

Abstract
A genome-wide association study (GWAS) for 10 yield and yield component traits was conducted using an association panel comprising 225 diverse spring wheat genotypes. The panel was genotyped using 10,904 SNPs and evaluated for three years (2016–2019), which constituted three environments (E1, E2 and E3). Heritability for different traits ranged from 29.21 to 97.69%. Marker-trait associations (MTAs) were identified for each trait using data from each environment separately and also using BLUP values. Four different models were used, which included three single trait models (CMLM, FarmCPU, SUPER) and one multi-trait model (mvLMM). Hundreds of MTAs were obtained using each model, but after Bonferroni correction, only 6 MTAs for 3 traits were available using CMLM, and 21 MTAs for 4 traits were available using FarmCPU; none of the 525 MTAs obtained using SUPER could qualify after Bonferroni correction. Using BLUP, 20 MTAs were available, five of which also figured among MTAs identified for individual environments. Using mvLMM model, after Bonferroni correction, 38 multi-trait MTAs, for 15 different trait combinations were available. Epistatic interactions involving 28 pairs of MTAs were also available for seven of the 10 traits; no epistatic interactions were available for GNPS, PH, and BYPP. As many as 164 putative candidate genes (CGs) were identified using all the 50 MTAs (CMLM, 3; FarmCPU, 9; mvLMM, 6, epistasis, 21 and BLUP, 11 MTAs), which ranged from 20 (CMLM) to 66 (epistasis) CGs. In-silico expression analysis of CGs was also conducted in different tissues at different developmental stages. The information generated through the present study proved useful for developing a better understanding of the genetics of each of the 10 traits; the study also provided novel markers for marker-assisted selection (MAS) to be utilized for the development of wheat cultivars with improved agronomic traits.
Supplementary Information
The online version contains supplementary material available at 10.1007/s11032-021-01240-1.
Keywords: Triticum aestivum L., Agronomic traits, GWAS, MTA, Epistasis, Candidate genes
Introduction
Wheat is the second most important staple food after maize (https://www.statista.com/statistics/263977/world-grain-production-by-type/), with a global production of ~ 764.39 Mt during 2019/2020 (http://www.worldagriculturalproduction.com/crops/wheat.aspx). In terms of food security also, wheat is the second most important food crop after rice. Also, more than 80% of the farmers world-wide depend on this crop for their livelihood (http://www.fao.org/3/y4011e/y4011e04.htm). Wheat is consumed by ~ 2.5 billion people across 89 countries providing ~ 20% of the total calories (https://wheat.org/wheat-in-the-world/) and ~ 20.4% of protein requirements of the growing human population across the world (https://www.wheatinitiative.org/). A part of the wheat grain is also utilized for industrial products like adhesives, paper additives, alcohol and several other products (Curtis BC; http://www.fao.org/3/y4011e04.htm).
According to some estimates, ~ 840 Mt of wheat will be required in 2050 to feed the ever-growing world population, suggesting that an additional ~ 80 Mt of wheat grain will be needed in 2050 over the present production of 759 Mt (https://www.statista.com/statistics/263977/world-grain-production-by-type/). At the national level, India achieved a record wheat production of 106.21 Mt in 2020 https://www.financialexpress.com/opinion/problem-of-plenty-wheat-rice-output-hits-a-record-high-concerns-over). This has placed India in a comfortable position to meet its current demand and also allowed some export of wheat grain. However, at the global level, the rate of annual growth of wheat grain yield has slowed down in recent years, resulting in the current annual growth of only 0.9%, relative to 3% annual growth recorded in earlier decades during and after two Green Revolutions (Ray et al. 2013; Yadav et al. 2019). There is also a realization that the cultivable land area is shrinking due to growing urbanization and industrialization. Therefore, increasing wheat productivity (yield) is necessary to meet the future demand for wheat grain and to ensure food security. The poor annual growth rate in wheat yield may be attributed to both a decline in the improvement in genetic yield potential and to increased sensitivity of current wheat cultivars to abiotic and biotic stresses that will occur more frequently in future due to climate change (Henry et al. 2016). Therefore, continuous improvement in the yield potential of wheat is needed to meet the future challenges of food security.
The grain yield and its contributing traits in wheat are complex and quantitative in nature and are each controlled by polygenes, each gene with small effect involving a significant component of epistasis. Most of these traits have low heritability and are also influenced by genotype x environment interaction (Kaya and Akcura 2014; Nehe et al. 2019). The QTLs for yield and related traits are known to be distributed on all the 21 chromosomes (see Gupta et al. 2020), making the study of the genetic architecture of these traits challenging. However, the availability of molecular markers (SSRs, AFLPs, DArT markers) and novel statistical tools over the past more than three decades has facilitated the detailed genetic dissection of grain yield and its component traits in wheat. This has also been facilitated by the recent availability of high throughput (HT) single nucleotide polymorphism (SNP) markers, which allow high-resolution genetic analysis and marker-assisted selection (MAS) in all crops including wheat (see Sun et al. 2020; Chu et al. 2020). Utilizing these molecular markers, several QTL interval mapping and genome-wide association mapping studies (GWAS) have been conducted to resolve the genetic control of yield and yield contributing traits in wheat (for a review see Gupta et al. 2020). During the past > 30 years, 750 QTLs from ~ 26 studies involving interval mapping and ~ 2000 MTAs from ~ 12 GWAS have been reported in wheat (for review see Gupta et al. 2020), and more QTLs/MTAs are being regularly added. However, most of these known QTLs are minor QTLs, each with small effect; many of these QTLs are also involved in epistasis and QTL x environment interactions. Therefore, there is a need to use novel genetic material and improved statistical tools to identify stable and large effect QTLs/MTAs with information about main effects and epistatic interactions. The availability of this information will help in designing suitable MAS strategies supplementing the classical approaches of wheat breeding for genetic improvement of grain yield and component traits.
We realize that the current available repertoire of QTLs/MTAs is not saturated and that there is still scope of finding out novel MTAs using additional germplasm. Keeping this in view, a GWAS was planned and executed using a novel association panel. Based on this study, in the present communication, we report the results of GWAS for 10 yield and yield component traits. The study involved the use of a panel of 225 diverse spring wheat genotypes that were genotyped using 10,904 SNPs and evaluated over three years (treated as three environments). Both main effect and epistatic QTLs/MTAs along with the underlying candidate genes for different traits were identified, and strategies are suggested for their use in future wheat breeding programmes.
Material and methods
Association panel and experimental data
The association panel used in the present study consisted of 225 diverse wheat genotypes, which is a subset of the original spring wheat reference set (SWRS) of 330 diverse wheat genotypes procured from CIMMYT; this panel was also used in our earlier studies (Kumar et al. 2018; Gahlaut et al. 2019). The original panel was SNP genotyped using DArT-seq; this genotyping work was outsourced by CIMMYT (under their “Seed for Discovery” project) to Diversity Array Technology Pvt Ltd, Australia. The subset used in the present study involved 10,904 SNPs out of 17,937 SNP markers that were made available for the original set of 330 genotypes; data for 3653 markers was missing (< 30%). Another set of 3380 markers had rare alleles (MAF < 0.05); these markers with rare alleles had to be excluded from the analysis for statistical reasons (Kumar et al. 2018; Table S1).
The above association panel of 225 genotypes was raised in a simple lattice design with two replications for three years at the same location (Agriculture Research Farm, Ch. Charan Singh University, Meerut, UP, India; 28.984644°N and 77.705956°E), thus providing for the following three environments: (i) 2016–2017 (E1), (ii) 2017–2018 (E2) and (iii) 2018–2019 (E3). Normal field management practices were followed for raising the crop (i.e., 200 kg/ha fertilizer; N:P:K = 8:8:8). Each genotype was represented by a plot of 3 rows of 1.5 m each, with a row-to-row distance of 0.25 m. Phenotypic data were recorded on the following 10 different agronomic traits: (i) days to heading (DTH; in days), (ii) days to anthesis (DTA; in days), (iii) days to maturity (DTM; in days), (iv) grain filling duration (GFD; in days), (v) plant height (PH; in cm), (vi) tiller number per plant (TNPP; in numbers), (vii) biological yield per plot (BYPP; in kg), (viii) grain number per spike (GNPS; in numbers), (ix) grain yield per plot (GYPP; in gram) and (x) 1000 grain weight (TGW; in gram).
Statistical analysis: frequency distribution, descriptive statistics, ANOVA and heritability
For each of the ten traits, violin plots were prepared to depict the distribution of phenotypic data for each of the three individual environments (E1, E2, and E3) and also using BLUP values (B). The BLUP values were generated using the ‘lme4’ package of the R programe (Bates et al. 2015). Descriptive statistics including mean, range, standard error, were estimated using SPSS v. 17.0 (SPSS Inc 2008). Agricolae package of R program was used for ANOVA using additive main effects and multiplicative interactions (AMMI) model. Broad sense heritability (H2) was calculated according to Allard (1999) from phenotypic variance (σ2p) and the genotypic variance (σ2g) using MS Excel 2010.
