

Abstract
Proteolysis-targeting chimeras (PROTACs) are heterobifunctional molecules consisting of one ligand that binds to a protein of interest (POI) and another that can recruit an E3 ubiquitin ligase. The chemically-induced proximity between the POI and E3 ligase results in ubiquitination and subsequent degradation of the POI by the ubiquitin-proteasome system (UPS). The event-driven mechanism of action (MOA) of PROTACs offers several advantages compared to traditional occupancy-driven small molecule inhibitors, such as a catalytic nature, reduced dosing and dosing frequency, a more potent and longer-lasting effect, an added layer of selectivity to reduce potential toxicity, efficacy in the face of drug-resistance mechanisms, targeting nonenzymatic functions, and expanded target space. Here, we highlight important milestones and briefly discuss lessons learned about targeted protein degradation (TPD) in recent years and conjecture on the efforts still needed to expand the toolbox for PROTAC discovery to ultimately provide promising therapeutics.
Graphical Abstarct
This review highlights important milestones in the evolution of PROTACs, briefly discusses recent lessons about targeted protein degradation, and presents future directions, such as expanding E3 ligase toolbox, mitigating cell permeability and drug resistance and targeting virus-related proteins or virus RNA for degradation.
Introduction
Many disease-related novel protein targets have been discovered as a result of the genomic revolution. However, classical strategies for small molecule inhibitor (SMI) development are unable to exploit all of them, due to the lack of suitable binding pockets that directly regulate protein function[1]. Although biologic modalities such as monoclonal antibodies and oligonucleotide therapies can provide opportunities to address such targets, a common major limitation is restricted delivery due to poor cell membrane permeability. Another limitation of SMIs is that high systemic exposure is usually required to achieve sufficient site occupancy to impact downstream biological process(es), which potentially increases the risk of off-target toxicity. Furthermore, because SMIs target the active site of proteins (e.g. kinases) this often limits their selectivity among homologous proteins due to the conserved functional domains. The more recently developed strategy of targeting specific proteins for post-translational degradation, rather than mere inhibition, holds the key to circumventing these therapeutic impasses.
Protein degradation provides a mechanism of quality control during protein folding, which is a normal process within the cell. Most proteins will undergo degradation through the ubiquitin-proteasome system (UPS)[2], wherein proteins are first labelled covalently with ubiquitin[3], a highly conserved protein containing 76 amino acids (8.6 kDa), and subsequently sent to 26S proteasome for degradation[4]. Ubiquitylation of proteins, a post-translational modification (PTM) process, is carried out by a cascade of three enzymes (Figure 1). In the first step, an E1 ubiquitin-activating enzyme activates ubiquitin by using ATP to produce an activated ubiquitin–adenylate, which is converted to a thioester intermediate by attaching to a catalytic cysteine in the E1 enzyme. In the second step, ubiquitin is transferred to the catalytic cysteine of an E2 ubiquitin-conjugation enzyme through a transthioesterification reaction. Finally, ubiquitin is transferred to the substrate protein via a ternary complex formed between the E2 enzyme, an E3 ubiquitin ligase and the substrate, leading to the formation of an isopeptide bond between the carboxy terminus of ubiquitin and the side chain of a lysine residue on the target protein surface. Depending on the E3 ligase class, ubiquitin can be transferred directly from E2 to the substrate or sequentially relayed from E2 to E3, and then to the substrate. This ubiquitination cascade can be repeated to generate a poly-ubiquitin chain on the substrate which will be recognized and degraded by the 26S proteasome.
Figure 1.

PROTACs hijack the UPS to induce targeted protein degradation. The figure was created with BioRender.com.
The PROTAC technology hijacks the UPS to degrade a specific protein of interest (POI). PROTACs consist of one ligand that binds to a POI and another that can recruit an E3 ubiquitin ligase, with both ligands connected to each other by a linker. This chemically-induced proximity between the POI and E3 ligase results in the ubiquitination and subsequent degradation of the POI by the UPS (Figure 1).
This review highlights important milestones in the evolution of PROTACs, briefly discusses recent lessons about targeted protein degradation (TPD) and presents future directions for this promising drug discovery approach.
Past:
Peptide-based PROTACs
In 2001, our laboratory, together with the Deshaies laboratory, reported the first proof-of-concept bifunctional molecules, termed PROTACs (for “PROteolysis TArgeting Chimeras”), that recruited the enzyme methionine aminopeptidase 2 (MetAP-2) to the E3 ligase complex F-box protein “β-transducin repeat-containing protein” (SCFβTRCP) and thereby promoted MetAP-2 ubiquitination and degradation[5]. One domain of this PROTAC was comprised of ovalicin, which covalently binds to MetAP-2; whereas the other structural component is the IκBα phosphopeptide that is recognized and bound by the SCFβTRCP. However, the dependency on phosphorylation of IκBα phosphopeptide for E3 ligase recruitment was limiting.
The first improvement in peptidic PROTACs was the recruiting of the von Hippel–Lindau (VHL) E3 ligase by using a seven-amino-acid recognition sequence derived from the ligase’s endogenous substrate, hypoxia-inducible factor 1α (HIF1α)[6]. An eight-D-arginine tag was included on the C-terminus of the peptide sequence to confer cell permeability and decrease nonspecific proteolysis, thereby circumventing the need for delivery of the PROTAC into cells by microinjection, as had been done with the SCFβTRCP-recruiting antecedents previously[7]. This was the first example of a synthesized molecule which alone could induce targeted protein degradation simply upon addition to cells.
As a new modality, PROTACs provides a novel approach to the study of protein function without genetically modifying the host cell. However, the potencies of these early peptidic PROTACs remained in the micromolar range. For instance, a 25 μM concentration of PROTAC was needed to induce GFP-AR degradation[6], probably due to its limited cell penetration. Another problem for these peptide-based PROTACs was the size of the chimeric molecule, which might in vivo be recognized by the immune system to produce antibodies that may neutralize the degradation effect.
Accordingly, much effort was increasingly focused on developing all small-molecule PROTACs. Once this was achieved, the strength of the PROTAC modality become fully realized and demonstrated.
Small-molecule PROTACs
The first all small molecule PROTAC was reported by our research group in 2008[8]. The mouse double minute 2 homologue (MDM2) E3 ligase was first recruited by employing nutlin-3a, a known MDM2-p53 protein–protein interaction (PPI) inhibitor, as the E3 ligand. The selective androgen receptor modulator (SARM) flutamide derivative was utilized as the androgen receptor (AR) ligand, which potently binds AR with a Ki of 4 nM. Although this study demonstrated that developing cell permeable PROTACs is feasible, the induced AR degradation was observed at a concentration of 10 μM (Figure 2, PROTAC 14).
Figure 2.

Early small-molecule PROTACs.
In 2010, the Hashimoto laboratory reported the first cellular inhibitor of apoptosis protein 1 (cIAP1)-recruiting PROTAC (Figure 2, PROTAC 4a-c). The E3 ligase, cIAP1, was recruited by using methyl bestatin as a ligand to induce cellular retinol- and retinoic acid-binding proteins (CRABP-I and II) for degradation[9, 10]. The all-trans retinoic acid was used as the targeting ligand of the PROTACs. However, cIAP1-based PROTACs can also promote the autoubiquitination and degradation[11, 12] of the E3 ligase itself, thus limiting the full potential of the technology.
In 2010, Ito et al. identified the E3 ligase cereblon (CRBN) as the primary target of the ligand, thalidomide and its analogs[13]. In 2014, Fischer et al. reported the structural basis of these Immunomodulatory imide drugs (IMiDs) (Figure 3) in binding with CRBN. IMiDs block endogenous substrates (MEIS2) from binding to CRL4CRBN while at the same time recruiting neosubstrates IKZF1 or IKZF3 for their contrived degradation[14]. This structural information paved the way for the application of IMiDs as E3 recruiting ligands for PROTAC development, which will be discussed later. Recently, several neosubstrates, such as IKZF1, IKZF3, casein kinase 1α (CK1α)[15-16], GSPT1[17] and SALL4[18-20], have been successfully degraded by way of IMiD-glued, CRBN-mediated ubiquitination.
Figure 3.
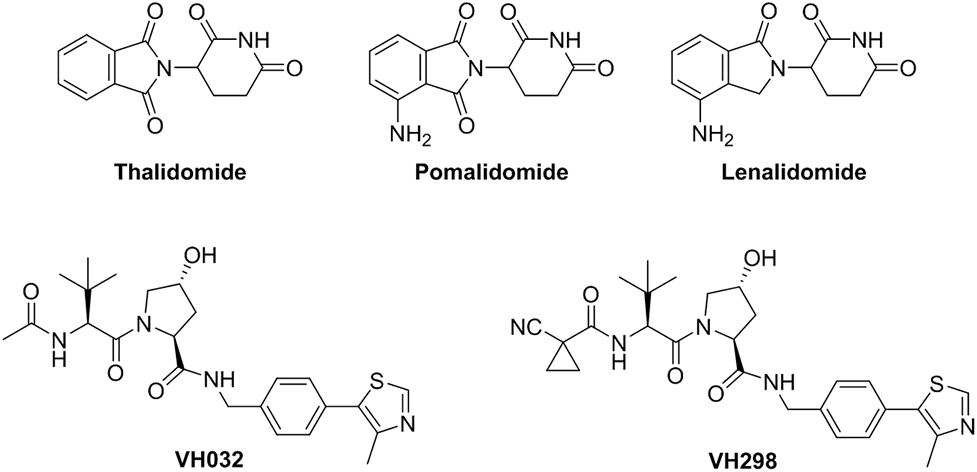
Representative IMiDs and VHL ligands.
In 2012, our group described the design and synthesis of the first small-molecule ligands for VHL through in silico and fragment-based screening[21-23]. These small-molecules inhibit the VHL/HIF-1α protein–protein interaction and possess submicromolar affinity for VHL. The crystal structure of VHL bound to the most potent inhibitor was obtained, thereby providing a starting point for further ligand optimization. A subsequent structure activity relationship (SAR) study of the VHL ligands was reported by the Ciulli laboratory in 2014, which identified a VHL ligand (VH032) with improved physical chemical properties and a Kd of 185 nM, similar to that of a 10-mer HIF1α peptide fragment (Kd = 0.2 μM)[24], and an even more potent VHL ligand (VH298, Figure 3) with Kd = 90 nM was subsequently published by the Ciulli laboratory[25-26]. VH298 engages VHL with high affinity and specificity. As a result, VH298 provides an attractive starting point for the development of potential PROTACs.
The small molecule based VHL E3 ligand was first employed to successfully degrade HaloTag7 fusion proteins[27]. HaloTag7 is a modified bacterial dehalogenase that has the ability to covalently react with linear chloroalkanes[28]. HaloPROTACs, consisting of the VHL ligand conjugated to various chloroalkanes successfully recruited VHL to promote the degradation of a green fluorescent protein (GFP)-HaloTag7 fusion protein. The most active compound, HaloPROTAC3 (Figure 2), induced 90% degradation of the target protein with a 19 nM DC50 (the concentration at which half of the maximal degradation (Dmax) is reached), making it one of the most potent PROTACs described at that time. Owning to its low toxicity, lack of effect on HIF stability, and the commercial availability of plasmids encoding 20,000 human proteins fused with HaloTag7[29], HaloPROTAC3 has proven to be a powerful probe for chemical genetic studies[30, 31].
In 2015, our group, in collaboration with GSK, published the first small molecule-based, VHL-recruiting PROTAC that induced endogenous protein degradation[32a]. These PROTACs were capable of specifically reducing levels of estrogen-related receptor-α (ERRα) and receptor-interacting serine/threonine protein kinase 2 (RIPK2) by >90% at nanomolar concentrations without affecting VHL’s endogenous target HIF1α function at concentrations up to 30 μM (Figure 2, PROTAC_ERRα and PROTAC_ RIPK2). Moreover, this study validated that PROTACs act via a sub-stoichiometric, catalytic mechanism whereby one molecule of PROTAC is able to induce multiple rounds of target protein ubiquitylation and degradation. Lastly, in an in vivo degradation study, we also demonstrated that PROTACs can reduce targeted protein in various tissues including solid tumors.
At almost the same time, the Ciulli laboratory reported the first selective BRD4 PROTAC constructed by using VH032 as the E3 recruiting element and JQ1, a pan-bromodomain and extra terminal (BET) selective bromodomain inhibitor[33], as the warhead for target engagement[34]. The optimized PROTAC, MZ1 (Figure 2), potently and rapidly induced reversible, long-lasting, and unexpectedly selective removal of BRD4 over BRD2 and BRD3. BRD4 degradation was achieved at a sufficiently low concentration of MZ1 that, similar to previous examples, did not induce HIF-1α stabilization. This result, together with our observations,[32a] suggested that VHL can be efficiently hijacked by PROTACs to allow endogenous POI degradation with a low potential to induce undesired effects (e.g. stabilization of the natural VHL substrate).
Considering the ability of thalidomide derivatives to bind CRBN, our group and others sought to utilize them as E3 ligase-recruiting elements to hijack CRBN for ubiquitination of proteins of interest. In 2015, we first reported ARV-825[35], a hetero-bifunctional PROTAC that recruits the E3 ubiquitin ligase CRBN to BRD4, leading to its fast, efficient, and prolonged degradation in all Burkitt’s lymphoma (BL) cell lines tested (Figure 2). Treatment of BL cell lines with ARV-825 resulted in almost complete BRD4 protein degradation, with picomolar DC50. Consequently, ARV-825 provided a more sustained suppression of MYC levels and downstream signaling than OTX015, the small molecule BRD4 inhibitor on which it is based, resulting in more effective anti-proliferative activity and apoptotic effects in BL cell lines.
At the same time, the Bradner laboratory appended the BRD4 inhibitor JQ1 to a phthalimide moiety to hijack the CRBN E3 ubiquitin ligase complex[36]. The resultant compound, dBET1 (Figure 2), induced highly selective CRBN-dependent BET protein degradation in vitro and in vivo. Compared with the effects of JQ1, pharmacologic destabilization of BRD4 by dBET1 resulted in improved antitumor efficacy in a human leukemia xenograft model. They also extended this strategy to induce FKBP12 degradation by conjugating a FKBP12-directed ligand (SLF) to the same phthalimide moiety (Figure 2, dFKBP-1).
Owning to the specific, biophysically validated binding affinities to their targeted E3 ligases, acceptable physicochemical profiles, and the well-characterized structural information of their binding modes, CRBN and VHL ligands are valuable elements in the chemical toolbox for PROTAC design. Unsurprisingly, their cognate E3 ligases are the top choices for recruitment for new POI degradation.
Present:
DC50/Dmax
To better describe and compare the degradation activity between different PROTACs, our group defined their maximal level of target degradation (Dmax) and their half-maximal degradation concentration (DC50) in the first report of VHL-recruiting all small-molecule PROTACs[27, 32a]. When targeting disease-causing proteins for degradation, we are aiming for a high Dmax (>90%) and a low DC50 (nM to pM range). From the development of our previous PROTACs, we learned that to improve their Dmax, one needs to address an important question: how stably can the POI and E3 ligase be held together? Ternary complex stability correlates with the binding affinity of both ligands used for PROTAC construction. With an appropriate linker length to facilitate ternary complex formation, the tighter the binding affinity of the ligands, the more stably the POI and E3 ligase can be held together, resulting in greater Dmax. However, it is possible to form too tight of a ternary complex and thereby lose the benefit of catalytic activity. In extreme cases, PROTAC can be degraded together with POI, similar with covalent PROTAC that covalently bind with POI[32b]. DC50 is determined by cell permeability to some extent, which can be optimized by fine-tuning the ligand physicochemical properties and linker length, composition and rigidity. Some other factors could also affect the Dmax and DC50, such as the accessibility to exposed POI lysines, target resynthesis or de-ubiquitination rate[32c].
Cooperativity
However, binary POI-ligand affinity or PROTAC-E3 affinity alone sometimes are not sufficient to guide PROTAC design. To understand whether there is any correlation between the target binding affinity and the PROTAC degradation profile, our lab generated one PROTAC based on the promiscuous kinase inhibitor, foretinib, which was itself confirmed to bind to 133 different kinases in a high-throughput competitive binding assay. This foretinib-based, VHL-recruiting PROTAC (Figure 4) could surprisingly degrade p38α to as much as 91%, with a 210 nM DC50, which is over 50-fold lower than the binary binding affinity of foretinib for p38α (Kd = 11 μM)[37]. Conversely, this same PROTAC could not induce SLK and Axl degradation despite the high binary binding affinity observed for foretinib to these kinases (Kd = 450 nM and 26 nM, respectively). It was found that the PROTAC mediated a thermodynamically-favorable ternary complex between the target protein, p38α, and the recruited E3 ligase, VHL, which compensated for the weak target-PROTAC affinity and thus led to successful degradation. On the contrary, unfavorable ternary complexes, such as those that induce steric clashes, might hamper target protein degradation despite the high-affinity target-PROTAC interaction. A similar result was observed by the Ciulli laboratory[38]. JQ1 (Kd for BRD4 ~ 100 nM)-based PROTACs exhibited positive cooperativities of ternary complex formation and showed more potent BRD4 degradation than I-BET726[39] (Kd ~ 4 nM)-based PROTACs, which actually showed negative cooperativities instead.
Figure 4.

