

Abstract
Recent advances in spatial transcriptomics (ST) enable gene expression measurements from a tissue sample while retaining its spatial context. This technology enables unprecedented in situ resolution of the regulatory pathways that underlie the heterogeneity in the tumor and its microenvironment (TME). The direct characterization of cellular co-localization with spatial technologies facilities quantification of the molecular changes resulting from direct cell-cell interaction, as occurs in tumor-immune interactions. We present SpaceMarkers, a bioinformatics algorithm to infer molecular changes from cell-cell interaction from latent space analysis of ST data. We apply this approach to infer molecular changes from tumor-immune interactions in Visium spatial transcriptomics data of metastasis, invasive and precursor lesions, and immunotherapy treatment. Further transfer learning in matched scRNA-seq data enabled further quantification of the specific cell types in which SpaceMarkers are enriched. Altogether, SpaceMarkers can identify the location and context-specific molecular interactions within the TME from ST data.
A record of this paper’s Transparent Peer Review process is included in the Supplemental Information.
Keywords: cell-cell interactions, spatial transcriptomics, latent space factorization, spatial analysis, tumor microenvironment, single-cell transcriptomics, transfer learning
eTOC blurb
Deshpande and Loth et al. present SpaceMarkers as an algorithm to identify molecular changes resulting from cell-cell interactions using latent space analysis of spatial transcriptomics. SpaceMarkers uses spatial co-localization of latent features as direct evidence of cellular interactions and applies the method to analyze tumor-immune interactions across tumor types.
Graphical Abstract
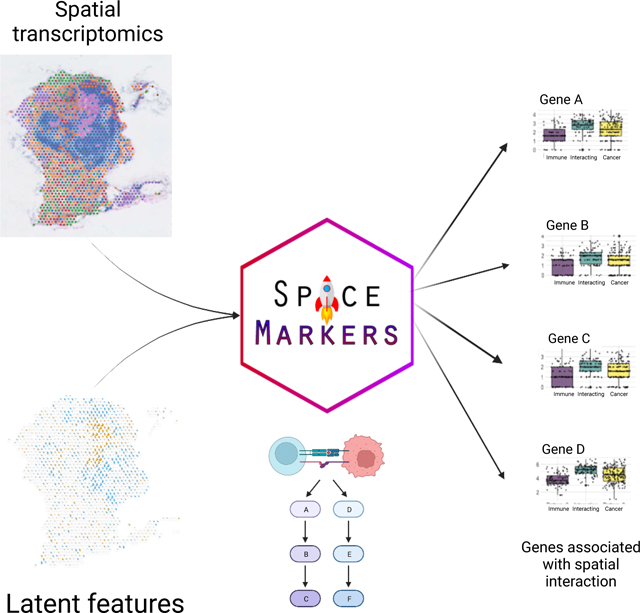
1. Introduction
The tumor microenvironment (TME) is the tissue region created and controlled by a tumor in its surroundings and plays a key role in tumorigenesis and therapeutic response in cancer36,8,21,44. The TME contains tumor cells, stroma, blood vessels, and immune cells as well as cells from the resident tissue44. A thorough understanding of the molecular profile of individual cells and the impact of inter-cellular interactions in the TME is crucial for distinguishing the determinants of tumor progression10,15,43 and precision medicine strategies21,6,25,33,39.
Advances in single-cell technologies have led to the development of spatially resolved transcriptomics (ST) which captures the transcriptome in situ34 and thus allows us to study the spatial relationship between the various cell populations within the TME as well as their relationship with the tumor cells. For example, the 10X Visium spatial transcriptomic technology allows us to resolve tissue heterogeneity at near single-cell resolution (from one to tens of cells per spot). The technique has been applied to characterize the cellular and molecular composition of tumors45,20,1. Robust analysis pipelines for cell-based analysis and cellular deconvolution have been proposed to model the cellular composition of spatial-transcriptomics data5,29,46,19,13 and cellular phenotypes within each spot28. While spot deconvolution methods can infer linear combinations of molecular markers that are reflective of cellular co-localization, new computational methods are needed to characterize the molecular changes resulting from cell-cell interaction at a genome-wide scale.
Many analysis pipelines for Visium ST rely on latent space methods for cellular deconvolution to overcome the mixture of cells at each spot. In this paper, we present the SpaceMarkers algorithm which leverages spatially interacting latent features to infer molecular changes resulting from interactions between cell types or biological processes represented by the features. SpaceMarkers uses a kernel-based smoothing approach to model the influence of a highly-expressed feature in a spot extending to its neighboring spots as well. Using latent features inferred from CoGAPS14, we demonstrate the broad utility of SpaceMarkers to inferring molecular changes resulting from cell-cell interactions in Visium samples from invasion to lymph node, pancreatic premalignant lesions, breast primary tumor, and immunotherapy treated cancer. We selected CoGAPS, a Bayesian non-negative matrix factorization approach, based on its robustness for single-cell RNA-seq data9,41. We also show the compatibility of SpaceMarkers with other latent space methods, using STdeconvolve29 as an example. Further extension of this approach to integrate Visium data with single-cell data through transfer learning also enables identification of the precise cell subtypes in which the molecular changes from cell-cell interactions are introduced. Altogether, our extension to latent space analysis enables us to simultaneously infer cellular architecture and model molecular changes resulting from spatially interacting biological processes.
2. Results
2.1. Interactions between overlapping latent features delineate inter-cellular interactions in ST data
Here we present SpaceMarkers, a bioinformatics algorithm for identifying genes associated with cell-cell interactions in ST data. SpaceMarkers is an extension of latent space analysis that leverages spatially overlapping latent features associated with distinct cellular signatures to infer the genes associated with their interaction (Figure 1). Fundamentally, this inference relies on estimation of spatially resolved latent features representative of cellular signatures in the ST data. That is, the latent feature information is characterized by continuous weights corresponding to each spatial coordinate in the ST data. We denote these continuous weights as the patterns in the ST data. The inputs to the SpaceMarkers algorithm are the ST data matrix and spatially resolved patterns learned through latent space analysis, and the output is a list of genes associated with the interaction between each pair of spatially overlapping patterns. The first stage of the algorithm involves the identification of each pattern’s region of influence and subsequently the region of pattern interaction (Figure 1A.; see also Methods). If a pattern has a nonzero value at a point, we hypothesize that its influence extends to its neighboring region but rapidly decreases with increasing distance. We model this by spatially smoothing the patterns using a Gaussian-kernel based approach (see Methods). Subsequently, we identify outlier values of smoothed patterns by testing it against a null-distribution obtained by identical smoothing of spatially permuted pattern values. We denote the region corresponding to these outlier values as the region of influence of the pattern. Furthermore, two patterns are deemed to be interacting in the region with overlapping influence from both patterns. We hypothesize that genes associated with the spatially overlapping influence from two patterns represent changes in molecular pathways due to the interaction between the biological features of the associated cells. Therefore, we devise the second stage of the SpaceMarkers algorithm to rank genes exhibiting higher activity levels in the interaction region relative to regions with exclusive influence from each pattern (Figure 1B.; see also Methods). To this end, we perform a non-parametric statistical test followed by posthoc analysis to identify these genes which constitute the SpaceMarkers output.
Figure 1.

SpaceMarkers identifies genes associated with cell-cell interaction using spatially overlapping patterns
A. Identifying interaction region: The input to the SpaceMarkers algorithm are spatially resolved latent features resulting from latent space analyses (e.g. CoGAPS patterns). The images on the left show the intensity levels of two spatially resolved CoGAPS patterns. For each pattern, the SpaceMarkers algorithm first identifies regions of influence (red and blue spots, respectively) using a Gaussian-kernel based outlier detection method. The patterns are deemed to be interacting in the region with overlapping influence (yellow spots) from both patterns. It also identifies regions with mutually exclusive influence from each pattern (red and blue spots).
B. Identifying SpaceMarkers genes: The second stage of the SpaceMarkers algorithm performs a non-parametric Kruskal-Wallis statistical test with posthoc analysis on the gene expression data in the three regions (pattern 1 only, pattern 2 only, and interaction region) to identify molecular changes due to cell-cell interaction. The output is a list of genes associated with the pattern interaction (see Methods).
In the examples demonstrated here, the spatial data is obtained using the spot-based 10x Visium spatial transcriptomics technology34 with 1–10 cells per spot. SpaceMarkers is readily applicable to spot-based ST data with regions of influence and interaction defined as sets of spots in which one or two patterns respectively have influence as identified by the Gaussian-kernel based approach. We use CoGAPS Bayesian nonnegative matrix factorization14,40 for identifying the latent features associated with cellular signatures. When two patterns have overlapping influence in the same region of the tissue, we assume an interaction between these patterns in this interaction region. We provide a differential expression (DE) mode for SpaceMarkers to quantify genes with enhanced expression in a region with overlapping influence from two patterns when compared to regions with exclusive influence from individual patterns. This DE mode allows for broad applicability across latent space methods, which we demonstrate by applications using CoGAPS and STDeconvolve29. Further we extend this approach to provide a “residual” mode — which identifies genes that have significantly higher residual error between the original ST data and its estimated fit from the CoGAPS model in the region with overlapping influence from two patterns when compared to the regions with exclusive influence from each pattern. We hypothesize that the residual mode detects the nonlinear effects of intercellular interaction more precisely by accounting for the underlying linear latent features to mitigate confounding effects from variations in the cell population density and cell types with common markers. Thus, the SpaceMarkers algorithm infers both simple molecular changes in the “DE” mode as well as more precise nonlinear molecular changes in the “residual” mode in regions with overlapping influence from patterns associated with different cell signatures. We denote such patterns with concurrent influence in a region as “spatially interacting” patterns. The reliance on latent space patterns from CoGAPS enables the further ability to integrate SpaceMarkers learned from ST data in corresponding single-cell data using transfer learning from projectR37,41 to refine the specific cells in which these molecular changes occur. While the examples in this paper use latent space patterns in ST data from CoGAPS or STdeconvolve to define cellular signatures, it is generally applicable to the output of any of a number of latent feature factorization approaches available in the literature.