Principal components, kinship matrix and MTAs
Principal component analysis (PCA) was conducted to address the problem of population structure. The principal components (PCs) (Q matrix) and kinship matrix (K matrix) were developed using tools available in Genomic Association and Prediction Integrated Tool (GAPIT) package (Lipka et al. 2012). The Q and K matrices were automatically generated (VanRaden 2008; Lipka et al. 2012) using genotypic data with the help of a default set of parameters. The first three PCs were used to produce a 3D scatter plot showing distribution of genotypes into sub-groups.
Four different models were used for identification of MTAs for each individual environment. BLUP values were also used separately for identification of MTAs. The four models used for GWAS included the following: (i) compressed mixed linear model (CMLM; Zhang et al. 2010); (ii) Fixed and random model Circulating Probability Unification (FarmCPU; Liu et al. 2016); (iii) Settlement of MLM Under Progressively Exclusive Relationship (SUPER; Wang et al. 2014); (iv) Matrix variate linear mixed model (mvLMM; Furlotte and Eskin 2015). The first three methods retain the computational advantage, increases statistical power and are freely available in Genomic Association and Prediction Integrated Tool (GAPIT) package (Lipka et al. 2012). The fourth model mvLMM was meant for multi-trait analysis leading to identification of MTAs associated each with two or more than two traits; this model is believed to be superior over other available models for multi-trait analysis (Furlotte and Eskin 2015). The epistatic interactions were identified using the function ‘interactionPval’ available in ‘SNPassoc’ package (González et al. 2007). MTAs obtained were subjected to Bonferroni correction to eliminate spurious associations that are common in GWAS due to occurrence of LD without linkage. P-value threshold involving Bonferroni correction was set up at − log p value = 6.0. Manhattan plots and quantile–quantile (Q-Q) plots were also generated using statistical tools available in GAPIT (Astle and Balding 2009).
Comparison of MTAs with known QTLs
The MTAs identified in the present study were also compared with known QTLs/MTAs. The physical positions for chromosome mapping of all MTAs detected in the present study were obtained using the data available in Ensembl Plants [version 50; https://plants.ensembl.org/Triticum_aestivum/Info/Index].
Identification of candidate genes
The MTAs detected after Bonferroni corrections were also used for identification of candidate genes (CGs) through alignment of DArT-seq to wheat IWGSC RefSeq v1.0 and v2.1 (IWGSC 2018; Zhu et al. 2021) available in the Ensembl Plants (http://www.ensembl.org/info/docs/tools/vep/index.html). Highly significant annotated genes were retrieved from a 200 kb window for each identified MTA. IWGSC website (http://www.wheatgenome.org/) was used for gene ontology (GO) annotation information of these CGs. In-silico gene expression analysis was also performed for CGs using RNA-Seq expression data from Wheat Expression Browser (http://www.wheat-expression.com/). Heatmaps for expression of CGs at different developmental stages in a variety of tissues were also generated.
Identification of important rare variants
During GWAS, markers each with rare allele (frequency < 0.5) are excluded from analysis due to statistical reasons. Each of these SNPs with rare allele was tested for its association with individual traits using “t-test” for their further use in future studies.
Results
Frequency distributions and descriptive statistics
The frequency distributions of phenotypic data based on BLUP values (Fig. 1) and for each of the three environments for all the 10 traits are presented in the form of violin plots (Fig. S1). The distributions indicated a high level of variability and the quantitative nature of each trait. Several traits lacked normal distribution and apparently exhibited either skewness (e.g., DTH, DTA, DTM and GFD) or/and kurtosis (e.g. GYPP; Fig. 1, Fig. S1 and Table S2).
Fig. 1.

Violin plots showing the frequency distribution of 10 yield-related traits using BLUP values of each trait. Shaded regions of the violin plots represent the frequency distribution of data. In each case, the vertical solid bar indicates the range of average values, and median is shown as a white circle, depicting the lower, medium and upper quartile
The results of Analysis of Variance (ANOVA) for all the 10 traits are presented in Table 1. It is apparent that variations due to genotypes, environments and G x E interaction were highly significant for seven of the 10 traits (except DTH, GYPP and GNPS for environmental effects, and except DTH, GYPP and TGW for g x e interactions). Broad sense heritability was generally high ranging from 55.65 (for PH) to 97.69% (for DTA) except for DTH (29.21%).
Table 1.
Analysis of variance (ANOVA) and heritability (H2) for 10 different traits in 225 genotypes tested in three different environments (E1:2016–2017, E2:2017–2018 and E3: 2018–2019)
| Source of variations | DF | Mean square | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Traits | |||||||||||
| DTH | DTA | DTM | GFD | PH | TNPP | BYPP | GYPP | GNPS | TGW | ||
| ENV | 2 | 54.21 | 680.63** | 1287.29** | 311.07* | 610.25** | 208.1 | 1.32* | 3768 | 215.07 | 703.14 * |
| REP (ENV) | 3 | 203.55 | 11.77 ** | 24.14 *** | 10.52* | 32.14* | 129.91*** | 0.08*** | 1033 | 688.87*** | 35.82 ** |
| GEN | 224 | 926.62 *** | 558.19*** | 388.94*** | 95.80*** | 425.36*** | 23.657*** | 1.52*** | 111,033*** | 619.11*** | 1297.17*** |
| GEN X EVN | 448 | 286.5 | 16.24 *** | 12.77*** | 19.88*** | 11.20** | 4.71*** | 0.04*** | 2605 | 6.31*** | 7.07 |
| Residuals | 672 | 266.57 | 2.19 | 2.39 | 3.79 | 3.24 | 1.45 | 0.01 | 2576 | 2.60 | 6.69 |
| Heritability (%) | 29.21 | 97.69 | 96.42 | 80.18 | 55.65 | 71.79 | 97.05 | 87.53 | 97.53 | 96.98 | |
***0.1% level of significance; **1% level of significance; *5% level of significance
Linkage disequilibrium (LD) and principal component analysis (PCA)
The LD-decay of the same association panel was reported in our earlier study (Kumar et al. 2018). During the present study, the results of PCA involving first three PCs (PC12, PC2 and PC3) are presented in Fig. 2, suggesting the presence of three sub-groups in the association panel. However, PC1, PC2 and PC3 accounted for only 6.0%, 4.55% and 3.44% of the variation respectively, suggesting low level of population structure in the association panel (Fig. 2 and Fig. S2).
Fig. 2.
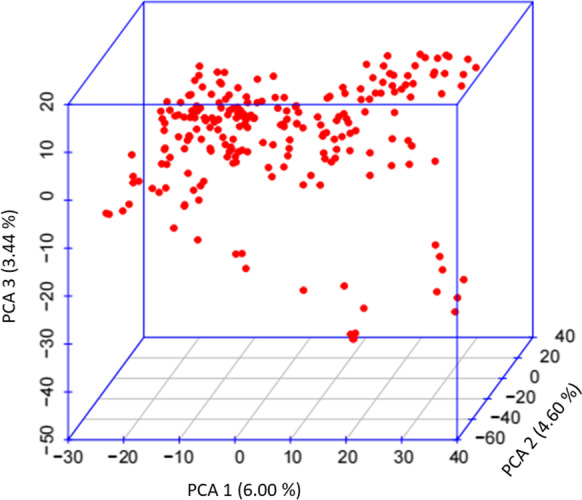
3D scatter plot (3D PCA) of 225 SWRS genotypes based on the first three principal components (PC1, PC2 and PC3). The horizontal and vertical coordinates represent PC1 (with variance explained 6.0%), PC2 (with variance explained 4.60%) and PC3 (with variance explained 3.44%). Each dot represents a genotype
MTAs from data for each of three individual environments and based on BLUP values
As many as 33 MTAs were available for three environments; an additional 20 MTAs using CMLM and FarmCPU were available from BLUP values (Figs. 3 and 4); these MTAs are listed in Table 2 and included the following: (i) Using CMLM, six MTAs for three traits (DTA, DTH and TNPP) were available, which included one common MTA (M1955-5A) for two traits, namely DTA (in E1-2017–18) and DTH (in E2-2017–18), the former in two different environments for DTA (in E2-2017–18 and E3-2018–19; Table 2 and Fig. S3). (ii) Using FarmCPU, 21 MTAs for four traits (DTH, DTA, DTM and TGW) were available; five MTAs were each available in more than one environment for the same trait; these MTAs included the following: (M12140-6B, and M11387-6B for TGW,; M5818-4D for DTH (E1), DTA (E2), DTH (E3); M11963-3B for DTH (E2) and DTA (E2) and M3072-3A for DTA (E1), DTM (E2) and DTM (E3; Table 2 and Fig. S4). (iii) Using mvLMM, a total of 38 multi-trait MTAs, involving 15 trait combinations, were identified, which included the following for each of the three environments: (i) 15 MTAs for 6 trait combinations in E1 (15 MTAs), (ii) 10 MTAs for 3 trait combinations in E2 and (iii) 13 MTAs for 6 trait combinations in E3; two of these multi-trait MTAs, both on chromosome 1A (M2375-1A and M3080-1A) were also available in more than one of the 15 trait combinations (involving DTM, GFD, PH and TNPP) in all the three environments (Table 2 and Fig. S5).