More recent small-molecule PROTACs.
The first crystal structure of a ternary complex (Figure 5) between the targeted Brd4 bromodomain (Brd4BD2), the PROTAC “MZ1” (Figure 2) and VHL was reported in 2017 by the Ciulli laboratory[40]. The structure revealed that MZ1 is ‘sandwiched’ within a bowl-shaped interface formed by extensive PPIs between Brd4BD2 and VHL. Biophysical binding studies in solution revealed marked isoform-specific cooperativity (α) of ternary complexes. Significant positively cooperative ternary complex formation was observed across all BET bromodomains tested, with the strongest cooperativity for Brd4BD2 (α = 17.6), followed by Brd3BD2 (α = 10.7). Besides, the oxygen atom in a PEG linker of MZ1 can also form a hydrogen bond with BD2-specific residue His437 at the ternary binding interface thus increasing positive cooperativity of ternary complex formation. Structure-guided design of a new PROTAC, AT1 (Figure 4), enabled highly selective Brd4 degradation over BRD2 and BRD3. This is the first example of a bifunctional molecule that could ‘fold on itself’ to facilitate recruitment of the POI and E3 into productive proximity, resulting in the formation of new PPIs that contributed to the high stability and cooperativity of the ternary complex. Other crystal structures of ternary complexes also have been utilized in structure-based PROTAC design[41-42, 68]. The PROTAC, GSK215[43], which targets focal adhesion kinase (Fak) was published recently (Figure 4). The ternary complex structure of Fak-GSK215-VHL-ElonginC-ElonginB (VCB) was successfully elucidated by X-ray crystallography (Figure 5). Many neo-interactions between Fak and VHL that occur at both ends of the PROTAC, such as H-bond between the sidechain of N67 on VHL and the backbone carbonyl of C427 on Fak, and a salt bridge between R69 on VHL and E430 on Fak explain the high ternary complex cooperativity induced by GSK215. The authors found that the target DC50 values correlate with cooperativity of complex formation by their panel of Fak-PROTACs, supporting the importance of positive cooperativity in achieving potent degradation.
Figure 5.

Representative ternary complex structures of POI-PROTAC-E3 ligase. The figure was created with ChimeraX.
In summary, the ability of a PROTAC to induce a stable ternary complex is more predictive of successful protein degradation than its affinity for the POI. PROTACs can stabilize PPIs between the POI and E3 and promote positive cooperativity in ternary complex formation.
Hook Effect
Since a single PROTAC molecule forms a ternary complex by simultaneous binding of the target protein and E3 ligase, saturating doses of PROTAC molecules that favour the binary binding of separate PROTAC molecules with either the target protein or E3 ligase will discourage the ternary complex and lead to lesser degradation of the target protein[44]. This phenomenon is known as the “Hook Effect”[45]. The Hook Effect is dependent on the specific target and E3 ligase recruited, and also on the different PROTAC utilized. Variables (e.g., POI concentration, E3 ligase concentration, PROTAC binary binding affinity and cooperativity) that influence ternary complex formation could affect the Hook Effect, which is not consistently observed. In 2017, Roy et al. mathematically described the Hook Effect and its behaviour under allosteric conditions. Their results indicated that positive cooperativity in ternary complex formation could mitigate the high-dose Hook Effect[46].
Catalytic Nature
As we discussed previously, PROTACs act as a sub-stoichiometric catalyst, whereby one molecule of PROTAC is able to induce multiple rounds of target protein ubiquitylation and degradation[32a]. Due to this catalytic mode of action, significantly lower concentrations of PROTACs (event-driven paradigm) are usually required than inhibitors in order to induce a desired pharmacological effect. PROTACs may therefore be less inclined than inhibitors to trigger the toxicities that are a result of the high target occupancy requirements of the latter (occupancy-driven paradigm). Because of this catalytic nature, PROTACs have additional pharmaceutic benefits, which we summarize below.
First, PROTACs have a prolonged duration of effect and a differentiated PK/PD profile compared to their parent SMIs. One extraordinary example that demonstrated the extended pharmacodynamic response of a PROTAC in vivo was reported by Mares et al. in 2020[47]. Receptor-Interacting Serine/Threonine Protein Kinase 2 (RIPK2) dysregulation is associated with several immune disorders. Protein turnover data in primary immune cells shows that RIPK2 has a prolonged half-life of about 50 h[48]. Optimization of IAP-recruiting PROTAC led to PROTAC 6 (Figure 4), which showed clearance in rats of 10 mL/min/kg and a volume of distribution using the steady-state method (Vdss) of 7.6 L/kg, along with half-life of 16 h. Significant in vivo potency was observed by using this optimized PROTAC: a single 0.5 mg/kg dose caused 78 ± 5% degradation of RIPK2 at 48 h post-administration, leading to 5 days of substantial inhibition (>70%) of TNFα level upon ex vivo whole blood L18-MDP challenge. Blood concentrations of PROTAC 6 remained measurable throughout the 5 days study at the 0.5 mg/kg dose. With once daily (QD) dosing of PROTAC 6 at 0.05 mg/kg/day, TNFα was inhibited by ~60% at 6 and 24 h and progressed to >90% inhibition by 54 h. Every 3 days (Q3D) dosing of 0.15mg/kg/day on the other hand, produced maximal inhibition of TNFα at 72 h post-administration, even though at 48 h post-administration the drug levels had dropped below the lower limit of quantification (LLQ).
The Q3D dosing strategy for PROTAC 6 provides the most compelling evidence for the extended PD response. Following the first dose, drug levels were maintained above the LLQ for 24 h. Notably, in the absence of measurable PROTAC 6 at the 72 h timepoint, prior to the administration of the second dose, there was clear evidence of persisting RIPK2 degradation and TNFα inhibition.
Another good example was published by the Gray lab in 2019[49]. Bruton’s Tyrosine Kinase (BTK)[50] plays a particularly important role in B-cell development and function. Inhibition of BTK kinase activity has been proven to be an important and practical way of treating B-cell malignancies. Ibrutinib[51] is a first-in-class, covalent BTK inhibitor that is able to bind C481 (cysteine 481) of BTK with 0.5 nM IC50. However, acquired resistance via BTKC481S mutation quickly arises that blocks covalent bond formation with ibrutinib and significantly reduces its potency. To address this issue, a series of PROTACs were made based on a previously reported selective BTK inhibitor, CGI1746[52]. One of them, DD-03-171 (Figure 4), resulted in significant degradation of BTK in vitro and in vivo, despite the fact that it has a half-life of about 2 h in C57Bl/6 mice. The efficacy of DD-03-171 was compared to ibrutinib and lenalidomide in DFBL-96069–engrafted mice model, which was derived from a patient who previously had failed multiple rounds of therapy, including chemotherapy and ibrutinib. After treating for 3 days, DD-03-171 demonstrated extensive BTK degradation in extracted DFBL-96069 cells; more importantly, significantly reduced tumor burden in the peripheral blood and extension of survival in mice was observed with treatment of DD-03-171 compared to the other 3 cohorts.
A second advantage of PROTACs is that only limited target engagement is needed to induce significant degradation. In 2018, our lab performed the first head-to-head comparison of an androgen receptor (AR) inhibitor (enzalutamide) and its PROTAC degrader counterpart (ARCC-4) (Figure 4) using cellular models of drug resistance[53]. ARCC-4 is able to achieve AR degradation with a DC50 of 5 nM and Dmax > 95%. ARCC-4 also shows much more anti-proliferation activity compared to both enzalutamide and an inactive variant of the PROTAC in VCaP cells. Montgomery et al. found that the prostate tumors themselves can produce testosterone that will compete with AR blockers for the active site of the drug[54]. Because of this competition, the occupancy rate of enzalutamide for AR drops, leading to the loss of the clinical benefit. We recapitulated this resistance mechanism by introducing increasing amounts of a synthetic androgen, R1881. When VCaP cells were treated with a fixed concentration of ARCC-4, enzalutamide or the inactive PROTAC (Figure 4) in the presence of increasing concentrations of R1881, ARCC-4 outperformed enzalutamide at maintaining antiproliferative and proapoptotic potency even at comparatively higher R1881 concentrations. These data clearly indicate that PROTACs can efficiently degrade POIs despite the limited target engagement possible when a competitor is being generated by the cells.
Linkerology
Regarding PROTAC optimization, “linkerology” (modification of linker length, composition and rigidity) plays a critical role in adjusting both the biological and physicochemical properties of PROTACs, such as target selectivity, cooperativity, biodistribution, metabolic stability, membrane permeability, and aqueous solubility. We will discuss the details in this section and the impact on cell permeability in the “Future” section.
First, linker length and tethering orientation can define selectivity. Epidermal growth factor receptor (EGFR), also known as HER1, has been implicated in a range of cancers[55]. Overexpression and/or activating mutations of EGFR are associated with poor prognosis, so significant effort has focused on targeting EGFR. Lapatinib is a SMI that targets EGFR and is also a potent binder to other HER family RTKs, like HER2. Our group explored the potential for EGFR and HER2 degradation by lapatinib-based, VHL recruiting PROTACs[56]. Immunoblotting analysis revealed that PROTAC 1, which contains a linker length of 2 PEG units, could concurrently degrade both EGFR and HER2 (Figure 6). However, PROTAC 5, with extension of the linker length to 3 PEG units, enabled the selective degradation of EGFR while sparing HER2. This observation suggests that PROTACs can provide an added layer of target specificity relative to the constituent inhibitor: off-target proteins are spared from degradation in spite of PROTAC binding, most likely due to the inability of the PROTAC to form productive ternary complexes with them.
Figure 6.

Linker length can define target selectivity.
Another excellent example of the impact of linkerology is the development of isoform-selective PROTACs for the p38 MAPK family using a single warhead (foretinib) and recruited E3 ligase (VHL), resulting in degradation of either p38α or p38δ[57]. SJFα, which contains a 13-atom linker length and a “left-hand” (lh)-VHL ligand[32a], degrades p38α with a DC50 of 7.16 nM and Dmax of 97.4%, but is far less effective at degrading the other p38 isoforms. SJFδ, which contains a 10-atom linker length and a “right-hand” (rh)-VHL ligand[27], degrades p38δ with a DC50 of 46.17 nM and Dmax of 99.4%, but does not degrade p38α, β, or γ (Figure 7). This observation shows that simply by changing linker length and tethering orientation one can gain selectivity.
Figure 7.

