

Abstract
Despite continuous advances in understanding the underlying pathogenesis of hyperexcitable networks and lowered seizure thresholds, the treatment of epilepsy remains a clinical challenge. Over one third of patients remain resistant to current pharmacological interventions. Moreover, even when effective in suppressing seizures, current medications are merely symptomatic without significantly altering the course of the disease. Much effort is therefore invested in identifying new treatments with novel mechanisms of action, effective in drug-refractory epilepsy patients, and with the potential to modify disease progression. Compelling evidence has demonstrated that the purines, ATP and adenosine, are key mediators of the epileptogenic process. Extracellular ATP concentrations increase dramatically under pathological conditions, where it functions as a ligand at a host of purinergic receptors. ATP, however, also forms a substrate pool for the production of adenosine, via the action of an array of extracellular ATP degrading enzymes. ATP and adenosine have assumed largely opposite roles in coupling neuronal excitability to energy homeostasis in the brain. This review integrates and critically discusses novel findings regarding how ATP and adenosine control seizures and the development of epilepsy. This includes purine receptor P1 and P2-dependent mechanisms, release and reuptake mechanisms, extracellular and intracellular purine metabolism, and emerging receptor-independent effects of purines. Finally, possible purine-based therapeutic strategies for seizure suppression and disease modification are discussed.
Keywords: Epilepsy, Purinergic signaling, Adenosine triphosphate, Adenosine, P1 receptors, P2 receptors
1. Epilepsy and purinergic signaling
Epilepsy encompasses a heterogeneous group of brain disorders characterized by the manifestation of spontaneous, unprovoked seizures, with an incidence of ~1%, affecting up to 70 million persons worldwide (Thijs et al. 2019). Along with unprovoked seizures, the quality of life of persons with epilepsy is further reduced by a risk of death 2–3 times higher than amongst the general population, and a 4-fold increased risk of additional comorbidities, such as anxiety or depression (LaFrance et al. 2008; Lin et al. 2012; Moshe et al. 2015). Epilepsy can result from genetic abnormalities such as polymorphisms, copy number variations, or de novo mutations. Epilepsy can also result from insults to the brain, such as traumatic brain injury (TBI), stroke, prolonged seizures, or infection (Scheffer et al. 2017; Klein et al. 2018).
Despite the heterogeneous nature of epilepsy, research on purinergic signaling and epilepsy has mainly focused on the study of acquired epilepsies, in particular temporal lobe epilepsy (TLE). Among the acquired epilepsies, TLE is the most common form which can result from a precipitating insult to the brain (Klein et al. 2018). TLE pathology includes structural, physiological, biochemical, and epigenetic alterations within the limbic system, including the amygdala and hippocampus (Engel, 1996; Klein et al. 2018). TLE typically involves hippocampal sclerosis, characterized by selective loss of neurons within the hilus and pyramidal neurons of the CA1 and CA3 hippocampal subfields, axonal sprouting and gliosis (Chang and Lowenstein, 2003). Epileptogenesis, triggered by a precipitating insult to the brain, is the process that turns a normal brain into a brain expressing spontaneous epileptic seizures. It is now understood, however, that pathological processes activated during epileptogenesis continue beyond the emergence of epilepsy and lead to an aggravation of the epileptic phenotype over time (Pitkanen and Engel, 2014). Epileptogenesis includes neurodegeneration, changes in structural and synaptic plasticity, increased permeability of the blood-brain barrier (BBB), aberrant neurogenesis, epigenetic changes (e. g., aberrant methylation of DNA, histone modifications) and the chronic activation of inflammatory processes (Henshall and Kobow, 2015; Pitkanen et al. 2015).
While several alternative therapeutic options are available for epilepsy, including surgical resection of the epileptic focus, use of the ketogenic diet, or vagus nerve or deep brain stimulation, for the majority of patients with epilepsy, anti-seizure drugs (ASDs) are the first line treatment (Moshe et al. 2015; Thijs et al. 2019). Current pharmacological interventions in epilepsy, however, remain mostly focused on targeting synaptic transmission or ion channels (Bialer and White, 2010). With over 30 % of patients not responding to current medication with a high burden of adverse effects (e.g., fatigue, irritability, dizziness), currently available medications are merely symptomatic, without noticeable impacts on disease progression. There is, therefore, a big need to better understand the processes that lead to epilepsy and its progression and to develop new therapies that go beyond seizure control (Loscher, 2020).
Over recent years, the role played by the purinergic signaling system in mediating the excitability of neuronal networks has become increasingly clear. It is now well established that neuronal excitability in the brain is determined not only by the glutamate/γ-aminobutyric acid (GABA) balance, but also by the ATP/adenosine balance. That adenosine functions as an endogenous anticonvulsant and seizure terminator has been known for decades (Dunwiddie and Hoffer, 1980; Dragunow and Goddard, 1984), while the excitatory role played by ATP during seizures has emerged more recently (Engel et al. 2012). The contribution of purines to the development and progression of epilepsy, however, is not restricted to their function at the synapse, but also in mediating other biological processes, such as inflammation (Beamer et al. 2016) or newly identified epigenetic functions of adenosine (Williams-Karnesky et al. 2013; Boison and Rho, 2019). New evidence has recently emerged, clarifying the cellular sources, initiating stimuli, and mechanisms of release of purines into the extracellular space following an insult (Beamer et al. 2019). Further, it is important to consider the role played by ectoenzymes in regulating the extracellular concentrations of both ATP and adenosine, and the interactions between extra- and intracellular adenosine metabolism, as well as transport (Plesner, 1995; Boison and Yegutkin, 2019). The two signaling systems of ATP and adenosine are intrinsically linked and a full understanding requires the purinergic signaling system to be considered as a whole. In the following section, we will provide a detailed discussion of recent advances in our understanding of purinergic signaling during seizures and epilepsy, including the release and degradation of purines and the contribution of the different purinergic receptor subtypes to disease pathology. We will also discuss how purinergic therapeutic strategies could be used to suppress seizures and prevent the development of epilepsy.
2. The role of purines in seizures and epilepsy
Over the past decades our understanding of the role played by purinergic signaling during seizures and epilepsy has increased substantially. While most efforts have been put on investigating a contribution of purines to status epilepticus and TLE using rodent models of chemically-induced status epilepticus [e.g., pilocarpine, kainic acid (KA)], other models mimicking a variety of different seizure types and epilepsies are also used including genetic models (e.g., Wistar Albino Glaxo/Rijswijk (WAG/Rij) rats) and electrostimulation models (Table 1). For a comprehensive and exhaustive description of seizure and epilepsy models please refer to other excellent reviews which were written on this topic [e.g., (Loscher, 2017; Campos et al. 2018; Loscher, 2020)].
Table 1.
In vivo approaches employed to investigate purinergic signaling during seizures and epilepsy using adult rodents (selected examples).
| Animal model | Type of Seizures / Epilepsy | Induction | Studies on purinergic signaling |
|---|---|---|---|
| PTZ seizure model | Acute generalized seizures | Intraperitoneal, intravenous, or subcutaneous injection of PTZ | P2X7R (mice) ( Fischer et al. 2016); P2X7R (mice) (Nieoczym et al. 2017) |
| PTZ kindling model | Partial seizures developing into secondarily generalized seizures | Continuous low-dose intraperitoneal administration of PTZ | A2AR (mice) (El Yacoubi et al. 2009); P2X7R (species not specified) (Soni et al. 2015); P2X7R (rats, mice) (Fischer et al. 2016); P2X3R (rats) (Xia et al. 2018) |
| Pilocarpine model of status epilepticus | Status epilepticus, TLE with hippocampal sclerosis | Intraperitoneal injection of pilocarpine | A2AR (mice) (El Yacoubi et al. 2009); P2X7R (rats) (Kim et al. 2009; Kim et al. 2010, 2011); P2X7R (mice) ( Kim and Kang, 2011); P2X7R (rats) (Amhaoul et al. 2016); A1R (rats) (Amorim et al. 2016); ATP release (rats) ( Dona et al. 2016; Lietsche et al. 2016); P2X7R (rats) (Rozmer et al. 2017) |
| Intraperitoneal KA model of status epilepticus | Status epilepticus, TLE with hippocampal sclerosis | Intraperitoneal injection of KA | P2X7R (mice) ( Kim and Kang, 2011); P2X4R (mice) (Ulmann et al. 2013); P2Y12R (mice) ( Eyo et al. 2014); P2X7R (rats) ( Amhaoul et al. 2016); P2Y1R (rats) (Simoes et al. 2018) |
| Intrahippocampal KA model of status epilepticus | Status epilepticus, TLE with hippocampal sclerosis | Intrahippocampal injection of KA | A1R (mice) ( Gouder et al. 2003; Fedele et al. 2006); ADK (mice) (Gouder et al. 2004; Fedele et al. 2005; Sandau et al. 2019); A1R (rats) ( Canas et al. 2018) |
| Intraamygdala KA model of status epilepticus | Status epilepticus, TLE with hippocampal sclerosis | Intraamygdala injection of KA | P2X7R (mice) ( Engel et al. 2012; Jimenez-Pacheco et al. 2013; Jimenez-Mateos et al. 2015, Jimenez-Pacheco et al. 2016); P2YR family (mice) ( Alves et al. 2017; Alves, De et al. 2019; Alves et al. 2019) |
| Controlled cortical impact (CCI) | Post-traumatic epilepsy, epileptogenesis, hippocampal sclerosis | Mechanical energy delivered to an intact dura exposed following a craniectomy | A1R (mice) ( Kochanek et al., 1997) |
| 6-Hz psychomotor seizures test | Acute partial seizures | Low-frequency (6 Hz) stimulation delivered through corneal electrodes | P2X7R (mice) ( Nieoczym et al. 2017) |
| Maximal electroshock seizure model | Acute generalized tonic-clonic seizures | Electrical stimulation via transcorneal or transauricular electrodes at an intensity sufficient to elicit tonic hind limb extension [50-mA (mice) or 150-mA (rats)] | ADK (rats) ( Ugarkar et al. 2000); P2X7R (mice) (Fischer et al. 2016); P2X7R (mice) ( Nieoczym et al. 2017) |
| Electrical kindling | Partial epilepsy, partial and secondarily generalized seizures, epileptogenesis | Electrical stimuli via depth electrode in basolateral amygdala, hippocampus, or performant path | P2Rs (rats) (Sun et al. 2018); A1R (rats) (Canas et al. 2018) |
| Seizure-prone mice | Audiogenic seizures | Inbred DBA/2 (D2) mice | ATP release ( Wieraszko and Seyfried, 1989) |
| Seizure-prone gerbils | From mild hypnotic/ cataleptic to full grand mal seizures | Seizure-sensitive (SS) gerbil | P2X2R and P2X4R (Kang et al. 2003) |
| Genetic model of absence epilepsy | Absence seizures | Glaxo/Rijswijk (WAG/Rij) rats | Uridine (Kovacs et al. 2013); P2X7R (Dogan et al. 2020) |
Abbreviations: ADK, Adenosine kinase; CCI, Controlled cortical impact; i.p., intraperitoneal; KA, Kainic acid; PTZ, Pentylenetetrazol; TLE, Temporal lobe epilepsy.
2.1. Release of purines during acute CNS insults and seizures
Evidence for purine release into the extracellular space (Fig. 1) during and following acute central nervous system (CNS) insults comes from in vitro preparations, in vivo models, and clinical measurements (Dale and Frenguelli, 2009). Overall, evidence for peri-insult spikes in extracellular adenosine is stronger than for ATP. Independent of the insult or assay, where both purines are measured simultaneously, increases in extracellular adenosine are invariably of a greater magnitude than that of ATP. Further, peri-insult spikes in extracellular adenosine have been demonstrated in a wider range of contexts than for ATP. The contribution of the extracellular pool of ATP to adenosine, via extracellular metabolism has long been debated, however, the consensus is that insult-induced spikes in extracellular adenosine are largely the result of direct release of adenosine from cells (Wall and Dale, 2008).
Fig. 1. Sources of extracellular purines.
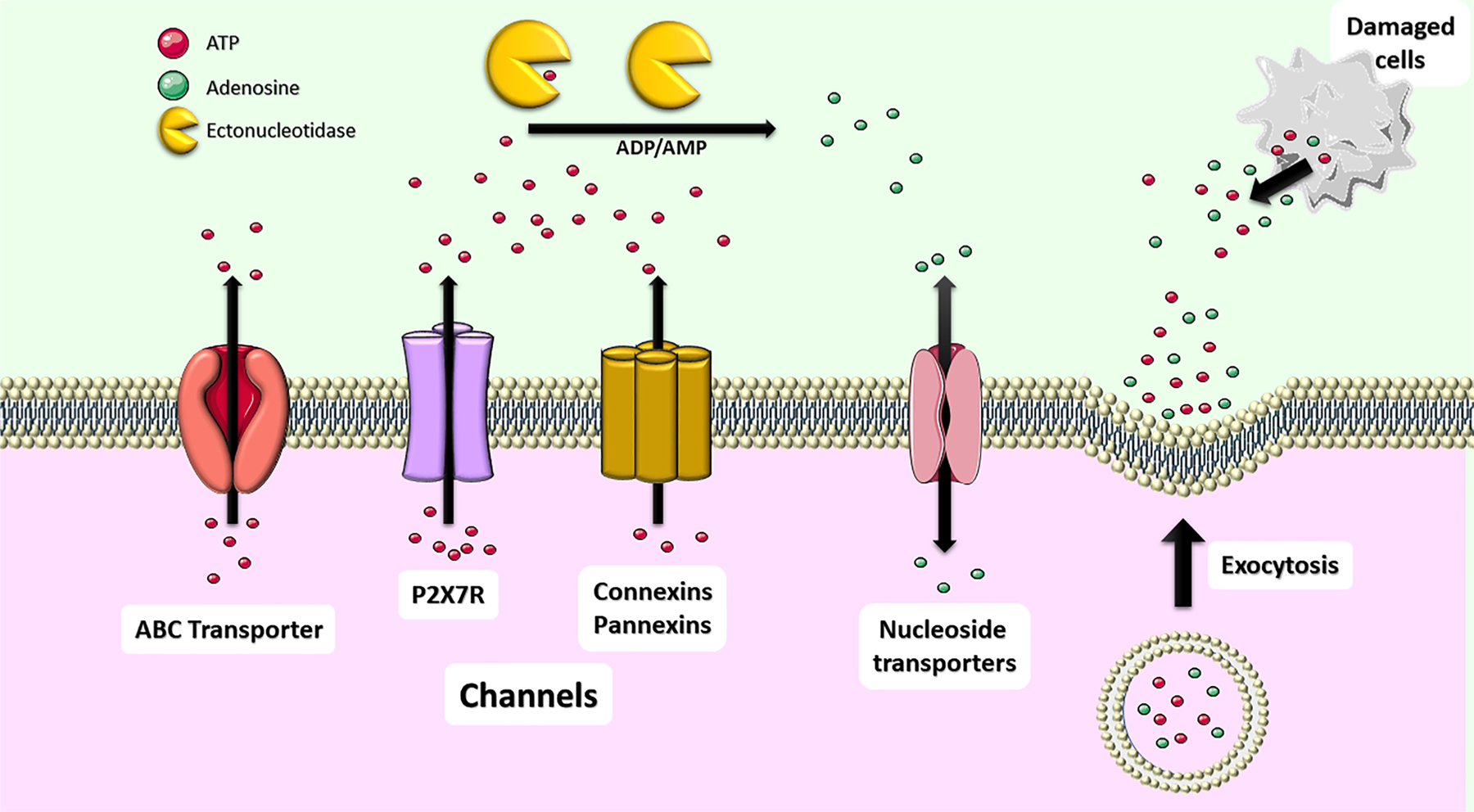
(A) Purines (ATP and adenosine) can be released into the extracellular space from both neurons and glia via exocytosis or non-exocytotic mechanisms including transporters (e.g., ATP-binding cassette (ABC) transporters for ATP and Nucleoside Transporters such as equilibrative nucleoside transporter (ENT) and concentrative nucleoside transporter (CNT) for adenosine) and membrane channels such as the P2X7R, and Pannexin-1 and Connexin-43 hemichannels. Purines are also released from dying cells escaping across a compromised lipid bilayer acting thereby as a danger signal. Once released, ATP is metabolized into different breakdown products including ADP, AMP and adenosine via the action of different ectonucleotidases as discussed in Chapter 5.
Enzyme-based sensors have been used to demonstrate release of both adenosine (Dale et al. 2000) and ATP (Frenguelli et al. 2007) in in vitro models of cerebral ischemia. The same sensors have been used to demonstrate adenosine release following in vitro seizure models (Diez et al. 2017). Similar studies investigating ATP release in the same context, however, have been mixed (Beamer et al. 2019) and appear to be dependent on the method of seizure induction employed. While seizure-like events induced by high K+ initiate ATP release (Heinrich et al. 2012), other methods, such as stimulation of the Schaffer collateral (Lopatar et al. 2011) found no increase in extracellular ATP. Diez et al. (Diez et al. 2017) found that ictal activity induced by low extracellular Ca2+ led to an increase in adenosine release through nucleoside transporters, but no change in extracellular ATP concentrations. The strongest in vitro evidence for ATP release during seizures comes from a study performed using post-operative brains from drug-resistant epilepsy patients (Dossi et al. 2018). In this study, extracellular ATP increased 80 % during high K+-induced ictal discharges. Similarly treated non-ictogenic surrounding tissue showed no ictal activity in response to high K+ and no ATP release. Further, ATP release, but not ictal activity, was blocked by pharmacological blockade of the Pannexin-1 hemichannel. It is not currently clear why high K+ in vitro seizure models induce ATP release, but other preparations do not.
Sims & Dale (Sims and Dale, 2014) report that adenosine is released from highly active cells as a consequence of increased activity of the Na+-K+-ATPase enzyme, necessary for the maintenance of ion homeostasis across the cell membrane. As the enzyme depletes ATP in order to drive ions against their concentration gradient, the intracellular concentration of adenosine increases, driving its transport across the cell membrane into the extracellular space through nucleoside transporters (Diez et al. 2017).
Following an insult, purines can enter the extracellular milieu via disruption of the integrity of cellular membranes, with ATP in particular driven by a very high intracellular-extracellular concentration gradient (Seminario-Vidal et al. 2009). Non-lytic routes of purine release, however, are many and varied. Wall & Dale (Wall and Dale, 2013) argue that under excitotoxic conditions, adenosine is released from neurons, while ATP is released from astrocytes, with the subsequent extracellular ATP pool then contributing to adenosine production via ectoenzymes. ATP release has been described via secretory granules from glia cells (Imura et al. 2013; Lalo et al. 2014), neuronal synaptic vesicles (Jo and Schlichter, 1999), diffusion through pannexin-1 hemichannels in neurons (Xia et al. 2012) and astrocytes (Bennett et al. 2012) and through connexin-43 hemichannels in astrocytes (Orellana et al. 2011). ATP is also released through the ATP-activated purinergic P2X7R (Suadicani et al. 2006). The first evidence for purine release from in vivo models of acute brain insult was produced by Berne et al. (Berne et al. 1974) demonstrating that interstitial adenosine increases 3-fold in the brains of dogs and rats within a minute of experimental ischemia. More recently, Ganesana & Venton (Ganesana and Venton, 2018) used fast-scan cyclic voltammetry to demonstrate an increased frequency of adenosine transients following experimental cerebral ischemia (bilateral common carotid artery occlusion) in the rat, with a 1.5-fold increase in cumulative concentration. Dona et al. (Dona et al. 2016), meanwhile, demonstrate that, in a rat model of pilocarpine-induced status epilepticus, adenosine is increased approximately 3-fold, alongside adenosine monophosphate (AMP) (2.5 fold) and adenosine diphosphate (ADP) (3.5-fold), with no measurable increase in ATP.
The direct measurement of ATP release in vivo is more challenging than adenosine, for a number of reasons. Firstly, it is present at extremely low baseline absolute extracellular concentrations, close to the threshold of detection (Dona et al. 2016). Secondly, the concentration gradient between the intracellular and extracellular compartments is around 1,000,000:1 (Schwiebert and Zsembery, 2003), meaning that cell damage resulting from invasive recording methods is likely to introduce experimental artifacts with a magnitude far in excess of the signal. Thirdly, ATP has a half-life in the extracellular milieu on the order of seconds (Picher et al. 2004), where it is swiftly degraded by a coordinated array of ectoenzymes (Yegutkin et al. 2006). Despite these challenges, convincing in vivo evidence has been presented for increases in interstitial ATP concentrations following a model of cerebral ischemia in the rat, with interstitial ATP increasing 1.3-fold (Melani et al. 2005). Evidence for ATP release during seizures, however, is more complex (Beamer et al. 2019).
Early in vivo evidence for seizure-induced ATP release comes from studies demonstrating a 30-fold spike in ATP measured in extracellular fluid collected in a cup from the cortex following direct electrical stimulation (Wu and Phillis, 1978). Haman & Attwell (Hamann and Attwell, 1996) suggest, however, that these findings may be an artefact of electroporation of cells. More recently, two studies have been performed using the lithium-pilocarpine model in rats (Dona et al. 2016; Lietsche et al. 2016). These studies found no change in extracellular concentrations of ATP in the hippocampus during or following status epilepticus. Lietsche et al. (Lietsche et al. 2016), however, found an 8-fold increase in extracellular ATP vs. controls, when an ectoATPase inhibitor was introduced with the microdialysis probe. This suggests that seizure activity increases both the cellular release of ATP into the extracellular space and the rate of its enzymatic degradation. Dona et al. (Dona et al. 2016), meanwhile, demonstrate, that while no ATP increase could be observed during acute, induced seizures, spontaneous seizures during the chronic phase of epilepsy were accompanied by a 4-fold increase in interstitial ATP concentration.
Clinical evidence for insult-driven purine release is restricted to adenosine and adenosine metabolites. Adenosine is elevated in the cerebrospinal fluid (CSF) in children following TBI (Robertson et al., 2001), correlating strongly with severity on the Glasgow Coma Scale. Bell et al. (Bell et al., 2001) report that interstitial concentrations of adenosine increase 3.1-fold following TBI in adults, along with concomitant increases in xanthine, hypoxanthine and cyclic AMP (cAMP). Dale et al. (Dale et al. 2019) report a 1.6-fold increase in adenosine concentration in the venous blood of stroke patients, within 4.5 h of symptom onset. During & Spencer (During and Spencer, 1992), meanwhile, recorded directly from the brain of an epilepsy patient during a seizure, using a deep electrode and microdialysis probe. Extracellular concentrations of adenosine increased 6 – 31-fold during a seizure and persisted into the post-ictal period, a concentration of adenosine the authors demonstrated to be sufficient for the suppression of ictal-like activity in in vitro models.
2.2. Purinergic receptors
2.2.1. P1 (Adenosine) receptors
The family of G protein-coupled purinergic P1 receptors, also known as adenosine receptors (ARs), is comprised of four subtypes: A1,R A2AR, A2BR, A3R, which have all been cloned and extensively characterized (Dunwiddie et al. 1997; B.B. Fredholm et al., 1999; Benarroch, 2008). ARs are widely distributed throughout the nervous, respiratory, cardiovascular, immune, gastrointestinal, and urogenital systems, and are also found in skin, bones, and eyes (Peleli et al. 2017). Each AR varies, not only in distribution across tissue and cell type, but also in terms of downstream effectors and physiological effects they mediate. A1Rs and A3Rs are coupled to inhibitory Gi/o proteins, whereas A2ARs and A2BRs are coupled to stimulatory Gs proteins (Borea et al. 2018). All ARs are activated by their endogenous ligand, adenosine. The affinity of each receptor, however, may vary depending on the assay systems used (Fredholm, 2014). Based on current consensus, adenosine has a high affinity for A1Rs, A2ARs, and A3Rs as compared to the A2BR subtype, with affinities of 1–10 nM, 30 nM, 100 nM and 1000 nM, respectively (Fredholm et al., 2011; Borea et al. 2018).
2.2.2. P2 (ATP) receptors
P2 receptors, activated by adenine and uridine nucleotides are divided into two groups based on their sequence homology, pharmacology and mechanism of action; the ionotropic P2X receptors (P2XR) and metabotropic P2Y receptors (P2YR) which are expressed and functional on both neurons and glial cells (Burnstock, 2016a,b). The fast-acting P2XRs, all activated by their main endogenous agonist, ATP, are a family of cation-selective channels, permeable to small cations including Na+, K+ and Ca2+. To date, seven mammalian subunits have been cloned (P2X1-7), ranging in length from 379 (P2X6) to 595 (P2X7) amino acids (Khakh and North, 2006). These subunits form either functional homo- or heterotrimers, depolarizing the cell membrane upon activation. P2XRs share a common topology with two transmembrane domains, a large extracellular loop and an intracellular amino and carboxyl terminus (Khakh and North, 2006). P2XRs are expressed throughout the brain and are present and functional on all cell types. Some controversy remains, however, regarding the cell type-specific expression and function of some receptors, in particular regarding the P2X7R subtype (Illes et al. 2017; Miras-Portugal et al. 2017). In the brain, the contribution of P2XRs to synaptic plasticity and, particularly, fast synaptic transmission has been well-established (Pankratov et al. 2009). The role of P2XRs is not limited, however, to the regulation of synaptic transmission. P2XRs have also been implicated in numerous other cellular processes such as proliferation, differentiation, maturation and survival, cell adhesion, migration and inflammation (Burnstock, 2016a,b).
