

Significance
The inositol 1,4,5-trisphosphate receptor (IP3R) Ca2+ release channel plays a crucial role in Ca2+ signaling and its regulation is fundamental for defining the appropriate Ca2+ release. Here, we focused on the regulation of IP3R by ER luminal oxidoreductases. We showed that two of the intraluminal cysteines of IP3R are involved in its tetramer formation and the other two in the regulation of IP3R by oxidation and reduction with ERp46 and ERdj5, respectively. Importantly, ERdj5 reciprocally regulates the Ca2+ channel (IP3R) and pump (SERCA2b) by sensing the intraluminal Ca2+ concentration. Thus, we clearly demonstrated that the cellular concentration of second messenger Ca2+ is finely tuned by the redox state in the ER, which is affected by cellular stress, aging, and disease.
Keywords: redox regulation, calcium homeostasis, PDI family members
Abstract
Inositol 1,4,5-trisphosphate receptors (IP3Rs) are one of the two types of tetrameric ion channels that release calcium ion (Ca2+) from the endoplasmic reticulum (ER) into the cytosol. Ca2+ released via IP3Rs is a fundamental second messenger for numerous cell functions. Disturbances in the intracellular redox environment resulting from various diseases and aging interfere with proper calcium signaling, however, the details are unclear. Here, we elucidated the regulatory mechanisms of IP3Rs by protein disulfide isomerase family proteins localized in the ER by focusing on four cysteine residues residing in the ER lumen of IP3Rs. First, we revealed that two of the cysteine residues are essential for functional tetramer formation of IP3Rs. Two other cysteine residues, on the contrary, were revealed to be involved in the regulation of IP3Rs activity; its oxidation by ERp46 and the reduction by ERdj5 caused the activation and the inactivation of IP3Rs activity, respectively. We previously reported that ERdj5 can activate the sarco/endoplasmic reticulum Ca2+-ATPase isoform 2b (SERCA2b) using its reducing activity [Ushioda et al., Proc. Natl. Acad. Sci. U.S.A. 113, E6055–E6063 (2016)]. Thus, we here established that ERdj5 exerts the reciprocal regulatory function for IP3Rs and SERCA2b by sensing the ER luminal Ca2+ concentration, which contributes to the calcium homeostasis in the ER.
Intracellular Ca2+ is one of the most crucial signaling ion in the cytosol, and it regulates various cellular functions, from fertilization to cell death (1). The cytosolic Ca2+ concentration ([Ca2+]i) is strictly controlled in both a time- and space-dependent manner and is amplified in signal transduction pathways (2, 3). Cytosolic Ca2+ is internalized from the extracellular space or released from intracellular Ca2+ stores, such as the endoplasmic reticulum (ER) (4). Disruption of intracellular calcium homeostasis is thought to be a cause of apoptosis and neurodegenerative disease (5–7). In a typical signal transduction pathway, a G protein-coupled receptor (GPCR) activates the production of inositol 1,4,5-trisphosphate (IP3) as a messenger. Inositol 1,4,5-trisphosphate receptors (IP3Rs) are Ca2+ release channels on the ER membrane that play crucial roles in generating Ca2+ waves and oscillations in the cytosol (8). The Ca2+ concentration in the ER ([Ca2+]ER) is maintained at a high level (approximately 10,000-fold higher than [Ca2+]i) by sarco-/endoplasmic reticulum Ca2+-ATPases (SERCAs) in an ATPase activity-dependent manner (9). Major ER-resident molecular chaperones are calcium-binding proteins that contribute to the maintenance of the total amount of calcium taken up by SERCAs. Interestingly, the binding of calcium to molecular chaperones and enzymes in the ER is required for their functions and enzymatic activities. In particular, binding immunoglobulin protein (BiP) and calreticulin (CRT) are known to undergo structural changes upon calcium binding and dissociation, and calcium binding is required for substrate binding that accelerates oxidative folding (10, 11). Protein disulfide isomerase (PDI) is known as a calcium-binding protein, and perturbation of the [Ca2+]ER diminishes its chaperone activity as well as its oxidoreductase activity, resulting in the perturbation of redox conditions in the ER (12, 13). These results suggest that the maintenance of Ca2+ homeostasis in the ER also significantly affects protein quality control. Notably, oxidative stress was reported to cause an abnormal increase in the calcium released from the ER through IP3Rs, which was measured by a single-channel recording system (14). In contrast, reduced and ER stress conditions caused a decrease in the amount of Ca2+ released from the ER (15–17). Thus, the activity of IP3Rs depends on the redox environment, but the effect of the redox state of the IP3Rs on their channel activity under physiological conditions has not been shown to date.
PDI is a typical member of a family of structurally related oxidoreductases in the ER, so-called PDI family proteins (PDIs), which catalyze disulfide bond formation, reduction, or isomerization of newly synthesized proteins in the ER. Nearly 20 members of the PDIs and ER oxidoreductin 1 (ERO1) constitute a redox network in the ER that is involved in maintaining the ER redox environment and oxidative protein folding (18). Recent studies have revealed that the redox status of proteins determines the function or activity of certain proteins involved in Ca2+ regulation in the ER. The activity of SERCA2b, an ER-resident Ca2+ pump, is regulated by the redox state of a luminal cysteine pair (19). The reductases ERdj5 and SEPN1 are involved in SERCA2b reduction, resulting in its activation. ERp57 and TMX1 have been implicated in the oxidation and inactivation of SERCA2b (20–23). However, direct evidence for the oxidation of SERCA2b has not been demonstrated. Furthermore, ERp57 is involved in the regulation of stromal interaction molecule 1 (STIM1), and calcium influx into the cytosol via store-operated Ca2+ entry (SOCE) may alter the overall calcium balance in cells (24). It has also been reported that ERp44 binds to reduced IP3R1 and negatively regulates its channel activity (16). However, since ERp44 shows no reductase activity (25), it is unclear how IP3R1 is reduced. Hence, the involvement of PDIs in ER calcium homeostasis has been reported in several studies, and yet the overall picture is still unclear.
All subtypes of IP3Rs (IP3R1–3) have an IP3-binding core in their large cytosolic region. The channel domain contains six transmembrane domains (TMDs), and three loops are exposed to the ER lumen (26). The third luminal loop contains four cysteine residues close to the channel pore. We focused on these four cysteine residues in the ER luminal site to elucidate the molecular mechanism of redox-mediated IP3Rs regulation. Here, we addressed three critical questions regarding the regulation of IP3Rs by the redox state; what is the role of the four cysteine residues in the third loop of the IP3Rs, which member(s) of the PDI family is involved in the regulation of the redox state of the IP3Rs in a cell, and finally, does the redox state of IP3Rs in the ER alter IP3R channel activity?
Results
ER-Resident Oxidoreductases Modulate IICR.
To identify the oxidoreductase of IP3Rs, we focused on soluble PDIs, which contain thioredoxin-like domains with redox-active motifs (Cys-X-X-Cys, CXXC) (Fig. 1A). As a strategy to identify substrates of these PDIs, immunoprecipitation of cell lysate was performed using mutant enzymes in which the second cysteine in the CXXC motif was converted to alanine (CXXA), which establishes an intermolecular, uncleavable disulfide bond with the cysteine residues of a substrate (27). Here, we adopted this approach to identify potential oxidoreductase(s) of IP3R1. We confirmed that the CA mutants of the indicated PDIs, except ERp72, bound to IP3R1 (Fig. 1B). Next, to evaluate the effects of the PDIs on IP3-induced calcium release (IICR), we used the CRISPR/Cas9 system to establish PDI-deficient cell lines (28). We confirmed the successful knockout (KO) of each PDI by immunoblotting with specific antibodies to determine each PDI expression level (SI Appendix, Fig. S1A). By performing immunoblotting, we excluded the possibility that a deleted PDI affected the expression and/or folding of the IP3Rs, bradykinin (BK) receptors, or SERCA2 (SI Appendix, Fig. S1A).