Fig. 3.

Manhattan plots (on the left) and Q-Q plots (on the right) for GWAS conducted using CMLM (based on BLUP values only) for only four traits, for which MTAs were available afer Bonferroni corrections (for the remaining six traits, no MTAs were available after Bonferroni corrections): significant MTAs on 21 chromosomes can be visualized in Manhattan plots on or above the blue line in each case; MTAs qualifying Bonferroni correction are shown on or above the red line; deviation of observed p values from expected p values (based on null hypothesis) can be visualized on Q-Q plots. ump; indicated unmapped SNP
Fig. 4.

Manhattan plots (on the left) and Q-Q plots (on the right) for GWAS conducted using FarmCPU (based on BLUP values only) for only six traits, for which MTAs were available after Bonferroni correction (for the remaining four traits, no MTAs were available after Bonferroni correction). Significant MTAs on 21 chromosomes can be visualized in Manhattan plots on or above the blue line in each case; MTAs qualifying Bonferroni correction are shown on or above the red line; deviation of observed p values from expected p values (based on null hypothesis) can be visualized on Q-Q plots. ump; indicated unmapped SNP
Table 2.
MTAs for different traits detected in three environments and BLUP using CMLM, FarmCPU and mvLMM approaches*
| SNP | Allele | Chromosome | Position |
|---|---|---|---|
| CMLM | |||
| DTH, (E2, B); DTA (E1, E3) | |||
| M1955 | A/G | NA | NA |
| TNPP (E3) | |||
| M11981 | C/T | 1B | 1.79 |
| M11668 | T/C | 3A | 0.61 |
| M7483 | T/C | 3B | 1.8 |
| TNPP (B) | |||
| M14817 | C/T | NA | NA |
| M11668 | T/C | 3A | 0.61 |
| GNPS (B) | |||
| M4839 | C/G | 2B | 1.51 |
| GYPP (B) | |||
| M13299 | A/G | 1A | 2.29 |
| M10606 | G/A | 2A | 1.26 |
| FarmCPU | |||
| DTH (E1) | |||
| M14066 | T/C | NA | NA |
| M13005 | G/T | 4A | 1.78 |
| M1727 | T/G | 5B | 0.9 |
| DTH (E2) | |||
| M5818 | A/G | 4D | 0.84 |
| M11963 | C/T | 3B | 1.33 |
| DTH (B) | |||
| M5921 | G/C | 2D | 2.17 |
| M4079 | C/G | NA | NA |
| M14066 | T/C | NA | NA |
| DTA (E1) | |||
| M3072 | T/G | 3A | 0.14 |
| M5818 | A/G | 4D | 0.84 |
| M1920 | T/G | 4A | 1.12 |
| DTA (E2) | |||
| M11963 | C/T | 3B | 1.33 |
| DTA (B) | |||
| M4079 | C/G | NA | NA |
| M1026 | G/A | NA | NA |
| M5818 | A/G | 4D | 0.84 |
| DTM (E1) | |||
| M4098 | A/G | 5A | 2.29 |
| M12308 | A/G | NA | NA |
| DTM (E2) | |||
| M3072 | T/G | 3A | 0.14 |
| M1156 | A/G | 2B | 1.48 |
| DTM (E3) | |||
| M3072 | T/G | 3A | 0.14 |
| M12889 | T/C | NA | NA |
| M5818 | A/G | 4D | 0.84 |
| DTM (B) | |||
| M3072 | T/G | 3A | 0.14 |
| M8641 | A/G | NA | NA |
| M1026 | G/A | NA | NA |
| M5818 | G/A | 4D | 0.84 |
| TNPP (B) | |||
| M14817 | T/C | NA | NA |
| GYPP (B) | |||
| M10606 | A/G | 2A | 1.26 |
| TGW (E1) | |||
| M12140 | G/T | 6B | 1.59 |
| M11387 | T/’C | 6B | 0.66 |
| TGW (E2) | |||
| M12140 | G/T | 6B | 1.59 |
| M11387 | T/C | 6B | 0.66 |
| M4710 | T/A | 3A | 2.62 |
| TGW (B) | |||
| M1935 | T/G | NA | NA |
| M11387 | T/’C | 6B | 0.66 |
| mvLMM | |||
| DTM, GFD, PH (E1) | |||
| M2375 | C/T | 1A | 1.55 |
| M3080 | C/T | 1A | 4.3 |
| M9267 | T/C | 3B | 1.58 |
| M274 | C/T | 1A | 1.75 |
| DTM, GFD, PH (E2) | |||
| M3080 | T/C | 1A | 4.3 |
| M2375 | C/T | 1A | 1.55 |
| M373 | T/G | 1A | 4.28 |
| DTM, GFD, PH (E3) | |||
| M3080 | C/T | 1A | 4.3 |
| M2375 | C/T | 1A | 1.55 |
| M1879 | C/G | 6B | 0.64 |
| M3482 | C/G | 6B | 0.84 |
| DTM, GFD, PH, TNPP (E1) | |||
| M3080 | C/T | 1A | 4.3 |
| M2375 | T/C | 1A | 1.55 |
| M9267 | T/C | 3B | 1.58 |
| DTM, GFD, PH, TNPP (E2) | |||
| M2375 | T/C | 1A | 1.55 |
| M3080 | T/C | 1A | 4.3 |
| DTM, GFD, PH, TNPP (E3) | |||
| M2375 | T/C | 1A | 1.55 |
| M3080 | C/T | 1A | 4.3 |
| M1879 | C/G | 6B | 0.64 |
| M3482 | C/G | 6B | 0.84 |
| M1396 | C/T | 1A | 1.57 |
| DTM, GFD, PH, TNPP, BYPP (E2) | |||
| M2711 | G/C | NA | NA |
| M2375 | C/T | 1A | 1.55 |
| M3080 | C/T | 1A | 4.3 |
| M13982 | T/C | 6A | 0.52 |
| M4870 | A/C | 7B | 0.89 |
| TNPP, BYPP, GNPS (E1) | |||
| M13982 | T/C | 6A | 0.52 |
| TNPP, BYPP, GNPS (E3) | |||
| M8971 | C/T | NA | NA |
| TNPP, BYPP, GNPS, GYPP (E1) | |||
| M8971 | T/C | NA | NA |
| M13982 | C/T | 6A | 0.52 |
| M12201 | C/T | 7D | 1.2 |
| TNPP, BYPP, GNPS, GYPP (E3) | |||
| M8971 | T/C | NA | NA |
| BYPP, GNPS, GYPP (E1) | |||
| M8971 | T/C | NA | NA |
| M12201 | T/C | 7D | 1.2 |
| BYPP, GNPS, GYPP (E3) | |||
| M8971 | C/T | NA | NA |
| BYPP, GNPS, GYPP, TGW (E1) | |||
| M8971 | T/C | NA | NA |
| M12201 | T/C | 7D | 1.2 |
| BYPP, GNPS, GYPP, TGW (E3) | |||
| M8971 | T/C | NA | NA |
*E1, environment1; E2, environment2; E3, environment3; B, BLUP
MTAs identified by more than one models
Among MTAs available from three GWAS models after Bonferroni correction, no common MTA from more than one model were available. however, because Bonferroni often leads to false negatives, MTAs based on BLUP values, identified prior to Bonferroni correction were examined for common MTAs; 34 common MTAs were identified using CMLM, FarmCPU and SUPER for nine of the ten traits (except DTM). The maximum numbers of common MTAs (8MTAs) were found for GYPP and minimum number of MTAs was found for DTH and DTA (one for each). The details of all these common MTAs are presented in the Table S3.
