

Summary
At active human genes, the +1 nucleosome is located downstream of the RNA polymerase II (RNA Pol II) pre-initiation complex (PIC). However, at inactive genes, the +1 nucleosome is found further upstream, at a promoter-proximal location. Here, we establish a model system to show that a promoter-proximal +1 nucleosome can reduce RNA synthesis in vivo and in vitro, and we analyze its structural basis. We find that the PIC assembles normally when the edge of the +1 nucleosome is located 18 base pairs (bp) downstream of the transcription start site (TSS). However, when the nucleosome edge is located further upstream, only 10 bp downstream of the TSS, the PIC adopts an inhibited state. The transcription factor IIH (TFIIH) shows a closed conformation and its subunit XPB contacts DNA with only one of its two ATPase lobes, inconsistent with DNA opening. These results provide a mechanism for nucleosome-dependent regulation of transcription initiation.
Keywords: transcription initiation, RNA polymerase II, pre-initiation complex, +1 nucleosome, promoter-proximal +1 nucleosome, gene regulation, transcription reduction
Graphical abstract
Highlights
-
•
Promoter proximity of the +1 nucleosome reduces transcription in vivo and in vitro
-
•
The +1 nucleosome impairs transcription by interfering with productive PIC assembly
-
•
TFIIH can bind to the PIC in its closed conformation, but in an inhibited state
-
•
Transcription regulation by the +1 nucleosome depends on its proximity to the TSS
Through functional assays and cryoelectron microscopy, Abril-Garrido et al. elucidated a mechanism by which the +1 nucleosome reduces transcription. When the +1 nucleosome is located in proximity to the gene promoter, the PIC assembles—albeit in an inhibited state incompatible with DNA opening.
Introduction
Gene promoters are flanked by nucleosomes that are referred to as −1 and +1 for the first upstream and downstream nucleosome, respectively.1,2,3,4,5,6 The +1 nucleosome is located downstream of the transcription start site (TSS), near the RNA polymerase II (RNA Pol II) pre-initiation complex (PIC).2,3,7,8,9,10,11,12 The position of the +1 nucleosome varies for genes that are expressed at different levels,3,13,14,15,16 but how the position of a nucleosome influences transcription initiation is not understood. At transcriptionally active mammalian genes, the edge of the +1 nucleosome is located around 40–60 base pairs (bp) downstream of the TSS.13,17,18,19,20 However, at inactive or weakly active genes, the +1 nucleosome is located further upstream, in close proximity to the TSS.14 This proximal nucleosome position is thought to interfere with PIC formation or function and to regulate gene activity. Reduction of transcription activity by the presence of a proximal nucleosome is consistent with in vivo studies in yeast21,22,23 and with the long-standing observation that transcription initiation in vitro is inhibited by a nucleosome located at the promoter.7,24,25,26
Very recently, the first structural studies of PICs in the presence of a +1 nucleosome were published.27,28 For human PICs, transcription factor IIH (TFIIH) and Mediator were found to form weak interactions with nucleosomes positioned at distal locations of 40 or 50 bp from the TSS,27 but proximal nucleosome locations were not investigated. For PICs of the yeast Saccharomyces cerevisiae (S. cerevisiae), a high-resolution cryoelectron microscopy (cryo-EM) study found that TFIIH establishes intimate interactions with the nucleosome and that its translocase subunit Ssl2 (XPB in human TFIIH) drives the rotation of the +1 nucleosome, leading to partial detachment of nucleosomal DNA.28 However, the yeast and human systems differ. In contrast to human RNA Pol II, yeast RNA Pol II does not undergo promoter-proximal pausing and instead scans DNA downstream before initiation.29,30,31,32,33
Whereas these published studies provided structural information on PIC-nucleosome interactions, they did not provide insights into how nucleosome positioning regulates transcription.27,28 It has long been known that compaction of chromatin correlates with a reduction in transcription activity.34,35,36 For example, critical biological processes such as heterochromatin maintenance and mitosis have been associated with a reduction in RNA synthesis.37,38 However, we do not understand the mechanisms by which nucleosomes near the promoter can cause such reduction in gene activity.
Here, we employed a combination of functional and structural studies to investigate the regulatory role of a promoter-proximal +1 nucleosome in mammalian transcription initiation. In particular, we describe how nucleosome proximity to the promoter can regulate transcription using biochemical and cell-based assays and we determine cryo-EM structures of two PIC-nucleosome complexes. We describe that a reduction of RNA synthesis correlates with an upstream shift of the +1 nucleosome toward the TSS in vivo. In addition, we observe that positioning a nucleosome at a TSS-proximal location in vitro downregulates transcription by affecting PIC assembly and altering the conformation of TFIIH, which impedes its translocase subunit XPB from fully engaging downstream promoter DNA. Our results suggest that positioning of the +1 nucleosome at a promoter-proximal location can interfere with productive PIC formation and transcription initiation.
Results
Promoter proximity of the +1 nucleosome reduces transcription
Various studies have related the proximal location to the TSS of the +1 nucleosome to low gene expression levels,13,14 but it was thus far not investigated whether genes that contain promoter-proximal nucleosomes indeed show low RNA synthesis rates. To address this, we analyzed transient transcriptome sequencing (TT-seq) and micrococcal nuclease sequencing (MNase-seq) data obtained from HEK293 cells (T. Velychko et al., unpublished data). We split genes into ten groups based on their RNA synthesis levels (inactive and RNA synthesis deciles q1–q10) and calculated the distance from the TSS to the +1 nucleosome edge for genes in each group. We observed that a reduction of RNA synthesis correlates with an upstream shift of the +1 nucleosome (Figure 1A). The median distance from the nucleosome edge to the TSS for moderate-to-highly (q3–q10), weakly (q1 and q2) active, and inactive (off) genes was 44–35, 30–20, and 12 bp, respectively. This suggests that gene activity is reduced when the nucleosome is proximally located to the promoter region in vivo.
Figure 1.

Promoter proximity of the +1 nucleosome reduces transcription
(A) Boxplots showing TSS to +1 nucleosome edge distances for gene groups with different RNA synthesis levels in HEK293 cells. Genes were split into inactive and deciles of RNA synthesis q1–q10 (low-high, see STAR Methods). Box limits are the first and third quartiles, and the band inside the box is the median. The ends of the whiskers extend the box by 1.5 times the interquartile range. Notches represent 95% confidence intervals for the median values. Statistical significance is denoted above the respective boxplots (∗∗p value < 0.01; ∗p value < 0.05). p values for off-q1, q1 and q2, and q2 and q3 are 3.2E−2, 4.0E−3, and 2.6E−2, respectively.
(B) Urea-PAGE of in vitro transcription assays where the edge of the nucleosome-positioning sequence is adjacent to the TSS, or 10 and 18 bp downstream of the TSS (PIC0W, PIC10W, and PIC18W, respectively). Transcription reactions were performed with and without TFIIH and the nucleosome. The expected full-length RNA product is indicated with an asterisk. Dashed rectangles denote the area used for quantifications.
(C) Quantifications of the transcription assays shown in (B) (see STAR Methods). Data are represented as mean over replicates (spheres).
(D) TFIIH-dependent transcription of nucleosome-reconstituted DNA templates shows that the proximity of the nucleosome regulates gene activity. Plotted intensities correspond to transcribing PIC complexes in the presence of a nucleosome located at different distances from the TSS. Statistical significance is shown (∗∗∗p value < 0.001; ∗∗p value < 0.01). p values for PIC-Nuc18W-PIC-Nuc10W, PIC-Nuc18W-PIC-Nuc0W, and PIC-Nuc10W-PIC-Nuc0W are 4.2E−3, 3.0E−4, and 1.8E−3, respectively (see STAR Methods).
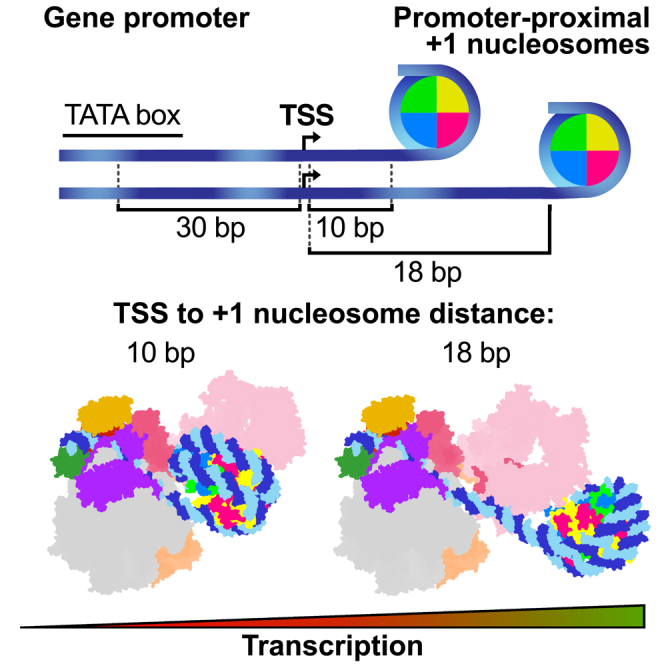
To corroborate these findings, we carried out in vitro transcription assays with highly purified, recombinant human initiation factors and endogenous Sus scrofa RNA Pol II, which is 99.9% identical to human RNA Pol II (Figure S1A). For these assays, we used DNA templates containing a +1 nucleosome positioned at increasing distances from the TSS (Figures 1B–1D and 2A). Promoter-dependent de novo transcription of nucleosome-containing DNA templates required TFIIH, providing a positive control (Figures 1B and 1C). We found that transcription was reduced ∼2.5-fold or ∼4.5-fold when templates were used that contained a nucleosome with its edge located either 18 or 10 bp from the TSS (PIC-Nuc18W and PIC-Nuc10W templates, respectively), when compared with the corresponding nucleosome-free DNA templates (Figure 1C). We further observed that positioning a nucleosome with its edge directly at the TSS (PIC-Nuc0W) fully inhibited transcription (Figure 1D), consistent with previous studies.7,24,25,26 In summary, shifting a +1 nucleosome from a downstream location to a more promoter-proximal position closer to the TSS reduces transcription activity in vitro and in vivo.
Figure 2.

Structures of PIC-nucleosome complexes
(A) Scheme of the nucleosome-containing DNA templates used for the structures shown in (B) and (C). Distances between the TSS and the edge of the nucleosome as well as from the TATA-box midpoint to the nucleosome dyad are indicated. Core promoter motifs and the nucleosome-positioning sequences are highlighted with solid lines and curves, respectively.