The slower-acting metabotropic P2YR family consists of eight G-protein coupled receptors including P2Y1, P2Y2, P2Y4, P2Y6, P2Y11, P2Y12, P2Y13 and P2Y14. P2YRs feature the typical characteristics of G-protein-coupled receptors, including an extracellular amino terminus, intracellular carboxyl terminus and seven transmembrane-spanning motifs. While P2Y1, P2Y2, P2Y4, P2Y6 and P2Y11Rs are coupled to Gq/G11 proteins, stimulating phospholipase C leading to the release of calcium from intracellular stores and the activation of protein kinase C (PKC), P2Y12, P2Y13 and P2Y14Rs are coupled to Gi/G0 proteins decreasing cAMP production via inhibition of adenylate cyclase. The P2Y11R is an exception as this receptor also couples to Gs, increasing the production of cAMP via stimulation of adenylate cyclase. Endogenous agonists of the P2YR family include the adenine nucleotides ATP (P2Y1R, P2Y2R and P2Y11R) and ADP (P2Y1R, P2Y12R and P2Y13R) and the uridine nucleotides uridine triphosphate (UTP) (P2Y2R and P2Y4R), uridine diphosphate (UDP) (P2Y6R and P2Y14R) and UDP-glucose (P2Y14R) (von Kugelgen and Hoffmann, 2016; von Kugelgen, 2019). As for P2XRs, P2YRs are expressed and functional on all brain cells and are implicated in numerous different cellular functions and pathological processes pertinent to epileptogenesis and epilepsy. These range from synaptic reorganization, changes in neurotransmitter release, cellular survival and neuroinflammation (Jacobson and Boeynaems, 2010; Guzman and Gerevich, 2016).
2.3. The role of adenosine P1 receptors in seizure modulation and epilepsy
2.3.1. A1 receptors
A1Rs interact with Gi and Go proteins, contributing to the suppression of adenyl cyclase and a consequent reduction in the production of cAMP, reduction in the activation of protein kinase A (PKA), and inhibition of GABA uptake into astrocytes (Fredholm et al., 2011; Cristovao-Ferreira et al. 2013). The A1R subtype is widely expressed in the CNS, mainly in pre- and post-synaptic excitatory synapses of the cortex, hippocampus, cerebellum, spinal cord, in glial cells, and autonomic nerve terminals (Chen et al. 2013). Activation of A1Rs mediates a variety of different functions including reduction of neuronal hyperexcitability, neuroprotection, seizure control, pain reduction, and control of sleep/-wakefulness (Stenberg et al. 2003; Ciruela et al. 2006; Gessi et al. 2011; Cunha, 2016; Sawynok, 2016). The role of A1Rs on physiological neurotransmission is through the blockade of N-type calcium channels and the induction of neuronal hyperpolarization through activation of G-protein coupled inwardly rectifying potassium (GIRK) channels (Wu and Saggau, 1994; Gundlfinger et al. 2007). This leads to a combined action of presynaptic inhibition of neurotransmitter release (glutamate) and decreased postsynaptic glutamate receptor (N-methyl-D-aspartate receptor (NMDA)) activation, thereby suppressing neuronal hyperexcitability and maintaining an A1R-dependent inhibitory tonus in the brain (Yoon and Rothman, 1991; Von Lubitz et al. 1994). The anti-convulsant effects of adenosine are largely mediated by A1Rs, because of this receptor’s high affinity for adenosine and the dominance of its expression in the seizure-prone limbic system (Hargus et al. 2012). In TLE patients, a 6 to 31-fold increase in adenosine levels and an increase in A1R expression was shown following a seizure when compared to baseline (During and Spencer, 1992). The increase in adenosine and consequent A1R activation is believed to be a protective feedback mechanism to limit seizure duration and the intensity or spread of focal seizures (Dulla et al. 2009; Ilie et al. 2012; Lovatt et al. 2012; Van Gompel et al. 2014). Furthermore, activation of A1Rs through selective receptor agonists effectively suppresses seizure activity, even in pharmaco-resistant epilepsy (Gouder et al. 2003; Vianna et al. 2005; Mares, 2010; Li and Zhang, 2011; Tosh et al. 2012; Li et al. 2013; Muzzi et al. 2013). Importantly, a local increase in adenosine resulting in enhanced A1R activation in the hippocampus is sufficient to exert potent anticonvulsant effects, while avoiding systemic side-effects associated with global A1R activation (Huber et al. 2001; Guttinger et al. 2005). The importance of A1Rs has been demonstrated in A1R-knock-out (KO) mice, which not only display spontaneous electrographic seizures (Masino et al. 2011), but also a generalization and spread of focal seizure activity after unilateral intrahippocampal KA-induced status-epilepticus (Fedele et al. 2006). A1R-KO mice also exhibited excessive neuronal cell loss when subjected to seizures (Fedele et al. 2006) and TBI (Kochanek et al. 2006). Findings from those studies highlight (i) the capability of endogenous adenosine to limit seizure activity to an epileptogenic focus and to prevent propagation of seizure activity to other brain regions and (ii) the neuroprotective effects associated with A1R activation (Fedele et al. 2006; Kochanek et al. 2006). In line with these findings, A1R antagonists increase seizure activity and decrease the potential benefits of standard ASDs (Chwalczuk et al. 2008; Fukuda et al. 2010). In spite of the anti-convulsant and neuroprotective properties of adenosine and A1R activation, it is important to note that the seizure-induced increase in endogenous adenosine might also be implicated in sudden unexpected death in epilepsy (SUDEP), because of the depressant effect of adenosine on brainstem respiratory centers (Vandam et al. 2008; Faingold et al. 2016; Reklow et al. 2019).
2.3.2. A2 and A3 receptors
In epilepsy, overexpression of A2ARs in the cerebral cortex was found in both amygdala-kindled rats and following systemic KA-induced status epilepticus in mice, with upregulation largely restricted, in both cases, to excitatory glutamatergic terminals (Rebola et al. 2005). In addition, there is a threefold increase in A2ARs in hippocampal astrocytes of patients with mesial temporal lobe epilepsy (MTLE), when compared to controls (Barros-Barbosa et al., 2016a). Interestingly, no A2AR upregulation was seen in GABAergic synapses, which would imply that A2ARs function largely through their mediation of excitatory neurotransmission (Canas et al. 2018).
A2ARs are coupled to Gs proteins and are associated with the activation of adenylate cyclase (Kull et al. 2000). Contrary to A1Rs, A2ARs are known for their excitatory effects through the enhancement of NMDA receptor function and increase of glutamate release at glutamatergic axon terminals (Lopes et al. 2002; Marchi et al. 2002; Rebola et al. 2008; Rebola et al. 2011). Although some studies have suggested a neuroprotective and anti-convulsive role of A2ARs (Adami et al. 1995; Vianna et al. 2005), the majority of available data demonstrates a proconvulsive and neurodegenerative role for A2ARs (Stockwell et al. 2017). In a recent study, A2ARs were shown to play a key role in neurodegeneration via the modulation of synaptic excitability following KA-induced seizures in rats and mice (Canas et al. 2018). The sequence of events observed are, an initial process of glutamate-induced excitotoxicity at 6 h, followed by synaptotoxicity at 12 h, and finally overt neurodegeneration at 24 h, suggesting a window of opportunity for intervention to achieve seizure suppression by using A2AR antagonists (Canas et al. 2018). In support of the pro-convulsant action of A2ARs, mice with a genetic inactivation of A2ARs were shown to have a reduced seizure susceptibility (El Yacoubi et al. 2001, 2008; El Yacoubi et al. 2009). The robust increase in A2AR- and decrease in A1R-density over time provides a rationale for a combined approach using A2AR antagonism and A1R agonism for seizure control (Rebola et al. 2005). The roles of A2B and A3Rs in epilepsy are not clearly characterized. Activation of A3Rs can counteract the inhibitory effects of A1Rs through heterologous desensitization of A1Rs (Dunwiddie et al. 1997) and A3R activation by endogenous adenosine increased epileptic activity in hippocampal slice cultures (Etherington and Frenguelli, 2004), possibly through interaction with A1Rs. On the contrary, A3R antagonism decreased seizure intensity and duration in vitro (Etherington and Frenguelli, 2004). Both A2B and A3R antagonists reduced GABA currents in membrane preparations derived from human epileptic hippocampi after transplantation into frog oocytes (Roseti et al. 2008).
Taken together, while the anticonvulsant effects of adenosine are well-established, we have now an increasing understanding of how adenosine receptors function during seizures and epilepsy, critical to develop adenosine-based therapeutic strategies for seizure control and epilepsy (Table 2).
Table 2.
Pharmacological approaches to modulate adenosine metabolism and signaling in epilepsy (selected examples).
| Receptor subtype / adenosine metabolism | Process | Epilepsy model | Strategy | Outcome | References |
|---|---|---|---|---|---|
| A 1 R | Status epilepticus | Intrahippocampal KA (50 nl of 20 mM solution) in mice (males and females) | A1R-KO | Lethal SE in A1R-KO mice. | (Fedele et al. 2006) |
| Status epilepticus | Theophylline-associated seizures (5, 10, and 25 mg/kg i.p) in rats | DPCPX (0.5 or 5 mg/kg i.p) | A1R antagonism led to increased seizure activity and duration. | (Fukuda et al. 2010) | |
| Epilepsy | i.p. pilocarpine (320 mg/kg) in rats (males) | A1R agonist RPia (25 μg/kg/day i.p); A1R antagonist DPCPX (50 μg/kg/day i.p) administered for 10 consecutive days, starting from 4 months post-SE | A1R agonist reduced seizure rate and hippocampal excitability; while the opposite effect was observed using A1R antagonists. | (Amorim et al. 2016) | |
| Brain injury | Controlled cortical impact (CCI) in mice (males) | A1R-KO | Lethal SE in A1R-KO mice. | (Kochanek et al. 2006) | |
| Pharmacoresistant epilepsy | Intrahippocampal injection of KA (200 ng in 50 nl) in mice (males) | A1R agonist CCPA (1.5 or 3 mg/ kg i.p) | A1R agonism led to suppression of drug resistant seizures in mice. | (Gouder et al. 2003) | |
| Pharmacoresistant seizures | Human temporal neocortical slices; SLEs e induced by perfusing slices with ACSF containing 8 mM K+ and 50 μM bicuculline methiodide | A1R agonist SDZ WAG 994 (1 μM); A1R antagonist DPCPX (1 μM) | A1R agonist completely suppressed SLEs in 73% of slices including slices derived from pharmacoresistant patients; A1R antagonism prevented suppression of SLEs. | (Klaft et al. 2016) | |
| A 2A R | Epileptogenesis | i.p KA injections (10 mg/kg); Amygdala kindling (1-s train at 50 Hz with pulses of 1 ms and 500 μA) in rats (males) | A2AR antagonist, SCH58261 (0.05 mg/kg i.p) administered 30 min before KA injections | A2AR antagonism led to suppression of epileptogenesis in amygdala kindled rats; in i.p KA rats, A2AR antagonism prevented seizure-induced neurodegeneration in the hippocampus. | (Canas et al. 2018) |
| Epileptogenesis | PTZ kindling (40 – 90 mg/kg, i. p.) and pilocarpine i.p. (350 mg/ kg, i.p.) in mice (sex not specified) | A2AR –KO | A2AR-KO mice are partially resistant to limbic seizures; A2ARs are involved in excitatory neurotransmission and seizure aggravation. | (El Yacoubi et al. 2009) | |
| A2B and A3R | Epilepsy | Human hippocampal slices | A2BR antagonist (MRS1706, 10 nM) or A3R antagonist (MRS1334, 30 nM) | Both A2B and A3R antagonists altered the stability of GABA. Reduced GABA-induced rundown currents in membranes. | (Roseti et al. 2008) |
| Adenosine production (CD73) | Epilepsy | Human TLE and MTLE hippocampus | NT5E measurement through Western blot analysis | Increase in NT5E in tissue from patients with epilepsy | (Barros-Barbosa et al. 2016) |
| Adenosine metabolism (ADK) | Epileptogenesis | Intrahippocampal injection of KA (400 ng in 200 nl) in mice (males) | ADK inhibitor 5-ITU, (1.6 mg/ kg, b.i.d) i.p for 5 days from day 3 to day 8 after SE | 5-ITU used transiently after status epilepticus suppress epileptogenesis. | (Sandau et al. 2019) |
| Epilepsy | MES-induced seizures in rats (60-Hz current of 150 mA for 0.2 s via corneal electrodes) (males) | 5-iodotubercidin, 5-deoxy-5-iodotubercidin and 5-amino-5-deoxyadenosine | ADK inhibitors showed suppression of seizure activity induced by electroshock. | (Ugarkar et al. 2000) | |
| Epilepsy | Intrahippocampal KA (1 nmol) in transgenic mice (males) | ADK inhibitor, 5ITU (3.1 mg/kg) i.p; Adktm1– /– -Tg(UbiAdk) (ADK overexpressing mice) | ADK overexpressed mice showed spontaneous seizure activity and exacerbation of seizures induced by KA, which was successfully treated by ADK inhibitor 5-ITU. | (Fedele et al. 2005) | |
| Pharmacoresistant epilepsy | Intrahippocampal injection of KA (200 ng in 50 nl) in mice (males) | ADK inhibitor 5-ITU, (3.1 mg/ kg), i.p | ADK inhibitors suppressed seizure activity including pharmacoresistant seizures. | (Gouder et al. 2004) |
Abbreviations: ADK, Adenosine kinase; ACSF, Artificial cerebrospinal fluid; CCPA, 2-chlorN6cyclopentyladenosine; DPCPX, 8-Cyclopentyl-1,3-dipropylxanthine; GABA, Gama amino butyric acid; i.p., intraperitoneal; KA, Kainic acid; KO, knock out; MES, Maximal electric shock; MRS1334, 1,4-Dihydro-2-methyl-6-phenyl-4-(phe-nylethynyl)-3,5-pyridinedicarboxylic acid 3-ethyl-5-[(3-nitrophenyl)methyl] ester; MRS1706, N-(4-Acetylphenyl)-2-[4-(2,3,6,7-tetrahydro-2,6-dioxo-1,3-dipropyl-1H-purin-8-yl)phenoxy]acetamide; MTLE, Mesial temporal lobe epilepsy; NT5E, Ecto-5’-nucleotidase; PTZ, Pentylenetetrazol; 5ITU, 5-indotubercidin; SE, Status epilepticus; SLE, Seizure-like events; TLE, Temporal lobe epilepsy.
2.4. The role of ATP P2 receptors in seizure modulation and epilepsy
Although the study of adenine and uracil nucleotide-activated P2 receptors in epilepsy is relatively new when compared to adenosine-activated P1 receptors, there has been substantial progress made over the past decade deciphering the role P2 receptors play, not only during seizure generation, but also during the development and maintenance of epilepsy (Engel et al. 2016; Rassendren and Audinat, 2016; Cieslak et al. 2017). Early evidence linking extracellular ATP to increased hyperexcitability stems from studies in a seizure-prone strain of mice, which presented increased extracellular ATP concentrations (Wieraszko and Seyfried, 1989) and from data showing motor seizures caused by microinjections of ATP into the prepiriform cortex (Knutsen and Murray, 1997). More recent evidence demonstrating a contribution of elevated extracellular ATP to brain hyperexcitability was provided by studies showing that direct injection of ATP, ADP or the ATP analog 2’, 3’-O-(4-benzoyl-benzoyl)ATP (BZATP) leads to high spiking on the electroencephalogram (EEG) or exacerbates seizure activity during intra-amygdala induced-status epilepticus (Engel et al. 2012; Sebastian-Serrano et al., 2016; Alves et al. 2017). In contrast to adenine nucleotides, however, treatment with the uridine nucleotide, UTP, reduced seizure severity during intra-amygdala induced status epilepticus in mice (Alves et al. 2017) and neuronal firing in Long Evans and Wistar Albino Glaxo/Rijswijk (WAG/Rij) rats, both strains modelling absence seizures (Kovacs et al. 2013).
2.4.1. Changes in P2 receptor expression
2.4.1.1. Acute seizures.
Among P2X receptors, changes in expression associated with seizures or a period of status epilepticus have been identified in P2X1R, P2X2R, P2X4R and P2X7R. These findings come from chemoconvulsant-induced status epilepticus models in mice. The P2X1R has been reported to be increased in the hippocampus 24 h following intraperitoneal KA in mice at the transcriptional level (Avignone et al. 2008). No changes in hippocampal P2X1R protein levels have, however, been found using the intra-amygdala KA mouse model (Engel et al. 2012). The P2X2R has been shown to be downregulated 24 h following status epilepticus in the intra-amygdala KA mouse model (Engel et al., 2012), while P2X4R expression was found to be increased in the hippocampus of mice 24 h following systemic administration of KA (Avignone et al. 2008; Ulmann et al. 2013), but not intra-amygdala KA- or pilocarpine-induced status epilepticus (Dona et al. 2009; Engel et al. 2012).
The P2XR which has been studied in most depth in association with seizures is the P2X7R. Under normal physiological conditions, P2X7R expression is mainly restricted to endothelial cells, microglia, oligodendrocytes and neural progenitor cells (Monif et al. 2009; Rozmer et al. 2017; Kaczmarek-Hajek et al. 2018). It is also suggested, however, that the P2X7R is expressed in neuronal presynaptic termini (Armstrong et al. 2002; Sperlagh et al. 2006). Following status epilepticus, P2X7R is consistently upregulated at both the transcriptional and expressional level. P2rx7 mRNA is elevated in the hippocampus following either intraperitoneal or intra-amygdala KA-induced status epilepticus in mice (Avignone et al. 2008; Engel et al. 2017). At the protein level, P2X7R is increased post-status epilepticus in the hippocampus and cortex of rats and mice subjected to pilocarpine- and KA-induced status epilepticus. While some studies using systemic KA-treated mice and pilocarpine-treated rats have shown that P2X7R is mainly up-regulated on microglia in both cortex and hippocampus shortly following status epilepticus (Rappold et al. 2006; Kim et al. 2009), thereby possibly contributing to microglia activation (Avignone et al. 2008), other studies have also suggested a neuronal upregulation post-intra-amygdala KA-induced status epilepticus (Dona et al. 2009; Engel et al. 2012). P2X7R expression was also found to be increased on neuronal progenitor cells following status epilepticus induced via systemic KA in mice (Rozmer et al. 2017), potentially impacting on status epilepticus-induced aberrant neurogenesis. Neuronal expression of the P2X7R has, however, recently been questioned using a P2X7R reporter mouse, where P2X7R is fused to the enhanced green fluorescent protein (EGFP) (Kaczmarek-Hajek et al. 2018). In this model P2X7-EGFP co-localized to microglia and oligodendrocytes post-intra-amygdala KA-induced status epilepticus in mice, however, no co-localization was observed for P2X7-EGFP with neurons or astrocytes (Morgan et al. 2020). Thus, while there is broad consent for P2X7R upregulation on microglia following status epilepticus, the question of whether the P2X7R increases on neurons is still a matter of debate. Progress has also been made in deciphering what drives P2X7R expression in the brain during seizures. Following intra-amygdala KA induced status epilepticus in mice, P2X7R transcription seems to be under the control of the specificity protein 1 (Sp1) (Engel et al. 2017) which is a member of the injury-activated transcription factor (IATF) family and highly expressed in the brain (Kiryu-Seo et al. 2008). Using the same intra-amygdala KA mouse model, P2X7R expression during status epilepticus also appears, however, to be controlled at the post-transcriptional level, involving the targeting of P2rx7 mRNA by microRNA-22, which is also under the control of Sp1 (Engel et al. 2017). Increased microRNA-22 expression under the control of Sp1 is highly sensitive to intracellular Ca2+ concentrations. Elevated Ca2+ concentrations associated with status epilepticus block Sp1 binding to the microRNA-22 promotor, disinhibiting the suppression of P2rx7 mRNA translation into protein (Engel et al. 2017). This suggests that, while ubiquitously expressed in all cell types, Sp1 is capable of modulating its activity to provide a stimulation and/or injury-specific transcriptional response, thereby possibly contributing to the cell type-specific expression profile of down-stream targets such as the P2X7R. Of note, Sp1 also contributes to the regulation of the Adk gene, providing a possible mechanisms to link P2X7R signaling to adenosine metabolism (Kiese et al. 2016).
One of the first studies analyzing changes in the expression of the P2YR family following status epilepticus was carried out using the intraperitoneal KA mouse model. Here the authors observed increased transcription of P2ry6, P2ry12 and P2ry13 in the hippocampus (Avignone et al. 2008). Rozmer et al. (Rozmer et al. 2017) showed the P2Y1R, similar to the P2X7R, to be upregulated on neuronal progenitor cells following pilocarpine-induced status epilepticus in mice. Using the intra-amygdala KA and intraperitoneal pilocarpine mouse model of status epilepticus, Alves et al. carried out the first comprehensive study analyzing the expression of the entire P2YR family post-status epilepticus (Alves et al. 2017). Analyzing the hippocampus at different time-points following status epilepticus, the authors found that while mRNA levels of the uridine-sensitive P2Y2R, P2Y4R and P2Y6R were increased, transcription of the adenine-sensitive P2Y1R, P2Y12R and P2Y13R was downregulated. On the other hand, at the protein level, P2YRs coupled to Gq showed an increase in their expression (P2Y1R, P2Y2R, P2Y4R and P2Y6R), while P2YRs coupled to Gi were either down-regulated (P2Y12R) or unchanged (Alves et al. 2017). These surprising results suggest that changes in P2YR transcription and expression in the hippocampus post-status epilepticus are dependent on P2YR agonists (e.g., uridine-sensitive receptors show increased transcription) and downstream signaling (e.g., P2Y receptors coupled to Gq show increased expression). In contrast to hippocampal P2YR expression, P2YR expression in the cortex was mainly upregulated following intra-amygdala KA in mice, with P2Y1R and P2Y4R showing the strongest increase (Alves et al., 2019b).
2.4.1.2. Epilepsy.
Unlike seizures or status epilepticus, clinical evidence is available for changes in P2 receptor expression during epilepsy, with tissue available from surgical resection from patients with drug resistant TLE and focal cortical dysplasia. Whereas most widely reported changes in receptor expression have been found for the P2X7R (Beamer et al. 2017), other P2XRs analyzed during epilepsy include the P2X2R, P2X3R and P2X4R. While both P2X2R and P2X4R expression was found to be decreased in the hippocampus of seizure-sensitive gerbils (Kang et al. 2003), P2X4R expression was also found to be decreased in the hippocampus of pilocarpine-treated epileptic rats (Dona et al. 2009). In contrast, P2X3R, has been found increased in the cortex of patients with TLE and in the hippocampus and cortex of epileptic pilocarpine-treated rats being mainly localized to the cell bodies and dendrites of neurons (Zhou et al. 2016). P2X7R is increased in the hippocampus and cortex during epilepsy in both experimental rodent models of TLE and patients with TLE (Dona et al. 2009; Jimenez-Pacheco et al. 2013; Barros-Barbosa et al., 2016b; Jimenez-Pacheco et al. 2016). Early studies analyzing changes in P2X7R expression during epilepsy using a pilocarpine rat model demonstrated an increase in the hippocampus, with strong immunoreactivity associated with microglia, mossy fibers and glutamatergic nerve terminals (Vianna et al. 2002; Dona et al. 2009). Similarly to status epilepticus, more recent studies seem to confirm a mainly microglial and neuronal P2X7R induction during epilepsy in the hippocampus and cortex using the intra-amygdala KA mouse model (Jimenez-Pacheco et al. 2013; Jimenez-Pacheco et al. 2016). These data were, however, challenged by a study using P2X7-EGFP reporter mice subjected to intra-amygdala KA showing P2X7R expression increases to be restricted to microglia and oligodendrocytes (Morgan et al. 2020). Increases in P2X7R expression were absent on astrocytes during experimental epilepsy (Jimenez-Pacheco et al. 2016; Morgan et al. 2020).
Much less is known regarding the expression of P2YRs during epilepsy. Using the intra-amygdala KA mouse model, Alves et al. (Alves et al. 2017) showed that, in contrast to status epilepticus, hippocampal P2YR transcription and expression is either increased (mRNA: P2ry1, P2ry2 and P2ry6; protein: P2Y1R, P2Y2R and P2Y12R) or remains unchanged during epilepsy, suggesting hippocampal P2YR upregulation to be the predominant response during experimental epilepsy. P2YR expression is also increased in resected tissue from patients with TLE with the only exception being the P2Y13R, which is downregulated (Alves et al. 2017). Further proof of P2YR upregulation in the hippocampus during epilepsy is provided in an investigation carried out by Sukigara et al. where the authors found increased levels of P2Y1R, P2Y2R and P2Y4R in hippocampal tissue from patients with intractable epilepsy associated with focal cortical dysplasia, with the main increase in astrocytes (Sukigara et al. 2014).
2.4.2. Targeting P2 receptors during seizures and epilepsy
2.4.2.1. Targeting P2XRs during acute seizures.
As mentioned in the previous paragraph, among the P2XRs, the P2X7R subtype has attracted most attention as a potential drug target in epilepsy (Beamer et al. 2017). Among the P2XR family, P2X7R has some unique structural and functional characteristics, making this receptor a particularly attractive therapeutic target. It has a relatively low affinity for ATP (EC50 ≥ 100 μM, activation threshold: 0.3–0.5 mM), suggesting P2X7R activation occurs mainly under pathological conditions of high ATP release. It also has slow desensitization dynamics, the ability to permeabilize the cell membrane to molecules up to 900 Daltons in size and is a key driver of inflammation via activation of the NLRP3 inflammasome (Beamer et al. 2016; Adinolfi et al. 2018). For a detailed review on P2X7R function in the brain see (Jimenez-Mateos et al. 2019; Kopp et al. 2019). The first data suggesting anticonvulsive properties of P2X7R antagonists during status epilepticus was provided using the intra-amygdala KA mouse model. Here, mice treated before and shortly following intra-amygdala KA with the P2X7R antagonists A-438079 and Brilliant Blue G (BBG) (pre-and post-treatment regime) experienced less severe seizures (EEG and behavioral) and a reduction in neurodegeneration (Engel et al. 2012; Jimenez-Pacheco et al. 2013). In the same study, seizure severity was also reduced in P2X7R KO mice (Solle et al. 2001) and by P2X7R antibodies delivered into the lateral ventricle (Engel et al., 2012). Critically, P2X7R antagonists, when given in combination with lorazepam at a later time-point during status epilepticus, when sensitivity to lorazepam was reduced, efficiently stopped seizures, suggesting the potential of P2X7R antagonists as adjunctive treatment for pharmaco-resistant status epilepticus (Engel et al. 2012). Treatment using three different P2X7R antagonists (BBG, A-438079, A-740003) reduced seizures also during coriaria lactone-induced status epilepticus in rats (Huang et al. 2017).