MTAs involved in epistasis
A total of 28 first-order epistatic interactions (QTL × QTL interactions) were available, which involved seven of the 10 traits (except GNPS, PH and BYPP); these interactions included the following (i) 7 interactions in E1, with a range of 1–2 interactions for each of the following 5 traits: DTH, DTA, DTM, TNPP, and TGW; (ii) 10 interactions in E2, with a range of 1–3 interactions for each the following 5 traits: DTH, DTA, DTM, GFD, and TNPP; (iii) 11 interactions in E3 with a range of 1–3 interactions for each of the following 7 traits: DTH, DTA, DTM, GFD, TNPP, GYPP and TGW (Table S4).
MTAs overlapping or occurring in the vicinity of known QTLs
When compared with known QTLs reported in earlier studies, MTAs were placed in the following three categories. (i) MTAs lying within QTL interval: nine MTAs for the following three different traits: GFD, GNPS & TGW: these MTAs included one MTA identified through FarmCPU, five MTAs identified through mvLMM, two MTAs exhibiting epistatic interactions and one identified through BLUP (Table 3). (ii) MTAs outside the interval, but close to flanking marker within a physical distance of 60 Mb: 12 MTAs (11 SNPs) for the following traits were available: DTA, DTH, DTM, PH, TNPP, GFD, GYPP & TGW. Out of these 12 MTAs, three MTAs were identified through FarmCPU, four through mvLMM and four showing epistatic interactions (Table 3). (iii) Novel MTAs: 24 novel MTAs included three MTAs each using CMLM and mvLMM, five MTAs using FarmCPU, 10 MTAs exhibiting epistatic interactions and three using BLUP (Table 4).
Table 3.
MTAs with corresponding QTLs (either overlapping known QTLs or located in the vicinity of known QTLs)
| MTA (Chromosome, Position) | Corresponding QTL (Chromosome, associated marker) | Reference |
|---|---|---|
| MTAs overlapping known QTLs | ||
| GFD | ||
| M2375# (1A, 341,154,353–341,154,421) | QGfd.nfcri-1A (1A, Xgwm357-Xbarc350) | Wang et al. 2009 |
| M3080# (1A, 574,215,287–574,215,355 | ||
| M274# (1A, 338,834,928–338,834,974) | ||
| M373# (1A, 573,920,780–573,920,835) | ||
| M1396# (1A, 320,519,538–320,519,606) | ||
| GNPS | ||
| M4839B (2B, 550,990,403–550,990,471) | QGns.saas-2B (Xbarc1027-Xwmc441) | Tang et al. 2011 |
| TGW | ||
| M6513* (2B, 539,373,679–539,373,747) | QTkw.ncl-2B.1 (2B, Xbarc55-Xbarc37) | Ramya et al. 2010 |
| M12140@ (6B, 650,012,151–650,012,168) | QKwps|Tkw-WY-6B.1 (6B, Xbarc178-Xwmc473) | Cui et al. 2013 |
| M2913* (7B, 630,554,745–630,554,813) | QTkw.dms-7B (7B, wPt1826-wPt8233) | Chen et al. 2015 |
| MTAs in the vicinity of known QTLs | ||
| DTH | ||
| M13005@ (4A, 646,730,830–646,730,898) | QHed.dms-4A (4A, wPt-9183-wPt-1161) | Chen et al. 2015 |
| DTA | ||
| M1920@ (4A, 616,986,080–616,986,148) | QFld.dms-4A (4A, wPt-9183-wPt-1161) | Chen et al. 2015 |
| DTM | ||
| M274# (1A, 338,834,928–338,834,974) | QMat.dms-1A (1A, wPt-4897-wPt-8016) | Chen et al. 2015 |
| M373# (1A, 573,920,780–573,920,835) | QMat.dms-1A (1A, wPt-8016-wPt-7339) | Chen et al. 2015 |
| M9267# (3B, 683,764,839–683,764,907) | QMat.crc-3B (3B, wmc827-N/A) | Cuthbert et al. 2008 |
| M3393# (5A, 536,221,003–536,221,071) | QMat.crc-5A (5A, barc151-N/A) | Cuthbert et al. 2008 |
| M10129# (7A, 38,327,805–38,327,873) | QMat.crc-7A (7A, gwm276-N/A) | Cuthbert et al. 2008 |
| GFD | ||
| M4870# (7B, 426,881,350–426,881,418) | QGft.crc-7B (7B, wmc723-N/A) | Cuthbert et al. 2008 |
| PH | ||
| M9267# (3B, 683,764,839–683,764,907) | QPht.nwu-3B (3B, Xbarc102-Xgwm533) | Daoura et al. 2014 |
| TNPP | ||
| M3080* (1A, 574,215,287–574,215,355) | QTp.ccsu-1A.1 (1A, Xmwg632-N/A) | Kumar et al. 2007 |
| GYPP | ||
| M15191* (1A, 514,115,832–514,115,900) | QYld.crc-1A (1A, wmc716-N/A) | Cuthbert et al. 2008 |
| TGW | ||
| M4710@ (3A, 745,517,190–745,517,258) | QTgw-3A1 (3A, Xgwm480-Xcfd2183) | Liu et al. 2014 |
@; MTAs identified using FarmCPU, #; MTAs identified using mvLMM, *; MTAs involved in epistatic interaction and B; MTAs identified using BLUP
Table 4.
Novel MTAs identified during the present study (never reported earlier either as QTL using interval mapping or MTA using GWAS)
| MTA | Chromosome | Location |
|---|---|---|
| DTH | ||
| M5921B | 2D | 572,257,407–572,257,475 |
| M4210* | 3A | 724,572,071–724,572,139 |
| M1727@ | 5B | 486,280,118–486,280,161 |
| DTH, DTA | ||
| M11963@ | 3B | 605,293,347–605,293,415 |
| DTH, DTA, DTM | ||
| M8127* | 2A | 751,840,515–751,840,583 |
| M5818@ | 4D | 336,833,286–336,833,354 |
| M13836* | 7B | 125,884,474–125,884,538 |
| DTA | ||
| M5818B | 4D | 336,833,286–336,833,354 |
| DTM | ||
| M9877* | 1A | 22,664,662–22,664,730 |
| M1156@ | 2B | 657,300,532–657,300,587 |
| M8239* | 3B | 707,906,591–707,906,651 |
| M13682* | 6B | 151,887,057–151,887,125 |
| DTM, GFD, PH, TNPP | ||
| M1879# | 6B | 613,595,484–613,595,523 |
| DTM, GFD, PH, GYPP | ||
| M3482* | 6B | 658,954,327–658,954,395 |
| DTM, GFD, PH, TNPP, BY, GNPS, GYPP | ||
| M13982# | 6A | 19,155,299–19,155,367 |
| GFD | ||
| M13252* | 2B | 669,232,955–669,233,023 |
| TNPP | ||
| M11981$ | 1B | 431,456,198–431,456,266 |
| M10707* | 1B | 418,056,499–418,056,567 |
| M11668$B | 3A | 42,066,916–42,066,962 |
| M7483$ | 3B | 745,290,131–745,290,199 |
| TNPP, BYPP, GNPS, GYPP, TGW | ||
| M12201# | 7D | 78,865,376–78,865,439 |
| BYPP | ||
| M420* | 5B | 448,467,016–448,467,084 |
| GYPP | ||
| M10606B | 2A | 94,163,381–94,163,449 |
| TGW | ||
| M11387@B | 6B | 559,513,056–559,513,124 |
$: MTAs identified using CMLM, @; MTAs identified using FarmCPU, #; MTAs identified using mvLMM, *; MTAs involved in epistatic interaction and B; MTAs identified using BLUP
MTAs for possible use in MAS
The MTAs identified using each of the three methods of GWAS and also those found to be involved in epistasis were subjected to further scrutiny in order to identify the most important MTAs, which could be recommended for MAS. There were 11 such MTAs, which were selected using one or more of the following criteria: (i) highest R2, (ii) lowest P value, (iii) common for two or more traits, (iv) stable (identified in all the three environments), (v) detected in earlier studies (interval mapping and/or GWAS). The selected MTAs are listed in Table 5.
Table 5.