Linker orientation can define target selectivity.
Linker rigidity can influence cooperativity of ternary complex formation. In 2019, Farnaby et al. took advantage of the crystal structure of a ternary complex and biophysical tools to guide rational and efficient PROTAC optimization, and developed the potent and cooperative degrader (ACBI1) for SMARCA2 and SMARCA4[42]. It was shown that proper rigidity in the linker can constrain a PROTAC in its bioactive conformation, which may lead to improved protein degradation[58, 59]. To increase conformational restraint, a phenyl ring was implanted in the linker to replace the PEG unit (Figure 8). This phenyl ring formed an additional pi-stacking interaction to Y98 of VHL and substantially reduced the polarity of the linker. In both fluorescence polarization (FP) and TR–FRET assays, ACBI1 demonstrated a cooperativity (α) of approximately 30. Correspondingly, ACBI1 demonstrated complete and potent degradation of SMARCA2 (DC50 of 6 nM) and SMARCA4 (DC50 of 11 nM) in cultured cells. Another attractive strategy to constrain a molecule in its bioactive conformation is macrocyclization. In 2020, Testa et al. designed and synthesized a macrocyclic PROTAC by imbedding a cyclic linker to the BET degrader, MZ1[60]. Despite a more than 10-fold loss of binary binding affinity for Brd4, macro-PROTAC-1 displayed high positive cooperativity (α = 20, compared with 17.6 for MZ1) by ITC and exhibited potent cellular activity comparable to MZ1.
Figure 8.

Linker rigidity can influence cooperativity.
Finally, linkerology can also be used to adjust physicochemical properties, like metabolic stability, membrane permeability, and aqueous solubility of PROTACs[48, 61]. Our group recently reported the discovery of MT802[65], a PROTAC that degrades BTK in cells at low nanomolar concentrations. However, the pharmacokinetic data of MT802 in mice, specifically its high clearance (1662 mL/min/kg) and quite short half-life (0.119 h), indicated it was not suitable for in vivo studies. With the goal of improving the pharmacokinetic properties of MT802, we have designed and made a more robust and metabolically stable PROTAC, SJF608[62] by replacing the amide of MT802 with an ether moiety between the linker and CRBN ligand while keeping the BTK ligand and linker length constant (Figure 9). SJF608 revealed >16 times slower clearance (102 mL/min/kg) and about 13 times longer half-life (1.62 h) after intravenous (IV) administration in mice. Although further optimizations are still needed to improve pharmacokinetic properties, the work to date showcases the critical rule of linker composition in adjusting physicochemical properties of PROTACs. In 2020, Goracci et al. reported a systematic study on the metabolic stability of 40 PROTACs in cryopreserved human hepatocytes[63]. Comprehensive analysis of the half-life values and metabolic soft spots revealed general trends in PROTAC metabolism, with the linker oftentimes being the most metabolically labile moiety in a PROTAC molecule. Indeed, the most unstable parts were located at the linker attachment points to the ligands, such that N-dealkylation and amide hydrolysis reactions helped to rationalize our observation of the MT802 short half-life. The study’s findings also provided the guidelines for linker choice, for example using PEG-like linkers versus aliphatic-based ones; using shorter aliphatic linkers; or using linkers endowed with cyclic moieties. Moreover, piperazinyl or pyridine groups[58] can be incorporated into a linker to improve not only rigidity but also PROTAC solubility.
Figure 9.

Linker composition can improve the PK properties.
Differential Biology
The event-driven PROTAC MOA can overcome inhibitor resistance mechanisms, including overexpression levels of the drug targets and resistance-conferring mutations[64], giving them a potentially much wider biological applicability over SMI.
As previously mentioned, ARV-825 can target BRD4 (Figure 2), leading to its fast, efficient, and prolonged degradation in all Burkitt’s lymphoma (BL) cell lines[35]. Interestingly, the use of occupancy-driven small molecule BRD4 inhibitors, like OTX015, causes BRD4 level to increase dramatically at high, therapeutically-relevant inhibitor concentrations, which leads to inefficient c-MYC suppression. When compensatory overexpression of the target protein occurs, more inhibitor is needed to saturate the protein and achieve the therapeutic effect. Usually, this kind of pharmacological “arms race” is difficult to win because cells are always able to produce more of the proteins, but the clinician may not be able to continually increase dosage to the patients due to toxicity. In contrast, our PROTAC ARV-825 resulted in almost complete BRD4 protein degradation, with 0.2 nM of DC50. That means the PROTAC degrades not only the pre-existing BRD4 but also the overexpressed proteins that cells are constantly making. Therefore, PROTAC induced degradation leads to a more profound suppression of downstream MYC.
Another common resistance mechanism is the mutation of the target protein in response to therapeutic inhibition. In addition to the CGI1746-based BTK-targeting PROTAC developed by the Gray lab[49], our lab[65] (Figure 9) and the Rao lab[66-67] also have developed BTK-targeting PROTACs, although these latter degraders are structurally based on a non-covalent derivative of the kinase inhibitor, ibrutinib, which has a reactive electrophile eliminated (Figure 10, P13I and L18I). These PROTACs can induce the degradation of both wildtype and mutant BTKs despite the fact that they are unable to covalently bind to the target. The above results suggest that the BTK degraders provide great potential of inhibiting the function of non-wild type BTK isoforms, which is especially beneficial in instances of cancers that acquire ibrutinib resistance. Various AR mutations (e.g., F876L, H874Y, M896V, T877A and L702H) have been observed in clinical samples from metastatic prostate cancer patients. Our lab found that ARCC-4 (Figure 4) can degrade all of the different clinically relevant AR mutants tested[53]. Some other drug resistance mutations, like G250E of BCR-ABL1 and L858R and/or T790M of EGFR, which had demonstrated a low clinical response rate to kinase inhibitors were successfully degraded by converting the inhibitor to PROTACs[56, 69]. These data suggest that protein degradation could have therapeutic benefits in mutational contexts that are not susceptible to inhibition.
Figure 10.

PROTACs derived from ibrutinib show both WT and C481S mutated BTK degradation.
In a recent study, PROTACs that degrade RTKs[56] and Fak[70] were able to eliminate not only their kinase activity but also target their scaffolding function (which current inhibitors do not). In addition to their capacity to eliminate the well-defined activities of mutated forms of their targets, PROTACs have the capability to eliminate all biological activities associated with a target protein -- some of which that may be beyond the reach of current SMIs. Focal adhesion kinase (Fak) is widely expressed in different species and plays a critical role in tumor invasion and metastasis through the coordination of both kinase-dependent enzyme functions and kinase-independent scaffolding functions with several signalling proteins, simultaneously. Accordingly, many essential functions mediated through Fak scaffolding remain beyond the reach of Fak kinase inhibitors, not to mention the drug resistance they can cause. To overcome the mechanistic shortcomings of Fak kinase inhibitors, our group designed highly selective, low nanomolar potency Fak degraders based on the SMI defactinib[70] (Figure 11). The most promising degrader, PROTAC-3, recruits the E3 ligase VHL and showed selective and potent Fak protein degradation in PC3 cells with DC50 = 3 nM and Dmax = 99% after 24 h of treatment. More importantly, low nanomolar concentrations of PROTAC-3 are sufficient to significantly impair cell migration and cell invasion of MDA-MB-231 cells, while defactinib shows no significant effect in either assay. Other groups[71-72] have since created several potent and selective Fak degraders -- of these PROTACs, GSK215 conspicuously stands out from the others for its inhibition of BT474 cells in 3-D proliferation assay[43] (Figure 4). More in vitro and in vivo experiments should be conducted using those Fak PROTACs to answer how the kinase-independent scaffolding functions of Fak are required for cell proliferation, which will provide better understanding of Fak biology.
Figure 11.

Fak and PCAF/GCN5 PROTACs.
P300/CBP-associated factor (PCAF) and general control nonderepressible 5 (GCN5) are epigenetic proteins, each containing an acetyltransferase domain and a bromodomain. Gene knock-out experiments have implicated PCAF in the production of inflammatory cytokines in the immune system, which suggested that PCAF might be a novel anti-inflammatory therapeutic target. GSK4027[73] is a potent small-molecule inhibitor targeting the bromodomains of PCAF and GCN5 and is highly selective for them over other bromodomain family members. However, GSK4027 caused no significant reduction of lipopolysaccharide (LPS)-induced cytokine production in human cells, suggesting that inhibition of the PCAF/GCN5 bromodomains is not sufficient to disrupt the pro-inflammatory function of these proteins. By converting the inhibitor into a degrader, the most potent PROTAC, GSK699[74], induced robust and concentration-dependent degradation of PCAF and GCN5 in both macrophages and dendritic cells (DCs) (Figure 11). Moreover, PROTAC-mediated degradation of these proteins markedly impaired the ability of macrophages and DCs to respond to LPS, significantly reducing the production of numerous inflammatory cytokines, such as IL-6, IL-12p70, IL-10, IL-1β, and IFN-γ. This differential biology between the SMI GSK4027 and the PROTAC GSK699 suggests that the preponderance of PCAF and GCN5 pro-inflammatory activity derive from another, heretofore unidentified functionality rather than their bromodomains.
In summary, PROTACs have the potential to control the enzymatic and non-enzymatic functions of proteins that are not easily addressed by traditional small-molecule inhibitors. The best PROTAC targets are those that can offer differential biology upon degradation.
Expanding Target Space for Protein Degradation
In the past several years, the targeted protein degradation field has expanded dramatically, with dozens of target proteins amenable to induced degradation and the major effort focused on oncogenic proteins. However, proteins for other diseases, like neurodegenerative diseases have also been widely pursued. Proteins targeted by PROTACs include: protein kinases, nuclear receptors, epigenetic readers, neurodegenerative disease-related proteins, regulatory proteins, enzymes, anti-apoptotic proteins, transcription factors, scaffolding proteins, cytokines[90], virus-related proteins and so on, most of them have been reviewed elsewhere[44, 75-77 and 94].
Some proteins have been termed “undruggable”, because of (1) their flat and featureless binding sites, (2) the lack of hydrogen-bond donors and acceptors, and (3) the need for adaptive changes in their conformation. Gratifyingly, PROTACs have shown the ability to target these proteins for degradation. Some of them (e.g. STAT3) have been reviewed elsewhere[79-83], here we discuss some more recently published examples.
1. Transcription factors (TFs)
FOXM1.
The Forkhead box M1 (FOXM1) protein is a transcription factor that regulates multiple proliferation-related genes through selective protein-DNA and protein–protein interactions and that is overexpressed in a variety of human cancers. Its increased transcriptional activity leads to tumorigenesis, cancer progression, metastasis, and chemotherapy resistance. However, identifying potent FOXM1 SMIs is still challenging, due to the lack of well-defined drug binding sites and a large interaction interface. Luo et al. reported[84] the development of FOXM1 degraders by conjugating FDI-6 -- a molecule that specific interferes with FOXM1-DNA interaction -- to the CRBN recruiting ligand, pomalidomide. The most potent compound, 17d (Figure 12) induced 93% FOXM1 degradation with a 1.96 μM DC50 value in MDA-MB-231 cells. At the same time, 17d produced significant downregulation of the downstream FOXM1 effectors CDC25B and cyclin B1, along with a remarkable decrease in nuclear accumulation of β-catenin, outperforming the parent inhibitor FDI-6. In vivo antitumor assay indicated that both 17d and FDI-6 significantly inhibited triple-negative breast cancer (TNBC) tumor growth (78.2% and 47.8%, respectively) without decreasing body weight of the xenograft implanted mice. Overall, these results demonstrate that PROTAC-induced degradation of the transcription factor FOXM1 could be an efficient strategy for the treatment of FOXM1-related diseases. Further structural optimization on the lead PROTAC 17d should be done to improve the degradation potency.
Figure 12.

FOXM1 PROTAC.
β-catenin.
Aberrant activation of Wnt/β-catenin signaling is highly implicated in various types of cancers. As β-catenin is the central player of canonical Wnt signaling and is frequently mutated in cancers, it has become one of the most attractive intracellular targets. In 2020, Liao et al. designed several peptide-based PROTACs[85] to induce efficient β-catenin degradation that manifested in strong inhibition of Wnt signaling in cancer cells and could effectively restrain tumor formation in BALB/C in vivo.
Other Oligonucleotide-based PROTACs Targeting TFs.
Development of small-molecule inhibitors for TFs is challenging due to their lack of enzymatic activity and ligandable pockets. Therefore, the development of alternative strategies to target such proteins is crucial. Taking advantage of the intrinsic TF DNA-binding ability, our group developed chimeric oligos incorporating both TF-specific DNA sequences and an E3 ligase-recruiting moiety, named TRAnscription Factor TArgeting Chimeras (TRAFTACs)[86]. TRAFTACs can recruit both a TF of interest (TOI) and the VHL E3 ligase via the intermediate protein dCas9HT7, and have induced degradation of disease-relevant TFs NF-κB and brachyury in the additional presence of a haloPROTAC (Figure 13A). Later, a similar TF-PROTAC[87] was reported by Liu et al. by using in-situ click reaction to conjugate a DNA oligonucleotide to an VHL ligand, which could selectively degrade the TF of interest. Two series of VHL-based TF-PROTACs were developed, NF-κB-PROTAC and E2F-PROTAC, which induced endogenous p65 and E2F1 protein degradation, respectively, in cells (Figure 13B). At the same time, an oligonucleotide-based PROTAC (O’PROTACs)[88], in which a TF-recognizing double-stranded oligonucleotide was incorporated directly to the E3 ligase ligand, was designed and utilized to induce in vitro and in vivo degradation of lymphoid enhancer-binding factor 1 (LEF1) and ETS-related gene (ERG), two highly cancer-related transcription factors. Recently, our lab designed and synthesized the second generation TRAFTACs, oligoTRAFTACs[89], by directly attaching the TF binding oligonucleotide to an E3 ligase-recruiting ligand and demonstrating their ability to induce the degradation of two oncogenic TFs, c-Myc and brachyury (Figure 13C).
Figure 13.