Other studies, however, only observed limited or no effect via P2X7R antagonism. Nieczym et al. (Nieoczym et al. 2017) only found a weak anticonvulsant potential via the P2X7R antagonist BBG in the intravenous PTZ seizure threshold, maximal electroshock seizure threshold and 6 Hz psychomotor seizure threshold tests. In line with this, Fischer et al. observed no anticonvulsant effects provided by P2X7R antagonism using four different antagonists (JNJ-47965567, AFC-5128, BBG, transhinone IIA sulfonate) in the maximal electroshock seizure threshold test and the PTZ seizure threshold test in mice, although when given in combination with carbamazepine, P2X7R antagonists JNJ-47965567 and AFC-5128 increased the threshold in the maximal electroshock seizure test (Fischer et al. 2016). In a recent study, Dogan et al. (Dogan et al. 2020) using WAG/Rij rats, a model of absence seizures, observed no anticonvulsive effects when treating rats with the P2X7R antagonist A-438079.
In contrast to a pro-convulsive function of the P2X7R during seizures, Kim et al. found that P2X7R deficiency exacerbated seizure severity in the intraperitoneal pilocarpine mouse model (Kim and Kang, 2011). Moreover, P2X7R antagonism via oxidized ATP (OxATP) and BBG protected against astroglial cell death (Kim et al. 2009) and reduced infiltration of neutrophils into the frontoparietal cortex in the pilocarpine rat model (Kim et al. 2010). The same group further showed that treatment with the P2X7R antagonists A-438079, A-740003 and OxATP increased hippocampal cell death in the pilocarpine rat model of status epilepticus (Kim et al. 2011). P2X7R deficiency did, however, not alter seizure severity in the systemic KA mouse model or the picrotoxin mouse model (Kim and Kang, 2011).
Taken together, while the P2X7R seems to play a role during prolonged, damaging seizures (i.e., status epilepticus) with P2X7R antagonists altering seizure severity and seizure-induced pathology, effects of P2X7R antagonism seem to be minor or absent during acute non-damaging seizures (e.g., genetic model and electrical induced seizures). This suggests that P2X7R-based treatment is most likely more effective in reducing seizures once pathological processes have been triggered (e.g., increased inflammation during drug-refractory status epileptics) rather than to be used as prophylactic anticonvulsant therapy. The reasons behind this difference remains to be established. A likely explanation is, however, an increased availability of extracellular ATP during status epilepticus due to seizure-induced neurodegeneration and/or an increased inflammatory response. The mechanisms how the P2X7R alters seizure severity are still to be determined. It is, however, tempting to speculate that this is mediated, at least in part, by the suppression of inflammatory processes. The P2X7R is highly expressed in microglia under normal control conditions and following status epilepticus (Kaczmarek-Hajek et al. 2018; Morgan et al. 2020) and the P2X7R has been shown to drive microglial activation (Monif et al. 2009). Moreover, blocking of the P2X7R during status epilepticus leads to decreased release of the pro-convulsant cytokine IL-1β in the hippocampus (Vezzani and Baram, 2007; Engel et al. 2012). Although not shown to be increased on astrocytes in experimental models of epilepsy, the P2X7R can also activate astrocytes (Khan et al. 2019), whether directly or indirectly. Astrocytes can reduce the seizure threshold via various mechanism including dysregulation of the extracellular ionic balance, impaired neurotransmitter reuptake, release of pro-inflammatory cytokines and purines (e.g., ATP and adenosine) (Bedner et al. 2015; Robel et al. 2015; Illes et al., 2019a) or an increase in the expression of adenosine kinase (ADK) with the subsequent decreased concentration of adenosine in the extracellular space (Boison, 2008). The P2X7R may, however, also impact on other pathological processes during seizures including changes in neurotransmitter release [e.g., GABA and glutamate as shown by Barros-Barbosa et al. (Barros--Barbosa et al., 2016b)]. Importantly, we still do not know why P2X7R antagonists elicit different responses according to the experimental model used. There are multiple possible explanations (e.g., different models (KA vs. pilocarpine) and P2X7R antagonists used, time-point and dose, species, genetic background, cell-type recruited during seizures/epilepsy etc.). Identifying the reasons behind these differences will be crucial to advance therapies based on the P2X7R towards a clinical application.
The only other P2XR for which a functional role during acute seizures has been described is the P2X4R subtype. Interestingly, the P2X4R is the receptor with which the P2X7R shares most similarities among the P2XR family (Craigie et al. 2013). Using the intraperitoneal KA mouse model, Ulman et al. (Ulmann et al. 2013) found that mice deficient in the P2X4R, despite experiencing no changes in seizure severity during status epilepticus, are partially protected from seizure-induced neuronal cell death possibly via regulating the activation of microglia.
2.4.2.2. The role of P2XRs during epileptogenesis and epilepsy.
While a role for P2XRs during epilepsy has emerged much more recently, we now have substantial data not only regarding changes in their expression but, more importantly, data on the impact of P2XR-trargeting drugs on epileptogenesis and epilepsy. Similar to status epilepticus, this data is mostly restricted to the P2X7R subtype with one study investigating also the role of the P2X3R.
The first evidence for antiepileptogenic effects provided by P2X7R antagonists were reported in a study using the PTZ kindling model in rats, a well-established model of epileptogenesis (Dhir, 2012). Using this model, Soni et al. (Soni et al. 2015) showed that the P2X7R antagonist BBG decreased the mean kindling score and restored cognitive deficits and motor coordination. These findings were confirmed in a more recent study in the same model using the P2X7R antagonists JNJ-47965567, AFC-5128 and, with more modest effects, BBG (Fischer et al. 2016). Further evidence for a pro-epileptogenic role for P2X7R was presented in a study in mice using inhibitors of a P2X7R-suppressing microRNA (microRNA-22). Intra-amygdala KA-injected mice treated with microRNA-22 inhibitors presented increased hippocampal expression of the P2X7R and developed a more severe epileptic phenotype accompanied with increased cytokine release and astrogliosis (Jimenez-Mateos et al. 2015). Using the pilocarpine rat model of status epilepticus, Amorim et al. (Amorim et al. 2017) showed that injection of P2X7R-targeting siRNA 6 h post-status epilepticus not only delayed the emergence of the first seizure, but also reduced the frequency and severity of seizures. In contrast to these findings, treatment with the P2X7R antagonists AZ10606120 and BBG post-pilocarpine-induced status epilepticus in mice resulted in the development of a more severe epileptic phenotype (Rozmer et al. 2017). Evidence for the efficacy of P2X7R antagonists in altering fully established epilepsy is presented by two studies both published in 2016 (Amhaoul et al. 2016; Jimenez-Pacheco et al. 2016). In the first study, Amhaoul et al. (Amhaoul, Ali et al. 2016) used the multiple low-dose i.p. KA model in rats. Epileptic rats treated for one week with the P2X7R antagonist JNJ-47965567, three months following KA-induced status epilepticus, experienced the same number of seizures, however, showed a significant reduction in seizure severity. In the second study, Jimenez-Pacheco et al. (Jimenez-Pacheco et al. 2016) used the intra-amygdala KA mouse model. Mice were treated with the same P2X7R antagonist JNJ-47965567 starting treatment 10-days post-status epilepticus for five days. In contrast to the previous study, P2X7R antagonism reduced the total number of seizures during treatment. Remarkably, this effect persisted during a one-week drug-washout period suggesting disease-modifying potential. Thus, in contrast to acute seizures, the P2X7R seems to play a more prominent role once pathological processes have been initiated. At this stage, inflammatory processes are more likely to play an important role, potentially resulting in increased P2X7R expression and function. In line with P2X7R antagonism suppressing epileptic seizures via blocking inflammation, epileptic mice treated with a P2X7R antagonist (JNJ-47965567) showed a strong reduction in both astrogliosis and microgliosis (Jimenez-Pacheco et al. 2016). While suppressing inflammatory pathways is the most likely explanation, it is, however, important to keep in mind, as mentioned before, that the P2X7R is involved in a myriad of different pathological conditions including BBB disruption, changes in neuro-transmitter levels, synaptic reorganization, neurogenesis to name just a few (Sperlagh and Illes, 2014).
To date, the only other P2XR studied beside the P2X7R during epilepsy is the P2X3R. Here, Xia et al. (Xia et al. 2018) showed by using a PTZ-induced kindling rat model, that the P2X3R antagonist NF110 reduced the mean kindling score and improved other pathological parameters such as memory deficits, motor activity, neuronal damage and hippocampal inflammation.
2.4.2.3. The metabotropic P2YR family as a therapeutic target in status epilepticus and epilepsy.
While among the P2Rs most focus has been put on the study of the fast-acting ionotropic P2XR family, data now also suggests a prominent role for the metabotropic P2YR family during seizures (Alves et al. 2018). These studies are, however, limited to models where status epilepticus was induced via KA or pilocarpine. The first demonstration of a role for P2YRs during status epilepticus stems from a study using the intraperitoneal KA-induced status epilepticus mouse model demonstrating that P2Y12R deficiency leads to a suppression of microglial morphological changes associated with status epilepticus and in an exacerbated seizure phenotype (Eyo et al. 2014; Avignone et al. 2015). Further support for a role for P2YRs in regulating microglia activation in epilepsy is presented in a recent study analyzing microglial motility in tissue slices from patients with TLE (Milior et al. 2020). Here, the authors show that while low doses of ADP application triggered microglial process extension, which were blocked via P2Y12R antagonists, high doses of ADP caused process retraction and membrane ruffling, blocked by the joint application of P2Y1R and P2Y13R antagonists. A recent study using a model of intracerebroventricular KA injection has now also shown the P2Y12R to be involved in post-status epilepticus-induced neurogenesis (Mo et al. 2019). Most studies, however, focused on the P2Y1R subtype. The first in vivo evidence demonstrating P2Y1R involvement during seizures was published by Simones et al. (Simoes et al. 2018) showing P2Y1R antagonism (MRS2179)-mediated neuroprotection following systemic KA and intrahippocampal quinolinic acid in rats. P2Y1R antagonism had no impact on seizures in their study. In contrast to these findings, Alves et al. showed in a study published one year later (Alves et al., 2019a), that P2Y1R-deficient mice presented a lower seizure threshold, experiencing more severe seizures during intra-amygdala KA-induced status epilepticus with the resulting increase in hippocampal neurodegeneration. In line with a genetic P2Y1R deletion, pre-treatment with the specific P2Y1R antagonists MRS2500 exacerbated seizure severity and cell death during status epilepticus and, conversely, mice pre-treated with the P2Y1R agonist MRS2365 had less severe seizures during status epilepticus and less seizure-induced neurodegeneration. Surprisingly, when the authors used a post-treatment regime, the same P2Y1R antagonist suppressed seizures and protected the brain from damage, whereas the P2Y1R agonist increased seizure severity, suggesting P2Y1R-based treatment to be highly depending on the time-point of intervention. The exact reason for this context-specific role of P2Y1R during seizures remains to be elucidated; however, cell-specific changes in the expression of the receptor may represent a possible explanation. In their study, the authors found P2Y1R to be mainly expressed on neurons during normal physiological conditions, however, once seizures were provoked and pathological changes initiated within the brain, P2Y1R expression increased on microglia. In line with P2Y1Rs driving inflammatory processes and thereby contributing to seizures during status epilepticus, treatment with the P2Y1R antagonist MRS2500 had no effect in mice pre-treated with the anti-inflammatory drug minocycline (Alves et al., 2019a). Using immunohistochemistry, the authors further showed P2Y1R to be undetectable on astrocytes. This is a surprising finding, as other studies have found the P2Y1R to be expressed and functional on astrocytes (Franke et al. 2004). The participation of P2Y1Rs in astrocyte-astrocyte signaling is well established and there is also evidence of the P2Y1R contributing to seizure generation and epilepsy via the mediation of astrocytic-Ca2+ oscillations. In line with this, Wellmann et al. showed that, in hippocampal slices from fully kindled rats, targeting the P2Y1R decreased the mean duration of astroglial Ca2+ oscillations by reducing the frequency of slow Ca2+ transients (Wellmann et al. 2018). Nikolic et al. meanwhile, demonstrate that the targeting of P2Y1Rs restored normal excitatory synaptic activity in the inflamed hippocampus via the blockade of TNF-α-induced Ca2+-dependent glutamate release from astrocytes (Nikolic et al. 2018).
The only study analyzing a functional role for P2YRs during epilepsy was carried out by Alves et al. (Alves et al., 2019a), using the intra-amygdala KA mouse model of status epilepticus. Here, treatment with the P2Y1R antagonist MRS2500 post-status epilepticus delayed the onset of epilepsy and treatment during epilepsy suppressed epileptic seizures. Seizure-suppressing effects, however, were transient and did not persist beyond termination of treatment.
In summary, we have now compelling evidence for a functional contribution not only for P1Rs but also for P2Rs (in particular regarding the P2X7R and P2Y1R subtype) (Table 3), to seizure generation, seizureinduced brain damage and development of epilepsy (Fig. 2).
Table 3.
Impact of P2 receptor-targeting during acute seizures, epileptogenesis and epilepsy (selected examples).
|
Receptor subtype |
Process |
Epilepsy model |
Strategy |
Outcome |
References |
| Ionotropic P2XRs | |||||
| P2X7R | Status epilepticus | i.p. KA (25 mg/kg), i.p. picrotoxin (5 mg/kg), i.p. pilocarpine (150, 175, 200, 225, or 250 mg/kg) in mice (males) | i.c.v. OxATP (5 mM), A-438079 (10 μM), A740003 (10 μM) delivered over 1 week via osmotic mini-pump before seizure induction; P2X7R KO mice | Increased seizure susceptibility via P2X7R antagonism; P2X7R antagonisms did not affect seizures in KA and picrotoxin model. | (Kim and Kang, 2011) |
| Status epilepticus | i.p. pilocarpine (380 mg/kg) in rats (males) | i.c.v. BZATP (5 mM), OxATP (5 mM), A-740003 (5 mM) and A-438079 (10 μM) via osmotic mini-pump | Reduced neurodegeneration via BzATP; increased neurodegeneration, reduced astroglial death and reduced infiltration of neutrophils mediated via P2X7R antagonism. | (Kim et al. 2009), (Kim et al. 2011), (Kim et al. 2010) | |
| Status epilepticus | i.a. KA (3 μg/2 μl) in mice (males) | P2X7R agonists: i.c.v. BZATP (0.1 nmol); P2X7R antagonists: i.c.v. A438079 (1.75 nmol), i.c.v. BBG (1 pmol); i.c.v. P2X7R antibody (APR-008, 0.7 mg/mL); P2X7R KO mice | P2X7R agonist-increased seizure severity; P2X7R antagonists / P2X7R-targeting antibodies / P2X7R KO leads to seizure reduction and neuroprotection in hippocampus and cortex. | (Engel et al. 2012), ( Jimenez-Pacheco et al. 2013) | |
| Status epilepticus | i.m. coriaria lactone (40 mg/kg) in rats (males) | Pre-treatment with P2X7R antagonists BBG (1, 5, 10 μg; i.c. v.), A438079 (10 μM, ic.v.) and A740003 (10 μM, i.c.v.) | P2X7R antagonism reduced inflammation, neuronal damage, astrogliosis and microgliosis, seizures and improved cognitive function. | (Huang et al. 2017) | |
| Acute seizures (focal, generalized and generalized tonic-clonic) | Timed i.v. PTZ infusion test (1% PTZ 2 ml/min) in mice (males); MES-T; 6 Hz electroshock-induced seizures (0.2 ms square pulse at 6 Hz for 3 s) in mice (males) | i.p. BBG 150 mg/kg for i.v. PTZ and MES-T test and i.p. BBG 50 mg/kg for 6 Hz test | Reduced seizures during 6 Hz test (focal seizure) via BBG; no significant anticonvulsive effects of BBG in i.v. PTZ and MES-T test (generalized and generalized tonic-clonic seizures). | (Nieoczym et al. 2017) | |
| Acute seizures (absence seizures) | WAG/Rij rats (males) (inbred strain of rats with genetic absence epilepsy) | i.c.v. BZATP (50 μg and 100 μg); i.c.v. A-438079 (20 μg and 40 μg) | No effects of P2X7R agonists or antagonists on spike-wave discharges (SWDs) | (Dogan et al. 2020) | |
| Acute seizures / Epileptogenesis | MES-T (inusoidal pulses 4–14 mA, 50 Hz, 0.2 s duration) and s.c. PTZ-T (87 mg/kg) in mice (males); i.p. PTZ kindling (35 mg/ kg) in rats (males) for 25 days | JNJ-47965567 (15 or 30 mg/ kg),AFC-5128 (25 or 50 mg/ kg), BBG (50 mg/kg), transhinone (30 mg/kg), all drugs injected i.p. | No effects on acute seizures alone; reduced seizure severity in combination with carbamazepine; reduced kindling development and glial activation. | (Fischer et al. 2016) | |
| Epileptogenesis | i.p. PTZ kindling (30 mg/kg) every second day for 27 days in rats (sex not specified) | BBG (15 and 30 mg/kg, i.p.) 30 min before PTZ injection | Reduced seizure score and improved motor performance and cognitive deficits. | (Soni et al. 2015) | |
| Epileptogenesis | i.a. KA (3 μg/2 μl)-induced epilepsy in mice (males) | i.c.v. injection of antagomir-22 24 h before induction of SE | Increased P2X7R; increased seizure frequency during epilepsy, increased neuroinflammation. | (Jimenez-Mateos et al. 2015) | |
| Epileptogenesis | i.p. pilocarpine (370 mg/kg,i.p.) in rats (males) | P2X7R antagonists AZ10606120 (3 μg/2 μl, i.c.v.) post-SE / BBG (50 mg/kg, i.p.) 1 injection per day for 4 days post-SE | Neuroprotection mediated via P2X7R antagonism post-SE; P2X7R antagonisms increased seizure number and seizure severity during epilepsy. | (Rozmer et al. 2017) | |
| Epileptogenesis | i.p. pilocarpine (370 mg/kg) in rats (males) | P2X7R-targeting siRNA (i.c.v.) 6 h post-SE | P2X7R antagonisms mediated neuroprotection in hippocampus, reduced edema, reduced mortality following SE, delayed seizure onset and seizure numbers during chronic epilepsy. | (Amorim et al. 2017) | |
| Epilepsy | Multiple low-dose i.p. KA-induced epilepsy in rats (males) (total KA = 22.2 ± 2.02 mg/kg) | JNJ-47965567 during 1 week via osmotic mini-pump (0.6 g/ kg/2 ml) | Decreased seizure severity without changes in total number of seizures, no change in inflammation. | (Amhaoul et al. 2016) | |
| Epilepsy | i.a. KA (3 μg/2 μl)-induced-epilepsy in mice (males) | JNJ-47965567 (30 mg/kg, i.p.) twice daily for 5 days during epilepsy | Reduced seizure frequency during treatment and during washout-period; decreased inflammation (astrogliosis and microgliosis). | (Jimenez-Pacheco et al. 2016) | |
| P2X3R | Epileptogenesis | i.p. PTZ (30 mg/kg) administered on alternate days for a maximum of up to 35 days to rats (males) | P2X3R antagonist i.c.v. delivery of P2X3 antagonist NF110 in 3 different doses (20 nM, 40 nM, and 60 nM) from day 1 until the end of the study (day 42) | P2X3R antagonisms improved impaired behavior, learning, memory, locomotion, motor activity, discrimination ability, neuronal damage, hippocampal inflammation, oxidative stress, and mitochondrial dysfunction | (Xia et al. 2018) |
| P2X4R | Status epilepticus | i.p. KA (8–22 mg/kg) in mice (sex not specified) | P2X4R KO mice | Impaired microglial function; neuroprotection in hippocampus. | (Ulmann et al. 2013) |
| Metabotropic P2Y receptors | |||||
| P2Y 1 R | Status epilepticus | i.p. KA (10 mg/kg) in rats (males); intrahippocampal injection of quinolinic acid (1 μl at the rate of 0.2 μL/min) in rats (males) | One single injection of MRS2500 (nmol, i.c.v.) | P2Y1R antagonist-mediated neuroprotection; no impact on seizure severity. | (Simoes et al. 2018) |
| Status epilepticus / Epileptogenesis / Epilepsy | i.a. KA (3 μg/2 μl) in mice; i.p. pilocarpine (340 mg/kg) in mice (males) | P2Y1R antagonist MRS2500 (1 nmol, i.c.v.) before and during SE, post-SE and during chronic epilepsy once daily for 5 days, P2Y1R KO mice; P2Y1R agonist MRS2365 (0.3 and 1 nmol, i.c. v.) before and during SE | P2Y1R KO mice: Increase in seizure severity during SE and neurodegeneration. Pre-treatment: P2Y1R agonist reduces seizure severity during SE and protects brain from damage; P2Y1R antagonism increases seizure severity and brain damage during SE. Treatment during SE: P2Y1R agonist increases seizure severity during SE; P2Y1R antagonism decreases seizure severity and brain damage (hippocampus and cortex). P2Y1R antagonism post-SE: Delay in epilepsy development. P2Y1R antagonism during epilepsy: Suppression of epileptic seizures. | (Alves et al. 2019); ( Alves et al. 2019) | |
| P2Y 12 R | Status epilepticus | i.p. KA (18–22 mg/kg) or i.c.v. KA (0.12–0.18 μg)and KA in mice (males and females) | P2Y12R KO mice Increased seizure phenotype; reduced hippocampal microglial processes. | (Eyo et al. 2014) | |
| Broad-spectrum targeting | |||||
| Status epilepticus | i.a. KA (3 μg/2 μl) in mice (males) | ADP and UTP (P2Y agonists) (9 nmol i.c.v.) | ADP exacerbates seizure severity; UTP reduces seizure severity and neuronal death. | (Alves, Gomez-Villafuertes et al. 2017) | |
| Epilepsy | Amygdala kindling in rats (males) (1 s, monophasic square-wave pulses) | Reactive Blue (P2YR antagonists, 20 μg) and PPADS (P2 antagonist, 10, 20, 30 μg) once daily i.c.v. | Reduced seizure severity. | (Sun et al. 2018) | |
Abbreviations: ADP, Adenosine diphosphate; BBG, brilliant blue G; BzATP, 2’(3’)-O-(4-Benzoylbenzoyl)adenosine-5’-triphosphate; i.a., intraamygdala; i.m. intra-muscular; i.p., intraperitoneal; i.c.v., intracerebroventricular; i.v., intravenous; KA, Kainic acid; KO, Knock out; MES-T, Maximal electroshock seizure threshold test; PPADS, Pyridoxalphosphate-6-azophenyl-2’,4’-disulfonic; PTZ-T, Pentylenetetrazol seizure threshold test; SE, Status epilepticus; UDP, Uridine diphosphate; UTP, Uridine triphosphate.
Fig. 2. Targeting ATP and adenosine signaling as potential treatment for epilepsy.

Epileptogenesis, the development of epilepsy, can be triggered via a precipitating injury to the brain (e.g., traumatic brain injury (TBI), status epilepticus) causing numerous pathological changes (e.g., inflammation, cell death, epigenetic changes) which eventually lead to the occurrence of spontaneous, epileptic seizures. Increasing evidence has demonstrated that ATP and adenosine-dependent signaling plays a key role during seizure generation and the development and maintenance of epilepsy. Targeting specific components of the purinergic signaling cascade can attenuate the precipitating injury, modify the epileptogenic process as such, and finally suppress seizures in epilepsy.
3. Purinergic signaling and comorbidities of epilepsy
Targeting of purinergic signaling has been suggested as possible treatment avenue for numerous pathological CNS diseases including the most common comorbidities associated with epilepsy [e.g., depression, schizophrenia (Burnstock, 2008; Illes et al., 2019b; Miras-Portugal et al. 2019)]. According to Feinstein et al. (Feinstein, 1970) comorbidities are defined as “any distinct additional entity that has existed or may occur during the clinical course of a patient who has the index disease (i.e. epilepsy) under study.” When referring to epilepsy in terms of comorbidities it is, however, important to keep in mind that epilepsy, rather than constituting a uniform entity, represents a highly heterogeneous condition with differences in etiology, clinical manifestations and treatment responses. Epilepsy, therefore, may be better referred to as a spectrum of disorders featuring increased network excitability resulting in the occurrence of epileptic seizures (Sirven, 2015). It therefore comes to no surprise that epilepsy has a particularly high burden of comorbidities with some conditions up to eight times more common in patients with epilepsy, compared to the general population (Lin et al. 2012). Psychiatric disorders (e.g., mood, anxiety, and psychotic disorders) are one of the most common comorbidities reported in epilepsy and are estimated to occur in 25 %–50 % of patients (LaFrance et al. 2008) and are particularly high in patients with TLE and drug-refractory epilepsy (Bragatti et al. 2010; Patel et al. 2017; Jansen et al. 2019). Other frequently diagnosed comorbidities in epilepsy include migraine, heart diseases, stroke, respiratory diseases (e.g., asthma), allergies and an increased mortality (Keezer et al. 2016). Demonstrating the importance of treating both primary condition (i.e., epilepsy) and associated comorbidities, comorbidities may be linked to poor seizure control (e.g., migraine, psychiatric disorder) or a reduction in the quality of life (e.g., depression) (Velioglu et al. 2005; Taylor et al. 2011). Comorbidities in epilepsy may be caused by direct effects of seizures, adverse effects of ASDs and other treatments or by common pathogenic mechanisms (Keezer et al. 2016), with the latter suggesting that drugs acting on shared pathological processes (e.g., inflammation) may provide the opportunity to target both primary disease and associated comorbidities. On the other hand and further supporting shared pathological pathway activation across different brain diseases, increased brain excitability and seizures are a common comorbidity of numerous other brain diseases including neurodegenerative [e.g., Alzheimer’s disease (Vossel et al. 2016; Lam et al. 2017), Huntington’s disease (Rodrigues and Wild, 2016)] and psychiatric diseases [e.g. schizophrenia (Cascella et al. 2009)] where they contribute to disease progression (Vossel et al. 2017; Kanner et al. 2018). Treating patients with non-epileptic diseases with anti-seizure medication may therefore not only reduce hyperexcitability in the brain but also the primary disease pathology. In line with this, ASDs are also in use for the treatment of various non-epileptic CNS conditions including psychiatric disorders (Bialer, 2012).
