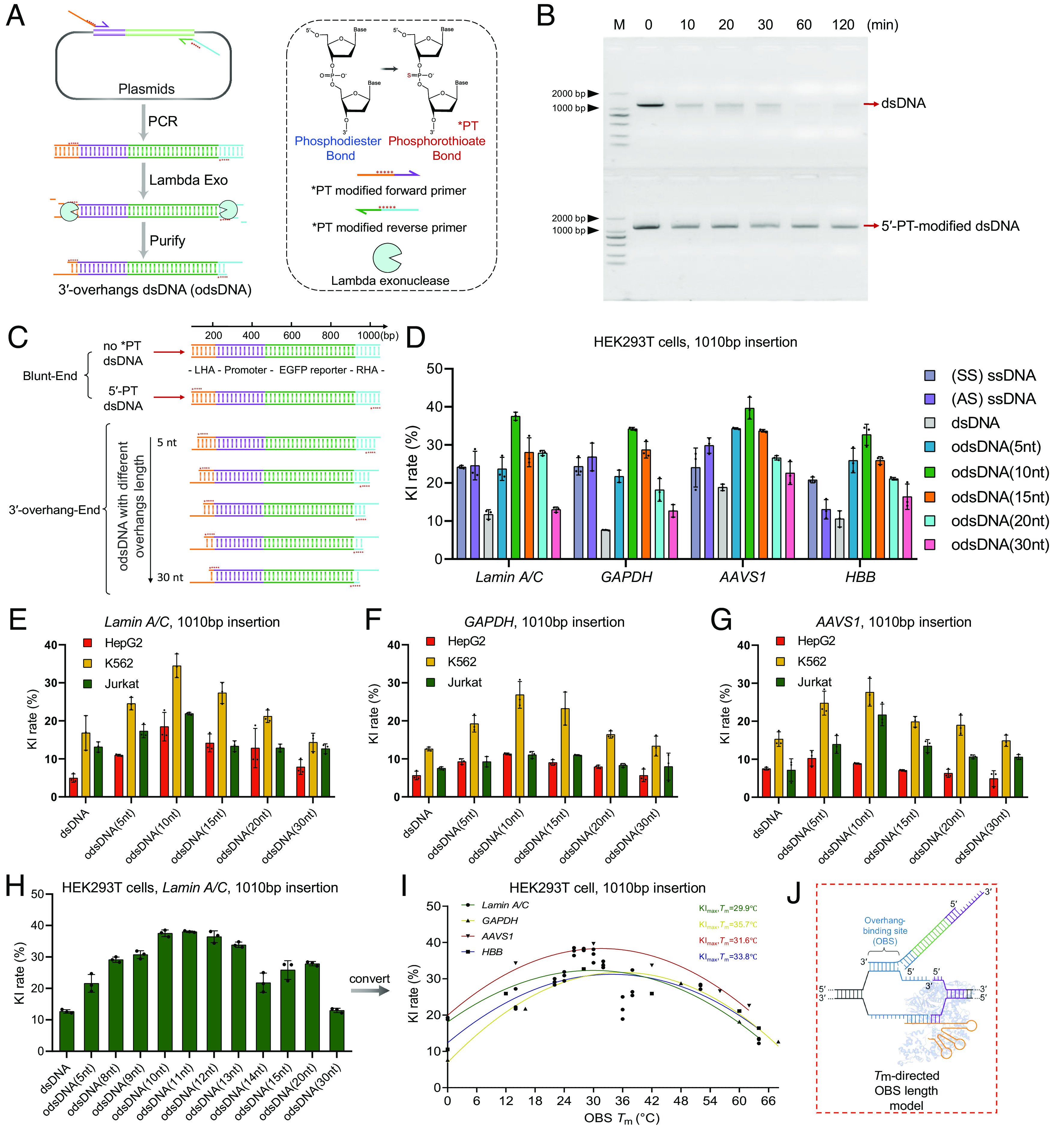
Preparation and determination of odsDNA donors with optimal 3′-overhang length as templates for large-fragment KI. (A) Schematic diagram for the generation of odsDNA donors. The odsDNA template was generated through common PCR amplification. Each primer was designed to comprise 50-nt homology arm sequence plus 20-nt base-paring sequence for specific KI template amplification. The 3′-overhang length of the odsDNA donor was specified by five consecutive PT (*) bond modifications, which prevent the 5′- to 3′- overdigestion of the odsDNA donor by Lambda exonuclease. Shown is a KI template consisting of promoter sequence (purple) and EGFP-coding sequence (green). The chemical structure in the figure were drawn by ChemDraw 19.0 software. (B) A representative agarose gel visualization showing the protective effect of PT modification against Lambda exonuclease. The 1,110 bp dsDNA fragments without (above) or with (bottom) five PT modifications were subjected to Lambda exonuclease digestion for varying time lengths as indicated on top. (C) Schematic illustration showing the preparation of 1,010 bp odsDNA donors (containing promoter and EGFP reporter sequences) with variable 3′-overhang lengths (5-, 10-, 15-, 20- and 30-nt). (D) A panel of four different genomic loci was selected for the 1,010 bp odsDNA donor KI in HEK293T cells. The sense ssDNA, antisense ssDNA and dsDNA donors were used as controls. Data were collected 15 d after nucleofection. At the GAPDH locus, the odsDNA donor with 10-nt overhang exhibited increased KI rate by up to 4.38-fold compared with dsDNA donor. (E–G) A side-by-side comparison of the 1,010 bp fragment KI rates using different donors as indicated, across three genomic loci (Lamin A/C, GAPDH and AAVS1) in HepG2 cells, K562 cells, and Jurkat cells, respectively. (H) Comparison of the KI rates using dsDNA and odsDNA donors with varying overhang lengths ranging from 8- to 15-nt at Lamin A/C locus in HEK293T cells. (I) A nonlinear quadratic fitting curve showing the correlation between the Tm of OBS and the KI rates across four loci in HEK293T cells. (J) A highlighted box showing that an optimal OBS Tm helps to raise the KI rate, presumably by stabilizing the duplex, the figure is adapted from figure 1c of Anzalone et al. (35). Data and error bars in D–I indicate the mean and SD of three independent biological replicates.
Nonetheless, whether the PT bonds located adjacent to the 5′-end of dsDNA can protect against the overdigestion of exonuclease has not been well studied. To this end, we designed a pair of PCR primers with five protective PT modifications at designated positions as indicated and performed PCR amplification on a target DNA template with 1.1 kb in length, followed by treatment with an overdose of lambda exonuclease for variable digestion times (Fig. 2B). This revealed that 1 μg (up to 3 µg in total) of total dsDNA (1,110 bp in length) was wholly digested in the presence of 5 U of Lambda exonuclease in 60 min (SI Appendix, Fig. S1). In contrast, the amount of 5′-PT-modified dsDNA retained almost constant levels over the entire digestion period (Fig. 2B), validating that 5′-PT-modification can be utilized to block exonuclease overdigestion.
Next, to prepare the odsDNA donor templates with varying lengths in the 3′-overhang (5 to 30-nt), we designed PCR primers (20-nt base paring with template plasmids plus a 50-nt HA) with five consecutive PT modifications in the HA located immediately adjacent to the junction between the overhang and the dsDNA. All these odsDNA templates were designed to harbor a 50-nt HA at both the left and right ends, based on i) they exhibiting the highest CRISPR-mediated HDR KI efficiency based on previous findings from our group (9) and ii) being readily incorporated into PCR primers at low cost. The internal nucleotides are composed of a promoter and an EGFP-coding sequence within the donor templates (Fig. 2C). After lambda exonuclease digestion and agarose gel visualization, the resultant odsDNA products at the predicted size were recovered for CRISPR KI assessment.
Determination of the Optimal 3′-Overhang Length for Efficient KI with odsDNA Donor Templates.
To exclude the background fluorescence produced by the spontaneous transcription and translation of the unintegrated donor templates in the cells, we first measured and determined the time point when the background fluorescent intensities declined to be close to the baseline autofluorescent levels in HEK293T cells via transfection with only donor templates. The flow cytometry recordings detailed that both dsDNA and odsDNA donors followed a similar declining trend, whereby the background signal disappeared at post-transfection day 15 and day 21 for the 1,110 bp and 2,600 bp donors, respectively (SI Appendix, Fig. S2). These time points were therefore chosen for subsequent flow cytometry analyses.