Oligonucleotide-based PROTACs Targeting TFs. (A) TRAFTAC recruits E3 ligase complex through dCas9-HT7 in the presence of haloPROTAC. (B) TF-PROTAC formed in-situ via a copper-free strain-promoted azide–alkyne cycloaddition (SPAAC) reaction to recruit E3 ligase complex. (C) O’PROTAC or oligoTRAFTAC with E3 ligand and oligonucleotide directly attached. The figure was created with BioRender.com.
Overall, these results suggest that oligonucleotide-based PROTACs that target TFs provide a generalizable platform to achieve selective degradation of many TFs and have the potential to target most “undruggable” proteins for anticancer drug development.
2. Cytokines
MIF.
The protein “macrophage migration inhibitory factor” (MIF) has been implicated in the pathogenesis of cancers. MIF exerts its functions mainly through protein-protein interactions with membrane-bound receptors such as cluster of differentiation 74 (CD74). Overexpression of MIF can stimulate proliferation of cancer cells through activation of the ERK/MAPK pathway and inhibition of the p53 pathway. Small molecules mainly target the tautomerase (enzyme) active site of MIF, however this is not enough to effectively interfere with MIF protein-protein interactions. Recently, Xiao et al. developed the first MIF-targeting PROTAC with improved potency compared to the parent SMI[90]. The most potent PROTAC, MD13 (Figure 14), could induce >90% MIF degradation with a DC50 of around 100 nM in A549 cells; however, only a moderate anti-proliferative effect in 2D-cell culture and inhibition of ERK signaling were observed. However, in a 3D spheroid model of proliferation, MD13 inhibited the growth of the spheroid volume by 81% upon treatment with 5 μM of the degrader. This is the first usage of a PROTAC in cytokine degradation, and while the knockdown was efficient it was also restricted to MIF in the cytoplasm. However, as a pro-inflammatory cytokine, a large portion of MIF is located extracellularly. Strategies that can simultaneously degrade cytoplasmic and extracellular cytokines may improve overall biological efficacy.
Figure 14.

MIF and SF3B1 PROTACs.
3. SF3B1
SF3B1.
Splicing factor 3B subunit 1 (SF3B1) is one of the fundamental components of the multiprotein complex spliceosome and is involved in 3’ splice site recognition at intron-exon junctions during RNA splicing[91]. The SF3B1 gene is frequently mutated in tumors, however the functions of individual components of the spliceosome are largely unknown, and there are only a limited number of SMIs for them. The first SF3B1-targeting PROTAC was reported recently by the Cheng lab[92] based on the bona fide SF3B1 activator, O4I2, which was discovered using high-throughput screening in their previous study[93]. PROTAC-O4I2 (Figure 14) showed selective degradation of SF3B1 and affected both the wild type and K700E mutant to the similar extent, leading to tumor growth inhibition in cells and in Drosophila intestinal tumor model. Taken together, their findings demonstrate that splicing factors can be targeted by PROTACs for degradation.
Degraders in Clinic Trials
The promise of induced protein degradation as a novel therapeutic modality has attracted the attention of both academia and the pharmaceutical industry[94, 95], especially after 2015 where our group and others showed VHL and CRBN can be recruited by using small molecules. The Targeted Protein Degradation (TPD) community, including many big pharma companies, biotechs and academic groups, is growing rapidly. The superiority of event-driven versus occupancy-driven pharmacology in preclinical models is encouraging and sets the stage for clinical applications. Since Arvinas’ first small molecule targeted protein degraders began clinic trials in 2019, nearly 15 PROTAC degraders have entered or are approaching clinical studies[96, 97]. Most of these programs are targeting cancer, except NX-5948 and KT-474, which are in Phase I clinical trials for treating patients with various autoimmune diseases. However, as more degraders enter the clinic, it is likely that encouraging therapeutic benefits will be obtained for a variety of indications in the future.
Non-conventional PROTACs and other TACs
1. Monobody-PROTAC Targeting RAS Mutants for Degradation
KRAS is mutated in ~20% of human cancers and has long been an attractive target for pharmacological modulation, despite it historically having been considered “undruggable”. The discovery of potent covalent inhibitors of the KRASG12C mutant in recent years has led to the development of a SMI drug, named sotorasib, which was recently given breakthrough approval by the FDA for use in non-small cell lung cancer. However, multiple treatment-emergent alterations were observed across 27/43 patients, such as KRASG12V or G13D, NRASQ61K or G13R, MRASQ71R and/or BRAFG596R, leading to a reduction in pharmacological efficacy of sotorasib[98]. Our group reported the first PROTAC capable of degrading endogenous KRASG12C by using a MRTX849 warhead, which covalently binds to KRASG12C, conjugated to a ligand that recruits the E3 ligase VHL[99]. However, there is still a lack of potent and selective inhibitors against other RAS mutants. Monobodies are synthetic binding proteins which are usually easy to be expressed in the functional form because of their lack of disulfide bonds. Monobody units also contain minimized Lysine residues, makes it stable in cells. So, the monobody platform has produced potent and selective inhibitors to diverse protein targets. Recently, Teng et al. reported a monobody-based PROTAC[100] that could selectively degrade KRAS mutants in the active state in a noncovalent manner and provide more extended suppression of mutant RAS activity than inhibition with the monobody alone. However, similar with the other functional biologics, the delivery of monobody-based PROTAC-like fusion proteins limits their application in drug development. Recently, a noncovalent, potent, and selective KRASG12D small molecule inhibitor, MRTX1133, was reported[101]. We can envision that, by appending this inhibitor to an E3 ligand, one might target KRASG12D for degradation and obtain a better pharmaceutical effect.
2. Antibody-PROTAC Conjugates
Dragovich et al. reported the conjugation of PROTACs to monoclonal antibodies using Antibody-Drug Conjugates (ADCs) technologies originally developed for cytotoxic payload delivery[102, 103]. By exploring the linker, BRD4-PROTAC loading, BRD4 recruiting element, the Antibody-PROTAC exhibits potent and antigen-dependent BRD4 degradation and antiproliferation activities in cells, and antitumor efficacy in mouse xenograft model. Their approach provides an alternate delivery option for PROTACs.
Nanobodies, also called single-domain antibodies, are heavy-chain-derived antibodies that possess remarkable stability and high permeability, which is particularly attractive for solid tumors. Zhang et al. reported a covalent nanobody-based PROTAC strategy[104]. By conjugation with a cell penetrating peptide (CPP) and a lysosomal-sorting sequence (LSS), the resulting nanobody-based PROTAC triggered the internalization and degradation of programmed death-ligand 1 (PD-L1) with high specificity and efficiency, which provides a new way for targeting membrane proteins for degradation.
3. Aptamer-PROTAC Conjugates
Aptamers are short, single-stranded DNA or RNA (ssDNA or ssRNA) molecules that can selectively bind to a specific target with affinity similar to or higher than that of antibodies. They usually are 10-fold smaller than antibodies and can be chemically-modified in a defined and precise way. They can be easily stored and delivered with reversible denaturation properties. Aptamer-PROTAC Conjugates[105] were developed by conjugating a BET-targeting PROTAC to the nucleic acid aptamer AS1411 via a cleavable linker. This aptamer binds to nucleolin, a cell surface protein that is enriched on tumor cells. The conjugation of the PROTAC with the aptamer led to improved tumor-specific targeting ability and in vivo antitumor potency compared to conventional PROTACs.
4. G4-PROTAC
G-quadruplexes (G4s) are four-stranded nucleic acid structures that can bind to proteins, such as DEAH-box helicase (RHAU), to regulate important biological processes. Patil et al. reported on several novel G4-targeting PROTACs they had created by combining a G4 warhead and pomalidomide or VH032 for CRBN or VHL recruitment, respectively[106]. The G4-pomalidomide PROTAC lead to near-complete depletion of RHAU at 25 nM, in a proteasomal-dependent manner.
5. Trivalent PROTACs
Due to the synergistic effects of drug combination, it will be beneficial to target two different proteins for degradation simultaneously, especially for tumors that acquire drug resistance by resorting to compensatory factors or switching signal pathways. Zheng et al. reported the first example of dual PROTAC[107] molecules that demonstrated the ability to degrade two completely different types of targets, EGFR and poly(ADP-ribose) polymerase (PARP), simultaneously (Figure 15, DP-C-1 and DP-V-4). However, their dual PROTACs did not show better antiproliferative activity compared to gefitinib, an existing EGFR inhibitor, alone. One possible explanation could be the poorer solubility and cell penetration. As we discussed before, linker composition is critical for PROTAC design. The authors used biocompatible natural amino acids as a linker to achieve the rapid synthesis but as a result also introduced two more amide bonds and a triazole, which might result in decreased cell permeability due to the high topological polar surface area (TPSA)[108]. As shown recently, one amide-to-ester substitution[109] is beneficial enough to drastically improve cell permeability.
Figure 15.

Trivalent PROTACs.
Later, Imaide et al. designed trivalent PROTACs[110] consisting of a bivalent BET inhibitor and an E3 ligand tethered via trimethylolethane (TME) together with extended PEG units. Compared to bivalent PROTACs, the best degrader SIM1 (Figure 15) demonstrated sustained and higher efficacy (low picomolar BET degradation, with preference for BRD2), which led to more potent anticancer activity. The biological and mechanistic investigation with SIM1 indicated that augmenting the valency of degraders has the chance to induce positive cooperativity, thereby enhancing ternary complex formation.
6. Conditional Activation of PROTACs
Our lab published on the first conditional PROTACs (PROTACs whose activity is dependent on a second, initiating stimulus) nearly ten years ago[78]. In that instance, activation of the degrader was induced by its phosphorylation. More recently, light-induced protein degradation with photocaged[111-114] and photoswitchable[115-117] PROTACs have been developed as an attractive way to activate PROTACs, although those will not be discussed here as they have been recently reviewed elsewhere[61, 118]. People have also taken advantage of protein expression level, environment or biomarkers of tumor cells for PROTAC design.
Folate-Caged PROTACs.
To control the on-target degradation activity of a PROTAC in a tissue-selective manner, Liu and Chen et al. developed a strategy for PROTACs by conjugating a folate group to the degraders[119, 120]. Because folate receptor α (FOLR1) is highly expressed in many cancer types compared to normal tissues, the folate-caged PROTAC allows the specific delivery of PROTACs into cancer cells to reduce the off-tissue, on-target toxicity. Three folate-PROTACs (folate-ARV-771 (Figure 16), a BRD-PROTAC; folate-MS432, a MEK-PROTAC; and folate-MS99, an ALK-PROTAC) were generated which demonstrated effective degradation of BRDs, MEK1/2 and ALK, respectively, in a FOLR1-dependent manner in cancer cells.
Figure 16.

Folate-Caged PROTAC.
Hypoxia-activated PROTACs.
During tumorigenesis, the blood vessel proliferation surrounding tumors cannot support the needs of the continuous oxygen consumption of the tumor. This insufficient oxygen supply results in a hypoxic environment. Recently Cheng et al. reported the first Hypoxia-activated PROTAC[121] by using a hypoxia-activated leaving groups (HALGs) to block the EGFRDel19-based PROTAC (Figure 17). Results showed that Hypoxia-activated PROTAC 13 induced significantly higher EGFRDel19 degradation in hypoxia than in normoxia in HCC4006 cells.
Figure 17.

Hypoxia-activated PROTAC.
7. Other TACs/bifunctional Molecules that Induce Target Degradation
New degrader technologies, like LYTAC[122], AUTAC[123] and ATTEC[124], have been established by harnessing the lysosomal degradation pathway, which greatly broadens the spectrum of targets, such as extracellular proteins and damaged organelles. These will not be discussed here as they have been reviewed recently[125].
RIBOTACs.
Strategies that recruit ribonuclease against an RNA of interest via chemically induced proximity for RNA degradation is a rapidly-developing area[126]. Recently, the Disney lab identified that dovitinib, a Receptor Tyrosine Kinase (RTK) Inhibitor, also binds to a functional site in the oncogenic microRNA precursor Pre-miR-21 with a Kd of 3 μM. They converted dovitinib into an RNA degrader[127], or ribonuclease targeting chimera (RIBOTAC), which is a chimeric small molecule that also recruits ribonuclease L (RNase L) by using a previously reported small-molecule activator[128]. The resulting RIBOTAC shifted the selectivity for pre-miR-21 by 2500-fold over canonical RTKs, despite the fact that dovitinib modulates the RTK more potently than the RNA target.
Future:
Either as a chemical biology tool or as a novel drug discovery paradigm, many potential applications of PROTACs have not yet been fully explored. Targeted protein degradation by using PROTAC presents both new opportunities and challenges.
Recruit a broader set of E3 ligases
Several years ago, our lab conducted a head-to-head comparison study of VHL- and CRBN-based PROTACs by utilizing the protein tyrosine kinase inhibitors bosutinib and dasatinib, targeting the chimeric breakpoint cluster region–Abelson tyrosine kinase (BCR–ABL) oncoprotein and c-ABL[129]. We discovered that changing the inhibitor warhead and the recruited E3 ligase determines which protein targets are susceptible to induced degradation.
Given the existence of more than 600 E3 ubiquitin ligases identified in the human proteome[130], which typically function as adaptor molecules that recognize substrates through PPIs in their role of dictating target specificity, our group explored whether the PROTAC approach can be more universally expanded to the larger broader set of E3 ligases in 2017[131]. We created six engineered E3 ligases representing the three major classes: U-box, HECT, and RING. The result indicated that five out of six E3 ligases were amenable to recruitment for target degradation. The one that did not work in our study, CHIP, has been shown to be recruited for targeted degradation in another approach[132]. Taken together, these two studies highlight the great opportunity for PROTAC development by expanding the accessibility of E3 ligases[76].
Target-based approaches, such as Fragment-Based Drug Design (FBDD) or cocrystal structures of E3 ligases with fragment peptides of substrate proteins, are a major and fruitful strategy for E3 ligase ligand discovery, e.g. cIAP and VHL. However, it takes a lot of resources and is time-consuming. High throughput screening (HTS) campaigns provide the opportunity to access millions of compounds. However, it is difficult to determine a proper functional readout to detect compound activity against a targeted E3 ligase. Moreover, targeting the substrate binding site of E3 ligases may require the disruption of protein–protein interactions, which are usually shallow or highly charged, making the ligand discovery more challenging. To date, only <5% E3 ligases have been recruited for PROTAC development, even taking into account that about 270 of the >600 human E3 ligases have been verified as being involved in the UPS[133]. While CRBN, VHL, IAP and MDM2 remain the most frequently used E3 ligases, recent reports have identified other ligands for recruitment of other E3 ligases, often by using a covalent fragment screening approach discussed elsewhere[79, 118, 134 and 135].
DNA-encoded libraries (DELs) are an attractive combinatorial approach for ligand discovery which allows the expansion of chemical space rapidly[136, 137]. Within months, one can easily generate billions of compounds. Information from DEL screening, such as the linker attachment point, and possible structure-binding activity of each cycle of building blocks are determined directly from the screen. DEL screening as a method for the identification of ligands, regardless of their biological function, is highly suited as an approach to screen new E3 ligase or target binders for PROTAC development. Recently, Disch et al. from X-chem reported an estrogen receptor alpha (ERα) PROTAC where the target ligand was generated by using the DEL approach[138]. First, they identified a small molecule binder (compound 2) by screening the in house ~120 billion DEL. Compound 2 was a potent binder of WT and mutant (S463P, Y537S, and D538G) ERα, but had modest cellular agonist activity (0.55 μM) in MCF7 cells. With the guidance of the DEL screening data and molecular docking, a small scale SAR study was performed, and an antagonist (compound 7) was obtained with improvements in potency (0.068 μM in MCF7 cells). Finally, degrader, compound 21 (Figure 18), was synthesized by converting compound 7 into a bifunctional PROTAC -- this was made easier because an inherently suitable linker attachment point already existed in the form of the solvent-exposed point at which the DNA is attached to compound 7. Compound 21 induced ERα degradation with a DC50 of 20 nM in MCF7 cells treated for 24 h with a dose-dependent reduction in expression of its downstream target progesterone receptor (EC50 90 nM). Subcutaneous administration of 10 mg/kg QD of compound 21 resulted in significant tumor growth inhibition (TGI) = 84.3% with no changes in average body weight in mice. These results demonstrated that compound 21 was an excellent tool compound for degradation of ERα both in vitro and in vivo.
Figure 18.