As mentioned in previous sections, among the P2R family, the ionotropic P2X7R has attracted most attention as possible therapeutic target in diseases of the CNS (Sperlagh and Illes, 2014). An emerging concept is that increased hyperexcitability and network changes are universal patho-mechanisms in numerous chronic brain diseases, caused and maintained by sustained glial activation (Kanner, 2012; Devinsky et al. 2013; Chitnis and Weiner, 2017; Bauer and Teixeira, 2019). The P2X7R has been described as a gatekeeper of inflammation driving the activation of microglia (Monif et al. 2009) whilst also regulating the release of neurotransmitters (e.g. glutamate, GABA) (Sperlagh et al. 2002). P2X7R may function, therefore, as a molecular link between glia-driven pro-inflammatory signaling and neuronal hyperexcitability (Henshall and Engel, 2015), thereby contributing to both primary disease pathology and associated comorbidities. In the same line, we recently proposed that a triad of synaptotoxicity, astrogliosis, and adenosine deficiency is a common pathological hallmark shared by epilepsy, Alzheimer’s disease, Parkinson’s disease and amyotrophic lateral sclerosis (Boison and Aronica, 2015). In line with this hypothesis, ADK overexpressing mice with decreased adenosinergic tone show impairment in multiple cognitive domains (Yee et al. 2007), whereas reconstitution of adenosine via the implantation of adenosine releasing cells into the hippocampus improved cognitive function (Shen et al. 2012).
Taken together, targeting of purinergic signaling during epilepsy may not only impact on seizures and the development of epilepsy, but also on associated comorbidities. Likewise, targeting the purinergic signaling system in other CNS diseases may also reduce brain hyperexcitability, the risk of seizures and their impact on disease progression. Despite data demonstrating a role for both ATP and adenosine-mediated signaling in different brain pathologies and diseases, a more systematic approach is needed to investigate how targeting of purinergic signaling impacts on primary disease and its associated comorbidities in the same experimental model.
4. Sudden unexpected death in epilepsy (SUDEP) and purinergic signaling
SUDEP remains a major concern for persons with epilepsy and their families. The incidence of SUDEP is estimated at 1–2.5 per 1000 patient years, however the underlying mechanisms are poorly understood. While increased adenosine suppresses neuronal activity as an innate mechanism to stop seizures (Lado and Moshe, 2008), it also suppresses cardiovascular and respiratory functions through increased activation of adenosine receptors in the brainstem (Evans et al. 1982; Barraco and Stefano, 1990; Herlenius et al. 1997). The seizure-induced adenosine surge as discussed above has been associated with the postictal state (Fisher and Schachter, 2000). The ‘adenosine hypothesis of SUDEP’ predicts that a seizure-induced adenosine surge originating from a seizure focus in combination with impaired metabolic clearance of adenosine from the brainstem can trigger lethal suppression of respiratory and/or cardiac functions. There is strong experimental and clinical evidence that the seizure-induced adenosine-surge (even when it originates from one focal area) affects the entire brain, including the brainstem, resulting in a brain-wide ‘post-ictal brain shutdown’ (During and Spencer, 1992; Fedele et al. 2006; Hirsch, 2010; Hesdorffer and Tomson, 2013). If excessive adenosine is a necessary trigger for SUDEP, then AR antagonists such as caffeine, or agents that enhance metabolic clearance of adenosine should prevent SUDEP. Indeed, an acute dose of caffeine was shown to extend life in a lethal seizure model (Shen et al. 2010) and prevent lethal apnea after TBI (Lusardi et al. 2012). These findings suggest that a commonly used psychoactive drug – caffeine – warrants closer attention and scrutiny within the context of epilepsy and SUDEP.
Despite the widespread use of caffeine and related methylxanthines in persons with epilepsy, dietary factors have received little attention in the study of SUDEP. In a study performed at Cleveland Clinic, only 20 % of 301 participants (all candidates for epilepsy surgery) did not consume caffeine, whereas 31 % drank up to 24 ounces of coffee per day, 45 % drank between 24 and 96 ounces, and 3% drank 100 ounces or more per day (Source: Epilepsy.com). It is thus fair to predict that, like the general population, a large proportion of adult persons with epilepsy regularly consume caffeine and related methylxanthines. While the acute use of methylxanthines may increase seizure risk (Boison, 2011), the same stimulants may also reduce risk for SUDEP. In contrast, chronic caffeine use may increase the brain’s vulnerability to SUDEP via upregulation of ARs, which renders the brain more susceptible to the effects of endogenous adenosine, a phenomenon termed ‘effect inversion’ (Jacobson et al. 1996; Fredholm, 1997). The widespread use of caffeine in persons with epilepsy warrants a detailed risk/benefit evaluation of chronic caffeination for SUDEP.
Although the seizure-induced adenosine-surge limits the spatial and temporal extent of a seizure (Fedele et al. 2006; Li et al. 2012), the resulting excess in adenosine needs to be metabolized effectively to avoid excessive postictal depression. As discussed above in the adult brain, the metabolic clearance of adenosine is largely under the control of ADK (Boison et al. 2010; Boison, 2012, 2013). We and others have demonstrated a tight spatial association of overexpressed ADK with epileptogenic brain activity (e.g., in the temporal lobe) (Aronica et al. 2011; Li, Lytle et al. 2012; Aronica et al. 2013). Importantly, over-expression of ADK is restricted to seizure generating brain areas, whereas lower ADK levels outside those areas maintain a protective adenosine tonus, thereby preventing the spread of seizures into adjacent or remote brain areas (Fedele et al. 2006; Li et al. 2012). Thus, reduced ADK expression in the brainstem would prevent seizure spread into the brainstem, but would, at the same time, increase SUDEP risk. Three lines of evidence support a critical role for adenosine and defects in its metabolic clearance for the development of lethal apnea: (i) A combination of a seizure with pharmacological disruption of metabolic adenosine clearance triggered death in normal mice (Shen et al. 2010). Conversely, a single acute dose of caffeine attenuated this outcome. (ii) Like seizures, severe TBI is associated with a surge in adenosine, and high CSF levels of adenosine correlated with lethal outcomes in human subjects (Clark et al. 1997). On the other hand, an acute bolus of caffeine given immediately after severe TBI in rats prevented apnea and mortality (Lusardi et al. 2012). (iii) SUDEP may share a similar etiology with sudden infant death syndrome (SIDS) (Hirsch, 2010; Tao et al. 2010), a condition that has been linked to global ADK deficiency in neonates (Boison et al. 2002). Deficiencies in the metabolic clearance of adenosine, therefore, constitute a candidate mechanism to trigger SUDEP.
5. Extracellular purine metabolism
5.1. Ectoenzymes
Changes in extracellular purine concentrations are not only dependent on the rate of release from cellular sources, but also on the rate of cellular reuptake (discussed in section 5.3) and of extracellular metabolism. With ATP and adenosine often acting as ligands for receptors mediating mutually antagonistic signalling pathways (Yegutkin et al. 2006), the rate at which ATP is broken down into adenosine in the extracellular space has a double impact: a decrease in the efficiency of ATP breakdown will lead, not only to excessive activation of P2 receptors, but also a reduced activity of P1-activated pathways – the ratio of extracellular purines is of critical importance and largely mediated by the activity of ectoenzymes (Zimmermann, 2000). ATP is metabolized to adenosine, often through a cascade of enzymes, with intermediate purine products, such as ADP and AMP also functioning as ligands for certain P2 receptors (discussed in section 2.2). ATP is a substrate for four principal families of ectoenzymes: the ectonucleoside triphosphate diphosphohydrolase family (E-NTPDases), the ectonucleotide pyrophosphatase/phosphodiesterase family (E-NNP), and the alkaline phosphatases (ALC) (Yegutkin et al. 2006).
Of the E-NTPDases, E-NTPDase 1 (CD39), 2, 3, and 8 are expressed on the extracellular cell surface, with CD39 both the most highly expressed and most extensively characterized (Robson et al., 2006). All the above E-NTPDases metabolize adenine and to a lesser extent, uridine nucleotides, with varying affinities. While CD39 has approximately equal affinity for ATP and ADP, E-NTPDase 2 is almost completely selective for ATP. E-NTPDase 3 and 8, meanwhile, have a higher affinity for ATP than ADP, but are less selective than E-NTPDase 2 (Robson, Sevigny et al. 2006). The E-NTPDase family all catalyse the degradation of ATP or ADP into AMP (or UTP or UDP to UMP) (Schetinger et al. 2007). Similarly, members of the E-NNP family cleave ATP to AMP (Masse et al. 2010). As well as acting as a ligand at certain P2 receptors, AMP forms the substrate for the ecto-5’nucleotidase (CD73)-dependent catalysis of adenosine (Strater, 2006). ALC enzymes, meanwhile, provide a rather non-selective cleavage route for the conversion of ATP into adenosine (Huizinga et al. 2012). The sequential action of CD39 and CD73 however is the major extracellular pathway responsible for the cleavage of ATP into adenosine (Cunha et al. 1992; Zimmermann, 2000; Koszalka et al. 2004; Fredholm et al., 2011; Augusto et al. 2013; Chu et al. 2014; Borea et al. 2018; Boison and Yegutkin, 2019).
While the concentration and activity of ectonucleotidases plays a role in determining the ratio of extracellular ATP to its metabolites in the extracellular space, with adenosine also released directly from the intracellular compartment, the extent to which the extracellular pool of ATP contributes to adenosine is not fully clear. While a study by Langer et al. (Langer et al. 2008) demonstrated that dephosphorylation of AMP to adenosine is largely restricted to the striatum and olfactory bulb, other studies have demonstrated CD73-dependent ATP metabolism to adenosine (Klyuch et al. 2012) in the cerebellum (Wall et al. 2007) and hippocampus (Wall and Dale, 2013). Experiments carried out on astrocyte/neuron co-cultures from CD73 KO mice, or with the use of CD73 inhibitors suggest that this enzyme contributes significantly to extracellular adenosine pools (Chu et al. 2014). Interestingly, CD73 is inhibited by ATP and ADP, allowing for the spatial and temporal separation of ATP release sites and sites of adenosine production (Dale, 1998, 2002; Masse and Dale, 2012).
Reduced activity of ectonucleotidases in genetic mouse models lowers the seizure threshold (Lanser et al. 2017), while following experimental status epilepticus, a substantial increase in CD73 in the hippocampus was observed in response to systemic pilocarpine or KA in –rats (Bonan et al. 2000). These results are consistent with human studies, in which CD73 was elevated in the hippocampus of TLE patients (Lie et al. 1999; Barros-Barbosa et al., 2016a). Further, genetic variations in CD73 are associated with greater risk of epilepsy development following TBI (Diamond et al. 2015). Theoretically, increasing the activity of CD73 might be a potential anti-epileptic treatment strategy (Schetinger et al. 2007; Sebastian-Serrano et al. 2015), however, pharmacological activators are challenging to design leaving gene therapy to overexpress CD73 as a potential option. Further, recent data from proximity ligation assays demonstrates a physical association between CD73 and excitatory A2ARs, implying functional coupling (Barros-Barbosa et al., 2016a). This suggests that activation of CD73 may have an excitatory effect.
5.2. Adenosine transporters
Extracellular adenosine is transported into the cell by the SLC29 family of equilibrative nucleoside transporters (ENTs) and concentrative nucleoside transporters (CNTs). The four ENT (1–4) isoforms are highly expressed throughout mammalian tissues with differences in their abundance (Table 4). ENT1 and ENT2 are highly expressed in the brain (Baldwin et al. 2004), whereas the pH-dependent transporters ENT3 and ENT4 are expressed in intracellular membranes and cardiac tissue, respectively (Baldwin et al. 2005; Barnes et al. 2006). In contrast to the wide expression of ENTs, the expression of CNTs is more limited (Jennings et al. 2001). Depending on the adenosine concentration, the four ENT (1–4) isoforms play a dynamic role in intra- and extracellular adenosine homeostasis, transporting nucleosides into or out of the cell. Simply stated, if intracellular adenosine levels are compromised, there is an influx of adenosine from the extracellular space through ENTs and vice versa (Boison, 2013). For this reason, ENT1 blockers such as dipyridamole increase the availability of extracellular adenosine as mechanism of action for their anticonvulsant effects (Kong et al. 2004; Carrier et al. 2006; Boison, 2008; Cieslak et al. 2017). The role of the three CNT (1–3) isoforms is to regulate the transport of adenosine using sodium ion gradients as a source of energy (Borea et al. 2018). Among the three nucleoside transporters, CNT2 is the major transporter of adenosine. Interestingly CNT2 activity is controlled by A1Rs and therefore contributes to the regulation of A1R signaling through an internal feedback loop (Aymerich et al. 2005). While the implications of CNTs in epilepsy are not yet established, the termination of extracellular adenosine signaling requires the synergistic action of both ENT and CNT
Table 4.
Table summarizing substrate specificity, Kms and tissue distribution of ENTs.
| ENTs | Km values (Adenosine) | Substrate specificity | Tissue distribution | References |
|---|---|---|---|---|
| ENT1 | 40 μM | Adenosine, purine and pyrimidines nucleosides | Ubiquitous. Predominant in adrenal gland, kidneys, stomach, small intestine. Also present in colon and ovaries. | (Griffith and Jarvis, 1996; Kong et al. 2004; Inoue, 2017) |
| ENT2 | 100 μM | Adenosine, purine and pyrimidines nucleosides, nucleobases | Ubiquitous. Highest in skeletal muscles. Also abundant in brain, neural tissues, heart, placenta, GI tract | (Griffith and Jarvis, 1996; Kong et al. 2004) |
| ENT3 | 2 mM | Adenosine, purine and pyrimidines nucleosides | Intracellular membranes, cerebral cortex, ovary, adrenal gland, and testis. | (Hyde et al. 2001; Baldwin et al. 2005) |
| ENT4 | 0.78 mM | Adenosine, serotonin | Abundant in heart, especially vascular endothelial cells. | (Baldwin et al. 2004; Barnes et al. 2006) |
Abbreviations: ENT, Equilibrative nucleotide transporter; GI, Gastrointestinal.
5.3. Intracellular adenosine metabolism
Adenosine is well known for its inhibitory effects in the brain and a large body of evidence is available regarding its receptor-dependent endogenous anticonvulsant effects (Boison, 2016). Intracellularly, adenosine mainly originates from the hydrolysis of S-adenosyl-L-homocysteine (SAH) and is metabolized by either ADK or adenosine deaminase (ADA). Between those two enzymes, ADK, as a low-capacity, low-Km enzyme, is the key metabolic regulator of intracellular adenosine under basal conditions (Boison, 2010). In the adult brain, both isoforms of ADK (nuclear ADK-L and cytoplasmic ADK-S) are predominantly expressed in astrocytes (Studer et al., 2006; Boison, 2013). Due to the existence of the ubiquitous equilibrative transporters for adenosine, a reduction in intracellular ADK activity increases both intra- and extracellular adenosine levels and consequently activation of A1Rs. In the following, “ADK” applies to both isoforms if not detailed otherwise. ADK-L has recently been identified as an epigenetic regulator of DNA methylation, an important new role, discussed below, implicated in epileptogenesis, neurogenesis, cell proliferation, and cancer (Williams-Karnesky et al. 2013; Boison and Rho, 2019; Gebril et al., 2020). In line with a role in the regulation of cell proliferation and plasticity, ADK-L is widely expressed in developing neurons (Studer et al., 2006) and its expression in adulthood is only maintained in the nuclei of dentate granule cells (Gebril et al., 2020) and granular neurons and Purkinje cells of the cerebellum (unpublished data). Cytoplasmic ADK-S, meanwhile, regulates the tissue tone of adenosine and AR activation (Boison, 2013). The two isoforms not only vary in their function, but also in their levels of expression at different developmental stages. During early postnatal brain development, under physiological conditions, there is a developmental shift in hippocampal ADK gene expression from predominantly neuronal at P4 to predominantly astrocytic at P14, as well as a shift in specific isoform expression where ADK-L is down-regulated in neurons and ADK-S upregulated in astrocytes during the same time window. While the exact mechanisms involved in these changes in transcription are unclear, it is possible that, as previously described for the P2X7R (Engel et al. 2017), these changes are also regulated by the transcription factor SP1, because of its binding to the ADK-L promoter in neurons at P4 (Kiese et al. 2016).
5.4. Adenosine receptor-independent, epigenetic antiepileptogenic effects of adenosine
Seizures can induce epigenetic alterations and thereby aggravate the epileptogenic condition (Kobow and Blumcke, 2011). Specifically, increased activity of DNA methylating enzymes and hypermethylated DNA has been associated with the development of human and experimental epilepsy (Kobow et al. 2009; Zhu et al. 2012; Kobow et al. 2013; Williams-Karnesky et al. 2013; Miller-Delaney et al. 2015). Interference with DNA methylation therefore offers novel opportunities for epilepsy prevention.
We recently discovered a novel epigenetic function of adenosine in epilepsy, where adenosine assumes a key role as regulator of DNA methylation (Williams-Karnesky et al. 2013). DNA methylation requires the donation of a methyl group from S-adenosylmethionine (SAM), catalyzed by DNA methyltransferases (DNMTs). The resulting product, S-adenosylhomocysteine (SAH) is converted into adenosine and homocysteine (HCY) by SAH hydrolase. This reaction will only proceed when adenosine and HCY are constantly removed (Kredich and Martin, 1977; Boison et al. 2002). If metabolic clearance of adenosine through ADK is impaired, SAH levels rise (Boison et al. 2002). SAH in turn inhibits DNMTs through product inhibition (James et al. 2002). Based on adenosine’s role as a product of DNA methylation, an increase in ADK and the resulting decrease in adenosine, as seen in epilepsy (Li et al. 2008; Masino et al. 2011), drives increased global DNA methylation in the brain. Adenosine augmentation reverses pathological DNA hypermethylation and thereby prevents epilepsy progression.
The antiepileptogenic potential of transient focal adenosine-delivery was directly tested in a rat model of systemic KA-induced progressive TLE (Williams-Karnesky et al. 2013). Young male rats received an acute dose of systemic KA to trigger acute convulsive status epilepticus. Once reaching a stable rate of 3–4 spontaneous recurrent seizures per week at 9 weeks post-KA, the rats were randomized and each received bilateral intraventricular adenosine-releasing silk-implants, silk-only implants, or a corresponding sham treatment. Adenosine implants were designed to transiently deliver a stable dose of 250 ng adenosine / ventricle / day restricted to 10 days of drug delivery (Szybala et al. 2009). 24 h post-surgery, continuous video monitoring was initiated, maintained for 4 weeks, and resumed after a 4 week hiatus for an additional 4 weeks. In the control groups seizures continued to increase both in number and severity. In contrast, adenosine-releasing implants almost completely suppressed seizures. Remarkably, reduced seizure activity was maintained far beyond expiration of adenosine-release from the polymer and at least 12 weeks post implantation. Even 12 weeks post implantation, seizure incidence was reduced by ≥ 70 %. In line with sustained seizure reduction, mossy fiber sprouting at 21 weeks post-KA was significantly attenuated in adenosine-treated rats. Importantly, transient delivery of adenosine restored normal DNA methylation long-term and epilepsy progression was halted.
6. Adenosine augmentation therapies
The anticonvulsant, antiepileptogenic and neuroprotective properties of adenosine are well-established (Boison, 2016). This section focuses on approaches to augment adenosine in the brain, and thereby increase the seizure threshold, prevent the emergence of epilepsy or arrest disease progression.
6.1. ADK inhibitors
The ADK hypothesis of epileptogenesis formulated in 2008 (Boison, 2008; Li et al. 2008) states that maladaptive overexpression of ADK is intrinsically linked to epileptogenesis and disease progression. It has been demonstrated that overexpression of ADK drives epileptogenesis through enhancing the rate of DNA-methylation (Williams-Karnesky et al. 2013). Because ADK progressively increases in line with epilepsy development and progression (Li et al., 2007a), and because epileptogenic activity originates from ADK-expressing brain areas (Li et al. 2008; Li et al. 2012), ADK is a biomarker to identify epileptogenic brain areas and to monitor epileptogenesis longitudinally. At the same time, ADK is a rational target for therapeutic intervention.
The first antiepileptogenic effect of ADK inhibitors was demonstrated by Sandau et al. in 2019 (Sandau et al. 2019) by showing that the transient use of the non-selective ADK inhibitor 5-ITU has potent antiepileptogenic effects in the intrahippocampal KA model of TLE. Three days after intrahippocampal KA-induced status epilepticus in mice, 5-ITU (1.6 mg/kg i.p. bid) or vehicle was injected for a restricted time span of 5 days. Six weeks after status epilepticus, hippocampal paroxysmal discharges (HPD) were quantified by intrahippocampal EEGs and brains were processed for histopathology. Most animals (16/17) from the KA/vehicle-injected control group developed a robust epilepsy phenotype by 6 weeks after the status epilepticus (23.5 ± 2.1 seizures / hour). In marked contrast, 59 % of all KA/5-ITU treated mice (10/17) were characterized with >95 % reduction of seizure rate and time spent in seizure. Importantly, 3 mice were completely seizure free. Histopathological analysis demonstrated characteristic granule cell dispersion in KA/vehicle-injected control animals that was prevented in KA/5-ITU-protected animals. These data demonstrate a potent anti-epileptogenic effect of the non-selective ADK-inhibitor 5-ITU given transiently during the latent period of epileptogenesis. Importantly, >95 % reduction of seizures in the majority of KA-injected animals was maintained 5 weeks after drug washout, indicating a lasting anti-epileptogenic effect.
ADK inhibitors can produce two distinct effects: (i) Blockade of cytoplasmic ADK-S raises extracellular adenosine, and thereby provides enhanced AR activation. Through this enhanced activation of A1Rs, ADK inhibitors suppress epileptic seizures (Wiesner et al. 1999; Gouder et al. 2004; McGaraughty et al. 2005), a focus of past drug development efforts. (ii) Blockade of nuclear ADK-L increases adenosine in the nucleus and thereby exerts epigenetic effects that are antiepileptogenic (Williams-Karnesky et al. 2013). In the past, differences between ADK-S and ADK-L had not been considered in drug development approaches. Because ADK-L has different (nuclear) protein interaction partners as compared to cytoplasmic ADK-S, there is a solid rationale to develop novel ADK inhibitors with enhanced epigenetic activity, mediated by ADK-L inhibition. In conclusion ADK inhibitors combine epigenetic with antiepileptogenic activity. ADK is a validated target for the prevention of epileptogenesis, which provides the premise to target ADK-L linked epigenetic alterations for antiepileptogenic therapy.
6.2. Gene and cell therapy
Systemic adenosine augmentation is associated with receptor-mediated, mostly cardiovascular, side effects. In an attempt to bypass adverse events of systemic adenosine augmentation, focal adenosine augmentation therapies were tested experimentally to enhance adenosine concentrations locally by using gene and cell therapy approaches. For this reason, local gene therapy designed to increase endogenous adenosine by inhibiting or knocking out the ADK gene, and restricted to an epileptogenic brain area with pathological overexpression of ADK, stands as a promising strategy for achieving seizure control in refractory epilepsy, with fewer or less severe side-effects (Huber et al. 2001; Guttinger et al. 2005). In line with this therapeutic rationale, injection of an astrocyte-specific adeno-associated virus vector (AVV), which expressed an Adk cDNA in antisense orientation into the CA3 subfield of the hippocampus of epileptic Adk-tg mice, reduced the seizure rate from 5.8 to 0.6 seizures per hour. Importantly, a gene therapy-induced decrease of only 3.1 % in ADK expression levels was found to be sufficient for seizure reduction (Lee et al. 2008; Boison, 2010; Theofilas et al. 2011). An example of a local cell therapy approach to treat seizures in modelled epilepsy is based on lentiviral RNAi-mediated downregulation of ADK. To achieve this, human mesenchymal stem cells (HMSC) were infected with a lentivirus that expresses an anti-Adk microRNA, which led to an 80 % decrease in ADK expression and an 8.5 ng/mL increase in adenosine levels in supernatants of the cultured cells. These HMSC cells were then transplanted into the mouse hippocampus one week prior to intra-amygdaloid KA injections, which in turn led to a 35 % decrease in seizure duration and 65 % decrease in seizure-induced hippocampal neuronal cell loss (Ren et al., 2007; Li et al. 2009). An alternative approach is the use of embryonic stem cells engineered to release adenosine by genetic disruption of ADK (Adk-/- cells), which can be differentiated into neural precursor cells prior to grafting into the intra-hippocampal fissures of rats and mice. This approach was shown to suppress epileptogenesis and seizure progression in kindled rats (Li et al., 2007b) and was demonstrated to prevent epileptogenesis in the mouse intra-amygdaloid KA model (Li et al. 2008). Together, those studies demonstrate the potential of focal adenosine augmentation therapies to suppress seizures in epilepsy, and to prevent the development of epilepsy.