Fig. 1.
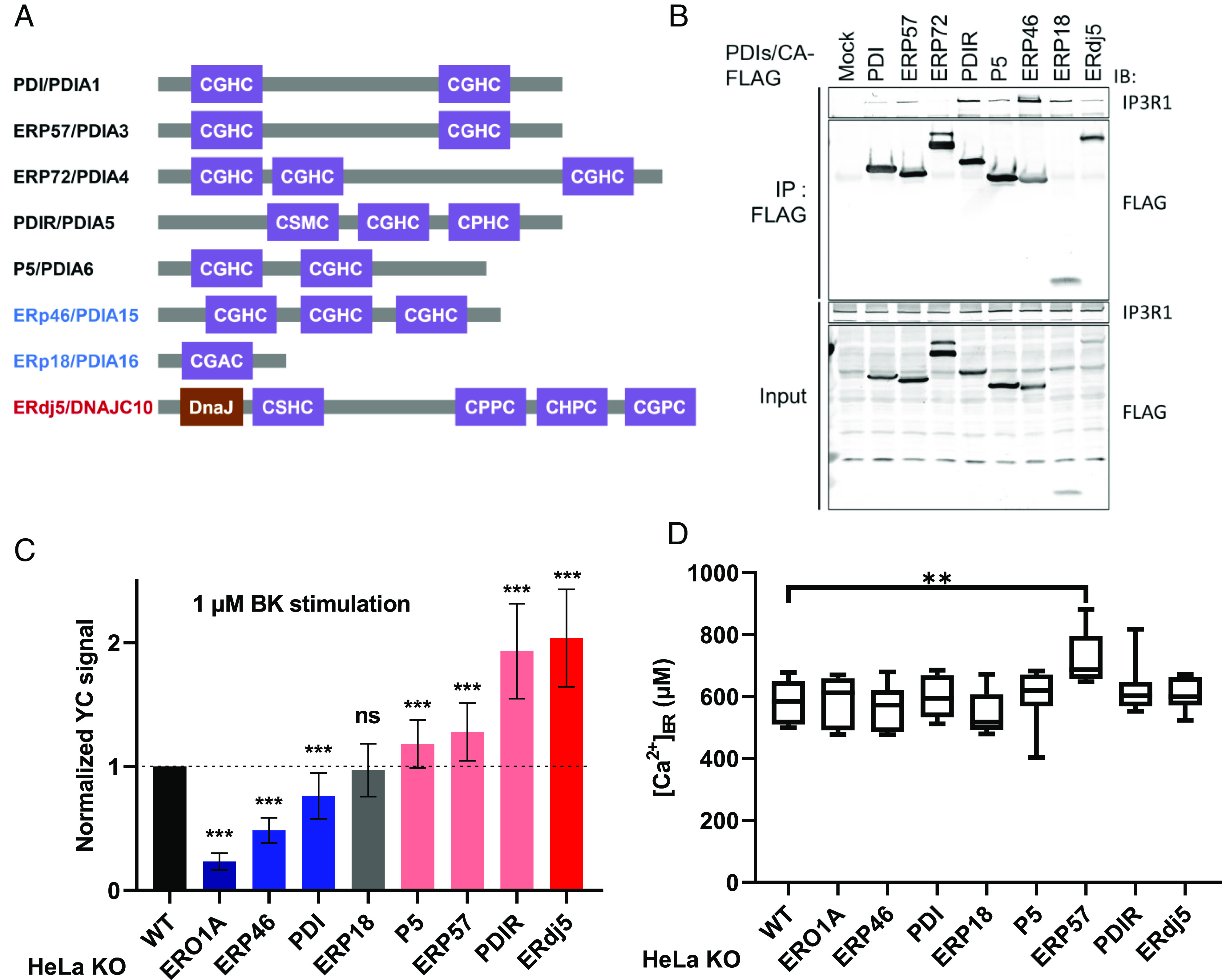
The modulation of IICR through PDI family proteins. (A) PDI family proteins (PDIs) examined in this work. The redox-active thioredoxin motif (CXXC) and DnaJ domain are shown. Black: oxidase, reductase, and isomerase. Blue: oxidase. Red: reductase. (B) The interaction of PDIs with IP3Rs. Immunoprecipitates were prepared from HEK-293T cells transiently expressing the indicated FLAG-tagged PDIs and detected by immunoblotting with the indicated antibodies. (C) Quantification of BK-induced IICR in HeLa WT cells or PDI-deficient cells. After transfection with YC3.6, the cells were stimulated with 1 μM BK, and the signal was normalized to the measured peak amplitude. The data present the means ± SD. See SI Appendix, Fig. S1 A and B for the results validating the PDI-deficient cells. (D) The absolute [Ca2+]ER in each KO cell line was calculated from measures of GEM-CEPIA1er ratio. The steady-state [Ca2+]ER in each KO line was determined by Ca2+ titration after sequential treatment with egtazic acid (EGTA) and CaCl2. The horizontal line within the box represents the median value, the upper and lower edges of the box represent 75 and 25% values, and the whiskers represent the total range. ns, not significant; **P < 0.01.
Next, we performed imaging with the fluorescent Ca2+ indicator Yellow Cameleon 3.6 (YC3.6) to examine whether the deletion of any of the PDIs affects Ca2+ release from the ER via IP3Rs. HeLa cells were treated with BK, a GPCR ligand, and BK-induced calcium release from the ER via IP3Rs was examined before and after the knockout of each PDI (Fig. 1C and SI Appendix, Fig. S1 A and B). IICR was the most profoundly decreased by the deletion of the oxidase ERp46 and moderately decreased by deletion of PDI. Disruption of ERO1, a PDI oxidation enzyme, also caused a decrease in IICR, which is consistent with a previous report (29). In contrast, IICR was increased by the deletion of reductase ERdj5 and PDIR.
To evaluate the regulation of IP3Rs by PDIs, however, it is necessary to ensure that IICR is independent of the changes in calcium concentration in the ER ([Ca2+]ER) in the steady state. Therefore, we examined changes in the stable [Ca2+]ER after the knockout of each PDI and found that the [Ca2+]ER was elevated only in ERp57-deficient cells (Fig. 1D and SI Appendix, Fig. S1A). Furthermore, we examined the effect of ERO1, which receives electrons from PDI and converts molecular oxygen to hydrogen peroxide (H2O2). The oxidative environment of the ER depends on the equilibrium of the redox states of PDIs and low molecular weight thiol compounds such as glutathione. ERO1-produced H2O2 also contributes to the maintenance of an oxidative environment in the ER. Deletion of ERO1 has been reported to inhibit IICR activity (29), which suggested that disruption of the oxidative ER state causes IICR insensitivity (30). To estimate the redox environments in the ER, we used the Forster resonance energy transfer (FRET)-based redox sensor CY-RL7 (31, 32) which has been fine-tuned to sense oxidative organelles in living cells. The relative oxidative ratio was calculated on the basis of the change in the diamide/dithiothreitol (DTT) ratio (SI Appendix, Fig. S1C). No significant change in the stable redox state of the ER was observed by the deletion of any of the PDIs except that of ERO1α. These results are consistent with the previous study (33). The results suggested that the reduction of IP3Rs disturbed the activity of Ca2+ release. Hence, we can conclude that ERp46 and ERdj5 regulate IICR activity in opposite ways without altering the ER redox environment.
Four Cysteine Residues in the Luminal Loop Are Involved in Both IP3R Channel Formation and Regulation.
