

Abstract
Gigaxonin is an adaptor protein for E3 ubiquitin ligase substrates. It is necessary for ubiquitination and degradation of intermediate filament (IF) proteins. Giant axonal neuropathy is a pathological condition caused by mutations in the GAN gene that encodes gigaxonin. This condition is characterized by abnormal accumulation of IFs in both neuronal and non‐neuronal cells; however, it is unclear what causes IF aggregation. In this work, we studied the dynamics of IFs using their subunits tagged with a photoconvertible protein mEOS 3.2. We have demonstrated that the loss of gigaxonin dramatically inhibited transport of IFs along microtubules by the microtubule motor kinesin‐1. This inhibition was specific for IFs, as other kinesin‐1 cargoes, with the exception of mitochondria, were transported normally. Abnormal distribution of IFs in the cytoplasm can be rescued by direct binding of kinesin‐1 to IFs, demonstrating that transport inhibition is the primary cause for the abnormal IF distribution. Another effect of gigaxonin loss was a more than 20‐fold increase in the amount of soluble vimentin oligomers in the cytosol of gigaxonin knock‐out cells. We speculate that these oligomers saturate a yet unidentified adapter that is required for kinesin‐1 binding to IFs, which might inhibit IF transport along microtubules causing their abnormal accumulation.
Keywords: giant axonal neuropathy, gigaxonin, intermediate filaments, kinesin‐1, microtubules, neurofilaments, vimentin
Giant Axonal Neuropathy, a neurodegenerative condition is caused by the loss of gigaxonin protein. Gigaxonin loss causes disorganization and aggregation of intermediate filaments in neurons and other cell types. In this work we have demonstrated that gigaxonin loss leads to inhibition of intermediate filament transport along microtubules by a microtubule motor kinesin‐1, causing their aggregation. We hypothesize that soluble form of intermediate filament proteins that is dramatically increased after gigaxonin loss inhibits kinesin binding to intermediate filaments required for transport.
Abbreviations
- GAN
giant axonal neuropathy
- IF
intermediate filament
- KO
knock out
- NF
neurofilament
- VIF
vimentin intermediate filament
1. INTRODUCTION
Gigaxonin is a ubiquitously expressed protein encoded by the GAN gene located on human chromosome 16q24.1. 1 , 2 The lack or loss of function of gigaxonin causes giant axonal neuropathy (GAN), an autosomal recessive disorder. The GAN gene has more than 50 distinct loss of function mutations that cause the disorder. 3 , 4 , 5 , 6 The loss of both central and peripheral neurons leads to gradual muscular atrophy in this pathological condition. Patients often begin displaying symptoms between the ages of 3 and 5 years old and die between the ages of 20 and 30. 7
Gigaxonin is a member of the BTB (Bric‐a‐brac, Tram track, and Broad)/Kelch domain superfamily. 8 E3 ligase cullin3 (Cul3), a member of the 26S proteasomal complex, binds to the BTB domain of gigaxonin, whereas intermediate filaments (IFs) interact with the Kelch domain. 9 Thus, gigaxonin functions as a Cul3 ligase adaptor, ubiquitinating IFs and regulating IF turnover via proteasomal degradation. 10 GAN gene loss causes an aberrant accumulation of neurofilaments (NFs) in neuronal axons, resulting in a giant axonal phenotype. 11 In addition to NFs, all other classes of IFs are disorganized and aggregated in non‐neuronal cells of GAN patients. 12
IFs are cytoskeletal structures required to stabilize and protect cells against both mechanical and non‐mechanical stress. 13 , 14 In the cytoplasm, IFs scaffold membrane organelles and connect with desmosomes and focal adhesions at the plasma membrane to build a network that holds the nucleus in place. Six types of IFs have been characterized based on their amino acid sequence. The structural organization of all IF proteins is similar, with a central rod domain and varying N‐ and C‐termini. 15 NFs are type IV IFs that form heteropolymers comprising three NF proteins (NFL, NFM, and NFH) as well as related IF proteins alpha‐internexin and peripherin. NFs stabilize neuronal morphology and participate in axonal development and other important neuronal processes. 17 Various neurodegenerative pathologies have been linked to disorganization and loss of functional NFs, 14 , 17 , 18 which result in reduced axonal development, neuronal caliber loss, and impaired transport of organelles and cargos. 19 , 20 Mutations in genes encoding NF proteins or their metabolism are widely accepted as the cause of disorganized NFs in neurodegenerative disorders. 16 GAN falls into the latter group since mutations in the GAN gene impede the degradation and organization of all IFs, including NFs. However, a molecular mechanism of IF aggregation in affected cells is unknown.
Normal distribution of IFs in a cell depends on cytoplasmic microtubules. 18 , 19 , 20 We and others have shown that IFs are transported along microtubules by the major plus‐end directed microtubule motor kinesin‐1. 19 , 21 In a mouse model, kinesin‐1 mutations promote inefficient anterograde transport of NF in neurons, as well as disordered accumulation of NFs in neuronal cell bodies. 22 Due to the absence of anterograde transport, IFs are disorganized and aggregate near the nucleus in kinesin‐1 knockout cells. 23 The similarities in IF aggregation and disorganization patterns between kinesin‐1 knockout and GAN suggest that IF aggregation in the cells of GAN patients could be triggered by impaired IF transport along microtubules. To address this possibility, we examined IF transport in GAN KO cells by applying fiduciary marks on the IF using a photoconversion approach. We observed that the loss of gigaxonin had a significant negative impact on the transport of IFs, while the motor transporting IFs was fully active and could move other cargoes. Abnormal IF distribution in GAN KO cells can be rescued by directly linking kinesin to IFs. We hypothesize that soluble oligomers of vimentin dominantly inhibit normal kinesin binding to IF thus blocking transport and disorganizing IF distribution.
2. MATERIALS AND METHODS
2.1. Plasmids
cDNA encoding vimentin tagged with mEOS3.2 photoconvertible tag was cloned in the pQCXIN vector as described. 20 To generate pQCXIN mEOS3.2_NFM, vimentin in pQCXIN mEOS3.2_vim was replaced with NFM insert using BamHI and EcoRI digestion and ligation. mEOS3.2_NFM was also cloned into pLVX lentiviral plasmid using XbaI and NotI digestion and ligation. For rescue experiment vimentin and 3XFlag tag with GCN4 peptide epitope fragments from mEOS‐vimentin and pcDNA4TO‐sfGCP‐24xGCN4_V1 were subcloned in the SpeI‐BamH1 and BamH1‐XbaI sites to generate pLVX‐vimentin‐3Xflag‐scFV GCN4 plasmid. Kinesin motor domain with stalk (1‐560 aa) and GCN4‐sfGFP with HA tag fragments from human kinesin‐emerald and pHR‐scFv GCN4‐sfGFP‐GB1‐NLS‐dWPRE plasmids were subcloned in the NotI‐PacI and BamH1‐EcoRI sites to generate pQCXIP‐hKIF5B (1‐560)‐GCN4‐sfGFP‐HA tag plasmid. Autophagosome‐specific marker mCherry‐hLC3B‐pcDNA3.1 was a gift from David Rubinsztein (Addgene plasmid # 40827; http://n2t.net/addgene:40827; RRID: Addgene_40 827).