6.3. Adenosine augmentation via ketogenic diet (KD)
The high fat, low carbohydrate ketogenic diet (KD), where 90 % of energy is derived from fats and 10 % from proteins and carbohydrates combined, has been used for almost 100 years for the treatment of drug resistant epilepsy, specifically in children (Freeman et al. 2009; Kossoff and Rho, 2009; Stafstrom and Rho, 2012). A systematic review on KD from 2018 outlined data from eleven randomized controlled trails including 712 children and 66 adult patients. In this review, an 85 % seizure reduction rate was observed at three months after initiation of KD therapy (Martin-McGill et al. 2018). In an attempt to make KD more tolerable and compliant, variations of KD such as modified Atkins diet (MAD), that includes daily calorie intake of 10 % carbohydrates with 60 % of fats and 30 % of protein’s was proposed (Cervenka et al. 2016). In a recent clinical study, adult patients, when treated for 24 weeks with a MAD, showed profound seizure reduction over a period of 12 weeks (Cervenka et al. 2016; de Souza Neves et al., 2020). The reason for the success of KD therapy is likely based on the fact that KD therapy combines several beneficial mechanisms, which include glucose restriction, free fatty acids and ketone bodies (Bough and Rho 2007). In addition, KD increases adenosine levels through: (i) Increases in acetyl-CoA which enhances mitochondrial ATP production and thereby adenosine production via ATP-degrading enzymes (Masino and Geiger, 2008, 2009). (ii) Reduced extracellular glucose which induces autocrine regulation of hippocampal CA3 neurons leading to the release of ATP and the consequent increase in extracellular adenosine concentration and activation of ATP-sensitive K+ channels (KATP) (Kawamura et al. 2010). (iii) Downregulation of ADK expression, an effect that is thought to be triggered via KD-induced metabolic stress (Masino et al. 2011; Lusardi et al. 2015). The KD-induced increase in adenosine levels from all three mechanisms leads to increased activation of A1Rs, mediating robust anticonvulsant effects (Masino et al. 2011; Masino et al. 2012).
An exciting new role of KD therapy is suppression of epileptogenesis and disease progression, which was demonstrated in two different rodent models of epilepsy, PTZ-kindled mice and pilocarpine-induced epilepsy in rats (Lusardi et al. 2015). This study demonstrated lasting antiepileptogenic effects even after diet reversal to a control diet. The observed antiepileptogenic effects were associated with the KD’s ability to restore normal adenosine levels in the epileptic brain and to reverse DNA hypermethylation (Lusardi et al. 2015). Taken together, those data demonstrate that adenosine contributes to the potent anticonvulsant, anti-epileptogenic, and disease-modifying properties of KD-based therapy.
7. Summary and outlook
There is now compelling evidence demonstrating the therapeutic potential of targeting the purinergic system during acute seizures, the development of epilepsy, and during epilepsy. This includes targeting of different receptor subtypes (including P1 and P2 receptors) and mechanisms of purine release and metabolism (Fig. 3). Of particular promise are the disease-modifying effects observed via targeting of the ATP-gated P2X7R or ADK (Jimenez-Pacheco et al. 2016; Sandau et al. 2019). The observed disease-modifying effects are even more remarkable as treatment of epileptic animals with anti-epileptic or anti-inflammatory drugs suppressed seizures during treatment but not during the wash-out period (Grabenstatter et al. 2007; Maroso et al. 2010; Klein et al. 2015). To date, approaches to identify drug targets based on purinergic signaling are restricted to the targeting of one specific pathway/receptor. The purinergic system is, however, a complex signaling system with one component affecting multiple targets. For example, inhibition of ATP release may prevent activation of pro-convulsive P2 receptors. Decreased ATP may, however, also lead to a decrease in extracellular concentrations of anticonvulsive adenosine. The targeting of several components of the purinergic system simultaneously may therefore be a better option, taking into account different aspects of the purinergic cascade. To take full advantage of the therapeutic possibilities provided by the purinergic system to treat epilepsy, several questions must be resolved before findings can be advanced beyond a preclinical stage.
Fig. 3. Targeting of the purinergic system to treat epilepsy.
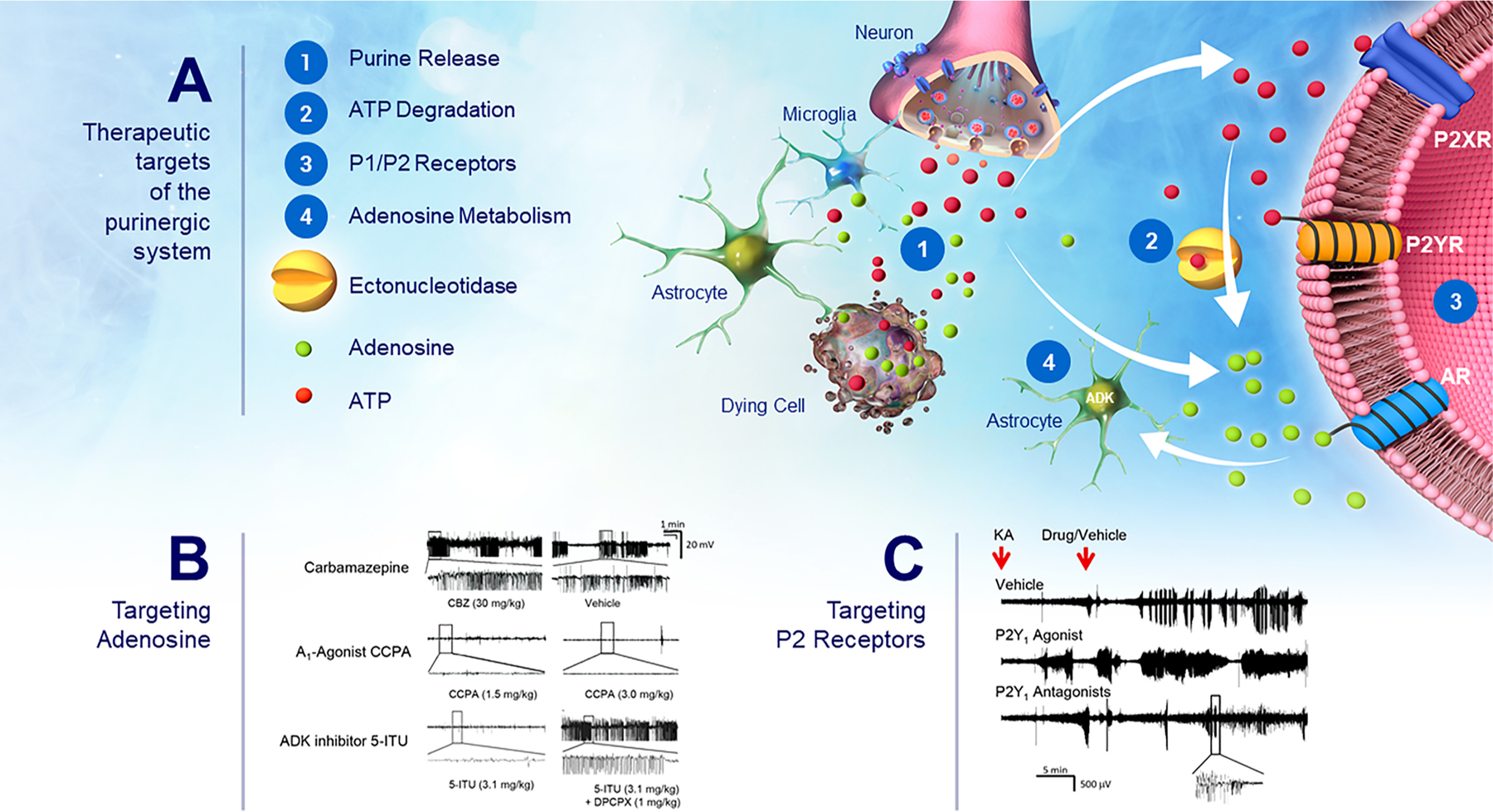
(A) Schematic illustrating possible intervention points targeting different components of the purinergic system to suppress seizures and to treat epilepsy. This includes: (1) Purine release mechanisms: Purines including ATP and adenosine are released in the brain from neurons and glial cells via active release mechanisms (e.g., via synaptic vesicles, pannexin-1 and connexin-43 hemichannels) or from dying cells. (2) ATP-metabolizing ectonucleotidases: Once release into the extracellular space, ATP is rapidly degraded into different breakdown products such as ADP, AMP and adenosine which often mediate opposing effects via their different receptors including P1 and P2 receptors. (3) Purinergic receptors: This includes P1 receptors (A1, A2A, A2B, A3) activated by extracellular adenosine and P2 receptors which are activated by extracellular adenine and uridine nucleotides (e.g., ATP) and are further subdivided into the ionotropic P2X receptor family and the metabotropic P2Y receptor family. (4) Adenosine kinase (ADK): ADK which is predominantly expressed in astrocytes catalyzes the phosphorylation of adenosine to AMP thereby removing extracellular adenosine. (B) Representative EEG traces depicting high amplitude high frequency spiking during intra-amygdala KA-induced status epilepticus. While treatment with the anti-seizure drug Carbamazepine had no effect on seizure severity during status epilepticus, the A1R agonist CCPA and the ADK inhibitor 5-ITU suppressed seizure activity. The seizure-suppressive effects provided via 5-ITU were reversed by treatment with DPCPX (A1R antagonists). (C) EEG traces during intra-amygdala KA-induced status epilepticus showing increased seizure severity when mice were treated with the P2Y1 agonist MRS2500 and seizure suppression when treated with the P2Y1 antagonists MRS2365. Each trace is from a different animal. Drugs/Vehicle was administered 15 min post-intra-amygdala KA injection.
Most findings to date are based on rodent models of TLE, but it will be important to investigate whether mechanisms implicated in TLE and its development are also applicable to other forms of epilepsy and at different developmental stages. For example, P2X7R antagonism has been shown to be anti-convulsive in mouse pups subjected to hypoxia-induced seizures (Rodriguez-Alvarez et al. 2017) and rat pups treated with intra-amygdala KA (Mesuret et al. 2014) suggesting anticonvulsive effects are independent on developmental stage (Menendez Mendez et al. 2020).
While slice work has shown increased extracellular ATP and adenosine concentrations during seizures, our understanding of the in vivo dynamics is limited. What are the mechanisms contributing to increased ATP release, and can these be targeted? What ectonucleotidases contribute to ATP degradation and what are the effects of targeting these enzymes? What about uridine nucleotides (e.g., UTP)? Are these also increased following seizures?
What receptors are recruited during seizures and epilepsy? Compelling evidence has demonstrated a role for the P2X7R and the P2Y1R and to some extent for the P2X4R and the P2Y12R. Do other P2 receptors also contribute to seizures and epilepsy development? While the contribution of A1R and A2AR to epilepsy seems to be well-established, the roles for A2BR and A3R remain incompletely understood.
For the majority of receptors we have now data showing expressional changes in different brain structures following acute seizures and during epilepsy (including data from patients). However, we still do not know the precise cell types (e.g., glia vs. neurons) and subcellular compartment (e.g., pre- and/or postsynaptic). While reporter mice exist for some of the receptors and highly specific nanobodies are developed [e.g., P2X7R and P2X4R (Kaczmarek-Hajek et al. 2018; Bergmann et al. 2019)], reliable detection tools are still missing for most of the purinergic receptors.
Purinergic receptors may elicit a different response according to cell type targeted [e.g., neuronal P2Y1R expression seems to be anticonvulsive whereas P2Y1R expressed on microglia may be proconvulsive (Alves et al., 2019a)]. Thus, cell type-specific KO/overexpression approaches should be undertaken.
Purinergic signaling has mainly been implicated in mediating inflammatory responses, in particular via P2 receptors. P1 and P2 signaling, however, also affects many more cellular process, therefore, future studies should establish signaling downstream of purinergic receptors during seizures and epilepsy.
To date, the majority of studies analyzing the role of purinergic signaling in epilepsy have been mainly limited to rodent models of acute seizures (e.g., PTZ, maximal electroshock) or models mimicking TLE (e.g., KA- or pilocarpine-induced status epilepticus) which has provided encouraging data. Epilepsy encompasses, however, a heterogeneous group of brain disorders with many different etiologies (e.g., brain damage, mutations) and underlying pathologies. Thus, to obtain a more complete picture of purinergic signaling during seizure-related disorders more models must be tested (e.g., genetic models) and, in the case of TLE, animal models should also be used which mimic closer the human condition [e.g., TBI model of epilepsy (Pitkanen et al. 2009)].
Are drugs targeting the purinergic system safe? Brain-penetrant P2X7R antagonists have recently been proven to be safe during clinical trials (Timmers et al. 2018); however, we do not know the effects of purinergic signaling-based long-term treatments, required for chronic brain diseases such as epilepsy. Purinergic receptors are expressed throughout the body (Burnstock, 2008). Are strategies targeting specifically brain cells better (e.g., viral delivery) than the simple administration of drugs available throughout the body?
The role of sex differences are increasingly recognized in brain functions and diseases of the CNS, including epilepsy (Christian et al. 2020). In general, epilepsy tends to be slightly more common in males than females. While the reasons for these differences are still a matter of debate, several potential mechanisms have been proposed such as differences in brain development (Ruigrok et al. 2014), hormonal and neurosteroidal levels (Chen et al. 2018) and glial function including astrocytes and microglia (Schwarz and Bilbo, 2012). Further demonstrating sex differences impacting on seizures and epilepsy, several models of epilepsy show differences in responses to pro-convulsant stimuli [e.g., male mice show reduced survival and greater seizure severity, cell loss and gliosis in the intrahippocampal KA mouse model of TLE (Li and Liu, 2019)]. Purine receptors have been shown to be expressed differentially according to sex (Crain et al. 2009). Importantly, data has shown dependency on sex of animals on treatments based on purinergic signaling previously [e.g., P2X7R antagonism was more effective in female mice in a mouse models of amyotrophic lateral sclerosis (Bartlett et al. 2017)]. While mixed sex groups have been used in some studies [e.g., (Eyo et al. 2014; Rozmer et al. 2017)] to our knowledge, whether targeting of the purinergic system during epilepsy leads to differences according to sex of the animals has not been addressed in a systematic manner to date (see also Tables 2 and 3) and should be investigated in the future.
Importantly, as mentioned previously, to take full advantage of the purinergic system, multi-targeting approaches should be carried out (e.g., activate A1Rs and at the same time block P2X7Rs).
While prior research efforts have focused on the anti-seizure mechanisms of purines, novel findings demonstrate a key role of adenosine-mediated epigenetic mechanisms in the epileptogenic process itself, and open up a new frontier for the development of novel disease-modifying therapeutics.
Finally, besides representing exciting therapeutic targets, the purinergic system provides also opportunities for biomarkers development. Blood-based inflammation markers may have a very strong diagnostic and prognostic potential in epilepsy (Vezzani et al. 2019). Purines are released in the brain and can be detected in the blood. Demonstrating the diagnostic potential of blood purine concentration changes, blood purine levels correlate closely with seizure severity during status epilepticus in mice and are elevated in patients with epilepsy (Beamer et al. 2021).
In summary, while there are still unanswered question which must be addressed in the future, we now have a substantial body of work demonstrating the therapeutic potential of targeting the purinergic system during epilepsy and its development.
Acknowledgements
This work was supported by funding from the National Institutes of Health (NIH) through grants NS103740, NS065957, the Health Research Board HRA-POR-2015-1243 and from Science Foundation Ireland [17/CDA/4708 and 16/RC/3948 (co-funded under the European Regional Development Fund and by FutureNeuro industry partners)]. We would like to thank Dr. Isabela Aparicio for her invaluable help in the design of our Figures. We would also like to apologize to those authors whose relevant work was not cited here.
Glossary
- 5-ITU
5-Iodotubercidin
- ADA
Adenosine deaminase
- ADK
Adenosine kinase
- AR
Adenosine receptor
- ASD
Anti-seizure drug
- BBB
Blood-brain barrier
- BBG
Brilliant Blue G
- BZATP
2’,3’-O-(4-benzoyl-benzoyl)ATP
- cAMP
cyclic adenosine monophosphate
- CD39
Ectonucleoside triphosphate diphosphohydrolase
- CD73
Ecto-5’-nucleotidase
- CNT
Concentrative nucleoside transporters
- DNMT
DNA methyltransferase
- EEG
Electroencephalogram
- EGFP
Enhanced green fluorescent protein
- ENT
Equilibrative nucleoside transporters
- GABA
γ-aminobutyric acid
- HMSC
Human mesenchymal stem cells
- HCY
Homocysteine
- IL-1β
Interleukin-1β
- KA
Kainic acid
- KD
Ketogenic diet
- KO
knock-out
- MAD
Modified Atkins diet
- MTLE
Mesial temporal lobe epilepsy
- NMDA
N-methyl-D-aspartate receptor
- NLRP3
NACHT, LRR and PYD domains-containing protein 3
- NT5E
ecto-5’-nucleotidase
- P2XR
P2X receptor
- P2YR
P2Y receptor
- PTE
post-traumatic epilepsy
- PTZ
Pentylenetetrazole
- SAH
S-adenosyl-L-homocysteine
- SAM
S-adenosylmethionine
- Sp1
Specificity protein 1
- SUDEP
Sudden unexpected death in epilepsy
- TBI
Traumatic brain injury
- TLE
Temporal lobe epilepsy
- UDP
Uridine diphosphate
- UTP
Uridine triphosphate.
Footnotes
Appendix A. The Peer Review Overview and Supplementary data
The Peer Review Overview and Supplementary data associated with this article can be found in the online version, at doi:https://doi.org/10.1016/j.pneurobio.2021.102105.