IP3Rs have a large cytosolic domain in the N terminus that contains the ligand-binding domain and six TMDs that contain the channel pore region near the C terminus. A major ER-luminal region located beside the channel pore has four cysteine residues (C1 to C4; Cys2505, Cys2513, Cys2536, Cys2542, Uniprot ID Q14643-1) (Fig. 2A). We constructed human IP3R1 mutants in which a cysteine was replaced with alanine (e.g., IP3R1/ACCC is the C1 mutation) and transfected these mutants into IP3R triple-knockout (IP3R TKO) cells to analyze channel activity. To verify the channel activity, we measured each IICR using YC3.6. Although IICR in the IP3R TKO cells was restored by the expression of IP3R1/wild type (WT), the mutation of C3 and C4 (CCAC and CCCA, respectively) and of both C3 and C4 (CCAA) in the internal helix of the channel pore domain failed to restore channel activity (Fig. 2B). On the other hand, transfection with the C1 and C2 mutant (AACC) significantly restored IICR activity, by approximately 70% compared with that of the WT (Fig. 2B). Since IP3Rs exert channel activity as a tetrameric complex, we examined whether any of the cysteine-substituted mutations in the luminal region affected tetramer formation. From the results of an analysis performed after sucrose density centrifugation, we confirmed that both endogenous IP3R1 and transiently expressed IP3R1 presented as homotetramers with a size of approximately 1.2 MDa. On the other hand, the immunoblot band of IP3R1/AAAA obtained through sucrose density centrifugation appeared with a peak in fractions 7 to 9, corresponding to the size of the dimer (approximately 0.6 MDa). IP3R1/AACC was deposited in the tetramer sediment fraction, whereas the IP3R1/CCAC and CCCA mutants were in the dimer fraction (Fig. 2C). Taken together, the results shown in Fig. 2B suggest that the loss-of-function of C3 and C4 in mutant IP3R1 was induced by the inability to form IP3R tetramers. On the other hand, the supposition that C1–C2 cysteine residues form intramolecular disulfide bonds has been supported by past structural analyses (34–36). Therefore, the role of the cysteine residues in the luminal loop can be divided into two different functional pairs.
Fig. 2.
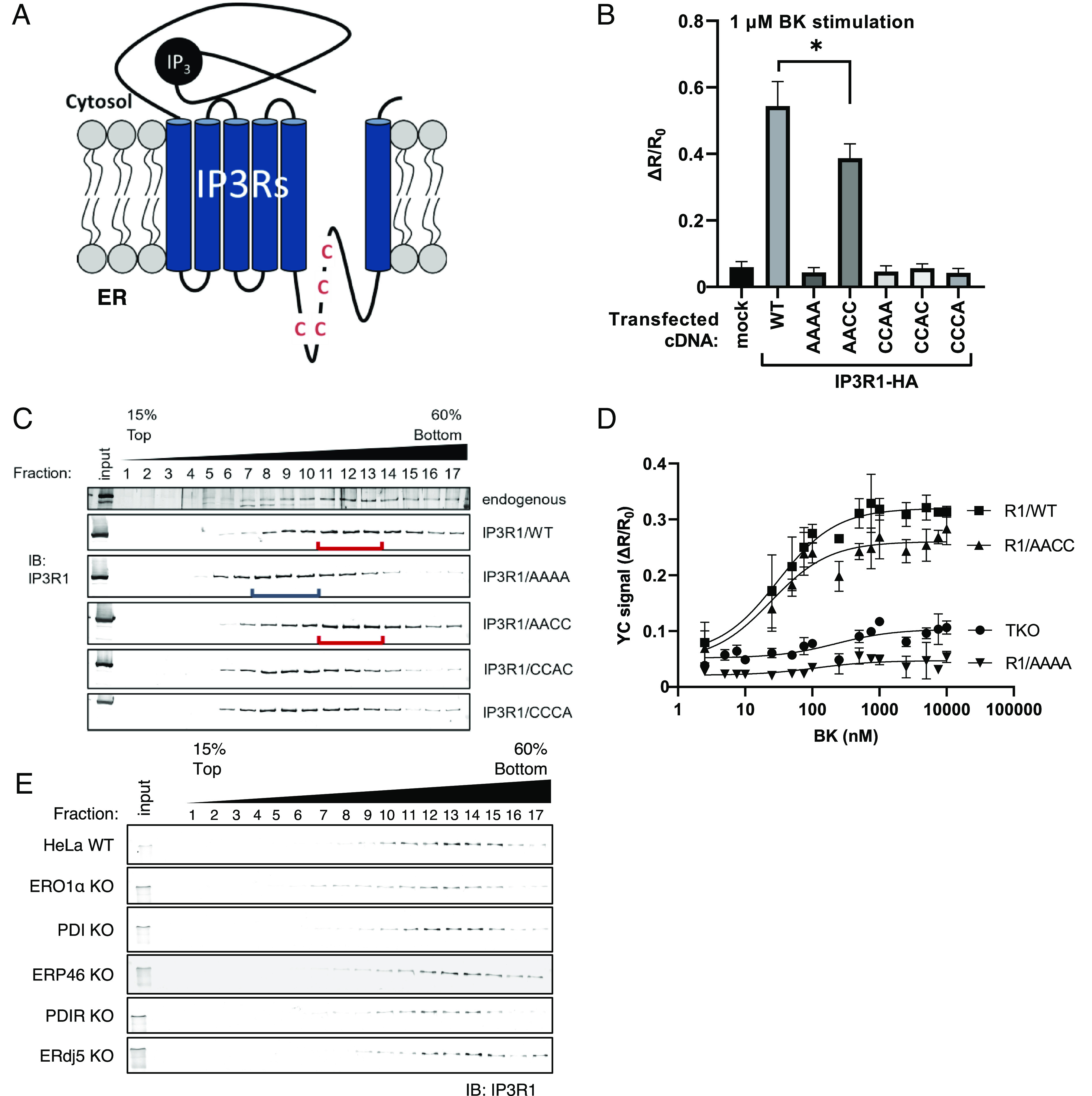
Cysteine residues in the luminal loop regulate tetrameric channel formation and channel activity. (A) IP3Rs contain an IP3-binding region in the cytoplasm, six TMDs, and four luminal cysteine residues in the vicinity of the channel pore. (B) IICR activity of each IP3R cysteine mutant. After transfection of the indicated cDNA into IP3R TKO cells, the cells were stimulated with 1 μM BK. IICR in each cell line was measured using Yellow Cameleon 3.6 (YC3.6) with a Varioskan LUX fluorescent plate reader. The average peak amplitudes of the calcium response are shown. The data present the means ± SD. *P < 0.05. (C) The fractionation of IP3R by the sucrose density gradient. After cell lysis with 1% Triton X-100, the cell lysates were fractionated by centrifugation on a 15 to 60% linear sucrose gradient. The proteins in each fraction were separated by SDS-PAGE and immunoblotted with an anti-IP3R1 antibody. Fraction nos. 7 to 9 have molecular weights corresponding to IP3R dimers, and fraction nos. 11 to 13 have molecular weights corresponding to IP3R tetramers. The red/blue lines indicate the peak fractions. (D) The dose–response curves of IICR after cell treatment with BK. The mean peak amplitudes of the YC3.6 signal (±SEM) after BK stimulation are shown. (E) The fractionation of endogenous IP3R in the indicated KO cells by sucrose density gradient. Sucrose density gradient fractionation and protein detection were performed with each KO cell under the same conditions as those described in C.
We next analyzed the role of the cysteine pairs with respect to calcium channel activity. Calcium release from the ER into the cytosol was measured in the presence of different concentrations of BK (Fig. 2D). While Ca2+ release in the TKO cells or the TKO cells transfected with the IP3R1/AAAA mutant was not observed, IP3R1/WT expressed in the IP3R TKO cells recovered Ca2+ release in a BK dose-dependent manner. However, the effect of IP3R1/AACC, a reduced form-mimic mutant, was lower than that of IP3R1/WT. As the IP3R1/AACC mutant was shown to form a tetramer, C1 and C2 were considered to be involved in the regulation of Ca2+ release. Notably, tetramer formation of IP3R1 was not affected by the knockout of any of the PDIs (Fig. 2E). Taken together with the results shown in Fig. 1C, it can be concluded that PDI family oxidoreductases are involved in the modulation of the redox state of C1 and/or C2 located in the ER luminal portion of IP3R1.