2.2. SpaceMarkers identifies molecular changes from tumor immune interactions associated with metastatic pancreatic cancer cells invading the lymph node
In the first example, we applied SpaceMarkers on Visium ST data from a pancreatic cancer metastasis to the lymph node in a patient who received neoadjuvant GVAX vaccination (see Figure 2). More specifically, this sample is characterized by the presence of metastatic PDAC, immune cell aggregates, and germinal centers of B-cell maturation (Figure 2A.). Analysis of the H&E imaging from the lymph node region used to generate the ST data identifies a region of the tissue in which the metastatic PDAC intersects the immune cells surrounding the germinal center. On factorizing this data using CoGAPS we obtain ten latent patterns based only on the expression data (Supplemental Figure S1, Methods). By matching pattern activity levels learned from the data with the independent histological annotations, we observe that CoGAPS can distinguish metastatic PDAC in Pattern 6 from immune cells in the surrounding lymph node tissue in Pattern 9 (Figure 2B.).
Figure 2.

SpaceMarkers identifies molecular changes associated with immune-metastatic pancreatic cancer interaction in the lymph node
A. H&E staining of a peritumoral pancreatic lymph node with metastasis from PDAC (arrow) and annotated germinal center and immune cells (dark lines).
B. Visualization of the relative activity in the CoGAPS patterns associated with metastatic PDAC (Pattern 6) and immune cells in the lymph node (Pattern 9). Each spot is represented as a pie chart with fractional gene expression at the location aggregated over the all genes for Pattern 6 (orange), Pattern 9 (blue), and all Other patterns put together (white).
C. Boxplots of the expression of selected genes showing higher expression levels in the interaction region of Pattern 6 and Pattern 9 compared to the regions with exclusive influence from Pattern 6 and Pattern 9 respectively.
D. Table showing Hallmark gene set pathways significantly overrepresented in the region of interaction between Pattern 6 and Pattern 9, with size of overlap and FDR value (see Table S2 for KEGG and Biocarta pathways).
We further analyzed the spatial activity of the metastatic PDAC (Pattern 6) and immune (Pattern 9) patterns to identify regions of overlapping influence to associate with metastasis-immune interaction. We represent the spatial variation in the activity levels of Pattern 6 and Pattern 9 in relation to all the other patterns in each spot (Figure 2B.). This proportional analysis of patterns enables us to observe a spatial overlap between the regions where Pattern 6 and Pattern 9 are active. However, we hypothesize that a pattern has influence in a spot even with zero pattern activity but high pattern activity levels in the neighboring spots. SpaceMarkers first identifies the region with spatially overlapping influence from these two patterns as their interaction region. Next, the SpaceMarkers algorithm identifies the gene expression changes that occur from metastasis-immune interaction in this interaction region (Data S1, Table S2). Due to the limited number of spots where the two patterns have overlapping influence, we define SpaceMarkers based upon differential expression. This analysis identifies 1442 genes which exhibit higher average expression in the interaction region with overlapping influence from the two patterns compared to spots where only metastatic PDAC in Pattern 6 or immune cells in Pattern 9 have exclusive influence (see Methods for details of the statistical test, Table S2 for complete gene list with the associated statistics). The SpaceMarkers optParams values are tabulated in Supplemental Table S1.
Supplemental Figure S1 shows the expression heatmap of the SpaceMarkers genes in spots belonging to regions with exclusive influence from the metastatic PDAC Pattern 6, exclusive influence from the immune cell Pattern 9, and overlapping influence from both patterns in metastasis-immune interaction. In all cases, the interactions are associated with changes in extracellular matrix genes, including notably genes associated with cytoskeleton regulation (TMSB10, TMSB4X, CFL1, MARCKSL1 ), the myosin pathway (MYL6, MYH9, MYL12B), actin regulation (ACTB, ACTN4, CAPG, LCP1, SPTBN1 ), the matrix metallopeptidase family (MMP9, MMP12 ), galectin genes (LGALS1, LGALS4, LGALS9, LGALS3BP), collagen (COL1A2, COL3A1, COL4A1, COL4A2, COL18A1, COL6A2 ), and cell adhesion (MSLN, ITGB4, ITGB6, ADRM1 ). The SpaceMarkers include genes reflecting cell death in the increased expression of ribosomal protein genes associated with immune response through expression of HLA family genes, immunogoblulins, interleukins, cytokines, chemokines, the interferon pathway IFITM2, and immune function. This immune response is counterbalanced by changes to pathways associated with enhanced invasion in cancer cells, including JUNB, JUND, VIM.
To further elucidate the molecular pathways associated with the metastasis-immune interaction in the lymph node, we performed gene set overrepresentation analysis (Figure 2D., Table S2) from the Hallmark, KEGG, and Biocarta molecular pathways using the Molecular Signatures Database (MSigDB)27,42,26. As seen in Figure 2D., Hallmark pathways related to allograft rejection, interferon gamma, and interferon alpha are all overrepresented in the pathway analysis for the SpaceMarkers genes, and hence in the region of overlap between the immune and metastatic PDAC patterns. This confirms activation of the immune response for tumor rejection at the interface between the metastatic PDAC and the immune cells in the lymph node observed at the gene level. Likewise, we observe overrepresentation in the epithelial to mesenchymal signaling and pathways consistent with the invasive process in the metastatic PDAC cells, further supported by the enrichment of the apical junction consistent with the changes to the extracellular matrix suggested by the gene-level SpaceMarkers analysis.
The DE mode of SpaceMarkers is applicable when the available latent features provide only a partial reconstruction of the original ST data matrix. However, the differential expression of a marker in the interaction region could occur because of cell-cell interactions or due to confounders such as variable cell populations in each spot and different co-localized cell types having common markers. In the examples to follow, we mitigate these confounding effects by using the residual error between the raw expression and its reconstruction from the CoGAPS patterns, which capture the effect of both variations in cell population density as well as variations in individual marker expression.
2.3. Confounding factors from unrelated cell populations can be mitigated by using SpaceMarkers in residuals mode
Using SpaceMarkers in the DE mode identifies genes that are enriched relative to two patterns, but the output is susceptible to confounding factors from cell types independent of the two patterns of interest. For example, if the interaction region between two patterns contains an independent cell type that is not significantly present in regions of exclusive influence of either pattern, we hypothesize that the cell-type-specific genes for the additional cell type will appear as space markers. To test this hypothesis, we applied SpaceMarkers to a sample of pancreatic intraepithelial neoplasia (PanIN)3, a premalignant lesion associated with PDAC (Figure 3). As described in our previous study of this sample, the H&E imaging provided with the Visium FFPE technology used to profile this sample enabled us to determine cell types within the slide at a single-cell resolution using CODA22, a deep learning classifier that annotates tissue regions based on their morphological features (Figure 3A.). This ground truth of cellular features also enables us to benchmark the latent space estimates of cellular features from CoGAPS. In this sample, we learned 10 transcriptional patterns from the PanIN using CoGAPS. Pattern 9 captures the PanIN on the tissue, and Pattern 6 captures a majority of stromal cells (Figure 3B.). The PanIN is surrounded by two large acini, which express high quantities of pancreatic enzymes that are not expressed elsewhere on the slide (Figure 3D.). The interaction region between Patterns 6 and 9 captures much of these acini. The SpaceMarkers analyses of these patterns in the DE mode results in several of the well-characterized pancreatic enzymes (Data S1) produced exclusively by acinar cells35. Pathway analysis reveals that pancreatic acinar cell gene set is the most over-represented gene set (Figure 3E., Supplementary Table S3).
Figure 3.

Residual mode can help to mitigate the confounding effects of other cell types present in the interaction region
A. Tissue regions annotated by CODA based on morphological features show clusters of acinar cells in close proximity to the neoplasia duct.
B. CoGAPS analysis reveals two patterns representing the stromal region (Pattern 6, orange), the neoplasia region (Pattern 9, blue) and all Other patterns (white).
C. Scatterpie chart showing the overlap between Pattern 6 and Pattern 9 also illustrated how the acinar cells coincide with the interaction region between the two patterns.
D. Two markers of acinar cells identified among the top SpaceMarkers of interaction between Patterns 6 and 9 also show overexpression in their interaction region.
E. Overrepresented pathways associated with neoplasia-stromal interactions identified by SpaceMarkers analysis in DE mode demonstrate overrepresentation of acinar cell markers.
F. Other relevant pathways are overrpresented in comparison to acinar markers with SpaceMarkers analysis in residual mode.
In the residuals mode, SpaceMarkers accounts for the gene signatures captured by CoGAPS patterns. Because Pattern 5 represents the acinar cells in our data set (Supplemental Figure S2), we hypothesize that the residuals mode attenuates the confounding factor due to the acinar cells (Data S1). Unlike the DE mode, the top pathway for residuals mode is no longer a pancreatic acinar cell pathway (Figure 3F., Supplementary Table S3). Residuals mode boosts the signal from pathways that are highly over-represented in the differential expression mode, while maintaining the significance of the acinar gene sets. Collectively, these results show that genes captured by differential expression mode can represent additional cell types that are not present in either patterns of interest. Additionally, if these cell type signatures are unique and strong enough to be captured as an independent transcriptional pattern, residuals mode is capable of attenuating the signal from this additional cell type relative to other expression changes present in the interaction region. The SpaceMarkers optParams values are tabulated in Supplemental Table S1.
2.4. SpaceMarkers identifies markers of tumor-immune interactions in invasive breast ductal carcinoma through residual space analysis
While providing a means to detect molecular changes from cellular interactions in limited interaction regions, using differential expression statistics for SpaceMarkers could confound nonlinear effects from cell-cell interactions with expression changes resulting from increased density of co-localized cell types with shared gene markers. In cases where the interaction region extends across a greater number of spots, these confounding effects can be mitigated by using the residual error between the raw expression and its estimated fit from the CoGAPS model for the SpaceMarkers. This estimated fit will capture the effect of both variations in cell population density as well as variations in individual marker expression to refine the estimates of the nonlinear effects from cell-cell interactions. We apply this approach to identify the molecular pathways associated with tumor cell and immune interactions in ST data from a breast cancer sample that contains multiple ductal carcinoma in situ (DCIS) lesions, an invasive carcinoma lesion, immune cells, and stroma (Figure 4A.).
Figure 4.