ERα PROTAC with ligand discovered from a DEL.
So far, no peer-reviewed publication has been reported using DEL technology to identify novel E3 ligase ligands for utilization in PROTAC development. However, as more open source or affordable DEL kits[139] become available to industry and academic researchers, we will undoubtedly see more and more E3 ligase ligands discovered through DEL in the near future.
Recently, as a proof-of-concept study, we reported the indirect recruitment of DCAF5 E3 ligase complex to the vicinity of our selected target proteins: FKBP12F36V and BRD2 by co-opting methyl-lysine reader protein L3MBTL3[159]. L3MBTL3-recruiting PROTACs induce nuclear FKBP12 and BRD2 protein degradation in a time, dose, L3MBTL3-, and proteasome-dependent manner (Figure 19, KL-4 and KL-7). We anticipate this will be a generalizable approach to utilize other ligandable reader protein: E3 ligase complexes to expand the E3 ligase repertoire and to achieve compartment- and disease-specific protein degradation in the future.
Figure 19.

L3MBTL3-recruiting PROTACs.
Mitigate Cell Permeability
Due to the relatively high molecular weight (MW > 800) and the presence of multiple hydrogen bond donors (HBDs) and acceptors (HBAs) compared to SMIs, PROTACs tend to have lower cell permeability, which leads to challenges in regard to achieving sufficient exposure in difficult-to-reach tissues, such as the central nervous system (CNS), to provide a therapeutic effect.
One simple and straightforward strategy is to improve the potency of the PROTACs, which bypasses the need for a large amount of PROTAC to enter the cell. One of the most promising PROTACs was published by the Wang group in 2018[140]. The most potent PROTAC 23 (Figure 20) induced BRD4 protein degradation at concentrations as low as 30 pM in the RS4;11 leukemia cell line; inhibited RS4;11 cell growth with an IC50 value of 51 pM; and induced rapid RS4;11 xenograft tumor regression in vivo. Mice bearing RS4;11 xenograft tumors were administered with a single dose of PROTAC 23 at 5 mg/kg intravenously. The concentrations of PROTAC 23 detected in plasma and RS4;11 tumor tissue indicated that the PROTAC can effectively penetrate the RS4;11 xenograft tumor tissue and has a concentration of 166.3, 98.5, and 35.8 ng/g in the tumor at 1, 3, and 6 h post-injection, respectively. This was far greater than the drug concentrations needed for effective BRD4 degradation by this PROTAC, despite its relatively large size (MW = 798.8).
Figure 20.

PROTAC with pM BRD4 degradation potency.
To better understand and compare PROTAC structure-permeability relationships, Klein et al. described a label-free method for detecting the permeability of several VH032-based PROTACs by integrating a parallel artificial membrane permeability assay (PAMPA) and a lipophilic permeability efficiency (LPE) metric[141]. The PAMPA permeability measured for PROTACs ranged from Pe = 0.006 × 10−6 cm/s to 0.6 × 10−6 cm/s, below the standard for “modest” permeability (Pe = 1 × 10−6 cm/s). As expected, the compounds that had lower MW, shorter linkers and fewer HBDs/HBAs showed significantly higher permeability. The authors believe that PROTAC cell permeability could also be fine-tuned by taking advantage of the intramolecular hydrogen bonds (IMHBs) and hydrogen bond donor (HBD)-shielding, which was confirmed in a later publication[142]. By using an NMR-based study of a VHL-recruiting PROTAC, Atilaw et al. found that PROTAC 1 (Figure 21) displayed elongated conformations in polar solutions, yet it adopted highly folded conformations and had a smaller polar surface in nonpolar solvent that mimic the cell membrane interior. Cell permeability of this PROTAC was improved by minimization of size and polarity, which benefitted from the formation of IMHBs, π−π interactions, and HBD-shielding despite the molecule having a high molecular weight, polarity and a large number of rotatable bonds.
Figure 21.

PROTAC displays folded conformations in nonpolar solvent.
However, collaborative efforts between academia and industry with medicinal chemistry expertise should be pursued to define ad hoc physicochemical descriptors[143] for new chemical modalities like PROTACs that lie in the Beyond the Rule of 5 (bRo5) chemical space[144, 145].
Another strategy that may improve the cell permeability of PROTACs is to take advantage of PROTACs that exhibit positive cooperativities that facilitate recruiting the POI and E3 into productive proximity and result in the formation of new PPIs. Using the cocrystal structure of the complexed POI, PROTAC and E3 to inform, theoretically one can make the PROTAC “smaller” by removing the unimportant moieties on both POI and E3 ligand that do not facilitate ternary complex formation. This should not drastically decrease the PROTACs affinity for its targets because the new PPI formed between the POI and E3 ligase can compensate for subtle losses of target affinity by the modified ligands. Using this strategy, researchers may be able to rationally re-design a PROTAC into a molecular glue[146], which are currently more often discovered serendipitously rather than through a rational designed process. This proved feasible, to some extent, by Han et al. in 2019[59] when they demonstrated that highly potent and efficient PROTAC degraders can be successfully obtained using an E3 ligase ligand with a micromolar binding affinity to the E3 ligase (Figure 22, ARD-266). This result echoed our previous finding that haloPROTAC10 could induce similar maximal degradation of GFP-HaloTag7 compared to HaloPROTAC3, though a micromolar VHL binder was utilized[27].
Figure 22.

Potent PROTAC degraders can be obtained using an E3 ligase ligand with moderate binding affinity.
Mechanisms of Resistance in Targeted Protein Degradation
As discussed previously, PROTACs have the ability to overcome mutation resistance of the POI. However, several publications have revealed resistance mechanisms to prolonged treatment with degraders in tumor cells, like reduced expression or loss of E2 and E3 ubiquitin ligase at the mRNA or protein level[147, 148], or point mutations at the molecular glue interface that can readily disrupt ternary complex formation or more distal mutations that impair maximal protein degradation (Dmax)[149-151]. These results highlight the importance and urgency of ligand discovery for different types of E3 ligases for the development of more and varied PROTACs. In the short term, sequential exposure to degraders containing different E3 ligases or combination strategies should be employed to slow resistance down. Upregulation of the drug efflux pump MDR1 has also been reported to mediate intrinsic and acquired resistance to PROTACs in cancer cells[152]. Co-treatment with MDR1 inhibitors and PROTACs resulted in durable POI degradation even in cells with upregulated MDR1 expression.
Targeting Virus-related Proteins/RNA
As with cancer chemotherapy, a major challenge in antiviral drug development is the emergence of drug resistance. PROTACs have several advantages compared to traditional enzymatic inhibitors for viral polymerases and proteases. The first and most important advantage of the PROTAC technology is the ability to overcome drug resistance. PROTACs require only a transient protein-binding event, rather than prolonged occupancy of a specific active site, providing more opportunity to target a much larger pathogenic proteome repertoire. Moreover, successful PROTAC design can be accomplished starting from abandoned target-binding ligands that alone have disqualifying low affinities, but upon conjugation may achieve significantly higher potency.
Hepatitis C virus (HCV) is the main causative pathogen that leads to liver diseases, including chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma. Telaprevir (VX-950)[153] is a first-generation peptidomimetic protease inhibitor that targets a serine-type NS3/4A protease that possess multiple essential functions during the infectious cycle. However, multiple resistant variants were rapidly generated after telaprevir exposure in patients. In 2019, Wispelaere et al. reported the development of the first series of PROTACs targeting the HCV NS3/4A protease by conjugating telaprevir to CRBN ligands[154]. The most potent degrader, DGY-08-097 (Figure 23), mediated on-target degradation of NS3 with a DC50 of 50 nM. Most interestingly, DGY-08-097 retained antiviral activity against mutant viruses that are resistant to telaprevir, underscoring that this is an attractive strategy for antivirals development. However, the overall antiviral activity of telaprevir is still better than DGY-08-097. This may be explained by the loss in permeability that comes with PROTAC development, which could be improved by further medicinal chemistry efforts.
Figure 23.