2.2. Cell culture, transfection, and stable cell lines
All cell lines were maintained at 37°C in 5% CO2. Human retinal pigment epithelial (RPE) cells were cultured in DMEM media (Sigma, #D5648) with 10% fetal bovine serum (FBS) (Neuromics, Minneapolis, MN, USA) supplemented with antibiotics (penicillin & streptomycin). RPE cells stably expressing mEOS3.2_vimentin (mEOS_vim) have been described previously. 23 CRISPR was used to generate GAN KO in the RPE mEOS_vim cell line. Following the manufacturer's protocol, the gigaxonin double nickase plasmid (Santa Cruz, #sc‐407 001‐NIC) was stably expressed in RPE cells using retrovirus. Puromycin antibiotic was used to select GAN KO pure clone cells. RPE vimentin KO cells were the gift from Dr. Stephen A Adam, and were described previously. 24
SH‐SY5Y neuroblastoma cells were cultured in a 1:1 mixture of EMEM (BD, #670086) and F12 (Gibco, #11765‐054), supplemented with 10% FBS and antibiotics (penicillin & streptomycin). Scrambled and GAN KO cell lines were generated by transducing SH‐SY5Y cells with lentivirus expressing the Mission pLKO.1 backbone for scrambled shRNA (Sigma, #SHC002) and GAN shRNA (Sigma, TRCN0000083861) respectively. The transduced cells were selected with puromycin for pure clonal cell generation. For differentiation of SH‐SY5Y cells into neurons, cells were incubated for 48 h with media that contained 1% FBS and 100 μM all‐trans retinoic acid (Thermo, #207341000). Lipofectamine™3000 transfection reagent (ThermoFischer, #3000‐008) was used for transient expression experiments.
Dorsal root ganglion (DRG) primary neuron culture was established as described in a previous report. 25 On day two DRGs were treated with lentiviral particles carrying either scrambled or GAN shRNA. Photoconvertible neurofilaments were expressed in the DRGs on day five using lentiviral particles containing mEOS_NFM and imaged on day seven for dynamics.
For whole cell lysate preparation, cells were lysed using RIPA buffer [50 mM Tris (pH 7.4), 150 mM NaCl, 1% TritonX‐100, 0.5% sodium deoxycholate, 0.1% SDS] supplemented with peptidase inhibitor (chymostatin, leupeptin, and pepstatin A, 20 μg/mL) and serine protease inhibitor (1 mM PMSF), then supernatant was collected after centrifugation at 10 000× g for 10 min. For soluble and insoluble fraction preparation, we followed the protocol described earlier. 26 Briefly, confluent 10 cm dishes were washed once with ice‐cold PBS and the cells were lysed in a buffer containing 0.5% Triton X‐100, 150 mM NaCl, 20 mM Tris·HCl pH 7.4, 2 mM EGTA, 2 mM EDTA, 1.5 mM sodium vanadate. Then cell lysates were ultracentrifuged for 30 min at 265 000× g and the pellet (insoluble fraction) and supernatant (soluble fraction) were collected. Acetone precipitation was used to concentrate the proteins in soluble fraction. In brief, two volumes of ice‐cold acetone were added to the soluble fraction and incubated at −20°C for 2 h. After centrifugation at 10 000× g for 10 min, the pellet was reconstituted in 5X Laemmli buffer and Western blot analysis was carried out. For the Western blot analysis, equal protein loads were maintained among samples. Antibodies used in these studies included chicken polyclonal anti‐vimentin (Biolegend, Cat #919101, 1:2000) and HRP‐conjugated mouse monoclonal GAPDH (Proteintech, cat HRP‐60004, 1:10 000). Blots were developed using WesternBright™ Quantum (Cat # K‐12042‐D20) and imaged using Li‐COR Odyssey Fc imaging instrument with Image studio software version 5.2.
2.3. Live cell imaging
Tokai‐Hit stage‐top incubator (Tokai‐Hit) and Okolab gas mixer (Okolab) were used to maintain temperature at 37°C and CO2 at 5% throughout the live imaging. Images were collected using a Nikon Eclipse Ti2 stage equipped with a W1 spinning disk confocal head (Yokogawa CSU with pinhole size 50 μm), 60X 1.49 N.A. or a 100X 1.45 N.A. oil immersion lenses. Photometrics Prime 95B sCMOS or Hamamatsu 29 ORCA‐Fusion Digital CMOS Camera driven by Nikon Elements software were used for image acquisition.
We have demonstrated previously that cytoskeletal dynamics can be studied efficiently by fluorescence microscopy of live cells using photoconversion of mEOS fluorescent protein fused with a cytoskeletal protein of interest. 20 Photoconversion of mEOS3.2 was achieved using 405 nm illumination from a Heliophor 89 North LED mounted in the epifluorescence pathway. A pinhole diaphragm was included in the light path for confinement of the photoconversion zone. For vimentin dynamics study, cells were plated on glass coverslips one day before imaging. Cells expressing mEOS_vim were photoconverted for 3 s and then timelapse sequences were acquired for 3 min with the interval of 15 s, using 561 nm laser excitation.
Neurofilament dynamics were studied in primary DRG neurons and differentiated SH‐SY5Y cells. On day seven of primary DRG neuron culture NFM dynamics was recorded for 30 min with interval of 0.5 or 1 min after 5 s photoconversion. Undifferentiated SH‐SY5Y cells were electroporated with mEOS_NFM in pQCXIN plasmid and plated onto glass coverslip. Neuronal differentiation was induced after eight hours of electroporation with differentiating medium. Post 48 h of differentiation, NFM dynamics was recorded for 30 min with interval of 0.5 or 1 min after photoconversion.
For mitochondrial and lysosomal motility 25 nM MitoTracker Red CMXRos (Invitrogen, #M7512), and 25 nM LysoTracker red (Invitrogen, #L7528) were used respectively. Live cells were incubated for 10 min with the respective dye and then imaged. For autophagosome analysis, cells were transiently transfected with the autophagosome‐specific marker protein LC3B tagged with mCherry. Autophagosome formation was induced 24 h post‐transfection using Earls Balanced Salt Solution (EBSS), pH 7.4 for 3 h. For organelles' motilities, randomly 15–20 fields were captured for a total time of 2–3 mins with intervals of 1–2 s and analyzed.
2.4. Image analysis
Fiji ImageJ plugins were used to analyze IF transport. 27 To determine the overall movement of photoconverted filaments, we used the curve tracing plugin. 28 Photoconverted filaments transported outside the initial photoconverted region were used for quantification and the original photoconversion zone was masked in all frames while quantification. To get the overall length of filaments transported over the duration of imaging, we calculated the total length of filaments transported for each frame and added all the values. Our plugins are deposited in GitHub, in the following URL (https://github.com/mkitti/CurveTracingUtils).
The analysis explorer package in Nikon Elements software (NIS elements AR, version 5.30.03) was used to quantify the motility of mitochondria, lysosomes, and autophagosomes. Preprocessing includes background correction and smoothing after which the particles were analyzed by the random motion method. Organelles that appeared in at least five consecutive frames were taken into consideration for analyzing the motility, and non‐motile organelles were excluded from the analysis. We had a velocity threshold of 200 nm/s for fast‐moving organelles.
In the rescue experiment, vimentin distribution was quantified using CellProfiler 4.2.1 29 module object intensity distribution. First, we determined the total cell area using HA staining against KIF5B (1‐560)‐HA. From the center to periphery, the cell area was divided into 5 radial zones, each of which was assigned a number from 1 to 5. Peripheral zones 4 & 5 were used for analysis. Fluorescence intensity of mEOS_vimentin in peripheral zones was summed up and quantified the vimentin distribution.
2.5. Immunostaining
Fixation and staining protocols were performed as described previously. 23 Cells were incubated with primary antibodies for 1 h at room temperature. Primary antibodies used in this study were microtubule (Dm1α mouse monoclonal, in‐house, 1:1000), Flag tag (proteintech, #66008‐1‐Ig), and HA tag (Genetex, GTX29110). Secondary antibodies used in this study were Rhodamine conjugated goat anti‐mouse and goat anti‐rabbit (Jackson ImmunoResearch, #115‐025‐003; 1:700, and #111‐025‐144, 1:500), Alexa‐Fluor 647‐conjugated donkey Anti‐Mouse (Jackson ImmunoResearch, #715–605‐151, 1:700).
2.6. Electron microscopy of platinum replicas
Cells were grown overnight on glass coverslips. The membrane and soluble cytosolic proteins were extracted with 1% Triton‐X 100 in PHEM buffer (80 mM Pipes, 1 mM EGTA, 1 mM MgCl2 at pH 6.8) for 5 min. Cells were treated with 0.6 M potassium chloride in PHEM buffer, for 10 min to remove microtubule and actin filaments from the cytoskeletons still attached to the coverslips. Platinum replica electron microscopy (PREM) was performed as previously described. 30 , 31 Briefly, samples were fixed with 2% glutaraldehyde in 0.1 M cacodylate buffer, tannic acid, and uranyl acetate; critical point dried; coated with platinum and carbon; and transferred onto electron microscopic grids for observation. Samples were imaged using a FEI Tecnai Spirit G2 transmission electron microscope (FEI Company, Hillsboro, OR) operated at 80 kV. Images were captured by Eagle 4k HR 200 kV CCD camera.
2.7. Statistical analysis
Data were analyzed using GraphPad Prism 9.3 (GraphPad Software Inc.) and are shown as mean with SEM. Statistical testing involved unpaired Student's t tests otherwise mentioned in the legend. p values and levels of significance are listed in figure legends.
3. RESULTS
3.1. GAN gene loss inhibits vimentin IF transport
IFs form a dense network extending from the nucleus toward the cell periphery and act as a mechanical stabilizer for the cell. IFs undergo constant rearrangement and transport along microtubules to facilitate the ongoing requirements of cellular activity. 32 We hypothesized that inhibition of normal IF dynamics causes IF disorganization in GAN. To test this hypothesis, we studied the transport of the vimentin IFs (VIF) in GAN knockout (GAN KO) cells using a photoconversion strategy. We used RPE cells stably expressing vimentin fused with a photoconvertible protein mEOS3.2 (mEOS‐vim). mEOS‐vim copolymerizes with endogenous vimentin and forms normal VIF networks in RPE cells (Figure 1A). Using CRISPR, we knocked out the GAN gene in RPE cells stably expressing mEOS‐vim. Consistent with previous studies, in GAN KO cells VIF organization was disrupted and the majority of VIFs formed aggregates in the juxtanuclear region, but some labeled filaments were still observed at the cell periphery (Figure 1B). This VIF disorganization is a typical phenotype of GAN KO cells. 33 , 34
FIGURE 1.
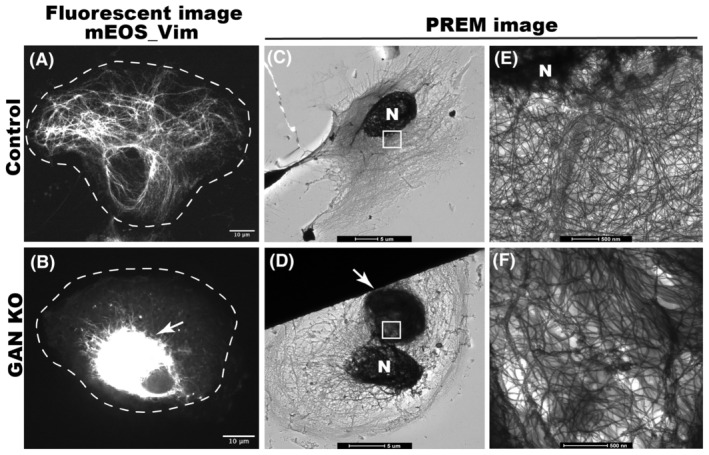
GAN KO affects organization of IFs. (A) shows the normal distribution of mEOS_vim in control RPE cell, (B) shows the mEOS_vim aggregation in GAN KO RPE cells. Scale bar 10 micrometers. Platinum replica electron microscope (PREM) images of control cell (C, E) and GAN KO cell (D, F). IF meshwork in the entire cell can be appreciated in lower magnification (C) and single filaments can be observed in higher magnification near nucleus (E). In GAN KO cells, juxtanuclear IF aggregates can be observed at lower magnification (D), single filaments can be observed in the IF aggregate at higher magnification of aggregated region (F). Nucleus is indicated by N, arrow points to IF aggregates and white box in C and D represent the magnified region for E and F respectively. Scale bar 5 μm for C and D; 500 nm for E and F images.
Platinum replica electron microscopy (PREM) was used for high‐resolution analysis of VIF organization and distribution in GAN KO cells. In control cells, we observed a fine meshwork of VIFs (Figure 1C). In GAN KO cells, we observed dense VIF aggregates in the juxtanuclear position (Figure 1D). At higher magnification, single filaments could be resolved in all regions of the control cell (Figure 1E). In GAN KO cell, VIF formed a dense interwoven network and bundles, yet single filaments could be observed within the aggregates (Figure 1F). The presence of single filaments in GAN KO aggregates motivated us to examine VIF transport in these cells.
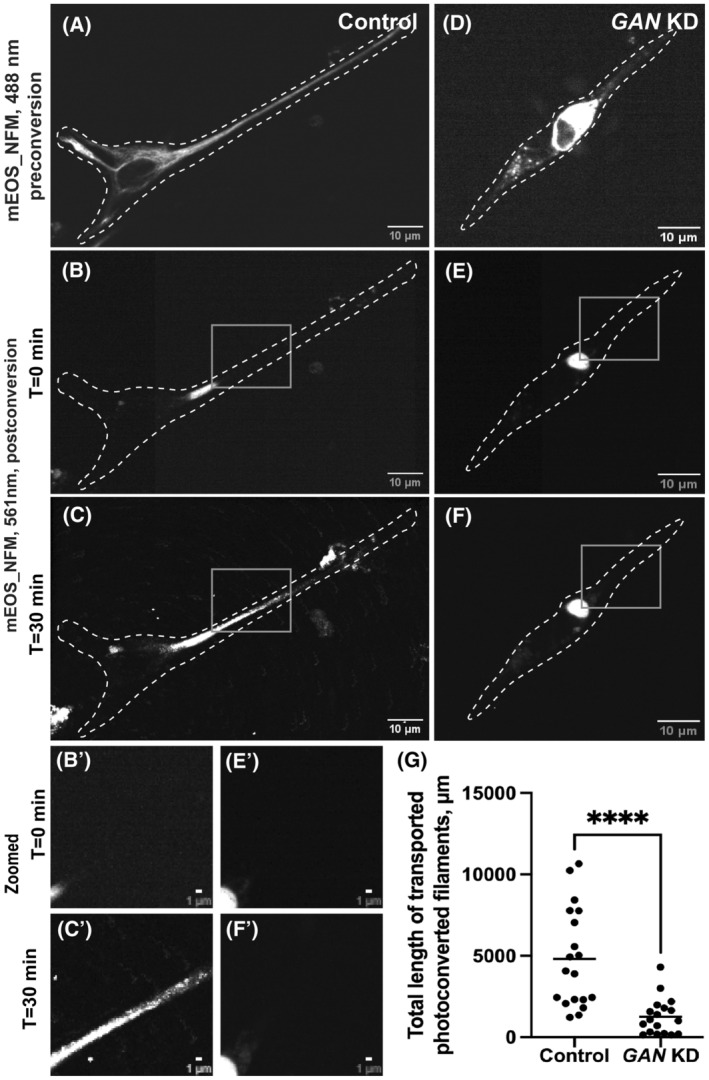
We have demonstrated previously that cytoskeletal dynamics can be studied efficiently by fluorescence microscopy of live cells using photoconversion of mEOS fluorescent protein fused with a cytoskeletal protein of interest. 20 Emission of mEOS can be switched from the green (488 nm) to the red (561 nm) channel by illumination with a 405 nm laser. Using a pinhole aperture, a small region of mEOS‐vim‐labeled filaments in a live cell was photoconverted and cells were imaged in the red channel. This technique showed that control cells display robust transport of VIFs away from the photoconverted zone. In control cells, several filaments were typically transported during the total imaging time (three minutes), demonstrating active VIF dynamics under normal conditions (Figure 2C,C′ and Video S1). However, mEOS‐vim in GAN KO cells behaved differently. All photoconverted filaments remained confined within the initial photoconverted zone throughout the duration of the experiment (Figure 2F,F′ and Video S2). These results showed that VIF transport was severely inhibited by the loss of GAN gene. It is possible that the inhibition of VIF transport could be a secondary effect of IF aggregation. To test this idea, we photoconverted a region of the cytoplasm in GAN KO cells where individual filaments could still be found. We found that even non‐aggregated single VIFs in GAN KO cells remained immotile (Video S3). We have quantified the total length of segments of VIF filaments transported outside of the initial photoconverted zone in control and GAN KO cells. This quantification showed dramatic inhibition of VIF transport after GAN KO (Figure 2G).
FIGURE 2.

GAN KO inhibits the transport of vimentin filament. Photoconversion of mEOS‐vimentin in RPE cells using spinning disk confocal microscopy in control (A–C) and GAN KO (D–F) cells. Panels (A) and (D) imaged under green channel (488 nm) before photoconversion. Panel (A) shows vimentin filament in a control cell, while panel D displays the aggregated vimentin filament in a GAN KO cell. mEOS‐vimentin was photoconverted from green to red at the specific region. Panels (B) and (E) were imaged under red channel (561 nm) at time 0 min after photoconversion. Panel (C) and (F) after 3 min of photoconversion. Gray box regions were zoomed in and shown below (B', C', E' and F'). Dotted lines mark the boundary of the cell. Scale bar 10 μm for full images and 1 μm for zoomed images. (G) Photoconverted vimentin IFs outside the conversion zone were quantified and segmented filaments were counted for each frame. Statistical significance was determined using Student's t‐test (n = 15 cells). ***p < 0.001.
To test whether this IF transport inhibition was specific for RPE cells, we used undifferentiated SH‐SY5Y (human neuroblastoma) cells to test VIF transport in control and GAN knockdown conditions. GAN gene expression was knocked down using stable expression of shRNA against the GAN gene (GAN KD) and cells expressing a scrambled shRNA served as a control. VIF transport was studied by transient expression of mEOS‐vim. Control cells displayed well‐developed and typically organized VIF meshwork (Figure S1A). GAN KD cells displayed classic juxtanuclear aggregation of VIFs (Figure S1D). VIF dynamics were measured by photoconversion of the EOS‐vim as described above. We found that control cells displayed a robust transport of filaments from the photoconversion zone (Figure S1C,C′ and Video S4). In contrast, VIF transport in GAN KD cells was severely inhibited (Figure S1F,F′ and Video S5). Therefore, gigaxonin loss causes inhibition of IFs transport and not restricted to RPE cells only.
3.2. Neurofilament transport is inhibited after GAN knock down
To understand whether the transport of neurofilaments was also inhibited in the absence of gigaxonin similar to VIFs, we used primary DRG neurons to study NF dynamics under gigaxonin knock down. As described above, photoconversion method was used to study NF transport in neurons. Control cells displayed proper NFM distribution (Figure 3A) in the cell body and neurites with active NF transport (Figure 3C,C′ and Video S6). Compared to control cells, GAN KD cells exhibited disorganized NFM and formed aggregates in the cell body (Figure 3D). Transport of NFs was severely impacted in GAN KD neuronal cells (Figure 3F,F′ and Video S7), as observed in the case of VIFs in RPE and SH‐SY5Y cells. Quantification revealed significant inhibition of NFM transport in GAN KD cells (Figure 3G). Overall, NFs dynamics were also severely inhibited in the absence of gigaxonin protein.
FIGURE 3.
Neurofilament transport is inhibited in GAN KD primary DRG neurons. Photoconverstion of mEOS_NFM in primary DRG neurons using spinning disk confocal microscopy in control (A–C) and GAN KD (D–F) cells. Panel (A) and (D) were imaged under the green channel (488 nm) before photoconversion. Panel (A) shows the NFM filament in a control cell while panel (D) displays the aggregated NFM filament in GAN KD cells. mEOS‐NFM was photoconverted from green to red at the specific region. Panel (B) and (E) were imaged under red channel at time 0 min after photoconversion. Panel (C) and (F) 30 min after photoconversion. Dotted lines mark the boundary of the cell. Gray box regions were zoomed in and shown below (B', C', E' and F'). Scale bar 10 μm for full images and 1 μm for zoomed images. (G) Photoconverted NFM IFs outside the conversion zone were quantified and segmented filaments were counted for each frame. Statistical significance was determined using Student's t‐test (n = 18 cells). ****p < 0.0001.
Besides primary neurons, we also tested the NF dynamics in differentiated neuroblastoma SH‐SY5Y cells. mEOS_NFM was expressed in SH‐SY5Y cells using transient transfection. Both control cells and GAN KD cells showed neurites 48 h after differentiation was induced. In control cells, NFM demonstrated active transport and proper distribution whereas its transport (Figure S2C,C′ and Video S8) was inhibited and aggregated in the GAN KD SH‐SY5Y cells (Figure S2E,E′ and Video S9). When gigaxonin is not present, NF transport is hindered in both cultured primary DRG neurons and differentiated SH‐SY5Y cells. Thus, gigaxonin is required not only for VIF but also for NF transport.
3.3. GAN KO increases the soluble fraction of vimentin
IF proteins exist as mature filaments and subunits/precursor oligomers, where the former is referred to as the insoluble, and the latter as the soluble fraction of cellular IFs. 26 , 35 In normal cells, the vast majority of vimentin is present in the insoluble filamentous form and only a small fraction is soluble. 36 Earlier reports showed that the total IF protein level in GAN KO cells is increased without changes in the mRNA level. 12 , 25 Here we analyzed separately the soluble (IF precursors) and insoluble (mature IF) fractions of vimentin in RPE cells. Our results confirm that in GAN KO cells, the vimentin level was increased both in the soluble and insoluble fractions. While in the insoluble fraction, mature vimentin IFs increased modestly, in agreement with the previous reports, 12 , 25 , 37 the amount of soluble vimentin oligomers was increased dramatically, about 20‐fold (Figure 4). In absence of gigaxonin, cell accumulated around ~0.5% vimentin in the soluble form whereas in control soluble vimentin is virtually undetectable.
FIGURE 4.
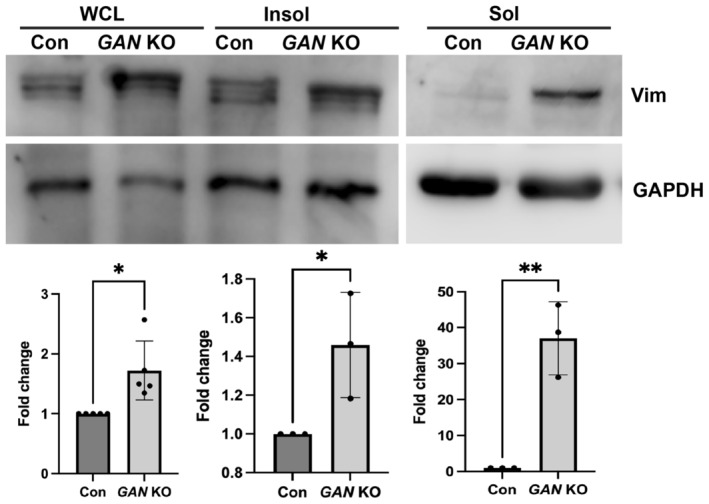
GAN KO cells show increased soluble vimentin. Immunoblotting analysis for vimentin in whole cell lysate (WCL), insoluble fraction (Insol), and soluble fraction (Sol). Vimentin protein level was increased both in WCL and insoluble fraction of GAN KO cells. Soluble fractions show dramatic increase in vimentin in GAN KO cells. GAPDH protein levels were used as loading control. Fold changes are calculated with GAPDH normalized values. *p < .05; **p < .01.
3.4. The kinesin‐1 motor is functional in GAN KO cells
Lack of IF motility in GAN KO cells raised the question of whether gigaxonin is required for the function of kinesin‐1 itself. We first tested if GAN KO affects expression of kinesin‐1. To test kinesin‐1 functionally we studied the motility of lysosomes and autophagosomes in GAN KO cells to check for the ability of kinesin to transport cargoes unrelated to IFs. We found that lysosome and autophagosome distributions in GAN KO cells were similar to their distributions in the control cells. Furthermore, after GAN KO there were no significant differences in the organelle motility between GAN KO cells and control cells (Figure 5A–D). Normal movement of membrane organelles (lysosomes and autophagosomes) and normal distribution of microtubules (Figure S3D) in GAN KO cells demonstrated that kinesin‐1 motor was present and was fully functional and the cells contained normal microtubule tracks.
FIGURE 5.
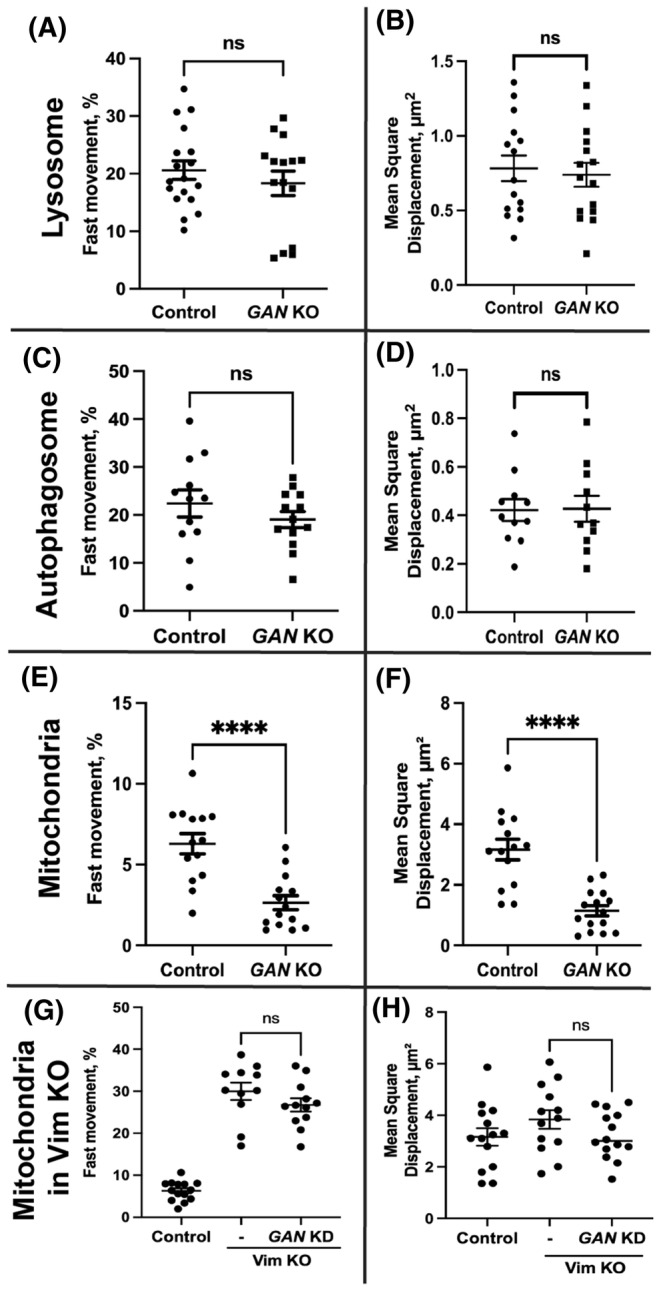
Membrane organelle motility under GAN KO and GAN KD. Lysosomes were visualized in live cell using lysotracker red. Panel (A) and (B) depict the fast movement and mean square displacement of lysosomes in control and GAN KO cells respectively. Autophagosomes were visualized in live cell using the autophagosome‐specific marker LC3 tagged with mCherry. Panel (C) and (D) depict the fast movement and mean square displacement of autophagosomes. Both lysosome and autophagosome motility are not affected in GAN KO cells. Mitochondria were visualized in live cell using Mitotracker red. Panel E shows that the fast mitochondrial movement is severely affected in GAN KO cells compared to control cells. (F) Mean square displacement of mitochondria in GAN KO is significantly lower than control cells. Silencing GAN expression in Vim KO cell has no effect on mitochondrial motility (G) and mean square displacement (H). Statistical significance was determined using Student's t‐test (n = 15 cells). ****p < .0001.
3.5. Vimentin accumulation traps mitochondria in GAN KO cells
Kinesin‐1 is required for anterograde transport of mitochondria. 38 , 39 Previous research has shown that the mitochondria in dorsal root ganglia neurons deficient of GAN gene are less motile and dysfunctional. 25 , 40 Here we studied the motility of mitochondria in GAN KO cells. In control cells, mitochondria were evenly distributed throughout the cell (Figure S4B), while in GAN KO cells their distribution and shape were severely altered (Figure S4D). They were rarely seen at the cell periphery and mostly were localized close/within the VIF aggregate. In GAN KO cells, mitochondrial motility was significantly lower than in control cells (Figure 5E). To quantify motility, we computed the mean square displacement, which revealed that mitochondria in GAN KO cells had significantly lower mean square displacement (Figure 5F). Loss of mitochondrial motility is consistent with the earlier studies. Although having functional kinesin‐1 motors, mitochondrial motility was strongly inhibited in GAN KO cells.
As kinesin‐1, the motor that moves mitochondria along microtubules is present and can move other membrane organelles, we hypothesize that the lack of mitochondria motility is explained by their anchoring to immotile IFs. To test this hypothesis, we combined KO of vimentin and silencing of GAN. Using sigma Mission pLKO.1 shRNA, GAN gene expression was silenced in Vim KO cells, and the mitochondrial dynamics were investigated as previously mentioned. GAN silencing had no effect on the distribution or motility of the mitochondria in Vim KO cells (Figure 5G,H). This result shows that the inhibition of mitochondria movement in GAN KO cells is a secondary effect of IF immobilization by VIF and the lack of VIF motility.
3.6. IF distribution can be rescued by direct binding of kinesin‐1
Even in the presence of active kinesin‐1, the absence of gigaxonin protein inhibits IF transport and causes aggregation. Therefore, we hypothesized that the loss of kinesin recruitment to IFs is the cause of IF aggregation. We decided to address this by direct linking of kinesin‐1 and IFs. We made use of the GCN4 antibody‐peptide pair reported by Worn et al. 41 We fused the GCN4 peptide epitope to vimentin and the GCN4 single‐chain variable fragment antibody to kinesin‐1. We also included reporter tags, such as a flag tag in the vimentin‐GCN4 construct and a HA‐tag in the kinesin‐1 GCN4‐scFV construct respectively. Control cells (Figure 6A) showed normal mEOS_vim distribution after expression of kinesin‐1 GCN4‐scFV alone. GAN KO cells (Figure 6C) displayed the juxtanuclear mEOS_vim aggregates and periphery of cell was mostly free of vimentin filaments. Expression of kinesin‐1 GCN4‐scFV alone has no effect on control cells and GAN KO cells. Expression of both the constructs (Vim‐GCN4 and kinesin‐1‐GCN4 scFV) in GAN KO cells resulted in redistribution of vimentin filaments; and their distribution stretching to the cell periphery (Figure 6E). Both flag‐tagged vimentin and mEOS‐vimentin can be found in the extended filaments, which shows that expressed GCN4‐tagged vimentin incorporates into VIF. The HA tagged kinesin‐1 was found colocalized with the flag tagged vimentin (Figure 6F). This colocalization indicates that vimentin filaments are indeed transported by the GCN4 scFV‐tagged kinesin‐1 through the interaction of GCN4 antibody and its peptide epitope. The fact that linking of kinesin‐1 motor to IFs rescues the defects of IF distribution shows that IF aggregation is caused by defects in transport. This strongly supports our hypothesis that the primary defect of IF distribution in GAN KO cells is caused by compromised interaction of kinesin with IFs.
FIGURE 6.
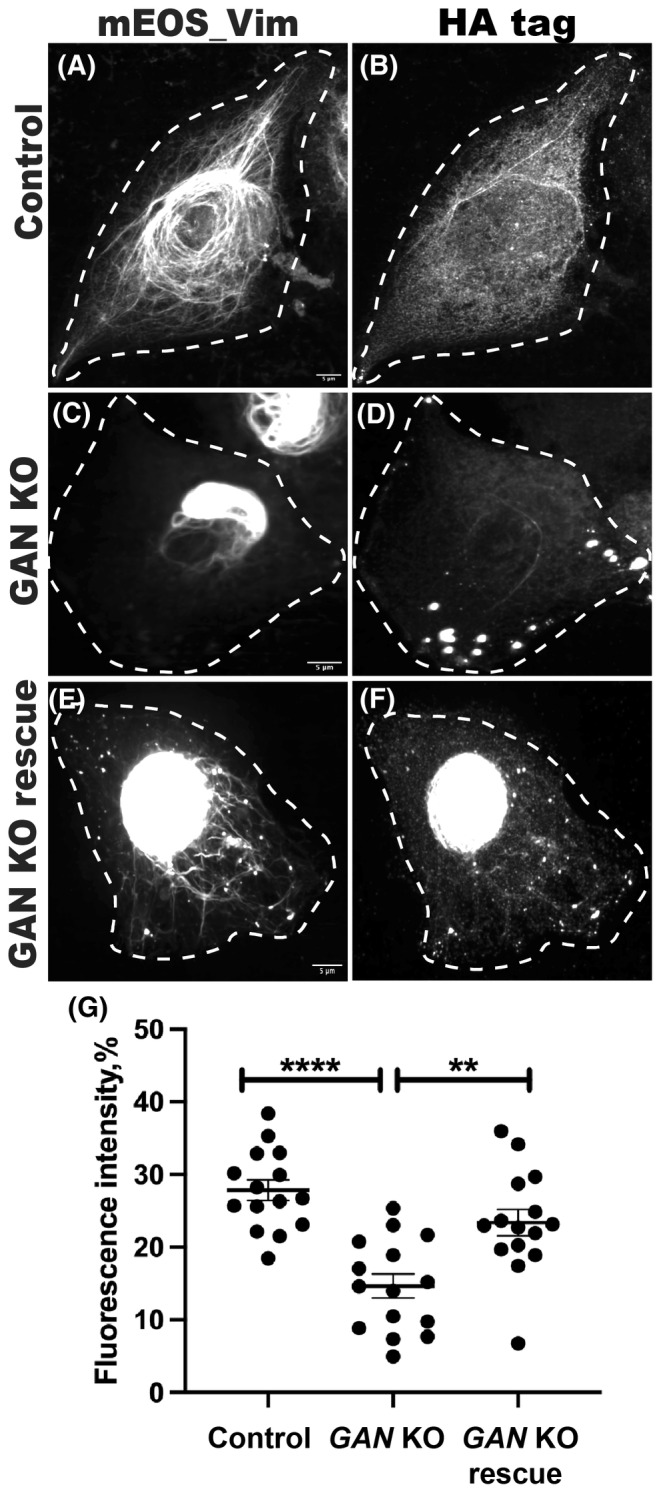
Vimentin distribution rescued by direct binding of vimentin filaments to kinesin‐1. Rescue experiment was done in RPE mEOS_vim (control and GAN KO) cells. Control (A, B) and GAN KO (C, D) cells treated with KIF5B (1‐560)‐scFV GCN4‐HA tag plasmid alone. Rescue condition (E, F), GAN KO cells received both vimentin‐GCN4‐flag tag and KIF5B (1‐560)‐scFV GCN4‐HA tag plasmid. mEOS_vim appeared normally distributed in control (A) and aggregated in GAN KO (C). Distribution of vimentin in control cells indicates that the expression of KIF5B (1‐560)‐scFV GCN4‐HA tag plasmid has no non‐specific effects. In rescue condition (E), mEOS_vim redistributed and stretched to the cell periphery besides the vimentin aggregate in the center. Filaments distribution was analyzed using Object Intensity Distribution module in CellProfiler. Cell was divided radially from the center to periphery and fluorescence intensity of mEOS_vim in each zone was calculated. (G) Intensity in the cell periphery was compared among the conditions. Rescue cells showed significantly higher vimentin filaments compared to the GAN KO condition. Statistical significance was determined using one‐way ANOVA and Tukey's multiple comparisons test (n = 15 cells). **p < .01; ****p < .0001.
4. DISCUSSION
Giant axonal neuropathy (GAN) is a rare autosomal recessive, progressive neurodegenerative disorder with onset in early childhood and death usually by the second/third decade of life. GAN is caused by an abnormal or complete loss of function of gigaxonin. 42 Aggregation of NFs has been reported in various neurodegenerative disorders like Parkinson's syndrome, Alzheimer's, and Amyotrophic Lateral Sclerosis. 43 , 44 However, GAN is a unique condition where all IFs undergo aggregation not only in neurons but all cell types including endothelial cells, epithelial cells, skin fibroblasts, muscle fibers, Schwann cells, and neurons. 3 , 26 , 28 This indicates that gigaxonin plays an important role in IF regulation and organization. IFs form a complex network in the cytoplasm of mammalian cells. This network undergoes constant reorganization by movement, severing, and annealing to accommodate the constant need for changing cell shape and cellular environment. 45 The organization of VIF and NFs is severely altered and filaments form juxtanuclear aggregates after the loss of the GAN gene as shown previously. 33 Examination of platinum replicas of GAN KO cells revealed that individual filaments with normal diameter are still present within this compact aggregate. This is in agreement with published data of GAN patient cells and cells with GAN KO in which individual NF and other IF filaments with normal diameter can be observed. 7 , 12 , 33 , 40 The presence of individual filaments in GAN aggregates shows that they could potentially be dynamic and transported. Hence, a failure of IF transport appears to be a possible explanation for their disorganization and aggregation in GAN.
In this work, IF transport was studied using the photoconversion technique. We observed that the dynamics of IFs (VIF and NF) were severely affected in the absence of gigaxonin. As IF anterograde transport is performed by the major microtubule motor kinesin‐1, 23 we tested the possibility that inhibited transport of IFs could be explained by the general failure of kinesin‐1. Strikingly, we found under GAN KO conditions, in addition to IF only mitochondrial motility was disrupted, whereas other membrane organelles such as lysosomes and autophagosomes were unaffected. Taken together it is clear that kinesin‐1 activity is not affected by the gigaxonin loss. However, to address the mitochondrial dysfunction in GAN disorder, importance of IFs in mitochondrial anchoring and positioning should be considered. 46 EM imaging showed mitochondria are associated with the IFs aggregate of GAN KO cells. 12 In GAN KO cells, mitochondria exhibit altered metabolism and elevated oxidative stress. 25 Loss of mitochondrial transport has been shown as the reason for distal axonal loss and atrophy in GAN. 25 Poor neuronal development in a number of neurodegenerative disorders is associated with defective motility and function of mitochondria. 47 In this study, we demonstrated that IF transport inhibition causes its meshwork collapse and aggregates in GAN. When IF becomes disorganized, mitochondrial activity and motility are impeded 40 ; moreover, IF traps the associated mitochondria in its aggregate. So, in GAN, the inhibited IF transport may lead to mitochondrial dysfunction and play a role in pathogenesis. Since kinesin‐1 is active in the GAN KO condition, there must be other reasons for inhibition of IF transport.
Since gigaxonin is necessary for the regulation and efficient turnover of IFs, its absence or loss of function results in accumulation of IFs. Proteomic and biochemical approaches have shown that gigaxonin can bind to IFs directly. 9 , 12 Ectopic overexpression of gigaxonin results in the complete clearance of IFs from the cell over time. 12 It has been proposed that mature IF filaments disassemble into soluble IF forms, which are gigaxonin substrates and are subsequently degraded. 12 , 48 Here we show dramatically increased levels of soluble vimentin oligomers with the loss of gigaxonin. Thus, the primary outcome of gigaxonin loss is accumulation of soluble IF oligomers due to poor turnover.
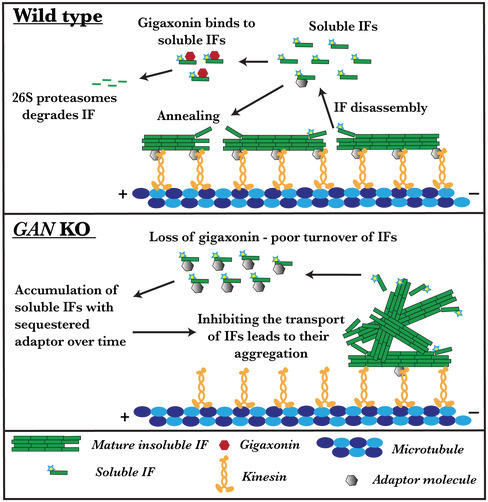
With gigaxonin loss, the kinesin motor is still functional and IFs have normal morphology; however, the transport of IF is inhibited. We demonstrated that distribution of IF can be restored by directly linking kinesin‐1 to IF. It is still not known whether kinesin‐1 binds to IF directly or through an adapter protein as the nature of this interaction is still elusive. Plausibly accumulating soluble IF oligomer in the absence of gigaxonin may sequester an unknown adaptor, linking kinesin‐1 to IFs. Thereby in a dominant‐negative way oligomers may block the IF interaction with kinesin‐1 motor and inhibit IF transport (Figure 7). Together, these facts suggest that a yet unknown adaptor protein likely plays a crucial part in the interaction between IFs and the kinesin‐1 motor. Consequently, the identification of the adapter that mediates the binding of kinesin‐1 to IFs will allow us to directly test this hypothesis.
FIGURE 7.
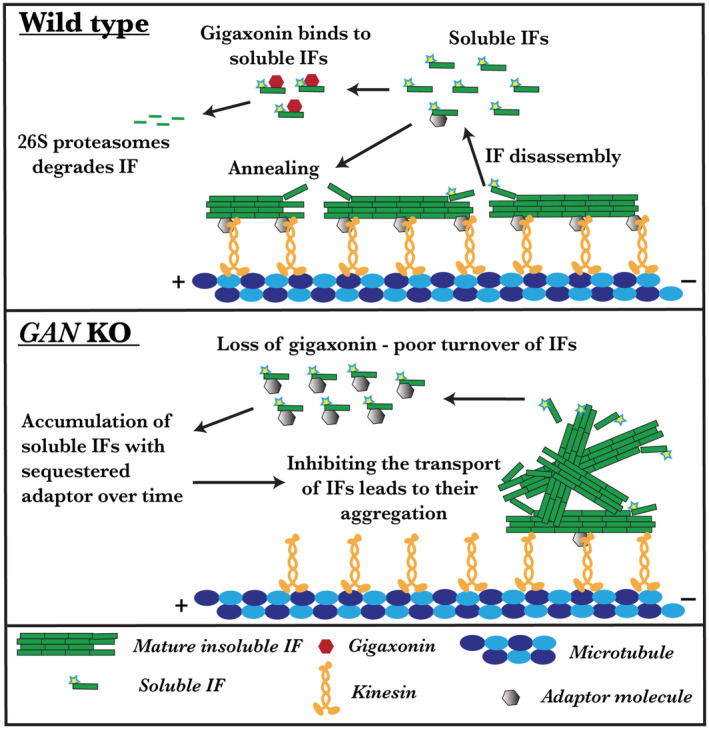
Inhibited IF transport causes aggregation in the absence of gigaxonin. In wild type, IF will be actively transported along microtubules by the kinesin‐1 motor. An unknown adaptor might aid the interaction between kinesin and IF. Besides transport, IF undergoes constant rearrangement by assembly and disassembly process by which soluble IF oligomers are released into the cytosol. Fraction of these soluble oligomers binds to gigaxonin and undergoes proteasomal degradation. Thereby, gigaxonin regulates the IFs turnover. Absence of gigaxonin protein leads to the accumulation of soluble IFs in the cytosol. Accumulated soluble IFs sequester the unknown adaptor which links kinesin to IF. Thereby, block the IF interaction with kinesin‐1 motor and inhibit IF transport in a dominant‐negative manner.
AUTHOR CONTRIBUTIONS
Vladimir I. Gelfand: Obtained the fund, experimental ideas, manuscript editing, and reviewing. Bhuvanasundar Renganathan: Experimental ideas, design and execution, data analysis, and wrote the manuscript. James P. Zewe, Yuan Cheng, Jean‐Michel Paumier, and Mark Kittisopikul: Involved in experimental tool preparation. Karen M. Ridge and Puneet Opal: Involved in experimental tool preparation and manuscript reviewing.
DISCLOSURES
The authors declare that they have no conflict of interest.
Supporting information
Figure S1.
Figure S2.
Figure S3.
Figure S4. \
Video S1.
Video S2.
Video S3.
Video S4.
Video S5.
Video S6.
Video S7.
Video S8.
Video S9.
Data S1
ACKNOWLEDGMENTS
The authors thank Dr. Stephen A Adam for stimulating discussions and critical reading of the manuscript and Dr. Farida Korobova, Center for Advanced Microscopy (Northwestern University) for PREM sample preparation and EM image acquisition. This work is supported by the National Institute of General Medical Sciences (NIH) under grants P01 GM096971 and R35 GM131752 to VIG. PO acknowledges grant support from NINDS (1R01NS127204‐01 and R01NS082351‐09) and in the past support from Hannah's Hope Fund (HHF). Center for Advanced Microscopy is supported by NCI CCSG P30 CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center.
Renganathan B, Zewe JP, Cheng Y, et al. Gigaxonin is required for intermediate filament transport. The FASEB Journal. 2023;37:e22886. doi: 10.1096/fj.202202119R
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available in the methods and material of this article.
REFERENCES
- 1. Cavalier L, BenHamida C, Amouri R, et al. Giant axonal neuropathy locus refinement to a < 590 kb critical interval. Eur. J. Hum. Genet. 2000;8(7):527‐534. [DOI] [PubMed] [Google Scholar]
- 2. Ben Hamida C, Cavalier L, Belal S, et al. Homozygosity mapping of giant axonal neuropathy gene to chromosome 16q24.1. Neurogenetics. 1997;1:129‐133. [DOI] [PubMed] [Google Scholar]
- 3. Bomont P, Cavalier L, Blondeau F, et al. The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat Genet. 2000;26:370‐374. [DOI] [PubMed] [Google Scholar]
- 4. Normendez‐Martínez MI, Monterde‐Cruz L, Martínez R, et al. Two novel mutations in the GAN gene causing giant axonal neuropathy. World J Pediatr. 2018;14:298‐304. [DOI] [PubMed] [Google Scholar]
- 5. Bomont P, Ioos C, Yalcinkaya C, et al. Identification of seven novel mutations in the GAN gene. Hum Mutat. 2003;21:446. [DOI] [PubMed] [Google Scholar]
- 6. Koop O, Schirmacher A, Nelis E, et al. Genotype‐phenotype analysis in patients with giant axonal neuropathy (GAN). Neuromuscul Disord. 2007;17:624‐630. [DOI] [PubMed] [Google Scholar]
- 7. Gambarelli D, Hassoun J, Pellissier JF, Livet MO, Pinsard N, Toga M. Giant axonal neuropathy—involvement of peripheral nerve, myenteric plexus and extra‐neuronal area. Acta Neuropathol. 1977;39:261‐269. [DOI] [PubMed] [Google Scholar]
- 8. Dhanoa BS, Cogliati T, Satish AG, Bruford EA, Friedman JS. Update on the Kelch‐like (KLHL) gene family. Hum Genomics. 2013;7:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Johnson‐Kerner BL, Garcia Diaz A, Ekins S, Wichterle H. Kelch domain of Gigaxonin interacts with intermediate filament proteins affected in Giant axonal neuropathy. PLoS One. 2015;10:e0140157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boizot A, Talmat‐Amar Y, Morrogh D, et al. The instability of the BTB‐KELCH protein Gigaxonin causes Giant axonal neuropathy and constitutes a new penetrant and specific diagnostic test. Acta Neuropathol Commun. 2014;2:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ganay T, Boizot A, Burrer R, Chauvin JP, Bomont P. Sensory‐motor deficits and neurofilament disorganization in gigaxonin‐null mice. Mol Neurodegener. 2011;6:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mahammad S, Prasanna Murthy SN, Didonna A, et al. Giant axonal neuropathy‐associated gigaxonin mutations impair intermediate filament protein degradation. J Clin Invest. 2013;123:1964‐1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lowery J, Kuczmarski ER, Herrmann H, Goldman RD. Intermediate filaments play a pivotal role in regulating cell architecture and function. J Biol Chem. 2015;290:17145‐17153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fuchs E, Cleveland DW. A structural scaffolding of intermediate filaments in health and disease. Science. 1998;279:514‐519. [DOI] [PubMed] [Google Scholar]
- 15. Hol EM, Capetanaki Y. Type III intermediate filaments Desmin, glial fibrillary acidic protein (GFAP), vimentin, and peripherin. Cold Spring Harb Perspect Biol. 2017;9:a021642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bomont P. The dazzling rise of neurofilaments: physiological functions and roles as biomarkers. Curr Opin Cell Biol. 2021;68:181‐191. [DOI] [PubMed] [Google Scholar]
- 17. Yuan A, Rao MV, Veeranna, Nixon RA. Neurofilaments at a glance. J Cell Sci. 2012;125:3257‐3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gyoeva FK, Gelfand VI. Coalignment of vimentin intermediate filaments with microtubules depends on kinesin. Nature. 1991;353:445‐448. [DOI] [PubMed] [Google Scholar]
- 19. Prahlad V, Yoon M, Moir RD, Vale RD, Goldman RD. Rapid movements of vimentin on microtubule tracks: kinesin‐dependent assembly of intermediate filament networks. J Cell Biol. 1998;143:159‐170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hookway C, Ding L, Davidson MW, Rappoport JZ, Danuser G, Gelfand VI. Microtubule‐dependent transport and dynamics of vimentin intermediate filaments. Mol Biol Cell. 2015;26:1675‐1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Navone F, Niclas J, Hom‐Booher N, et al. Cloning and expression of a human kinesin heavy chain gene: interaction of the COOH‐terminal domain with cytoplasmic microtubules in transfected CV‐1 cells. J Cell Biol. 1992;117:1263‐1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xia CH, Roberts EA, Her LS, et al. Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain KIF5A. J Cell Biol. 2003;161:55‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Robert A, Tian P, Adam SA, et al. Kinesin‐dependent transport of keratin filaments: a unified mechanism for intermediate filament transport. FASEB J. 2019;33:388‐399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gan Z, Ding L, Burckhardt CJ, et al. Vimentin intermediate filaments template microtubule networks to enhance persistence in cell polarity and directed migration. Cell Syst. 2016;3:252‐263.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Israeli E, Dryanovski DI, Schumacker PT, et al. Intermediate filament aggregates cause mitochondrial dysmotility and increase energy demands in giant axonal neuropathy. Hum Mol Genet. 2016;25:2143‐2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Robert A, Rossow MJ, Hookway C, Adam SA, Gelfand VI. Vimentin filament precursors exchange subunits in an ATP‐dependent manner. Proc Natl Acad Sci USA. 2015;112:E3505‐E3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schindelin J, Arganda‐Carreras I, Frise E, et al. Fiji: an open‐source platform for biological‐image analysis. Nat Methods. 2012;9:676‐682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Katrukha EA, Jurriens D, Salas Pastene DM, Kapitein LC. Quantitative mapping of dense microtubule arrays in mammalian neurons. eLife. 2021;10:e67925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stirling DR, Swain‐Bowden MJ, Lucas AM, Carpenter AE, Cimini BA, Goodman A. CellProfiler 4: improvements in speed, utility and usability. BMC Bioinform. 2021;22:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Svitkina T. Imaging cytoskeleton components by electron microscopy. Methods Mol Biol. 2009;586:187‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Korobova F, Svitkina T. Arp2/3 complex is important for filopodia formation, growth cone motility, and neuritogenesis in neuronal cells. Mol Biol Cell. 2008;19:1561‐1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yoon M, Moir RD, Prahlad V, Goldman RD. Motile properties of vimentin intermediate filament networks in living cells. J Cell Biol. 1998;143:147‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Prineas JW, Ouvrier RA, Wright RG, Walsh JC, McLeod JG. Gian axonal neuropathy—a generalized disorder of cytoplasmic microfilament formation. J Neuropathol Exp Neurol. 1976;35:458‐470. [DOI] [PubMed] [Google Scholar]
- 34. Opal P, Goldman RD. Explaining intermediate filament accumulation in giant axonal neuropathy. Rare Dis. (Austin, Tex.). 2013;1:e25378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Blikstad I, Lazarides E. Vimentin filaments are assembled from a soluble precursor in avian erythroid cells. J Cell Biol. 1983;96:1803‐1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Soellner P, Quinlan RA, Franke WW. Identification of a distinct soluble subunit of an intermediate filament protein: tetrameric vimentin from living cells. Proc Natl Acad Sci USA. 1985;82:7929‐7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dequen F, Bomont P, Gowing G, Cleveland DW, Julien JP. Modest loss of peripheral axons, muscle atrophy and formation of brain inclusions in mice with targeted deletion of gigaxonin exon 1. J Neurochem. 2008;107:253‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pilling AD, Horiuchi D, Lively CM, Saxton WM. Kinesin‐1 and dynein are the primary Motors for Fast Transport of mitochondria in drosophila motor axons. Mol Biol Cell. 2006;17:2057‐2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tanaka Y, Kanai Y, Okada Y, et al. Targeted disruption of mouse conventional kinesin heavy chain kif5B, results in abnormal perinuclear clustering of mitochondria. Cell. 1998;93:1147‐1158. [DOI] [PubMed] [Google Scholar]
- 40. Lowery J, Jain N, Kuczmarski ER, et al. Abnormal intermediate filament organization alters mitochondrial motility in giant axonal neuropathy fibroblasts. Mol Biol Cell. 2016;27:608‐616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wörn A, Auf der Maur A, Escher D, Honegger A, Barberis A, Plückthun A. Correlation between in vitro stability and in vivo performance of anti‐GCN4 intrabodies as cytoplasmic inhibitors. J Biol Chem. 2000;275:2795‐2803. [DOI] [PubMed] [Google Scholar]
- 42. Pérez‐Ollé R, López‐Toledano MA, Goryunov D, et al. Mutations in the neurofilament light gene linked to Charcot‐Marie‐tooth disease cause defects in transport. J Neurochem. 2005;93:861‐874. [DOI] [PubMed] [Google Scholar]
- 43. Douglas PM, Dillin A. Protein homeostasis and aging in neurodegeneration. J Cell Biol. 2010;190:719‐729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Soto C, Pritzkow S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat Neurosci. 2018;21:1332‐1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Herrmann H, Bär H, Kreplak L, Strelkov SV, Aebi U. Intermediate filaments: from cell architecture to nanomechanics. Nat Rev Mol Cell Biol. 2007;8:562‐573. [DOI] [PubMed] [Google Scholar]
- 46. Nekrasova OE, Mendez MG, Chernoivanenko IS, et al. Vimentin intermediate filaments modulate the motility of mitochondria. Mol Biol Cell. 2011;22:2282‐2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sheng Z‐H, Cai Q. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci. 2012;13:77‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lin N‐H, Huang Y‐S, Opal P, Goldman RD, Messing A, Perng M‐D. The role of gigaxonin in the degradation of the glial‐specific intermediate filament protein GFAP. Mol Biol Cell. 2016;27:3980‐3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Figure S2.
Figure S3.
Figure S4. \
Video S1.
Video S2.
Video S3.
Video S4.
Video S5.
Video S6.
Video S7.
Video S8.
Video S9.
Data S1
Data Availability Statement
The data that support the findings of this study are available in the methods and material of this article.