Oxidation of IP3R1 Enhances IICR.
As shown in Fig. 1C, some ER-resident oxidoreductases are suggested to be involved in the regulation of IP3Rs. In particular, the deletion of the oxidases ERp46, PDI, and ERO1 profoundly suppressed the activity of IP3Rs. A previous report has revealed an electron cascade between proteins in which ERp46 takes electrons from a substrate and transfers them to a PDI-ERO1 hub complex (37). To confirm the regulation of IP3Rs by this oxidative cascade, ERp46, Ero1, and PDI were overexpressed in IP3R TKO cells stably expressing IP3R1, and IICR was measured by YC 3.6 analysis. Consistent with the results shown in Fig. 1C, IICR was elevated by the overexpression of ERO1, PDI, or ERp46 (Fig. 3A and SI Appendix, Fig. S2A). Next, to investigate whether the elevation of IICR by these oxidases depends on the C1-C2 pair in the luminal loop of IP3R1, we established a cell line that stably expressing IP3R1/AACC in IP3R TKO cells. In contrast to the effect of the stable IP3R1/WT-expressing cells, the effect of the overexpression of these oxidases on IICR was not observed in the IP3R1/AACC stably expressing cells (Fig. 3B and SI Appendix, Fig. S2B). Additionally, we confirmed that the [Ca2+]ER was not changed by the overexpression of any of these oxidases by observing the calcium leakage from the ER in the cells treated with thapsigargin (Tg) (Fig. 3 A and B).
Fig. 3.

ER-resident oxidases activate the calcium-releasing activity of IP3R1. (A and B) The effect of ER oxidoreductin 1 (ERO1), PDI, and ERp46 on IP3-IICR. We established stable IP3 receptor 1 (IP3R1)/WT (A) or IP3R1/AACC (B) cell lines in IP3R TKO cells. After the transfection of the indicated FLAG-tagged PDIs, the cells were stimulated with 2 μM BK or 1 μM thapsigargin (Tg). IICR was measured by DeltaVision Elite with Yellow Cameleon 3.6 (YC3.6). Bar graph showing the mean amplitude (±SD) of the BK-induced Ca2+ peak (Upper) or Tg-stimulated Ca2+ leakage (Lower). *P < 0.05 and ***P < 0.001. (C) After transfection of the indicated FLAG-tagged PDIs, immunoprecipitates were prepared from lysates of cells stably expressing hemagglutinin (HA)-tagged IP3R1, and proteins were detected by immunoblotting using an antibody against the indicated peptide tags. (D) Schematic model of ERp46-mediated activation of IP3Rs through the ERO1-PDI hub complex. Red lines indicate the direction of electron transfer. Broken lines indicate a weak interaction or presumed electron transport.
Then, we examined the contribution of the C1–C2 pair in IP3R1 to receptor binding to PDI or ERp46. Through an immunoprecipitation analysis, the C1–C2 pair was found to be essential for the interaction of IP3R1 with ERp46 but not with PDI (Fig. 3C). Previous reports suggest that the PDI-ERO1 hub complex efficiently receives electrons from ERp46 (37, 38), causing the oxidation of ERp46, which subsequently oxidizes the C1–C2 pair of IP3R1. From these results, we concluded that ERp46 is a major oxidase for the activation of IP3R1. Thus, we propose a model in which the ER redox cascade composed of ERO1, PDI and ERp46 reinforces the activation of IP3R1 channel activity (Fig. 3D).
ERdj5 Is a Noncompetitive Negative Regulator of IP3R1.
The Fig. 1C shows that the disruption of two oxidoreductases, ERdj5 and PDIR, caused the upregulation of IP3Rs activity. While disruption of PDIR increased IICR activity, we concluded that PDIR is not a direct regulator of IICR because it interacted with IP3R1 independently of its regulatory cysteine residues (SI Appendix, Fig. S3A). Our previous report revealed that ERdj5 regulates the SERCA2b calcium pump through its reductase activity in response to a decrease in the [Ca2+]ER and contributes to the maintenance of calcium homeostasis in the ER (20). In this study, we verified the possibility that ERdj5 function as a negative regulator of IP3R1 by reducing the C1–C2 pair of IP3R1 that had been oxidized by ERp46.
To confirm the effect of ERdj5 on IICR, BK-induced IICR was calculated on the basis of a dose-response curve. As shown in Fig. 4A, BK activated the IICR of HeLa cells in a dose-dependent manner, and the activation of IICR was clearly upregulated upon the deletion of ERdj5, suggesting that ERdj5 negatively regulated the IICR. In this process, ERdj5 did not alter the ligand-channel affinity; no change in the half-maximal effective concentration (EC50) was observed, but there was a change in the maximum response.
Fig. 4.
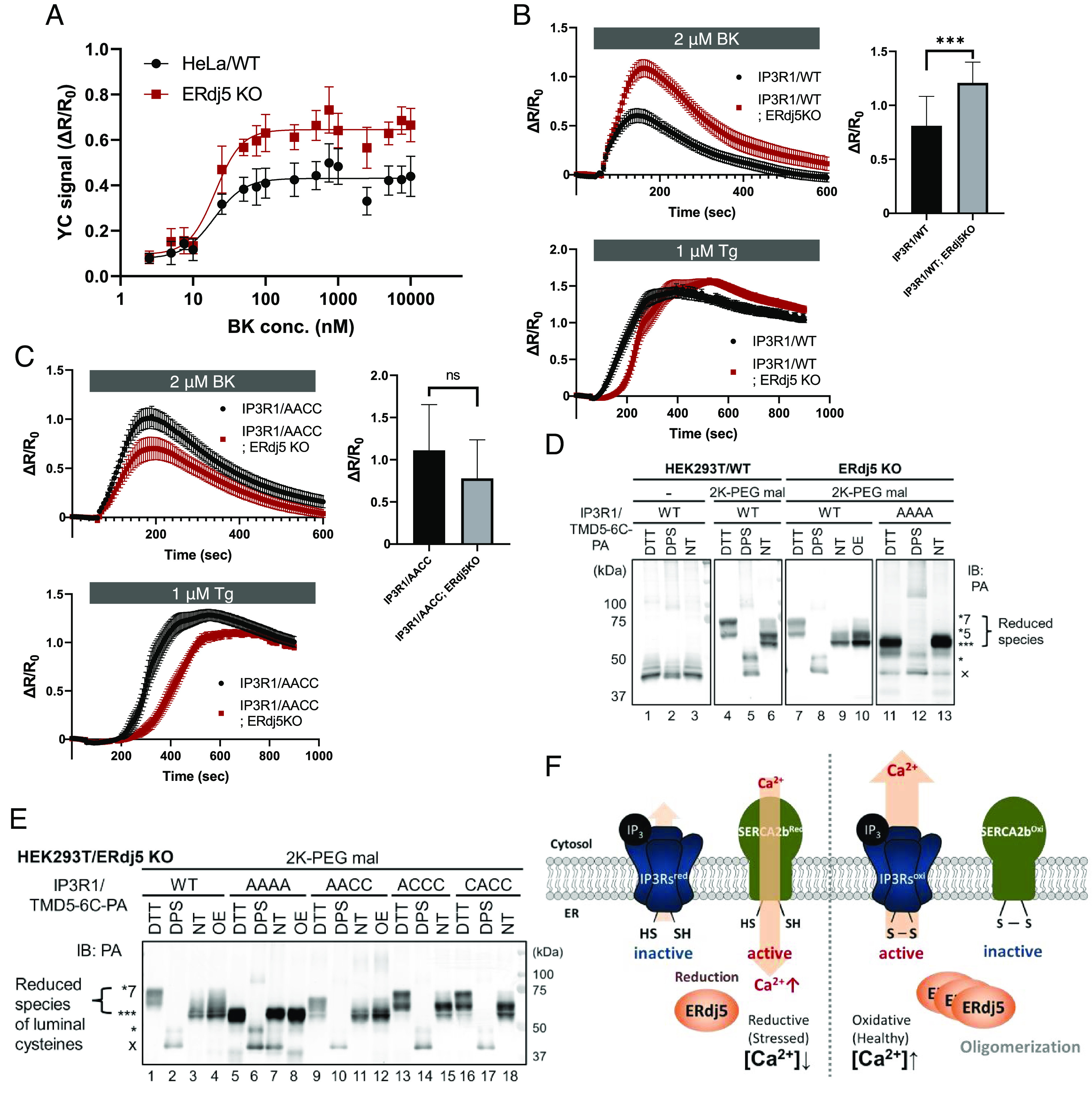
ERdj5 is a noncompetitive modulator of IP3Rs. (A) Effect of endogenous ERdj5 on IP3-IICR. After transfection with Yellow Cameleon 3.6 (YC 3.6) into WT or ERdj5-deficient HeLa cells, the cells were stimulated with BK, and the signal was normalized to the measured peak amplitude. Dose-response curves of the mean peak amplitudes are shown. (B and C) Effect of ERdj5 in IP3 receptor 1 (IP3R1)/WT- or IP3R1/AACC mutant-expressing cells on BK-induced Ca2+ release (Upper) or thapsigargin (Tg)-stimulated Ca2+ leakage (Lower). Each tracing represents the mean ± SEM of three independent experiments. ns, not significant; ***P < 0.001. (D and E) Redox state of PA-tagged IP3R1/TMD5-6C. After transfection with the indicated cDNA into HEK-293T cells, the cells were incubated in the presence or absence of 10 mM DTT or 1 mM dipyridyl disulfide (DPS) after fixing the redox reaction with trichloroacetic acid (TCA) and solubilized in the presence or absence of 10 mM maleimide-polyethylene glycol (2K-PEG mal). Proteins were separated by SDS-PAGE, and immunoblotting was performed to detect IP3R1/TMD5-6 with an anti-PA antibody. OE: overexpression of ERdj5. (F) ERdj5-mediated maintenance of Ca2+ homeostasis in the ER.