Targeting virus-related proteins/RNA.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has rapidly spread around the world and resulted in the large-scale COVID-19 pandemic, which has wreaked havoc on our healthcare systems and our daily lives[155]. Though several highly effective vaccines were approved, effective methods for the treatment of severe COVID-19 are still urgently needed because of the rapid emergence of mutant strains, like Delta and Omicron. Again, targeted protein degradation strategy could be one of the choices. In 2020, Chatterjee et al. reported on their development of the first anti-COVID (peptide-based) PROTAC[156] by use of computational design. Their PROTAC demonstrated robust viral spike protein receptor binding domain (RBD) degradation capabilities in human cells and the ability to inhibit infection-competent viral production. Recently Desantis et al. revealed the first indomethacin-based small molecular PROTAC[157] as a pan-coronavirus antiviral agent that targets a host protein (prostaglandin E synthase type 2) rather than a viral one for degradation with the advantage of overcoming resistance issues. In 2020, Haniff et al. have designed a ribonuclease targeting chimera (RIBOTAC)[158] that recruits a cellular ribonuclease to degrade the viral genome leading to inhibition of SARS-CoV-2 frameshifting. The RNA degrader, C5-RIBOTAC (Figure 23), had improved bioactivity at least 10-fold compared to the parent small molecule C5. This approach suggests that RIBOTACs can indeed engage in the functional site of the SARS-CoV-2 genome, however further study should be conducted to determine their effects on live virus.
Targeting virus-related proteins or virus RNA for degradation provides a versatile and novel therapeutic platform to combat COVID-19, future emergent viral threats, and numerous infectious diseases. Together with the discovery of highly potent SMIs or binders, these approaches will support the common goal of eliminating infectious diseases and ultimately improve the health for everyone.
Conclusions
Targeted protein degradation induced by PROTACs, and related approaches, work through a unique event-driven MOA that confers this new modality with several advantages compared to traditional occupancy-driven SMIs. These include the catalytic nature of PROTACs, their potent and longer-lasting effect, an added layer of selectivity, reduced toxicity, overcoming resistance mechanisms, targeting nonenzymatic functions, and expanding target space. However, due to the bRo5 characteristics of PROTACs, drug development based on this technology usually suffers from low cell permeability and challenging oral bioavailability. Because PROTAC-mediated cellular activities are regulated by multiple factors including E3 activity, binary and ternary binding affinity, cooperativity, cell permeability, and compound solubility and stability, medicinal chemistry efforts should continue to be conducted that are tailored to improving physiochemical properties to make the PROTACs more drug like. Acquired resistance mechanisms involving the E3 ligase/ubiquitylation or POI and E3 ligase protein-protein interface following treatment with degraders highlights the urgency of ligand discovery for different types of E3 ligases[76], or for E3 recruitment by using indirect strategies[159] published by our group recently. Sequential exposure strategy with degraders containing different E3 ligases should be taken into consideration to slow down resistance. Choosing a POI that drives synthetic lethality, recruiting E3s essential for and highly expressed in tumor cells/tissues or employing conditional activation of PROTACs, such as antibody-PROTAC, folate-caged PROTAC and hypoxia-activated PROTAC may be beneficial for solving the on-target off-tissue toxicity issue associated with both PROTACs and the SMIs that comprise them. The ongoing phase 1/2 clinical trial of CFT7455 is a recent lesson given its neutropenia side effect, a known on-target toxicity associated with this IKZF1/3 degraders[160]. One can mimic the strategy of DT2216, which selectively degrades BCL-XL for the treatment of cancer with reduced platelet toxicity by recruiting an E3 ligase that is minimally expressed in platelets[161]. Global proteomic studies should be performed after PROTAC treatment to evaluate off-target degradation and the potential accumulation of intrinsic E3 substrates before any clinical trials. Binary and ternary complex binding constants should be evaluated to mitigate the high-dose hook effect, if possible, by increasing cooperativity. Recent advances using in silico approaches, like Rosetta[162], PRosettaC[163], PROTAC-Model[164] and those discussed in Delinker[165] et al., will enable the rationalizing of PROTAC-mediated ternary complex formation and improve PROTAC development efficiency.
Despite the many advantages of PROTACs outlined in this review, not all the targets are suitable for PROTAC mediated degradation. Therefore, validation and filtering procedures[166] could enable efficient PROTAC drug candidate prioritization and sorting. Together with other novel approaches that induce degradation based on proximity, like LYTAC, AUTAC, ATTEC and RIBOTAC, the target degradation community will open up new possibilities for drug discovery and provide tangible therapeutic benefits.
Acknowledgements
We would like to thank Dr. John Hines, Dr. Michael J. Bond and Dr. Yue Wang for their insightful comments on this manuscript. C.M.C. gratefully acknowledges NIH funding from the NCI (R35 CA197589 and R01 CA238570) and is supported by an American Cancer Research Professorship.
Footnotes
Conflicts of Interest
C. M. C. is a shareholder and consultant to Halda Therapeutics and Siduma Therapeutics. He is also a shareholder in Arvinas Inc.
References
- 1.Hopkins AL and Groom CR, Nat. Rev. Drug Discov, 2002, 1, 727–730. [DOI] [PubMed] [Google Scholar]
- 2.Nandi D, Tahiliani P, Kumar A and Chandu D, J. Biosci, 2006, 31, 137–155. [DOI] [PubMed] [Google Scholar]
- 3.Kumar SV, Bugg CE and Cook WJ, J. Mol. Biol, 1987, 194, 531–544. [DOI] [PubMed] [Google Scholar]
- 4.Livneh I, Cohen-Kaplan V, Cohen-Rosenzweig C, Avni N and Ciechanover A, Cell Res., 2016, 26, 869–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM and Deshaies RJ, Proc. Natl. Acad. Sci. U.S.A, 2001, 98, 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schneekloth JS, Fonseca FN, Koldobskiy M, Mandal A, Deshaies RJ, Sakamoto K and Crews CM, J. Am. Chem. Soc, 2004, 126, 3748–3754. [DOI] [PubMed] [Google Scholar]
- 7.Sakamoto K, Kim KB, Verma R, Rasnick A, Stein B, Crews CM and Deshaies RJ, Mol. Cell. Proteomics, 2003, 2, 1350–1358. [DOI] [PubMed] [Google Scholar]
- 8.Schneekloth AR, Pucheault M, Tae HS and Crews CM, Bioorg. Med. Chem. Lett, 2008, 18, 5904–5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Itoh Y, Ishikawa M, Naito M and Hashimoto Y, J. Am. Chem. Soc, 2010, 132, 5820–5826. [DOI] [PubMed] [Google Scholar]
- 10.Sekine K, Takubo K, Kikuchi R, Nishimoto M, Kitagawa M, Abe F, Nishikawa K, Tsuruo T and Naito M, J. Biol. Chem, 2008, 283, 8961–8968. [DOI] [PubMed] [Google Scholar]
- 11.Demizu Y, Shibata N, Hattori T, Ohoka N, Motoi H, Misawa T, Shoda T, Naito M and Kurihara M, Bioorg. Med. Chem. Lett, 2016, 26, 4865–4869. [DOI] [PubMed] [Google Scholar]
- 12.Shibata N, Nagai K, Morita Y, Ujikawa O, Ohoka N, Hattori T, Koyama R, Sano O, Imaeda Y, Nara H, Cho N and Naito M, J. Med. Chem, 2018, 61, 543–575. [DOI] [PubMed] [Google Scholar]
- 13.Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, Yamaguchi Y and Handa H, Science, 2010, 327, 1345–1350. [DOI] [PubMed] [Google Scholar]
- 14.Fischer ES, Bohm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, Nagel J, Serluca F, Acker V, Lingaraju GM, Tichkule RB, Schebesta M, Forrester WC, Schirle M, Hassiepen U, Ottl J, Hild M, Beckwith REJ, Harper JW, Jenkins JL and Thoma NH, Nature, 2014, 512, 49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kronke J, Fink EC, Hollenbach PW, MacBeth KJ, Hurst SN, Udeshi ND, Chamberlain PP, Mani DR, Man HW, Gandhi AK, Svinkina T, Schneider RK, McConkey M, Jaras M, Griffiths E, Wetzler M, Bullinger L, Cathers BE, Carr SA, Chopra R and Ebert BL, Nature, 2015, 523, 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petzold G, Fischer ES and Thomä NH, Nature, 2016, 532, 127–132. [DOI] [PubMed] [Google Scholar]
- 17.Matyskiela ME, Lu G, Ito T, Pagarigan B, Lu C, Miller K, Fang W, Wang N, Nguyen D, Houston J, Carmel G, Tran T, Riley M, Nosaka L, Lander GC, Gaidarova S, Xu S, Ruchelman AL, Handa H, Carmichael J, Daniel TO, Cathers BE, Lopez-Girona A and Chamberlain PP, Nature, 2016, 535, 252–261. [DOI] [PubMed] [Google Scholar]
- 18.Matyskiela ME, Clayton T, Zheng X, Mayne C, Tran E, Carpenter A, Pagarigan B, McDonald J, Rolfe M, Hamann LG, Lu G and Chamberlain PP, Nat. Struc. Mol. Biol, 2020, 27, 319–322. [DOI] [PubMed] [Google Scholar]
- 19.Furihata H, Yamanaka S, Honda T, Miyauchi Y, Asano A, Shibata N, Tanokura M, Sawasaki T and Miyakawa T, Nat. Comm, 2020, 11, 4578–4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Belair DG, Lu G, Waller LE, Gustin JA, Collins ND and Kolaja KL, Scientific Reports, 2020, 10, 2864–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buckley DL, Molle IV, Gareiss PC, Tae HS, Michel J, Noblin DJ, Jorgensen WL, Ciulli A and Crews CM, J. Am. Chem. Soc, 2012, 134, 4465–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buckley DL, Gustafson JL, Molle IV, Roth AG, Tae HS, Gareiss PC, Jorgensen WL, Ciulli A and Crews CM, Angew. Chem. Int. Ed, 2012, 51, 11463–11467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Molle IV, Thomann A, Buckley DL, So EC, Lang S, Crews CM and Ciulli A, Chem. Biol, 2012, 19, 1300–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galdeano C, Gadd MS, Soares P, Scaffidi S, Molle IV, Birced I, Hewitt S, Dias DM and Ciulli A, J. Med. Chem, 2014, 57, 8657–8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frost J, Galdeano C, Soares P, Gadd MS, Grzes KM, Ellis L, Epemolu O, Shimamura S, Bantscheff M, Grandi P, Read KD, Cantrell DA, Rocha S and Ciulli A, Nat. Comm, 2016, 7, 13312–13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soares P, Gadd MS, Frost J, Galdeano C, Ellis L, Epemolu O, Rocha S, Read KD and Ciulli A, J. Med. Chem, 2018, 61, 599–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buckley DL, Raina K, Darricarrere N, Hines J, Gustafson JL, Smith IE, Miah AH, Harling JD and Crews CM, ACS Chem. Biol, 2015, 10, 1831–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohana RF, Encell LP, Zhao K, Simpson D, Slater MR, Urh M and Wood KV, Protein Expression Purif., 2009, 68, 110–120. [DOI] [PubMed] [Google Scholar]
- 29.HaloTag Technology, Expression-ready ORF cDNA Clone Collection∣GeneCopeia. http://www.genecopoeia.com/tech/halo-tag/. [Google Scholar]
- 30.Caine EA, Mahan SD, Johnson RL, Nieman AN, Lam N, Warren CR, Riching KM, Urh M and Daniels DL, Current Protocols in Pharmacology, 2020, 91, 81–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tovell H, Testa A, Maniaci C, Zhou H, Prescott AR, Macartney T, Ciulli A and Alessi DR, ACS Chem. Biol, 2019, 14, 882–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32a).Bondeson DP, Mares A, Smith ID, Ko E, Campos S, Miah AH, Mulholland KE, Routly N, Buckley DL, Gustafson JL, Zinn N, Grandi P, Shimamura S, Bergamini G, Savitski MF, Bantscheff M, Cox C, Gordon DA, Willard RR, Flanagan JJ, Casillas LN, Votta BJ, Besten W, Famm K, Kruidenier L, Carter PS, Harling JD, Churcher I and Crews CM, Nat. Chem. Biol, 2015, 11, 611–620. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tinworth CP, Lithgow H, Dittus L, Bassi ZI, Hughes SE, Muelbaier M, Dai H, Smith IED, Kerr WJ, Burley GA, Bantscheff M and Harling JD, ACS Chem. Biol 2019, 14, 342–347. [DOI] [PubMed] [Google Scholar]; c) Crew AP, Raina K, Dong H, Qian Y, Wang J, Vigil D, Serebrenik YV, Hamman BD, Morgan A, Ferraro C, Siu K, Neklesa TK, Winkler JD, Coleman KG and Crews CM, J. Med. Chem 2018, 61, 583–598. [DOI] [PubMed] [Google Scholar]