A summary of most important MTAs recommended for MAS on the basis of present study
| MTA | Chr | Pos | Allele* | Description |
|---|---|---|---|---|
| M1955 | NA | NA | G/A* | Detected using CMLM: Associated with DTH and DTA with highest R2 (20%) |
| M5818 | 4D | 0.84 | A/G | Detected using FarmCPU: Associated with DTH, DTA and DTM |
| M3072 | 3A | 0.14 | T/G | Detected using FarmCPU: Associated with DTA and DTM (lowest p value after Bonferroni) |
| M12140 | 6B | 1.59 | G/T | Detected using FarmCPU: Associated with TGW and reported in previous study for TGW (Cui et al. 2013) |
| M2375 | 1A | 1.55 | C/T | Detected using mvLMM: Associated with DTM, GFD, PH and TNPP (stable across the 3 environments), and reported in previous study for GFD and TNPP (Wang et al. 2009) |
| M3080 | 1A | 4.3 | C/T | |
| M1396 | 1A | 1.57 | C/T | Detected using mvLMM: Associated with DTM, GFD, PH and TNPP, and reported in previous study for GFD (Wang et al. 2009) |
| M274 | 1A | 1.75 | C/T | Detected using mvLMM: Associated with DTM, GFD and PH, and reported in previous study for GFD (Wang et al. 2009) |
| M373 | 1A | 4.28 | T/G | |
| M6513 | 2B | 1.48 | A/G | Detected using epistasis: Associated with TGW and reported for TGW in previous study (Ramya et al. 2010; Chen et al. 2015) |
| M2913 | 7B | 1.40 | G/A |
Chr, chromosome; Pos, position (cM) and *: desirable allele
Identification of putative candidate genes
As many as 164 CGs involving 50 MTAs (CMLM, 3; FarmCPU, 9; mvLMM, 6 and epistasis, 21 and BLUP, 11) were identified, each within a window of 200 Kb. For 29 MTAs, no CG was available (Table S5, S6, S7, S8 and S9). Of the 164 CGs, 20 CGs were available for MTAs obtained using CMLM, 27 CGs were available from MTAs obtained using FarmCPU, 21 CGs belonged to MTAs obtained using mvLMM, 66 CGs were available from MTAs involved in epistasis and 30 CGs were identified using BLUP MTAs. Results of in-silico gene expression analysis for all 164 CGs are presented in Figs. 5, 6, 7, 8. Of 164 CGs, 15 important CGs were selected based on their involvement in different biological processes relevant to traits under study (Table 6).
Fig. 5.

Results of in-silico expression analysis of different candidate genes (CGs) for MTAs identified using GWAS based on CMLM for only three traits (no CGs were available for other traits); against each CG, the trait, the identity of MTA to which CG belongs and the environment (in parentheses), in which MTA was detected are indicated; the expression of only relevant CGs (selected using corresponding protein domains) was examined in different tissues (shown on the X-axis)
Fig. 6.

Results of in-silico expression analysis of different candidate genes (CGs) for MTAs identified using FarmCPU for only five traits (no CGs were available for other traits); against each CG, the trait, the identity of MTA to which CG belongs and the environment (in parentheses), in which MTA was detected are indicated; the expression of only relevant CGs (selected using corresponding protein domains) was examined in different tissues (shown on the X-axis)
Fig. 7.

Results of in-silico expression analysis of different candidate genes (CGs) for multi-trait MTAs identified using GWAS based on mvLMM for only five combinations of multi-traits (no CGs were available for MTAs identified for 10 other multi-trait combinations); against each CG, the trait, the identity of MTA to which CG belongs and the environment (in parentheses), in which MTA was detected are indicated; the expression of only relevant CGs (selected using corresponding protein domains) was examined in different tissues (shown on the X-axis)
Fig. 8.

Results of in-silico expression analysis of different candidate genes (CGs) for MTAs involved in epistatic interactions, identified using GWAS; against each CG, the identity of traits (some CGs, each affected more than one trait) and MTA to which CG belongs and the environment (in parentheses), in which MTA was detected are indicated; the expression of only relevant CGs (selected using corresponding protein domains) was examined in different tissues (shown on the X-axis); some of the CGs affected more than one traits
Table 6.
A summary of of selected candidate genes (CGs) and their functions
| Trait; MTA | Candidate gene (CG) | Protein domain | Description |
|---|---|---|---|
| DTA and DTH; M1955 | TraesCS5A02G417200 | F-box domain | F-box protein plays an important role in growth and development. For example TaFBA1 gene in wheat encodes F-box protein that regulates abscisic acid (ABA) involved in a variety of processes during growth and development (Verslues and Zhu 2005; Finkelstein 2006; Fujii et al. 2007; An et al. 2019) |
| TNPP; M7483 | TraesCS3B02G500900 | ||
| TGW; M11387 | TraesCS6B02G312400 | ||
| DTA; M1920 | TraesCS4A02G333000 | ||
| DTM, GFD, PH and TNPP; M9267 | TraesCS3B02G443100 | ||
| TNPP, BYPP, GNPS, GYPP, TGW; M12201 | TraesCS7D02G126300 | ||
| TraesCS7D02G126400 | |||
| TGW; M4710 | TraesCS3A02G532500 | ||
| TraesCS3A02G532600 | CytP450 | Cytochrome P450 proteins are involved in several developmental processes through the biosynthesis and/or catabolism of phytohormones and secondary compounds (Li and Wei 2020) | |
| DTH; M13005 | TraesCS4A02G372100 | ABC transporter | The ABC transporter (e.g. TaABC3) plays an important role in grain formation, ripening and in mycotoxin tolerance in wheat (Lemmens et al. 2005; Walter et al. 2015) |
| DTA; M1920 | TraesCS4A02G333700 | AP2/ERF domain | APETALA2/Ethylene-responsive factor (AP2/ERF) is involved in regulation of tolerance to several abiotic stresses (Gahlaut et al. 2019) and regulates growth and development (Zhang et al. 2020) |
| TGW; M14981 | TraesCS2B02G054100 | ||
| DTM; M3393 | TraesCS5A02G325800 | GRAS | GRAS proteins are involved in regulation of processes like photosynthesis, growth, senescence and tolerance to photooxidative stress (Chen et al. 2015; Niu et al. 2019) |
| GFD (E2); M13252 (1,123,031): 2B | TraesCS2B02G472600 | Heat shock protein DnaJ | Heat shock protein DnaJ is involved in regulation of a variety of processes like response to heat stress, during grain filling (Kumar et al. 2020) |
| DTH and DTA; M5047 | TraesCS6B02G159400 | MADS-box | MADS box proteins regulate processes like inflorescence development, flowering time, floral organ identity, and also seed development (Richards 2000; Schilling et al. 2018) |
Identification of important rare variants
As many as nine SNPs with rare alleles were found to be associated with only three of the 10 traits (GNPS, TGW and GYPP). For the remaining seven traits, no significant association of markers with rare alleles was observed (Table S10).
Discussion
During the last three decades, a variety of molecular markers and statistical tools have been developed and utilized for thousands of studies involving interval mapping and association mapping (GWAS) in all major crops. In wheat alone, thousands of QTLs/MTAs have been identified. A recent survey conducted by us for developing a wheat QTL database revealed that > 30,000 QTLs for different traits are already available in wheat and more are being regularly added to this list, suggesting that we did not reach a saturation point (Singh et al. 2021 and our unpublished results). Thus, additional genetic studies are warranted using different materials. The present study is one such study, which added several novel MTAs to the ever-increasing list of markers associated with yield and related traits, although some of the MTAs detected during the present study may correspond to the MTAs/QTLs reported in earlier studies including the latest comprehensive GWAS from CIMMYT, which reported > 800 MTAs for grain yield alone (Juliana et al. 2021).
During the present study, a diverse panel of genotypes, exhibiting significant variation in phenotypes involving 10 different traits, was used for GWAS. This is apparent from the representative violin plots (based on BLUP values) presented in Fig. 1. These results exhibit significant variation for each of the 10 traits suggesting their suitability for conducting GWAS. However, the pattern of variation differs for the different traits although, for an individual trait, the patterns in the three environments are largely similar (Fig. S1). A similar pattern of variation was also observed in two of our earlier studies conducted using association panels comprising 246 genotypes in one study (Kumar et al. 2018), and 320 genotypes in another study, both drawn from the same original set of 330 diverse genotypes procured from CIMMYT (Gahlaut et al. 2019). For six of the 10 traits (including DTH, DTA, DTM, TNPP, GYPP and GFD), the variation for different traits exhibited skewness and/or kurtosis, particularly for traits like DTH, DTA and DTM (Figs. 1, S1). The violin plots suggest that majority of genotypes are early maturing associated with long duration for grain filling, which is a desirable trait. However, a small fraction of genotypes exhibited undesirable traits including long duration of maturity and short grain-filling period. It will be interesting to find the association of desirable features of days to maturity and grain filling duration with desirable patterns of the remaining agronomic traits. The four-grain yield-related traits showed a wide range of variability showing almost normal distribution. However, the pattern of variation for GNSP and TGW differed from the variation for BYPP and GYPP, although MAS would be useful in the segregating populations derived from crosses involving parents with higher values for these traits. Further, the results of ANOVA showed no significant variation due to environment for three traits including DTH, GYPP and GNPS; no significant genotype × environment interaction for three other traits, namely DTH, GYPP and TGW. These observations may have implications on the results of GWAS that are discussed.