Then, we examined whether the suppression of IP3Rs channel activity is caused by the ERdj5-induced reduction of the C1–C2 disulfide bond in IP3R1 by disrupting ERdj5 in cells stably expressing IP3R1/WT or the AACC mutant (SI Appendix, Fig. S3B). BK-induced IICR in the IP3R1/WT- or IP3R1/AACC-expressing cells was monitored in the presence or absence of ERdj5. As expected, the deletion of ERdj5 significantly increased the activation of IICR in the IP3R1/WT-expressing cells (Fig. 4B, Upper graph). However, the effect was not due to a decrease in releasable Ca2+ in the ER because the treatment of these cells with Tg, an inhibitor of SERCA2b, did not affect calcium leakage (Fig. 4 B, Lower graph). In contrast, the activation of IICR by ERdj5-KO was not observed in the IP3R1/AACC-expressing cells (Fig. 4 C, Upper graph). Considering the amount of released Ca2+ in the ER observed after Tg treatment, it can be concluded that there was no change in calcium release activity (Fig. 4 C, Lower graph). Our findings suggest that ERdj5 is a negative regulator of IP3R1, and its effect is mediated through the reduction of the regulatory C1–C2 pair.
Finally, we assessed the redox state of all luminal cysteine residues (C1, C2, C3, and C4) in IP3R1 by constructing a PA-tagged deletion mutant in which only the channel domain (IP3R1/TMD5-6C-PA) was retained (SI Appendix, Fig. S3C). A previous report confirmed that this deletion mutant forms a tetramer and shows channel activity (39). This mutant has seven cysteine residues, among which four cysteine residues (C1 to C4) are located in the luminal loop in the ER. The redox state of these cysteine residues was evaluated by band shifts caused by the modification of the free thiol groups with polyethylene glycol (PEG)-conjugated maleimide. In addition, the bands corresponding to the luminal cysteine residues were evaluated in comparison with a mutant in which all cysteine residues in the lumen were replaced with alanine residues (TMD5-6C/AAAA).
All three thiol groups of the TMD5-6C/AAAA mutant were modified with PEG, and no effect was observed when the cells carrying this mutant were treated with the reducing agent DTT (Fig. 4D, Lanes 11 and 13). This result clearly shows that the three cysteine residues that are not in the luminal loop are always in a reduced state. This finding is consistent with the fact that these three cysteine residues are oriented toward the cytosol, which is regarded as a reducing environment. Three or five thiol group modifications were observed in TMD5-6C/WT (Fig. 4D, Lane 6), suggesting that the luminal cysteine residues are usually in the oxidized state (they are in oxidized form) or one disulfide bond pair is in the reduced state (in reduced form). On the other hand, in ERdj5-deficient cells, the number of pairs in reduced form was found to be significantly decreased (Fig. 4D, Lane 9). Moreover, it was confirmed that the overexpression of ERdj5 rescued the reduced form in ERdj5-deficient cells (Fig. 4D, Lane 10).
To examine the effect on the redox states of C1 and C2, we compared each redox state of C1 and/or C2 mutants by performing a gel shift assay (Fig. 4E). IP3R1/AAAA and IP3R1/AACC tended to be in the oxidized form in ERdj5-KO cells (Fig. 4E, Lanes 7 and 11), and the redox state of these mutants was not changed by the overexpression of ERdj5 (Fig. 4E, Lanes 8 and 12). In contrast, in cells expressed the IP3R1/ACCC and IP3R1/CACC mutants, four cysteine residues (three cysteine residues in the cytosolic domain and one cysteine residue in the luminal portion) were found to be reduced (Fig. 4E, Lanes 15 and 18). These observations clearly suggested that ERdj5 is required to reduce the C1-C2 disulfide bond of IP3R1.
Discussion
Our study demonstrated a fundamental regulatory mechanism of IP3Rs in which simple modifications such as disulfide bond formation directly changed calcium release activity. Using the CRISPR/Cas9 system, we established cell lines in which each of the 8 PDI family proteins was knocked out and evaluated the IICR in each cell line, taking into account the steady-state [Ca2+]ER and redox environments (Fig. 1 and SI Appendix, Fig. S1). Remarkably, the effect on IP3Rs activity was variable, while the steady-state [Ca2+]ER was almost unchanged except when ERp57 was deleted. Since ERp57 may act as a negative regulator of SERCA2, the increased [Ca2+]ER in the ERp57-KO cells is consistent with a previous study (22); hence, the slight increase in IICR may have been caused by the increase in the [Ca2+]ER in the ERp57-KO cells. In contrast, the effects of other PDIs on IICR may have had multiple causes. P5 is known to function cooperatively with miR-322, and when P5 expression is suppressed, the calcium uptake through SOCE tends to be increased (40). This cooperation might trigger SOCE earlier in P5-KO cells than in WT cells. PDIR is involved in the functional regulation of ATF6 (41), and suppression of its expression attenuates the induction of genes downstream of ATF6 (42). Calcium is more likely to leak from the ER in ATF6-deficient C. elegans due to the decreased expression of CRT (43). It is possible that PDIR deficiency causes the loss of ATF6 function and that the IICR activity may be increased because of the decreased expression of CRT.
The detailed mutant analysis of the cysteine residues (C1 to C4; Cys2505, Cys2513, Cys2536, Cys2542) in IP3R1 revealed the role of two functionally independent cysteine pairs. In particular, it was found, for the first time, that the C3–C4 disulfide bond is an essential element for the formation of the IP3R1 tetramer (Fig. 2). In the structural analysis, this disulfide bond was found to be formed within the subunit molecule, but further analysis is needed to determine how this disulfide bond contributes to the complex formation of the four subunits of IP3R1.