Next, to interrogate the effect of 3′-overhang lengths on KI efficiencies, we synthesized a series of PT-modified PCR primers to amplify a 1,010 bp donor template, resulting in the generation of 5′-terminally PT-modified dsDNA, and 5-, 10-, 15-, 20-, 30-, 40-, 50-nt overhangs of odsDNA donors after lambda exonuclease digestion (Fig. 2C). These donors were each concurrently electroporated with Cas9 RNPs to target four genomic loci, including Lamin A/C, GAPDH, AAVS1, and HBB, in HEK293T cells, respectively (editing site information in SI Appendix, Table S1). The percentages of cells with EGFP expression served as a surrogate for KI efficiencies with different donors. Apparently, 5′-terminally PT-modified dsDNA displayed increased KI efficiencies across all four selected genomic loci, which is in line with the protective role of PT modification (SI Appendix, Fig. S3). Noticeably, odsDNA donors with different lengths of 3′-overhang exhibited variable KI efficiencies, with the 10-nt overhang showing the highest KI efficiency, equivalent to an average of 3.1-fold increase across all four targeted genomic sites compared to dsDNA donors (Fig. 2D). In contrast, odsDNA donors with longer overhangs (40-nt and 50-nt) displayed much lower KI frequencies, implying that an optimum length of 3′-overhang is required to achieve the highest KI efficiency (Fig. 2D and SI Appendix, Fig. S4). As a comparison, we synthesized and tested the KI rates for both sense (SS) and antisense (AS) ssDNA donors with 50-nt HAs. This revealed that ssDNA donors in general displayed higher KI rates than dsDNA, but exhibited lower rates than odsDNA donors with 10-nt overhang, across four loci in HEK293T cell. The average values for the highest efficiencies of the four loci are 11.75%:24.5%:36% (dsDNA vs. ssDNA vs. odsDNA) (Fig. 2D and SI Appendix, Fig. S5). To further test the generalizability of this conclusion, we performed a similar co-transfection of Cas9 RNPs with dsDNA or odsDNA harboring variable 3′-overhang lengths in three other mammalian cell types, including HepG2, K562, and Jurkat cells. Strikingly, odsDNA donors consistently resulted in higher KI efficiencies than dsDNA donors by an average of 1.5-fold in HepG2 cells, twofold in K562 cells, and 1.8-fold in Jurkat cells across all tested loci (Fig. 2 E–G). In agreement with previous findings, odsDNA donors with 10-nt 3′-overhangs consistently displayed the highest KI efficiencies among various genomic loci and cell lines. As a comparison, we also interrogated the KI rates using odsDNA donors with shorter HAs (40 bp and 20 bp). This result unraveled that the odsDNA donors with 10-nt overhang are still advantageous for improving the KI rates regardless of the HA lengths, and that longer HA lengths of the odsDNA donors displayed a propensity for higher KI rates (SI Appendix, Fig. S6).
Prior studies have demonstrated the difference in KI efficiencies for ssDNA donors with asymmetric lengths of HA overhangs, and the ssDNA donors with shorter non-PAM end but longer PAM end displayed the maximum KI efficiency (29). To explore whether the odsDNA donors comply with this rule, we synthesized the odsDNA donors with variable overhang lengths at both the PAM and non-PAM ends present in an asymmetric way (SI Appendix, Fig. S7). It revealed that, in support of our previous findings, the odsDNA donors with 10-nt overhangs exhibited the highest KI efficiencies, while asymmetric arraying of the HA overhangs offers no extra benefits (SI Appendix, Fig. S7).
Moreover, we performed fluorescence microscopy imaging of HEK293T, HepG2, K562 and Jurkat cells following KI at the AAVS1 safe harbor locus, located in intron 1 of the PPP1R12C gene (SI Appendix, Fig. S8). As revealed by flow cytometry, the odsDNA donors had a slightly higher cell viability than dsDNA donors (36.6% vs. 33.7%) (SI Appendix, Fig. S9). However, longer 3′-overhangs (>30-nt) exhibited decreased KI rates, which is presumably due to the formation of secondary structures, or the elicitation of intrinsic exonuclease activity owing to excess long 3′-overhangs (36) (SI Appendix, Fig. S4). Together, these results verified that odsDNA donors displayed higher KI efficiencies than conventional dsDNA donors in mammalian cells.
Optimization of the Tm of the 3′-Overhang Improves the KI Rate.
Previous studies have shown that the optimal hybrid melting temperature (Tm) between the prime binding site (PBS) sequence and an RT RNA template is crucial for improving the editing efficiency of the CRISPR-mediated prime editor (PE) by stabilizing duplex base pairing (35, 36). In addition, the maximum KI efficiencies of odsDNA donors were achieved with 10-nt 3′-overhang as described above but were diminished with either shorter or longer 3′-overhangs, suggesting that further optimization of the Tm of the odsDNA 3′-overhang might improve the general KI rates.
To test this hypothesis, we further synthesized primers for odsDNA production with a series of variable lengths of 3′-overhangs, as indicated in Fig. 2H and performed electroporation with Cas9 RNPs in HEK293T cells. This revealed that the KI efficiencies were magnified in odsDNA donors with 3′-overhangs in lengths ranging from 10 to 12 nt, and most intriguingly, the lengths corresponded to a Tm value at ~33 °C on average when calculating the hybrid temperature after a nonlinear quadratic fit across four loci in HEK293T cells (Fig. 2I). This reminds us the similar results attained in PE system in plants (37) and animals (38), indicating that there is an optimal Tm for the binding of the DSB-induced, free 3′-terminal overhang to the odsDNA overhang within the cellular milieu. To exclude cell- or genomic site-context-dependent effects, we refitted the KI rates in HEK293T, HepG2, K562 and Jurkat cells across the Lamin A/C, GAPDH, AAVS1, and HBB loci with the variable Tm values of the 3′-overhang of odsDNA, and the results all corroborated the optimal Tm values at 33 °C (SI Appendix, Fig. S10 A–D). Altogether, we propose a model wherein the maximum odsDNA-mediated KI efficiencies are leveraged through the 3′-overhang binding site (OBS)-directed Tm at approximately 33 °C via an average length of 10-nt 3′-overhang (Fig. 2J).
KI with odsDNA Donors Exhibited Lower Indel Rates Other than High HDR Efficiency.
In addition to high KI efficiency, it is known that the ssDNA donors exhibit lower on-target indel rates and lower off-target integration than dsDNA donors (15). After verifying the optimal 3′-overhang design, we next evaluated how odsDNA KI behaves with regard to the indel rate on the targeted editing sites. We performed similar Cas9 RNPs co-electroporation with dsDNA or odsDNA at the Lamin A/C, GAPDH, AAVS1 and HBB loci in HEK293T, HepG2, K562 and Jurakt cells, and conducted PCR amplification (SI Appendix, Fig. S11A). As visualized by agarose electrophoresis, all samples electroporated with any kind of donor templates harbored a full-length, donor template-integrated PCR band at the predicted size, which was absent in the non-donor transfection WT samples, as expected (SI Appendix, Fig. S11B). To investigate the integration accuracy of the donor templates, we carried out the PCR amplification of both the 5′ and 3′ junctions at the AAVS1 genomic loci and subjected them to Sanger sequencing (SI Appendix, Figs. S12 and S13). Bioinformatic sequencing analyses by the TIDE pipeline showed that, compared with dsDNA (no PT), odsDNA with a 10-nt overhang consistently displayed lower indel rates at both the 5′ and 3′ junctions (odsDNA vs. dsDNA: 5% vs. 9% for 5′ junction and 4.75% vs. 6.88% for 3′ junction) (Fig. 3A). Of note, it appeared that the indel rates for odsDNA vary, and there is no a conclusive relationship existing between the lengths of odsDNA 3′-overhang and resultant indel rates (Fig. 3A).
Fig. 3.

KI with odsDNA donors exhibited enhanced HDR frequencies with low off-target integration. (A) Comparative analyses for the on-target indel rates at 5′ and 3′ Junctions after 1,010 bp fragment KI using different donors by TIDE measurement at AAVS1 locus among HEK293T cells, HepG2 cells, K562 cells, and Jurkat cells. (B) High throughput amplicon-seq analyses for the on-target editing events as determined using dsDNA, odsDNA and ssDNA donors. The amplicon-seq library was constructed with specifically designed PCR primers spanning the junctions. (C) Schematic diagram showing the Genome-wide insertion site sequencing (GIS-seq) library construction pipeline for genome-wide off-target detection. The individual 1,010 bp dsDNA or 1,010 bp odsDNA donor was transduced into HEK293T cells through nucleofection. Following the sonication of the genomic DNA, the library was constructed with a specific primer (forward or reverse) against the donor and an adaptor primer. (D) Representative GIS-seq result showing plus/minus read distribution at on-target locus. (E) GIS-seq revealed that odsDNA donors had similar or even lower off-target integration at Lamin A/C locus than dsDNA donors in two biological experiments. Data and error bars in A and B indicate the mean and SD of three independent biological replicates.