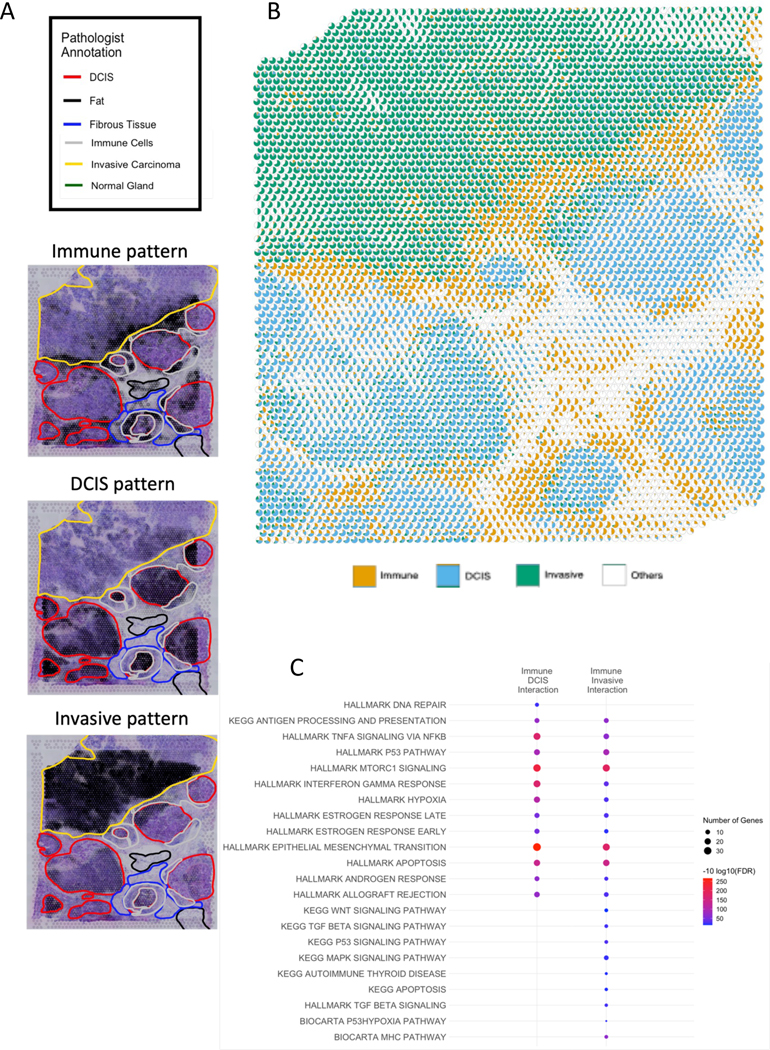
Low-resolution CoGAPS and SpaceMarkers analysis identifies markers of interaction between broad patterns in breast cancer tissue
A. Images of the breast cancer tissue showing activity levels of the immune, DCIS, and invasive carcinoma patterns respectively overlaid on annotated H&E slides showing regions with invasive carcinoma, DCIS lesions, immune cells and stroma.
B. Scatterpie visualization shows the relative activity levels and overlap between the invasive carcinoma (green), immune (orange), DCIS (blue) and all Other patterns combined (white).
C. Overrepresented pathways associated with DCIS-immune interactions and cancer-immune interactions (FDR < 0.05).
The visualization in Figure 4B. shows widespread spatial regions of interactions between immune and tumor cells at the boundaries of both the invasive carcinoma and the DCIS lesions, as well as some isolated spots of immune activity in the interior of the invasive tumor. However, the immune activity in these spots is not significantly over the threshold to be create substantial immune influence in the neighborhood. Thus, the immune-invasive cancer interaction is largely contained near the boundary of the the tumor. Whereas the pancreatic cancer sample in Figure 2 covered a smaller area with fewer spots (< 300) having tumor and immune influence respectively, we identify much larger regions (> 1000 spots) of influence from the immune, invasive carcinoma and DCIS cells (Figure 4B.). This larger number of spots enables us to estimate SpaceMarkers from CoGAPS residuals to distinguish the molecular changes in the invasive carcinoma from the DCIS lesions. Similar to our analysis of the metastatic pancreatic cancer data, we obtain latent features of the ST data from this breast sample using CoGAPS factorization. These latent features reveal histological annotations of invasive carcinoma, DCIS lesions, immune, and stromal regions estimated from the H&E stain (Figure 4A.).
Computing SpaceMarkers based upon the CoGAPS residuals identifies 461 genes associated with interaction between the immune and invasive carcinoma patterns and 413 markers of immune and DCIS pattern interaction (Data S1), compared to up to 3736 immune-invasive carcinoma and 3036 immune-DCIS genes identified from applying a similar analysis based upon differential expression for the same FDR value (Data S1). This reduction in the number of markers through the analysis of CoGAPS residuals relative to inference of SpaceMarkers through differential expression analysis is consistent with the isolation of specific nonlinear changes resulting from interactions between the cellular processes measured in the CoGAPS patterns using this mode. We note that 85 of the SpaceMarkers were associated with immune cell interactions in both the invasive carcinoma and DCIS regions. The SpaceMarkers optParams values are tabulated in Supplemental Table S1. To further determine the molecular pathways activated through immune and tumor cell interactions in both regions, we performed gene set overrepresentation analysis from the Hallmark, Kegg, and Biocarta molecular pathways using the Molecular Signatures Database (MSigDB), with a selection of the pathways presented in Figure 4C. (see Table S4 for the complete list of pathways). We find that while certain pathways were enriched in both interactions (e.g., antigen processing and presentation, p53 pathway, Tnf-alpha signaling, mTorc1 signaling, epithelial to mesenchymal transition, Interferon Gamma response, hypoxia, and estrogen response early/late), others were enriched exclusively in Immune-DCIS (DNA repair) and Immune-Invasive (WNT signaling, MapK signaling, and TGF beta signaling) respectively. Note that a pathway enriched in both Immune-DCIS and Immune-Invasive Carcinoma interactions may have distinct gene subsets associated with each interaction. For example, it is readily evident that the Hallmark interferon gamma response gene set has a greater overlap with the SpaceMarkers of the Immune-DCIS interaction compared to the Immune-Invasive interaction.
2.5. Using SpaceMarkers with high-resolution CoGAPS reveals greater heterogeneity in intercellular interactions within the TME
In all cases presented, the SpaceMarkers inferred fundamentally depend on the resolution of the cellular processes inferred in the CoGAPS latent space analysis. Indeed, nonlinear interactions in interacting regions at a low resolution analysis may be further refined by increasing the dimensionality of the factorization on the ST data consistent with recent advances to multi-resolution matrix factorization30. We further performed a higher resolution CoGAPS analysis of the breast cancer data to test if the interaction region between two patterns and the associated SpaceMarkers genes are identified by increasing the dimensionality of the latent space analysis. In this higher dimensional analysis, CoGAPS identifies 16 distinct patterns associated with the diverse biological processes in the TME. The activity levels of a selection of the patterns overlaid on an H&E stained slide of the sample are shown in Figure 5A. (also see Supplemental Figure S3). Although the higher number of patterns reveal greater heterogeneity of the biological processes in the sample by further resolving patterns identified in the low resolution analysis, it does not identify patterns specific to the interactions identified between the lower dimension patterns.
Figure 5.
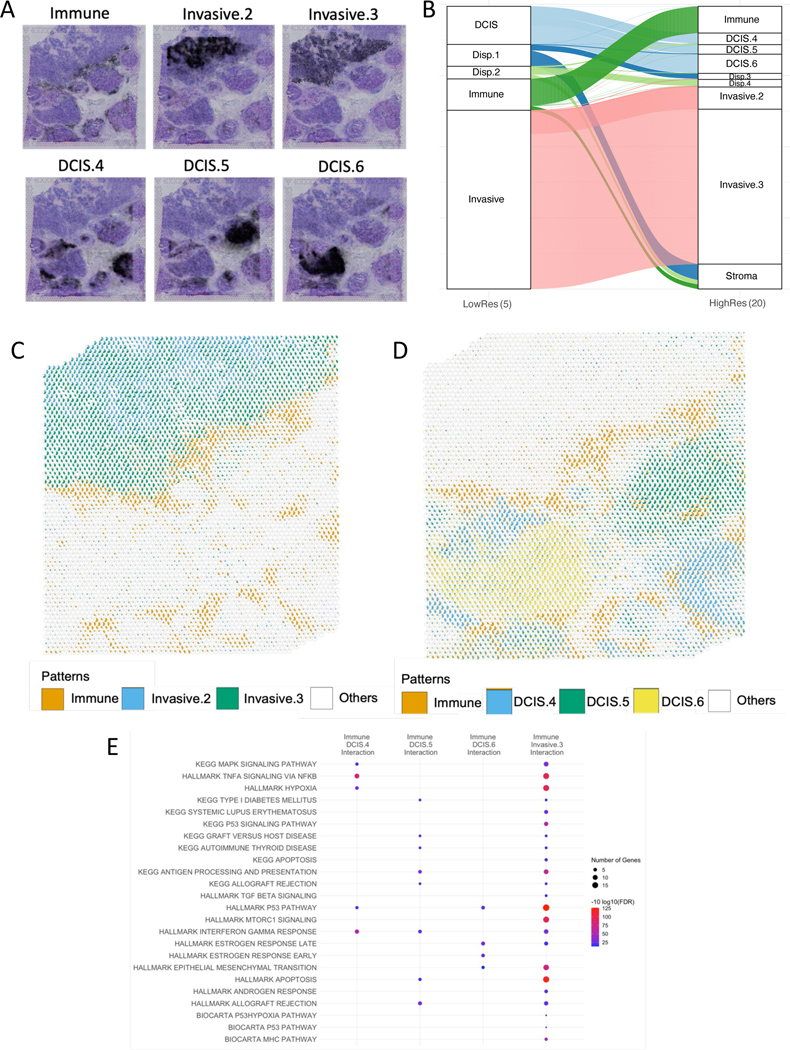
High-resolution CoGAPS and SpaceMarkers analysis of breast cancer tissue reveal greater heterogeneity in intercellular interactions (see Supplemental Figure S4 for SpaceMarkers with STdeconvolve).
A. Multiple patterns associated with invasive carcinoma and DCIS regions identified in higher-resolution CoGAPS analysis with 16 patterns highlights the heterogeneity in the tumor and TME by further resolving the underlying pathology (see Supplemental Figure S3 for remaining patterns).