In two earlier studies involving GWAS using related association panels, MTAs were identified in two locations including a drought-prone area (Powerkeda) and an irrigated site (Meerut); a subset of these association panels was used in the present study for three years. The results of our two earlier studies have been compared with the results of the present study (Kumar et al. 2018; Gahlaut et al. 2019).
Despite two earlier studies involving relatively larger panels, each panel including the present subset, a number of novel MTAs were identified, suggesting that new MTAs can be identified by repeating the study using the same association mapping panel at different time points. The association panel used during the present study and also those used in two earlier studies exhibited a low level of population structure comprising only three sub-populations. However, in a number of earlier GWAS, the number of subpopulations ranged from 3 (Wang et al. 2017; Rahimi et al. 2019) to 5 (Qaseem et al. 2018; Jamil et al. 2019). The absence of population structure or low level of population structure, as observed in three GWAS (including present study) is a desirable feature for conducting GWAS.
As indicated earlier in this paper, in the present GWA study, we used four different methods which included CMLM, FarmCPU, SUPER and mvLMM. The number of MTAs obtained without applying Bonferroni correction when compared with those available after applying Bonferroni corrections indicated drastic reduction in MTAs. In particular, all the 525 MTAs identified using SUPER disappeared after application of Bonferroni correction. Also, MTAs for only three traits were available using CMLM and for only four of 10 traits were available following FarmCPU suggesting that no MTAs were available for seven traits in CMLM and six traits following FarmCPU (Table 2 and S11). This reduction/disappearance of MTAs due to Bonferroni correction calls for caution because it is widely known that the Bonferroni correction while reducing the number of false positives (a desirable feature) also gives a large number of false negatives (undesirable feature). Therefore, it has been emphasized in several studies that Bonferroni correction is a trade-off (Chen et al. 2017; Wilson 2019). It has also been emphasized, therefore, that the MTAs, which disappear after the application of Bonferroni correction should not be entirely ignored. Therefore, we examined MTAs obtained prior to Bonferroni correction for the identification of MTAs that were obtained by more than one method (Table S11). The problem of false-negative MTAs due to Bonferroni correction has been widely discussed in published literature. Among these earlier studies, the problem was discussed initially by Narum (2006) and more recently by White et al. (2019) and Cortés et al. (2020).
Among the four methods used in the present study, FarmCPU (involving multi-locus analysis) gave a maximum of 21 MTAs. When, these results are compared with those obtained due to CMLM, it is apparent that the number of MTAs increased in FarmCPU, presumably due to inherent multilocus analysis in FarmCPU. Another important observation is that MTAs for only three (DTA, DTH, GNPP) of 10 traits were available using CMLM; no MTAs were available for the remaining seven traits. From this, it is also apparent that CMLM is relatively not as efficient as FarmCPU. The superiority of FarmCPU has also been demonstrated in some earlier studies in wheat (Kaur et al. 2017; Ward et al. 2019; Gahlaut et al. 2019; Muhammad et al. 2020) and other crops like maize (Zhang et al. 2019) and rice (Gyawali et al. 2019). Another important feature of the present study was equal number of MTAs detected in all the three environments. However, the number of MTAs in individual environments differed, when examined for different methods; MTAs for E1 using FarmCPU and in E3 using CMLM exceeded those detected in E2 using the same methods. This suggested different levels of efficiency of GWAS methods in detecting QTLs exhibiting QTL x environment interaction.
An important result of the present study is the detection of 38 multi-trait MTAs (using mvLMM) for 15 different combinations of traits, each combination involving 3–5 traits. This is in sharp contrast to the use of MTMM (Korte et al. 2012), which allows detection of MTAs for only two traits at a time (Jaiswal et al. 2016; Kumar et al. 2018). These results suggest pleiotropy or close linkage. Two of the 38 MTAs, located on chromosome 1A, (M2375-1A and M3080-1A) were, however, associated with more than one of the 15 trait combinations in all the three environments. Therefore, these two MTAs should prove useful for simultaneous improvement of more than one trait using MAS. Several earlier studies also used multi-trait analysis, however, mvLMM involving more than two traits has been used only sparingly (Furlotte and Eskin 2015; Kumar et al. 2018; Deng et al. 2018; Gao et al. 2021; Chen et al. 2021).
The availability of 28 first order epistatic interactions suggests that epistatic interactions were not uncommon. Two of these interactions, one each for GYPP and TGW were available in all the three environments and should be suitable candidates for MAS. Therefore, based on the present study, it is concluded that MTAs involved in epistatic interactions, contribute significantly to trait variation and should be utilized for MAS. Epistatic interactions in wheat using GWAS have also been reported in previous studies for flowering time (Reif et al. 2011; Langer et al. 2014), stem rust resistance (Yu et al. 2011), and agronomic traits (Sehgal et al. 2017). In two recent studies from our laboratory, 63 epistatic interactions involving 13 different traits and 73 epistatic interactions for six micronutrient and yield traits in wheat were reported using GWAS (Jaiswal et al. 2016; Kumar et al. 2018). However, MTAs involved in epistatic interactions have been sparingly used in MAS for crop improvement (Reif et al. 2011; Kao et al. 1999; Langer et al. 2014; Jaiswal et al. 2016; Sehgal et al. 2017; Kumar et al. 2018). Hence results of epistatic interactions from the previous and present study may also be useful to understand the genetic architecture of traits that are relevant for wheat improvement.
Another important aspect is the observation that as many as 9 MTAs overlapped the previously reported MTAs/QTLs and another 12 MTAs occurred in the vicinity of known MTAs/QTLs (Cui et al. 2013; Wang et al. 2009; Kumar et al. 2007; Ramya et al. 2010; Chen et al. 2015; Cuthbert et al. 2008; Liu et al. 2014; Daoura et al. 2014; Wang et al. 2009; Kumar et al. 2007; Table 3). In addition, 24 novel MTAs identified in the present study add to the knowledge about the genetic architecture of the traits examined in the present study (Table 4). Overall, 11 MTAs were found to be relatively more important, using several criteria listed in the results (Table 5). These MTAs may be used in MAS.
It may also be recalled that as many as 15 of the 164 candidate genes identified during the present study, were selected based on their involvement in relevant biological processes for traits of interest (Verslues and Zhu 2005; Finkelstein 2006; Fujii et al. 2007; An et al. 2019; Table 6). These candidate genes provide a resource for future studies.
Availability of variants associated with rare alleles is not an exception, but a rule, in all GWAS studies. Markers with rare alleles were also identified in previous studies in wheat (Jaiswal et al. 2016) and rice (Hu et al. 2015). These rare variants have been considered to be important because of their associations with rare desirable phenotypes because common variants have already been the focus of selection in plant breeding. In the present study, only nine of the 3380 SNPs with rare alleles were found to be associated with three of the 10 traits including GNPS, TGW and GYPP. Since all these three traits are important components of GY, these SNPs with rare alleles should be subjected to validation using suitable methods like the following: (i) use of biparental mapping population (derived from genotypes with rare alleles); (ii) joint linkage-association mapping (JLAM); (iii) use of large population; (iv) separate analysis for common variants (CWAS) and rare variants (RVAS; Zuka et al. 2014), (v) advanced statistical tests like burden test, variance component test, combined omnibus test (Lee et al. 2014; Gupta et al. 2014; Jaiswal et al. 2016). Thus, we feel that rare variants should not be ignored during GWAS, since some of the rare alleles may be responsible for important traits.
Conclusions
The important MTAs involving main effect and epistatic QTLs identified during the present study may be further validated using post-GWAS or joint linkage and association mapping (JLAM; Gupta et al. 2019; Gahlaut et al. 2019) and used for MAS in wheat breeding programmes targeted towards yield improvement. The information of the CGs may also be useful for the development of CG-based functional markers and for further basic research including CG-based association mapping and functional genomics approaches.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The material for association panel and the genotypic data for this work was received from CIMMYT, Mexico. Thanks are due to the Department of Biotechnology (DBT), Govt of India for providing funds in the form of research projects awarded to Shailendra Sharma (SS). The authors are thankful to Ch. Charan Singh University, Meerut for providing laboratory and field facilities. HSB was awarded positions of INSA Senior Scientist/Honorary Scientist during the course of the study by INSA, New Delhi.
Author contribution
SS (Shailendra Sharma) conceived and conducted or directed the experiments. PM, JK, SS1(SahadevSingh), SS2 (Shiveta Sharma) performed the experiments. JK, PKM, MS analysed the data. JKR, PKS, HSB, PKG and SS participated in manuscript writing. SS finalized the manuscript.