To further decipher the editing events in more detail, we conducted amplicon-sequencing on the junctions of donor integration, which was easier to fulfill using shorter, synthesized dsDNA, odsDNA and ssDNA donors with the same HA length but only containing a 6 bp insertion (Fig. 3B). To fairly evaluate the editing events, we performed the electroporation at the same low concentrations of donors to minimize the impact by the varied cellular toxicities from donors. Targeted sequencing analyses uncovered that the precise HDR KI rates were comparable between odsDNA and ssDNA donors and were approximately 2.5-fold higher than those of dsDNA donors across the Lamin A/C and GAPDH loci in HEK293T cells (Fig. 3 B, Right). Consequently, the indels editing events for odsDNA and ssDNA donors were significantly lower than that of dsDNA (dsDNA:ssDNA:odsDNA = 42.2%:36.7%:37.3%), and the relative HDR/indel ratio for ssDNA or odsDNA was also higher than that of dsDNA donor (dsDNA:ssDNA:odsDNA = 0.1:0.23:0.24) (Fig. 3 B, Right).
We next asked whether odsDNA donors could reduce the genome-wide off-target editing events. To this end, we adopted a modified genome-wide insertion site sequencing (GIS-seq) (39) procedure to obtain all on-target and off-target genome-editing events for dsDNA and odsDNA donors (Fig. 3 C and D). When applying GIS-seq to Lamin A/C and GAPDH loci in HEK293T cells and including sites with high confidence based on maximum likelihood estimation, this unveiled that odsDNA donors had low off-target editing activity (two similar off-target sites aligned to two chromosomes), which resembled that of dsDNA donors (two distinct off-target sites) (Fig. 3E). Together, these data validated that the application of odsDNA donors for large-fragment KI is highly efficient as ssDNA donors but with lower on-target indel rates compared with dsDNA donors.
The Cas9-PCV2 Fusion Protein Tethers odsDNA Through a Short PCV2 Linker to Enable Highly Efficient Large-Fragment KI.
The overarching goal for optimizing genome-editing tools is to raise the precise target integration while mitigating its potential off-target effect. The HUH endonuclease domain of Porcine Circovirus 2 (PCV2) Rep protein is a short peptide (13.4 kD) that has been shown to directly recognize the DNA replication Ori region, make an excision, and covalently attach to a single-strand DNA sequence (SI Appendix, Fig. S14) (40, 41). This feature was initially explored to improve HDR-mediated target insertion using ssDNA donors. However, the recognition and covalent tagging by PCV2 at the 5′-terminal of ssDNA might interfere with homology-directed 5′ junction repair, resulting in increased indels. To conquer this issue and to avoid the short stretch of optimal 3′-overhang nucleotides (10-nt) based on our optimization strategy for odsDNA donors as described above, we thereby devised a short PCV2-recognized linker that presumably tethers the odsDNA donor to the editing site via base paring. The PCV2 domain was C-terminally fused to the Cas9 protein to attain the fusion protein Cas9-PCV2, which was expressed and purified in E. coli BL21 (SI Appendix, Fig. S15 A and B). An in vitro cleavage assay showed similar cleavage activities on the same dsDNA substrates between Cas9 and Cas9-PCV2 (SI Appendix, Fig. S15C).
The FAM-Quench ssDNA was designed to detect the activity of the purified Cas9-PCV2 fusion protein (Fig. 4A). At a dilution of concentrations (0.02 μM, 0.06 μM and 0.1 μM), Cas9-PCV2 was able to rapidly cleave the ssDNA strand and release fluorescence, reaching reaction equilibrium within 30 min at low concentrations (Fig. 4A). Next, we probed whether Cas9-PCV2 can specifically bind to odsDNA donors. The short synthesized PCV2 linker is composed of two parts: one side harboring the sequence for annealing to the odsDNA donor via base pairing, and the other side being the substrate sequence covalently recognized by PCV2. Herein, to attain maximum KI efficiency, we opted to exploit the 10-nt overhang odsDNA for comparisons. The PCV2 linker was allowed to anneal with the odsDNA donor, followed by incubation with Cas9-PCV2 at variable ratios at 37 °C for 30 min. This study revealed that, at a molar ratio of 3 to 1 (Cas9-PCV2 vs odsDNA), the substrate odsDNA donor was fully bound to the PCV2 linker, which was covalently attached to Cas9-PCV2 (Fig. 4B).
Fig. 4.
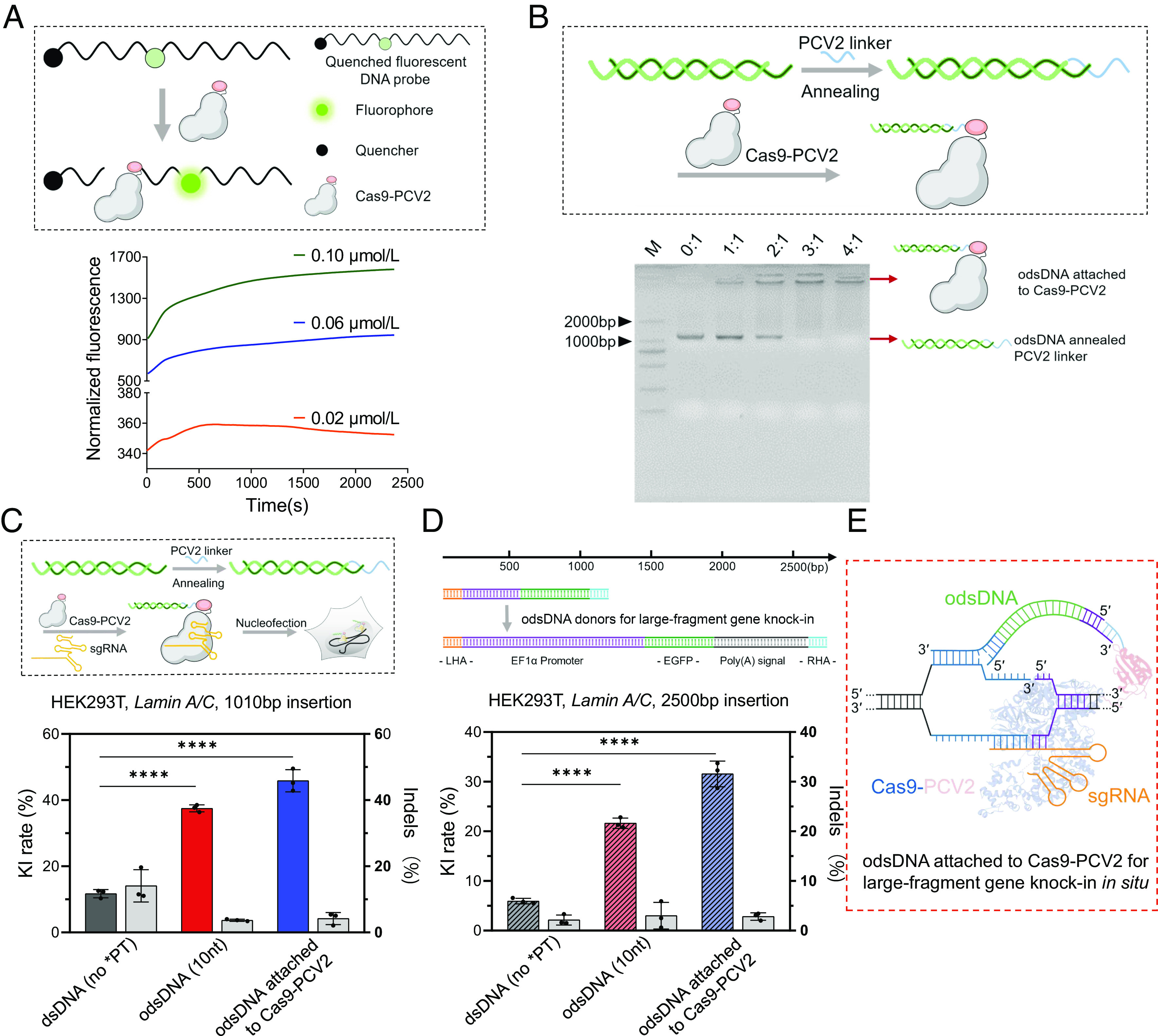
Cas9-PCV2 Covalently Tethers odsDNA Donors for Enhanced Gene-Sized KI. (A) Schematic diagram showing the cleavage of FAM-Quenched fluorescent ssDNA probe by Cas9-PCV2 fusion protein, releasing the fluorescent signal (Upper). Kinetic recording of fluorescence curves for the reaction between Cas9-PCV2 and F-Q ssDNA at 0.02 μM, 0.06 μM and 0.1 μM as indicated (Lower). (B) Efficient tethering of odsDNA donors to Cas9-PCV2 through base-pair annealing with variable molar ratios (Cas9-PCV2: pre-annealed odsDNA/linker) as visualized by agarose gel electrophoresis. (C) A schematic diagram illustrating the assembly of the RNPs complex consisting of Cas9/Cas9-PCV2, sgRNA, and odsDNA/linker, for nucleofection (above). The 1,010 bp donor templates, including Cas9-bound dsDNA and 10-nt odsDNA as well as Cas9-PCV2-bound 10-nt odsDNA, were electroporated to target the Lamin A/C locus in HEK293T cells. Both the KI rates and the indel occurrences were plotted for side-by-side comparison (Bottom). On average, the KI frequencies increased up to 3.19-fold and 3.9-fold in odsDNA (10-nt) and Cas9-PCV2 attached odsDNA (10-nt) groups, respectively, as compared with dsDNA donor (Gray bar indicates Indels rate as plotted on the right y axis). (D) A larger gene-sized fragment (2,600 bp, 50 bp HAs on both ends) composed of promoter, EGFP and poly(A) sequences was exploited for electroporation, as performed above at Lamin A/C locus in HEK293T, for examination of insertion-sized effect on KI and indel rates. On average, the KI frequencies increased up to 3.63-fold and 5.29-fold in odsDNA (10-nt) and Cas9-PCV2 attached odsDNA (10-nt) groups, respectively, as compared with dsDNA donor (Gray bar indicates Indels rate as plotted on the right y axis). (E) The dashed box illustrates a putative model where Cas9-PCV2-tethered odsDNA facilitates the duplex annealing between the 3′-overhang of odsDNA and the released genomic DNA strand around the breakage site, the figure is adapted from figure 1c of Anzalone et al. (35). P value was calculated by two-tailed unpaired t test, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Data and error bars in C and D indicate the mean and SD of three independent biological replicates.