B. Alluvial plot showing the most dominant pattern associated with each spot using low-resolution and high-resolution CoGAPS respectively. Spots dominated by low resolution DCIS pattern are dominated by three distinct DCIS-related patterns associated with different lesions in the high-resolution analysis. Invasive pattern in low resolution resolves into three invasive carcinoma related patterns associated with varying levels of immune infiltration in the high-resolution analysis. For alluvial plot with all 16 patterns, see Supplemental Figure S4A..
C. Relative activity levels of immune patterns with two invasive patterns reveals that the immune (orange) and Invasive.2 (blue) patterns have no overlap, hence do not interact. Immune interaction with Invasive Carcinoma (green) is captured through the overlap between Immune and Invasive.3 pattern. White represents all other patterns combined.
D. Relative activity levels of immune pattern (orange) with three DCIS patterns (blue, green, and yellow) associated with separate lesions reveals distinct overlapping regions associated with each interaction. White represents all other patterns combined.
E. SpaceMarkers of Immune-DCIS and Immune-Invasive interactions reveal functional heterogeneity of the enriched pathways mirroring the spatial heterogeneity revealed in 5C. and 5D. (FDR < 0.05). (See Table S4 for complete list of gene sets).
Although we do not associate each Visium spot with solely one pattern, studying the most dominant pattern in spots informs us of the dominant biological process at that location in the tissue as inferred by CoGAPS. Consequently, the same spots are associated with broader biological processes at the lower resolution and with more specific processes at a higher resolution. The alluvial plot in Figure 5B. shows the relationship between the most dominant low resolution and high resolution patterns at each spot.
For example, the single DCIS-related pattern in Figure 4A. resolves into multiple DCIS patterns, some of which are associated with individual DCIS lesions. Even within the single invasive carcinoma lesion, the low resolution invasive carcinoma pattern resolves into two distinct patterns, one of which is isolated to the interior of the invasive carcinoma and one which spans to the tumor-immune boundary. While the DCIS lesions and invasive carcinoma have universally high ERBB2 and ESR1 expression, evaluating the genes associated with the distinct patterns identifies heterogeneity in growth factor signaling pathways with enhanced IGFBP3 expression in the DCIS.5 pattern, FGFR4 expression in the DCIS.6 pattern, and FGFR1 expression in the Invasive.2 carcinoma pattern (Supplemental Figure S3, Table S5) We also see spots previously associated with the immune pattern or with dispersed patterns at the low resolution now being associated with a dominant pattern which can be associated with the stromal region. To further compare the enhanced resolution intra-tumor heterogeneity to tumor-immune interactions in the high resolution factorization, Figure 5C. shows relative pattern weights and overlap between the immune pattern and the two invasive patterns. It is clear that only one of the invasive patterns overlaps with the immune pattern, thus contributing to the tumor-immune interaction. Still, both of these interacting patterns contain a substantial numbers of spots that are isolated to the immune and invasive carcinoma region, respectively, suggesting that increasing the resolution of the factorization does not compensate for the estimation of nonlinear effects through the interaction statistic. Similarly, Figure 5D. shows relative pattern weights and overlap between the immune pattern and the three DCIS patterns. It logically follows that the overlapping regions of the distinct DCIS patterns are also distinct, and hence correspond to different molecular alterations from DCIS-immune interactions that will impact subsequent outgrowth of these distinct lesions.
For these interactions involving the immune pattern, we identify SpaceMarkers genes associated with the inter-pattern interactions as the genes having higher CoGAPS residuals in the interaction region compared to regions with exclusive influence from the individual patterns (Data S1). The SpaceMarkers optParams values are tabulated in Supplemental Table S1. Upon identification of statistically significant (FDR < 0.05) signaling pathways (see Supplementary Table S4) pertaining to interaction of the immune pattern with invasive carcinoma and DCIS patterns in the high-dimensional CoGAPS results and comparing them to those found in 5 dimensions, we find pathways common to all interactions and unique to specific pattern interactions. For example, we find 59 signaling pathways enriched due to immune-invasive carcinoma interaction in 5 dimensions as well as 16 dimensions. These include but are not limited to pathways related to epithelial-mesenchymal transition, apoptosis, antigen processing and presentation, hypoxia, p53 signaling, interferon alpha and gamma responses, and lastly targets of the oncogene MYC. However, the higher resolution analysis also reveals unique pathways relevant to specific immune-invasive carcinoma pattern interactions. We found pathways related to the cancer-immune interactions including those related to IL-5 and IL6 signaling, KRAS signaling, Toll-like receptor signaling and the CDC25 pathway exclusively when the dominant invasive carcinoma pattern (Invasive.3) interacts with the immune cells. Similarly, the distinct Immune-DCIS interactions reveal a heterogeneity in the enriched pathways which were not evident with a single DCIS pattern using low-resolution CoGAPS. Among the immune interactions with different DCIS lesions, the MapK signaling, Tnf alpha signaling, and hypoxia pathways, known to be mechanisms of resistance to endocrine and immunotherapies, are enriched exclusively in the Immune-DCIS.4 interaction, antigen processing, allograft rejection and autoimmunity related pathways are enriched exclusively in Immune-DCIS.5, and EMT pathway, and estrogen response early/late are exclusively enriched in the Immune DCIS.6 interaction. These pathways are consistent with the heterogeneity of subsequent outgrowth of these DCIS lesions, with successful activation of pathways associated with immune attack in DCIS.5 relative to the invasive processes observed in both DCIS.4 and DCIS.6.
Finally, in addition to the SpaceMarkers analysis of interacting CoGAPS patterns, we also performed cell deconvolution using STdeconvolve29 to identify cell populations abundant in the invasive carcinoma and DCIS lesions respectively as well as the immune cells (Supplemental Figure S4). We used SpaceMarkers to identify the markers of interaction between immune cells and the cell populations found to be spatially interacting with them (Data S1). The SpaceMarkers optParams values are tabulated in Supplemental Table S1.
2.6. Integrated ST and single-cell RNA-seq analysis identifies cell type specific molecular changes from immunotherapy treatment in hepatocellular carcinoma
In the examples so far, the SpaceMarkers statistic revealed molecular changes associated with intercellular interactions. Since SpaceMarkers relies on spot-based colocalization, it limits the ability to identify the cell subtypes in which these molecular changes were induced. Transfer learning allows us project new data into learned latent spaces, subsequently associating samples from the new data with known biology. We first factorize the ST data collected from a resected hepatocellular carcinoma (HCC) tumor after administration of a neoadjuvant cabozantinib and nivolumab therapy to obtain 9 CoGAPS patterns. Figure 6A. shows the individual tumor and immune associated patterns overlaid on an H&E stained image of the HCC tumor sample. As in the other examples, these tumor and immune patterns are spatially overlapping (Figure 6B.), and are deemed to be interacting in regions where they have overlapping influence. This analysis identifies two distinct tumor cell patterns, one of which spans all malignant regions in the sample (Pattern 2) and the other isolated to a specific region (Pattern 1) that has less co-localization of the immune cells (Pattern 8). The interaction between the immune cells and each of the tumor patterns learned through SpaceMarkers identifies enhanced expression of hepatocyte markers (KRT18, SERPIN family genes, APOC2, CD24), immune markers (CD63, HLA genes), and cell death markers (TNF pathway associated genes, ribosomal genes, ANXA2) consistent with killing of tumor cells through immune cells in the interaction between Patterns 2 and 8 (Data S1). In contrast, SpaceMarkers genes of the interaction between Patterns 1 and 8 identify fibroblast markers (Collagen coding genes, MYL9, TAGLN) consistent with a lack of successful immune attack and infiltration in this portion of the tumor. The SpaceMarkers optParams values are tabulated in Supplemental Table S1.
Figure 6.
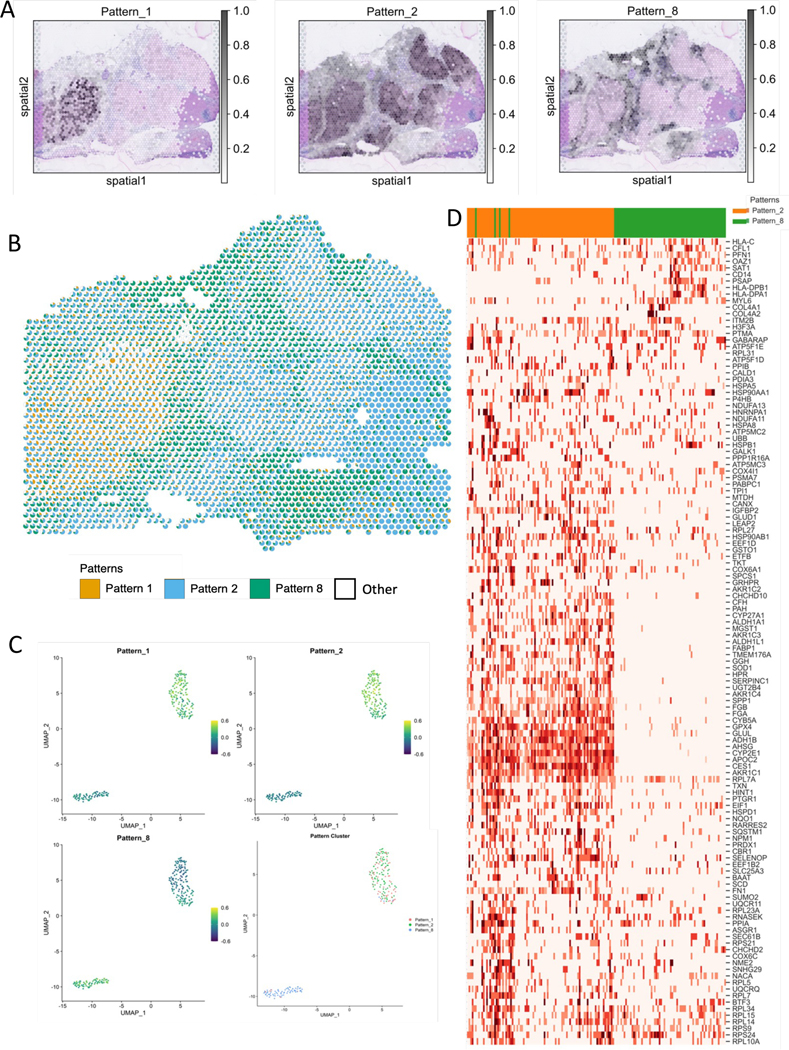
Contextualizing scRNAseq data using SpaceMarkers and transfer learning from matched ST-scRNAseq data in HCC (see also Supplemental Figures S5 and S6).