(B) Model of the mammalian PIC-nucleosome18W complex (PIC-Nuc18W) shown as cartoon-sphere in side view.
(C) Model of the mammalian PIC-nucleosome10W complex (PIC-Nuc10W) shown as cartoon-sphere in side view. Major conformational changes between PIC-Nuc18W and PIC-Nuc10W are indicated with arrowheads. The MAT1 helical region is highlighted with a dashed line (black color denotes ordered and red disordered).
Mammalian PIC-nucleosome structure
To investigate how the position of the +1 nucleosome influences transcription initiation, we performed cryo-EM analyses of reconstituted PIC-nucleosome complexes. We first designed a DNA promoter containing the edge of the +1 nucleosome 18 bp downstream of the TSS (Figure 2A). This design is consistent with TSS-nucleosome distances we and others found in vivo for weakly expressed genes (Figure 1A).13,14 We reconstituted the PIC on this nucleosome template and determined the structure by cryo-EM analysis at an overall resolution of 4.0 Å (PIC-Nuc18W) (Figures 2A, 2B, and S1A–S1D). Signal subtraction and focused refinement strategies improved the resolution of the XPB-containing part of TFIIH, the XPD-containing part of TFIIH, the TFIIH subcomplex CDK-activating kinase module (CAK), the core PIC (cPIC), and the nucleosome to 3.9, 4.7, 3.3, 3.0, and 3.6 Å, respectively (Figures S1D and S2).
In the resulting structure, we observed the canonical conformation of the PIC,39,40 and only minor movements in the upstream complex containing TBP, TFIIA, and TFIIB (Figures 2B and S3A), consistent with its previously reported flexibility.40 TFIIH adopts an open conformation, as observed in all previous PIC structures (Figures 3A and S3A),27,39,40,41,42,43,44 allowing for the complete engagement of both ATPase lobes with DNA (Figure 4A). This is facilitated by the distal position of the nucleosome, which renders the downstream DNA-binding region of XPB nucleosome-free (Figure S3B). This downstream region is slightly bent when compared with the structure of the PIC on a nucleosome-free DNA (Figure S3A).40 In this PIC-nucleosome structure, about two turns of nucleosomal DNA are detached from the histone octamer at superhelical location (SHL) −5 to SHL −7 (Figure 4B).
Figure 3.

Nucleosome proximity alters TFIIH conformation
(A) A distal nucleosome (PIC-Nuc18W) induces an open state of TFIIH. Solid and dashed arrowheads denote rearrangements from the closed state, the red dashed oval indicates the region of XPB-XPD contacts.
(B) A proximal nucleosome (PIC-Nuc10W) induces a closed state of TFIIH. Solid and dashed arrowheads denote rearrangements from the open state, the red dashed oval indicates the region of XPB-XPD contacts.
Figure 4.

Nucleosome position alters PIC-DNA contacts
(A and C) Comparison of XPB binding to (A) a distal (PIC-Nuc18W) and (C) proximal nucleosome (PIC-Nuc10W) by superimposition on XPB and the nucleosome. The position of the nucleosome alters the DNA binding of the ATPase lobes of the TFIIH subunit XPB. The color code is provided at the bottom legend. Numbers on DNA denote the distance from the TSS.
(B) Cartoon representation of the different nucleosomes determined (PIC-bound), showing distinct nucleosomal DNA wrapping states. Top-left legend specifies the color used for the models. Model superimposition was carried out by aligning on the nucleosome. SHL, superhelical location.
Nucleosome proximity alters PIC conformation
To investigate the structural basis of transcription reduction by a promoter-proximal +1 nucleosome, we also determined the structure of a PIC-nucleosome complex where the +1 nucleosome was positioned at a more proximal location, with the edge of the nucleosome located 10 bp downstream of the TSS (PIC-Nuc10W) (Figures 2A and 2C). We obtained an overall resolution of 4.1 Å for this complex (Figures S4A–S4D). With the use of signal subtraction and focused refinement, we improved the resolution of the reconstructions for the cPIC to 3.1–3.2 Å, for TFIIH to 4.5 Å, for the nucleosome to 3.2–3.5 Å and for the CAK to 3.8 Å (Figures S4D and S5).
In the obtained structure, the conformation of the cPIC (lacking TFIIH) does not deviate from that observed in the PIC-Nuc18W structure and is also similar to that observed in the absence of a nucleosome (Figure S6A).40 However, in contrast to all known PIC structures, TFIIH adopts a closed conformation that had only been observed previously for free TFIIH (Figures 3B and S6B).39,40,41,42,43,44,45,46 In this closed conformation, the two ATPase subunits, XPB and XPD, contact each other. The TFIIH subunit MAT1 favors this arrangement by contacting the XPB damage recognition domain (DRD) and its helical region stabilizes the closed TFIIH conformation. In summary, these observations show that the more TSS-proximal location of the +1 nucleosome led to an alternative PIC conformation with a closed state of TFIIH.
Closed state of TFIIH is incompatible with DNA opening
In the observed closed state of TFIIH, the XPB translocase subunit, which is essential for opening DNA, cannot fully engage the promoter. Instead, the ATPase lobe 1 binds nucleosomal DNA approximately 88 bp downstream of the TSS, at SHL +0.5, whereas lobe 2 is located ∼30 Å away from the DNA (Figure 4C). The observed PIC conformation and PIC-DNA contacts are thus incompatible with promoter DNA opening and transcription initiation. The closed TFIIH conformation was induced by the proximal location of the nucleosome because it is the only difference in the two structure determination experiments. We animated the conformational transition from the closed to the open TFIIH state in Video S1.
Upon transition from PIC-Nuc10W to PIC-Nuc18W, the proximal +1 nucleosome at the promoter region locates further downstream, TFIIH adopts an alternative conformation, TFIIH subunit XPB fully engages with promoter DNA and ∼2 turns of DNA are unwrapped from the proximal nucleosome.
During 3D classifications, we could also resolve the cPIC-nucleosome complex lacking TFIIE and TFIIH (cPIC-Nuc10W) and refined its structure at 3.8 Å resolution (Figures 5A, S4D, and S5). In this structure, the nucleosome overlaps with the location of TFIIH in a PIC complex as it is shifted by ∼20 Å and tilted by ∼25°, compared with the complete PIC-Nuc10W structure (Figure 5B; Video S2). This is consistent with a proximally positioned +1 nucleosome interfering with complete promoter DNA engagement by TFIIH. We, therefore, suggest that the PIC with the closed TFIIH conformation represents an inhibited state.
Figure 5.

Structural transition of the nucleosome upon binding or release of TFIIE and TFIIH on the cPIC-Nuc10W
(A) Top views of the cPIC-Nuc10W (lacking TFIIE and TFIIH, top panel) and PIC-Nuc10W (bottom panel).
(B) Overlay of both structural models shown in (A), showing a conformational change of the nucleosome depending on the presence of TFIIE and TFIIH. Arrowheads describe the direction of such movements. Superimposition of the models was performed by aligning on RNA Pol II. Histone octamers are depicted in different shades of the gray surface. Colors of different protein subunits or DNA strands are depicted in the figure or at the bottom legend.
A promoter-proximal +1 nucleosome located 10 bp downstream of the TSS (cPIC-Nuc10W) is displaced ∼20 Å and tilted by ∼25° by association of TFIIE and TFIIH to the cPIC (yielding PIC-Nuc10W).
Altered TFIIH-nucleosome contacts
TFIIH bridges the cPIC to the +1 nucleosome in both structures we determined. The comparison of these two structures shows that the rotational position of the nucleosome with respect to TFIIH changes (Figures 2B and 2C). As a consequence, TFIIH engages differently with promoter DNA, and its contacts with the nucleosome are altered. In the PIC-Nuc18W structure (Figure 6A), the TFIIH subunit p52 establishes most of the nucleosome contacts, with mostly basic residues within the α13-α14 loop (residues 274–278) binding nucleosomal DNA at SHL −1, and acidic residues of the p52 middle domain interacting with basic amino acids of the C terminus of H2A and H3 α1. The N-terminal region of XPB contacts the N-terminal region of histone H3. Conversely, in the PIC-Nuc10W structure (Figure 6B), the p8 subunit contacts the H2B acidic patch of the nucleosome and the N-terminal regions of H3 and H4. Basic residues of XPB ATPase lobe 1 bind to nucleosomal DNA at SHL +0.5, forming a major TFIIH-nucleosome interface.
Figure 6.

TFIIH-nucleosome contacts
(A) Cartoon representation showing interactions between TFIIH and the nucleosome in PIC-Nuc18W. Residues T51, K52, D54, and Q64 of the XPB N terminus (interface 1) interact mainly electrostatically with the N-terminal tail (NTT) of H3 (residues 37–41). p52 acidic residues (E189, E192, interface 2) contact basic residues from both H2A α3 (K74) and H3 α1 (R52, K56), and its residues 168–170 (interface 3) establish backbone interactions with residues 118–119 of H2A C terminus. In addition, mostly basic residues of the p52 α13-α14 loop (274–278, interface 4) bind to the minor groove adjacent to the nucleosome dyad (SHL −1). SHL, superhelical location.
(B) Cartoon representation showing interactions between TFIIH and the nucleosome in PIC-Nuc10W. Residues K31-N27 (interface 1), R56 (interface 2), and Q63 (interface 3) of the p8 subunit make electrostatic contacts to the H2B acidic patch, and the N-terminal regions of H3 (D81) and H4 (R23), respectively. XPB ATPase lobe 1 residues 416–420 and K449 (interface 4) engulf the DNA major groove adjacent to the nucleosome dyad (SHL +0.5). SHL, superhelical location. The positions of the C-α atoms of interacting residues are shown as spheres.
TFIIH kinase module and RPB6 NTT
We also observed the TFIIH kinase module (CAK) in our structures (Figures 3 and S7A). The CAK interacts with MED6 and the hook of Mediator in previously determined Mediator-PIC complexes, positioning CAK far from the RNA Pol II surface.41,42,44 In our structures, however, the CAK docks between the RPB1 foot and the RNA Pol II stalk (RPB4–RPB7), using its subunit cyclin H (CycH) to form a wedge between them. The N-terminal region of the CAK CycH subunit contacts the RPB1 linker helix connecting to the C-terminal domain (CTD) and the RPB7 α1-α2 loop, whereas the C-terminal helix of CycH mostly establishes charge-based interactions with the RPB1 foot helix α28 and α31 (Figure S7B). This location had been previously observed with different levels of confidence in yeast47 and humans (Figure S7C).43,48
Finally, in our structures, we observed an extra density in the RNA Pol II cleft (Figure S7D). Based on the similar location found for the yeast N-terminal tail (NTT) of the RNA Pol II subunit Rpb6,28 we suggest that this density is due to the mammalian counterpart in RPB6 (Figures S7E and S7F). Whereas the S. cerevisiae NTT is located over the RNA Pol II bridge helix, the density of the putative mammalian NTT locates closer to RPB1 helix α37 (Figure S7F). Consistent with findings in yeast,28 the cryo-EM density shows clashes between the putative NTT and a modeled DNA template in the active center (Figure S7G), indicating that the NTT must be released from the cleft upon formation of an open complex.
Discussion
Here, we combine structural and functional studies to show that a promoter-proximal +1 nucleosome can reduce transcription activity and provide a mechanism for such +1 nucleosome-dependent gene regulation (Figure 7). When the nucleosome is located with its edge ∼2 turns of DNA downstream of the TSS, the PIC assembles normally and TFIIH adopts an open conformation that is fully engaged with promoter DNA, as required for DNA opening. However, when the nucleosome is positioned with its edge only ∼1 turn of DNA downstream of the TSS, TFIIH adopts a closed conformation and can only partially engage with DNA, consistent with a reduction in transcription activity.
Figure 7.

Model of transcription reduction by a +1 nucleosome
The proximity of a downstream nucleosome to the promoter region of a gene distinctly reduces RNA Pol II-mediated RNA synthesis by changing the binding of TFIIH to promoter DNA. The model of PIC-Nuc40W belongs to PDB: 8GXS.27 Different gene categories and a gradient showing decreasing RNA synthesis are denoted on the left (green, high synthesis; red, no synthesis).
When the closed conformation of TFIIH was first observed, it was thought to be a feature that is specific to the PIC-unbound, free TFIIH complex.45,46 However, our results show that TFIIH can adopt its closed conformation also within the PIC, where it may serve to regulate TFIIH activity. Superposition of our PIC-nucleosome structures onto a cPIC structure with open promoter DNA,40 shows that, upon TFIIH-independent DNA opening, the nucleosome would sterically clash with the RNA Pol II clamp and the TFIIE winged helix domain (Figure S8), providing a possible explanation for the requirement of TFIIH for transcription of nucleosomal templates. Overall, this suggests that a promoter-proximal nucleosome can have an inhibitory role in transcription initiation by interfering with productive PIC formation, altering DNA binding of TFIIH and its conformation, thus impairing promoter DNA opening and subsequent RNA synthesis.
Structural work on Mediator-PIC complexes revealed that the presence of Mediator positions the TFIIH kinase module (CAK) in close vicinity of its hook region and MED6 subunit,41,42,44 suggesting Mediator is a key player in relocating the CAK module to make accessible the RNA Pol II CTD for CDK7-mediated phosphorylation. On the contrary, in our structures, we observe the CAK located between the RPB1 foot and the RNA Pol II stalk (RPB4–RPB7), without evidence for the presence of the RNA Pol II CTD. This alternative location of the CAK had been previously observed, albeit at a low level of confidence (∼15 Å resolution), in yeast,47 proposed in a molecular dynamics study for the human TFIID-free PIC,48 and recently determined in the presence of TFIID in a PIC.43 Here, we therefore provide evidence that the position of the kinase is conserved from yeast to human and confirm that it is TFIID-independent and Mediator-dependent.
Comparison of our structures with the recently published yeast PIC-nucleosome complex28 shows that even in our productive PIC structure (PIC-Nuc18W), the nucleosome locates closer to the PIC and TFIIH (Figure S9). Whereas ∼2 turns of DNA are detached from the histone octamer in our PIC-Nuc18W structure, only ∼1 turn of DNA is detached in the previous yeast structure.28 We suggest that the different rotational positions of the nucleosome in these structures lead to different degrees of nucleosomal DNA detachment, consistent with the observation that the parts of TFIIH contacting the nucleosome are not highly conserved.49 Consequently, whereas nucleosome contacts are formed by p52 in our PIC-Nuc18W structure, they are formed by the p52-p8 dimerization domain in the yeast structure.
Finally, our work complements recently published human PIC-nucleosome structures that used much larger TSS-nucleosome distances that are found at active genes (Figure S9).27 In the published work, the PIC adopts the canonical conformation, where TFIIH and Mediator form contacts with the nucleosome that are suggested to enhance transcription.27 By contrast, we show that shifting the +1 nucleosome to a TSS-proximal location, representative of inactive genes, leads to alternative TFIIH-nucleosome contacts, induces a closed TFIIH conformation and reduces transcription activity. Thus, these studies are complementary and it emerges that the +1 nucleosome can regulate transcription in various ways, dependent on its relative location to the TSS and the PIC.
Limitations of the study
Cryo-EM data processing aimed at obtaining intermediates at the highest possible resolution, and low-resolution conformational states may have been filtered out during processing. The lack of Mediator or TFIID in our studies does not alter our conclusions, but future work should aim at including such coactivators. The nucleosome has a certain degree of flexibility, and using a strong positioning sequence helps in obtaining stable intermediate complexes that are attainable to cryo-EM structural studies.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| E.coli BL21 CodonPlus (DE3) RIL | Agilent | Cat#230245 |
| E. coli LOBSTR-BL21(DE3)-RIL | Kerafast | Cat# EC1002 |
| E.coli DH10EMBacY | Geneva Biotech | N/A |
| E.coli XL-1 Blue | Agilent | Cat#200249 |
| Biological samples | ||
| Sus scrofa thymus | Locally sourced | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Sus scrofa RNA polymerase II | Vos et al.50 | N/A |
| Homo sapiens TBP | Aibara et al.40 | N/A |
| Homo sapiens TFIIB | Aibara et al.40 | N/A |
| Homo sapiens TFIIA | Aibara et al.40 | N/A |
| Homo sapiens TFIIF | Aibara et al.40 | N/A |
| Homo sapiens TFIIE | Aibara et al.40 | N/A |
| Homo sapiens TFIIH-core | This study | N/A |
| Homo sapiens CAK | Kokic et al.51 | N/A |
| Xenopus laevis histones H3, H4, H2A, H2B | Dyer et al.52 | N/A |
| 8WG16 (αRPB1 CTD) antibody | Hu et al.53 | RRID: AB_10013665 |
| Glutaraldehyde 25% | EMS | Cat#16200 |
| Poly(vinyl alcohol) | Sigma-Aldrich | Cat#P8136 |
| RNasin Plus Ribonuclease Inhibitor | Promega | Cat#N2611 |
| Proteinase K | New England Biolabs | Cat#P8107S |
| Invitrogen UltraPure 0.5M EDTA, pH 8.0 | Thermo Fisher Scientific | Cat#15575020 |
| SDS 10% | Thermo Fisher Scientific | Cat#AM9822 |
| DNAseI (RNase-free) | New England Biolabs | Cat#M0303S |
| Invitrogen Ambion Sodium Acetate (3M), pH 5.5 | Thermo Fisher Scientific | Cat#AM9740 |
| NTP Set, 100 mM Solution | Thermo Fisher Scientific | Cat#R0481 |
| SYBR™ Gold Nucleic Acid Gel Stain | Thermo Fisher Scientific | Cat#S11494 |
| BSA-Molecular Biology Grade | New England Biolabs | Cat#B9000S |
| Urea (RNase-free) | Panreac AppliChem | Cat#A1049 |
| 2x RNA Loading Dye | New England Biolabs | Cat#B0363S |
| DL-Dithiothreitol solution, 1M | Sigma-Aldrich | Cat#43816 |
| 40% Acrylamide/bis-acrylamide 19:1 | Sigma-Aldrich | Cat#A9926 |
| TRIS borate-EDTA buffer solution (10x) | Sigma-Aldrich | Cat#93290 |
| Phusion DNA Polymerase | House sourced | N/A |
| Phusion® HF Buffer Pack | New England Biolabs | Cat#B0518S |
| dNTP Set, 100 mM Solutions | Thermo Fisher Scientific | Cat#R0186 |
| DMSO | Sigma-Aldrich | Cat#D8418 |
| Deposited data | ||
| cPIC-nucleosome10W cryo-EM globally refined map | This study | EMD: 16335 |
| Core PICcPIC-Nuc10W cryo-EM focused refined map | This study | EMD: 16336 |
| NucleosomecPIC-Nuc10W cryo-EM focused refined map | This study | EMD: 16337 |
| cPIC-nucleosome10W cryo-EM composite map | This study | EMD: 16338 |
| cPIC-nucleosome10W model | This study | PDB: 8BZ1 |
| PIC-nucleosome10W cryo-EM globally refined map | This study | EMD: 16331 |
| Core PICPIC-Nuc10W cryo-EM focused refined map | This study | EMD: 16339 |
| NucleosomePIC-Nuc10W cryo-EM focused refined map | This study | EMD: 16340 |
| TFIIHPIC-Nuc10W cryo-EM focused refined map | This study | EMD: 16342 |
| CAKPIC-Nuc10W cryo-EM focused refined map | This study | EMD: 16341 |
| PIC-nucleosome10W cryo-EM composite map | This study | EMD: 16343 |
| PIC-nucleosome10W model | This study | PDB: 8BYQ |
| PIC-nucleosome18W cryo-EM globally refined map | This study | EMD: 16274 |
| Core PICPIC-Nuc18W cryo-EM focused refined map | This study | EMD: 16365 |
| NucleosomePIC-Nuc18W cryo-EM focused refined map | This study | EMD: 16366 |
| TFIIHPIC-Nuc18W cryo-EM focused refined map | This study | EMD: 16367 |
| XPB-containing TFIIHPIC-Nuc18W cryo-EM focused refined map | This study | EMD: 16367, add. |
| XPD-containing TFIIHPIC-Nuc18W cryo-EM focused refined map | This study | EMD: 16367, add. |
| CAKPIC-Nuc18W cryo-EM focused refined map | This study | EMD: 16368 |
| PIC-nucleosome18W cryo-EM composite map | This study | EMD: 16369 |
| PIC-nucleosome18W model | This study | PDB: 8BVW |
| Experimental models: cell lines | ||
| Sf9 Cells | Thermo Fisher Scientific | Cat#11496015 |
| High Five Cells | Thermo Fisher Scientific | Cat#B85502 |
| Oligonucleotides | ||
| Widom 601 template: 5’ – ATC GGA TGT ATA TAT CTG ACA CGT GCC TGG AGA CTA GGG AGT AAT CCC CTT GGC GGT TAA AAC GCG GGG GAC AGC GCG TAC GTG CGT TTA AGC GGT GCT AGA GCT GTC TAC GAC CAA TTG AGC GGC CTC GGC ACC GGG ATT CTC GAT – 3’ | This work | Integrated DNA Technologies |
| Widom 601 non-template 5’ – ATC GAG AAT CCC GGT GCC GAG GCC GCT CAA TTG GTC GTA GAC AGC TCT AGC ACC GCT TAA ACG CAC GTA CGC GCT GTC CCC CGC GTT TTA ACC GCC AAG GGG ATT ACT CCC TAG TCT CCA GGC ACG TGT CAG ATA TAT ACA TCC GAT – 3’ | This work | Integrated DNA Technologies |
| AdML0W cloning forward primer 5’ – TCG AGG TAC CGG ATC CGA TAT CCG GGT GTT CCT GAA GGG GGG CTA TAA AAG GGG GTG GGG GCG CGT TCG TCC TCA ATC GAG AAT CCC GGT GCC GAG G – 3’ | This work | Sigma-Aldrich |
| AdML10W cloning forward primer 5’ – TCG AGG TAC CGG ATC CGA TAT CCG GGT GTT CCT GAA GGG GGG CTA TAA AAG GGG GTG GGG GCG CGT TCG TCC TCA CTC TCT TCC GAT CGA GAA TCC CGG TGC CGA GG – 3’ | This work | Sigma-Aldrich |
| AdML18W cloning forward primer 5’ – TCG AGG TAC CGG ATC CGA TAT CCG GGT GTT CCT GAA GGG GGG CTA TAA AAG GGG GTG GGG GCG CGT TCG TCC TCA CTC TCT TCC GCA TCG CTG ATC GAG AAT CCC GGT GCC GAG G – 3’ | This work | Sigma-Aldrich |
| Widom 601 cloning reverse primer 5’ – CGA AGA TCT GAT ATC ATC GGA TGT ATA TAT CTG ACA CGT GCC TGG AGAC – 3’ | This work | Sigma-Aldrich |
| AdMLW PCR forward primer 5’ – CGG GTG TTC CTG AAG GGG GGC TAT AAA AGG GGG TG – 3’ | This work | Sigma-Aldrich |
| Widom 601 PCR reverse primer 5’ – ATC GGA TGT ATA TAT CTG ACA CGT GCC TGG AGA CTA GGG AG – 3’ | This work | Sigma-Aldrich |
| Recombinant DNA | ||
| 438A-hTBP | Aibara et al.40 | N/A |
| pOPINF-hTFIIB | Aibara et al.40 | N/A |
| 438A-hTFIIA | Aibara et al.40 | N/A |
| pETDuet-1-hTFIIE | Aibara et al.40 | N/A |
| pAHS3C-hTFIIF | Aibara et al.40 | N/A |
| 438C-XPD-p52-p34-p8-p62-p44-XPD (hTFIIH-core) | This study | N/A |
| 438B-CCNH-CDK7-MAT1 (CAK) | Kokic et al.51 | N/A |
| pUC119-AdML0W | This study | N/A |
| pUC119-AdML10W | This study | N/A |
| pUC119-AdML18W | This study | N/A |
| Software and algorithms | ||
| SerialEM 4.0 | Mastronarde54 | https://bio3d.colorado.edu/SerialEM/#Source |
| cryoSPARC 3.2.0 | Punjani et al.55 | https://cryosparc.com/ |
| RELION 3.1 | Scheres56 and Zivanov et al.57 | https://github.com/3dem/relion |
| Warp 1.0.9 | Tegunov and Cramer58 | http://www.warpem.com |
| PHENIX 1.19.2 | Afonine et al.59 | http://www.phenix-online.org |
| PyMol 2.5.0 | Schrodinger and Delano60 | http://www.pymol.org |
| UCSF Chimera | Pettersen et al.61 | https://www.cgl.ucsf.edu/chimera/ |
| UCSF Chimera X-1.4 | Goddard et al.62 | https://www.cgl.ucsf.edu/chimerax/ |
| ISOLDE 1.3 | Croll63 | https://isolde.cimr.cam.ac.uk/ |
| Coot 0.9.6 | Emsley et al.64 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| ImageJ 2.1.0 | Schindelin et al.65 | https://imagej.nih.gov/ij/index.html |
| Prism 9.1.0 | GraphPad Software Inc (California, USA) | https://www.graphpad.com/ |
| Other | ||
| Glacios | Thermo Fisher Scientific | N/A |
| Falcon-III Direct Electron Detector | Thermo Fisher Scientific | N/A |
| Titan Krios G2 | FEI/Thermo Fisher Scientific | N/A |
| QuantumLS energy filter | Gatan | N/A |
| K3 Summit Direct Electron Detector | Gatan | N/A |
| Typhoon™ 9500 FLA imager | GE Healthcare Life Sciences | N/A |
| BioComp Gradient Master 108 | BioComp Instruments | N/A |
| Model 491 Prep Cell | Bio-Rad | Cat#1702927 |
| Slide-A-Lyzer™ MINI Dialysis Devices (3.5 kDa MWCO) | Thermo Fisher Scientific | Cat# 69552 |
| Slide-A-Lyzer™ MINI Dialysis Devices (20 kDa MWCO) | Thermo Fisher Scientific | Cat# 69590 |
| Amicon Millipore 15 ml (50 kDa MWCO) | MERCK Milipore | Cat# UFC9050 |
| Quantifoil™ R3.5/1, copper, mesh 200 | Quantifoil | N/A |
Resource availability
Lead contact
Correspondence and requests for materials should be addressed to P.C. (pcramer@mpinat.mpg.de).
Materials availability
Materials are available from Patrick Cramer upon request under a material transfer agreement with the Max Planck Society.
Method details
Nucleosome reconstitution
Adenoviral major late promoter (AdMLP) DNA scaffolds containing a 147 bp Widom-601 sequence, which was located at different distances from the TSS, were inserted into pUC119 vectors (STAR Methods). DNA templates were amplified by PCR from these vectors and purified through Resource Q 6 ml (GE Healthcare), using a gradient of 0-50% TE high-salt buffer (10 mM Tris pH 8.0, 1 mM EDTA pH 8.0, 2 M NaCl). Eluates where ethanol-precipitated and resuspended in TE buffer (10 mM Tris pH 8.0, 1 mM EDTA pH 8.0).
Histones preparation and nucleosome reconstitution was performed as described.52,66 Briefly, X. laevis histones were purified from Escherichia coli (E. coli) BL21(DE3)-RIL, assembled into histone octamers using a HiLoad 16/600 Superdex 200 pg (Cytiva) and then used for nucleosome reconstitution with the above-mentioned templates through salt-gradient dialysis (SGD). Before assembling the PIC-nucleosome complexes, these nucleosomes were further purified over 4% polyacrylamide gels (0.2x TBE) using a Model 491 PrepCell (Bio-Rad), and subsequently dialysed into PIC-buffer (20 mM HEPES pH 7.5, 100 mM KCl, 2.5% glycerol, 2 mM MgCl2, 1 mM TCEP) overnight at 4°C. Reconstitutions were checked on 1% agarose gels (0.5x TBE running buffer). Nucleosomes were concentrated on Amicon Millipore 15 ml 50,000 MWCO centrifugal concentrator to a final concentration of 7-9 μM and their concentration was monitored by measuring their absorbance at 260 nm.
Assembly of mammalian PIC-nucleosome complexes
Human TBP, TFIIA, TFIIB, TFIIE, TFIIF and TFIIH were expressed and purified as described previously.40,51 Whereas TFIIA and TBP were independently expressed in insect cells, TFIIB, TFIIF and TFIIE were expressed in LOBSTR-BL21(DE3)-RIL E. coli and BL21-Codon Plus(DE3)-RIL E. coli cells, respectively. All these general transcription factors (GTFs) were purified first using GE HisTrap HP (5 ml), subsequently followed by ion exchange and size exclusion chromatography steps.
TFIIH was cloned in two different insect cell expression vectors: XPD, p8, p52, p44, p62, p34 and XPB in one (7-core TFIIH) and the CAK module subunits MAT1, CDK7 and cyclin H together in a second vector. While the first vector contained N-terminal 6xHis-TEV tags on p44 and p62 and a N-terminal 6xHis-MBP-TEV tag on XPD, all subunits of the CAK module were tagged N-terminally with 6xHis-TEV tags. Hi5 cells expressing the 7-core TFIIH were lysed with an EmulsiFlex-C5 cell disruptor (Avestin) supplemented with DNase I in lysis buffer (25 mM KOH-HEPES pH 7.6, 400 mM KCl, 20% glycerol, 5 mM TCEP, 0.284 μg ml−1 leupeptin, 1.37 μg ml−1 pepstatin A, 0.17 mg ml−1 PMSF and 0.33 mg ml−1 benzamidine). Lysate was loaded on a self-packed XK16/20 column (Cytiva) with 25 ml of amylose resin (New England Biolabs) in buffer A1 (25 mM KOH-HEPES pH 7.6, 400 mM KCl, 10% glycerol and 2 mM DTT) and eluted with buffer B1 (25 mM KOH-HEPES pH 7.6, 400 mM KCl, 10% glycerol, 2 mM DTT and 100 mM maltose) into a GE HiTrap Q HP (5 ml) column, pre-equilibrated with buffer A1. Elution of the anion exchange step was performed from 0-100% buffer HB1 (25 mM KOH-HEPES pH 7.6, 2000 mM KCl, 10% glycerol and 2 mM DTT), flow-through fractions collected, cleaved with 2.5 mg TEV protease for 8 hr at 4°C and loaded into a GE HiTrap Heparin HP (1 ml) column, pre-equilibrated with buffer HA1 (25 mM KOH-HEPES pH 7.6, 300 mM KCl, 10% glycerol and 2 mM DTT). Elution was performed using 0-100% buffer B1, fractions collected and further purified using a Superose 6 Increase 10/300 GL in buffer GF (25 mM KOH-HEPES pH 7.6, 300 mM KCl, 10% glycerol and 3 mM TCEP). Stochiometric 7-core TFIIH was concentrated with Vivaspin 6 50,000 MWCO (GE Healthcare), flash-frozen in liquid nitrogen and stored at -70°C. The kinase module of TFIIH was purified as described with minor modifications.67 In brief, Hi5 cells expressing the kinase were lysed with an EmulsiFlex-C5 cell disruptor (Avestin) supplemented with DNase I in lysis buffer (25 mM KOH-HEPES pH 7.6, 400 mM KCl, 15% glycerol, 1 mM MgCl2, 10 μM ZnCl2, 30 mM imidazole pH 8.0, 3 mM TCEP, 0.284 μg ml−1 leupeptin, 1.37 μg ml−1 pepstatin A, 0.17 mg ml−1 PMSF and 0.33 mg ml−1 benzamidine). The clarified lysate was loaded onto a GE HisTrap HP (5 ml) column, pre-equilibrated with lysis buffer, subsequently washed with buffer A1 HisTrap (25 mM KOH-HEPES pH 7.6, 100 mM KCl, 10% glycerol, 30 mM imidazole pH 8.0 and 1 mM DTT) and eluted with a linear gradient of 0-100% of buffer B1 HisTrap (25 mM K-HEPES, pH 7.6, 100 mM KCl, 10% glycerol, 500 mM imidazole pH 8.0 and 1 mM DTT) in 12 CV. Peak fractions were pooled, diluted with buffer A1 IEX (25 mM K-HEPES, pH 7.6, 100 mM KCl, 5% glycerol and 1 mM DTT), incubated with 2.5 mg of TEV protease at 4°C for 8 hr and loaded onto a GE HiTrap Q HP (5 ml), pre-equilibrated with buffer A1 IEX. The column was washed with 10 CV of buffer A1 IEX and eluted with a linear gradient 0-30% buffer B1 IEX (25 mM K-HEPES, pH 7.6, 2000 mM KCl, 5% glycerol and 1 mM DTT) for 80 CV, a step elution with 50% buffer B1 IEX for 2 CV and a final step with 100% buffer B1 IEX for 2 CV. Stochiometric TFIIH kinase trimer was pooled and concentrated using Vivaspin 20 10,000 MWCO (GE Healthcare) and loaded onto a HiLoad 16/600 Superdex 200 pg (Cytiva) which was eluted with buffer GF2 (25 mM K-HEPES, pH 7.6, 100 mM KCl, 5% glycerol and 2 mM TCEP). Stochiometric fractions were pooled, concentrated with Vivaspin 20 10,000 MWCO (GE Healthcare), flash-frozen in liquid nitrogen and stored at -70°C.
The 12-subunit RNA Pol II was purified endogenously from Sus scrofa thymus tissue as previously reported.50,53,68 In summary, S. scrofa thymus tissue was homogenized in a 2 L blender (Waring) for 3 minutes at 4°C. The homogenized tissue was filtered through two layers of Miracloth, mixed with polyethyleneimine (final concentration 0.04%) and stirred for 30 minutes at 4°C. The solution was centrifuged at maximum speed and pellets resuspended in 0.4 M HepR buffer (25 mM Tris-HCl pH 7.9, 400 mM (NH4)2SO4, 1 mM EDTA, 10 μM ZnCl2, 10% glycerol, 0.284 μg ml−1 leupeptin, 1.37 μg ml−1 pepstatin A, 0.17 mg ml−1 PMSF and 0.33 mg ml−1 benzamidine), centrifuged once more and supernatant adjusted to the conductivity of 0.2 M HepR buffer (25 mM Tris-HCl pH 7.9, 200 mM (NH4)2SO4, 1 mM EDTA, 10 μM ZnCl2, 10% glycerol, 0.284 μg ml−1 leupeptin, 1.37 μg ml−1 pepstatin A, 0.17 mg ml−1 PMSF and 0.33 mg ml−1 benzamidine) with 0 M HepR buffer (25 mM Tris-HCl pH 7.9, 1 mM EDTA, 10 μM ZnCl2, 10% glycerol, 0.284 μg ml−1 leupeptin, 1.37 μg ml−1 pepstatin A, 0.17 mg ml−1 PMSF and 0.33 mg ml−1 benzamidine). This lysate was loaded on a 225-ml MacroPrepQ column, pre-equilibrated in 0.2 M HepR buffer, the column was washed with 0.2 M HepR buffer (supplemented with 1 mM DTT) and eluted with 0.4 M HepR buffer (supplemented with 1 mM DTT). Eluates were pooled, precipitated by addition of (NH4)2SO4 to 50% saturation, stirred at 4°C for 1 hr, centrifuged and pellets resuspended in 0 M HepR2 buffer (25 mM Tris-HCl pH 7.9, 1 mM EDTA, 10 μM ZnCl2, 10% glycerol, 1 mM sodium metabisulfite, 0.25 mM PMSF and 1 mM benzamidine). The conductivity of the solution was adjusted on ice to that of 0.15 M HepR buffer (25 mM Tris-HCl pH 7.9, 150 mM (NH4)2SO4, 1 mM EDTA, 10 μM ZnCl2, 10% glycerol, 1 mM sodium metabisulfite, 0.25 mM PMSF and 1 mM benzamidine), loaded at 4°C on a 5-ml gravity flow column of 8WG16 (αRPB1 CTD) antibody-coupled sepharose, pre-equilibrated in 0.15 M HepR buffer. The antibody column was washed with five column volumes of 0.5 M HepR buffer (25 mM Tris-HCl pH 7.9, 500 mM (NH4)2SO4, 1 mM EDTA, 10 μM ZnCl2, 10% glycerol, 1 mM sodium metabisulfite, 0.25 mM PMSF and 1 mM benzamidine) at 4°C, and eluted at room temperature with 0.5 M HepR2 buffer (25 mM Tris-HCl pH 7.9, 500 mM (NH4)2SO4, 1 mM EDTA, 10 μM ZnCl2, 50% glycerol, 1 mM sodium metabisulfite, 0.25 mM PMSF and 1 mM benzamidine). Eluted fractions were immediately 5-fold diluted with Pol II dilution buffer (25 mM Tris-HCl pH 7.9, 1 mM EDTA, 10 μM ZnCl2 and 2 mM DTT). Diluted fractions were pooled, centrifuged and the supernatant loaded to a UnoQ1 column (Bio-Rad), pre-equilibrated with 0.1 M HepR buffer (25 mM Tris-HCl pH 7.9, 100 mM (NH4)2SO4, 1 mM EDTA, 10 μM ZnCl2 and 2 mM DTT), the column washed and eluted with a 20 CV linear gradient 20-100% from 0.1 M HepR buffer to 0.5 M HepR3 buffer (25 mM Tris-HCl pH 7.9, 500 mM (NH4)2SO4, 1 mM EDTA, 10 μM ZnCl2 and 2 mM DTT). GDOWN1-free RNA Pol II fractions were pooled, concentrated using an Amicon 100,000 MWCO Ultra Centrifugal Filter (Merck Millipore), buffer exchanged with Pol II final buffer (10 mM HEPES pH 7.5, 150 mM NaCl, 10% glycerol, 10 μM ZnCl2 and 1 mM DTT), flash-frozen in liquid nitrogen and stored at −70 °C.
The PIC-nucleosome complexes were prepared identically for both AdMLP templates (PIC-Nuc10W and PIC-Nuc18W), following the previously established protocol.40,69,70 In short, the 7-subunit core TFIIH (480 pmol) was mixed with the 3-subunit kinase module (480 pmol) to reconstitute the complete 10-subunit TFIIH at 25°C for 10 min. At the same time, RNA Pol II (240 pmol) was pre-incubated with TFIIF (1.2 nmol) at 25°C for 10 min. Subsequently, TFIIH was incubated with TFIIE (480 pmol), the KCl concentration was immediately adjusted to 150 mM with 0-salt buffer (20 mM HEPES pH 7.5, 2.5% glycerol, 2 mM MgCl2, 1 mM TCEP) and the subcomplex incubated at 25°C for 5 min. In the meantime, the PIC upstream complex was formed by adding TBP (1.2 nmol), TFIIA (2.4 nmol) and TFIIB (1.2 nmol) to the nucleosomal scaffolds (300 pmol) and incubating it at 25°C for 5 min. Afterwards, both the upstream complex and TFIIH-TFIIE were combined with RNA Pol II-TFIIF and sample salt concentration was decreased to 100 mM KCl by adding 0-salt buffer. This reaction was incubated at ∼400 rpm, 25°C for 90 – 120 min. Once the reaction was finished, the sample was centrifuged at 21,130g for 10-15 min and further purified by gradient ultracentrifugation. 30% of the sample was purified in a gradient for analytical purposes, whereas the remaining 70% was purified by GraFix71 and used for structural studies. The gradient was prepared from 15% to 40% sucrose in a buffer with 20 mM HEPES pH 7.5, 100 mM KCl, 2.5% glycerol, 2 mM MgCl2, 1 mM TCEP, via a BioComp Gradient Master 108 (BioComp Instruments). Sample preparations for cryo-EM were complemented with 0.2% glutaraldehyde in the 40% sucrose solution. The ultracentrifugation step was carried out at 175,000g for 16 h at 4°C. Subsequently, the gradient was fractionated in 200 μl aliquots, checked by SDS-PAGE and Coomassie staining (analytical gradient) or Native-PAGE and SYBR Gold and Coomassie staining. GraFix samples were immediately quenched after fractionation with a cocktail of pH-adjusted 10 mM lysine and 40 mM aspartate. The stochiometric crosslinked PIC-nucleosome complexes were dialysed for 6-7 hr at 4°C into PIC-dialysis buffer (20 mM HEPES pH 7.5, 75 mM KCl, 1% glycerol, 2 mM MgCl2, 1 mM TCEP) in Slide-A-Lyzer MINI Dialysis Devices (0.1 ml, 20 kDa MWCO) (Thermo Fisher Scientific) for sucrose removal.
Cryo-electron microscopy
Both PIC-nucleosome complexes (∼130 μl) were incubated on a floating ∼3.0 nm continuous carbon support for 7 min, after which the carbon film was attached to a holey carbon grid (Quantifoil R3.5/1, copper, mesh 200), washed with 4 μl of PIC-dialysis buffer and placed in a Vitrobot Mark IV (FEI/Thermo Fisher Scientific) under 100% humidity at 4°C. Under these conditions, samples were blotted with force 5 for 2 s and plunged frozen into liquid ethane. Optimal samples were identified using a Glacios transmission-electron microscope (Thermo Fisher Scientific) operated at 200 keV and equipped with a Falcon-III direct-electron detector (Thermo Fisher Scientific). Data was then collected using SerialEM 4.054 on a Titan Krios G2 transmission-electron microscope (FEI/Thermo Fisher Scientific) operated at 300 keV, with 20 eV slit width of a QuantumLS energy filter (Gatan), and equipped with a K3 summit direct detector. Imaging was performed at a nominal magnification of 81,000x (corresponding to a pixel size of 1.05 Å/pixel), with 3 s exposure in counting mode and a total dose of 41.58 and 50.45 e- per Å2, over 40 and 50 frames for PIC-Nuc18W and PIC-Nuc10W, respectively, at a defocus range from 0.5-1.5 μm. A total of 41517 and 36478 micrographs were collected for PIC-nucleosome18W and PIC-nucleosome10W, respectively.
Data processing
Motion correction, CTF-estimation, dose-weighting and particle-picking was performed in Warp 1.0.9.58 Micrographs were filtered by resolution and motion estimation, yielding a total of 39399 and 29668, on which Warp auto-picking resulted in 4,667,603 and 4,606,320 initial particles for PIC-nucleosome18W and PIC-nucleosome10W, respectively.
For PIC-nucleosome18W, 4,667,603 particles were extracted with a binning factor of 4. The data was initially classified in cryoSPARC 3.2.055 through 4 rounds of 2D and 3D classification where, for the latter, an ab initio model was generated in order to sort out falsely picked particles, ice contamination and aggregated particles. After the initial cleaning of the datasets, all subsequent image processing steps were performed using RELION 3.1.0 (Figure S1).56,57 1,725,420 particles containing cPIC were merged and unbinned, after which 2 rounds of CTF refinement, masked 3D refinement and Bayesian polishing on cPIC were done to reconstruct the cPIC at 2.6 Å. From these particles, after a series of signal subtraction, where a spherical mask was initially applied to keep the signal coming between the RNA Pol II stalk and foot, and focused masked 3D focused classifications with or without image alignment, we identified a final set of 147,341 particles which contained the CAK module at 3.3 Å. Subsequently, global 3D classifications with alignment were carried out to keep particles containing both TFIIH and the nucleosome. After signal subtracting and classifying these 1,462,564 particles with a spherical mask covering TFIIH and the nucleosome, the processing was split in order to yield the highest achievable resolution for both TFIIH and nucleosome. Regarding TFIIH, signal subtraction and 2 rounds of masked 3D focused classification yielded a reconstruction of TFIIH at 4.3 Å (188,832 particles). Masked 3D focused refinements led us to obtain XPD-containing TFIIH at 4.7 Å and XPB-containing TFIIH at 3.9 Å. In addition, reverting signal subtraction on these particles and performing either a masked or global refinement on cPIC or PIC-Nuc18W generated maps at 3.0 Å and 4.0 Å, respectively. On the other hand, this same type of classification procedure was employed in order to identify those particles of highest resolution for the nucleosome in 3 subsequent rounds. 246,363 particles containing the nucleosome were then refined to 3.6 Å.
Data processing for PIC-nucleosome10W was performed similarly to PIC-nucleosome18W (Figure S4). Briefly, after initial cleaning of data, and performing 2 rounds of CTF refinement, masked 3D refinement and Bayesian polishing, we obtained 1,415,094 particles containing cPIC at 2.4 Å. The CAK module could be resolved at 3.8 Å by following a strategy similar as described above for PIC-Nuc18W. Secondly, a global 3D classification with alignment was carried out to sort particles that did not contain TFIIH (cPIC-Nuc) from the ones that did (PIC-Nuc and PIC-like). The 705,995 particles containing TFIIH or TFIIH and nucleosome were exhaustively 3D classified, yielding a set of 376,063 particles, whereas the cPIC-Nuc reconstruction was generated from 668,443 particles. For these sets of particles, the cPIC signal was subtracted by applying a spherical mask on the TFIIH-Nuc region, subsequently performing a masked 3D focused classification. In the cPIC-Nuc scheme, sorting was aimed at keeping particles with highest resolution details for the nucleosome, whereas for PIC-Nuc we classified for those having the highest occupancy for TFIIH-Nuc. A last round of masked 3D focused classification without alignment was performed on both schemes and the TFIIH and the nucleosome of highest resolution were selected for further processing. Lastly, signal subtraction was reverted on the final set of particles for both reconstructions, and masked global 3D refinement was applied on cPIC-Nuc10W (3.8 Å) and PIC-Nuc10W (4.1 Å). For the former, the focused maps of cPIC and the nucleosome were reconstructed at 3.1 Å and 3.2 Å, respectively. As for PIC-Nuc10W, focused maps of cPIC, TFIIH and the nucleosome were obtained at 3.2 Å, 4.5 Å and 3.5 Å.
The resolution of the reconstructions was determined following the gold-standard Fourier shell correlation (cut-off at 0.143). Sharpening of maps was performed with the postprocessing tool of RELION 3.1.0, which automatically calculated the reported B-factors (Table S1). Local resolution was estimated in RELION 3.1.0 using the previously calculated B-factors. For the overall and cPIC maps, however, local resolution maps with B-factor of 0 Å were determined in RELION 3.1.0 and subsequently used in PHENIX 1.19.259 for map-sharpening. Density map figures were made in UCSF ChimeraX-1.4.62
Model building and refinement
Previously built and published structural models (PDB: 7NVS, 6NMI, 7NVW, 7OHC, 6XBZ, 7EGB, 7ZSB)28,40,43,46,72,73 were rigid-body fitted into the cryo-EM density maps obtained with highest resolution using UCSF Chimera.61 Iterative rounds of real-space refinement and manual adjustments were performed using ISOLDE 1.363 and PHENIX 1.19.2,59 whereas de novo building was performed in COOT 0.9.6.64 Merging of the refined structural models was done in COOT 0.9.6,64 and ISOLDE 1.363 was used to flexibly fit the linker DNA between cPIC and the nucleosome. Validation statistics from Molprobity74 showed good geometry and stereochemistry for the final refined models (Table S1). Atomic model figures were made in PyMOL 2.5.060 and UCSF ChimeraX-1.4,62 where the color assigned for every component is consistent throughout the manuscript.
In vitro transcription assay
Transcription initiation assays were performed in vitro with reconstituted components as described previously,40 albeit with minor modifications. DNA templates and nucleosomes were prepared as generated for cryo-EM studies (STAR Methods). DNA was stored at -20°C in TE buffer and nucleosomes were used right after reconstitution.
Briefly, we assembled the PIC step-wise at 25°C on both nucleosome free- and nucleosome-reconstituted DNA templates, as described above. Per reaction replicate, 3.7 pmol of DNA or nucleosome, 4.6 pmol RNA Pol II, 23 pmol TFIIF and TFIIA, 6.9 pmol TFIIE, TFIIH and CAK, 11.5 pmol TBP and TFIIB were used. Replicates were performed in a final volume of 23.8 μl, with final buffer conditions of 3 mM HEPES pH 7.9, 20 mM Tris-HCl pH 7.9, 60 mM KCl, 8 mM MgCl2, 2% (w/v) PVA, 3% glycerol, 0.5 mM DTT, 0.5 mg/ml BSA and 20 units RNase inhibitor. After assembling the PIC for 30 min, 1.25 μl of 10 mM NTP solution (Thermo Fisher Scientific) was added to each reaction (final concentration 0.5 mM/NTP) and incubated at 30°C for 60 min. Transcription reactions were stopped with 116 μl Stop buffer (10 mM Tris-HCl pH 7.5, 300 mM NaCl, 0.5 mM EDTA, 1% SDS and 4 μg proteinase K (New England Biolabs)) and incubated for 30-60 minutes at 37 °C. Nucleic acids were then precipitated with isopropanol, in presence of 300 mM sodium acetate and 0.5 mg/ml GlycoBlue (Thermo Fisher Scientific), on ice for 60 minutes. After resuspending nucleic acids, they were immediately supplemented with 1 unit DNase I (New England Biolabs) and incubated at 37°C for 60 min to digest the DNA template. A second nucleic acid isolation was performed by precipitating with isopropanol overnight at -20°C. Samples were then resuspended in 10 μl of water. RNA samples were diluted with 2x RNA Loading Dye (New England Biolabs), loaded into urea gels (2 M urea, 1x TBE, 6% acrylamide:bis-acrylamide 19:1) and separated by electrophoresis in 1x TBE buffer running buffer for 33 minutes at 180 V. Low Range ssRNA Ladder (New England Biolabs) was used for size reference, gels were stained for 10 min with SYBR™ Gold (Invitrogen) and RNA was visualized with a Typhoon 9500 FLA imager (GE Healthcare Life Sciences).
TT-seq and MNase-seq data analysis
TT-seq labeled and total RNA data (raw and processed) and MNase-seq data (DANPOS3 called nucleosome dyad positions) were taken from T. Velychko et al. (unpublished data). Protein-coding genes (RefSeq GCF_000001405.39, NM) were split into groups based on their RNA synthesis levels as follows: active genes were defined as the genes that are contained in the major transcript isoform annotation taken from T. Velychko et al., which is based on the total RNA expression data. Active genes were further split deciles q1-q10 based on their RNA synthesis level (TT-seq labeled RNA RPKM). Moreover, active genes were selected to have labeled RNA RPKM ≥ 0.01 and inactive (off) genes were defined by having RPKM < 0.01 for both labeled and total RNA samples. This cutoff was determined by plotting densities of replicate-averaged log2 (TT-seq labeled RNA RPKM) values over all genes and selecting a suitable cutoff in the valley between the two peaks of the bimodal distribution. To determine the correct TSS for each of the inactive genes we selected genes with only one RefSeq annotated isoform. For each gene, the +1 nucleosome dyad position was defined by considering nucleosome dyad position(s) falling within the region from TSS to 200 bp downstream. If two nucleosome dyads overlapped this region, the one closer to the TSS was defined as +1 nucleosome, and if no nucleosome dyad was called in this region, the gene was excluded from further analysis. The final gene sets contained 2680 inactive genes and 9970 active genes (997 per RNA synthesis decile), of which 2261 (off), 892 (q1), 812 (q2), 854 (q3), 837 (q4), 875 (q5), 898 (q6), 903 (q7), 913 (q8), 933 (q9) and 956 (q10) had an annotated +1 nucleosome. TSS to +1 nucleosome edge distances were calculated by subtracting 73 bp from the TSS-to-dyad distances.
Quantification and statistical analysis
For in vivo data analysis, significance between deciles was determined by calculating p-values by two-sided Mann–Whitney U-test.
For the in vitro transcription assays, intensity values of gels were quantified using ImageJ 2.1.0,65 subtracted against the background and normalized to the signal of the corresponding reaction of DNA templates without nucleosomes reconstituted. To facilitate comparisons between different nucleosome distances, signals were scaled to the normalized intensity of Nuc18W. Statistical analysis was performed using one-way ANOVA tests with Welch’s correction to obtain statistical significance (p values). All statistical analysis and diagrams were generated using RStudio or GraphPad Prism 9.1.0.
Acknowledgments
We thank U. Steuerwald for maintaining the cryo-EM infrastructure; U. Neef and P. Rus for maintenance of the insect cell culture facility; M. Klein for maintenance of the computing infrastructure; E. Mohammad for pre-processing TT-seq, RNA-seq, and MNase-seq data; E. Oberbeckmann for critically reading the manuscript; and S. Schilbach, S. Aibara, J. Walshe, I. Fianu, and Y. Zhan for discussions. P.C. was supported by the ERC Advanced Investigator Grant CHROMATRANS (grant agreement no. 882357).
Author contributions
J.A.-G. carried out all experiments and data analysis unless stated otherwise. J.A.-G. and C.D. collected cryo-EM data. J.A.-G. purified RNA Pol II, histones, and prepared nucleosomes, and F.G. assisted with the protein purification of initiation factors. T.V. collected and M.L. analyzed TT-seq, RNA-seq, and MNase-seq data. H.W. and P.C. supervised research. J.A.-G., H.W., and P.C. interpreted the data and wrote the manuscript with input from all authors.
Declaration of interests
The authors declare no competing interests.
Published: May 5, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.molcel.2023.04.011.
Contributor Information
Haibo Wang, Email: haibo.wang@zju.edu.cn.
Patrick Cramer, Email: pcramer@mpinat.mpg.de.
Supplemental information
Data and code availability
-
•
Cryo-EM density maps have been deposited in the Electron Microscopy Data Bank (EMDB) and coordinates within the Protein Data Bank (PDB) for cPIC-nucleosome10W (accession codes EMDB: EMD-16335and PDB: 8BZ1, and EMDB: EMD-16336, EMD-16337, and EMD-16338 for focused refinement maps of cPIC, nucleosome, and composite map, respectively ), PIC-nucleosome10W (accession codes EMDB: EMD-16331 and PDB: 8BYQ, and EMDB: EMD-16339, EMD-16340, EMD-16341, EMD-16342, and EMD-16343 for focused refinement maps of cPIC, nucleosome, CAK, TFIIH, and composite map, respectively) and PIC-nucleosome18W (accession codes EMDB: EMD-16274 and PDB: 8BVW, and EMDB: EMD-16365, EMD-16366, EMD-16367, EMD-16368, and EMD-16369 for focused refinement maps of cPIC, nucleosome, TFIIH, CAK, and composite map, respectively).
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Hughes A.L., Rando O.J. Mechanisms underlying nucleosome positioning in vivo. Annu. Rev. Biophys. 2014;43:41–63. doi: 10.1146/annurev-biophys-051013-023114. [DOI] [PubMed] [Google Scholar]
- 2.Jiang C., Pugh B.F. Nucleosome positioning and gene regulation: advances through genomics. Nat. Rev. Genet. 2009;10:161–172. doi: 10.1038/nrg2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Serizay J., Dong Y., Jänes J., Chesney M., Cerrato C., Ahringer J. Distinctive regulatory architectures of germline-active and somatic genes in C. elegans. Genome Res. 2020;30:1752–1765. doi: 10.1101/gr.265934.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kornberg R.D., Lorch Y. Primary role of the nucleosome. Mol. Cell. 2020;79:371–375. doi: 10.1016/j.molcel.2020.07.020. [DOI] [PubMed] [Google Scholar]
- 5.Lai W.K.M., Pugh B.F. Understanding nucleosome dynamics and their links to gene expression and DNA replication. Nat. Rev. Mol. Cell Biol. 2017;18:548–562. doi: 10.1038/nrm.2017.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Struhl K., Segal E. Determinants of nucleosome positioning. Nat. Struct. Mol. Biol. 2013;20:267–273. doi: 10.1038/nsmb.2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li G., Margueron R., Hu G., Stokes D., Wang Y.H., Reinberg D. Highly compacted chromatin formed in vitro reflects the dynamics of transcription activation in vivo. Mol. Cell. 2010;38:41–53. doi: 10.1016/j.molcel.2010.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santana J.F., Collins G.S., Parida M., Luse D.S., Price D.H. Differential dependencies of human RNA polymerase II promoters on TBP, TAF1, TFIIB and XPB. Nucleic Acids Res. 2022;50:9127–9148. doi: 10.1093/nar/gkac678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shao W., Zeitlinger J. Paused RNA polymerase II inhibits new transcriptional initiation. Nat. Genet. 2017;49:1045–1051. doi: 10.1038/ng.3867. [DOI] [PubMed] [Google Scholar]
- 10.Tramantano M., Sun L., Au C., Labuz D., Liu Z., Chou M., Shen C., Luk E. Constitutive turnover of histone H2A.Z at yeast promoters requires the preinitiation complex. eLife. 2016;5 doi: 10.7554/eLife.14243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rhee H.S., Pugh B.F. Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature. 2012;483:295–301. doi: 10.1038/nature10799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rossi M.J., Kuntala P.K., Lai W.K.M., Yamada N., Badjatia N., Mittal C., Kuzu G., Bocklund K., Farrell N.P., Blanda T.R., et al. A high-resolution protein architecture of the budding yeast genome. Nature. 2021;592:309–314. doi: 10.1038/s41586-021-03314-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schones D.E., Cui K., Cuddapah S., Roh T.Y., Barski A., Wang Z., Wei G., Zhao K. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132:887–898. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valouev A., Johnson S.M., Boyd S.D., Smith C.L., Fire A.Z., Sidow A. Determinants of nucleosome organization in primary human cells. Nature. 2011;474:516–520. doi: 10.1038/nature10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwartz U., Németh A., Diermeier S., Exler J.H., Hansch S., Maldonado R., Heizinger L., Merkl R., Längst G. Characterizing the nuclease accessibility of DNA in human cells to map higher order structures of chromatin. Nucleic Acids Res. 2019;47:1239–1254. doi: 10.1093/nar/gky1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guenther M.G., Levine S.S., Boyer L.A., Jaenisch R., Young R.A. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weber C.M., Ramachandran S., Henikoff S. Nucleosomes are context-specific, H2A.Z-modulated barriers to RNA polymerase. Mol. Cell. 2014;53:819–830. doi: 10.1016/j.molcel.2014.02.014. [DOI] [PubMed] [Google Scholar]
- 18.Žumer K., Maier K.C., Farnung L., Jaeger M.G., Rus P., Winter G., Cramer P. Two distinct mechanisms of RNA polymerase II elongation stimulation in vivo. Mol. Cell. 2021;81:3096–3109.e8. doi: 10.1016/j.molcel.2021.05.028. [DOI] [PubMed] [Google Scholar]
- 19.Albert I., Mavrich T.N., Tomsho L.P., Qi J., Zanton S.J., Schuster S.C., Pugh B.F. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature. 2007;446:572–576. doi: 10.1038/nature05632. [DOI] [PubMed] [Google Scholar]
- 20.Mavrich T.N., Jiang C., Ioshikhes I.P., Li X., Venters B.J., Zanton S.J., Tomsho L.P., Qi J., Glaser R.L., Schuster S.C., et al. Nucleosome organization in the Drosophila genome. Nature. 2008;453:358–362. doi: 10.1038/nature06929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klein-Brill A., Joseph-Strauss D., Appleboim A., Friedman N. Dynamics of chromatin and transcription during transient depletion of the RSC chromatin remodeling complex. Cell Rep. 2019;26:279–292.e5. doi: 10.1016/j.celrep.2018.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kubik S., Bruzzone M.J., Challal D., Dreos R., Mattarocci S., Bucher P., Libri D., Shore D. Opposing chromatin remodelers control transcription initiation frequency and start site selection. Nat. Struct. Mol. Biol. 2019;26:744–754. doi: 10.1038/s41594-019-0273-3. [DOI] [PubMed] [Google Scholar]
- 23.Korber P., Barbaric S. The yeast PHO5 promoter: from single locus to systems biology of a paradigm for gene regulation through chromatin. Nucleic Acids Res. 2014;42:10888–10902. doi: 10.1093/nar/gku784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lorch Y., LaPointe J.W., Kornberg R.D. Nucleosomes inhibit the initiation of transcription but allow chain elongation with the displacement of histones. Cell. 1987;49:203–210. doi: 10.1016/0092-8674(87)90561-7. [DOI] [PubMed] [Google Scholar]
- 25.Knezetic J.A., Luse D.S. The presence of nucleosomes on a DNA template prevents initiation by RNA polymerase II in vitro. Cell. 1986;45:95–104. doi: 10.1016/0092-8674(86)90541-6. [DOI] [PubMed] [Google Scholar]
- 26.Workman J.L., Roeder R.G. Binding of transcription factor TFIID to the major late promoter during in vitro nucleosome assembly potentiates subsequent initiation by RNA polymerase II. Cell. 1987;51:613–622. doi: 10.1016/0092-8674(87)90130-9. [DOI] [PubMed] [Google Scholar]
- 27.Chen X., Wang X., Liu W., Ren Y., Qu X., Li J., Yin X., Xu Y. Structures of +1 nucleosome-bound PIC-Mediator complex. Science. 2022;378:62–68. doi: 10.1126/science.abn8131. [DOI] [PubMed] [Google Scholar]
- 28.Wang H., Schilbach S., Ninov M., Urlaub H., Cramer P. Structures of transcription preinitiation complex engaged with the +1 nucleosome. Nat. Struct. Mol. Biol. 2023;30:226–232. doi: 10.1038/s41594-022-00865-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fishburn J., Galburt E., Hahn S. Transcription start site scanning and the requirement for ATP during transcription initiation by RNA polymerase II. J. Biol. Chem. 2016;291:13040–13047. doi: 10.1074/jbc.M116.724583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qiu C., Jin H., Vvedenskaya I., Llenas J.A., Zhao T., Malik I., Visbisky A.M., Schwartz S.L., Cui P., Čabart P., et al. Universal promoter scanning by Pol II during transcription initiation in Saccharomyces cerevisiae. Genome Biol. 2020;21:132. doi: 10.1186/s13059-020-02040-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giardina C., Lis J.T. DNA melting on yeast RNA polymerase II promoters. Science. 1993;261:759–762. doi: 10.1126/science.8342041. [DOI] [PubMed] [Google Scholar]
- 32.Core L.J., Lis J.T. Transcription regulation through promoter-proximal pausing of RNA polymerase II. Science. 2008;319:1791–1792. doi: 10.1126/science.1150843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rougvie A.E., Lis J.T. The RNA polymerase II molecule at the 5′ end of the uninduced hsp70 gene of D. melanogaster is transcriptionally engaged. Cell. 1988;54:795–804. doi: 10.1016/s0092-8674(88)91087-2. [DOI] [PubMed] [Google Scholar]
- 34.Eberharter A., Becker P.B. Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep. 2002;3:224–229. doi: 10.1093/embo-reports/kvf053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kornberg R.D., Lorch Y. Chromatin and transcription: where do we go from here. Curr. Opin. Genet. Dev. 2002;12:249–251. doi: 10.1016/s0959-437x(02)00293-9. [DOI] [PubMed] [Google Scholar]
- 36.Steger D.J., Workman J.L. Remodeling chromatin structures for transcription: what happens to the histones? BioEssays. 1996;18:875–884. doi: 10.1002/bies.950181106. [DOI] [PubMed] [Google Scholar]
- 37.Palozola K.C., Donahue G., Liu H., Grant G.R., Becker J.S., Cote A., Yu H., Raj A., Zaret K.S. Mitotic transcription and waves of gene reactivation during mitotic exit. Science. 2017;358:119–122. doi: 10.1126/science.aal4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Penagos-Puig A., Furlan-Magaril M. Heterochromatin as an important driver of genome organization. Front. Cell Dev. Biol. 2020;8:579137. doi: 10.3389/fcell.2020.579137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He Y., Yan C., Fang J., Inouye C., Tjian R., Ivanov I., Nogales E. Near-atomic resolution visualization of human transcription promoter opening. Nature. 2016;533:359–365. doi: 10.1038/nature17970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aibara S., Schilbach S., Cramer P. Structures of mammalian RNA polymerase II pre-initiation complexes. Nature. 2021;594:124–128. doi: 10.1038/s41586-021-03554-8. [DOI] [PubMed] [Google Scholar]
- 41.Abdella R., Talyzina A., Chen S., Inouye C.J., Tjian R., He Y. Structure of the human Mediator-bound transcription preinitiation complex. Science. 2021;372:52–56. doi: 10.1126/science.abg3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rengachari S., Schilbach S., Aibara S., Dienemann C., Cramer P. Structure of the human Mediator-RNA polymerase II pre-initiation complex. Nature. 2021;594:129–133. doi: 10.1038/s41586-021-03555-7. [DOI] [PubMed] [Google Scholar]
- 43.Chen X., Qi Y., Wu Z., Wang X., Li J., Zhao D., Hou H., Li Y., Yu Z., Liu W., et al. Structural insights into preinitiation complex assembly on core promoters. Science. 2021;372 doi: 10.1126/science.aba8490. [DOI] [PubMed] [Google Scholar]
- 44.Chen X., Yin X., Li J., Wu Z., Qi Y., Wang X., Liu W., Xu Y. Structures of the human Mediator and Mediator-bound preinitiation complex. Science. 2021;372 doi: 10.1126/science.abg0635. [DOI] [PubMed] [Google Scholar]
- 45.Greber B.J., Nguyen T.H.D., Fang J., Afonine P.V., Adams P.D., Nogales E. The cryo-electron microscopy structure of human transcription factor IIH. Nature. 2017;549:414–417. doi: 10.1038/nature23903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Greber B.J., Toso D.B., Fang J., Nogales E. The complete structure of the human TFIIH core complex. eLife. 2019;8 doi: 10.7554/eLife.44771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsai K.L., Yu X.D., Gopalan S., Chao T.C., Zhang Y., Florens L., Washburn M.P., Murakami K., Conaway R.C., Conaway J.W., Asturias F.J. Mediator structure and rearrangements required for holoenzyme formation. Nature. 2017;544:196–201. doi: 10.1038/nature21393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yan C.L., Dodd T., He Y., Tainer J.A., Tsutakawa S.E., Ivanov I. Transcription preinitiation complex structure and dynamics provide insight into genetic diseases. Nat. Struct. Mol. Biol. 2019;26:397–406. doi: 10.1038/s41594-019-0220-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rimel J.K., Taatjes D.J. The essential and multifunctional TFIIH complex. Protein Sci. 2018;27:1018–1037. doi: 10.1002/pro.3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vos S.M., Farnung L., Urlaub H., Cramer P. Structure of paused transcription complex Pol II-DSIF-NELF. Nature. 2018;560:601–606. doi: 10.1038/s41586-018-0442-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kokic G., Chernev A., Tegunov D., Dienemann C., Urlaub H., Cramer P. Structural basis of TFIIH activation for nucleotide excision repair. Nat. Commun. 2019;10 doi: 10.1038/s41467-019-10745-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dyer P.N., Edayathumangalam R.S., White C.L., Bao Y., Chakravarthy S., Muthurajan U.M., Luger K. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 2004;375:23–44. doi: 10.1016/s0076-6879(03)75002-2. [DOI] [PubMed] [Google Scholar]
- 53.Hu X., Malik S., Negroiu C.C., Hubbard K., Velalar C.N., Hampton B., Grosu D., Catalano J., Roeder R.G., Gnatt A. A Mediator-responsive form of metazoan RNA polymerase II. Proc. Natl. Acad. Sci. USA. 2006;103:9506–9511. doi: 10.1073/pnas.0603702103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mastronarde D.N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 2005;152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 55.Punjani A., Rubinstein J.L., Fleet D.J., Brubaker M.A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods. 2017;14:290–296. doi: 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- 56.Scheres S.H.W. Amyloid structure determination in RELION-3.1. Acta Crystallogr. D Struct. Biol. 2020;76:94–101. doi: 10.1107/S2059798319016577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zivanov J., Nakane T., Forsberg B.O., Kimanius D., Hagen W.J., Lindahl E., Scheres S.H. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife. 2018;7 doi: 10.7554/eLife.42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tegunov D., Cramer P. Real-time cryo-electron microscopy data preprocessing with Warp. Nat. Methods. 2019;16:1146–1152. doi: 10.1038/s41592-019-0580-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Afonine P.V., Poon B.K., Read R.J., Sobolev O.V., Terwilliger T.C., Urzhumtsev A., Adams P.D. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Struct. Biol. 2018;74:531–544. doi: 10.1107/S2059798318006551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schrödinger L., Delano W. 2020. PyMOL v2.5.0.http://www.pymol.org/pymol [Google Scholar]
- 61.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 62.Goddard T.D., Huang C.C., Meng E.C., Pettersen E.F., Couch G.S., Morris J.H., Ferrin T.E. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 2018;27:14–25. doi: 10.1002/pro.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Croll T.I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D Struct. Biol. 2018;74:519–530. doi: 10.1107/S2059798318002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang H., Farnung L., Dienemann C., Cramer P. Structure of H3K36-methylated nucleosome-PWWP complex reveals multivalent cross-gyre binding. Nat. Struct. Mol. Biol. 2020;27:8–13. doi: 10.1038/s41594-019-0345-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Boehning M., Dugast-Darzacq C., Rankovic M., Hansen A.S., Yu T., Marie-Nelly H., McSwiggen D.T., Kokic G., Dailey G.M., Cramer P., et al. RNA polymerase II clustering through carboxy-terminal domain phase separation. Nat. Struct. Mol. Biol. 2018;25:833–840. doi: 10.1038/s41594-018-0112-y. [DOI] [PubMed] [Google Scholar]
- 68.Bernecky C., Herzog F., Baumeister W., Plitzko J.M., Cramer P. Structure of transcribing mammalian RNA polymerase II. Nature. 2016;529:551–554. doi: 10.1038/nature16482. [DOI] [PubMed] [Google Scholar]
- 69.Plaschka C., Hantsche M., Dienemann C., Burzinski C., Plitzko J., Cramer P. Transcription initiation complex structures elucidate DNA opening. Nature. 2016;533:353–358. doi: 10.1038/nature17990. [DOI] [PubMed] [Google Scholar]
- 70.Schilbach S., Hantsche M., Tegunov D., Dienemann C., Wigge C., Urlaub H., Cramer P. Structures of transcription pre-initiation complex with TFIIH and Mediator. Nature. 2017;551:204–209. doi: 10.1038/nature24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kastner B., Fischer N., Golas M.M., Sander B., Dube P., Boehringer D., Hartmuth K., Deckert J., Hauer F., Wolf E., et al. GraFix: sample preparation for single-particle electron cryomicroscopy. Nat. Methods. 2008;5:53–55. doi: 10.1038/nmeth1139. [DOI] [PubMed] [Google Scholar]
- 72.Greber B.J., Perez-Bertoldi J.M., Lim K., Iavarone A.T., Toso D.B., Nogales E. The cryoelectron microscopy structure of the human CDK-activating kinase. Proc. Natl. Acad. Sci. USA. 2020;117:22849–22857. doi: 10.1073/pnas.2009627117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang H., Xiong L., Cramer P. Structures and implications of TBP-nucleosome complexes. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2108859118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen V.B., Arendall W.B., 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Upon transition from PIC-Nuc10W to PIC-Nuc18W, the proximal +1 nucleosome at the promoter region locates further downstream, TFIIH adopts an alternative conformation, TFIIH subunit XPB fully engages with promoter DNA and ∼2 turns of DNA are unwrapped from the proximal nucleosome.
A promoter-proximal +1 nucleosome located 10 bp downstream of the TSS (cPIC-Nuc10W) is displaced ∼20 Å and tilted by ∼25° by association of TFIIE and TFIIH to the cPIC (yielding PIC-Nuc10W).
Data Availability Statement
-
•
Cryo-EM density maps have been deposited in the Electron Microscopy Data Bank (EMDB) and coordinates within the Protein Data Bank (PDB) for cPIC-nucleosome10W (accession codes EMDB: EMD-16335and PDB: 8BZ1, and EMDB: EMD-16336, EMD-16337, and EMD-16338 for focused refinement maps of cPIC, nucleosome, and composite map, respectively ), PIC-nucleosome10W (accession codes EMDB: EMD-16331 and PDB: 8BYQ, and EMDB: EMD-16339, EMD-16340, EMD-16341, EMD-16342, and EMD-16343 for focused refinement maps of cPIC, nucleosome, CAK, TFIIH, and composite map, respectively) and PIC-nucleosome18W (accession codes EMDB: EMD-16274 and PDB: 8BVW, and EMDB: EMD-16365, EMD-16366, EMD-16367, EMD-16368, and EMD-16369 for focused refinement maps of cPIC, nucleosome, TFIIH, CAK, and composite map, respectively).
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.