After verifying the capacity of tethering the odsDNA donor by the fused Cas9-PCV2 through the PCV2 linker, we next assessed the performance of Cas9-PCV2-guided large-fragment KI in conjunction with odsDNA donors. The Cas9 or Cas9-PCV2 RNP was co-transfected with either dsDNA or 10-nt overhang odsDNA donors with a 1,010 bp length targeted to the Lamin A/C locus in HEK293T cells. Compared with the 12% KI rate in the Cas9/dsDNA group, there was a significant improvement in the KI rate being reached at 37% using Cas9/10-nt odsDNA, and even higher at 45% with Cas9-PCV2/10-nt odsDNA (Fig. 4C and SI Appendix, Fig. S16), reminiscent of an up to 3.75-fold improvement in the KI rate.
To further corroborate this finding, we prepared a series of longer, 2,500 bp-containing odsDNA donor templates with variable lengths of 3′-overhangs (5 to 20-nt) and conducted the co-transfection with Cas9 RNP or Cas9-PCV2. Consistent with previous findings, whereas the non-PT dsDNA donors exerted a KI rate at 6%, the 10-nt overhang odsDNA donor gave rise to a higher KI rate, reaching 21% among all non-tethered odsDNA donors with or without 3′-overhang in the Cas9 RNP group (Fig. 4D and SI Appendix, Fig. S17). By comparison, Cas9-PCV2-tethered 10-nt odsDNA donors robustly increased the KI rate as high as 31%, corresponding to 5.2-fold improvement (Fig. 4D and SI Appendix, Fig. S17). Not surprisingly, odsDNA donor-mediated KI with either Cas9 RNPs or Cas9-PCV2 constantly displayed lower indel rates than non-PT-modified dsDNA donors, regardless of the insertion size (Fig. 4 C and D). Taken together, this evidence concluded that the application of the optimized Cas9-PCV2/linker/odsDNA strategy by tethering donor templates to the editing sites significantly improved the large-fragment KI rates by more than a fivefold increase (Fig. 4E), while maintaining lower indel events compared with common dsDNA donors.
Tethering the odsDNA Donors with 3′-Extended sgRNA (esgRNA) Enhances the Gene-Sized KI.
The crystal structure of Cas9/sgRNA/target DNA revealed that, other than the exposed tetraloop and stem loop2, the 3′-terminal end of sgRNA also protrude outside of the bilobed ribonucleoprotein architecture (42). Indeed, this free 3′-terminal end has been leveraged in the prime editing gRNA (pegRNA)-mediated PE system by serving as an RT template for accurate insertion, deletion, and replacement of short stretches of nucleotides (35). Inspired by the versatile applications of pegRNA, we reasoned that tethering the odsDNA donor through the base-pairing between the odsDNA 3′-overhang and the artificially extended sequence at the 3′-end of sgRNA, termed esgRNA, would help increase the editing efficiency (SI Appendix, Fig. S18). To examine this hypothesis, we first prepared esgRNA with appended 30-nt nucleotides (spaced region plus base-pairing sequences) through in vitro transcription. Agarose gel visualized that esgRNA could stably bind the odsDNA donors in vitro (Fig. 5A). The co-electroporation of fluorescein-labeled Cas9 (green) and esgRNA/odsDNA (red) demonstrated that the odsDNA donor was effectively trafficked and co-localized within the nuclei of HEK293T cells (Fig. 5B). After validation of the effective tethering of the odsDNA donors by esgRNA/Cas9 RNPs, we next interrogated whether esgRNA/Cas9 RNP-tethered odsDNA donors could increase large-fragment KI integration. Likewise, we executed the co-electroporation of esgRNA/Cas9 RNPs with a 2,500 bp odsDNA donor template. Compared with the KI rate (~10%) for the dsDNA donor, there was a marked increase in the KI rate (38%) in the combinatorial esgRNA/Cas9/odsDNA group by up to fourfold (Fig. 5C). Consistently, esgRNA/Cas9/odsDNA samples also showed lower indel editing than common dsDNA donors (Fig. 5C).
Fig. 5.
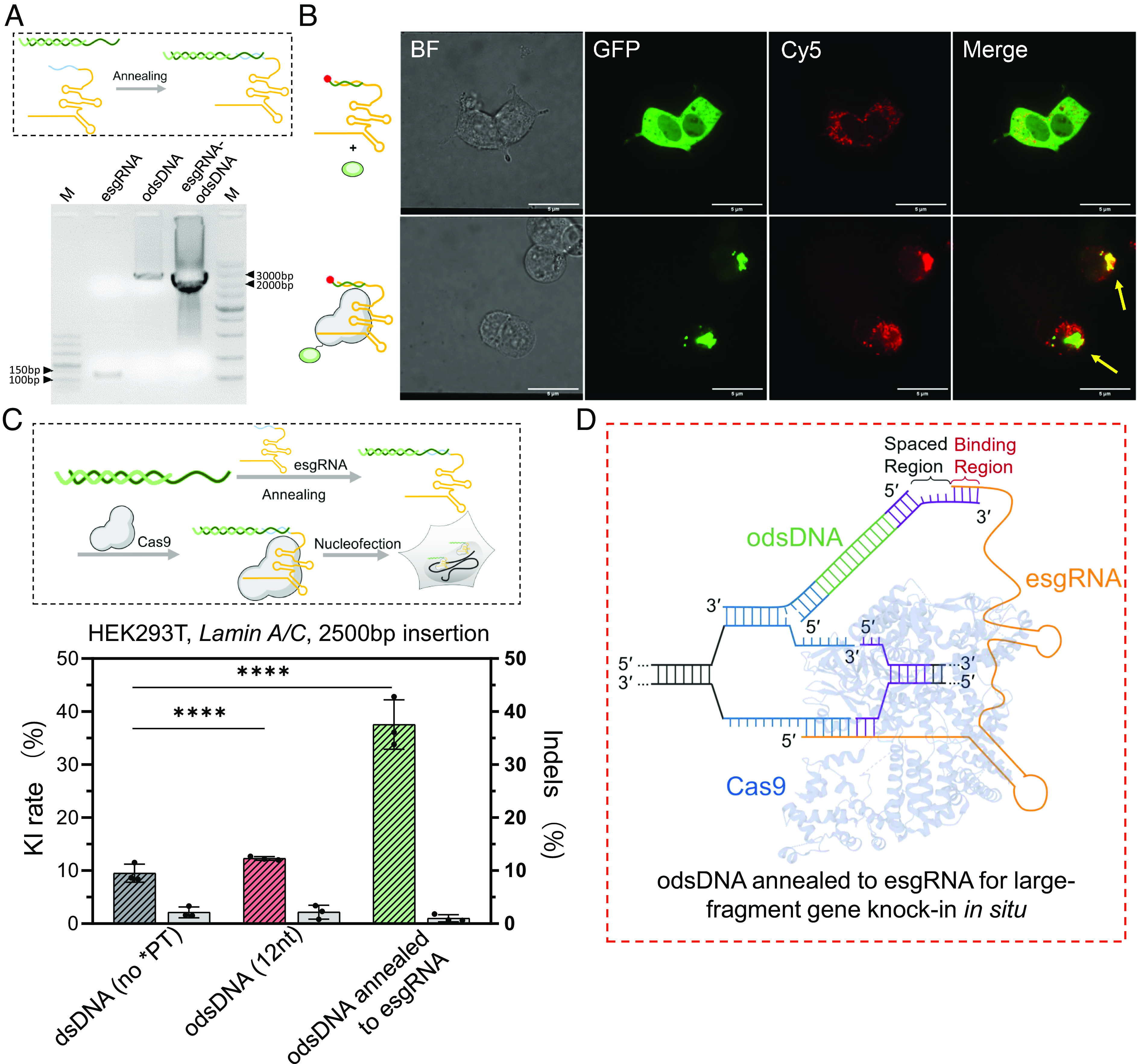
An optimized design of esgRNA efficiently tethers odsDNA donor for improved gene-sized insertion. (A) A schematic drawing illustrates a short stretch of ribonucleotides was added to the 3′-terminal of sgRNA, referred to as esgRNA (as inspired by the pegRNA), which was employed for strand-annealing with the 3′-overhang of odsDNA for tethering (above). Above, schematic of customized odsDNA annealed to esgRNA. The annealing efficiency of equimolar amounts of odsDNA and esgRNA were visualized by agarose gel (Bottom). (B) Cas9-EGFP (Lower), but not EGFP alone (Upper), co-localized with Cy5-labelled esgRNA-odsDNA complexes in the nuclei of HEK293T cells. Arrows point to the co-localization. Bar = 5 µm. (C) Schematic diagram showing the RNP assembly complex, which is comprised of Cas9, esgRNA and the odsDNA donor (Upper). A 2,500 bp donor fragment was employed for KI test at Lamin A/C locus in HEK293T cells. The KI/indel frequencies were plotted for side-by-side comparison (Lower). On average, the KI frequencies increased up to 1.29-fold and 3.94-fold in odsDNA (12-nt) and esgRNA attached odsDNA (12-nt) groups, respectively, as compared with dsDNA donor. P value was calculated by two-tailed unpaired t test, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Data and error bars reflect the mean and SD of three independent biological replicates (Gray bar indicates Indels rate as plotted on the right y axis). (D) The dashed box illustrates a working model where the free 3′-terminal end of Cas9-bound esgRNA facilitates the tethering of odsDNA donor to the DSB site through base-pair annealing with 3′-overhang of odsDNA (which divides the 3′-overhang of odsDNA into spaced region and binding region, as indicated), the figure is adapted from figure 1c of Anzalone et al. (35).
To reduce the cost and facilitate practical application, we designed a fixed esgRNA sequence containing a total of 30-nt nucleotides; thus the base-pairing counterpart with varying lengths in the odsDNA donors could be easily introduced by appending additional nucleotides to the primers during preparation of the templates. We next evaluated the effect of the variable lengths for base-pairing and spaced region (9 to 16-nt) within a total length of 30-nt on KI rates. Flow cytometry revealed that the lengths of the spaced region had no direct relationship with the KI rates, presumably implying less steric hindrance due to the relative flexibility of the free extended 3′-end of esgRNA (SI Appendix, Fig. S19). In summary, this evidence demonstrated that exploiting the esgRNA/Cas9/odsDNA system is an effective method for precise large-fragment donor KI (Fig. 5D).
Discussion
Despite the recent advances in the accurate genetic corrections by base editors and prime editing strategies, improved approaches for gene-sized genome modification are urgently needed for a broad range of applications (1). Short ssDNAs have been widely utilized as donor templates owing to the ease of direct chemical synthesis and the exceptionally low cellular toxicity (13, 14). Moreover, when applied at equimolar amounts, ssDNA exhibits enhanced KI and decreased off-target integration compared with dsDNA donors (15). These advantages initially inspired us to devise a hybrid “odsDNA” approach as described in this study, which we show could be readily prepared with low-cost and time-saving procedures. In a host of mammalian cell lines and among varying genomic loci, we demonstrate that odsDNA consistently outperformed the dsDNA donors for precise gene-sized DNA insertion with enhanced KI frequencies and low off-target events. Moreover, since these experiments were validated using 50-nt HA that can be easily incorporated into the PCR primers, we thus anticipate that odsDNA will have significant potential applications in either basic biomedical research or biotechnological innovations that likely substitute dsDNA or lssDNA as HDR donors in the future.
Recent high-throughput sequencing evidence suggests that the repair outcome of Cas9-induced DSB is not random but is predictable (22, 24). Strikingly, while cNHEJ is traditionally considered the predominant pathway for error-prone DSB repair, more recent data have revealed that non-canonical repair pathways, such as the MMEJ and SSA pathways, are most prominent in repairing Cas9 cut sites (4, 22, 23). Both MMEJ and SSA execute the DSB repair through stable strand-annealing of complementary 3′-overhangs revealed by end resection to varying extents, rather than by direct ligation of the DSB ends. It is therefore not surprising to observe that the 3′-overhangs residing at both ends of odsDNA are intrinsically tentative to form stable DNA duplexes for efficient HDR-mediated DNA repair (4, 43). However, this advantage of 3′-overhangs is unique to odsDNA but not for ssDNA or dsDNA (Fig. 1).
Mechanistically, the enhanced KI efficiency and low off-target integration for odsDNA donors could be explained by the known strand or polarity bias of ssDNA annealing and the pathway choice of Cas9-induced DSB repair. Based on the synthesis-dependent strand annealing (SDSA) model, the 3′-overhangs are exposed through resection on both ends of the DSB (26). The 3′-overhang of ssDNA preferentially anneals to the donor 3′-overhang, followed by strand extension via new synthesis, known as the SDSA pathway. Upon dissociation from the donor template, the newly synthesized strand would anneal back to the genome and undergo accurate recombination through HDR at the 5′-end of ssDNA. Therefore, the polarity of the 5′-3′ direction of ssDNA dictates the discrepant mechanisms for homologous recombination: HDR occurs effectively between the DSB 3′-overhang and the 3′-overhang of ssDNA owing to direct complementarity via strand-annealing, which requires a shorter length of HA; however, owing to the parallel polarity/direction between ssDNA and DSB overhang, repair of the 5′-end of ssDNA is dependent on the SDSA pathway, which requires a longer HA length and takes a longer time. This explanation is well corroborated by recent evidence showing that i) asymmetric ssDNA donors with longer 5′ HA complementary to the non-target strand exhibited the highest KI efficiency (29). ii) Cas9-induced DSB repair is locally polarity-sensitive, relying on SDSA (25, 44). iii) a donor dsDNA carrying a sticky 5′ end complementary to Cas12a-generated 5′-overhang as well as a longer HA at the other end exhibited the most precise DSB repair (45).
In summary, we optimized and provided an efficient and affordable approach, termed LOCK, for gene-sized donor KI by utilizing specifically designed odsDNA templates. We anticipate that the remarkably improved KI efficiency along with the low off-target activity makes the LOCK method suitable for a wide range of large-fragment genome-editing applications where the base editors or the prime editing could not fulfill.
Materials and Methods
Generation of Plasmids, sgRNA, esgRNA, Cas9, and Cas9-PCV2.
All the 1,010 bp and 2,500 bp gene fragments were synthesized and cloned into plasmids (SI Appendix, Table S2) (General Biol), including promoter region and EGFP sequence. Using the plasmids as templates, KI donors at different loci were obtained by PCR amplification with specific primers harboring HA sequences (SI Appendix, Table S3).
A panel of four human genomic loci was selected for targeted KI (SI Appendix, Table S1). The sgRNAs were designed by the online pipeline CHOPCHOP (46). CRISPR sgRNAs and esgRNAs used for nucleofection were prepared using the GeneArt Precision gRNA Synthesis Kit (Thermo Fisher Scientific) with respective primers (SI Appendix, Table S4). All RNA products were purified with the GeneArt gRNA Clean-up Kit (Thermo Fisher Scientific). The template sequence of esgRNA (SI Appendix, Table S4) was achieved by gene synthesis (Sangon Biotech).
For expression and purification of Cas9 protein, the Cas9 plasmid pET-28b-3NLS-Cas9-6His (addgene, no. 47327) was transformed into Escherichia coli Rosetta (DE3) strain. A single clone was inoculated into 10 mL LB media from a selective plate at 37 °C and 220 rpm for 12 h, which was further expanded into 1 L TB media for protein expression. Protein induction was conducted with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at 16 °C for 20 h. Next, the cells were harvested by centrifugation (7,000 rpm, 40 min) and resuspended in lysis buffer (500 mM NaCl, 20 mM Tris-HCl, 0.05% β-mercaptoethanol, pH 8.0). For extraction of the Cas9 protein, cells were lysed by Q125 Sonicator (QSONICA) on ice, and the supernatant was collected for purification by Ni-NTA affinity chromatography using elution buffer (250 mM imidazole, 500 mM NaCl, pH 8.0). Finally, Cas9 protein was concentrated by centrifugal filter devices (Amicon Ultra, Millipore), desalted by PD-10 desalting columns (GE Healthcare), and kept in the storage solution (20 mM Tris-HCl, 200 mM KCl, 10 mM MgCl2 and 20% glycerol, pH 8.0). The purity of the purified product was determined using SDS-PAGE gel (4 to 20%). The final protein concentration was measured by A280 absorbance and the aliquots were stored at −80 °C. The extraction method of Cas9-PCV2 protein was the same as the above, PCV2 gene was synthesized (Sangon Biotech) and Cas9-PCV2 plasmid (sequence in SI Appendix, Table S9) was assembled by Gibson assembly. GenCRISPR Cas9 v1.2 (Genscript).
Assays for Cas9, Cas9-PCV2, sgRNA, and esgRNA Activity.
The cleavage activities of the purified Cas9 and Cas9-PCV2 proteins were tested in vitro. In brief, 130 ng sgRNA (esgRNA), 0.3 μg Cas9 (or Cas9-PCV2) and 100 ng substrate DNA were complexed in digestion buffer (15 mM KCl, 1 mM MgCl2, 0.05 mM DTT, 0.01 mM EDTA, 2 mM HEPES, pH 7.5) at 37 °C for 30 min, the cleaved bands were visualized by 3% agarose gel electrophoresis. For measurement of the enzymatic activity of PCV2, a fluorescently quenched ssDNA strand containing the recognition sequence of PCV2 protein was co-incubated with Cas9-PCV2 at 37 °C, and the fluorophore intensity was detected using 0.02 μM Cas9-PCV2 protein along with varying concentrations of F-Q ssDNA (SI Appendix, Table S8) (0.02 μM, 0.06 μM, and 0.10 μM). The kinetic course of fluorescence was determined by Fluorescence Spectrophotometer F-7000 (HITACHI). The pure TrueCut Cas9 Protein v2 (Thermo Fisher Scientific) was used as a control.
Preparation and Purification of ssDNA, dsDNA, and odsDNA Donors.
All primers (Including phosphorylated primers and five consecutive PT bond-modified primers) were commercially synthesized (Sangon Biotech) and diluted to 10 μM (The primers used for the different KI donors are listed in SI Appendix, Table S3). To produce the ssDNA strand of interest, the phosphorylated strand of the PCR product was degraded by treatment with two enzymes, Strandase Mix A and Strandase Mix B, for 5 min/kb at 37 °C, respectively. Enzymes were deactivated by a 5 min incubation at 80 °C. A more detailed protocol for the Guide-it Long ssDNA Production System (Takara, 632666) can be found on the manufacturer’s website. The dsDNA and five-consecutive PT bond-modified dsDNA were obtained by PCR amplification from plasmids using specific primers. The sequences of KI donors are listed in SI Appendix, Table S2. To produce the 3′-overhang, the PCR-amplified dsDNA products with five-consecutive PT bond-modified nucleotides were digested with Lambda Exonuclease (New England Biolabs) at 37 °C for 60 min to obtain odsDNA. All the dsDNA donors and odsDNA donors were finally clean-up by GeneJET PCR Purification Kit (Thermo Fisher Scientific). The quantity and quality of the DNA products were examined by NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific).
Calculation of Tm for 3′-Overhang Binding Site (OBS).
The algorithm for computing the OBS Tm of the odsDNA was adapted from the Oligo Analysis Tool (https://www.eurofinsgenomics.eu/en/ecom/tools/oligo-analysis/). The formula used is as follows:
where NG:C and NA:T are the numbers of G:C and A:T base pairs in the OBS sequence, respectively, and N is the sum of NG:C and NA:T (SI Appendix, Table S10).
Covalent Attachment of odsDNA Donors to Cas9-PCV2.
The PCV2 linker was mixed with odsDNA donor in a molar ratio of 2:1 (SI Appendix, Table S7). To form a stable duplex between the PCV2 linker and the 3′-overhang of odsDNA, the strand-annealing protocol started with a heat denaturation step at 65 °C to remove any undesired secondary structure, followed by a gradual temperature drop to 20 °C within 1 h. To form the Cas9-PCV2 RNPs complex, this duplex was incubated with 30 pmol of Cas9-PCV2 protein at 37 °C for 30 min, followed by the addition of 90 pmol sgRNA with an extended incubation for 10 min. The cells were immediately electroporated with freshly prepared RNPs complex each time.
The Strand Annealing of esgRNA to the odsDNA Donor for Preparation of Cas9/esgRNA RNPs Complex.
A total of 5 μg odsDNA was annealed with 90 pmol esgRNA (SI Appendix, Table S5) in nucleofection buffer with an annealing temperature course ranging from 65 °C to 25 °C in a PCR Thermocycler. A 30 pmol of Cas9 was added and incubated at 37 °C for 10 min. The cells were electroporated with freshly prepared RNPs complex. For co-localization assay, 90 pmol of esgRNA was annealed with an equal amount of 5′-Cy5-modified ssDNA (SI Appendix, Table S8) from 65 °C to 25 °C, and electroporated alone or together with Cas9-EGFP.
Culture Conditions for Mammalian Cell Lines.
HEK293T (ATCC CRL-3216), HepG2 (ATCC HB-8065), K562 (ATCC CCL-243), and Jurkat (ATCC TIB-152) cells were purchased from the American Type Culture Collection (ATCC). They were cultured and passaged in Dulbecco′s modified Eagle′s medium (DMEM, Gibco), McCoy′s 5A Medium (Gibco), RPMI 1640 Medium (Gibco), and RPMI 1640 Medium (Gibco), respectively, each supplemented with 10% (vol/vol) fetal bovine serum (FBS, Gibco, qualified). All cell types were maintained and cultured at 37 °C with 5% CO2. Each cell line was authenticated by its respective supplier and tested negative for mycoplasma.
Cell Nucleofection.
To achieve high KI rates, all cell cultures used for nucleofection were maintained for less than five generations. Cell nucleofection was performed using the Amaxa Nucleofector IIb device (Lonza) with the settings as below: Cell Line Nucleofector kit V (Lonza) and program D-032 for HEK293T cells, program T-028 for HepG2 cells, program T-016 for K562 cells and program X-005 for Jurkat cells, according to the manufacturer′s protocol. The adherent cells were expanded to 70 to 90% confluence and were dissociated using TrypLE Express (Gibco). The K562 and Jurkat cells were directly washed twice with 1×DPBS to remove the medium. The Cas9/sgRNA RNPs complex was prepared by mixing Cas9 (300 nM) and sgRNA (900 nM) with a molar ratio of 1:3, and was incubated in 10 μL nucleofection solution for 10 min. The RNPs complex was added to the resuspended 1 × 106 cells in 90 μL of nucleofection solution. Lower or higher cell numbers may influence nucleofection results. The donor templates were added to the tubes before the nucleofection. In this study, we uniformly used 5 μg (1,110 bp, 70 nM) dsDNA or odsDNA for nucleofection. A higher input amount will have a higher KI rate, but would likely cause the rise in cell mortality. For the Cas9-PCV2 tethering system, the purified Cas9-PCV2 fusion protein was firstly incubated with odsDNA that had been annealed with the PCV2 linker at 37 °C for 30 min. The sgRNA was added and incubated at 37 °C for another 10 min, followed by electroporation. For 3′-extended sgRNA (esgRNA) tethering system, the nucleofection was conducted similarly as described above, except that the esgRNA was pre-annealed to the odsDNA donors prior to incubation at 37 °C for 10 min.
Fluorescent Imaging.
Inverted microscope IX71 (Olympus) was used to assess cell growth and GFP expression. Confocal inverted microscope IX81 (Olympus) was used to scan the intracellular fluorescence signal distribution. The 640 nm excitation wavelength can excite Cy5 (emission wavelength is 670 nm), and the 488 nm excitation wavelength can excite EGFP (emission wavelength is 507 nm). GenCrispr NLS-Cas9-EGFP Nuclease (Genscript).
Assessment of KI Rates by Flow Cytometry.
The cells (HEK293T and HepG2) grown in monolayer culture were washed with 1×PBS, prior to dissociation by TrypLE Express (Gibco). An equal volume of DMEM medium with 10% FBS was added to terminate digestion. The K562 and Jurkat cells can be directly sampled and analyzed. For each experiment, 15,000–30,000 cells were analyzed using a BD Accuri C6 flow cytometer (BD Biosciences), with optical filter FL1 (530/30 nm) and FL1 (530/30 99%) for EGFP fluorescence mode, and the raw data were analyzed using FlowJo Analytical Software (Tree Star, Inc.).
Genomic DNA PCR and DNA Sequencing.
In all cases, cells were cultured three days following nucleofection before genomic DNA extraction. About 1 × 105 HEK293T cells were collected and extracted using ONE-4-ALL Genomic DNA Mini-Preps Kit (Sangon Biotech) following the manufacturer′s instructions. For HepG2, K562, and Jurkat cells, genomic DNA was extracted from 1 × 106 cells with an extended final incubation step at 98 °C to 2 min using QuickExtract DNA (Epicentre) kit following manufacturer′s instructions. The resulting genomic DNA was stored in 50 μL QuickExtract solution at −20 °C. PCR was performed using KOD FX DNA polymerase (TOYOBO) with 100 ng of genomic DNA (or 5 μL supernatant of cell lysates) as template in a 50 μL reaction supplemented with the primers (SI Appendix, Table S6). Both control cells and genome-edited cells were exploited to conduct PCR for examination of full-length insertion, 5′ and 3′ junction regions surrounding gene KI sites. The bands were visualized through 2% agarose gel electrophoresis. The PCR products were finally verified by sanger sequencing (Sangon Biotech).
Analysis of Indels by TIDE.
The PCR products for 5′ and 3′ junction regions were purified using GeneJET PCR Purification Kit (Thermo Fisher Scientific) and subjected to DNA Sanger sequencing (Sangon Biotech). Each raw chromatogram data sequence was analyzed by the pipeline TIDE (47), along with the sequence without genome-editing as a reference.
Library Preparation for Amplicon Sequencing.
The specific primers used for donor preparation for amplicon-sequencing were designed and synthesized (SI Appendix, Table S11). The dsDNA donor was prepared by the strand-annealing of two complementary synthesized ssDNAs, with a 6-base insertion in the middle and 50-base homology arms on both ends (106-nt in total). The odsDNA donor was formed by asymmetric annealing of two ssDNAs (containing phosphorothioate modifications at the 5′-end), with a 6-base cleavage site by a restriction enzyme in the middle, and 40-base homology arms on both ends (a total of 96-nt with 10-nt overhangs). A total of 5 pmol dsDNA, odsDNA, and ssDNA donors were electroporated, respectively, into HEK293T cells at Lamin A/C and GAPDH loci along with Cas9 RNPs. On the third day, the cells were collected and the genomic DNA was extracted. A total of 200 ng genomic DNA was used for the amplicon-seq library preparation. For each gene, the junction sites were amplified using specific primers (SI Appendix, Table S12) in the first-round PCR reaction by Ultra II Q5 Master Mix (New England Biolabs). Illumina adaptors and index barcodes were introduced with a second-round PCR using the primers listed in SI Appendix, Table S4. The PCR products were purified by gel electrophoresis (2%) using a GeneJET Gel Extraction Kit (Thermo Fisher Scientific). The purified products were examined and quantified by using Qubit 4.0 Fluorometer (Thermo Fisher Scientific) and Qubit dsDNA HS assay kit (Thermo Fisher Scientific), and subjected to high-throughput sequencing (PE150) on an Illumina Novaseq 6000 system (Novogene).
High-Throughput Amplicon-Sequencing Data Analysis.
The processed (demultiplexed, trimmed, and merged) sequencing reads were analyzed to determine the editing outcomes using CRISPResso2 (48) by aligning the sequenced amplicons to the reference and the expected HDR amplicons. The quantification window was set to 10 bp surrounding the expected cut site to better capture diverse editing outcomes but substitutions were ignored to avoid the inclusion of sequencing errors. Only clean reads without mismatches to the expected amplicon were considered for HDR quantification; Clean reads containing indels that partially matched the expected amplicons were included in the overall reported indel frequency.
GIS-seq for Off-Target Detection.
Genome-wide, unbiased off-target analysis for dsDNA or odsDNA KI was performed following a modified GIS-seq protocol (39) (SI Appendix, Fig. S20). In brief, for all samples, the HEK293T cells were transfected with Cas9 RNPs with 1,110 bp dsDNA and 1,110 bp odsDNA donors, respectively, as detailed in the nucleofection protocol above. We exploited the beads-based method for genomic DNA extraction (ZYMO Research) on the third day of transfection. Following the instructions of NGS OnePot DNA Library Prep Kit for Illumina (Yeasen Biotech), 500 ng genomic DNA was enzymatically fragmented for 12 min by smearase fragmentase to attain 200 to 500 bp fragments. The adaptor primers were custom-synthesized and pre-annealed (SI Appendix, Table S13), and were ligated to the fragmented genomic cDNA using T4 ligase. First-round PCR was performed using a pair of primers against the adaptor on one end and the KI fragment on the other end (with plus and minus for 5′ and 3′ junctions respectively, primers sequences are provided in the SI Appendix, Table S13), similar to the process described in GUIDE-seq/iGUIDE-seq (49, 50). The first-round PCR products ranging between 200~500 bp were purified from a 2% agarose gel using a GeneJET Gel Extraction Kit (Thermo Fisher Scientific). A second-round PCR was carried out to remove nonspecific amplification and to introduce the barcoded sequencing primers. Final libraries were sequenced (PE150) using Illumina Hiseq Xten (Novogene). Sequencing data was analyzed to identify any off-target insertion events, and all analysis codes were stored on Github (detailed in the Data, Materials, and Software Availability section).
Statistical Analysis.
GraphPad Prism v.8.0 software and the R package (v3.1) were used to analyze the data. Unless otherwise stated, all experiments were conducted with biological triplicates. The student’s t test was used for calculation of significance and data were aggregated for display and analysis. Source data are available online.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
This work was supported by the Ministry of Science and Technology of China (2019YFA0802600 and 2020YFA0710700), the National Natural Science Foundation of China (Nos. 21991132, 52033010, 52021002, 31970793 and 32170856).
Author contributions
W.H., J.B., and H.L. designed research; W.H., Z.L., Y.G., K.H., W.L., and L.G. performed research; W.H., M.H., X.Y., J.Z., C.L., and D.Y. contributed new reagents/analytic tools; W.H. and C.X. analyzed data; and W.H., J.B., and H.L. wrote the paper.
Competing interests
The authors have submitted a patent application based on the results reported in this paper.
Footnotes
This article is a PNAS Direct Submission. B.H. is a guest editor invited by the Editorial Board.
Contributor Information
Jianqiang Bao, Email: jqbao@ustc.edu.cn.
Haojun Liang, Email: hjliang@ustc.edu.cn.
Data, Materials, and Software Availability
All data supporting the findings of this study are publicly available. All next-generation sequencing data, including data from the targeted endogenous genome loci profiling and on/off-target analysis, are deposited to the NCBI Sequence Read Archive database with accession code PRJNA905048 (51).
Supporting Information
References
- 1.Chen P. J., Liu D. R., Prime editing for precise and highly versatile genome manipulation. Nat. Rev. Genet. 24, 161–177 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nambiar T. S., Baudrier L., Billon P., Ciccia A., CRISPR-based genome editing through the lens of DNA repair. Mol. Cell 82, 348–388 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verma P., Greenberg R. A., Noncanonical views of homology-directed DNA repair. Genes Dev. 30, 1138–1154 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van de Kooij B., Kruswick A., van Attikum H., Yaffe M. B., Multi-pathway DNA-repair reporters reveal competition between end-joining, single-strand annealing and homologous recombination at Cas9-induced DNA double-strand breaks. Nat. Commun. 13, 5295 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Savic N., et al. , Covalent linkage of the DNA repair template to the CRISPR-Cas9 nuclease enhances homology-directed repair. Elife 7, e33761 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma M., et al. , Efficient generation of mice carrying homozygous double-floxp alleles using the Cas9-Avidin/Biotin-donor DNA system. Cell Res. 27, 578–581 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cruz-Becerra G., Kadonaga J. T., Enhancement of homology-directed repair with chromatin donor templates in cells. Elife 9, e55780 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gu B., Posfai E., Rossant J., Efficient generation of targeted large insertions by microinjection into two-cell-stage mouse embryos. Nat. Biotechnol. 36, 632–637 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Yu Y., et al. , An efficient gene knock-in strategy using 5’-modified double-stranded DNA donors with short homology arms. Nat. Chem. Biol. 16, 387–390 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu Y., et al. , Targeted, efficient sequence insertion and replacement in rice. Nat. Biotechnol. 38, 1402–1407 (2020). [DOI] [PubMed] [Google Scholar]
- 11.Renaud J. B., et al. , Improved genome editing efficiency and flexibility using modified oligonucleotides with TALEN and CRISPR-Cas9 nucleases. Cell Rep. 14, 2263–2272 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Gutierrez-Triana J. A., et al. , Efficient single-copy HDR by 5’ modified long dsDNA donors. Elife 7, e39468 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leonetti M. D., Sekine S., Kamiyama D., Weissman J. S., Huang B., A scalable strategy for high-throughput GFP tagging of endogenous human proteins. Proc. Natl. Acad. Sci. U.S.A. 113, E3501–E3508 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang Y., Ren J., Li C. C., Establishment of a GFP::LMNB1 knockin cell line (CSUi002-A-1) from a dystonia patient-specific iPSC by CRISPR/Cas9 editing. Stem Cell Res. 55, 102505 (2021). [DOI] [PubMed] [Google Scholar]
- 15.Shy B. R., et al. , High-yield genome engineering in primary cells using a hybrid ssDNA repair template and small-molecule cocktails. Nat. Biotechnol. 41, 521–531 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quadros R. M., et al. , Easi-CRISPR: A robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biol. 18, 92 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roth T. L., et al. , Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 559, 405–409 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanca O., et al. , An efficient CRISPR-based strategy to insert small and large fragments of DNA using short homology arms. Elife 8, e51539 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Q., et al. , Catalytic DNA-assisted mass production of arbitrary single-stranded DNA. Angew. Chem. Int. Ed. Engl. 62, e202212011 (2023). [DOI] [PubMed] [Google Scholar]
- 20.Minev D., et al. , Rapid in vitro production of single-stranded DNA. Nucleic Acids Res. 47, 11956–11962 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mabuchi A., et al. , ssDNA is not superior to dsDNA as long HDR donors for CRISPR-mediated endogenous gene tagging in human diploid cells. bioRxiv [Preprint] (2022), 10.1101/2022.06.01.494308 (Accessed 1 June 2022). [DOI] [PMC free article] [PubMed]
- 22.Xue C., Greene E. C., DNA repair pathway choices in CRISPR-Cas9-mediated genome editing. Trends Genet. 37, 639–656 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sartori A. A., et al. , Human CtIP promotes DNA end resection. Nature 450, 509–514 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sfeir A., Symington L. S., Microhomology-mediated end joining: A back-up survival mechanism or dedicated pathway? Trends Biochem. Sci. 40, 701–714 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Allen F., et al. , Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol. 37, 64–72 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paix A., et al. , Precision genome editing using synthesis-dependent repair of Cas9-induced DNA breaks. Proc. Natl. Acad. Sci. U.S.A. 114, E10745–E10754 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Canaj H., et al. , Deep profiling reveals substantial heterogeneity of integration outcomes in CRISPR knock-in experiments. bioRxiv [Preprint] (2019), 10.1101/841098 (Accessed 13 November 2019). [DOI]
- 28.Liang X., Potter J., Kumar S., Ravinder N., Chesnut J. D., Enhanced CRISPR/Cas9-mediated precise genome editing by improved design and delivery of gRNA, Cas9 nuclease, and donor DNA. J. Biotechnol. 241, 136–146 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Richardson C. D., Ray G. J., DeWitt M. A., Curie G. L., Corn J. E., Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 34, 339–344 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Yeh C. D., Richardson C. D., Corn J. E., Advances in genome editing through control of DNA repair pathways. Nat. Cell Biol. 21, 1468–1478 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Ohle C., et al. , Transient RNA-DNA hybrids are required for efficient double-strand break repair. Cell 167, 1001–1013 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Liu S., et al. , RNA polymerase III is required for the repair of DNA double-strand breaks by homologous recombination. Cell 184, 1314–1329 (2021). [DOI] [PubMed] [Google Scholar]
- 33.Kan Y., Ruis B., Takasugi T., Hendrickson E. A., Mechanisms of precise genome editing using oligonucleotide donors. Genome Res. 27, 1099–1111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsai S. Q., et al. , GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33, 187–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anzalone A. V., et al. , Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nelson J. W., et al. , Engineered pegRNAs improve prime editing efficiency. Nat. Biotechnol. 40, 402–410 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin Q., et al. , High-efficiency prime editing with optimized, paired pegRNAs in plants. Nat. Biotechnol. 39, 923–927 (2021). [DOI] [PubMed] [Google Scholar]
- 38.Mathis N., et al. , Predicting prime editing efficiency and product purity by deep learning. Nat. Biotechnol. 10.1038/s41587-022-01613-7 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang C., et al. , Microbial single-strand annealing proteins enable CRISPR gene-editing tools with improved knock-in efficiencies and reduced off-target effects. Nucleic Acids Res. 49, e36 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aird E. J., Lovendahl K. N., St Martin A., Harris R. S., Gordon W. R., Increasing Cas9-mediated homology-directed repair efficiency through covalent tethering of DNA repair template. Commun. Biol. 1, 54 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lovendahl K. N., Hayward A. N., Gordon W. R., Sequence-directed covalent protein-DNA linkages in a single step using HUH-Tags. J. Am. Chem. Soc. 139, 7030–7035 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nishimasu H., et al. , Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156, 935–949 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sakuma T., Nakade S., Sakane Y., Suzuki K. T., Yamamoto T., MMEJ-assisted gene knock-in using TALENs and CRISPR-Cas9 with the PITCh systems. Nat. Protoc. 11, 118–133 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Davis L., Maizels N., Homology-directed repair of DNA nicks via pathways distinct from canonical double-strand break repair. Proc. Natl. Acad. Sci. U.S.A. 111, E924–E932 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao Z., Shang P., Sage F., Geijsen N., Ligation-assisted homologous recombination enables precise genome editing by deploying both MMEJ and HDR. Nucleic Acids Res. 50, e62 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Labun K., et al. , CHOPCHOP v3: Expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 47, W171–W174 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brinkman E. K., et al. , Easy quantification of template-directed CRISPR/Cas9 editing. Nucleic Acids Res. 46, e58 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clement K., et al. , CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 37, 224–226 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Malinin N. L., et al. , Defining genome-wide CRISPR-Cas genome-editing nuclease activity with GUIDE-seq. Nat. Protoc. 16, 5592–5615 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nobles C. L., et al. , iGUIDE: An improved pipeline for analyzing CRISPR cleavage specificity. Genome Biol. 20, 14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Han, et al. , HEK293T cells Raw sequence reads-The genome of HEK293T cells after gene knock-in. NCBI BioProject. https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA905048. Deposited 24 November 2022. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All data supporting the findings of this study are publicly available. All next-generation sequencing data, including data from the targeted endogenous genome loci profiling and on/off-target analysis, are deposited to the NCBI Sequence Read Archive database with accession code PRJNA905048 (51).