A. CoGAPS factorization reveals spatial patterns associated with tumor annotations of tumor and immune cells. (see Supplemental Figure S5).
B. Scatterpie visualization shows the relative pattern activity levels associated with the spatially overlapping tumor (orange) and immune (blue) patterns in each Visium spot using a pie chart (white represents activity from all other patterns). SpaceMarkers are genes exhibiting nonlinear effects in the residual space of the CoGAPS patterns in the region with tumor-immune overlap.
C. Transfer learning of Patterns 1, 2, and 8 from ST data to matched scRNAseq data. Scatter plot shows projections of the spatial patterns onto individual cells in the scRNAseq data. Individual cells in the scRNAseq data are associated with the pattern having the highest projection in the cell.
D. Expression heatmap of SpaceMarkers in tumor and immune cells from matched single-cell data from the same tumor provide the spatial context of the individual cells.
While the SpaceMarkers analysis of ST data suggests molecular changes associated with cell-cell interactions, this analysis alone does not pinpoint the precise cells in which these molecular changes occur. By transfer learning37,41 of these latent features into matched single-cell RNA-seq data from the same tumor, we can associate individual cells with specific patterns corresponding to tumor and immune signatures (Figure 6C.). This association can both identify whether a SpaceMarkers gene’s expression changes in tumor or immune cells, and also whether we can also predict the precise subpopulations of tumor and immune cells involved in intercellular interactions by observing the gene expression changes of the relevant SpaceMarkers in individual cells. From Figure 6D., we observe that changes in the expression of genes SERPINC1, APOC2 and ADH1B, are induced in a subset of the cancer cells attributed to Pattern 2, whereas expression changes in gene PFN1 and CD14 are induced in a subset of the immune cells. A further subset of both Pattern 2 tumor cells and immune cells co-express HSP90AA1 and ribosomal genes. Based on these gene expression patterns of the respective SpaceMarkers, we hypothesize that these individual cells are sourced from the tumor-immune boundary. Note that although the analysis in this section demonstrated the interaction between the dominant patterns (1,2,8), some of the less dominant patterns could represent rare cell types or minor biological processes which are essential to the tumor progression and immune response. Accordingly, users should include such patterns for SpaceMarkers analysis in their workflow if needed.
3. Discussion
We demonstrate how co-localization of multiple cellular processes in spatial transcriptomics data can be leveraged as an asset to infer molecular changes resulting from cell-cell interactions. Specifically, this inference is enabled through SpaceMarkers, an algorithm for identifying genes associated with pairs of spatially interacting latent features which represent distinct cellular processes. We accomplish this by first identifying a region of influence for each latent feature in the vicinity of spots with high feature activity. Two features are deemed to be interacting in spots where they have concurrent influence. The SpaceMarkers algorithm can estimate molecular changes from spatially interacting cellular processes in two ways — a default residual mode and a differential expression (DE) mode. We demonstrate that the DE mode is able to identify genes with significantly higher expression in the region where two latent features overlap. However, the DE mode is subject to confounding factors such as variable cell populations and marker association with multiple cell types. We mitigate these confounding effects in the residual mode, where we identify genes with significantly higher residual error between the original data and its reconstruction in the region of overlap between two latent features. However, this statistic requires a greater number of spots for robust analysis than the DE method. While we found that this requirement limited the application of the residual model in the case with the smaller lymph node sample with PDAC metastasis, it was generally applicable to the other tumor-immune interactions in our sample cohort. While the examples used in this paper use spot-based technologies, we note that SpaceMarkers is readily applicable to alternative imaging-based ST technologies that achieve single-cell resolutions. Consequently, increased spatial resolution of the ST characterization or multi-omics methods for inferring cellular boundaries4,32 will enable broader application of SpaceMarkers for cell-cell interactions.
We validated the SpaceMarkers output against independent tissue classification algorithm22,3 and note that if a cell type is entirely occurring within the interaction region, its marker genes will be inferred as a marker of spatial interaction through SpaceMarkers (Figure 3). While not a direct molecular change in the input cell states, this co-localization of the cell type exclusively in the interaction region may be a biological effect induced through the TME state induced by the inter-cellular interactions. We also note that this effect is mitigated to an extent, but not completely removed in the residual mode if some of the learned patterns are associated with that cell type. Ultimately, we leave it to the user to determine which inferences from SpaceMarkers merit further investigation. Future work can also include follow-up experimental studies using in-vitro 2D/3D cocultures or in-vivo depletion studies of cell types found in the interaction region to validate the SpaceMarkers output.
Although SpaceMarkers is not optimized for specific cancer types, we notice that the analysis pipeline performs better when inferring cell-cell interactions for the larger volume of cancer cells in breast and liver tumors (Figures 4, 5, and 6) as compared to smaller density of tumor cells surrounding the duct in the pancreatic samples (Figure 2 and Figure 3). We hypothesize that this difference in performance could be due to a combination of factors including the fact that spot-based Visium technology does not capture the minute details of diffuse tumors and their microenvironment, smaller samples resulting in fewer spots for SpaceMarkers analysis. Future work will focus on the application and optimization of SpaceMarkers to spatial data with single-cell or subcellular-level resolution and to extend its performance for cancers with different types of tumor structures.
Due to our focus on tumor-immune cell interactions in our biological analyses, the current version of the SpaceMarkers algorithm admits only two overlapping latent features as input. However, this approach is generally applicable to cell-cell inference from ST data across biological contexts and to features associated with any cell subtype or cellular feature defined through the latent space analysis. For example, this approach also enables analysis of the molecular changes from cell-cell interactions between immune and stromal cells in the breast cancer tissue (Supplemental Figure S3, Data S1) and between additional cell types in the PanIN sample (Figure 3). In many cases, multiple latent features are co-localized at the same spot. This could result in the same genes being associated with multiple interaction types, although we did not observe such effects in our case studies. Furthermore, many critical intercellular interactions such as cancer-associated fibroblast (CAF)-driven immunosuppression31 result from possible colocalization of multiple cell phenotypes. To address this, future work should extend the application of SpaceMarkers to identify genes associated with multiple overlapping latent features.
We note that our inference of interactions between cellular processes is performed directly from latent space analyses of the ST data, without the need for additional reference datasets for single-cell resolution5 or direct estimates of cellular deconvolution46. While our approach is generally applicable to linear latent space estimation methods, the results of our algorithm fundamentally depend on the latent space method selected for analysis of the ST data. We demonstrate the application of SpaceMarkers to 10x Visium ST data from different cancers and we identify markers associated with the interaction between latent features associated with different biological processes. In all cases, we observe that the Bayesian matrix factorization method CoGAPS41,14,38 can learn latent features that distinguish regions with tumor and immune cells directly from the ST data without reliance on prior knowledge of marker genes, histology annotations, or spatial coordinates. Because CoGAPS uses high-dimensional features to define cellular phenotypes, it can go beyond the discrete cell types learned from H&E through pathology, and enables deconvolution of spots into a more nuanced mix of biological patterns (Figure 4B.). Moreover, pathology annotations from H&E imaging can be limited on flash-frozen OCT samples (Figure 4), as they do not preserve cellular morphology7. In the case of formalin-fixed paraffin-embedded (FFPE) samples, automated machine-learning based pathology annotations can be used for cell-type identification3. Creating higher resolution CoGAPS analysis by increasing the number of latent features inferred from the ST data is able to further resolve the biological signatures, revealing the tissue heterogeneity. These higher dimensional patterns are independent of the interaction regions between the latent features inferred with SpaceMarkers at a lower dimension. This observation suggests that our approach indeed isolates effects due to inter-cellular interactions rather than unresolved latent features associated with specific cellular processes.
To demonstrate the compatibility of SpaceMarkers with other latent space methods, we have provided an example of its application in DE mode to the output of STdeconvolve (Supplemental Figure S4). Future work could extend the SpaceMarkers algorithm to additional latent space methods emerging for ST data and include nonlinear regression with terms involving combinations of patterns to supplement the available SpaceMarkers modes. Still, we note that the current modes for SpaceMarkers can readily be applied to nonlinear latent space methods, provided that the low-dimensional features they infer can be associated with a set of weights for each cell as through linearization.
The use of SpaceMarkers on the spot-based 10x Visium technology limits direct inference of the specific cell subtypes in which interactions induce molecular alterations. We demonstrate that transfer learning37,41 of the latent features inferred from CoGAPS analysis of the ST data into matched single-cell RNA-seq data enables us to define the precise cellular subpopulations with gene expression changes in each SpaceMarkers gene. Other approaches mitigate the need for paired data by coordinated expression changes between annotated pairs of ligands and receptors in both spatial and non-spatial single-cell data. While these approaches directly model the signaling process, they rely on the correspondence between gene expression and protein function and databases of ligand-receptor pairs12. Coupling spatial data with newer single-cell technologies that isolate interacting cells16 can further enhance this inference.
Ultimately, the results of SpaceMarkers depends on the patterns inferred from the latent space method. Biological robustness of the SpaceMarkers statistic relies on the use of patterns associated with significant activity levels as well as a spatial overlap with other patterns of interest. For example, we analyzed the interaction of immune cells with one invasive carcinoma pattern out of the three invasive carcinoma patterns learned using high resolution CoGAPS analysis. We did not analyze the other two patterns because one was isolated away from the immune pattern and hence had no interactions, and although the other pattern had a spatial overlap with the immune pattern, it had much lower activity levels. For the residual mode to be effectively used, it is important not just to resolve the ST data into biologically meaningful latent features, but also to provide a good fit between the original ST data and its reconstruction from the latent features. In the absence of a good fit, the residual errors contain not just the effects attributable to inter-feature interaction and the measurement error, but also the estimation errors resulting from an overly constrained factorization. In such cases, we recommend using the SpaceMarkers in the DE mode. Similarly, the utility of SpaceMarkers is diminished if the learned latent features do not correspond to individual cell phenotypes, or if markers of essential cell types are not represented by any of the learned latent features. Future work can overcome this limitation through semi-supervised learning methods that use cell-type marker expression as a proxy for the latent feature input in the DE mode for SpaceMarkers.