References
- Adami M, Bertorelli R, Ferri N, Foddi MC, Ongini E, 1995. “Effects of repeated administration of selective adenosine A1 and A2A receptor agonists on pentylenetetrazole-induced convulsions in the rat.”. Eur. J. Pharmacol 294 (2–3), 383–389. [DOI] [PubMed] [Google Scholar]
- Adinolfi E, Giuliani AL, De Marchi E, Pegoraro A, Orioli E, Di Virgilio F, 2018. “The P2X7 receptor: a main player in inflammation.”. Biochem. Pharmacol 151, 234–244. [DOI] [PubMed] [Google Scholar]
- Alves M, Gomez-Villafuertes R, Delanty N, Farrell MA, O’Brien DF, Miras-Portugal MT, Hernandez MD, Henshall DC, Engel T, 2017. “Expression and function of the metabotropic purinergic P2Y receptor family in experimental seizure models and patients with drug-refractory epilepsy.”. Epilepsia 58 (9), 1603–1614. [DOI] [PubMed] [Google Scholar]
- Alves M, Beamer E, Engel T, 2018. “The metabotropic purinergic P2Y receptor family as novel drug target in epilepsy.”. Front. Pharmacol 9, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves M, De Diego Garcia L, Conte G, Jimenez-Mateos EM, D’Orsi B, Sanz-Rodriguez A, Prehn JHM, Henshall DC, Engel T, 2019a. “Context-Specific switch from Anti- to pro-epileptogenic function of the P2Y1 receptor in experimental epilepsy.”. J. Neurosci 39 (27), 5377–5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves M, Smith J, Engel T, 2019b. “Differential expression of the metabotropic P2Y receptor family in the cortex following status epilepticus and neuroprotection via P2Y1 antagonism in mice.”. Front. Pharmacol 10, 1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amhaoul H, Ali I, Mola M, Van Eetveldt A, Szewczyk K, Missault S, Bielen K, Kumar-Singh S, Rech J, Lord B, Ceusters M, Bhattacharya A, Dedeurwaerdere S, 2016. "P2X7 receptor antagonism reduces the severity of spontaneous seizures in a chronic model of temporal lobe epilepsy.". Neuropharmacology 105, 175–185. [DOI] [PubMed] [Google Scholar]
- Amorim BO, Hamani C, Ferreira E, Miranda MF, Fernandes MJS, Rodrigues AM, de Almeida AG, Covolan L, 2016. “Effects of A1 receptor agonist/antagonist on spontaneous seizures in pilocarpine-induced epileptic rats.”. Epilepsy Behav 61, 168–173. [DOI] [PubMed] [Google Scholar]
- Amorim RP, Araujo MGL, Valero J, Lopes-Cendes I, Pascoal VDB, Malva JO, da Silva Fernandes MJ, 2017. “Silencing of P2X7R by RNA interference in the hippocampus can attenuate morphological and behavioral impact of pilocarpine-induced epilepsy.”. Purinergic Signal 13 (4), 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong JN, Brust TB, Lewis RG, MacVicar BA, 2002. “Activation of presynaptic P2X7-like receptors depresses mossy fiber-CA3 synaptic transmission through p38 mitogen-activated protein kinase.”. J. Neurosci 22 (14), 5938–5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronica E, Zurolo E, Iyer A, de Groot M, Anink J, Carbonell C, van Vliet EA, Baayen JC, Boison D, Gorter JA, 2011. "Upregulation of adenosine kinase in astrocytes in experimental and human temporal lobe epilepsy.". Epilepsia 52 (9), 1645–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronica E, Sandau US, Iyer A, Boison D, 2013. “Glial adenosine kinase–a neuropathological marker of the epileptic brain.”. Neurochem. Int 63 (7), 688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augusto E, Matos M, Sevigny J, El-Tayeb A, Bynoe MS, Muller CE, Cunha RA, Chen JF, 2013. “Ecto-5’-nucleotidase (CD73)-mediated formation of adenosine is critical for the striatal adenosine A2A receptor functions.”. J. Neurosci 33 (28), 11390–11399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avignone E, Ulmann L, Levavasseur F, Rassendren F, Audinat E, 2008. “Status epilepticus induces a particular microglial activation state characterized by enhanced purinergic signaling.”. J. Neurosci 28 (37), 9133–9144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avignone E, Lepleux M, Angibaud J, Nagerl UV, 2015. “Altered morphological dynamics of activated microglia after induction of status epilepticus.”. J. Neuroinflammation 12, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aymerich I, Duflot S, Fernandez-Veledo S, Guillen-Gomez E, Huber-Ruano I, Casado FJ, Pastor-Anglada M, 2005. “The concentrative nucleoside transporter family (SLC28): new roles beyond salvage?”. Biochem. Soc. Trans 33 (Pt 1), 216–219. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, K.B., Holmén, J., Nehlig, A., E.E, 1999. “Actions of caffeine in the brain with special reference to factors that contribute to its widespread use.”. Pharmacol. Rev 51, 83–133. [PubMed] [Google Scholar]
- Baldwin SA, Beal PR, Yao SY, King AE, Cass CE, Young JD, 2004. “The equilibrative nucleoside transporter family, SLC29.”. Pflugers Arch 447 (5), 735–743. [DOI] [PubMed] [Google Scholar]
- Baldwin SA, Yao SY, Hyde RJ, Ng AM, Foppolo S, Barnes K, Ritzel MW, Cass CE, Young JD, 2005. “Functional characterization of novel human and mouse equilibrative nucleoside transporters (hENT3 and mENT3) located in intracellular membranes.”. J. Biol. Chem 280 (16), 15880–15887. [DOI] [PubMed] [Google Scholar]
- Barnes K, Dobrzynski H, Foppolo S, Beal PR, Ismat F, Scullion ER, Sun L, Tellez J, Ritzel MW, Claycomb WC, Cass CE, Young JD, Billeter-Clark R, Boyett MR, Baldwin SA, 2006. “Distribution and functional characterization of equilibrative nucleoside transporter-4, a novel cardiac adenosine transporter activated at acidic pH.”. Circ. Res 99 (5), 510–519. [DOI] [PubMed] [Google Scholar]
- Barraco RA, Stefano GB, 1990. “Pharmacological evidence for the modulation of monoamine release by adenosine in the invertebrate nervous system.”. J. Neurochem 54 (6), 2002–2006. [DOI] [PubMed] [Google Scholar]
- Barros-Barbosa AR, Ferreirinha F, Oliveira A, Mendes M, Lobo MG, Santos A, Rangel R, Pelletier J, Sevigny J, Cordeiro JM, Correia-de-Sa P, 2016a. “Adenosine A2A receptor and ecto-5’-nucleotidase/CD73 are upregulated in hippocampal astrocytes of human patients with mesial temporal lobe epilepsy (MTLE).”. Purinergic Signal 12 (4), 719–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros-Barbosa AR, Fonseca AL, Guerra-Gomes S, Ferreirinha F, Santos A, Rangel R, Lobo MG, Correia-de-Sa P, Cordeiro JM, 2016b. "Up-regulation of P2X7 receptor-mediated inhibition of GABA uptake by nerve terminals of the human epileptic neocortex.". Epilepsia 57 (1), 99–110. [DOI] [PubMed] [Google Scholar]
- Bartlett R, Sluyter V, Watson D, Sluyter R, Yerbury JJ, 2017. "P2X7 antagonism using Brilliant Blue G reduces body weight loss and prolongs survival in female SOD1(G93A) amyotrophic lateral sclerosis mice.". PeerJ 5, e3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer ME, Teixeira AL, 2019. “Inflammation in psychiatric disorders: what comes first?”. Ann. N. Y. Acad. Sci 1437 (1), 57–67. [DOI] [PubMed] [Google Scholar]
- Beamer E, Goloncser F, Horvath G, Beko K, Otrokocsi L, Kovanyi B, Sperlagh B, 2016. “Purinergic mechanisms in neuroinflammation: an update from molecules to behavior.”. Neuropharmacology 104, 94–104. [DOI] [PubMed] [Google Scholar]
- Beamer E, Fischer W, Engel T, 2017. “The ATP-Gated P2X7 receptor As a target for the treatment of drug-resistant epilepsy.”. Front. Neurosci 11, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beamer E, Conte G, Engel T, 2019. “ATP release during seizures - A critical evaluation of the evidence.”. Brain Res. Bull 151, 65–73. [DOI] [PubMed] [Google Scholar]
- Beamer E, Lacey A, Alves M, Conte G, Tian F, de Diego-Garcia L, Khalil M, Rosenow F, Delanty N, Dale N, El-Naggar H, Henshall DC, Engel T, 2021. "Elevated blood purine levels as a biomarker of seizures and epilepsy.". Epilepsia [DOI] [PubMed] [Google Scholar]
- Bedner P, Dupper A, Huttmann K, Muller J, Herde MK, Dublin P, Deshpande T, Schramm J, Haussler U, Haas CA, Henneberger C, Theis M, Steinhauser C, 2015. "Astrocyte uncoupling as a cause of human temporal lobe epilepsy.". Brain 138 (Pt 5), 1208–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell MJ, Robertson CS, Kochanek PM, Goodman JC, Gopinath SP, Carcillo JA, Clark RS, Marion DW, Mi Z, Jackson EK, 2001. “Interstitial brain adenosine and xanthine increase during jugular venous oxygen desaturations in humans after traumatic brain injury.”. Crit. Care Med 29 (2), 399–404. [DOI] [PubMed] [Google Scholar]
- Benarroch EE, 2008. "Adenosine and its receptors. Multiple modulatory functions and potential therapeutic targets for neurologic disease.". Neurology 70, 231–236. [DOI] [PubMed] [Google Scholar]
- Bennett MV, Garre JM, Orellana JA, Bukauskas FF, Nedergaard M, Saez JC, 2012. “Connexin and pannexin hemichannels in inflammatory responses of glia and neurons.”. Brain Res 1487, 3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann P, Garcia de Paco E, Rissiek B, Menzel S, Dubberke G, Hua J, Rassendren F, Ulmann L, Koch-Nolte F, 2019. “Generation and characterization of specific monoclonal antibodies and nanobodies directed against the ATP-Gated channel P2X4.”. Front. Cell. Neurosci 13, 498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berne RM, Rafael R, Curnish RR, 1974. “Effect on cerebral vascular resistance and incorporation into cerebral adenine nucleotides.”. Circ. Res 35, 262–271. [Google Scholar]
- Bialer M, 2012. "Why are antiepileptic drugs used for nonepileptic conditions?". Epilepsia 53 (Suppl 7), 26–33. [DOI] [PubMed] [Google Scholar]
- Bialer M, White HS, 2010. “Key factors in the discovery and development of new antiepileptic drugs.”. Nat. Rev. Drug Discov 9 (1), 68–82. [DOI] [PubMed] [Google Scholar]
- Boison D, 2008. “The adenosine kinase hypothesis of epileptogenesis.”. Prog. Neurobiol 84 (3), 249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, 2010. "Inhibitory RNA in epilepsy: research tools and therapeutic perspectives.". Epilepsia 51 (9), 1659–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, 2011. “Methylxanthines, seizures, and excitotoxicity.”. Handb. Exp. Pharmacol (200), 251–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, 2012. "Adenosine dysfunction in epilepsy.". Glia 60 (8), 1234–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, 2013. “Adenosine kinase: exploitation for therapeutic gain.”. Pharmacol. Rev 65 (3), 906–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, 2016. "Adenosinergic signaling in epilepsy.". Neuropharmacology 104, 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, Aronica E, 2015. “Comorbidities in Neurology: is adenosine the common link?”. Neuropharmacology 97, 18–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, Rho JM, 2019. “Epigenetics and epilepsy prevention: the therapeutic potential of adenosine and metabolic therapies.”. Neuropharmacology, 107741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, Yegutkin GG, 2019. “Adenosine metabolism: emerging concepts for cancer therapy.”. Cancer Cell 36 (6), 582–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, Scheurer L, Zumsteg V, Rulicke T, Litynski P, Fowler B, Brandner S, Mohler H, 2002. “Neonatal hepatic steatosis by disruption of the adenosine kinase gene.”. Proc. Natl. Acad. Sci. U. S. A 99 (10), 6985–6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, Chen JF, Fredholm BB, 2010. “Adenosine signaling and function in glial cells.”. Cell Death Differ 17 (7), 1071–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonan CD, Walz R, Pereira GS, Worm PV, Battastini AM, Cavalheiro EA, Izquierdo I, Sarkis JJ, 2000. “Changes in synaptosomal ectonucleotidase activities in two rat models of temporal lobe epilepsy.”. Epilepsy Res 39 (3), 229–238. [DOI] [PubMed] [Google Scholar]
- Borea PA, Gessi S, Merighi S, Vincenzi F, Varani K, 2018. “Pharmacology of adenosine receptors: the state of the art.”. Physiol. Rev 98 (3), 1591–1625. [DOI] [PubMed] [Google Scholar]
- Bragatti JA, Torres CM, Londero RG, Assmann JB, Fontana V, Martin KC, Hidalgo MP, Chaves ML, Bianchin MM, 2010. “Prevalence of psychiatric comorbidities in temporal lobe epilepsy: the value of structured psychiatric interviews.”. Epileptic Disord 12 (4), 283–291. [DOI] [PubMed] [Google Scholar]
- Burnstock G, 2008. “Purinergic signalling and disorders of the central nervous system.”. Nat. Rev. Drug Discov 7 (7), 575–590. [DOI] [PubMed] [Google Scholar]
- Burnstock G, 2016a. "An introduction to the roles of purinergic signalling in neurodegeneration, neuroprotection and neuroregeneration.". Neuropharmacology 104, 4–17. [DOI] [PubMed] [Google Scholar]
- Burnstock G, 2016b. “P2X ion channel receptors and inflammation.”. Purinergic Signal 12 (1), 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos G, Fortuna A, Falcao A, Alves G, 2018. “In vitro and in vivo experimental models employed in the discovery and development of antiepileptic drugs for pharmacoresistant epilepsy.”. Epilepsy Res 146, 63–86. [DOI] [PubMed] [Google Scholar]
- Canas PM, Porciuncula LO, Simoes AP, Augusto E, Silva HB, Machado NJ, Goncalves N, Alfaro TM, Goncalves FQ, Araujo IM, Real JI, Coelho JE, Andrade GM, Almeida RD, Chen JF, Kofalvi A, Agostinho P, Cunha RA, 2018. “Neuronal adenosine A2A receptors are critical mediators of neurodegeneration triggered by convulsions.”. eNeuro 5 (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrier EJ, Auchampach JA, Hillard CJ, 2006. “Inhibition of an equilibrative nucleoside transporter by cannabidiol: a mechanism of cannabinoid immunosuppression.”. Proc. Natl. Acad. Sci. U. S. A 103 (20), 7895–7900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascella NG, Schretlen DJ, Sawa A, 2009. “Schizophrenia and epilepsy: is there a shared susceptibility?”. Neurosci. Res 63 (4), 227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervenka MC, Patton K, Eloyan A, Henry B, Kossoff EH, 2016. “The impact of the modified Atkins diet on lipid profiles in adults with epilepsy.”. Nutr. Neurosci 19 (3), 131–137. [DOI] [PubMed] [Google Scholar]
- Chang BS, Lowenstein DH, 2003. “Epilepsy.”. N. Engl. J. Med 349 (13), 1257–1266. [DOI] [PubMed] [Google Scholar]
- Chen JF, Eltzschig HK, Fredholm BB, 2013. “Adenosine receptors as drug targets–what are the challenges?”. Nat. Rev. Drug Discov 12 (4), 265–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Wu CC, Huang YC, Hung CF, Wang LJ, 2018. “Gender differences in the relationships among neurosteroid serum levels, cognitive function, and quality of life.”. Neuropsychiatr. Dis. Treat 14, 2389–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitnis T, Weiner HL, 2017. “CNS inflammation and neurodegeneration.”. J. Clin. Invest 127 (10), 3577–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian CA, Reddy DS, Maguire J, Forcelli PA, 2020. “Sex differences in the epilepsies and associated comorbidities: implications for use and development of pharmacotherapies.”. Pharmacol. Rev 72 (4), 767–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu S, Xiong W, Parkinson FE, 2014. “Effect of ecto-5’-nucleotidase (eN) in astrocytes on adenosine and inosine formation.”. Purinergic Signal 10 (4), 603–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chwalczuk K, Rubaj A, Swiader M, Czuczwar SJ, 2008. "[Influence of the antagonist of adenosine A1 receptors, 8-cyclopentyl-1, 3-dipropylxanthine, upon the anticonvulsant activity of antiepileptic drugs in mice].". Przegl Lek 65 (11), 759–763. [PubMed] [Google Scholar]
- Cieslak M, Wojtczak A, Komoszynski M, 2017. “Role of the purinergic signaling in epilepsy.”. Pharmacol. Rep 69 (1), 130–138. [DOI] [PubMed] [Google Scholar]
- Ciruela F, Casado V, Rodrigues RJ, Lujan R, Burgueno J, Canals M, Borycz J, Rebola N, Goldberg SR, Mallol J, Cortes A, Canela EI, Lopez-Gimenez JF, Milligan G, Lluis C, Cunha RA, Ferre S, Franco R, 2006. “Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers.”. J. Neurosci 26 (7), 2080–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RS, Carcillo JA, Kochanek PM, Obrist WD, Jackson EK, Mi Z, Wisneiwski SR, Bell MJ, Marion DW, 1997. "Cerebrospinal fluid adenosine concentration and uncoupling of cerebral blood flow and oxidative metabolism after severe head injury in humans.". Neurosurgery 41 (6), 1284–1292 discussion 1292–1283. [DOI] [PubMed] [Google Scholar]
- Craigie E, Birch RE, Unwin RJ, Wildman SS, 2013. “The relationship between P2X4 and P2X7: a physiologically important interaction?”. Front. Physiol 4, 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crain JM, Nikodemova M, Watters JJ, 2009. “Expression of P2 nucleotide receptors varies with age and sex in murine brain microglia.”. J. Neuroinflammation 6, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristovao-Ferreira S, Navarro G, Brugarolas M, Perez-Capote K, Vaz SH, Fattorini G, Conti F, Lluis C, Ribeiro JA, McCormick PJ, Casado V, Franco R, Sebastiao AM, 2013. “A1R-A2AR heteromers coupled to Gs and G i/0 proteins modulate GABA transport into astrocytes.”. Purinergic Signal 9 (3), 433–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha RA, 2016. “How does adenosine control neuronal dysfunction and neurodegeneration?”. J. Neurochem 139 (6), 1019–1055. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Sebastiao AM, Ribeiro JA, 1992. “Ecto-5’-nucleotidase is associated with cholinergic nerve terminals in the hippocampus but not in the cerebral cortex of the rat.”. J. Neurochem 59 (2), 657–666. [DOI] [PubMed] [Google Scholar]
- Dale N, 1998. “Delayed production of adenosine underlies temporal modulation of swimming in frog embryo.”. J. Physiol 511 (Pt 1), 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale N, 2002. “Resetting intrinsic purinergic modulation of neural activity: an associative mechanism?”. J. Neurosci 22 (23), 10461–10469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale N, Frenguelli BG, 2009. “Release of adenosine and ATP during ischemia and epilepsy.”. Curr. Neuropharmacol 7 (3), 160–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale N, Pearson T, Frenguelli BG, 2000. “Direct measurement of adenosine release during hypoxia in the CA1 region of the rat hippocampal slice.”. J. Physiol 526 (Pt 1), 143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale N, Tian F, Sagoo R, Phillips N, Imray C, Roffe C, 2019. “Point-of-care measurements reveal release of purines into venous blood of stroke patients.”. Purinergic Signal 15 (2), 237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza Neves G, Dos Santos Lunardi M, Papini Gabiatti M, Kurrle Rieger Venske D, Ribeiro LC, Lin K, Dubois Moreira J, 2020. “Cardiometabolic risk and effectiveness of the modified Atkins Ketogenic Diet for adult patients with pharmacoresistant epilepsies in a middle-income country.”. Epilepsy Res 160, 106280. [DOI] [PubMed] [Google Scholar]
- Devinsky O, Vezzani A, Najjar S, De Lanerolle NC, Rogawski MA, 2013. “Glia and epilepsy: excitability and inflammation.”. Trends Neurosci 36 (3), 174–184. [DOI] [PubMed] [Google Scholar]
- Dhir A, 2012. “Pentylenetetrazol (PTZ) kindling model of epilepsy.”. Curr. Protoc. Neurosci Chapter 9: Unit9 37. [DOI] [PubMed] [Google Scholar]
- Diamond ML, Ritter AC, Jackson EK, Conley YP, Kochanek PM, Boison D, Wagner AK, 2015. "Genetic variation in the adenosine regulatory cycle is associated with posttraumatic epilepsy development.". Epilepsia 56 (8), 1198–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez R, Richardson MJE, Wall MJ, 2017. “Reducing extracellular Ca(2 ) induces adenosine release via equilibrative nucleoside transporters to provide negative feedback control of activity in the Hippocampus.”. Front. Neural Circuits 11, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan E, Aygun H, Arslan G, Rzayev E, Avci B, Ayyildiz M, Agar E, 2020. “The role of NMDA receptors in the effect of purinergic P2X7 receptor on spontaneous seizure activity in WAG/Rij rats with genetic absence epilepsy.”. Front. Neurosci 14, 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dona F, Ulrich H, Persike DS, Conceicao IM, Blini JP, Cavalheiro EA, Fernandes MJ, 2009. “Alteration of purinergic P2X4 and P2X7 receptor expression in rats with temporal-lobe epilepsy induced by pilocarpine.”. Epilepsy Res 83 (2–3), 157–167. [DOI] [PubMed] [Google Scholar]
- Dona F, Conceicao IM, Ulrich H, Ribeiro EB, Freitas TA, Nencioni AL, da Silva Fernandes MJ, 2016. “Variations of ATP and its metabolites in the hippocampus of rats subjected to pilocarpine-induced temporal lobe epilepsy.”. Purinergic Signal 12 (2), 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dossi E, Blauwblomme T, Moulard J, Chever O, Vasile F, Guinard E, Le Bert M, Couillin I, Pallud J, Capelle L, Huberfeld G, Rouach N, 2018. “Pannexin-1 channels contribute to seizure generation in human epileptic brain tissue and in a mouse model of epilepsy.”. Sci. Transl. Med 10 (443). [DOI] [PubMed] [Google Scholar]
- Dragunow M, Goddard GV, 1984. “Adenosine modulation of amygdala kindling.”. Exp. Neurol 84 (3), 654–665. [DOI] [PubMed] [Google Scholar]
- Dulla CG, Frenguelli BG, Staley KJ, Masino SA, 2009. “Intracellular acidification causes adenosine release during states of hyperexcitability in the hippocampus.”. J. Neurophysiol 102 (3), 1984–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV, Hoffer BJ, 1980. “Adenine nucleotides and synaptic transmission in the in vitro rat hippocampus.”. Br. J. Pharmacol 69 (1), 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV, Diao L, Kim HO, Jiang JL, Jacobson KA, 1997. “Activation of hippocampal adenosine A3 receptors produces a desensitization of A1 receptor-mediated responses in rat hippocampus.”. J. Neurosci 17 (2), 607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- During MJ, Spencer DD, 1992. “Adenosine: a potential mediator of seizure arrest and postictal refractoriness.”. Ann. Neurol 32 (5), 618–624. [DOI] [PubMed] [Google Scholar]
- El Yacoubi M, Ledent C, Parmentier M, Bertorelli R, Ongini E, Costentin J, Vaugeois JM, 2001. “Adenosine A2A receptor antagonists are potential antidepressants: evidence based on pharmacology and A2A receptor knockout mice.”. Br. J. Pharmacol 134 (1), 68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Yacoubi M, Ledent C, Parmentier M, Costentin J, Vaugeois JM, 2008. "Evidence for the involvement of the adenosine A(2A) receptor in the lowered susceptibility to pentylenetetrazol-induced seizures produced in mice by long-term treatment with caffeine.". Neuropharmacology 55 (1), 35–40. [DOI] [PubMed] [Google Scholar]
- El Yacoubi M, Ledent C, Parmentier M, Costentin J, Vaugeois JM, 2009. “Adenosine A2A receptor deficient mice are partially resistant to limbic seizures.”. Naunyn Schmiedebergs Arch. Pharmacol 380 (3), 223–232. [DOI] [PubMed] [Google Scholar]
- Engel J Jr., 1996. “Introduction to temporal lobe epilepsy.”. Epilepsy Res 26 (1), 141–150. [DOI] [PubMed] [Google Scholar]
- Engel T, Gomez-Villafuertes R, Tanaka K, Mesuret G, Sanz-Rodriguez A, Garcia-Huerta P, Miras-Portugal MT, Henshall DC, Diaz-Hernandez M, 2012. “Seizure suppression and neuroprotection by targeting the purinergic P2X7 receptor during status epilepticus in mice.”. FASEB J 26 (4), 1616–1628. [DOI] [PubMed] [Google Scholar]
- Engel T, Alves M, Sheedy C, Henshall DC, 2016. "ATPergic signalling during seizures and epilepsy.". Neuropharmacology 104, 140–153. [DOI] [PubMed] [Google Scholar]
- Engel T, Brennan GP, Sanz-Rodriguez A, Alves M, Beamer E, Watters O, Henshall DC, Jimenez-Mateos EM, 2017. “A calcium-sensitive feed-forward loop regulating the expression of the ATP-gated purinergic P2X7 receptor via specificity protein 1 and microRNA-22.”. Biochim. Biophys. Acta Mol Cell Res 1864 (2), 255–266. [DOI] [PubMed] [Google Scholar]
- Etherington LA, Frenguelli BG, 2004. “Endogenous adenosine modulates epileptiform activity in rat hippocampus in a receptor subtype-dependent manner.”. Eur. J. Neurosci 19 (9), 2539–2550. [DOI] [PubMed] [Google Scholar]
- Evans DB, Schenden JA, Bristol JA, 1982. “Adenosine receptors mediating cardiac depression.”. Life Sci 31 (22), 2425–2432. [DOI] [PubMed] [Google Scholar]
- Eyo UB, Peng J, Swiatkowski P, Mukherjee A, Bispo A, Wu LJ, 2014. “Neuronal hyperactivity recruits microglial processes via neuronal NMDA receptors and microglial P2Y12 receptors after status epilepticus.”. J. Neurosci 34 (32), 10528–10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faingold CL, Randall M, Kommajosyula SP, 2016. “Susceptibility to seizure-induced sudden death in DBA/2 mice is altered by adenosine.”. Epilepsy Res 124, 49–54. [DOI] [PubMed] [Google Scholar]
- Fedele DE, Gouder N, Guttinger M, Gabernet L, Scheurer L, Rulicke T, Crestani F, Boison D, 2005. "Astrogliosis in epilepsy leads to overexpression of adenosine kinase, resulting in seizure aggravation.". Brain 128 (Pt 10), 2383–2395. [DOI] [PubMed] [Google Scholar]
- Fedele DE, Li T, Lan JQ, Fredholm BB, Boison D, 2006. “Adenosine A1 receptors are crucial in keeping an epileptic focus localized.”. Exp. Neurol 200 (1), 184–190. [DOI] [PubMed] [Google Scholar]
- Feinstein AR, 1970. “The pre-therapeutic classification of Co-morbidity in chronic disease.”. J. Chronic Dis 23 (7), 455–468. [DOI] [PubMed] [Google Scholar]
- Fischer W, Franke H, Krugel U, Muller H, Dinkel K, Lord B, Letavic MA, Henshall DC, Engel T, 2016. “Critical evaluation of P2X7 receptor antagonists in selected seizure models.”. PLoS One 11 (6), e0156468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RS, Schachter SC, 2000. “The postictal state: a neglected entity in the management of epilepsy.”. Epilepsy Behav 1 (1), 52–59. [DOI] [PubMed] [Google Scholar]