- 33.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, Thangue NL, French CA, Wiest O, Kung AL, Knapp S and Bradner JE, Nature, 2010, 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zengerle M, Chan KH and Ciulli A, ACS Chem. Biol, 2015, 10, 1770–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, Hines J, Winkler JD, Crew AP, Coleman K and Crews CM, Chem. Biol, 2015, 22, 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Paganon SD and Bradner JE, Science, 2015, 348, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bondeson DP, Smith BE, Burslem GM, Buhimschi AD, Hines J, Figueroa SJ, Wang J, Hamman BD, Ishchenko A and Crews CM, Cell Chem. Biol, 2018, 25, 78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chan KH, Zengerle M, Testa A and Ciulli A, J. Med. Chem, 2018, 61, 504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gosmini R, Nguyen VL, Toum J, Simon C, Brusq J-MG, Krysa G, Mirguet O, Riou-Eymard AM, Boursier EV, Trottet L, Bamborough P, Clark H, Chung C-WL, Cutler, Demont EH, Kaur R, Lewis AJ, Schilling MB, Soden PE, Taylor S, Walker AL, Walker MD, Prinjha RK and Nicodeme E, J. Med. Chem, 2014, 57, 8111–8131. [DOI] [PubMed] [Google Scholar]
- 40.Gadd MS, Testa A, Lucas X, Chan KH, Chen W, Lamont DJ, Zengerle M and Ciulli A, Nat. Chem. Biol, 2017, 13, 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nowak RP, DeAngelo SL, Buckley D, He Z, Donovan KA, An J, Safaee N, Jedrychowski MP, Ponthier CM, Ishoey M, Zhang T, Mancias JD, Gray NS, Bradner JE and Fischer ES, Nat. Chem. Biol, 2018, 14, 706–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Farnaby W, Koegl M, Roy MJ, Whitworth C, Diers E, Trainor N, Zollman D, Steurer S, Oezguer JK, Riedmueller C, Gmaschitz T, Wachter J, Dank C, Galant M, Sharps B, Rumpel K, Traxler E, Gerstberger T, Schnitzer R, Petermann O, Greb P, Weinstabl H, Bader G, Zoephel A, Puxbaum AW, Wölfer KE, Wöhrle S, Boehmelt G, Rinnenthal J, Arnhof H, Wiechens N, Wu M, Hughes TO, Ettmayer P, Pearson M, McConnell DB and Ciulli A, Nat. Chem. Biol, 2019, 15, 672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Law RP, Nunes J, Chung C, Bantscheff M, Buda K, Dai H, Evans JP, Flinders A, Klimaszewska D, Lewis AJ, Muelbaier M, Stevens PS, Stacey P, Tame CJ, Watt GF, Zinn N, Queisser MA, Harling JD and Benowitz AB, Angew. Chem. Int. Ed, 2021, 60, 23327–23334. [DOI] [PubMed] [Google Scholar]
- 44.Pettersson M and Crews CM, Drug Disco. Today Technol, 2019, 31, 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Douglass EF, Miller CJ, Sparer G, Shapiro H and Spiegel DA. J. Am. Chem. Soc, 2013, 135, 6092–6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roy RD, Rosenmund C and Stefan MI, BMC Sys. Biol, 2017, 11, 74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mares A, Miah AH, Smith IED, Rackham M, Thawani AR, Cryan J, Haile PA, Votta BJ, Beal AM, Capriotti C, Reilly MA, Fisher DT, Zinn N, Bantscheff M, MacDonald TT, Vossenkamper A, Dace P, Churcher I, Benowitz AB, Watt G, Denyer J, Stevens PS and Harling JD, Commun. Biol, 2020, 3, 140–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mathieson T, Franken H, Kosinski J, Kurzawa N, Zinn N, Sweetman G, Poeckel D, Ratnu VS, Schramm M, Becher I, Steidel M, Noh KM, Bergamini G, Beck M, Bantscheff M and Savitski MM, Nat. Commun, 2018, 9, 689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dobrovolsky D, Wang ES, Morrow S, Leahy C, Faust T, Nowak RP, Donovan KA, Yang G, Li Z, Fischer ES, Treon SP, Weinstock DM and Gray NS, Blood, 2019, 133, 952–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mohamed AJ, Yu L, Backesjo CM, Vargas L, Faryal R, Aints A, Christensson B, Berglof A, Vihinen M, Nore BF and Smith CIE, Immunol. Rev, 2009, 228, 58–73. [DOI] [PubMed] [Google Scholar]
- 51.Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA and Buggy JJ, Proc. Natl. Acad. Sci. U.S.A, 2010, 107, 13075–13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paolo JAD, Huang T, Balazs M, Barbosa J, Barck KH, Bravo BJ, Carano RAD, Darrow J, Davies DR, DeForge LE, Diehl L, Ferrando R, Gallion SL, Giannetti AM, Gribling P, Hurez V, Hymowitz SG, Jones R, Kropf JE, Lee WP, Maciejewski PM, Mitchell SA, Rong H, Staker BL, Whitney JA, Yeh S, Young WB, Yu C, Zhang J, Reif K and Currie KS, Nat. Chem. Biol, 2011, 7, 41–50. [DOI] [PubMed] [Google Scholar]
- 53.Salami J, Alabi S, Willard RR, Vitale NJ, Wang J, Dong H, Jin M, McDonnell DP, Crew AP, Neklesa TK and Crews CM, Commun. Biol, 2018, 1, 100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, True LD and Nelson PS, Cancer Res., 2008, 68, 4447–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Konecny GE, Pegram MD, Venkatesan N, Finn R, Yang G, Rahmeh M, Untch M, Rusnak DW, Spehar G, Mullin RJ, Keith BR, Gilmer TM, Berger M, Podratz KC and Slamon DJ, Cancer Res., 2006, 66, 1630–1639. [DOI] [PubMed] [Google Scholar]
- 56.Burslem GM, Smith BE, Lai AC, Figueroa SJ, McQuaid DC, Bondeson DP, Toure M, Dong H, Qian Y, Wang J, Crew AP, Hines J and Crews CM, Cell Chem. Biol, 2018, 25, 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith BE, Wang SL, Figueroa SJ, Harbin A, Wang J, Hamman BD and Crews CM, Nat. Commun, 2019, 10, 131–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Han X, Wang C, Qin C, Xiang W, Salas EF, Yang CY, Wang M, Zhao L, Xu T, Chinnaswamy K, Delproposto J, Stuckey J and Wang S, J. Med. Chem, 2019, 62, 941–964. [DOI] [PubMed] [Google Scholar]
- 59.Han X, Zhao L, Xiang W, Qin C, Miao B, Xu T, Wang M, Yang CY, Chinnaswamy K, Stuckey J and Wang S, J. Med. Chem, 2019, 62, 11218–11231. [DOI] [PubMed] [Google Scholar]
- 60.Testa A, Hughes SJ, Lucas X, Wright JE and Ciulli A, Angew. Chem. Int. Ed, 2020, 59, 1727–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hu Z and Crews CM, Chem. Bio. Chem, 2022, 23, e202100270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Figueroaa SJ, Buhimschia AD, Tourea M, Hinesa J, Crews CM, Bioorg. Med. Chem. Lett, 2020, 30, 126877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Goracci L, Desantis J, Valeri A, Castellani B, Eleuteri M, Cruciani G, J. Med. Chem, 2020, 63, 11615–11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang X and Zhang H, Chen X, Cancer Drug Resist., 2019, 2, 141–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Buhimschi AD, Armstrong HA, Toure M, Figueroa SJ, Chen TL, Lehman AM, Woyach JA, Johnson AJ, Byrd JC and Crews CM, Biochemistry, 2018, 57, 3564–3575. [DOI] [PubMed] [Google Scholar]
- 66.Sun Y, Zhao X, Ding N, Gao H, Wu Y, Yang Y, Zhao M, Hwang J, Song Y, Liu W and Rao Y, Cell Research, 2018, 28, 779–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sun Y, Ding N, Song Y, Yang Z, Liu W, Zhu J and Rao Y, Leukemia, 2019, 33, 2105–2110. [DOI] [PubMed] [Google Scholar]
- 68.Schiemer J, Horst R, Meng Y, Montgomery JI, Xu Y, Feng X, Borzilleri K, Uccello DP, Leverett C, Brown S, Che Y, Brown MF, Hayward MM, Gilbert AM, Noe MC and Calabrese MF, Nat. Chem. Biol, 2021, 17, 152–160. [DOI] [PubMed] [Google Scholar]
- 69.Burslem GM, Schultz AR, Bondeson DP, Eide CA, Stevens SLS, Druker BJ and Crews CM, Cancer Res., 2019, 79, 4744–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cromm PM, Samarasinghe KTG, Hines J and Crews CM, J. Am. Chem. Soc, 2018, 140, 17019–17026. [DOI] [PubMed] [Google Scholar]
- 71.Popow J, Arnhof H, Bader G, Berger H, Ciulli A, Covini D, Dank C, Gmaschitz T, Greb P, Özguer JK, Koegl M, McConnell DB, Pearson M, Rieger M, Rinnenthal J, Roessler V, Schrenk A, Spina M, Steurer S, Trainor N, Traxler E, Wieshofer C, Zoephel A and Ettmayer P, J. Med. Chem, 2019, 62, 2508–2520. [DOI] [PubMed] [Google Scholar]
- 72.Gao H, Wu Y, Sun Y, Yang Y, Zhou G and Rao Y, ACS Med. Chem. Lett, 2020, 11, 1855–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Humphreys PG, Bamborough P, Chung C, Craggs PD, Gordon L, Grandi P, Hayhow TG, Hussain J, Jones KL, Lindon M, Michon AM, Renaux JF, Suckling CJ, Tough DF and Prinjha RK, J. Med. Chem, 2017, 60, 695–709. [DOI] [PubMed] [Google Scholar]
- 74.Bassi ZI, Fillmore MC, Miah AH, Chapman TD, Maller C, Roberts EJ, Davis LC, Lewis DE, Galwey NW, Waddington KE, Parravicini V, Jones ALM, Gongora C, Humphreys PG, Churcher I, Prinjha RK and Tough DF, ACS Chem. Biol, 2018, 13, 2862–2867. [DOI] [PubMed] [Google Scholar]
- 75.Lai AC and Crews CM, Nat. Rev. Drug Discov, 2017, 16, 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schapira M, Calabrese MF, Bullock AN and Crews CM, Nat. Rev. Drug Discov, 2019, 18, 949–963. [DOI] [PubMed] [Google Scholar]
- 77.Verma R, Mohl D and Deshaies RJ, Molecular Cell, 2020, 77, 446–460. [DOI] [PubMed] [Google Scholar]
- 78.Hines J, Gough JD, Corson TW and Crews CM, Proc. Natl. Acad. Sci. U.S.A, 2013, 110, 8942–8947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bond MJ and Crews CM, RSC Chem. Biol, 2021, 2, 725–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hu B, Zhou Y, Sun D, Yang Y, Liu Y, Li X, Li Hua and Chen L, Eur. J. Med. Chem, 2020, 207, 112698–112710. [DOI] [PubMed] [Google Scholar]
- 81.Bai L, Zhou H, Xu R, Zhao Y, Chinnaswamy K, McEachern D, Chen J, Yang C, Liu Z, Wang M, Liu L, Jiang H, Wen B, Kumar P, Meagher JL, Sun D, Stuckey JA and Wang S, Cancer Cell, 2019, 36, 498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhou H, Bai L, Xu R, Zhao Y, Chen J, McEachern D, Chinnaswamy K, Wen B, Dai L, Kumar P, Yang C, Liu Z, Wang M, Liu L, Meagher JL, Yi H, Sun D, Stuckey JA and Wang S, J. Med. Chem, 2019, 62, 11280–11300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhou H, Bai L, Xu Renqi, McEachern D, Chinnaswamy K, Li R, Wen B, Wang M, Yang C, Meagher JL, Sun D, Stuckey JA and Wang S, ACS Med. Chem. Lett, 2021, 12, 996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Luo G, Lin X, Medina AV, Xiao M, Li G, Wei H, Martínez CAV and Xiang H, J. Med. Chem, 2021, 64, 17098–17114. [DOI] [PubMed] [Google Scholar]
- 85.Liao H, Li X, Zhao L, Wang Y, Wang X, Wu Y, Zhou X, Fu W, Liu L, Hu H and Chen Y, Cell Disc., 2020, 6, 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Samarasinghe KTG, Figueroa SJ, Burgess M, Nalawansha DA, Dai K, Hu Z, Bebenek A, Holley SA and Crews CM, Cell Chem. Biol, 2021, 28, 648–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liu J, Chen H, Kaniskan HÜ, Xie L, Chen X, Jin J and Wei W, J. Am. Chem. Soc, 2021, 143, 8902–8910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shao J, Yan Y, Ding D, Wang D, He Y, Pan Y, Yan W, Kharbanda A, Li H and Huang H, Adv. Sci, 2021, 8, 2102555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Samarasinghe KTG, An E, Genuth MA, Chu L, Holley SA and Crews CM, bioRxiv, 2021, doi: 10.1101/2021.12.20.473482. [DOI] [Google Scholar]
- 90.Xiao Z, Song S, Chen D, Merkerk R, Wouden PE, Cool RH, Quax WJ, Poelarends GJ, Melgert BN and Dekker FJ, Angew. Chem. Int. Ed, 2021, 60, 17514–17521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Golas MM, Sander B, Will CL, Luhrmann R, Stark H, Science, 2003, 300, 980–984. [DOI] [PubMed] [Google Scholar]
- 92.Brambila RAG, Chen J, Zhou J, Tascher G, Munch C and Cheng X, Cell Chem. Biol, 2021, 28, 1616–1627. [DOI] [PubMed] [Google Scholar]
- 93.Cheng X, Yoshida H, Raoofi D, Saleh S, Alborzinia H, Wenke F, Göhring A, Reuter S, Mah N, Fuchs H, Navarro MAA, Adjaye J, Gul S, Utikal J, Mrowka R and Wölf S, J. Med. Chem, 2015, 58, 5742–5750. [DOI] [PubMed] [Google Scholar]
- 94.Zou Y, Ma D and Wang Y, Cell Biochem. Funct, 2019, 37, 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sun X, Gao H, Yang Y, He M, Wu Y, Song Y, Tong Y and Rao Y, Signal Transduction Targeted Therapy, 2019, 4, 64–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mullard Asher, Nat. Rev. Drug Disc, 2021, 20, 247–250. [DOI] [PubMed] [Google Scholar]
- 97.Békés M, Langley DR and Crews CM, Nat. Rev. Drug Disc, 2022, 21, 181–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhao Y, Goroff YRM, Xue JY, Ang A, Lucas J, Mai TT, Paula AFC, Saiki AY, Mohn D, Achanta P, Sisk AE, Arora KS, Roy RS, Kim D, Li C, Lim LP, Li M, Bahr A, Loomis BR, Stanchina E, Filho JSR, Weigelt B, Berger M, Riely G, Arbour KC, Lipford JR, Li BT and Lito P, Nature, 2021, 599, 679–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bond MJ, Chu L, Nalawansha DA, Li K and Crews CM, ACS Cent. Sci, 2020, 6, 1367–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100a).Teng KW, Tsai ST, Hattori T, Fedele C, Koide A, Yang C, Hou X, Zhang Y, Neel BG, O’Bryan JP and Koide S, Nature Commun., 2021, 12, 2656–2668. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sha F, Gencer EB, Georgeon S, Koide A, Yasui N, Koide S and Hantschel O, Proc. Natl. Acad. Sci. U.S.A, 2013, 110, 14924–14929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang X, Allen S, Blake JF, Bowcut V, Briere DM, Calinisan A, Dahlke JR, Fell JB, Fischer JP, Gunn RJ, Hallin J, Laguer J, Lawson JD, Medwid J, Newhouse B, Nguyen P, O’Leary JM, Olson P, Pajk S, Rahbaek L, Rodriguez M, Smith CR, Tang TP, Thomas NC, Vanderpool D, Vigers GP, Christensen JG and Marx MA, J. Med. Chem, 2022, 65, 3123–3133. [DOI] [PubMed] [Google Scholar]