When genes associated with cell-surface interactions and cytokine secretions are grouped together in a latent feature, the assignment of a single kernel-width parameter to the latent feature in the SpaceMarkers algorithm is inconsistent with the varying distances associated with these two types of intercellular interactions. Identification of intercellular interactions in such scenarios requires a mathematical framework for spatially resolved causal inference which models distinct cell types, varying ranges and gradients of influence for cytokine-secretions and surface interactions, and spatially resolved expression of individual genes. One such example is MESSI24, which uses mixture-of-experts and multi-task learning approaches to predict the gene expression in a particular cell type with the help of signaling genes in neighboring cells. Future work integrating these methods with latent features in place of individual genes will both reduce the computational complexity and enhance the biological interpretability of these spatially aware network inference methods.
4. STAR Methods
RESOURCE AVAILABILITY
Lead Contact:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Elana J Fertig (ejfertig@jhmi.edu)
Materials Availability:
This study did not generate new materials.
Data and Code Availability:
Processed 10x Visium data from the PDAC lymph node, PanIN, and HCC samples have been deposited at the Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
The original code for the SpaceMarkers package is available at www.github.com/FertigLab/SpaceMarkers under MIT license and archived on Zenodo (https://doi.org/10.5281/zenodo.7621285). The scripts used for the analysis presented in this paper is available at www.github.com/atuldeshpande/SpaceMarkers-paper and archived on Zenodo (https://doi.org/10.5281/zenodo.7621291).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited Data | ||
| Processed spatial and single-cell transcriptomics data. | This paper. | GEO:GSE22441 |
| Software and Algorithms | ||
| SpaceMarkers v0.81 | This paper. | doi:10.5281/zenodo.7621285 |
| SpaceMarkers analysis scripts. | This paper. | doi:10.5281/zenodo.7621291 |
| STdeconvolve | MIller et al., 2022 | https://github.com/JEFworks-Lab/STdeconvolve |
| CoGAPS v3.15.2 | Sherman et al., 2020 | doi:10.18129/B9.bioc.CoGAPS |
| projectR v1.6.0 | Sharma et al., 2022 | doi:10.18129/B9.bioc.projectR |
| GSEA | Subramanian et al., 2005; https://www.gsea-msigdb.org/ | GSEA v4.2.3 |
| Seurat v4.1.0 | Hao*, Hao*, et al., 2021 | Version 4.1.0 |
| Other | ||
| Gene sets | www.msigdb.com | MSigDB v7.5.1 (Hallmark, Biocarta, and Kegg) |
| High resolution figures | This paper. | doi:10.5281/zenodo.7622690 |
High Resolution Figures:
High resolution versions of the figures in this manuscript are available on Zenodo (https://doi.org/10.5281/zenodo.7622690).
METHOD DETAILS
4.1. Sample collection, preparation, and storage
Invasive breast ductal carcinoma:
The fresh frozen invasive breast ductal carcinoma was collected in 2011 and obtained from BioIVT. The tumor was stage IIA, ER Positive, PR Negative, Hercep Test 2+. The RNA quality of the sample, as measured with Bioanalyzer (Agilent) was RIN = 9.26. The sample was embedded in optimal cutting temperature (OCT) compound and immediately frozen. Cryosections of 10 μm were placed on Visium Gene Expression slides (10x Genomics).
PDAC metastatic lymph node:
The PDAC peritumoral lypmh node was surgically resected during curative surgery at the Johns Hopkins University. The lymph node was embedded in OCT and immediately frozen. Pathological examination of an H&E stained cryosection identified a PDAC metastasis to the lymph node. A cryosection of 10 μm were placed on a Visium Gene Expression slide (10x Genomics).
PanIN sample:
The PanIN sample was a surgical specimen from a collection obtained during 2016 to 2018 available in the Johns Hopkins University School of Medicine Department of Pathology archives under Institutional Review Board approval (IRB00274690) under a waiver of consent.
HCC sample:
The HCC sample was surgically obtained as part of a clinical trial (NTC03299946) for neoadjuvant cabozantinib and nivolumab previously described18. The surgical specimen was immediately embedded in OCT, frozen and a 10 μm cryosection was placed in a Visium Gene Expression slide (10x Genomics).
4.2. ST library preparation
Briefly, following tissue permeabilization optimization, according to 10x Genomics instructions, samples were fixed in methanol, stained (H&E) and imaged. Sequencing libraries were prepared using the Visium Spatial Gene Expression Reagent Kit (10x Genomics), following manufacturer’s instructions, and sequenced on an Illumina NovaSeq.
4.3. SpaceMarkers algorithm
Here we describe the SpaceMarkers algorithm to identify genes associated with nonlinear effects of latent feature interactions. To facilitate exposition, we will refer to the spatial component of the latent features as ”patterns”.
Modeling pattern interactions in the residual space:
We assume a generic latent space representation model where the ST data matrix is factorized into two low-rank matrices and . Consequently, the matrix product is a low-rank approximation of the high-dimensional spatial RNAseq data, accounting for all linear combinations of the latent patterns such that
where measurement noise are independent and normally distributed with zero mean (see14 for the CoGAPS-specific model). However, this assumption associates the CoGAPS residuals purely with measurement noise, disregarding any molecular changes resulting from inter-pattern interactions. To that end, we introduce an additional term which represents the unknown molecular changes due to pattern interactions such that
where the measurement noise are independent and normally distributed with zero mean and variance . Thus, we hypothesize that the residuals represent both measurement noise and the molecular changes from inter-pattern interactions. Within the scope of this paper, we seek to only identify genes which exhibit higher residual effects associated with two interacting patterns. To this end, we use CoGAPS with the default settings and analyze the residual space of the CoGAPS factorization results. That is, we use the CoGAPS residuals as an estimate of such that
in regions where two patterns interact (i.e., have overlapping influence) versus regions where each pattern has exclusive influence. To identify the genes associated with the nonlinear interactions between a given pair of patterns, we first identify hotspots of pattern influence for each pattern. If both patterns have overlapping influence in a spot, they are deemed to be interacting in that spot. The CoGAPS residuals are computed in the interacting regions as well as in regions where each pattern is individually active. When the null hypothesis of non-interaction between the patterns is true, the residuals have no dependence on underlying regions (interacting or exclusive). On the other hand, genes associated with higher CoGAPS residuals in the interacting regions compared with the regions with exclusive pattern influence from either pattern show a strong dependence on spatial overlap between the patterns, and thus reject the null hypothesis. These genes constitute the SpaceMarkers, markers of spatial interaction between the two patterns in question. Focusing on strictly higher residuals avoids the confounding factors from decreased gene expression due to heterogeneous spot populations compared to homogeneous ones.
Identifying regions of pattern influence and pattern interaction:
For each spatially resolved pattern, we identify its region of influence by using a Gaussian kernel-based spatial smoothing approach. Through the spatial smoothing, we model a pattern’s influence extending beyond a spot to its neighboring spots as well. Given the pattern intensity associated with a -th spot in the sample, we calculate the spatially smoothed pattern intensities by using the leave-one-out method
with the spatial Gaussian kernel
where is the distance between the -th and -th spots, and is the kernel width. We used the Smooth.ppp function in the R package spatstat2 to perform the smoothing. We obtain a null-distribution by applying the kernel-based smoothing to spatially permuted pattern values (by pseudorandomly assigning spot locations ()). This null-distribution is assumed to be normal, and we obtain the sample mean and standard deviation for each pattern. We identify the pattern’s region of influence as the set of spots with outliers
where is the outlier threshold for the pattern. The optimal values of the kernel width and outlier threshold are the arguments that minimize the spatial autocorrelation (Moran’s I) of the residuals
The optimal kernel width for each pattern is the value which minimizes the Moran’s I in the residuals over all spots in the sample. Subsequently, the optimal outlier threshold minimizes spatial autocorrelation of the residuals over the spots contained in the resulting region of pattern influence. If a spot is influenced by two or more patterns, these patterns are said to be interacting in such a spot. For each pattern pair of interest, the set of all such spots is defined as their interacting region.
nPattern values and number of learned patterns for different CoGAPS runs. The values shown in boldface are used in further analysis.
| Sample | # genes | # spots | numPatterns (Learned Patterns) |
|---|---|---|---|
|
| |||
| PDAC metastatic lymph node | 18418 | 1351 | 5(5), 8(10), 15(21) |
| PanIN | 16,954 | 1,872 | 5(5), 10(10) |
| Invasive breast ductal carcinoma | 24228 | 4898 | 5(5), 10(9), 15(14), 20(16) |
| HCC | 20423 | 3006 | 5(4), 10(7), 15(9), 20(10), 30(18) |
Statistical test to identify genes associated with pattern interactions:
For a given pair of patterns and with a substantial regions of exclusive pattern influence and pattern interaction, we define three subregions characterized by
The spots with influence and no influence.
The spots with influence with no influence.
The spots with overlapping influence from both and .
The elements from each row of corresponding to the subregions described above denote the CoGAPS residuals in the respective subregions. For each gene (row) , we perform a non-parametric Kruskal-Wallis test23 for stochastic dominance of the CoGAPS residuals in at least one of the three subregions, with a posthoc Dunn’s test11 to ascertain the relative dominance between the respective subregions. Of particular interest to us are the genes which have statistically significantly higher CoGAPS residuals (FDR¡0.05) in the interacting region relative to the other two subregions as well as genes which exhibit statistically significantly higher CoGAPS residuals exclusively in the interacting region compared to at least one of the two other subregions.
4.4. Multi-resolution CoGAPS analysis
The ST genes by spot counts data for each sample was filtered to remove genes and spots with no or constant signal and then log2 normalized. The final matrix size of the input data matrix D are noted in the table below. The element represents the expression of the -th gene in the -th spot. The CoGAPS (version 3.5.8) 38 algorithm was run using the filtered and normalized counts data as input. Additionally, default CoGAPS parameters were used except for nIterations = 50,000, sparseOptimization = TRUE, distributed = single-cell, and nSets = 4. CoGAPS factorization results in two lower-dimensional matrices: an amplitude matrix (A) containing gene weights and a pattern matrix (P) containing corresponding spot weights estimated for a pre-specified number of latent features (nPatterns). On each of the input datasets, the algorithm was tested for a range of nPatterns.