- Franke H, Krugel U, Grosche J, Heine C, Hartig W, Allgaier C, Illes P, 2004. "P2Y receptor expression on astrocytes in the nucleus accumbens of rats.". Neuroscience 127 (2), 431–441. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, 1997. “Adenosine and neuroprotection.”. Int. Rev. Neurobiol 40, 259–280. [PubMed] [Google Scholar]
- Fredholm BB, 2014. “Adenosine–a physiological or pathophysiological agent?”. J. Mol. Med 92 (3), 201–206. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, AP IJ, Jacobson KA, Linden J, Muller CE, 2011. “International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors–an update.”. Pharmacol. Rev 63 (1), 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman JM, Vining EP, Kossoff EH, Pyzik PL, Ye X, Goodman SN, 2009. "A blinded, crossover study of the efficacy of the ketogenic diet.". Epilepsia 50 (2), 322–325. [DOI] [PubMed] [Google Scholar]
- Frenguelli BG, Wigmore G, Llaudet E, Dale N, 2007. “Temporal and mechanistic dissociation of ATP and adenosine release during ischaemia in the mammalian hippocampus.”. J. Neurochem 101 (5), 1400–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M, Suzuki Y, Hino H, Kuzume K, Morimoto T, Ishii E, 2010. "Adenosine A1 receptor blockage mediates theophylline-associated seizures.". Epilepsia 51 (3), 483–487. [DOI] [PubMed] [Google Scholar]
- Ganesana M, Venton BJ, 2018. "Early changes in transient adenosine during cerebral ischemia and reperfusion injury.". PLoS One 13 (5), e0196932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebril Hoda M., Gesese Raey, R.M.R., Emond, Martine P., Huo, Yuqing, Aronica, Eleonora, Boison, Detlev, 2020. “Adenosine kinase inhibition promotes proliferation of neural stem cells after traumatic brain injury.”. Brain Commun [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessi S, Merighi S, Fazzi D, Stefanelli A, Varani K, Borea PA, 2011. “Adenosine receptor targeting in health and disease.”. Expert Opin. Investig. Drugs 20 (12), 1591–1609. [DOI] [PubMed] [Google Scholar]
- Gouder N, Fritschy JM, Boison D, 2003. "Seizure suppression by adenosine A1 receptor activation in a mouse model of pharmacoresistant epilepsy.". Epilepsia 44 (7), 877–885. [DOI] [PubMed] [Google Scholar]
- Gouder N, Scheurer L, Fritschy JM, Boison D, 2004. “Overexpression of adenosine kinase in epileptic hippocampus contributes to epileptogenesis.”. J. Neurosci 24 (3), 692–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabenstatter HL, Clark S, Dudek FE, 2007. "Anticonvulsant effects of carbamazepine on spontaneous seizures in rats with kainate-induced epilepsy: comparison of intraperitoneal injections with drug-in-food protocols.". Epilepsia 48 (12), 2287–2295. [DOI] [PubMed] [Google Scholar]
- Griffith DA, Jarvis SM, 1996. “Nucleoside and nucleobase transport systems of mammalian cells.”. Biochim. Biophys. Acta 1286 (3), 153–181. [DOI] [PubMed] [Google Scholar]
- Gundlfinger A, Bischofberger J, Johenning FW, Torvinen M, Schmitz D, Breustedt J, 2007. “Adenosine modulates transmission at the hippocampal mossy fibre synapse via direct inhibition of presynaptic calcium channels.”. J. Physiol 582 (Pt 1), 263–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttinger M, Padrun V, Pralong WF, Boison D, 2005. “Seizure suppression and lack of adenosine A1 receptor desensitization after focal long-term delivery of adenosine by encapsulated myoblasts.”. Exp. Neurol 193 (1), 53–64. [DOI] [PubMed] [Google Scholar]
- Guzman SJ, Gerevich Z, 2016. “P2Y receptors in synaptic transmission and plasticity: therapeutic potential in cognitive dysfunction.”. Neural Plast 2016, 1207393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann M, Attwell D, 1996. “Non-synaptic release of ATP by electrical stimulation in slices of rat hippocampus, cerebellum and habenula.”. Eur. J. Neurosci 8 (7), 1510–1515. [DOI] [PubMed] [Google Scholar]
- Hargus NJ, Jennings C, Perez-Reyes E, Bertram EH, Patel MK, 2012. "Enhanced actions of adenosine in medial entorhinal cortex layer II stellate neurons in temporal lobe epilepsy are mediated via A(1)-receptor activation.". Epilepsia 53 (1), 168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich A, Ando RD, Turi G, Rozsa B, Sperlagh B, 2012. “K depolarization evokes ATP, adenosine and glutamate release from glia in rat hippocampus: a microelectrode biosensor study.”. Br. J. Pharmacol 167 (5), 1003–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henshall DC, Engel T, 2015. “P2X purinoceptors as a link between hyperexcitability and neuroinflammation in status epilepticus.”. Epilepsy Behav 49, 8–12. [DOI] [PubMed] [Google Scholar]
- Henshall DC, Kobow K, 2015. “Epigenetics and Epilepsy.”. Cold Spring Harb. Perspect. Med 5 (12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlenius E, Lagercrantz H, Yamamoto Y, 1997. “Adenosine modulates inspiratory neurons and the respiratory pattern in the brainstem of neonatal rats.”. Pediatr. Res 42 (1), 46–53. [DOI] [PubMed] [Google Scholar]
- Hesdorffer DC, Tomson T, 2013. "Sudden unexpected death in epilepsy. Potential role of antiepileptic drugs.". CNS Drugs 27 (2), 113–119. [DOI] [PubMed] [Google Scholar]
- Hirsch LJ, 2010. “Is sudden unexpected death in epilepsy due to postictal brain shutdown?”. Ann. Neurol 68 (6), 773–775. [DOI] [PubMed] [Google Scholar]
- Huang C, Chi XS, Li R, Hu X, Xu HX, Li JM, Zhou D, 2017. “Inhibition of P2X7 receptor ameliorates nuclear factor-kappa B mediated neuroinflammation induced by status epilepticus in rat Hippocampus.”. J. Mol. Neurosci 63 (2), 173–184. [DOI] [PubMed] [Google Scholar]
- Huber A, Padrun V, Deglon N, Aebischer P, Mohler H, Boison D, 2001. “Grafts of adenosine-releasing cells suppress seizures in kindling epilepsy.”. Proc. Natl. Acad. Sci. U. S. A 98 (13), 7611–7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huizinga R, Kreft KL, Onderwater S, Boonstra JG, Brands R, Hintzen RQ, Laman JD, 2012. “Endotoxin- and ATP-neutralizing activity of alkaline phosphatase as a strategy to limit neuroinflammation.”. J. Neuroinflammation 9, 266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde RJ, Cass CE, Young JD, Baldwin SA, 2001. “The ENT family of eukaryote nucleoside and nucleobase transporters: recent advances in the investigation of structure/function relationships and the identification of novel isoforms.”. Mol. Membr. Biol 18 (1), 53–63. [PubMed] [Google Scholar]
- Ilie A, Raimondo JV, Akerman CJ, 2012. “Adenosine release during seizures attenuates GABAA receptor-mediated depolarization.”. J. Neurosci 32 (15), 5321–5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illes P, Khan TM, Rubini P, 2017. “Neuronal P2X7 receptors revisited: do they really exist?”. J. Neurosci 37 (30), 7049–7062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illes P, Burnstock G, Tang Y, 2019a. “Astroglia-Derived ATP modulates CNS neuronal circuits.”. Trends Neurosci 42 (12), 885–898. [DOI] [PubMed] [Google Scholar]
- Illes P, Verkhratsky A, Tang Y, 2019b. “Pathological ATPergic signaling in major depression and bipolar disorder.”. Front. Mol. Neurosci 12, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imura Y, Morizawa Y, Komatsu R, Shibata K, Shinozaki Y, Kasai H, Moriishi K, Moriyama Y, Koizumi S, 2013. "Microglia release ATP by exocytosis.". Glia 61 (8), 1320–1330. [DOI] [PubMed] [Google Scholar]
- Inoue K, 2017. “Molecular basis of nucleobase transport systems in mammals.”. Biol. Pharm. Bull 40 (8), 1130–1138. [DOI] [PubMed] [Google Scholar]
- Jacobson KA, Boeynaems JM, 2010. “P2Y nucleotide receptors: promise of therapeutic applications.”. Drug Discov. Today 15 (13–14), 570–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, von Lubitz DK, Daly JW, Fredholm BB, 1996. “Adenosine receptor ligands: differences with acute versus chronic treatment.”. Trends Pharmacol. Sci 17 (3), 108–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James SJ, Melnyk S, Pogribna M, Pogribny IP, Caudill MA, 2002. “Elevation in S-adenosylhomocysteine and DNA hypomethylation: potential epigenetic mechanism for homocysteine-related pathology.”. J. Nutr 132 (8 Suppl), 2361S–2366S. [DOI] [PubMed] [Google Scholar]
- Jansen C, Francomme L, Vignal JP, Jacquot C, Schwan R, Tyvaert L, Maillard L, Hingray C, 2019. “Interictal psychiatric comorbidities of drug-resistant focal epilepsy: prevalence and influence of the localization of the epilepsy.”. Epilepsy Behav 94, 288–296. [DOI] [PubMed] [Google Scholar]
- Jennings LL, Hao C, Cabrita MA, Vickers MF, Baldwin SA, Young JD, Cass CE, 2001. "Distinct regional distribution of human equilibrative nucleoside transporter proteins 1 and 2 (hENT1 and hENT2) in the central nervous system.". Neuropharmacology 40 (5), 722–731. [DOI] [PubMed] [Google Scholar]
- Jimenez-Mateos EM, Arribas-Blazquez M, Sanz-Rodriguez A, Concannon C, Olivos-Ore LA, Reschke CR, Mooney CM, Mooney C, Lugara E, Morgan J, Langa E, Jimenez-Pacheco A, Silva LF, Mesuret G, Boison D, Miras-Portugal MT, Letavic M, Artalejo AR, Bhattacharya A, Diaz-Hernandez M, Henshall DC, Engel T, 2015. “microRNA targeting of the P2X7 purinoceptor opposes a contralateral epileptogenic focus in the hippocampus.”. Sci. Rep 5, 17486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Mateos EM, Smith J, Nicke A, Engel T, 2019. “Regulation of P2X7 receptor expression and function in the brain.”. Brain Res. Bull 151, 153–163. [DOI] [PubMed] [Google Scholar]
- Jimenez-Pacheco A, Mesuret G, Sanz-Rodriguez A, Tanaka K, Mooney C, Conroy R, Miras-Portugal MT, Diaz-Hernandez M, Henshall DC, Engel T, 2013. "Increased neocortical expression of the P2X7 receptor after status epilepticus and anticonvulsant effect of P2X7 receptor antagonist A-438079.". Epilepsia 54 (9), 1551–1561. [DOI] [PubMed] [Google Scholar]
- Jimenez-Pacheco A, Diaz-Hernandez M, Arribas-Blazquez M, Sanz-Rodriguez A, Olivos-Ore LA, Artalejo AR, Alves M, Letavic M, Miras-Portugal MT, Conroy RM, Delanty N, Farrell MA, O’Brien DF, Bhattacharya A, Engel T, Henshall DC, 2016. “Transient P2X7 receptor antagonism produces lasting reductions in spontaneous seizures and Gliosis in experimental temporal lobe epilepsy.”. J. Neurosci 36 (22), 5920–5932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo YH, Schlichter R, 1999. “Synaptic corelease of ATP and GABA in cultured spinal neurons.”. Nat. Neurosci 2 (3), 241–245. [DOI] [PubMed] [Google Scholar]
- Kaczmarek-Hajek K, Zhang J, Kopp R, Grosche A, Rissiek B, Saul A, Bruzzone S, Engel T, Jooss T, Krautloher A, Schuster S, Magnus T, Stadelmann C, Sirko S, Koch-Nolte F, Eulenburg V, Nicke A, 2018. "Re-evaluation of neuronal P2X7 expression using novel mouse models and a P2X7-specific nanobody.". Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TC, An SJ, Park SK, Hwang IK, Won MH, 2003. “P2X2 and P2X4 receptor expression is regulated by a GABA(A) receptor-mediated mechanism in the gerbil hippocampus.”. Brain Res. Mol. Brain Res 116 (1–2), 168–175. [DOI] [PubMed] [Google Scholar]
- Kanner AM, 2012. “Can neurobiological pathogenic mechanisms of depression facilitate the development of seizure disorders?”. Lancet Neurol 11 (12), 1093–1102. [DOI] [PubMed] [Google Scholar]
- Kanner AM, Ribot R, Mazarati A, 2018. “Bidirectional relations among common psychiatric and neurologic comorbidities and epilepsy: do they have an impact on the course of the seizure disorder?”. Epilepsia Open 3 (Suppl Suppl 2), 210–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura M Jr., Ruskin DN, Masino SA, 2010. “Metabolic autocrine regulation of neurons involves cooperation among pannexin hemichannels, adenosine receptors, and KATP channels.”. J. Neurosci 30 (11), 3886–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keezer MR, Sisodiya SM, Sander JW, 2016. “Comorbidities of epilepsy: current concepts and future perspectives.”. Lancet Neurol 15 (1), 106–115. [DOI] [PubMed] [Google Scholar]
- Khakh BS, North RA, 2006. "P2X receptors as cell-surface ATP sensors in health and disease.". Nature 442 (7102), 527–532. [DOI] [PubMed] [Google Scholar]
- Khan MT, Deussing J, Tang Y, Illes P, 2019. “Astrocytic rather than neuronal P2X7 receptors modulate the function of the tri-synaptic network in the rodent hippocampus.”. Brain Res. Bull 151, 164–173. [DOI] [PubMed] [Google Scholar]
- Kiese K, Jablonski J, Boison D, Kobow K, 2016. “Dynamic regulation of the adenosine kinase gene during early postnatal brain development and maturation.”.Front. Mol. Neurosci 9, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JE, Kang TC, 2011. “The P2X7 receptor-pannexin-1 complex decreases muscarinic acetylcholine receptor-mediated seizure susceptibility in mice.”. J. Clin. Invest 121 (5), 2037–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JE, Kwak SE, Jo SM, Kang TC, 2009. “Blockade of P2X receptor prevents astroglial death in the dentate gyrus following pilocarpine-induced status epilepticus.”. Neurol. Res 31 (9), 982–988. [DOI] [PubMed] [Google Scholar]
- Kim JE, Ryu HJ, Yeo SI, Kang TC, 2010. “P2X7 receptor regulates leukocyte infiltrations in rat frontoparietal cortex following status epilepticus.”. J. Neuroinflammation 7, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JE, Ryu HJ, Kang TC, 2011. “P2X7 receptor activation ameliorates CA3 neuronal damage via a tumor necrosis factor-alpha-mediated pathway in the rat hippocampus following status epilepticus.”. J. Neuroinflammation 8, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiryu-Seo S, Kato R, Ogawa T, Nakagomi S, Nagata K, Kiyama H, 2008. “Neuronal injury-inducible gene is synergistically regulated by ATF3, c-Jun, and STAT3 through the interaction with Sp1 in damaged neurons.”. J. Biol. Chem 283 (11), 6988–6996. [DOI] [PubMed] [Google Scholar]
- Klaft ZJ, Hollnagel JO, Salar S, Caliskan G, Schulz SB, Schneider UC, Horn P, Koch A, Holtkamp M, Gabriel S, Gerevich Z, Heinemann U, 2016. "Adenosine A1 receptor-mediated suppression of carbamazepine-resistant seizure-like events in human neocortical slices.". Epilepsia 57 (5), 746–756. [DOI] [PubMed] [Google Scholar]
- Klein S, Bankstahl M, Loscher W, 2015. "Inter-individual variation in the effect of antiepileptic drugs in the intrahippocampal kainate model of mesial temporal lobe epilepsy in mice.". Neuropharmacology 90, 53–62. [DOI] [PubMed] [Google Scholar]
- Klein P, Dingledine R, Aronica E, Bernard C, Blumcke I, Boison D, Brodie MJ, Brooks-Kayal AR, Engel J Jr., Forcelli PA, Hirsch LJ, Kaminski RM, Klitgaard H, Kobow K, Lowenstein DH, Pearl PL, Pitkanen A, Puhakka N, Rogawski MA, Schmidt D, Sillanpaa M, Sloviter RS, Steinhauser C, Vezzani A, Walker MC, Loscher W, 2018. “Commonalities in epileptogenic processes from different acute brain insults: do they translate?”. Epilepsia 59 (1), 37–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klyuch BP, Dale N, Wall MJ, 2012. “Deletion of ecto-5’-nucleotidase (CD73) reveals direct action potential-dependent adenosine release.”. J. Neurosci 32 (11), 3842–3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutsen JE, Murray T, 1997. Adenosine and ATP in epilepsy. In: Jakobson KA, Jarvis MF (Eds.), Purinergic Approaches in Experimental Therapeutics), pp. 432–447. [Google Scholar]
- Kobow K, Blumcke I, 2011. "The methylation hypothesis: do epigenetic chromatin modifications play a role in epileptogenesis?". Epilepsia 52 (Suppl 4), 15–19. [DOI] [PubMed] [Google Scholar]
- Kobow K, Jeske I, Hildebrandt M, Hauke J, Hahnen E, Buslei R, Buchfelder M, Weigel D, Stefan H, Kasper B, Pauli E, Blumcke I, 2009. “Increased reelin promoter methylation is associated with granule cell dispersion in human temporal lobe epilepsy.”. J. Neuropathol. Exp. Neurol 68 (4), 356–364. [DOI] [PubMed] [Google Scholar]
- Kobow K, Kaspi A, Harikrishnan KN, Kiese K, Ziemann M, Khurana I, Fritzsche I, Hauke J, Hahnen E, Coras R, Muhlebner A, El-Osta A, Blumcke I, 2013. “Deep sequencing reveals increased DNA methylation in chronic rat epilepsy.”. Acta Neuropathol 126 (5), 741–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochanek PM, Clark RS, Obrist WD, Carcillo JA, Jackson EK, Mi Z, Wisniewski SR, Bell MJ, Marion DW, 1997. “The role of adenosine during the period of delayed cerebral swelling after severe traumatic brain injury in humans.”. Acta Neurochir. Suppl 70, 109–111. [DOI] [PubMed] [Google Scholar]
- Kochanek PM, Vagni VA, Janesko KL, Washington CB, Crumrine PK, Garman RH, Jenkins LW, Clark RS, Homanics GE, Dixon CE, Schnermann J, Jackson EK, 2006. “Adenosine A1 receptor knockout mice develop lethal status epilepticus after experimental traumatic brain injury.”. J. Cereb. Blood Flow Metab 26 (4), 565–575. [DOI] [PubMed] [Google Scholar]
- Kong W, Engel K, Wang J, 2004. “Mammalian nucleoside transporters.”. Curr. Drug Metab 5 (1), 63–84. [DOI] [PubMed] [Google Scholar]
- Kopp R, Krautloher A, Ramirez-Fernandez A, Nicke A, 2019. “P2X7 interactions and signaling - making head or tail of it.”. Front. Mol. Neurosci 12, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kossoff EH, Rho JM, 2009. "Ketogenic diets: evidence for short- and long-term efficacy.". Neurotherapeutics 6 (2), 406–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koszalka P, Ozuyaman B, Huo Y, Zernecke A, Flogel U, Braun N, Buchheiser A, Decking UK, Smith ML, Sevigny J, Gear A, Weber AA, Molojavyi A, Ding Z, Weber C, Ley K, Zimmermann H, Godecke A, Schrader J, 2004. “Targeted disruption of cd73/ecto-5’-nucleotidase alters thromboregulation and augments vascular inflammatory response.”. Circ. Res 95 (8), 814–821. [DOI] [PubMed] [Google Scholar]
- Kovacs Z, Slezia A, Bali ZK, Kovacs P, Dobolyi A, Szikra T, Hernadi I, Juhasz G, 2013. “Uridine modulates neuronal activity and inhibits spike-wave discharges of absence epileptic Long Evans and Wistar Albino Glaxo/Rijswijk rats.”. Brain Res. Bull 97, 16–23. [DOI] [PubMed] [Google Scholar]
- Kredich NM, Martin DV Jr., 1977. "Role of S-adenosylhomocysteine in adenosinemediated toxicity in cultured mouse T lymphoma cells.". Cell 12 (4), 931–938. [DOI] [PubMed] [Google Scholar]
- Kull B, Svenningsson P, Fredholm BB, 2000. “Adenosine A(2A) receptors are colocalized with and activate g(olf) in rat striatum.”. Mol. Pharmacol 58 (4), 771–777. [DOI] [PubMed] [Google Scholar]
- Lado FA, Moshe SL, 2008. "How do seizures stop?". Epilepsia 49 (10), 1651–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFrance WC Jr., Kanner AM, Hermann B, 2008. “Psychiatric comorbidities in epilepsy.”. Int. Rev. Neurobiol 83, 347–383. [DOI] [PubMed] [Google Scholar]
- Lalo U, Palygin O, Rasooli-Nejad S, Andrew J, Haydon PG, Pankratov Y, 2014. “Exocytosis of ATP from astrocytes modulates phasic and tonic inhibition in the neocortex.”. PLoS Biol 12 (1), e1001747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam AD, Deck G, Goldman A, Eskandar EN, Noebels J, Cole AJ, 2017. “Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer’s disease.”. Nat. Med 23 (6), 678–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer D, Hammer K, Koszalka P, Schrader J, Robson S, Zimmermann H, 2008. “Distribution of ectonucleotidases in the rodent brain revisited.”. Cell Tissue Res 334 (2), 199–217. [DOI] [PubMed] [Google Scholar]
- Lanser AJ, Rezende RM, Rubino S, Lorello PJ, Donnelly DJ, Xu H, Lau LA, Dulla CG, Caldarone BJ, Robson SC, Weiner HL, 2017. "Disruption of the ATP/adenosine balance in CD39(-/-) mice is associated with handling-induced seizures.". Immunology 152 (4), 589–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Messing A, Su M, Brenner M, 2008. "GFAP promoter elements required for region-specific and astrocyte-specific expression.". Glia 56 (5), 481–493. [DOI] [PubMed] [Google Scholar]
- Li F, Liu L, 2019. “Comparison of kainate-induced seizures, cognitive impairment and hippocampal damage in male and female mice.”. Life Sci 232, 116621. [DOI] [PubMed] [Google Scholar]
- Li T, Quan Lan J, Fredholm BB, Simon RP, Boison D, 2007a. “Adenosine dysfunction in astrogliosis: cause for seizure generation?”. Neuron Glia Biol 3 (4), 353–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Steinbeck JA, Lusardi T, Koch P, Lan JQ, Wilz A, Segschneider M, Simon RP, Brustle O, Boison D, 2007b. "Suppression of kindling epileptogenesis by adenosine releasing stem cell-derived brain implants.". Brain 130 (Pt 5), 1276–1288. [DOI] [PubMed] [Google Scholar]
- Li T, Ren G, Lusardi T, Wilz A, Lan JQ, Iwasato T, Itohara S, Simon RP, Boison D, 2008. “Adenosine kinase is a target for the prediction and prevention of epileptogenesis in mice.”. J. Clin. Invest 118 (2), 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Ren G, Kaplan DL, Boison D, 2009. “Human mesenchymal stem cell grafts engineered to release adenosine reduce chronic seizures in a mouse model of CA3-selective xt”. Epilepsy Res 84 (2–3), 238–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Lytle N, Lan JQ, Sandau US, Boison D, 2012. "Local disruption of glial adenosine homeostasis in mice associates with focal electrographic seizures: a first step in epileptogenesis?". Glia 60 (1), 83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Kang R, Shi J, Liu G, Zhang J, 2013. "Anticonvulsant activity of B2, an adenosine analog, on chemical convulsant-induced seizures.". PLoS One 8 (6), e67060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Zhang JJ, 2011. "[Sedative, hypnotic and anticonvulsive effects of an adenosine analogue WS090501].". Yao Xue Xue Bao 46 (6), 742–746. [PubMed] [Google Scholar]
- Lie AA, Blumcke I, Beck H, Wiestler OD, Elger CE, Schoen SW, 1999. “5’-Nucleotidase activity indicates sites of synaptic plasticity and reactive synaptogenesis in the human brain.”. J. Neuropathol. Exp. Neurol 58 (5), 451–458. [DOI] [PubMed] [Google Scholar]
- Lietsche J, Imran I, Klein J, 2016. “Extracellular levels of ATP and acetylcholine during lithium-pilocarpine induced status epilepticus in rats.”. Neurosci. Lett 611, 69–73. [DOI] [PubMed] [Google Scholar]
- Lin JJ, Mula M, Hermann BP, 2012. "Uncovering the neurobehavioural comorbidities of epilepsy over the lifespan.". Lancet 380 (9848), 1180–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopatar J, Dale N, Frenguelli BG, 2011. “Minor contribution of ATP P2 receptors to electrically-evoked electrographic seizure activity in hippocampal slices: evidence from purine biosensors and P2 receptor agonists and antagonists.”. Neuropharmacology 61 (1–2), 25–34. [DOI] [PubMed] [Google Scholar]
- Lopes LV, Cunha RA, Kull B, Fredholm BB, Ribeiro JA, 2002. "Adenosine A(2A) receptor facilitation of hippocampal synaptic transmission is dependent on tonic A (1) receptor inhibition.". Neuroscience 112 (2), 319–329. [DOI] [PubMed] [Google Scholar]
- Loscher W, 2017. “Animal models of seizures and epilepsy: past, present, and future role for the discovery of antiseizure drugs.”. Neurochem. Res 42 (7), 1873–1888. [DOI] [PubMed] [Google Scholar]
- Loscher W, 2020. “The holy grail of epilepsy prevention: preclinical approaches to antiepileptogenic treatments.”. Neuropharmacology 167, 107605. [DOI] [PubMed] [Google Scholar]
- Lovatt D, Xu Q, Liu W, Takano T, Smith NA, Schnermann J, Tieu K, Nedergaard M, 2012. “Neuronal adenosine release, and not astrocytic ATP release, mediates feedback inhibition of excitatory activity.”. Proc. Natl. Acad. Sci. U. S. A 109 (16), 6265–6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusardi TA, Lytle NK, Szybala C, Boison D, 2012. “Caffeine prevents acute mortality after TBI in rats without increased morbidity.”. Exp. Neurol 234 (1), 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusardi TA, Akula KK, Coffman SQ, Ruskin DN, Masino SA, Boison D, 2015. "Ketogenic diet prevents epileptogenesis and disease progression in adult mice and rats.". Neuropharmacology 99, 500–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi M, Raiteri L, Risso F, Vallarino A, Bonfanti A, Monopoli A, Ongini E, Raiteri M, 2002. “Effects of adenosine A1 and A2A receptor activation on the evoked release of glutamate from rat cerebrocortical synaptosomes.”. Br. J. Pharmacol 136 (3), 434–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mares P, 2010. “Anticonvulsant action of 2-chloroadenosine against pentetrazol-induced seizures in immature rats is due to activation of A1 adenosine receptors.”. J. Neural Transm. (Vienna) 117 (11), 1269–1277. [DOI] [PubMed] [Google Scholar]
- Maroso M, Balosso S, Ravizza T, Liu J, Aronica E, Iyer AM, Rossetti C, Molteni M, Casalgrandi M, Manfredi AA, Bianchi ME, Vezzani A, 2010. “Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures.”. Nat. Med 16 (4), 413–419. [DOI] [PubMed] [Google Scholar]
- Martin-McGill KJ, Jackson CF, Bresnahan R, Levy RG, Cooper PN, 2018. “Ketogenic diets for drug-resistant epilepsy.”. Cochrane Database Syst. Rev 11, Cd001903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masino SA, Geiger JD, 2008. “Are purines mediators of the anticonvulsant/ neuroprotective effects of ketogenic diets?”. Trends Neurosci 31 (6), 273–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masino SA, Geiger JD, 2009. "The ketogenic diet and epilepsy: is adenosine the missing link?". Epilepsia 50 (2), 332–333. [DOI] [PubMed] [Google Scholar]