- 102.Dragovich PS, Pillow TH, Blake RA, Sadowsky JD, Adaligil E, Adhikari P, Bhakta S, Blaquiere N, Chen J, Chuh JC, Gascoigne KE, Hartman SJ, He M, Kaufman S, Kleinheinz T, Kozak KR, Liu L, Liu L, Liu Q, Lu Y, Meng F, Mulvihill MM, O’Donohue A, Rowntree RK, Staben LR, Staben ST, Wai J, Wang J, Wei B, Wilson C, Xin J, Xu Z, Yao H, Zhang D, Zhang H, Zhou H and Zhu X, J. Med. Chem, 2021, 64, 2534–2575. [DOI] [PubMed] [Google Scholar]
- 103.Dragovich PS, Pillow TH, Blake RA, Sadowsky JD, Adaligil E, Adhikari P, Chen J, Corr N, Chuh JC, Rosario GD, Fullerton A, Hartman SJ, Jiang F, Kaufman S, Kleinheinz T, Kozak KR, Liu L, Lu Y, Mulvihill MM, Murray JM, O’Donohue A, Rowntree RK, Sawyer WS, Staben LR, Wai J, Wang J, Wei B, Wei W, Xu Z, Yao H, Yu S, Zhang D, Zhang H, Zhang S, Zhao Y, Zhou H and Zhu X, J. Med. Chem, 2021, 64, 2576–2607. [DOI] [PubMed] [Google Scholar]
- 104.Zhang H, Han Y, Yang Y, Lin F, Li K, Kong L, Liu H, Dang Y, Lin J and Chen PR, J. Am. Chem. Soc, 2021, 143, 16377–16382. [DOI] [PubMed] [Google Scholar]
- 105.He S, Gao F, Ma J, Ma H, Dong G and Sheng C, Angew. Chem. Int. Ed, 2021, 60, 23299–23305. [DOI] [PubMed] [Google Scholar]
- 106.Patil KM, Chin D, Seah HL, Shi Q, Lim KW and Phan AT, Chem. Commun, 2021, 57, 12816–12819. [DOI] [PubMed] [Google Scholar]
- 107.Zheng M, Huo J, Gu X, Wang Y, Wu C, Zhang Q, Wang W, Liu Y, Liu Y, Zhou X, Chen L, Zhou Y and Li H, J. Med. Chem, 2021, 64, 7839–7852. [DOI] [PubMed] [Google Scholar]
- 108.Maple HJ, Clayden N, Baron A, Stacey C and Felix R, Med. Chem. Commun, 2019, 10, 1755–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Klein VG, Bond AG, Craigon C, Lokey RS and Ciulli A, J. Med. Chem, 2021, 64, 18082–18101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Imaide S, Riching KM, Makukhin N, Vetma V, Whitworth C, Hughes SJ, Trainor N, Mahan SD, Murphy N, Cowan AD, Chan K, Craigon C, Testa A, Maniaci C, Urh M, Daniels DL and Ciulli A, Nat. Chem. Biol, 2021, 17, 1157–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Xue G, Wang K, Zhou D, Zhong H and Pan Z, J. Am. Chem. Soc, 2019, 141, 18370–18374. [DOI] [PubMed] [Google Scholar]
- 112.Naro Y, Darrah K and Deiters A, J. Am. Chem. Soc, 2020, 142, 2193–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kounde CS, Shchepinova MM, Saunders CN, Muelbaier M, Rackham MD, Harling JD and Tate EW, Chem. Commun, 2020, 56, 5532–5535. [DOI] [PubMed] [Google Scholar]
- 114.Liu J, Chen H, Ma L, He Z, Wang D, Liu Y, Lin Q, Zhang T, Gray N, Kaniskan HÜ, Jin J, Wei W, Sci. Adv, 2020, 6, 5154–5164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pfaff P, Samarasinghe KTG, Crews CM and Carreira EM, ACS Cent. Sci, 2019, 5, 1682–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jin Y, Lu M, Wang Y, Shan W, Wang X, You Q and Jiang Z, J. Med. Chem, 2020, 63, 4644–4654. [DOI] [PubMed] [Google Scholar]
- 117.Reynders M, Matsuura BS, Bérouti M, Simoneschi D, Marzio A, Pagano M, Trauner D, Sci. Adv, 2020, 6, 5064–5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nalawansha DA and Crews CM, Cell Chem. Biol, 2020, 27, 998–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Liu J, Chen H, Liu Y, Shen Y, Meng F, Kaniskan HÜ, Jin J and Wei W, J. Am. Chem. Soc, 2021, 143, 7380–7387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chen H, Liu J, Kaniskan HÜ, Wei W and Jin J, J. Med. Chem, 2021, 64, 12273–12285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cheng W, Li S, Wen X, Han S, Wang S, Wei H, Song Z, Wang Y, Tian X and Zhang X, Chem. Commun, 2021, 57, 12852–12855. [DOI] [PubMed] [Google Scholar]
- 122.Banik SM, Pedram K, Wisnovsky S, Ahn G, Riley NM and Bertozzi CR, Nature, 2020, 584, 291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Takahashi D, Moriyama J, Nakamura T, Miki E, Takahashi E, Sato A, Akaike T, Nakama KI and Arimoto H, Mol. Cell, 2019, 76, 797–810. [DOI] [PubMed] [Google Scholar]
- 124.Li Z, Wang C, Wang Z, Zhu C, Li J, Sha T, Ma L, Gao C, Yang Y, Sun Y, Wang J, Sun X, Lu C, Difiglia M, Mei Y, Ding C, Luo S, Dang Y, Ding Y, Fei Y and Lu B, Nature, 2019, 575, 203–209. [DOI] [PubMed] [Google Scholar]
- 125.Ding Y, Fei Y and Lu B, Trends Pharm. Sci, 2020, 41, 464–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Baisden JT, Disney JLC, Ryan LS and Disney MD, Curr. Opin. Chem. Biol, 2021, 62, 119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhang P, Liu X, Abegg D, Tanaka T, Tong Y, Benhamou RI, Baisden J, Crynen G, Meyer SM, Cameron MD, Chatterjee AK, Adibekian A, Disney JLC and Disney MD, J. Am. Chem. Soc, 2021, 143, 13044–13055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Costales MG, Aikawa H, Li Y, Disney JLC, Abegg D, Hoch DG, Velagapudi SP, Nakai Y, Khan T, Wang KW, Yildirim I, Adibekian A, Wang ET and Disney MD, Proc. Natl. Acad. Sci. U. S. A, 2020, 117, 2406–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lai AC, Toure M, Hellerschmied D, Salami J, Figueroa SJ, Ko E, Hines J and Crews CM, Angew. Chem. Int. Ed, 2016, 55, 807–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Clague MJ, Heride C and Urbe S, Trends Cell Biol., 2015, 25, 417–426. [DOI] [PubMed] [Google Scholar]
- 131.Ottis P, Toure M, Cromm PM, Ko E, Gustafson JL and Crews CM, ACS Chem. Biol, 2017, 12, 2570–2578. [DOI] [PubMed] [Google Scholar]
- 132.Wang Q, Ru Y, Zhong D, Zhang J, Yao L and Li X, Oncotarget, 2014, 5, 4945–4958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Liu L, Damerell DR, Koukouflis L, Tong Y, Marsden BD, Schapira M, Bioinformatics, 2019, 35, 2882–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ishida T and Ciulli A, SLAS Discovery, 2021, 26, 484–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gabizon R and London N, Curr. Opin. Chem. Biol, 2021, 62, 24–33. [DOI] [PubMed] [Google Scholar]
- 136.Brenner S and Lerner RA, Proc. Natl. Acad. Sci. U.S.A, 1992, 89, 5381–5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.A Clark M, Acharya RA, Muendel CCA, Belyanskaya SL, Benjamin DR, Carlson NR, Centrella PA, Chiu CH, Creaser SP, Cuozzo JW, Davie CP, Ding Y, Franklin GJ, Franzen KD, Gefter ML, Hale SP, Hansen NJV, Israel DI, Jiang J, Kavarana MJ, Kelley MS, Kollmann CS, Li F, Lind K, Mataruse S, Medeiros PF, Messer JA, Myers P, O’Keefe H, Oliff MC, Rise CE, Satz AL, Skinner SR, Svendsen JL, Tang L, Vloten K, Wagner RW, Yao G, Zhao B and Morgan BA, Nat. Chem. Biol, 2009, 5, 647–654. [DOI] [PubMed] [Google Scholar]
- 138.Disch JS, Duffy JM, Lee ECY, Gikunju D, Chan B, Levin B, Monteiro MI, Talcott SA, Lau AC, Zhou F, Kozhushnyan A, Westlund NE, Mullins PB, Yu Y, Rechenberg M, Zhang J, Arnautova YA, Liu Y, Zhang Y, McRiner AJ, Keefe AD, Kohlmann A, Clark MA, Cuozzo JW, Huguet C and Arora S, J. Med. Chem, 2021, 64, 5049–5066. [DOI] [PubMed] [Google Scholar]
- 139.See the selected DEL opensource/kits here: https://www.hitgen.com/en/capabilities-details-21.html; https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/chemistry-and-synthesis/lead-discovery/fragment-based-dna-encoded-library-for-drug-discovery; https://hits.wuxiapptec.com/delopen.
- 140.Zhou B, Hu J, Xu F, Chen Z, Bai L, Salas EF, Lin M, Liu L, Yang C, Zhao Y, McEachern D, Przybranowski S, Wen B, Sun D and Wang S, J. Med. Chem, 2018, 61, 462–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Klein VG, Townsend CE, Testa A, Zengerle M, Maniaci C, Hughes SJ, Chan K, Ciulli A and Lokey RS, ACS Med. Chem. Lett, 2020, 11, 1732–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Atilaw Y, Poongavanam V, Nilsson CS, Nguyen D, Giese A, Meibom D, Erdelyi M and Kihlberg J, ACS Med. Chem. Lett, 2021, 12, 107–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ermondi G, Jimenez DG, Sebastiano MR and Caron G, ACS Med. Chem. Lett, 2021, 12, 1056–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Caron G, Kihlberg J, Goetz G, Ratkova E, Poongavanam V and Ermondi G, ACS Med. Chem. Lett, 2021, 12, 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Poongavanam V, Doak BC and Kihlberg J, Curr. Opin. in Chem. Biol, 2018, 44, 23–29. [DOI] [PubMed] [Google Scholar]
- 146.For selected molecular glue reviews, see: a) Buhimschi AD and Crews CM, Biochemistry, 2019, 58, 861–864. [DOI] [PubMed] [Google Scholar]; b) Schreiber SL, Cell, 2021, 184, 3–9. [DOI] [PubMed] [Google Scholar]; c) Ege N, Bouguenina H, Tatari M and Chopra R, Cell Chem. Biol, 2021, 28, 283–299. [DOI] [PubMed] [Google Scholar]; d) Dong G, Ding Y, He S and Sheng C, J. Med. Chem, 2021, 64, 15, 10606–10620. [DOI] [PubMed] [Google Scholar]; e) Kozicka Z and Holger Thoma N, Cell Chem. Biol, 2021, 28, 1032–1047. [DOI] [PubMed] [Google Scholar]; f) Geiger TM, Schafer SC, Dreizler JK, Walz M and Hausch F, Curr. Research in Chem. Biol, 2022, 2, 100018. [Google Scholar]
- 147.Ottis P, Palladino C, Thienger P, Britschgi A, Heichinger C, Berrera M, Laferriere AJ, Roudnicky F, Thong TK, Bischoff JR, Martoglio B and Pettazzoni P, ACS Chem. Biol, 2019, 14, 2215–2223. [DOI] [PubMed] [Google Scholar]
- 148.Shirasaki R, Matthews GM, Gandolfi S, Simoes RM, Buckley DL, Vora JR, Sievers QL, Bruggenthies JB, Dashevsky O, Poarch H, Tang H, Bariteau MA, Sheffer M, Hu Y, Kopyscinski SLD, Hengeveld PJ, Glassner BJ, Dhimolea E, Ott CJ, Zhang T, Kwiatkowski NP, Laubach JP, Schlossman RL, Richardson PG, Culhane AC, Groen RWJ, Fischer ES, Vazquez F, Tsherniak A, Hahn WC, Levy J, Auclair D, Licht JD, Keats JJ, Boise LH, Ebert BL, Bradner JE, Gray NS and Mitsiades CS, Cell Rep., 2021, 34, 108532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sievers QL, Gasser JA, Cowley GS, Fischer ES and Ebert BL, Blood, 2018, 132, 1293–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gooding S, Pour NA, Towfic F, Estevez MO, Chamberlain PP, Tsai KT, Flynt E, Hirst M, Rozelle D, Dhiman P, Neri P, Ramasamy K, Bahlis N, Vyas P and Thakurta A, Blood, 2021, 137, 232–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gosavi PM, Ngan KC, Yeo M, Su C, Li J, Lue NZ, Hoenig SM and Liau BB, ACS Cent. Sci 2022, 8, 417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kurimchak AM, Montávez CH, Montserrat S, Araiza D, Hu J, Jin J and Duncan JS, bioRxiv, 2021, doi: 10.1101/2021.12.02.470920. [DOI] [Google Scholar]
- 153.Perni RB, Almquist SJ, Byrn RA, Chandorkar G, Chaturvedi PR, Courtney LF, Decker CJ, Dinehart K, Gates CA, Harbeson SL, Heiser A, Kalkeri G, Kolaczkowski E, Lin K, Luong Y, Rao BG, Taylor WP, Thomson JA, Tung RD, Wei Y, Kwong AD and Lin C, Antimicrob. Agents Chemother, 2006, 50, 899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wispelaere M, Du G, Donovan KA, Zhang T, Eleuteri NA, Yuan JC, Kalabathula J, Nowak RP, Fischer ES, Gray NS and Yang PL, Nat. Commun, 2019, 10, 3468–3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Schröder I, ACS Chem. Health Saf, 2020, 27, 160–169. [DOI] [PubMed] [Google Scholar]
- 156.Chatterjee P, Ponnapati M, Kramme C, Plesa AM, Church GM and Jacobson JM, Commun. Biol, 2020, 3, 715–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Desantis J, Mercorelli B, Celegato M, Croci F, Bazzacco A, Baroni M, Siragusa L, Cruciani G, Loregian A and Goracci L, Eur. J. Med. Chem, 2021, 226, 113814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Haniff HS, Tong Y, Liu X, Chen JL, Suresh BM, Andrews RJ, Peterson JM, O’Leary CA, Benhamou RI, Moss WN and Disney MD, ACS Cent. Sci, 2020, 6, 1713–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Nalawansha DA, Li K, Hines J, Crews CM, J. Am. Chem. Soc, 2022, 144, 5594–5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. https://www.globenewswire.com/news-release/2022/04/08/2419449/0/en/C4-Therapeutics-Presents-Clinical-Data-from-Cohort-A-of-the-Ongoing-Phase-1-2-Clinical-Trial-of-CFT7455-a-Novel-IKZF1-3-Degrader.html.
- 161.Khan S, Zhang X, Lv D, Zhang Q, He Y, Zhang P, Liu X, Thummuri D, Yuan Y, Wiegand JS, Pei J, Zhang W, Sharma A, McCurdy CR, Kuruvilla VM, Baran N, Ferrando AA, Kim Y, Rogojina A, Houghton PJ, Huang G, Hromas R, Konopleva M, Zheng G and Zhou D, Nat. Med 2019, 25, 1938–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Bai N, Miller SA, Andrianov GV, Yates M, Kirubakaran P and Karanicolas J, J. Chem. Inf. Model, 2021, 61, 1368–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zaidman D, Prilusky J and London N, J. Chem. Inf. Model, 2020, 60, 4894–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Weng G, Li D, Kang Y and Hou T, J. Med. Chem, 2021, 64, 16271–16281. [DOI] [PubMed] [Google Scholar]
- 165.Imrie F, Bradley AR, Schaar M and Deane CM, J. Chem. Inf. Model, 2020, 60, 1983–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Schneider M, Radoux CJ, Hercules A, Ochoa D, Dunham I, Zalmas LP, Hessler G, Ruf S, Shanmugasundaram V, Hann MM, Thomas PJ, Queisser MA, Benowitz AB, Brown K and Leach AR, Nat. Rev. Drug Disc, 2021, 20, 789–797. [DOI] [PubMed] [Google Scholar]