Moreover, we successfully identified the ER-resident oxidase ERp46 as an activator of IP3R1 (Fig. 3) and the reductase ERdj5 as a direct negative regulator of IP3R1 (Fig. 4). In our previous report, we demonstrated that ERdj5 activates the SERCA2 Ca2+ pump through its reductase activity (20). In addition, we clearly demonstrated that ERdj5 directly reduced the disulfide bond formed between C1 and C2 in the ER luminal portion of IP3R1, and this reduction of the disulfide bond led to IICR suppression (Fig. 4D). This redox regulation of IP3R1 appears to be an independent mechanism from the regulation of IP3R1 by phosphorylation on the cytosolic domain (SI Appendix, Fig. S3D). Thus, the four cysteine residues located in the loop between TMD5 and TMD6 in IP3R1 can be divided into two groups: one consisting of C1 and C2 is involved in the regulation of IP3R1 channel activity, and the other consisting of C3 and C4 is involved in subunit formation. Here, only the homotetramer of IP3R1 was analyzed, but the cysteine residues (C1 to C4) are also conserved in other subtypes. The reason we focused on IP3R1 is that regulation by ERp44 is an IP3R1-specific phenomenon (16), and we started our research with the possibility that redox regulation might only occur in IP3R1. Since IP3Rs can form a heterotetramer, in this paper we used IP3R TKO as a parental cell line and collected data in an experimental system with stable expression to focus only on homotetramers including cysteine mutations. By simplifying the experimental system, we were able to clearly verify the mechanism of redox regulation in the ER luminal portion of IP3R1, while it remains to be investigated whether redox regulation also exists in IP3R2/3 and how it is affected as a heterotetramer.
We had previously demonstrated the feedback regulatory mechanism of ERdj5 activity in terms of the activation of the SERCA2b calcium pump, showing that ERdj5 is inactivated after forming oligomers, when the [Ca2+]ER becomes high enough, and the uptake of calcium ions from the cytosol into the ER lumen is suppressed by the oxidation of the disulfide bond of SERCA2b. By incorporating the results shown here, we propose a reciprocal regulatory program of ERdj5 in terms of IP3R1 and SERCA2b regulation (Fig. 4F). Under the condition where the [Ca2+]ER is low, ERdj5 exerts reductive activity and reduces both IP3R1 and SERCA2b, which causes the activation of SERCA2 and the inactivation of IP3R1. Thus, calcium uptake is accelerated, and release from the ER is suppressed. In contrast, ERdj5 is inactivated by oligomer formation when the [Ca2+]ER is sufficiently high, and then, IP3R1 and SERCA2b are active and inactive, respectively. Under these conditions, the ER luminal calcium ions can be actively released from the ER by activated IP3R1, but the uptake of calcium ions by SERCA2b is suppressed. This reciprocal regulatory system contributes to maintain the calcium homeostasis in the ER.
Materials and Methods
Cell Lines.
HeLa cells and HEK-293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (11995040; Gibco) supplemented with 10% fetal bovine serum (FBS) (172012–500; Sigma-Aldrich), penicillin (100 U ml–1) and streptomycin (100 U ml–1) (25030–081; Nacalai Tesque) in a 5% CO2 incubator. For the measurement of fluorescent probes, cells were cultured in FluoroBrite DMEM (A1896701; Gibco) supplemented with 10% FBS and GlutaMAX (35050061; Thermo Fisher Scientific) in a 5% CO2 incubator.
Plasmids.
cDNAs encoding human IP3R1, PDI, ERP57, ERP72, PDIR, P5, ERP46, ERP18, DNAJC10, and ERO1A were inserted into pcDNA3.1(+), pcDNA3.1(−), or pcDNA3.1(+)/Hygro plasmids (Thermo Fisher Scientific) (37, 44). DNAs encoding a hemagglutinin (HA) tag (YPYDVPDYA), PA tag (GVAMPGAEDDVV), or FLAG tag (DYKDDDDK) were used for tagging. Truncated constructs were prepared by PCR-mediated site-directed mutagenesis. Single-guide RNAs (sgRNAs) targeting Human IP3R1 (5′-GTACGCGGAGGGATCGACAAA-3′), Human IP3R2 (5′- GACTCGGCCCAGAAGCAGTAC-3′), Human IP3R3 (5′-TATGTATTTCTGGTTAACAC-3′), Human PDI (5′-CCCTGGTGCGCGCCGACGCCCCC-3′), Human ERP57 (5′-CCCGCCTCGCCGCTGCCTCCGAC-3′), Human PDIA5/PDIR (5′-CCGGGCCGGCGTGGCTGCTGCTG-3′), Human PDIA6/P5 (5′-TGTGTGGGTGTGCCGACCACTGG-3′), Human ERP46 (5′-GTACACGGCCGACATGTTCA-3′), Human ERP18 (5′-GGAGACGCGGCCTCGTCTCGGGG-3′), Human DNAJC10/ERdj5 (5′-TAAGAAAGAGAATGGGAGTC-3′), and Human ERO1A (5′-GCCGGAGCTGCAATGGGCCG-3′) were cloned into pSpCas9(BB)-2A-puro (a gift from F. Zhang, Broad Institute of Massachusetts Institute of Technology; Addgene #48138). CY-RL7er was prepared as a FRET probe for mSECFP and moxVenus using the same sequence as the original linker sequence (32). YC3.6 was a gift from Takeharu Nagai (Osaka University) (45). GEM-CEPIA1er was purchased from Addgene (#58217) (46).
Generation of Stable Cell Lines.
Cells were transfected with polyethylenimine (PEI) max (24765; Polysciences), and stable transfectants were selected with hygromycin (ant-hg; InvivoGen).
Establishment of PDI-KO and IP3R-TKO Cells.
Cells were transfected with pX459-encoding sgRNAs targeting PDI family genes using PEI max. Transfectants were selected with puromycin (A1113803; Gibco), and single clones were obtained by the limiting dilution technique. Clones with mutations in both alleles were identified by immunoblotting.
Immunoprecipitation and Immunoblotting.
Cell lysates were prepared in lysis buffer [50 mM Tris-HCl, pH 7.5; 150 mM NaCl; 10 mM N-ethylmaleimide; 1% Triton X-100; and EDTA-free protease inhibitors (03969; Nacalai Tesque)]. After centrifugation at 14,000 × g for 20 min, the supernatants were subjected to immunoprecipitation using anti-FLAG M2 affinity gel (A2220; Sigma-Aldrich) or anti-HA antibody (561; MBL) with Ab-Capcher MAG2 (P-052; ProteNova). The precipitated immunocomplexes were washed three times with washing buffer (50 mM Tris-HCl, pH 7.5; 300 mM NaCl; and 1% Triton X-100) and boiled in sample buffer (46.7 mM Tris-HCl, pH 6.8; 5% glycerol; 1.67% sodium dodecyl sulfate (SDS); 1.55% DTT, and 0.02% bromophenol blue). Samples were subsequently separated by SDS-PAGE and transferred to Amersham Protran nitrocellulose membranes (10600042; Cytiva). Immunoblotting analysis was performed with the indicated antibodies. SIGMAFASTBCIP/NBT (B5655; Sigma) was used to visualize the signals. Contrast and brightness adjustment and quantification were performed using Fiji software (ImageJ; NIH) (47) and Photoshop CS6 (Adobe).
