

Abstract

Cyclic porphyrin oligomers have been studied as models for photosynthetic light-harvesting antenna complexes and as potential receptors for supramolecular chemistry. Here, we report the synthesis of unprecedented β,β-directly linked cyclic zinc porphyrin oligomers, the trimer (CP3) and tetramer (CP4), by Yamamoto coupling of a 2,3-dibromoporphyrin precursor. Their three-dimensional structures were confirmed by nuclear magnetic resonance (NMR) spectroscopy, mass spectrometry, and single-crystal X-ray diffraction analyses. The minimum-energy geometries of CP3 and CP4 have propeller and saddle shapes, respectively, as calculated using density functional theory. Their different geometries result in distinct photophysical and electrochemical properties. The smaller dihedral angles between the porphyrin units in CP3, compared with CP4, result in stronger π-conjugation, splitting the ultraviolet–vis absorption bands and shifting them to longer wavelengths. Analysis of the crystallographic bond lengths indicates that the central benzene ring of the CP3 is partially aromatic [harmonic oscillator model of aromaticity (HOMA) 0.52], whereas the central cyclooctatetraene ring of the CP4 is non-aromatic (HOMA –0.02). The saddle-shaped structure of CP4 makes it a ditopic receptor for fullerenes, with affinity constants of (1.1 ± 0.4) × 105 M–1 for C70 and (2.2 ± 0.1) × 104 M–1 for C60, respectively, in toluene solution at 298 K. The formation of a 1:2 complex with C60 is confirmed by NMR titration and single-crystal X-ray diffraction.
Introduction
Covalent cyclic porphyrin oligomers have been widely studied since the 1970s as models for photosynthetic systems,1,2 as multitopic receptors for molecule recognition,3−6 as catalysts,7 and as models for exploring electronic delocalization and aromaticity.8 Most of these macrocycles have “spacers” bridging between the porphyrin units, such as alkynes,9 butadiynes,10 thiophenes,11 or para-phenylenes.12 Directly linked cyclic porphyrin oligomers (i.e., with no spacers) can exhibit fascinating cooperative behavior as a result of the tight proximity between neighboring porphyrin chromophores,13 but they are difficult to synthesize. Osuka and co-workers reported the first meso,meso (5,10) directly linked cyclic porphyrin oligomers, with various ring sizes, through an oxidative coupling strategy14 (Figure 1a). The same team also prepared β,β (2,12)-linked oligomers15 (Figure 1b), via the formation of PtII complexes followed by reductive elimination, as well as β,β (3,7)-linked rings, via Suzuki–Miyaura coupling.16 These porphyrin arrays show unusual photophysical properties, such as size-dependent excitation energy transfer.2,13,15 Here, we report the synthesis of two unprecedented β,β (2,3) directly linked cyclic porphyrin oligomers (Figure 1c), trimer CP3 and tetramer CP4, via one-step Yamamoto coupling of a 2,3-dibromo-10,15-bis-(3,5-di-tert-butylphenyl)porphyrin(Zn) 1 (Figure 1c and Scheme 1). These two porphyrin oligomers feature a central aromatic benzene or nonaromatic cyclooctatetraene (COT) ring, respectively. Their structures have been unambiguously characterized by a combination of nuclear magnetic resonance (NMR) spectroscopy, mass spectrometry, and X-ray single-crystal diffraction analysis.
Figure 1.

Representative examples of directly linked cyclic porphyrin oligomers: (a) meso,meso (5,10)-linked; (b) β,β (2,12)-linked; and (c) β,β (2,3)-linked trimer CP3 and tetramer CP4 in this work.
Scheme 1. Synthesis of β,β-Linked Cyclic Porphyrin Trimer (CP3) and Tetramer (CP4).

Ar = 3,5-di-tert-butylphenyl; COD = 1,5-cyclooctadiene; DDQ = 2,3-dichloro-5,6-dicyano-1,4-benzoquinone; TFA = CF3CO2H; and DMF = N,N-dimethylformamide.
The difference in geometry between CP3 and CP4 has a striking effect on their optical properties, electronic structures, and affinities for fullerenes. Trimer CP3 is almost flat; its density functional theory (DFT)-calculated geometry is D3 propeller-shaped (Figure 1c), with a small energy difference of 4.6 kJ/mol with respect to other conformers, making it conformationally dynamic. In contrast, CP4 has a saddle-shaped conformation with a high barrier to saddle inversion. The almost planar geometry of CP3 allows efficient π-conjugation, reducing the highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) gap and shifting the UV–vis absorption and fluorescence spectra to longer wavelengths, whereas absorption and fluorescence spectra of CP4 are more like those of a porphyrin monomer, with weak electronic coupling between the porphyrin units. While CP3 shows no interaction with fullerenes, the saddle-shaped geometry of CP4 makes it a good ditopic receptor, with association constants of (1.1 ± 0.4) × 105 M–1 for C70 and (2.2 ± 0.1) × 104 M–1 for C60 in toluene solution at 298 K. This tetramer is the first directly linked cyclic porphyrin oligomer with a high affinity for fullerenes.