Data and materials availability
Not applicable.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors consented to publication.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Parveen Malik and Jitendra Kumar equal contribution
References
- Allard RW (1999) Principles of Plant Breeding. Wiley and Sons, Inc., New York, p 485
- An J, Li Q, Yang J, et al. Wheat F-box protein TaFBA1 positively regulates plant drought tolerance but negatively regulates stomatal closure. Front Plant Sci. 2019;10:1–20. doi: 10.3389/fpls.2019.01242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astle W, Balding DJ. Population structure and cryptic relatedness in genetic association studies. Stat Sci. 2009;24:451–471. doi: 10.1214/09-STS307. [DOI] [Google Scholar]
- Bates D, Mächler M, Bolker B, Walker S (2015) Fitting linear mixed-effects models using lme4. J Stat Softw 67:1–48
- Chen K, Li H, Chen Y et al (2015) TaSCL14, a novel wheat (Triticum aestivum L.) GRAS gene, regulates plant growth, photosynthesis, tolerance to photooxidative stress, and senescence. J Genet Genomics 42:21–32 [DOI] [PubMed]
- Chen SY, Feng Z, Yi X. A general introduction to adjustment for multiple comparisons. J Thorac Dis. 2017;9:1725. doi: 10.21037/jtd.2017.05.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wu H, Yang W et al (2021) Multivariate linear mixed model enhanced the power of identifying genome-wide association to poplar tree heights in a randomized complete block design. G3 Genes Genomes Genet. 10.1093/g3journal/jkaa053 [DOI] [PMC free article] [PubMed]
- Chu J, Zhao Y, Beier S, et al. Suitability of single-nucleotide polymorphism arrays versus genotyping-by-sequencing for genebank genomics in wheat. Front Plant Sci. 2020;11:1–12. doi: 10.3389/fpls.2020.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortés J, Mahecha M, Reichstein M, Brenning A. Accounting for multiple testing in the analysis of spatio-temporal environmental data. Environ Ecol Stat. 2020;27:293–318. doi: 10.1007/s10651-020-00446-4. [DOI] [Google Scholar]
- Cui F, Zhao C, Li J, et al. Kernel weight per spike: what contributes to it at the individual QTL level? Mol Breed. 2013;31:265–278. doi: 10.1007/s11032-012-9786-8. [DOI] [Google Scholar]
- Cuthbert JL, Somers DJ, Brûlé-Babel AL, et al. Molecular mapping of quantitative trait loci for yield and yield components in spring wheat (Triticum aestivum L.) Theor Appl Genet. 2008;117:595–608. doi: 10.1007/s00122-008-0804-5. [DOI] [PubMed] [Google Scholar]
- Daoura BG, Chen L, Du Y, Hu YG (2014) Genetic effects of dwarfing gene Rht-5 on agronomic traits in common wheat (Triticum aestivum L.) and QTL analysis on its linked traits. F Crop Res 156:22–29
- Deng X, Wang B, Fisher V, et al. Genome-wide association study for multiple phenotype analysis. BMC Proc. 2018;12:139–144. doi: 10.1186/s12919-018-0135-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelstein RR. Studies of abscisic acid perception finally flower. Plant Cell. 2006;18:786–791. doi: 10.1105/tpc.106.041129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii H, Verslues PE, Zhu JK. Identification of two protein kinases required for abscisic acid regulation of seed germination, root growth, and gene expression in Arabidopsis. Plant Cell. 2007;19:485–494. doi: 10.1105/tpc.106.048538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlotte NA, Eskin E. Efficient multiple-trait association and estimation of genetic correlation using the matrix-variate linear mixed model. Genetics. 2015;200:59–68. doi: 10.1534/genetics.114.171447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahlaut V, Jaiswal V, Singh S, et al. Multi-locus genome wide association mapping for yield and its contributing traits in hexaploid wheat under different water regimes. Sci Rep. 2019;9:1–15. doi: 10.1038/s41598-019-55520-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B, Yang C, Liu J, Zhou X. Accurate genetic and environmental covariance estimation with composite likelihood in genome-wide association studies. PLoS Genet. 2021;17:1–25. doi: 10.1371/journal.pgen.1009293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González JR, Armengol L, Solé X, et al. SNPassoc: an R package to perform whole genome association studies. Bioinformatics. 2007;23:644–645. doi: 10.1093/bioinformatics/btm025. [DOI] [PubMed] [Google Scholar]
- Gupta PK, Kulwal PL, Jaiswal V. Association mapping in crop plants: opportunities and challenges. Adv Genet. 2014;85:109–147. doi: 10.1016/B978-0-12-800271-1.00002-0. [DOI] [PubMed] [Google Scholar]
- Gupta PK, Kulwal PL, Jaiswal V. Association mapping in plants in the post-GWAS genomics era. Adv Genet. 2019;104:75–154. doi: 10.1016/bs.adgen.2018.12.001. [DOI] [PubMed] [Google Scholar]
- Gupta PK, Balyan HS, Sharma S, Kumar R. Genetics of yield, abiotic stress tolerance and biofortification in wheat (Triticum aestivum L.) Theor Appl Genet. 2020;133:1569–1602. doi: 10.1007/s00122-020-03583-3. [DOI] [PubMed] [Google Scholar]
- Gyawali A, Shrestha V, Guill KE, et al. Single-plant GWAS coupled with bulk segregant analysis allows rapid identification and corroboration of plant-height candidate SNPs. BMC Plant Biol. 2019;19:1–15. doi: 10.1186/s12870-019-2000-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry RJ, Rangan P, Furtado A. Functional cereals for production in new and variable climates. Curr Opin Plant Biol. 2016;30:11–18. doi: 10.1016/j.pbi.2015.12.008. [DOI] [PubMed] [Google Scholar]
- Hu J, Wang Y, Fang Y, et al. A rare allele of GS2 enhances grain size and grain yield in rice. Mol Plant. 2015;8:1455–1465. doi: 10.1016/j.molp.2015.07.002. [DOI] [PubMed] [Google Scholar]
- IWGSC Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science. 2018;345:1251788. doi: 10.1126/science.aar7191. [DOI] [PubMed] [Google Scholar]
- Jaiswal V, Gahlaut V, Meher PK, et al. Genome wide single locus single trait, multi-locus and multi-trait association mapping for some important agronomic traits in common wheat (T. aestivum L.) PLoS ONE. 2016;11:1–25. doi: 10.1371/journal.pone.0159343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamil M, Ali A, Gul A, et al. Genome-wide association studies of seven agronomic traits under two sowing conditions in bread wheat. BMC Plant Biol. 2019;19:1–18. doi: 10.1186/s12870-019-1754-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliana P, Singh RP, Poland J, et al. Elucidating the genetics of grain yield and stress-resilience in bread wheat using a large-scale genome-wide association mapping study with 55,568 lines. Sci Rep. 2021;11:1–15. doi: 10.1038/s41598-021-84308-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao CH, Zeng ZB, Teasdale RD. Multiple interval mapping for quantitative trait loci. Genetics. 1999;152:1203–1216. doi: 10.1093/genetics/152.3.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S, Zhang X, Mohan A, et al. Genome-wide association study reveals novel genes associated with culm cellulose content in bread wheat (Triticum aestivum, L.) Front Plant Sci. 2017;8:1–7. doi: 10.3389/fpls.2017.01913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya Y, Akcura M. Effects of genotype and environment on grain yield and quality traits in bread wheat (T. aestivum L.) Food Sci Technol. 2014;34:386–393. doi: 10.1590/fst.2014.0041. [DOI] [Google Scholar]
- Korte A, Vilhjálmsson BJ, Segura V, et al. A mixed-model approach for genome-wide association studies of correlated traits in structured populations. Nat Genet. 2012;44:1066–1071. doi: 10.1038/ng.2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N, Kulwal PL, Balyan HS, Gupta PK. QTL mapping for yield and yield contributing traits in two mapping populations of bread wheat. Mol Breed. 2007;19:163–177. doi: 10.1007/s11032-006-9056-8. [DOI] [Google Scholar]
- Kumar J, Saripalli G, Gahlaut V, et al. Genetics of Fe, Zn, β-carotene, GPC and yield traits in bread wheat (Triticum aestivum L.) using multi-locus and multi-traits GWAS. Euphytica. 2018;214:1–17. doi: 10.1007/s10681-018-2284-2. [DOI] [Google Scholar]
- Kumar A, Sharma S, Chunduri V, et al. Genome-wide identification and characterization of heat shock protein family reveals role in development and stress conditions in Triticum aestivum L. Sci Rep. 2020;10:1–12. doi: 10.1038/s41598-020-64746-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer SM, Longin CFH, Würschum T. Flowering time control in European winter wheat. Front Plant Sci. 2014;5:1–12. doi: 10.3389/fpls.2014.00537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HS, Jung JU, Kang CS, et al. Mapping of QTL for yield and its related traits in a doubled haploid population of Korean wheat. Plant Biotechnol Rep. 2014;8:443–454. doi: 10.1007/s11816-014-0337-0. [DOI] [Google Scholar]