The pattern weights for each spot were plotted over the tissue to show association between a pattern and a tissue region. In highRes Breast cancer analysis, genes were assigned to the pattern they were most strongly associated with using the patternMarker function in CoGAPS (version) in R (version). The genes for each pattern were submitted to the Molecular Signatures Database and searched within the BIOCARTA, KEGG, and HALLMARK pathways27,42,26. Pathways were considered significant if FDR < 0.05.
4.5. Scatterpie visualizations
We use the A and P matrices in the CoGAPS result to represent each Visium spot as a combination of overlapping latent patterns. To this end, we calculate the fractional gene expression in pattern at spot as
where is the gene index. We use the ‘vizAllTopics’ function from the ‘STdeconvolve’ package29 to visualize each spot as a pie chart showing the fractional gene expression in each pattern.
4.6. ProjectR analysis with matched single-cell RNAseq data
For the HCC sample in Figure 6, we have matched single-cell RNAseq data from the same patient. This scRNAseq data was preprocessed using the ‘sctransform’ package17, a normalization and variance stabilization method based on regularized negative binomial regression method, available in Seurat package in R. The transfer learning method, ProjectR, was used to project the spatial patterns from the HCC sample onto matched scRNAseq data from the same patient. Although the Visium data for CoGAPS and single-cell datasets use different normalization methods, our previous studies have shown that projectR can identify related cellular attributes across various data types and modalities in spite of batch effects41. The R package projectR (version 1.6.0) was used to project the A matrix of the CoGAPS result into the target dataset. The CoGAPS result object and the counts data from the matched scRNAseq dataset were used as input where FULL = TRUE. Each individual cell in the scRNAseq dataset is associated with the pattern with the highest projection. We limit the pattern association to the dominant patterns in the spatial data, namely Patterns 1,2, and 8.
4.7. Gene Set Enrichment Analysis using MsigDB
For each gene list query corresponding to SpaceMarkers for pairs of patterns, we compute their overlaps with gene sets belonging to the HALLMARK, BIOCARTA and KEGG pathways in MsigDB27,26,42, and report statistically significant overlaps (FDR<0.05).
Supplementary Material
Table S2: Genes and MSigDB genesets in lymph node from Figure 2.
Table S3: MsigDB genesets in PanIN with SpaceMarkers DE and residual mode from Figure 3.
Table S4: MsigDB genesets in lowRes breast cancer from Figure 4.
Table S5: MsigDB genesets in highRes breast cancer from Figure 5.
Table S6: PatternMarkers of CoGAPS patterns in highRes breast cancer from 5.
Highlights.
Latent space analyses of spatial transcriptomics show spatially varying cellular activity
SpaceMarkers identifies genes associated with spatially interacting latent features
SpaceMarkers identifies molecular changes from tumor-immune interaction in various tumors
7. Acknowledgements
We thank Jennifer Durham for facilitating the sample acquisition and data archival process, and Ana Cordova for providing valuable inputs for improving the visualizations. This work was supported by an NCI F31CA268724-01 (to D.N.S), NIH R01CA138264 (to A.V.F.), R50CA243627 (to L.D.), R01CA138264 (to A.S.P.), P01-CA247886 (to E.M.J.), P30CA006973 (to E.M.J.), U01CA212007 (to E.J.F. and A.S.P.), U01CA253403 (to E.J.F.), U54CA274371 (to E.J.F. and L.D.W), U01CA271273 (to E.J.F. and L.D.W.), U01AG060903 (to D.W.), U54CA268083 (to D.W., L.D.W., and A.K.), U54AR081774 (to D.W.), U54CA143868 (to D.W.), P50CA062924-24A1 (to E.M.J, E.J.F., L.T.K, L.Z., and L.D.W.); NIH K99 NS122085 from BRAIN Initiative in partnership with the National Institute of Neurological Disorders (to G.S-O’B); SU2C/AACR DT-14-14 (to E.M.J.); Lustgarten Foundation Translational Convergence Program Grant (to E.M.J.); Lustgarten Foundation grant (to L.D.W.); The Sol Goldman Pancreatic Cancer Research Center grant (to L.T.K. and L.D.W.); the Emerson Cancer Research Fund (to E.M.J., E.J.F., W.J.H); an Allegheny Health Network (AHN) grant (to E.J.F. and W.J.H.); Kavli NDS Distinguished Postdoctoral Fellowship (to G.S-O’B); Johns Hopkins Provost Postdoctoral Fellowship (to G.S-O’B); and the JHU Discovery Award (to E.J.F. and L.D.W.).
8. Declaration of Interests
EJF is on the Scientific Advisory Board of Viosera Therapeutics and is a paid consultant for Merck and Mestag Therapeutics. WJH reports royalties from Rodeo/Amgen; grants from Sanofi and NeoTX; and consulting fees from Exelixis outside the submitted work. EMJ reports other support from Abmeta, personal fees from Achilles, personal fees from DragonFly, other support from Parker Institute, grants and other support from Lustgarten, personal fees from Carta and Bluedot, grants and other support from Genentech, grants, and grants and other support from Break Through Cancer outside the submitted work. SRW, CRU, JC, AH, and ZWB are equity stockholders and employees of 10x Genomics. LZ receives grant support from Bristol-Meyer Squibb, Merck, AstraZeneca, iTeos, Amgen, NovaRock, Inxmed, and Halozyme. LZ is a paid consultant/Advisory Board Member at Biosion, Alphamab, NovaRock, Ambrx, Akrevia/Xilio, QED, Natera, Novagenesis, Snow Lake Captials, Tempus, Amberston, and Mingruizhiyao. LZ holds shares at Alphamab and Mingruizhiyao. MY reports grants and research support from: Bristol-Myers Squibb, Incyte, Genentech, and honoraria from Genentech, Exelixis, Eisai, AstraZeneca, Replimune, Hepion. CSM reports research funding from Pfizer, Astrazeneca, BMS, GSK/Tesaro, and serves on the advisory boards of Bristol Myers Squibb (not paid), Seattle Genetics, Genomic Health, Athenex. RAA receives research support from Bristol Myers Squibb, RAPT therapeutics, Stand up to Cancer, and the National Institutes of Health and serves on the advisory boards for Bristol Myers Squibb, Merck SD, and AstraZeneca. ASP is a consultant to AsclepiX Therapeutics and CytomX Therapeutics; he is the founder and Chief Scientific Advisor of AsclepiX Therapeutics; he receives research grants from AstraZeneca and Boehringer Ingelheim. The terms of these arrangements are being managed by the Johns Hopkins University in accordance with its conflict-of-interest policies.
9. Inclusion and Diversity
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Andersson A, Larsson L, Stenbeck L, Salmén F, Ehinger A, Wu SZ, Al-Eryani G, Roden D, Swarbrick A, Borg Å et al. (2021). Spatial deconvolution of HER2-positive breast cancer delineates tumor-associated cell type interactions. Nat. Commun. 12: 6012, doi: 10.1038/s41467-021-26271-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baddeley A, Rubak E and Turner R (2015). Spatial Point Patterns: Methodology and Applications with R. London: Chapman and Hall/CRC Press. [Google Scholar]
- 3.Bell AT, Mitchell JT, Kiemen AL, Fujikura K, Fedor H, Gambichler B, Deshpande A, Wu P-H, Sidiropoulos DN, Erbe R et al. (2022). PanIN and CAF Transitions in Pancreatic Carcinogenesis Revealed with Spatial Data Integration. Preprint at bioRxiv. doi: 10.1101/2022.07.16.500312. [DOI] [PMC free article] [PubMed]
- 4.Biancalani T, Scalia G, Buffoni L, Avasthi R, Lu Z, Sanger A, Tokcan N, Vanderburg CR, Segerstolpe Å, Zhang M et al. (2021). Deep learning and alignment of spatially resolved single-cell transcriptomes with Tangram. Nat. Methods 18: 1352–1362, doi: 10.1038/s41592-021-01264-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cable DM, Murray E, Zou LS, Goeva A, Macosko EZ, Chen F and Irizarry RA (2021). Robust decomposition of cell type mixtures in spatial transcriptomics. Nat. Biotechnol. 40: 517–526, doi: 10.1038/s41587-021-00830-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chaudhary B and Elkord E (2016). Regulatory T cells in the tumor microenvironment and cancer progression: Role and therapeutic targeting. Vaccines 4: 28, doi: 10.3390/vaccines4030028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cox ML, Schray CL, Luster CN, Stewart ZS, Korytko PJ, M Khan KN, Paulauskis JD and Dunstan RW (2006). Assessment of fixatives, fixation, and tissue processing on morphology and RNA integrity. Exp. Mol. Pathol. 80: 183–191, doi: 10.1016/j.yexmp.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 8.Davis-Marcisak EF, Deshpande A, Stein-O’Brien GL, Ho WJ, Laheru D, Jaffee EM, Fertig EJ and Kagohara LT (2021). From bench to bedside: single-cell analysis for cancer immunotherapy. Cancer Cell 39: 1062–1080, doi: 10.1016/j.ccell.2021.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davis-Marcisak EF, Fitzgerald AA, Kessler MD, Danilova L, Jaffee EM, Zaidi N, Weiner LM and Fertig EJ (2021). Transfer learning between preclinical models and human tumors identifies a conserved NK cell activation signature in anti-CTLA-4 responsive tumors. Genome Med. 13: 129, doi: 10.1186/s13073-021-00944-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dhanasekaran R, Baylot V, Kim M, Kuruvilla S, Bellovin DI, Adeniji N, Rajan Kd A, Lai I, Gabay M, Tong L et al. (2020). MYC and twist1 cooperate to drive metastasis by eliciting crosstalk between cancer and innate immunity. Elife 9: e50731, doi: 10.7554/eLife.50731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dunn OJ (1964). Multiple comparisons using rank sums. Technometrics 6: 241–252, doi: 10.2307/1266041. [DOI] [Google Scholar]
- 12.Efremova M, Vento-Tormo M, Teichmann SA and others (2020). CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat. Protoc. 15: 1484–1506, doi: 10.1038/s41596-020-0292-x. [DOI] [PubMed] [Google Scholar]