Results and Discussion
Synthesis and NMR Spectroscopy
The key intermediate in the synthesis of CP3 and CP4 is the 2,3-dibromo-10,15-diarylporphyrin 1. This monomer was synthesized via [3 + 1] condensation of tripyrane 5 and 3,4-dibromopyrrole 4 (Scheme 1). First, twofold hydroxymethylation of pyrrole with paraformaldehyde gave 2,5-bis(hydroxymethyl)pyrrole 2 in 15% yield.17 Then, oxidation of 2 with activated MnO2 gave 2,5-diformylpyrrole 3 in 56% yield.18 Treatment with bromine in the presence of pyridine converts 3 to 3,4-dibromo-2,5-diformylpyrrole (4) in 95% yield.19 Meanwhile, tripyrrane 5 was synthesized in 17% yield by condensation of pyrrole and 3,5-di-tert-butylbenzaldehyde catalyzed by trifluoroacetic acid.20 Condensation of tripyrrane 5 and 3,4-dibromo-2,5-diformylpyrrole (4), followed by oxidation with DDQ and metalation with zinc acetate, provided porphyrin 1 in 13% yield over two steps.21 The structure of this dibromoporphyrin was confirmed by X-ray crystallography (Scheme 1, CCDC: 2253581). Finally, Yamamoto coupling of 1 using Ni(COD)2 in DMF gave a mixture of linear and cyclic porphyrin oligomers. After silica gel column chromatography, CP3 and CP4 were isolated in 12 and 9% yields, respectively. Molecular ions were observed at m/z: 2238.92 for CP3 (calcd for C144H150N12Zn3, 2239.00 [M]+) and m/z: 2985.88 for CP4 (calcd for C192H200N16Zn4, 2985.33 [M]+) by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) and the isotopic distribution patterns agree well with simulations (Figures S52 and S53). We also detected larger oligomeric byproducts by MS (Figure S48), but they were not isolated or fully characterized.
1H NMR spectra of CP3 and CP4 exhibit simple patterns, reflecting their high symmetries (Figure 2). Distinct sets of signals can be recognized consisting of one singlet associated with the meso-protons (Ha), two doublets coupled to each other corresponding to the β-positions (Hb and Hc), and one singlet from the other β-positions (Hd), similar to that of precursor porphyrin 1 (Figure 2c). The meso-protons (Ha) of CP3 resonate at an extremely high chemical shift (δ = 12.9 ppm) compared with that of 1 (δ = 9.7 ppm), reflecting the proximity between the porphyrin units that puts Ha in the deshielding region of two porphyrins. However, the meso-protons (Ha) of CP4 is only slightly downfield shifted to 11.0 ppm. 1H NMR signals of the 3,5-di-tert-butylphenyl groups show that desymmetrization occurs for He and Hg, which appear as two triplets (Figure 2b). The splitting of these signals implies CP4 adopts a stable D2d symmetric saddle shape, which cannot convert to other conformers (Figure 3). In contrast, the simple spectrum of CP3 implies either that the three porphyrin units are coplanar or that the different conformers interconvert rapidly on the NMR time scale. Even when the 1H NMR spectrum of CP3 was recorded at −90 °C (Figure S46), no splitting of 1H NMR signals of He could be seen, reflecting the low barrier to conformation exchange. By contrast, for CP4, no coalescence of 1H NMR peaks He and Hg was observed, even at 100 °C (Figure S46), demonstrating the high barrier to saddle inversion.
Figure 2.

Aromatic regions of the 1H NMR spectra of (a) CP3, (b) CP4 and (c) 2,3-dibromo-10,15-bis-(3,5-di-tert-butylphenyl)porphyrin(Zn) (1) recorded in CDCl3 (1% v/v of pyridine-d5 was added for CP3 to suppress aggregation, see Figure S47 for spectra without pyridine-d5) at 298 K, 400 MHz.
Figure 3.

Structure of CP3 from single-crystal X-ray diffraction studies (only one enantiomer is shown, hydrogen atoms and solvent molecules are omitted for clarity): (a) top view; (b) front view; (c) bond lengths in the central benzene ring and NICS(1) (in red); (d,e) optimized structure of CP4 by DFT on the B3LYP/6-31G(d,p) level of theory; (f) bond lengths of central COT and NICS(1) (in red); and (g) dihedral angle of unsubstituted COT reported in the literature (CCDC 1320377).22
Crystallography and Computational Modeling
Crystals of CP3 suitable for X-ray analysis were grown by diffusion of methanol vapor into a solution in 1,2-dichloroethane at room temperature. The structure determination revealed a star-shaped geometry (Figure 3a,b). The dihedral angles around the central benzene ring (measured Cα–Cβ–Cβ′–Cα′) are 21.8(19), −18.1(18), and −10.4(17)° (Figure 3a); two of the porphyrin units are twisted, relative to the central benzene, whereas the third is slightly folded. In contrast, DFT calculations predicted that the D3-symmetric propeller-shaped conformation has the lowest energy in the gas phase. (All DFT calculations in this study were performed with B3LYP/6-31G(d,p), Gaussian 16/A.03; see the Supporting Information for details). The calculated energy difference between the conformation in the crystal and the DFT-optimized geometry of CP3 is only 4.6 kJ/mol, and coordination of solvent molecules (methanol) to the zinc centers may influence the preferred conformation. In the crystals, two enantiomers were found (P,M,M and P,P,M, where P and M denote right-handed and left-handed helixes, respectively), which form homochiral dimers and these dimers pack alternatively along the c-axis (Figure S26).