- Lemmens M, Scholz U, Berthiller F, et al. The ability to detoxify the mycotoxin deoxynivalenol colocalizes with a major quantitative trait locus for fusarium head blight resistance in wheat. Mol Plant-Microbe Interact. 2005;18:1318–1324. doi: 10.1094/MPMI-18-1318. [DOI] [PubMed] [Google Scholar]
- Li Y, Wei K. Comparative functional genomics analysis of cytochrome P450 gene superfamily in wheat and maize. BMC Plant Biol. 2020;20:1–22. doi: 10.1186/s12870-020-2288-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipka AE, Tian F, Wang Q, et al. GAPIT: genome association and prediction integrated tool. Bioinformatics. 2012;28:2397–2399. doi: 10.1093/bioinformatics/bts444. [DOI] [PubMed] [Google Scholar]
- Liu G, Jia L, Lu L, et al. Mapping QTLs of yield-related traits using RIL population derived from common wheat and Tibetan semi-wild wheat. Theor Appl Genet. 2014;127:2415–2432. doi: 10.1007/s00122-014-2387-7. [DOI] [PubMed] [Google Scholar]
- Liu X, Huang M, Fan B, et al. Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLOS Genet. 2016;12:1–24. doi: 10.1371/journal.pgen.1005767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhammad A, Hu W, Li Z, et al. Appraising the genetic architecture of Kernel traits in hexaploid wheat using GWAS. Int J Mol Sci. 2020;21:1–21. doi: 10.3390/ijms21165649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narum SR. Beyond Bonferroni: less conservative analyses for conservation genetics. Conserv Genet. 2006;7:783–787. doi: 10.1007/s10592-005-9056-y. [DOI] [Google Scholar]
- Nehe A, Akin B, Sanal T, et al. Genotype x environment interaction and genetic gain for grain yield and grain quality traits in Turkish spring wheat released between 1964 and 2010. PLoS ONE. 2019;14:1–18. doi: 10.1371/journal.pone.0219432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu X, Chen S, Li J et al (2019) Genome-wide identification of GRAS genes in Brachypodium distachyon and functional characterization of BdSLR1 and BdSLRL1. BMC Genomics 20:1–18 [DOI] [PMC free article] [PubMed]
- Qaseem MF, Qureshi R, Muqaddasi QH, et al. Genome-wide association mapping in bread wheat subjected to independent and combined high temperature and drought stress. PLoS ONE. 2018;13:1–22. doi: 10.1371/journal.pone.0199121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahimi Y, Bihamta MR, Taleei A, et al. Genome-wide association study of agronomic traits in bread wheat reveals novel putative alleles for future breeding programs. BMC Plant Biol. 2019;19:1–19. doi: 10.1186/s12870-019-2165-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramya P, Chaubal A, Kulkarni K, et al. QTL mapping of 1000-kernel weight, kernel length, and kernel width in bread wheat (Triticum aestivum L.) J Appl Genet. 2010;51:421–429. doi: 10.1007/BF03208872. [DOI] [PubMed] [Google Scholar]
- Ray DK, Mueller ND, West PC, Foley JA. Yield trends are insufficient to double global crop production by 2050. PLoS ONE. 2013;8:1–8. doi: 10.1371/journal.pone.0066428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reif JC, Maurer HP, Korzun V, et al. Mapping QTLs with main and epistatic effects underlying grain yield and heading time in soft winter wheat. Theor Appl Genet. 2011;123:283–292. doi: 10.1007/s00122-011-1583-y. [DOI] [PubMed] [Google Scholar]
- Richards RA. Selectable traits to increase crop photosynthesis and yield of grain crops. J Exp Bot. 2000;51:447–458. doi: 10.1093/jexbot/51.suppl_1.447. [DOI] [PubMed] [Google Scholar]
- Schilling S, Pan S, Kennedy A, Melzer R. MADS-box genes and crop domestication: the jack of all traits. J Exp Bot. 2018;69:1447–1469. doi: 10.1093/jxb/erx479. [DOI] [PubMed] [Google Scholar]
- Sehgal D, Autrique E, Singh R, et al. Identification of genomic regions for grain yield and yield stability and their epistatic interactions. Sci Rep. 2017;7:1–12. doi: 10.1038/srep41578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K, Batra R, Sharma S et al (2021) Wheat QTL db: A QTL database for wheat. Mol Genet Genomics (in press) [DOI] [PubMed]
- SPSS Inc (2008) Released, SPSS Statistics for Windows, Version 17.0. Chicago: SPSS Inc
- Sun C, Dong Z, Zhao L, et al. The Wheat 660K SNP array demonstrates great potential for marker-assisted selection in polyploid wheat. Plant Biotechnol J. 2020;18:1354–1360. doi: 10.1111/pbi.13361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanRaden PM. Efficient methods to compute genomic predictions. J Dairy Sci. 2008;91:4414–4423. doi: 10.3168/jds.2007-0980. [DOI] [PubMed] [Google Scholar]
- Verslues PE, Zhu JK. Before and beyond ABA: upstream sensing and internal signals that determine ABA accumulation and response under abiotic stress. Biochem Soc Trans. 2005;33:375–379. doi: 10.1042/BST0330375. [DOI] [PubMed] [Google Scholar]
- Walter S, Kahla A, Arunachalam C, et al. A wheat ABC transporter contributes to both grain formation and mycotoxin tolerance. J Exp Bot. 2015;66:2583–2593. doi: 10.1093/jxb/erv048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RX, Hai L, Zhang XY, et al. QTL mapping for grain filling rate and yield-related traits in RILs of the Chinese winter wheat population Heshangmai x Yu8679. Theor Appl Genet. 2009;118:313–325. doi: 10.1007/s00122-008-0901-5. [DOI] [PubMed] [Google Scholar]
- Wang S, Wong D, Forrest K, et al. Characterization of polyploid wheat genomic diversity using a high-density 90 000 single nucleotide polymorphism array. Plant Biotechnol J. 2014;12:787–796. doi: 10.1111/pbi.12183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SX, Zhu YL, Zhang DX, et al. Genome-wide association study for grain yield and related traits in elite wheat varieties and advanced lines using SNP markers. PLoS ONE. 2017;12:1–14. doi: 10.1371/journal.pone.0188662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward BP, Brown-Guedira G, Kolb FL, et al. Genome-wide association studies for yield-related traits in soft red winter wheat grown in Virginia. PLoS ONE. 2019;14:1–28. doi: 10.1371/journal.pone.0208217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White T, van der Ende J, Nichols TE. Beyond Bonferroni revisited: concerns over inflated false positive research findings in the fields of conservation genetics, biology, and medicine. Conserv Genet. 2019;20:927–937. doi: 10.1007/s10592-019-01178-0. [DOI] [Google Scholar]
- Wilson DJ. The harmonic mean p-value for combining dependent tests. Proc Natl Acad Sci. 2019;116:1195–1200. doi: 10.1073/pnas.1814092116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav AK, Carroll AJ, Estavillo GM, et al. Wheat drought tolerance in the field is predicted by amino acid responses to glasshouse-imposed drought. J Exp Bot. 2019;70:4931–4947. doi: 10.1093/jxb/erz224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu LX, Lorenz A, Rutkoski J, et al. Association mapping and gene-gene interaction for stem rust resistance in CIMMYT spring wheat germplasm. Theor Appl Genet. 2011;123:1257–1268. doi: 10.1007/s00122-011-1664-y. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Ersoz E, Lai CQ, et al. Mixed linear model approach adapted for genome-wide association studies. Nat Genet. 2010;42:355–360. doi: 10.1038/ng.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YM, Jia Z, Dunwell JM (2019) Editorial: The applications of new multi-locus GWAS methodologies in the genetic dissection of complex traits. Front Plant Sci 10:1–6 [DOI] [PMC free article] [PubMed]
- Zhang L, Liu P, Wu J et al (2020) Identification of a novel ERF gene, TaERF8, associated with plant height and yield in wheat. BMC Plant Biol 20:1–12 [DOI] [PMC free article] [PubMed]
- Zhu T, Wang L, Rimbert H et al (2021) Optical maps refine the bread wheat Triticum aestivum cv. Chinese Spring genome assembly. Plant J 1–12 [DOI] [PMC free article] [PubMed]
- Zuka O, Schaffner SF, Samocha K, et al. Searching for missing heritability: designing rare variant association studies. Proc Natl Acad Sci. 2014;111:E455–E464. doi: 10.1073/pnas.1322563111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not applicable.