- 13.Elosua-Bayes M, Nieto P, Mereu E, Gut I and Heyn H (2021). SPOTlight: seeded NMF regression to deconvolute spatial transcriptomics spots with single-cell transcriptomes. Nucleic Acids Res. 49: e50, doi: 10.1093/nar/gkab043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fertig EJ, Ding J, Favorov AV, Parmigiani G and Ochs MF (2010). CoGAPS: an R/C++ package to identify patterns and biological process activity in transcriptomic data. Bioinformatics 26: 2792–2793, doi: 10.1093/bioinformatics/btq503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fu L-Q, Du W-L, Cai M-H, Yao J-Y, Zhao Y-Y and Mou X-Z (2020). The roles of tumor-associated macrophages in tumor angiogenesis and metastasis. Cell. Immunol. 353: 104–119, doi: 10.1016/j.cellimm.2020.104119. [DOI] [PubMed] [Google Scholar]
- 16.Giladi A, Cohen M, Medaglia C, Baran Y, Li B, Zada M, Bost P, Blecher-Gonen R, Salame T-M, Mayer JU et al. (2020). Dissecting cellular crosstalk by sequencing physically interacting cells. Nat. Biotechnol. 38: 629–637, doi: 10.1038/s41587-020-0442-2. [DOI] [PubMed] [Google Scholar]
- 17.Hafemeister C and Satija R (2019). Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20: 296, doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho WJ, Zhu Q, Durham J, Popovic A, Xavier S, Leatherman J, Mohan A, Mo G, Zhang S, Gross N et al. (2021). Neoadjuvant cabozantinib and nivolumab convert locally advanced hepatocellular carcinoma into resectable disease with enhanced antitumor immunity. Nature Cancer 2: 891–903, doi: 10.1038/s43018-021-00234-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu J, Li X, Coleman K, Schroeder A, Ma N, Irwin DJ, Lee EB, Shinohara RT and Li M (2021). SpaGCN: Integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat. Methods 18: 1342–1351, doi: 10.1038/s41592-021-01255-8. [DOI] [PubMed] [Google Scholar]
- 20.Ji AL, Rubin AJ, Thrane K, Jiang S, Reynolds DL, Meyers RM, Guo MG, George BM, Mollbrink A, Bergenstråhle J et al. (2020). Multimodal analysis of composition and spatial architecture in human squamous cell carcinoma. Cell 182: 497–514, doi: 10.1016/j.cell.2020.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Juengpanich S, Topatana W, Lu C, Staiculescu D, Li S, Cao J, Lin J, Hu J, Chen M, Chen J and Cai X (2020). Role of cellular, molecular and tumor microenvironment in hepatocellular carcinoma: Possible targets and future directions in the regorafenib era. Int. J. Cancer 147: 1778–1792, doi: 10.1002/ijc.32970. [DOI] [PubMed] [Google Scholar]
- 22.Kiemen AL, Braxton AM, Grahn MP, Han KS, Babu JM, Reichel R, Jiang AC, Kim B, Hsu J, Amoa F et al. (2022). CODA: quantitative 3D reconstruction of large tissues at cellular resolution. Nat. Methods 19: 1490–1499, doi: 10.1038/s41592-022-01650-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kruskal WH and Wallis WA (1952). Use of ranks in One-Criterion variance analysis. J. Am. Stat. Assoc. 47: 583–621, doi: 10.2307/2280779. [DOI] [Google Scholar]
- 24.Li D, Ding J and Bar-Joseph Z (2020). Identifying signaling genes in spatial single cell expression data. Bioinformatics 37: 968–975, doi: 10.1093/bioinformatics/btaa769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li T, Liu T, Zhu W, Xie S, Zhao Z, Feng B, Guo H and Yang R (2021). Targeting MDSC for Immune-Checkpoint blockade in cancer immunotherapy: Current progress and new prospects. Clin. Med. Insights Oncol. 15, doi: 10.1177/11795549211035540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1: 417–425, doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P and Mesirov JP (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics 27: 1739–1740, doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luca BA, Steen CB, Matusiak M, Azizi A, Varma S, Zhu C, Przybyl J, Espín-Pérez A, Diehn M, Alizadeh AA et al. (2021). Atlas of clinically distinct cell states and ecosystems across human solid tumors. Cell 184: 5482–5496.e28, doi: 10.1016/j.cell.2021.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller BF, Huang F, Atta L, Sahoo A and Fan J (2021). Reference-free cell-type deconvolution of multi-cellular pixel-resolution spatially resolved transcriptomics data. Nat. Commun. 13: 2339, doi: 10.1101/2021.06.15.448381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mohammadi S, Davila-Velderrain J and Kellis M (2020). A multiresolution framework to characterize single-cell state landscapes. Nat. Commun. 11: 5399, doi: 10.1038/s41467-020-18416-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monteran L and Erez N (2019). The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 10: 1835, doi: 10.3389/fimmu.2019.01835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pham DT, Tan X, Xu J, Grice LF, Lam PY, Raghubar A and others (2020). stLearn: integrating spatial location, tissue morphology and gene expression to find cell types, cell-cell interactions and spatial trajectories within undissociated tissues. Preprint at bioRxiv. doi: 10.1101/2020.05.31.125658v1. [DOI]
- 33.Place AE, Jin Huh S and Polyak K (2011). The microenvironment in breast cancer progression: biology and implications for treatment. Breast Cancer Res. 13: 227, doi: 10.1186/bcr2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rao N, Clark S and Habern O (2020). Bridging genomics and tissue pathology. Genetic Engineering & Biotechnology News 40: 50–51, doi: 10.1089/gen.40.02.16. [DOI] [Google Scholar]
- 35.Segerstolpe Å, Palasantza A, Eliasson P, Andersson E-M, Andréasson A-C, Sun X, Picelli S, Sabirsh A, Clausen M, Bjursell MK et al. (2016). Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab. 24: 593–607, doi: 10.1016/j.cmet.2016.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seo HR (2015). Roles of tumor microenvironment in hepatocelluar carcinoma. Curr. Med. Chem. 11: 82–93, doi: 10.2174/1573394711666151022203313. [DOI] [Google Scholar]
- 37.Sharma G, Colantuoni C, Goff LA, Fertig EJ and Stein-O’Brien G (2020). projectR: an R/Bioconductor package for transfer learning via PCA, NMF, correlation and clustering. Bioinformatics 36: 3592–3593, doi: 10.1093/bioinformatics/btaa183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sherman TD, Gao T and Fertig EJ (2020). CoGAPS 3: Bayesian non-negative matrix factorization for single-cell analysis with asynchronous updates and sparse data structures. BMC Bioinformatics 21: 453, doi: 10.1186/s12859-020-03796-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soysal SD, Tzankov A and Muenst SE (2015). Role of the tumor microenvironment in breast cancer. Pathobiology 82: 142–152, doi: 10.1159/000430499. [DOI] [PubMed] [Google Scholar]
- 40.Stein-O’Brien GL, Arora R, Culhane AC, Favorov AV, Garmire LX, Greene CS, Goff LA, Li Y, Ngom A, Ochs MF et al. (2018). Enter the matrix: Factorization uncovers knowledge from omics. Trends Genet. 34: 790–805, doi: 10.1016/j.tig.2018.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stein-O’Brien GL, Clark BS, Sherman T, Zibetti C, Hu Q, Sealfon R, Liu S, Qian J, Colantuoni C, Blackshaw S et al. (2019). Decomposing cell identity for transfer learning across cellular measurements, platforms, tissues, and species. Cell Systems 8: 395–411.e8, doi: 10.1016/j.cels.2019.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102: 15545–15550, doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wei C, Yang C, Wang S, Shi D, Zhang C, Lin X, Liu Q, Dou R and Xiong B (2019). Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol. Cancer 18: 64, doi: 10.1186/s12943-019-0976-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Whiteside TL (2008). The tumor microenvironment and its role in promoting tumor growth. Oncogene 27: 5904–5912, doi: 10.1038/onc.2008.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu SZ, Al-Eryani G, Roden DL, Junankar S, Harvey K, Andersson A, Thennavan A, Wang C, Torpy JR, Bartonicek N et al. (2021). A single-cell and spatially resolved atlas of human breast cancers. Nat. Genet. 53: 1334–1347, doi: 10.1038/s41588-021-00911-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao E, Stone MR, Ren X, Guenthoer J, Smythe KS, Pulliam T, Williams SR, Uytingco CR, Taylor SEB, Nghiem P et al. (2021). Spatial transcriptomics at subspot resolution with BayesSpace. Nat. Biotechnol. 39: 1375–1384, doi: 10.1038/s41587-021-00935-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2: Genes and MSigDB genesets in lymph node from Figure 2.
Table S3: MsigDB genesets in PanIN with SpaceMarkers DE and residual mode from Figure 3.
Table S4: MsigDB genesets in lowRes breast cancer from Figure 4.
Table S5: MsigDB genesets in highRes breast cancer from Figure 5.
Table S6: PatternMarkers of CoGAPS patterns in highRes breast cancer from 5.
Data Availability Statement
Processed 10x Visium data from the PDAC lymph node, PanIN, and HCC samples have been deposited at the Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
The original code for the SpaceMarkers package is available at www.github.com/FertigLab/SpaceMarkers under MIT license and archived on Zenodo (https://doi.org/10.5281/zenodo.7621285). The scripts used for the analysis presented in this paper is available at www.github.com/atuldeshpande/SpaceMarkers-paper and archived on Zenodo (https://doi.org/10.5281/zenodo.7621291).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited Data | ||
| Processed spatial and single-cell transcriptomics data. | This paper. | GEO:GSE22441 |
| Software and Algorithms | ||
| SpaceMarkers v0.81 | This paper. | doi:10.5281/zenodo.7621285 |
| SpaceMarkers analysis scripts. | This paper. | doi:10.5281/zenodo.7621291 |
| STdeconvolve | MIller et al., 2022 | https://github.com/JEFworks-Lab/STdeconvolve |
| CoGAPS v3.15.2 | Sherman et al., 2020 | doi:10.18129/B9.bioc.CoGAPS |
| projectR v1.6.0 | Sharma et al., 2022 | doi:10.18129/B9.bioc.projectR |
| GSEA | Subramanian et al., 2005; https://www.gsea-msigdb.org/ | GSEA v4.2.3 |
| Seurat v4.1.0 | Hao*, Hao*, et al., 2021 | Version 4.1.0 |
| Other | ||
| Gene sets | www.msigdb.com | MSigDB v7.5.1 (Hallmark, Biocarta, and Kegg) |
| High resolution figures | This paper. | doi:10.5281/zenodo.7622690 |