We were not able to grow suitable single crystals of CP4 for X-ray analysis, despite screening many solvent systems, although the crystal structure of fullerene complex CP4·2C60 was determined, as discussed below. According to the DFT calculations, the most stable conformation of CP4 is saddle-shaped having stereochemistry of P,M,P,M. The lowest energy geometry has a C1 symmetry, but it is only 0.09 kJ/mol below the idealized D2d saddle conformation, and these two conformers are more stable than the other diastereoisomers by over 252 kJ/mol (Figure S7). In the lowest energy C1 symmetric geometry of CP4, the angles between the planes of neighboring porphyrin cores (planes each calculated for 24 core C and N atoms) are ∼57.3°, as shown in Figure 3d,e. The angle between the mean plane of each porphyrin and that of the COT ring is 42.7° (Figure 3e). The distance of the outermost β carbon atoms from the mean plane of the central COT is 6.21 Å. The folding angle of the COT unit, defined as the angle between the mean plane of one C–C=C–C unit and the mean plane of the whole COT is 42.2°, which is similar to that in unsubstituted COT (mean 41.9°).22
The local aromaticity of the benzene ring at the core of CP3 can be evaluated from the C–C bond lengths using the harmonic oscillator model of aromaticity (HOMA), which compares the bond lengths around a ring to those in benzene (Ropt = 1.388 Å).23 This analysis gives HOMA values of 0.52(11) and 0.81, from the crystallographic and DFT coordinates, respectively, which indicates that this benzene core has partial aromatic characters (see the Supporting Information for details). The nucleus-independent chemical shift calculated at a position of 1.0 Å above the center of this ring is NICS(1) = −7.5 ppm (for the D3 geometry of CP3), which should be compared with a value of −10.2 ppm for benzene, confirming the partial aromatic character.24 The central COT ring in CP4 has strong bond-length alternation (Figure 3f), giving HOMA values of +0.21 and −0.02(18), from the DFT coordinates and the crystal structure of CP4·2C60, respectively. This shows that there is no significant aromatic or antiaromatic delocalization in this core, as found in COT [HOMOA = −0.18(3)].22 The NICS(1) for the center of CP4 is +5.2 ppm, which confirms that it is nonaromatic.
Photophysics and Electrochemistry
UV–vis absorption and fluorescence spectra of CP3 and CP4 are compared with those of monomer P1, as shown in Figure 4 (all spectra recorded in toluene). Absorption spectra of both CP3 and CP4 show broader Soret bands and red-shifted Q-bands, compared with P1. The absorption bands of CP3 are broader and more red-shifted than those of CP4, reflecting stronger electronic coupling and π-conjugation between the porphyrin units in CP3. Fluorescence spectra of CP3 and CP4 also red shift with respect to P1 and their predominant emission peaks appear at 681 and 600 nm, respectively. Fluorescence quantum yields are 0.040 and 0.068 in non-deaerated toluene using zinc tetraphenylporphyrin (Φ = 0.029 in toluene) as a standard25 and are slightly higher than P1 (Φ = 0.031).
Figure 4.

(a) UV–vis absorption and normalized fluorescence spectra of P1, CP3, and CP4 measured in toluene (concentration 1 μM); for fluorescence measurements, excitation wavelengths are 417 nm (P1), 416 nm (CP3), and 415 nm (CP4); (b) cyclic and square wave voltammograms of P1, CP3, and CP4 measured in dichloromethane with 0.1 M n-Bu4NPF6 at 298 K, scan rate 50 mV/s.
The electrochemistry of P1, CP3, and CP4 was investigated by cyclic voltammetry (CV) and square wave voltammetry (SWV). The CV trace of P1 exhibits two reversible oxidation peaks at half-wave potentials of 0.41 and 0.71 V and two reduction peaks with half-wave potentials of −1.89 and −2.13 V versus Fc/Fc+. CP3 shows three reversible oxidation peaks at 0.30, 0.46, and 0.71 V as well as one observable reduction peak at −1.84 V. The narrower HOMO–LUMO energy gap of CP3 reflects the strong π-conjugation between the constituted porphyrins. Two reversible oxidation and reduction peaks located at 0.50, 0.91, −1.66, and −1.93 V were observed for CP4 within the measured potential range. The fact that the measured redox potentials of CP4 are close to P1 reflects the weak electronic communication between the porphyrin units. It is surprising that the first oxidation potential of CP4 (0.50 V) is higher than that of P1 (0.41 V); a possible explanation for this effect is that the more sterically congested environment around each porphyrin unit in CP4 hinders solvation of the positive charge when one of the four porphyrin units becomes oxidized to the radical cation.
Fullerene Binding
The design and synthesis of molecular receptors for fullerenes has been a focus of research because of their application in the separation and regioselective functionalization of fullerenes.4,26 Although many porphyrin-based fullerene receptors have been reported, all of them have spacers linking the porphyrin units.4−6 The saddle-shaped geometry of CP4 and the cavity between two cofacial porphyrins inspired us to investigate its application as a host for fullerenes. The formation of a CP4·2C60 complex was first detected by UV–vis titrations in toluene solution (Figure 5a). The Soret band absorption maxima of CP4 red-shifted from 415 to 424 nm with the addition of C60. The binding constant was measured to be (2.2 ± 0.1) × 104 M–1 by fitting the titration data to a 1:1 binding isotherm (Table S3). Similar behavior was observed for C70, which shows a higher binding constant of (1.1 ± 0.4) × 105 M–1. Most porphyrin-based fullerene receptors bind C70 more strongly than C60 and this selectivity has been used for separating fullerenes.4,27 The strong binding of CP4 with fullerenes was also confirmed by MALDI-TOF MS measurements using trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) as a matrix, which show peaks of the complexes with C60 (Figure S23).