- Masino SA, Li T, Theofilas P, Sandau US, Ruskin DN, Fredholm BB, Geiger JD, Aronica E, Boison D, 2011. “A ketogenic diet suppresses seizures in mice through adenosine A(1) receptors.”. J. Clin. Invest 121 (7), 2679–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masino SA, Kawamura M Jr., Ruskin DN, Geiger JD, Boison D, 2012. “Purines and neuronal excitability: links to the ketogenic diet.”. Epilepsy Res 100 (3), 229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masse K, Dale N, 2012. “Purines as potential morphogens during embryonic development.”. Purinergic Signal 8 (3), 503–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masse K, Bhamra S, Allsop G, Dale N, Jones EA, 2010. “Ectophosphodiesterase/nucleotide phosphohydrolase (Enpp) nucleotidases: cloning, conservation and developmental restriction.”. Int. J. Dev. Biol 54 (1), 181–193. [DOI] [PubMed] [Google Scholar]
- McGaraughty S, Cowart M, Jarvis MF, Berman RF, 2005. “Anticonvulsant and antinociceptive actions of novel adenosine kinase inhibitors.”. Curr. Top. Med. Chem 5 (1), 43–58. [DOI] [PubMed] [Google Scholar]
- Melani A, Turchi D, Vannucchi MG, Cipriani S, Gianfriddo M, Pedata F, 2005. “ATP extracellular concentrations are increased in the rat striatum during in vivo ischemia.”. Neurochem. Int 47 (6), 442–448. [DOI] [PubMed] [Google Scholar]
- Menendez Mendez A, Smith J, Engel T, 2020. “Neonatal seizures and purinergic signalling.”. Int. J. Mol. Sci 21 (21). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesuret G, Engel T, Hessel EV, Sanz-Rodriguez A, Jimenez-Pacheco A, Miras-Portugal MT, Diaz-Hernandez M, Henshall DC, 2014. “P2X7 receptor inhibition interrupts the progression of seizures in immature rats and reduces hippocampal damage.”. CNS Neurosci. Ther 20 (6), 556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milior G, Morin-Brureau M, Chali F, Le Duigou C, Savary E, Huberfeld G, Rouach N, Pallud J, Capelle L, Navarro V, Mathon B, Clemenceau S, Miles R, 2020. “Distinct P2Y receptors mediate extension and retraction of microglial processes in epileptic and peri-tumoral human tissue.”. J. Neurosci [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller-Delaney SF, Bryan K, Das S, McKiernan RC, Bray IM, Reynolds JP, Gwinn R, Stallings RL, Henshall DC, 2015. "Differential DNA methylation profiles of coding and non-coding genes define hippocampal sclerosis in human temporal lobe epilepsy.". Brain 138 (Pt 3), 616–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miras-Portugal MT, Sebastian-Serrano A, de Diego Garcia L, Diaz-Hernandez M, 2017. “Neuronal P2X7 receptor: involvement in neuronal physiology and pathology.”. J. Neurosci 37 (30), 7063–7072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miras-Portugal MT, Queipo MJ, Gil-Redondo JC, Ortega F, Gomez-Villafuertes R, Gualix J, Delicado EG, Perez-Sen R, 2019. “P2 receptor interaction and signalling cascades in neuroprotection.”. Brain Res. Bull 151, 74–83. [DOI] [PubMed] [Google Scholar]
- Mo M, Eyo UB, Xie M, Peng J, Bosco DB, Umpierre AD, Zhu X, Tian DS, Xu P, Wu LJ, 2019. “Microglial P2Y12 receptor regulates seizure-induced neurogenesis and immature neuronal projections.”. J. Neurosci 39 (47), 9453–9464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monif M, Reid CA, Powell KL, Smart ML, Williams DA, 2009. “The P2X7 receptor drives microglial activation and proliferation: a trophic role for P2X7R pore.”. J. Neurosci 29 (12), 3781–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan J, Alves M, Conte G, Menendez-Mendez A, de Diego-Garcia L, de Leo G, Beamer E, Smith J, Nicke A, Engel T, 2020. “Characterization of the expression of the ATP-Gated P2X7 receptor following status epilepticus and during epilepsy using a P2X7-EGFP reporter mouse.”. Neurosci. Bull [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moshe SL, Perucca E, Ryvlin P, Tomson T, 2015. "Epilepsy: new advances.". Lancet 385 (9971), 884–898. [DOI] [PubMed] [Google Scholar]
- Muzzi M, Coppi E, Pugliese AM, Chiarugi A, 2013. “Anticonvulsant effect of AMP by direct activation of adenosine A1 receptor.”. Exp. Neurol 250, 189–193. [DOI] [PubMed] [Google Scholar]
- Nieoczym D, Socala K, Wlaz P, 2017. “Evaluation of the anticonvulsant effect of brilliant blue g, a selective P2X7 receptor antagonist, in the iv PTZ-, maximal electroshock-, and 6 hz-induced seizure tests in mice.”. Neurochem. Res 42 (11), 3114–3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolic L, Shen W, Nobili P, Virenque A, Ulmann L, Audinat E, 2018. "Blocking TNFalpha-driven astrocyte purinergic signaling restores normal synaptic activity during epileptogenesis.". Glia 66 (12), 2673–2683. [DOI] [PubMed] [Google Scholar]
- Orellana JA, Froger N, Ezan P, Jiang JX, Bennett MV, Naus CC, Giaume C, Saez JC, 2011. “ATP and glutamate released via astroglial connexin 43 hemichannels mediate neuronal death through activation of pannexin 1 hemichannels.”. J. Neurochem 118 (5), 826–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankratov Y, Lalo U, Krishtal OA, Verkhratsky A, 2009. "P2X receptors and synaptic plasticity.". Neuroscience 158 (1), 137–148. [DOI] [PubMed] [Google Scholar]
- Patel RS, Elmaadawi A, Mansuri Z, Kaur M, Shah K, Nasr S, 2017. “Psychiatric comorbidities and outcomes in epilepsy patients: an insight from a nationwide inpatient analysis in the United States.”. Cureus 9 (9), e1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peleli M, Fredholm BB, Sobrevia L, Carlstrom M, 2017. “Pharmacological targeting of adenosine receptor signaling.”. Mol. Aspects Med 55, 4–8. [DOI] [PubMed] [Google Scholar]
- Picher M, Burch LH, Boucher RC, 2004. “Metabolism of P2 receptor agonists in human airways: implications for mucociliary clearance and cystic fibrosis.”. J. Biol. Chem 279 (19), 20234–20241. [DOI] [PubMed] [Google Scholar]
- Pitkanen A, Engel J Jr., 2014. "Past and present definitions of epileptogenesis and its biomarkers.". Neurotherapeutics 11 (2), 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkanen A, Immonen RJ, Grohn OH, Kharatishvili I, 2009. "From traumatic brain injury to posttraumatic epilepsy: what animal models tell us about the process and treatment options.". Epilepsia 50 (Suppl 2), 21–29. [DOI] [PubMed] [Google Scholar]
- Pitkanen A, Lukasiuk K, Dudek FE, Staley KJ, 2015. “Epileptogenesis.”. Cold Spring Harb. Perspect. Med 5 (10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plesner L, 1995. “Ecto-ATPases: identities and functions.”. Int. Rev. Cytol 158, 141–214. [DOI] [PubMed] [Google Scholar]
- Rappold PM, Lynd-Balta E, Joseph SA, 2006. “P2X7 receptor immunoreactive profile confined to resting and activated microglia in the epileptic brain.”. Brain Res 1089 (1), 171–178. [DOI] [PubMed] [Google Scholar]
- Rassendren F, Audinat E, 2016. “Purinergic signaling in epilepsy.”. J. Neurosci. Res 94 (9), 781–793. [DOI] [PubMed] [Google Scholar]
- Rebola N, Porciuncula LO, Lopes LV, Oliveira CR, Soares-da-Silva P, Cunha RA, 2005. "Long-term effect of convulsive behavior on the density of adenosine A1 and A 2A receptors in the rat cerebral cortex.". Epilepsia 46 (Suppl 5), 159–165. [DOI] [PubMed] [Google Scholar]
- Rebola N, Lujan R, Cunha RA, Mulle C, 2008. "Adenosine A2A receptors are essential for long-term potentiation of NMDA-EPSCs at hippocampal mossy fiber synapses.". Neuron 57 (1), 121–134. [DOI] [PubMed] [Google Scholar]
- Rebola N, Simoes AP, Canas PM, Tome AR, Andrade GM, Barry CE, Agostinho PM, Lynch MA, Cunha RA, 2011. “Adenosine A2A receptors control neuroinflammation and consequent hippocampal neuronal dysfunction.”. J. Neurochem 117 (1), 100–111. [DOI] [PubMed] [Google Scholar]
- Reklow RJ, Alvares TS, Zhang Y, Miranda Tapia AP, Biancardi V, Katzell AK, Frangos SM, Hansen MA, Toohey AW, Cass CE, Young JD, Pagliardini S, Boison D, Funk GD, 2019. “The purinome and the preBotzinger complex - a menage of unexplored mechanisms that may Modulate/Shape the hypoxic ventilatory response.”. Front. Cell. Neurosci 13, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren G, Li T, Lan JQ, Wilz A, Simon RP, Boison D, 2007. “Lentiviral RNAi-induced downregulation of adenosine kinase in human mesenchymal stem cell grafts: a novel perspective for seizure control.”. Exp. Neurol 208 (1), 26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robel S, Buckingham SC, Boni JL, Campbell SL, Danbolt NC, Riedemann T, Sutor B, Sontheimer H, 2015. “Reactive astrogliosis causes the development of spontaneous seizures.”. J. Neurosci 35 (8), 3330–3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson CL, Bell MJ, Kochanek PM, Adelson PD, Ruppel RA, Carcillo JA, Wisniewski SR, Mi Z, Janesko KL, Clark RS, Marion DW, Graham SH, Jackson EK, 2001. “Increased adenosine in cerebrospinal fluid after severe traumatic brain injury in infants and children: association with severity of injury and excitotoxicity.”. Crit. Care Med 29 (12), 2287–2293. [DOI] [PubMed] [Google Scholar]
- Robson SC, Sevigny J, Zimmermann H, 2006. “The E-NTPDase family of ectonucleotidases: structure function relationships and pathophysiological significance.”. Purinergic Signal 2 (2), 409–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues FB, Wild EJ, 2016. “Psychogenic non-epileptic seizures in early Huntington’s disease.”. Pract. Neurol 16 (6), 452–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Alvarez N, Jimenez-Mateos EM, Engel T, Quinlan S, Reschke CR, Conroy RM, Bhattacharya A, Boylan GB, Henshall DC, 2017. "Effects of P2X7 receptor antagonists on hypoxia-induced neonatal seizures in mice.". Neuropharmacology 116, 351–363. [DOI] [PubMed] [Google Scholar]
- Roseti C, Martinello K, Fucile S, Piccari V, Mascia A, Di Gennaro G, Quarato PP, Manfredi M, Esposito V, Cantore G, Arcella A, Simonato M, Fredholm BB, Limatola C, Miledi R, Eusebi F, 2008. “Adenosine receptor antagonists alter the stability of human epileptic GABAA receptors.”. Proc. Natl. Acad. Sci. U. S. A 105 (39), 15118–15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozmer K, Gao P, Araujo MGL, Khan MT, Liu J, Rong W, Tang Y, Franke H, Krugel U, Fernandes MJS, Illes P, 2017. “Pilocarpine-Induced status epilepticus increases the sensitivity of P2X7 and P2Y1 receptors to nucleotides at neural progenitor cells of the juvenile rodent Hippocampus.”. Cereb. Cortex 27 (7), 3568–3585. [DOI] [PubMed] [Google Scholar]
- Ruigrok AN, Salimi-Khorshidi G, Lai MC, Baron-Cohen S, Lombardo MV, Tait RJ, Suckling J, 2014. “A meta-analysis of sex differences in human brain structure.”. Neurosci. Biobehav. Rev 39, 34–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandau US, Yahya M, Bigej R, Friedman JL, Saleumvong B, Boison D, 2019. "Transient use of a systemic adenosine kinase inhibitor attenuates epilepsy development in mice.". Epilepsia 60 (4), 615–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawynok J, 2016. "Adenosine receptor targets for pain.". Neuroscience 338, 1–18. [DOI] [PubMed] [Google Scholar]
- Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, Hirsch E, Jain S, Mathern GW, Moshe SL, Nordli DR, Perucca E, Tomson T, Wiebe S, Zhang YH, Zuberi SM, 2017. “ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology.”. Epilepsia 58 (4), 512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schetinger MR, Morsch VM, Bonan CD, Wyse AT, 2007. "NTPDase and 5’-nucleotidase activities in physiological and disease conditions: new perspectives for human health.". Biofactors 31 (2), 77–98. [DOI] [PubMed] [Google Scholar]
- Schwarz JM, Bilbo SD, 2012. “Sex, glia, and development: interactions in health and disease.”. Horm. Behav 62 (3), 243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwiebert EM, Zsembery A, 2003. “Extracellular ATP as a signaling molecule for epithelial cells.”. Biochim. Biophys. Acta 1615 (1–2), 7–32. [DOI] [PubMed] [Google Scholar]
- Sebastian-Serrano A, de Diego-Garcia L, Martinez-Frailes C, Avila J, Zimmermann H, Millan JL, Miras-Portugal MT, Diaz-Hernandez M, 2015. Tissue-nonspecific alkaline phosphatase regulates purinergic transmission in the central nervous system during development and disease. Comput. Struct. Biotechnol. J 13, 95–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian-Serrano A, Engel T, de Diego-Garcia L, Olivos-Ore LA, Arribas-Blazquez M, Martinez-Frailes C, Perez-Diaz C, Millan JL, Artalejo AR, Miras-Portugal MT, Henshall DC, Diaz-Hernandez M, 2016. Neurodevelopmental alterations and seizures developed by mouse model of infantile hypophosphatasia are associated with purinergic signalling deregulation. Hum. Mol. Genet 25 (19), 4143–4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seminario-Vidal L, Lazarowski ER, Okada SF, 2009. Assessment of extracellular ATP concentrations. Methods Mol. Biol 574, 25–36. [DOI] [PubMed] [Google Scholar]
- Shen HY, Li T, Boison D, 2010. A novel mouse model for sudden unexpected death in epilepsy (SUDEP): role of impaired adenosine clearance. Epilepsia 51 (3), 465–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HY, Singer P, Lytle N, Wei CJ, Lan JQ, Williams-Karnesky RL, Chen JF, Yee BK, Boison D, 2012. Adenosine augmentation ameliorates psychotic and cognitive endophenotypes of schizophrenia. J. Clin. Invest 122 (7), 2567–2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoes AP, Silva CG, Marques JM, Pochmann D, Porciuncula LO, Ferreira S, Oses JP, Beleza RO, Real JI, Kofalvi A, Bahr BA, Lerma J, Cunha RA, Rodrigues RJ, 2018. Glutamate-induced and NMDA receptor-mediated neurodegeneration entails P2Y1 receptor activation. Cell Death Dis 9 (3), 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims RE, Dale N, 2014. Activity-dependent adenosine release may be linked to activation of Na( )-K( ) ATPase: an in vitro rat study. PLoS One 9 (1), e87481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirven JI, 2015. Epilepsy: a Spectrum disorder. Cold Spring Harb. Perspect. Med 5 (9), a022848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solle M, Labasi J, Perregaux DG, Stam E, Petrushova N, Koller BH, Griffiths RJ, Gabel CA, 2001. Altered cytokine production in mice lacking P2X(7) receptors. J. Biol. Chem 276 (1), 125–132. [DOI] [PubMed] [Google Scholar]
- Soni N, Koushal P, Reddy BV, Deshmukh R, Kumar P, 2015. Effect of GLT-1 modulator and P2X7 antagonists alone and in combination in the kindling model of epilepsy in rats. Epilepsy Behav 48, 4–14. [DOI] [PubMed] [Google Scholar]
- Sperlagh B, Illes P, 2014. P2X7 receptor: an emerging target in central nervous system diseases. Trends Pharmacol. Sci 35 (10), 537–547. [DOI] [PubMed] [Google Scholar]
- Sperlagh B, Kofalvi A, Deuchars J, Atkinson L, Milligan CJ, Buckley NJ, Vizi ES, 2002. Involvement of P2X7 receptors in the regulation of neurotransmitter release in the rat hippocampus. J. Neurochem 81 (6), 1196–1211. [DOI] [PubMed] [Google Scholar]
- Sperlagh B, Vizi ES, Wirkner K, Illes P, 2006. P2X7 receptors in the nervous system. Prog. Neurobiol 78 (6), 327–346. [DOI] [PubMed] [Google Scholar]
- Stafstrom CE, Rho JM, 2012. The ketogenic diet as a treatment paradigm for diverse neurological disorders. Front. Pharmacol 3, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenberg D, Litonius E, Halldner L, Johansson B, Fredholm BB, Porkka-Heiskanen T, 2003. Sleep and its homeostatic regulation in mice lacking the adenosine A1 receptor. J. Sleep Res 12 (4), 283–290. [DOI] [PubMed] [Google Scholar]
- Stockwell J, Jakova E, Cayabyab FS, 2017. Adenosine A1 and A2A receptors in the brain: current research and their role in neurodegeneration. Molecules 22 (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strater N, 2006. Ecto-5’-nucleotidase: structure function relationships. Purinergic Signal 2 (2), 343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer FE, Fedele DE, Marowsky A, Schwerdel C, Wernli K, Vogt K, Fritschy JM, Boison D, 2006. Shift of adenosine kinase expression from neurons to astrocytes during postnatal development suggests dual functionality of the enzyme. Neuroscience 142 (1), 125–137. [DOI] [PubMed] [Google Scholar]
- Suadicani SO, Brosnan CF, Scemes E, 2006. P2X7 receptors mediate ATP release and amplification of astrocytic intercellular Ca2 signaling. J. Neurosci 26 (5), 1378–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukigara S, Dai H, Nabatame S, Otsuki T, Hanai S, Honda R, Saito T, Nakagawa E, Kaido T, Sato N, Kaneko Y, Takahashi A, Sugai K, Saito Y, Sasaki M, Goto Y, Koizumi S, Itoh M, 2014. Expression of astrocyte-related receptors in cortical dysplasia with intractable epilepsy. J. Neuropathol. Exp. Neurol 73 (8), 798–806. [DOI] [PubMed] [Google Scholar]
- Sun H, Ma L, Zhang Y, Pan X, Wang C, Zhang J, Zhang X, Sun H, Wang Q, Zhu W, 2018. A purinergic P2 receptor family-mediated increase in Thrombospondin-1 bolsters synaptic density and epileptic seizure activity in the amygdala-kindling rat model. Front. Cell. Neurosci 12, 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szybala C, Pritchard EM, Lusardi TA, Li T, Wilz A, Kaplan DL, Boison D, 2009. Antiepileptic effects of silk-polymer based adenosine release in kindled rats. Exp. Neurol 219 (1), 126–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao JX, Qian S, Baldwin M, Chen XJ, Rose S, Ebersole SH, Ebersole JS, 2010. SUDEP, suspected positional airway obstruction, and hypoventilation in postictal coma. Epilepsia 51 (11), 2344–2347. [DOI] [PubMed] [Google Scholar]
- Taylor RS, Sander JW, Taylor RJ, Baker GA, 2011. Predictors of health-related quality of life and costs in adults with epilepsy: a systematic review. Epilepsia 52 (12), 2168–2180. [DOI] [PubMed] [Google Scholar]
- Theofilas P, Brar S, Stewart KA, Shen HY, Sandau US, Poulsen D, Boison D, 2011. Adenosine kinase as a target for therapeutic antisense strategies in epilepsy. Epilepsia 52 (3), 589–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thijs RD, Surges R, O’Brien TJ, Sander JW, 2019. Epilepsy in adults. Lancet 393 (10172), 689–701. [DOI] [PubMed] [Google Scholar]
- Timmers M, Ravenstijn P, Xi L, Triana-Baltzer G, Furey M, Van Hemelryck S, Biewenga J, Ceusters M, Bhattacharya A, van den Boer M, van Nueten L, de Boer P, 2018. Clinical pharmacokinetics, pharmacodynamics, safety, and tolerability of JNJ-54175446, a brain permeable P2X7 antagonist, in a randomised single-ascending dose study in healthy participants. J. Psychopharmacol 32 (12), 1341–1350. [DOI] [PubMed] [Google Scholar]
- Tosh DK, Paoletta S, Deflorian F, Phan K, Moss SM, Gao ZG, Jiang X, Jacobson KA, 2012. Structural sweet spot for A1 adenosine receptor activation by truncated (N)-methanocarba nucleosides: receptor docking and potent anticonvulsant activity. J. Med. Chem 55 (18), 8075–8090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugarkar BG, DaRe JM, Kopcho JJ, Browne CE 3rd, Schanzer JM, Wiesner JB, Erion MD, 2000. Adenosine kinase inhibitors. 1. Synthesis, enzyme inhibition, and antiseizure activity of 5-iodotubercidin analogues. J. Med. Chem 43 (15), 2883–2893. [DOI] [PubMed] [Google Scholar]
- Ulmann L, Levavasseur F, Avignone E, Peyroutou R, Hirbec H, Audinat E, Rassendren F, 2013. Involvement of P2X4 receptors in hippocampal microglial activation after status epilepticus. Glia 61 (8), 1306–1319. [DOI] [PubMed] [Google Scholar]
- Van Gompel JJ, Bower MR, Worrell GA, Stead M, Chang SY, Goerss SJ, Kim I, Bennet KE, Meyer FB, Marsh WR, Blaha CD, Lee KH, 2014. "Increased cortical extracellular adenosine correlates with seizure termination.". Epilepsia 55 (2), 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandam RJ, Shields EJ, Kelty JD, 2008. “Rhythm generation by the pre-Botzinger complex in medullary slice and island preparations: effects of adenosine A(1) receptor activation.”. BMC Neurosci 9, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velioglu SK, Boz C, Ozmenoglu M, 2005. "The impact of migraine on epilepsy: a prospective prognosis study.". Cephalalgia 25 (7), 528–535. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Baram TZ, 2007. “New roles for interleukin-1 Beta in the mechanisms of epilepsy.”. Epilepsy Curr 7 (2), 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Balosso S, Ravizza T, 2019. “Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy.”. Nat. Rev. Neurol 15 (8), 459–472. [DOI] [PubMed] [Google Scholar]
- Vianna EP, Ferreira AT, Naffah-Mazzacoratti MG, Sanabria ER, Funke M, Cavalheiro EA, Fernandes MJ, 2002. "Evidence that ATP participates in the pathophysiology of pilocarpine-induced temporal lobe epilepsy: fluorimetric, immunohistochemical, and Western blot studies.". Epilepsia 43 (Suppl 5), 227–229. [DOI] [PubMed] [Google Scholar]
- Vianna EP, Ferreira AT, Dona F, Cavalheiro EA, da Silva Fernandes MJ, 2005. "Modulation of seizures and synaptic plasticity by adenosinergic receptors in an experimental model of temporal lobe epilepsy induced by pilocarpine in rats.". Epilepsia 46 (Suppl 5), 166–173. [DOI] [PubMed] [Google Scholar]
- von Kugelgen I, 2019. “Pharmacology of P2Y receptors.”. Brain Res. Bull 151, 12–24. [DOI] [PubMed] [Google Scholar]
- von Kugelgen I, Hoffmann K, 2016. "Pharmacology and structure of P2Y receptors.". Neuropharmacology 104, 50–61. [DOI] [PubMed] [Google Scholar]
- Von Lubitz DK, Paul IA, Ji XD, Carter M, Jacobson KA, 1994. “Chronic adenosine A1 receptor agonist and antagonist: effect on receptor density and N-methyl-D-aspartate induced seizures in mice.”. Eur. J. Pharmacol 253 (1–2), 95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Ranasinghe KG, Beagle AJ, Mizuiri D, Honma SM, Dowling AF, Darwish SM, Van Berlo V, Barnes DE, Mantle M, Karydas AM, Coppola G, Roberson ED, Miller BL, Garcia PA, Kirsch HE, Mucke L, Nagarajan SS, 2016. “Incidence and impact of subclinical epileptiform activity in Alzheimer’s disease.”. Ann. Neurol 80 (6), 858–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Tartaglia MC, Nygaard HB, Zeman AZ, Miller BL, 2017. “Epileptic activity in Alzheimer’s disease: causes and clinical relevance.”. Lancet Neurol 16 (4), 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall M, Dale N, 2008. “Activity-dependent release of adenosine: a critical reevaluation of mechanism.”. Curr. Neuropharmacol 6 (4), 329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall MJ, Dale N, 2013. “Neuronal transporter and astrocytic ATP exocytosis underlie activity-dependent adenosine release in the hippocampus.”. J. Physiol 591 (16), 3853–3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall MJ, Atterbury A, Dale N, 2007. “Control of basal extracellular adenosine concentration in rat cerebellum.”. J. Physiol 582 (Pt 1), 137–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellmann M, Alvarez-Ferradas C, Maturana CJ, Saez JC, Bonansco C, 2018. “Astroglial Ca(2 )-Dependent hyperexcitability requires P2Y1 purinergic receptors and Pannexin-1 channel activation in a chronic model of epilepsy.”. Front. Cell. Neurosci 12, 446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieraszko A, Seyfried TN, 1989. “Increased amount of extracellular ATP in stimulated hippocampal slices of seizure prone mice.”. Neurosci. Lett 106 (3), 287–293. [DOI] [PubMed] [Google Scholar]
- Wiesner JB, Ugarkar BG, Castellino AJ, Barankiewicz J, Dumas DP, Gruber HE, Foster AC, Erion MD, 1999. “Adenosine kinase inhibitors as a novel approach to anticonvulsant therapy.”. J. Pharmacol. Exp. Ther 289 (3), 1669–1677. [PubMed] [Google Scholar]
- Williams-Karnesky RL, Sandau US, Lusardi TA, Lytle NK, Farrell JM, Pritchard EM, Kaplan DL, Boison D, 2013. “Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis.”. J. Clin. Invest 123 (8), 3552–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu PH, Phillis JW, 1978. “Distribution and release of adenosine triphosphate in rat brain.”. Neurochem. Res 3 (5), 563–571. [DOI] [PubMed] [Google Scholar]
- Wu LG, Saggau P, 1994. "Adenosine inhibits evoked synaptic transmission primarily by reducing presynaptic calcium influx in area CA1 of hippocampus.". Neuron 12 (5), 1139–1148. [DOI] [PubMed] [Google Scholar]
- Xia J, Lim JC, Lu W, Beckel JM, Macarak EJ, Laties AM, Mitchell CH, 2012. “Neurons respond directly to mechanical deformation with pannexin-mediated ATP release and autostimulation of P2X7 receptors.”. J. Physiol 590 (10), 2285–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Wang H, Zhang Q, Han Z, 2018. “Modulation of P2X purinoceptor 3 (P2X3) in pentylenetetrazole-induced kindling epilepsy in rats.”. Med. Sci. Monit 24, 6165–6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee BK, Singer P, Chen JF, Feldon J, Boison D, 2007. “Transgenic overexpression of adenosine kinase in brain leads to multiple learning impairments and altered sensitivity to psychomimetic drugs.”. Eur. J. Neurosci 26 (11), 3237–3252. [DOI] [PubMed] [Google Scholar]
- Yegutkin GG, Mikhailov A, Samburski SS, Jalkanen S, 2006. “The detection of micromolar pericellular ATP pool on lymphocyte surface by using lymphoid ectoadenylate kinase as intrinsic ATP sensor.”. Mol. Biol. Cell 17 (8), 3378–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon KW, Rothman SM, 1991. “Adenosine inhibits excitatory but not inhibitory synaptic transmission in the hippocampus.”. J. Neurosci 11 (5), 1375–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Ma LM, Xiong Y, Huang H, Yuan JX, Li RH, Li JN, Chen YM, 2016. “Upregulated P2X3 receptor expression in patients with intractable temporal lobe epilepsy and in a rat model of epilepsy.”. Neurochem. Res 41 (6), 1263–1273. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Wang L, Zhang Y, Zhao FH, Luo J, Xiao Z, Chen GJ, Wang XF, 2012. “Increased expression of DNA methyltransferase 1 and 3a in human temporal lobe epilepsy.”. J. Mol. Neurosci 46 (2), 420–426. [DOI] [PubMed] [Google Scholar]
- Zimmermann H, 2000. “Extracellular metabolism of ATP and other nucleotides.”. Naunyn Schmiedebergs Arch. Pharmacol 362 (4–5), 299–309. [DOI] [PubMed] [Google Scholar]