The antibodies with the indicated dilutions were as follows: anti-PDI (Cell Signaling Technology, 3501S, RRID:AB_2156433; 1:1,000), anti-ERP57 (GeneTex, GTX113719, RRID:AB_10720538; 1:1,000), anti-PDIA5 (ATLAS Antibody, HPA030355, RRID:AB_10602200; 1:1,000), anti-PDIA6 (Abcam, ab154820, RRID:AB_2714011; 1:1,000), anti-ERP46 (GeneTex, GTX106914, RRID:AB_11170188; 1:1,000), anti-ERP19 (Santa Cruz Biotechnology, sc376410, RRID:AB_11150289; 1:500), anti-DNAJC10 (Abnova, H00054431M01, RRID:AB_534841; 1:1,000), anti-ERO1A (Merck Millipore, MABT376; 1:1,000), anti-IP3R1 (ATLAS Antibodies, HPA014765, RRID:AB_1851843; 1:1,000), anti-IP3R2 (Santa Cruz Biotechnology, sc-398434; 1:500), anti-IP3R3 (BD Bioscience, 610313, RRID:AB_397705; 1:1,000), anti-Phospho-IP3 Receptor (Ser1756) (Cell Signaling Technology, 8548; RRID:AB_10949506; 1:1,000), anti-Phospho-IP3 Receptor 1 (Ser1588, Ser1598) (Thermo Fisher Scientific, PA5-38628; RRID:AB_ 2555223; 1:1,000), anti-Phospho-IP3 Receptor 1 (Tyr353) (Thermo Fisher Scientific, PA5-64735; RRID:AB_ 2662549; 1:500), anti-SERCA2 (Cell Signaling Technology, 9580; 1:1,000), anti-B2R (Santa Cruz Biotechnology, sc-136216, RRID:AB_2064180; 1:500), anti-FLAG (Sigma-Aldrich, F3165, RRID:AB_259529; 1:1,000), anti-FLAG (ROCKLAND antibodies & assays, 600-401-383, RRID:AB_219374; 1:1,000), anti-PA (FUJIFILM Wako, 012-25863; 1:1,000), anti-HA (MBL, 561, RRID:AB_591844; 1:1,000), anti-HA (MBL, M180-3, RRID:AB_10951811; 1:1,000), anti-α-tubulin (MBL, PM054, RRID:AB_10598496; 1:1,000), anti-BiP (BD Bioscience, 610978, RRID:AB_398291; 1:1,000), AP-conjugated goat polyclonal anti-rabbit IgG (Jackson ImmunoResearch Laboratories, 111-055-144, RRID:AB_2337953; 1:1,000), AP-conjugated goat polyclonal anti-mouse IgG (Jackson ImmunoResearch Laboratories, 115-055-146, RRID:AB_2338538; 1:1,000), AP-conjugated goat polyclonal anti-rat IgG (Jackson ImmunoResearch Laboratories, 112-055-003, RRID:AB_2338148; 1:1,000), HRP-conjugated goat polyclonal anti-rabbit IgG (Thermo Fisher Scientific, 65-6120, RRID:AB_2533967; 1:2,000).
Detection of the Redox States of IP3R1.
Cells in suspension were either untreated or treated with 10 mM DTT or 1 mM DPS and then precipitated with 10% (vol/vol) TCA on ice. After centrifugation at 12,000 × g, cell pellets were washed three times with acetone and dried completely. To determine the protein redox state of the cells, proteins in pellets were lysed and modified by incubation for 30 min at room temperature in buffer containing 10 mM mPEG2000-mal (SUNBRIGHT ME-020MA; Nichiyu) and 1% SDS. Samples were adjusted to contain 1× Laemmli buffer for SDS-PAGE and subjected to immunoblotting with the anti-PA tag antibody NZ-1 (4548995036537; FUJIFILM, Wako).
Detection of the Phosphorylated IP3R1.
Cell lysates from HA-tagged IP3R1-expressing cells were prepared in lysis buffer supplemented with phosphatase inhibitors (07575-51; Nacalai Tesque). After centrifugation at 14,000 × g for 20 min, the supernatants were subjected to immunoprecipitation with anti-HA antibody (M180-3; MBL) using Ab-Capcher MAG2. The precipitated immunocomplexes were washed three times with washing buffer and boiled in sample buffer. Dephosphorylated samples were prepared by adding protein phosphatase (P0753; NEB) to immunoprecipitate solubilized in lysis buffer without phosphatase inhibitors. The samples were then separated by SDS-PAGE and transferred to nitrocellulose membranes. Immunoblotting analysis was performed with the indicated antibodies. Amersham ECL Prime (RPN2232; Cytiva) was used for signal visualization with LAS-3000 system (Fujifilm, Tokyo, Japan).
Calcium Indicator Imaging.
HeLa cells expressing calcium indicators were plated on 96-well plates (165305; Thermo Fisher Scientific), and HEK-293T cells were plated on poly-L-lysin-coated glass-bottom dishes (D11131H; Matsunami glass).
For measurements of IICR, cells expressing YC 3.6 were cultured in FluoroBrite DMEM and used to measure the fluorescence brightness of cyan fluorescent protein [CFP; excitation (ex) wavelength, 435 nm; emissions wavelength (em), 480 nm], yellow fluorescent protein (YFP; ex, 510 nm; em, 535 nm), and FRET (ex, 435 nm; em, 535 nm) using a VARIOSKAN LUX fluorescent plate reader (Thermo Fisher Scientific). FRET signals were calculated by the following equation:
where R = FRET/CFP, R0 = average signal before stimulation.
Then, normalized YC signals were calculated as the ratio of the peak amplitudes of KO cells to those of WT cells.
For the estimation of [Ca2+]ER based on ratiometric measurements using GEM-CEPIA1er (#58217; Addgene), [Ca2+]ER was obtained by the following equation:
where R = (F at 466 nm)/(F at 510 nm), n = 1.37 and Kd= 558 μM.
Rmin was measured after treatment with 20 μM digitonin, 15 μM ionomycin and 10 mM EGTA.
Rmax was obtained as 100 mM CaCl2 was added continuously after the Rmin measurement.
Time-lapse images of YC3.6 in HEK-293T cells were taken using a DeltaVision Elite fluorescence microscope (GE Healthcare Life Sciences) equipped with a 20× U Apo 340 (1-UB765; Olympus, NA0.75) at a rate of one frame every 3 or 5 s. Images were taken with the following sets of excitation and emission filters: 438 ± 24 nm and 475 ± 24 nm for enhanced CFP (ECFP) and 513 ± 17 nm and 548 ± 22 nm for Venus.
Redox Imaging.
CY-RL7er-expressing cells seeded in 96-well plates were used to measure the fluorescence brightness of CFP (ex, 435 nm; em, 480 nm), YFP (ex, 510 nm; em, 535 nm), and FRET (ex, 435 nm; em, 535 nm) using a VARIOSKAN LUX fluorescent plate reader. The relative amount of oxidized CY-RL7er was calculated according to the following equation:
where R is the ratio of FRET at steady state to CFP fluorescence. Rox and Rred are normalized FRET signals of fully oxidized and reduced CY-RL7 that were determined by incubating cells with 5 mM diamide and 10 mM DTT, respectively.
Sucrose Density Gradient Fractionation.
Cell lysates were applied to a 15 to 60% linear sucrose gradient in gradient buffer (50 mM Tris-HCl, pH 7.5, and 150 mM NaCl), and a gradient was generated using Gradient Master (BioComp). Lysates were centrifuged at 36,000 × g for 16 h. Each 250 μL fraction collected from the top was adjusted to contain 1× Laemmli buffer for separation by SDS-PAGE.
Quantification and Statistical Analysis.
Two groups of data were evaluated by unpaired two-tailed Student’s t test, and multiple comparison tests were performed by one-way ANOVA followed by Sidak’s multiple comparison test. A P value less than 0.05 was considered statistically significant. The distribution of the data was assumed to be normal, but this assumption was not formally tested.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
This work was supported by a Grant-in-Aid for Scientific Research from Ministry of Education, Culture, Sports, Science and Technology, Japan (Grant Numbers 24227009, JP18H04002 to K.N., JP18K06140, JP21H05268, JP22H02622 to R.U.), from the Japan Science and Technology Agency, Core Research for Evolutional Science and Technology (Grant Number JPMJCR13M6 to K.N.) and from the Japan Agency for Medical Research and Development (Grant Number JP21GM1410008 to R.U.). This work was also supported by the Takeda Science Foundation (to K.N.)
Author contributions
S.F., R.U., and K.N. designed research; S.F. performed research; S.F. analyzed data; and S.F., R.U., and K.N. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission. M.S. is a guest editor invited by the Editorial Board.