Figure 5.

(a) UV–vis absorption spectra change of CP4 (c = 1.65 μM) with the addition of C60 (c = 0–0.47 mM) in toluene at 298 K, 10 mm path length. Spectra were corrected by subtracting C60 absorption; the inset shows plots of A421–A605 against the concentration of C60, circles indicate the experimental data; (b) plots of chemical shift changes of protons (Ha, He, and Hg) on CP4 (c = 0.49 mM) with the addition of C60 (400 MHz, d8-toluene, 298 K).
The stoichiometry of binding was checked by the 1H NMR titration of CP4 with C60. As shown in Figure 5b, the protons (Ha) on the meso-positions of porphyrin and the ortho-position of the aryl groups (He) all shifted to the lower field region, while the protons on the other ortho-position of the aryl groups (Hg) shifted to the high-magnetic field region with the addition of C60. The chemical shift changes approach saturation at around [C60]/CP4 = 2:1. 1H NMR titration data fitted well to a 1:1 binding isotherm with a concentration of receptor of 0.98 mM, which is exactly twice the actual concentration of CP4, implying that both sides of the receptor bind C60 independent of each other, i.e., with no cooperativity.
Single crystals of complex CP4·2C60 were obtained by slow diffusion of methanol vapor into a solution of CP4 and excess C60 in o-dichlorobenzene. Single-crystal X-ray diffraction studies revealed the 1:2 complex (Figure 6), which crystalized in the triclinic space group P1̅. The asymmetric unit was found to contain two molecules of CP4 and four molecules of C60. Examination of the structure suggested the presence of pseudo symmetry, which was thought to potentially be caused by a phase transition. For this reason, the sample was re-examined to explore whether data collected on a high temperature phase would give better results. Despite numerous attempts, the original triclinic phase was not seen again, instead a new orthorhombic phase was repeatedly found. This also revealed a 1:2 complex of CP4 with C60. The data and refinement for this new polymorph were markedly better than the first triclinic phase so this is the one primarily discussed here (though the triclinic polymorph is included in the Supporting Information).28
Figure 6.
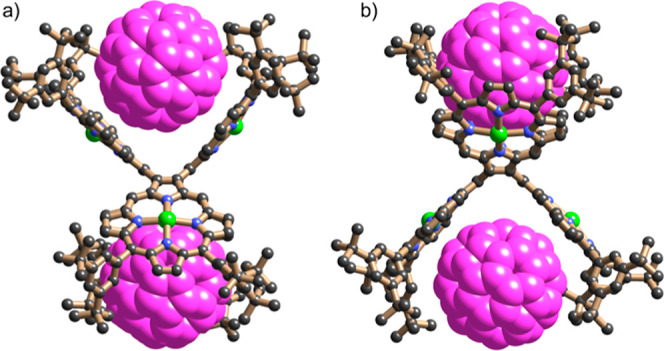
X-ray structure of CP4·2C60 (orthorhombic phase). Two orthogonal views of the molecular unit; solvent molecules, hydrogen atoms, and disorder omitted for clarity.
All three crystallographically distinct CP4·2C60 complexes seen in both crystal structure determinations have essentially the same geometry with methanol molecules coordinated to the central zinc atoms from the side that does not interact with fullerenes. Two fullerene molecules are captured by the cavity between two porphyrins located above and below the central COT ring. The shortest fullerene carbon to zinc distances are in the range of 3.15–3.29 Å, which is shorter than the sum of the van der Waals radi,29 indicating an attractive interaction between fullerene and porphyrins. Each porphyrin skeleton adopts a saddle distortion, with a concave face towards a bound C60 molecule. The geometry of the CP4 unit in this crystal of the C60 complex is similar to the DFT-calculated structure of CP4 (Figure 3), which is consistent with the non-cooperative binding behavior deduced from UV–vis absorption and 1H NMR titration data.
Conclusions
We have reported the synthesis of two 2,3-linked cyclic porphyrin oligomers CP3 and CP4, with benzene and COT cores, via Yamamoto coupling of 2,3-dibromoporphyrin. The Yamamoto coupling of vicinal dibromides has previously been applied to link together various other conjugated π-systems, such as helicenes,30 acepleiadylene,31 and tetracenes,32 but in most cases, this reaction gives just the benzene-centered cyclic trimer, not the COT-centered cyclic tetramer. The different numbers of porphyrin units in CP3 and CP4 result in different 3D geometries, leading to distinct optoelectronic properties. Although the minimum-energy geometry of CP3 has a D3 propeller-shaped structure, its crystal structure adopts a lower symmetry P,M,M/M,P,P configuration, whereas CP4 exists in a stable saddle-shaped conformation with stereochemistry of P,M,P,M. π-Conjugation in CP3 results in splitting and red shift of the absorption bands. By contrast, the saddle-shaped CP4 shows similar optical and electrochemical properties to the monomer, due to the large dihedral angles between porphyrin units. The saddle-shaped geometry of CP4 makes it a good receptor for fullerene and high binding constants of (1.1 ± 0.4) × 105 and (2.2 ± 0.1) × 104 M–1 were obtained for C70 and C60, respectively, in toluene solution at 298 K. The 1:2 fullerene binding mode of CP4 could be useful for holding two paramagnetic endohedral fullerenes at a fixed distance for quantum information processing.5b,33 This work also provides new insights into the design and synthesis of directly linked cyclic porphyrin arrays with applications in photophysics and supramolecular chemistry.
Acknowledgments
We thank the ERC for financial support (grant 885606 ARO-MAT). Q.C. is grateful to the German Research Foundation for a Walter Benjamin fellowship (grant number CH 2577/1-1). We gratefully thank Diamond Light Source for an award of beamtime on I19 (MT20876) and the EPSRC for a Strategic Equipment grant (EP/V208995/1).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c03549.
Experimental details, characterization data, 1H NMR, 13C NMR, and MS spectra, and calculation details (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Ogoshi H.; Sugimoto H.; Yoshida Z.-i. Cyclophane porphyrin-III. Double porphyrins. Tetrahedron Lett. 1977, 18, 169–172. 10.1016/s0040-4039(01)92579-1. [DOI] [Google Scholar]; b Kagan N. E.; Mauzerall D.; Merrifield R. B. strati-Bisporphyrins. A novel cyclophane system. J. Am. Chem. Soc. 1977, 99, 5484–5486. 10.1021/ja00458a045. [DOI] [PubMed] [Google Scholar]; c Nakamura Y.; Aratani N.; Osuka A. Cyclic porphyrin arrays as artificial photosynthetic antenna: synthesis and excitation energy transfer. Chem. Soc. Rev. 2007, 36, 831–845. 10.1039/B618854K. [DOI] [PubMed] [Google Scholar]; d Parkinson P.; Kondratuk D. V.; Menelaou C.; Gong J. Q.; Anderson H. L.; Herz L. M. Chromophores in molecular nanorings—When is a ring a ring?. J. Phys. Chem. Lett. 2014, 5, 4356–4361. 10.1021/jz5022153. [DOI] [PubMed] [Google Scholar]
- Aratani N.; Kim D.; Osuka A. Discrete cyclic porphyrin arrays as artificial light-harvesting antenna. Acc. Chem. Res. 2009, 42, 1922–1934. 10.1021/ar9001697. [DOI] [PubMed] [Google Scholar]
- a Anderson H. L.; Sanders J. K. M. Synthesis of a cyclic porphyrin trimer with a semi-rigid cavity. J. Chem. Soc., Chem. Commun. 1989, 1714–1715. 10.1039/c39890001714. [DOI] [Google Scholar]; b Hamilton A.; Lehn J. M.; Sessler J. L. Coreceptor molecules. Synthesis of metalloreceptors containing porphyrin subunits and formation of mixed substrate supermolecules by binding of organic substrates and of metal ions. J. Am. Chem. Soc. 2002, 108, 5158–5167. 10.1021/ja00277a021. [DOI] [Google Scholar]; c Hogben H. J.; Sprafke J. K.; Hoffmann M.; Pawlicki M.; Anderson H. L. Stepwise effective molarities in porphyrin oligomer complexes: preorganization results in exceptionally strong chelate cooperativity. J. Am. Chem. Soc. 2011, 133, 20962–20969. 10.1021/ja209254r. [DOI] [PubMed] [Google Scholar]
- a Tashiro K.; Aida T. Metalloporphyrin hosts for supramolecular chemistry of fullerenes. Chem. Soc. Rev. 2007, 36, 189–197. 10.1039/B614883M. [DOI] [PubMed] [Google Scholar]; b Durot S.; Taesch J.; Heitz V. Multiporphyrinic cages: Architectures and functions. Chem. Rev. 2014, 114, 8542–8578. 10.1021/cr400673y. [DOI] [PubMed] [Google Scholar]; c Mondal P.; Rath S. P. Cyclic metalloporphyrin dimers: Conformational flexibility, applications and future prospects. Coord. Chem. Rev. 2020, 405, 213117. 10.1016/j.ccr.2019.213117. [DOI] [Google Scholar]
- a Tashiro K.; Aida T.; Zheng J.-Y.; Kinbara K.; Saigo K.; Sakamoto S.; Yamaguchi K. A cyclic dimer of metalloporphyrin forms a highly stable inclusion complex with C60. J. Am. Chem. Soc. 1999, 121, 9477–9478. 10.1021/ja992416m. [DOI] [Google Scholar]; b Gil-Ramirez G.; Karlen S. D.; Shundo A.; Porfyrakis K.; Ito Y.; Briggs G. A.; Morton J. J.; Anderson H. L. A cyclic porphyrin trimer as a receptor for fullerenes. Org. Lett. 2010, 12, 3544–3547. 10.1021/ol101393h. [DOI] [PubMed] [Google Scholar]; c Shi Y.; Cai K.; Xiao H.; Liu Z.; Zhou J.; Shen D.; Qiu Y.; Guo Q.-H.; Stern C.; Wasielewski M. R.; Diederich F.; Goddard W. A. III; Stoddart J. F. Selective extraction of C70 by a tetragonal prismatic porphyrin cage. J. Am. Chem. Soc. 2018, 140, 13835–13842. 10.1021/jacs.8b08555. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Gsänger S.; Minameyer M. B.; Imaz I.; Maspoch D.; Shyshov O.; Schwer F.; Ribas X.; Drewello T.; Meyer B.; von Delius M. Highly strained, radially pi-conjugated porphyrinylene nanohoops. J. Am. Chem. Soc. 2019, 141, 18500–18507. 10.1021/jacs.9b08584. [DOI] [PubMed] [Google Scholar]
- a Hamilton A. D. Synthesis of tetrameric and hexameric cyclo-porphyrins. J. Chem. Soc., Chem. Commun. 1987, 293–295. 10.1039/C39870000293. [DOI] [Google Scholar]; b Walter C. J.; Anderson H. L.; Sanders J. K. M. exo-Selective acceleration of an intermolecular Diels–Alder reaction by a trimeric porphyrin host. J. Chem. Soc., Chem. Commun. 1993, 458–460. 10.1039/c39930000458. [DOI] [Google Scholar]; c Anderson H. L.; Sanders J. K. M. Enzyme mimics based on cyclic porphyrin oligomers: strategy, design andexploratory synthesis. J. Chem. Soc., Perkin Trans. 1 1995, 1, 2223–2229. 10.1039/p19950002223. [DOI] [Google Scholar]; d Mackay L. G.; Wylie R. S.; Sanders J. K. M. Catalytic acyl transfer by a cyclic porphyrin trimer: efficient turnover without product inhibition. J. Am. Chem. Soc. 2002, 116, 3141–3142. 10.1021/ja00086a061. [DOI] [Google Scholar]; e Totten R. K.; Ryan P.; Kang B.; Lee S. J.; Broadbelt L. J.; Snurr R. Q.; Hupp J. T.; Nguyen S. T. Enhanced catalytic decomposition of a phosphate triester by modularly accessible bimetallic porphyrin dyads and dimers. Chem. Commun. 2012, 48, 4178–4180. 10.1039/c2cc17568a. [DOI] [PubMed] [Google Scholar]
- a Peeks M. D.; Claridge T. D. W.; Anderson H. L. Aromatic and Antiaromatic Ring Currents in a Molecular Nanoring. Nature 2017, 541, 200–203. 10.1038/nature20798. [DOI] [PubMed] [Google Scholar]; b Ren L.; Gopalakrishna T. Y.; Park I.-H.; Han Y.; Wu J. Porphyrin/quinoidal-bithiophene-based macrocycles and their dications: Template-free synthesis and global aromaticity. Angew. Chem., Int. Ed. 2020, 59, 2230–2234. 10.1002/anie.201911269. [DOI] [PubMed] [Google Scholar]; c Jirásek M.; Anderson H. L.; Peeks M. D. From macrocycles to quantum rings: Does aromaticity have a size limit?. Acc. Chem. Res. 2021, 54, 3241–3251. 10.1021/acs.accounts.1c00323. [DOI] [PubMed] [Google Scholar]
- a Yu C.; Long H.; Jin Y.; Zhang W. Synthesis of cyclic porphyrin trimers through alkyne metathesis cyclooligomerization and their host–guest binding study. Org. Lett. 2016, 18, 2946–2949. 10.1021/acs.orglett.6b01293. [DOI] [PubMed] [Google Scholar]; b Haver R.; Tejerina L.; Jiang H.-W.; Rickhaus M.; Jirasek M.; Grübner I.; Eggimann H. J.; Herz L. M.; Anderson H. L. Tuning the circumference of six-porphyrin nanorings. J. Am. Chem. Soc. 2019, 141, 7965–7971. 10.1021/jacs.9b02965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Anderson S.; Anderson H. L.; Sanders J. K. M. Expanding Roles for Templates in Synthesis. Acc. Chem. Res. 1993, 26, 469–475. 10.1021/ar00033a003. [DOI] [Google Scholar]; b Bols P. S.; Anderson H. L. Template-directed synthesis of molecular nanorings and cages. Acc. Chem. Res. 2018, 51, 2083–2092. 10.1021/acs.accounts.8b00313. [DOI] [PubMed] [Google Scholar]
- Song J.; Aratani N.; Shinokubo H.; Osuka A. A β-to-β 2,5-thienylene-bridged cyclic porphyrin tetramer: its rational synthesis and 1:2 binding mode with C60. Chem. Sci. 2011, 2, 748–751. 10.1039/c0sc00605j. [DOI] [Google Scholar]
- Jiang H. W.; Tanaka T.; Kim T.; Sung Y. M.; Mori H.; Kim D.; Osuka A. Synthesis of [n]cyclo-5,15-porphyrinylene-4,4’-biphenylenes displaying size-dependent excitation-energy hopping. Angew. Chem., Int. Ed. Engl. 2015, 54, 15197–15201. 10.1002/anie.201507822. [DOI] [PubMed] [Google Scholar]
- Kim D.; Osuka A. Directly linked porphyrin arrays with tunable excitonic interactions. Acc. Chem. Res. 2004, 37, 735–745. 10.1021/ar030242e. [DOI] [PubMed] [Google Scholar]
- a Nakamura Y.; Hwang I. W.; Aratani N.; Ahn T. K.; Ko D. M.; Takagi A.; Kawai T.; Matsumoto T.; Kim D.; Osuka A. Directly meso-meso linked porphyrin rings: Synthesis, characterization, and efficient excitation energy hopping. J. Am. Chem. Soc. 2005, 127, 236–246. 10.1021/ja045254p. [DOI] [PubMed] [Google Scholar]; b Nakamura Y.; Aratani N.; Shinokubo H.; Takagi A.; Kawai T.; Matsumoto T.; Yoon Z. S.; Kim D. Y.; Ahn T. K.; Kim D.; Muranaka A.; Kobayashi N.; Osuka A. A directly fused tetrameric porphyrin sheet and its anomalous electronic properties that arise from the planar cyclooctatetraene core. J. Am. Chem. Soc. 2006, 128, 4119–4127. 10.1021/ja057812l. [DOI] [PubMed] [Google Scholar]
- Jiang H.-W.; Tanaka T.; Mori H.; Park K. H.; Kim D.; Osuka A. Cyclic 2,12-porphyrinylene nanorings as a porphyrin analogue of cycloparaphenylenes. J. Am. Chem. Soc. 2015, 137, 2219–2222. 10.1021/ja513102m. [DOI] [PubMed] [Google Scholar]
- Cai H.; Fujimoto K.; Lim J. M.; Wang C.; Huang W.; Rao Y.; Zhang S.; Shi H.; Yin B.; Chen B.; Ma M.; Song J.; Kim D.; Osuka A. Synthesis of direct β-to-β linked porphyrin arrays with large electronic interactions: Branched and cyclic oligomers. Angew. Chem., Int. Ed. 2014, 53, 11088–11091. 10.1002/anie.201407032. [DOI] [PubMed] [Google Scholar]
- Katritzky A. R.; Law K. W. A 13C study of hydroxymethyl derivatives of five-membered ring heterocycles. Magn. Reson. Chem. 1988, 26, 129–133. 10.1002/mrc.1260260209. [DOI] [Google Scholar]
- Sudhakar G.; Kadam V. D.; Bayya S.; Pranitha G.; Jagadeesh B. Total synthesis and stereochemical revision of acortatarins A and B. Org. Lett. 2011, 13, 5452–5455. 10.1021/ol202121k. [DOI] [PubMed] [Google Scholar]
- Cadamuro S.; Degani I.; Fochi R.; Gatti A.; Piscopo L. Convenient route for the synthesis of 3-substituted and 3,4-disubstituted pyrrole-2,5-dicarbaldehydes. J. Chem. Soc., Perkin Trans. 1 1996, 1, 2365–2369. 10.1039/p19960002365. [DOI] [Google Scholar]
- Gałȩzowski M.; Gryko D. T. Synthesis of locked meso-β-substituted chlorins via 1,3-dipolar cycloaddition. J. Org. Chem. 2006, 71, 5942–5950. 10.1021/jo060545x. [DOI] [PubMed] [Google Scholar]
- Gryko D. T.; Gałȩzowski M. Simple approach to “locked” chlorins. Org. Lett. 2005, 7, 1749–1752. 10.1021/ol050327a. [DOI] [PubMed] [Google Scholar]
- Claus K. H.; Krüger C. Structure of cyclooctatetraene at 129 K. Acta Crystallogr., Sect. C: Struct. Chem. 1988, 44, 1632–1634. 10.1107/s0108270188005840. [DOI] [Google Scholar]
- a Kruszewski J.; Krygowski T. M. Definition of aromaticity basing on the harmonic oscillator model. Tetrahedron Lett. 1972, 13, 3839–3842. 10.1016/s0040-4039(01)94175-9. [DOI] [Google Scholar]; b Makino M.; Nishina N.; Aihara J. Critical evaluation of HOMA and MBL as local aromaticity indices. J. Phys. Org. Chem. 2018, 31, e3783 10.1002/poc.3783. [DOI] [Google Scholar]
- Chen Z.; Wannere C. S.; Corminboeuf C.; Puchta R.; Schleyer P. Nucleus-independent chemical shifts (NICS) as an aromaticity criterion. Chem. Rev. 2005, 105, 3842–3888. 10.1021/cr030088+. [DOI] [PubMed] [Google Scholar]
- a Wurth C.; Grabolle M.; Pauli J.; Spieles M.; Resch-Genger U. Relative and absolute determination of fluorescence quantum yields of transparent samples. Nat. Protoc. 2013, 8, 1535–1550. 10.1038/nprot.2013.087. [DOI] [PubMed] [Google Scholar]; b Taniguchi M.; Lindsey J. S.; Bocian D. F.; Holten D. Comprehensive review of photophysical parameters (ε, Φf, τs) of tetraphenylporphyrin (H2TPP) and zinc tetraphenylporphyrin (ZnTPP)—Critical benchmark molecules in photochemistry and photosynthesis. J. Photochem. Photobiol., C 2021, 46, 100401. 10.1016/j.jphotochemrev.2020.100401. [DOI] [Google Scholar]
- a Ubasart E.; Borodin O.; Fuertes-Espinosa C.; Xu Y.; García-Simón C.; Gómez L.; Juanhuix J.; Gándara F.; Imaz I.; Maspoch D.; von Delius M.; Ribas X. A three-shell supramolecular complex enables the symmetry-mismatched chemo- and regioselective bis-functionalization of C60. Nat. Chem. 2021, 13, 420–427. 10.1038/s41557-021-00658-6. [DOI] [PubMed] [Google Scholar]; b Huang N.; Wang K.; Drake H.; Cai P.; Pang J.; Li J.; Che S.; Huang L.; Wang Q.; Zhou H. C. Tailor-made pyrazolide-based metal-organic frameworks for selective catalysis. J. Am. Chem. Soc. 2018, 140, 6383–6390. 10.1021/jacs.8b02710. [DOI] [PubMed] [Google Scholar]
- a Fang X.; Zhu Y. Z.; Zheng J. Y. Clawlike tripodal porphyrin trimer: ion-controlled on-off fullerene binding. J. Org. Chem. 2014, 79, 1184–1191. 10.1021/jo4026176. [DOI] [PubMed] [Google Scholar]; b Zhang C.; Wang Q.; Long H.; Zhang W. A highly C70 selective shape-persistent rectangular prism constructed through one-step alkyne metathesis. J. Am. Chem. Soc. 2011, 133, 20995–21001. 10.1021/ja210418t. [DOI] [PubMed] [Google Scholar]
- a Single-crystal X-ray diffraction data for the triclinic polymorph were collected using beamline I19 at Diamond Light Source;Allan D. R.; et al. A novel dual air-bearing fixed-χ diffractometer for small-molecule single-crystal X-ray diffraction on beamline I19 at Diamond Light Source. Crystals 2017, 7, 336. 10.3390/cryst7110336. [DOI] [Google Scholar]; b Processed using the XIA2 software;Winter G. xia2: An expert system for macromolecular crystallography data reduction. J. Appl. Cryst. 2010, 43, 186–190. 10.1107/S0021889809045701. [DOI] [Google Scholar]; c Data for the orthorhombic phase were collected using a Rigaku Synergy DW diffractometer. Structure solution and refinement were carried out within the CRYSTALS software suite;Palatinus L.; Chapuis G. SUPERFLIP—a computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Cryst. 2007, 40, 786–790. 10.1107/S0021889807029238. [DOI] [Google Scholar]; d Parois P.; Cooper R. I.; Thompson A. L. Crystal structures of increasingly large molecules: meeting the challenges with CRYSTALS software. Chem. Cent. J. 2015, 9, 30. 10.1186/s13065-015-0105-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Disorder in the fullerenes was partially modelled using hollow spheres;Schröder L.; Watkin D. J.; Cousson A.; Cooper R. I.; Paulus W. CRYSTALS enhancements: refinement of atoms continuously disordered along a line, on a ring or on the surface of a sphere. J. Appl. Cryst. 2004, 37, 545–550. 10.1107/S0021889804009847. [DOI] [Google Scholar]; f Diffuse solvent in the void region was treated with SQUEEZE;van der Sluis P.; Spek A. L. BYPASS: an effective method for the refinement of crystal structures containing disordered solvent regions. Acta Cryst. 1990, A46, 194–201. 10.1107/S0108767389011189. [DOI] [Google Scholar]
- a Rowland R. S.; Taylor R. Intermolecular nonbonded contact distances in organic crystal structures: Comparison with distances expected from van der waals radii. J. Phys. Chem. 1996, 100, 7384–7391. 10.1021/jp953141+. [DOI] [Google Scholar]; b Alvarez S. A cartography of the van der Waals territories. Dalton Trans. 2013, 42, 8617–8636. 10.1039/c3dt50599e. [DOI] [PubMed] [Google Scholar]
- Roy M.; Berezhnaia V.; Villa M.; Vanthuyne N.; Giorgi M.; Naubron J.-V.; Poyer S.; Monnier V.; Charles L.; Carissan Y.; Hagebaum-Reignier D.; Rodriguez J.; Gingras M.; Coquerel Y. Stereoselective syntheses, structures, and properties of extremely distorted chiral nanographenes embedding hextuple helicenes. Angew. Chem., Int. Ed. 2020, 59, 3264–3271. 10.1002/anie.201913200. [DOI] [PubMed] [Google Scholar]
- Liu P.; Chen X.-Y.; Cao J.; Ruppenthal L.; Gottfried J. M.; Müllen K.; Wang X.-Y. Revisiting acepleiadylene: Two-step synthesis and π-extension toward nonbenzenoid nanographene. J. Am. Chem. Soc. 2021, 143, 5314–5318. 10.1021/jacs.1c01826. [DOI] [PubMed] [Google Scholar]
- Rüdiger E. C.; Porz M.; Schaffroth M.; Rominger F.; Bunz U. H. F. Synthesis of soluble, alkyne-substituted trideca- and hexadeca-starphenes. Chem.—Eur. J. 2014, 20, 12725–12728. 10.1002/chem.201403697. [DOI] [PubMed] [Google Scholar]
- Farrington B. J.; Jevric M.; Rance G. A.; Ardavan A.; Khlobystov A. N.; Briggs G. A.; Porfyrakis K. Chemistry at the nanoscale: synthesis of an N@C60-N@C60 endohedral fullerene dimer. Angew. Chem., Int. Ed. 2012, 51, 3587–3590. 10.1002/anie.201107490. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.