Contributor Information
Ryo Ushioda, Email: ryo3ussy3@cc.kyoto-su.ac.jp.
Kazuhiro Nagata, Email: nagata@cc.kyoto-su.ac.jp.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Berridge M. J., Inositol trisphosphate and calcium signalling. Nature 361, 315–325 (1993). [DOI] [PubMed] [Google Scholar]
- 2.Berridge M. J., The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium 32, 235–249 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Patterson R. L., Boehning D., Snyder S. H., Inositol 1,4,5-trisphosphate receptors as signal integrators. Annu. Rev. Biochem. 73, 437–465 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Monteith G. R., Prevarskaya N., Roberts-Thomson S. J., The calcium-cancer signalling nexus. Nat. Rev. Cancer 17, 367–380 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Orrenius S., Zhivotovsky B., Nicotera P., Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 4, 552–565 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Mattson M. R., Calcium and neurodegeneration. Aging Cell 6, 337–350 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Berridge M. J., The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol. Rev. 96, 1261–1296 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Prole D. L., Taylor C. W., Structure and function of ip3 receptors. Cold Spring Harb. Perspect. Biol. 11, a035063 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vandecaetsbeek I., Vangheluwe P., Raeymaekers L., Wuytack F., Vanoevelen J., The Ca2+ pumps of the endoplasmic reticulum and Golgi apparatus. Cold Spring Harb. Perspect. Biol. 3, 1–24 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michalak M., Groenendyk J., Szabo E., Gold L. I., Opas M., Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem. J. 417, 651–666 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Preissler S., et al. , Calcium depletion challenges endoplasmic reticulum proteostasis by destabilising bip-substrate complexes. Elife 9, 1–36 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lucero H. A., Kaminer B., The role of calcium on the activity of ERcalcistorin/protein-disulfide isomerase and the significance of the c-terminal and its calcium binding: A comparison with mammalian protein-disulfide isomerase. J. Biol. Chem. 274, 3243–3251 (1999). [DOI] [PubMed] [Google Scholar]
- 13.Avezov E., et al. , Retarded PDI diffusion and a reductive shift in poise of the calcium depleted endoplasmic reticulum. BMC Biol. 13, 1–15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thrower E. C., Duclohier H., Lea E. J. A., Molle G., Dawson A. P., The inositol 1,4,5-trisphosphate-gated Ca2+ channel: Effect of the protein thiol reagent thimerosal on channel activity. Biochem. J. 318, 61–66 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayashi T., Su T. P., Sigma-1 receptor chaperones at the ER- mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 131, 596–610 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Higo T., et al. , Subtype-specific and ER lumenal environment-dependent regulation of inositol 1,4,5-trisphosphate receptor type 1 by ERp44. Cell 120, 85–98 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Higo T., et al. , Article mechanism of ER stress-induced brain damage by IP(3) receptor. Neuron 68, 865–878 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Zhang L., et al. , Different interaction modes for protein-disulfide isomerase (PDI) as an efficient regulator and a specific substrate of endoplasmic reticulum oxidoreductin-1α (Ero1α). J. Biol. Chem. 289, 31188–31199 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Appenzeller-Herzog C., Simmen T., ER-luminal thiol/selenol-mediated regulation of Ca 2+ signalling. Biochem. Soc. Trans. 44, 452–459 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Ushioda R., et al. , Redox-assisted regulation of Ca2+ homeostasis in the endoplasmic reticulum by disulfide reductase ERdj5. Proc. Natl. Acad. Sci. U.S A. 113, E6055–E6063 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marino M., et al. , SEPN1, an endoplasmic reticulum-localized selenoprotein linked to skeletal muscle pathology, counteracts hyperoxidation by means of redox-regulating SERCA2 pump activity. Hum. Mol. Genet. 24, 1843–1855 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Li Y., Camacho P., Ca2+-dependent redox modulation of SERCA 2b by ERp57. J. Cell Biol. 164, 35–46 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raturi A., TMX1 determines cancer cell metabolism as a thiol-based modulator of ER–mitochondria Ca2+ flux. J. Cell Biol. 214, 433–444 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prins D., Groenendyk J., Touret N., Michalak M., Modulation of STIM1 and capacitative Ca2+ entry by the endoplasmic reticulum luminal oxidoreductase ERp57. EMBO Rep. 12, 1182–1188 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang L., et al. , Crystal structure of human ERp44 shows a dynamic functional modulation by its carboxy-terminal tail. EMBO Rep. 9, 642–647 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fan G., et al. , Gating machinery of InsP3R channels revealed by electron cryomicroscopy. Nature 527, 336–341 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hatahet F., Ruddock L. W., Substrate recognition by the protein disulfide isomerases. FEBS J. 274, 5223–5234 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Ran F. A., et al. , Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li G., et al. , Role of ERO1-α-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell Biol. 186, 783–792 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anelli T., et al. , Ero1α regulates Ca 2+ fluxes at the endoplasmic reticulum-mitochondria interface (MAM). Antioxidants Redox Signal. 16, 1077–1087 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Kolossov V. L., et al. , Development of a high-dynamic range, GFP-based FRET probe sensitive to oxidative microenvironments. Exp. Biol. Med. 236, 681–691 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kolossov V. L., et al. , Förster resonance energy transfer-based sensor targeting endoplasmic reticulum reveals highly oxidative environment. Exp. Biol. Med. 237, 652–662 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang J., et al. , Secretory kinase Fam20C tunes endoplasmic reticulum redox state via phosphorylation of Ero1α. EMBO J. 37, 1–16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fan G., et al. , Cryo-EM reveals ligand induced allostery underlying InsP3R channel gating. Cell Res. 28, 1158–1170 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kang S., et al. , Effects of redox potential and Ca2+ on the inositol 1,4,5-trisphosphate receptor L3–1 loop region: Implications for receptor regulation. J. Biol. Chem. 283, 25567–25575 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Paknejad N., Hite R. K., Structural basis for the regulation of inositol trisphosphate receptors by Ca2+ and IP3. Nat. Struct. Mol. Biol. 25, 660–668 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Araki K., et al. , Ero1-α and pdis constitute a hierarchical electron transfer network of endoplasmic reticulum oxidoreductases. J. Cell Biol. 202, 861–874 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kojima R., et al. , Radically different thioredoxin domain arrangement of ERp46, an efficient disulfide bond introducer of the mammalian PDI family. Structure 22, 431–443 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Parker A. K. T., Gergely F. V., Taylor C. W., Targeting of inositol 1,4,5-trisphosphate receptors to the endoplasmic reticulum by multiple signals within their transmembrane domains. J. Biol. Chem. 279, 23797–23805 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Groenendyk J., et al. , Interplay between the oxidoreductase PDIA6 and microRNA-322 controls the response to disrupted endoplasmic reticulum calcium homeostasis. Sci. Signal. 7, 1–15 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oka O. B., et al. , ER p18 regulates activation of ATF 6α during unfolded protein response. EMBO J. 38, 1–18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Higa A., et al. , Endoplasmic reticulum stress-activated transcription factor ATF6α requires the disulfide isomerase PDIA5 to modulate chemoresistance. Mol. Cell. Biol. 34, 1839–1849 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burkewitz K., et al. , Atf-6 regulates lifespan through ER-mitochondrial calcium homeostasis. Cell Rep. 32, 108125 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ushioda R., Hoseki J., Araki K., Jansen G., ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science 307, 569–573 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Nagai T., Yamada S., Tominaga T., Ichikawa M., Miyawaki A., Expanded dynamic range of fluorescent indicators for Ca2+ by circularly permuted yellow fluorescent proteins. Proc. Natl. Acad. Sci. U.S.A. 101, 10554–10559 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suzuki J., et al. , Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat. Commun. 5, 1–13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schindelin J., et al. , Fiji: an open-source platform for biological-image analysis. Nat. Methods, 9, 676–682 (2012). 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix.