

Abstract
Influenza is a seasonal respiratory illness that kills hundreds of thousands of people every year. Currently, neuraminidase inhibitors and endonuclease inhibitors are used in antiviral therapy. However, both drug types have encountered drug-resistant influenza strains in the human body. Fortunately, there is currently no resistance to endonuclease inhibitors in wild strains of influenza. We obtained the molecules with endonuclease inhibitor activity independent of the existing drug-resistant strains through computer-aided drug design, and we hope the obtained results can lay a theoretical foundation for the development of high-activity endonucleases. Combining a traditional fragment-based drug discovery approach with AI-directed fragment growth, we selected and designed a compound that achieved antiviral activity on drug-resistant strains by avoiding mutable residues and drug-resistant residues. We predicted the related properties using an ADMET model. Finally, we obtained a compound similar to baloxavir in terms of binding free energy but not affected by baloxavir resistance.
Graphical abstract

Supplementary Information
The online version contains supplementary material available at 10.1007/s11030-023-10659-x.
Keywords: Virtual screening, FBDD, Molecular docking, Fragment growth
Introduction
Due to the influence of the COVID-19 epidemic, the personal protection level of the population is relatively high, and the death toll due to influenza has greatly decreased compared with previous years. But a “retaliatory rebound” can be expected with the end of COVID-19. Influenza is prevented and treated by vaccines and small-molecule drugs. However, the efficacy of vaccines is directly related to the accuracy of virus strain prediction by the national centers for disease control and prevention. For example, in 2017, the average efficacy of the vaccine recommended by the World Health Organization was only 20% [1]. Neuraminidase inhibitors are the main anti-influenza drugs throughout the world. For example, zanamivir, peramivir, and oseltamivir are the antiviral drugs used in China. Influenza viruses that have been pandemic in recent years have shown no resistance to neuraminidase inhibitors, but oseltamivir resistance was demonstrated in influenza in 2008 and 2011 [2–4]. Oseltamivir has been used for 20 years as an oral antiviral drug [5], and peramivir injection [6], the most recently approved treatment for clinical use, has been used for 10 years. Therefore, we need to develop new drugs to prevent and treat influenza.
Influenza virus endonuclease inhibitors are effective anti-influenza inhibitors. Such inhibitors achieve antiviral effects by inhibiting the endonuclease activity of the polymerase acidic (PA) subunit located on viral RNA-dependent RNA polymerase (RdRp) [7]. Among them, baloxavir was recommended for antiviral treatment by the Centers for Disease Control and Prevention in the United States immediately after its release to the market. The main problem with this drug is that drug-resistant strains quickly appear in patients treated with baloxavir (Xofluza), and there is a possibility of severe drug resistance after long-term treatment with Xofluza [8].
Endonuclease inhibitors can affect the replication and transcription of viral RNA, and the conserved region of virus is the region of action. Due to different mechanisms, these drugs work well against strains that are resistant to neuraminidase inhibitors. By occupying the viral splice site, endonuclease inhibitors prevent viral RNA from obtaining pre-mRNA in the host, which prevents the normal replication of the virus. However, the baloxavir clinical data showed that the use of this drug could easily lead to an I38 mutation, resulting in drug resistance [9, 10]. This mutant strain was found in 2.2 and 9.7% of patients in phase II (H1N1pdm99) and phase III (H3N2) trials of the drug, respectively [11]. At present, clinical research has been completed only on Xofluza in this field, and its overall development has been backward. Moreover, most of the activity inhibitors are constantly optimized and improved for the ligand structure, and there is a lack of adaptation research for proteins [12, 13].
In the present study, we reported on a compound based on fragment growth and molecular docking that was active against an Xofluza-resistant strain. We probed the accessible residues in the binding pocket using fragment growth and finally generated the molecular library. With molecular docking, the R groups that could effectively increase the activity of the endonuclease inhibitor were identified. By connecting this part of the structure, compounds that had good activity and were not affected by the existing drug resistance were obtained.
Results and discussion
Binding site analysis
Our primary objective is to design a potent inhibitor that exhibits high activity against the influenza A virus while also possessing the ability to target as many influenza virus subtypes as possible. To accomplish this, we utilized a computational approach to identify the consensus sequence (Supplementary Fig. S1) of the PA protein in influenza viruses, based on the frequency of amino acid occurrence. Subsequently, we generated a homology model of the PA protein using the consensus sequence as a template. The identity of the template protein was 90.40% compared to consensus sequences. Both the missing and differential residues were remote from the binding site. To facilitate our analysis, we divided the entire pocket into five regions based on the reported accessible amino acids of the inhibitors (Fig. 1).
Fig. 1.
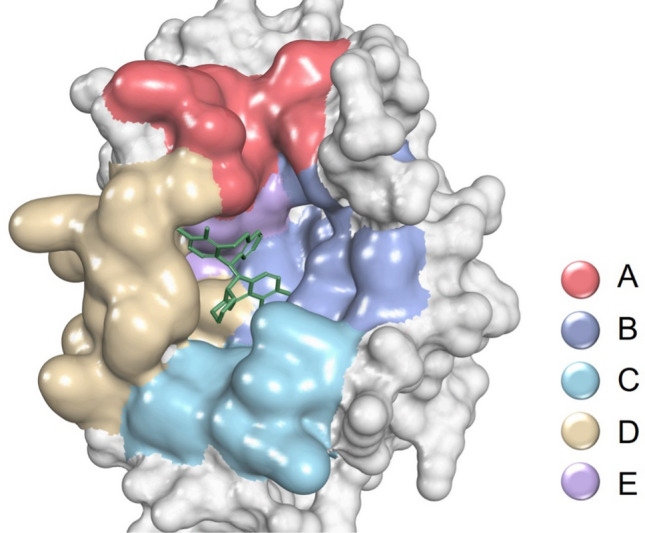
Partition of PA protein binding sites. Surface: protein; molecule: baloxavir acid
Humans are primarily infected with the influenza A, B, and C viruses, while the D influenza virus mainly infects cattle. Considering the substantial disparity in the number of subtype A in the protein sequences analyzed compared to the other subtypes, we amplified the number of subtypes B and C proportionally to better assess the variability of influenza viruses. Following the identification of the variable regions of the protein, we observed that region A exhibited the highest degree of variability, indicating significant differences in this region among different subtypes. This observation suggests that region A may not be suitable for developing broad-spectrum antiviral drugs. In support of this point, we note that the inhibitor developed by Joseph et al. [14], which targets the binding region that predominantly covers the A region, is effective in inhibiting influenza A virus but not influenza B virus. The regions B, C, and D were very conservative. Region E was relatively conservative, and the I38 residue mutation in this region was responsible for the development of resistance to current PA inhibitors (Fig. 2). This study involved research on the three regions (B, C, and D) to avoid the current drug resistance problem and obtain the active lead compound.
Fig. 2.
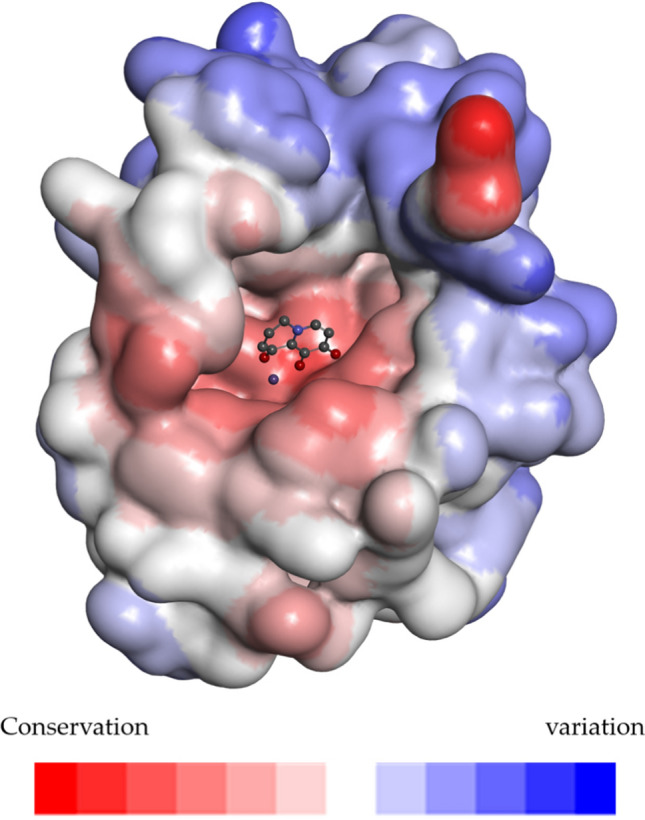
The surface of influenza virus is colored according to variability. Conservation-variation: red-white-blue; stick model: the location of the parent ring
Re-docking verification
At first, to validate the molecular docking protocols, re-docking of the co-crystallized small-molecule inhibitor Baloxavir(B factor = 17.061) into a protein (PDB: 6FS8) was performed [15]. The results showed that the docking pose of molecules was consistent with the crystal pose (RMSD = 0.3946 nm), and Vina could effectively predict the binding conformation of the molecules at this binding site.
Molecular docking
The Single Pharmacophore Fragments Library provided by Enamine Ltd. (KIEV, Ukraine) and the growth fragment library of Deepfrag were docked with the proteins. The docking results (8022 fragments) were used to find accessible residues in the pocket.
To develop novel PA inhibitors, we performed fragment growth on the key structures of the three PA inhibitors. For the accuracy of the results, we optimized the structure of the resulting molecules and docked them back into the protein. The docking results have been evaluated, removing compounds that scored lower than the initial structure score for fragment growth. Finally, three compounds were obtained from the three growth results of the parent rings (Fig. 3).
Fig. 3.
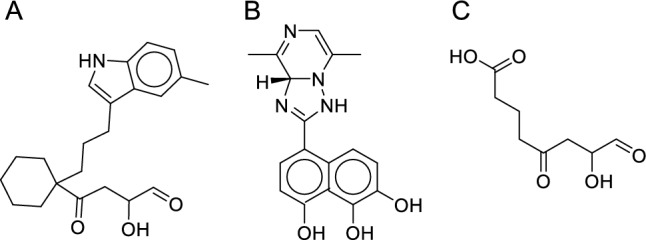
Compound with the highest score among the three key structure
Prediction of accessible residues
The binding of Mn2+ to the multidentate ligand involves four residues: E80, D108, E119, and I120. These residues were avoided in our later design. The docking of the fragment library showed that the residues in the binding sites that could easily interact with 10% of the small fragments effectively included A20, Y24, A37, I38, H41, E80, E119, V122, Y130, and K134.
According to the statistics of the docking results of the growth molecular libraries, the residues that were easy to form and interact with each other included A20, Y24, A37, I38, H41, E80, D108, E119, I120, V122, Y130, and K134 (Table 1).
Table 1.
Analysis of growth molecular library
| Grown parent ring | Residues that interact with more than 2000 molecules | Residues that interact with more than 1000 molecules |
|---|---|---|
| A | Y24, I38, H41 and E119 | A20, A37, E80, D108 and K134 |
| B | A37, H41 and V122 | I38, E119, I120, Y130 and K134 |
| C | V122 and K134 | H41, D108 and E119 |
Molecular design
Parent ring selection
Existing PA inhibitor studies have shown that an effective combination with metal ions is the key to generating drug activity. Increasingly stable binding results in increased drug activity with sufficient specificity. According to the study by Benjamin et al. [16] on the metal binding pharmacophore of a PA inhibitor, we selected structure 1 with the most potential for improvement as the basis. This structure is also included in Xofluza [15]. We made additional improvements to structure 1. And through cyclization, we ensured that the designed compound would not change the conformation of the multidentate structure due to steric hindrance. To find the possible planar conformation, we added N atoms to the ring, and finally, five different parent ring structures were designed (Fig. 4).
Fig. 4.
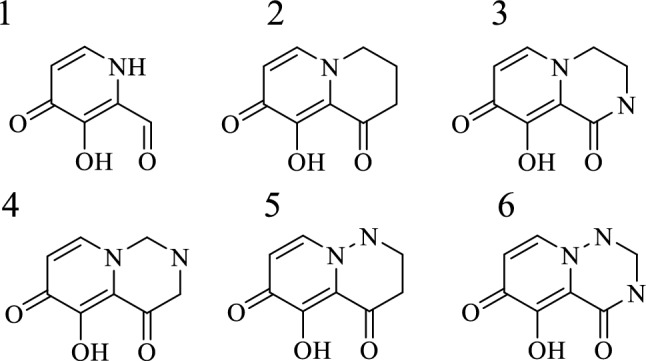
Structural of parent ring. 1: the best measured activity; 2, 3, 4, 5, 6: designed parent ring structure
Region B design
Molecular docking indicated that the accessible residues in region B included H41, V122, and Y130. The primary mode of action of H41 was conjugate stacking, but the parent ring could not continue to claim an appropriate group parallel to H41. V122 was achieved after the parent ring passed more than four carbon atoms, but it did not form a strong hydrophobic interaction. Y130 was usually bound by hydrogen bonds. However, the groups extending from the parent ring had relatively poor hydrogen bond formation angles. Therefore, we did not modify region B to preserve the designability space for the molecule (Fig. 5).
Fig. 5.
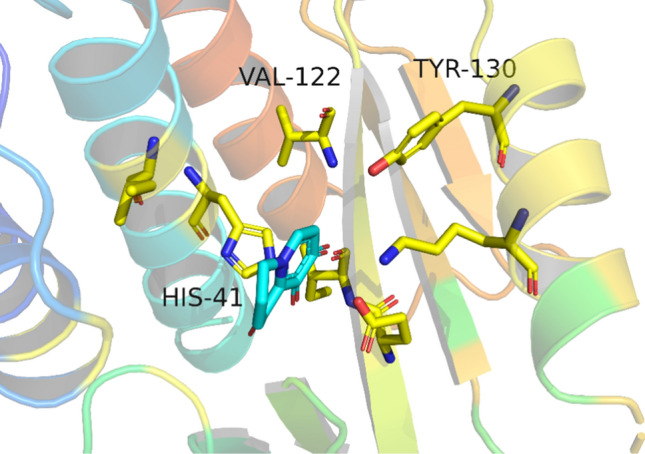
Accessible amino acid in region B. Stick: residue, line: female ring
Region C design
There was an inward hole in region C, which was reflected in the molecular docking results. The results of structural scoring and resolution showed that the molecule binding force could not be significantly improved by extending into the hole. We believed that excessive pursuit of binding sites accessible by large flexible structures was not conducive to the actual development, so this type of structure was not selected (Fig. 6).
Fig. 6.
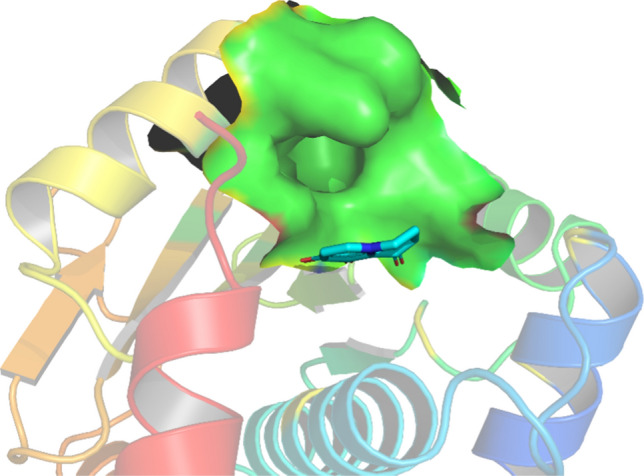
Region C hole location. The protein surface in region C: green
The binding of region C does not often occur in molecular docking. According to the scoring results, we selected the structure with the highest score as the design result for region C (Fig. 7). This structure could have a hydrophobic interaction with F105.
Fig. 7.
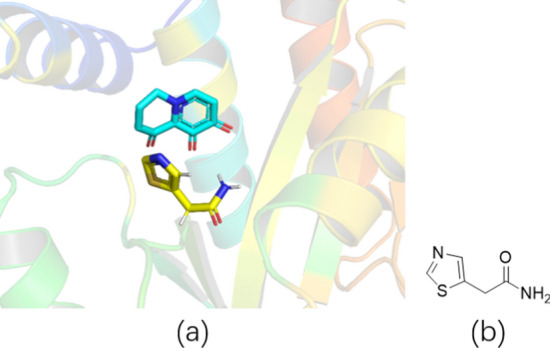
Designed fragment. a 3D structure displays parent ring and design fragment; b 2D display fragment
Region D design
The accessible amino acids in region D included A20, Y24, and E80. E80 was too close to the parent ring, and the extension structure that was not conducive to the design was discarded (see Fig. 8). According to the position–structure relationship between A20 and Y24, we designed two different structures (Fig. 9). Structure 1 was normally conjugated with Y24, and parent ring accessibility was achieved through the cyclopropane structure. Structure 2 was a hydrophobic five-membered ring that was combined by hydrophobic interaction with A20 and Y24.
Fig. 8.
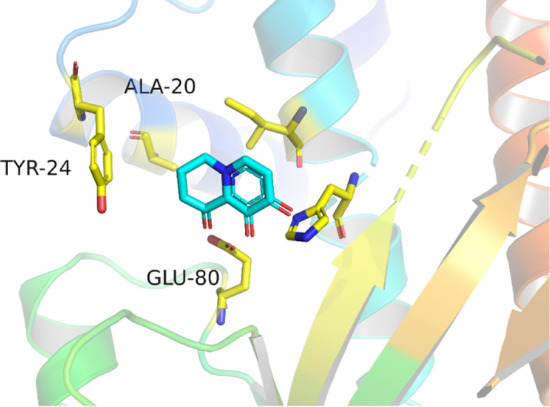
Accessible amino acids in region D. Stick: residue, line: female ring
Fig. 9.
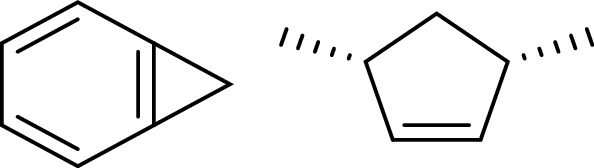
Fragment designed in region D
Combination and screening of molecules
The designed fragment was combined with five parent ring structures, and the intermediate connecting structure was completed by FragRep (Fig. 10).
Fig. 10.
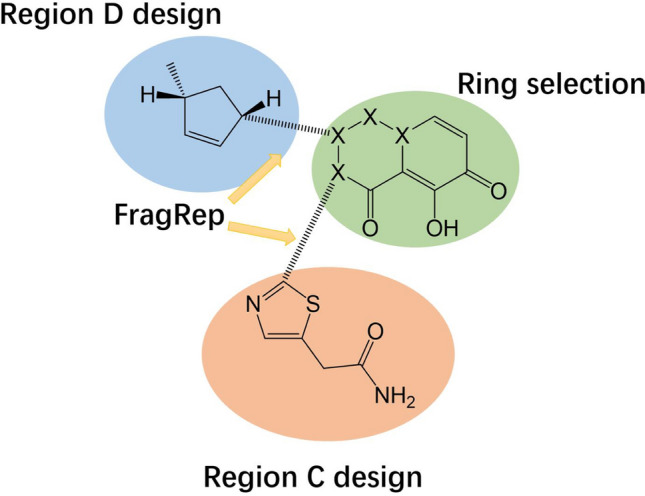
Composition method of compounds
Finally, the molecules were scored by the Vinardo scoring function after energy minimization and re-docked by VINA. The purpose of these operations was to compare the binding energy with that of baloxavir acid. Compound 3 (Fig. 11), with the highest score, was ultimately selected as the design result and had a similar binding energy to baloxavir acid. The synthetic accessibility score (SA) is a crucial metric for predicting a compound's ease of synthesis. In this study, we assessed the SA scores of three compounds: compound 3, Baloxavir acid, and Baloxavir marboxil. We found that compound 3 has an SA score of 38.466, while Baloxavir acid and Baloxavir marboxil have SA scores of 55.358 and 38.543, respectively. Although the SA rating of compound 3 is comparable to that of Baloxavir marboxil, its score is relatively lower in the lead compound. This lower score can be attributed to our simplistic approach to connect different regions, which may have resulted in a lower SA rating for compound 3. To facilitate the subsequent synthesis process, we recommend incorporating easy-to-synthesize groups to connect of different regions for improved synthetic accessibility.
Fig. 11.
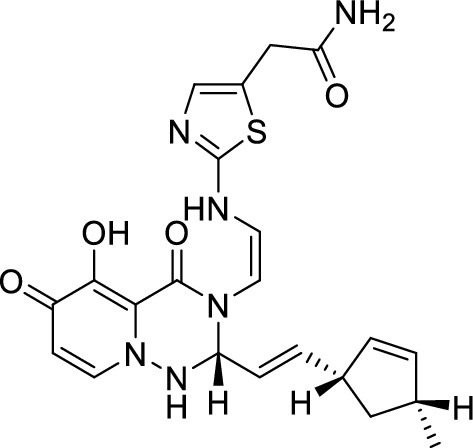
The screened molecule (Compound 3)
In view of the possible existence of different binding configurations of this compound, we verified, using 10 parallel re-docking experiments, that the dominant conformation in the pocket was the design conformation and not another binding conformation. We compared the screened compounds with baloxavir acid, and the results showed that the binding ability of the screened compounds was close to that of baloxavir acid in the two docking scoring methods (Table 2).
Table 2.
Compound 3 was compared with baloxavir acid in the molecular docking model
| Compound | Vinardo scores | re-docking scores |
|---|---|---|
| Compound 3 | – 4.32 kcal/mol | – 9.358 kcal/mol |
| Baloxavir acid | – 4.96 kcal/mol | – 10.393 kcal/mol |
Simulation of molecular dynamics
Molecular dynamics simulations of compound 3 were performed for up to 100 ns to further validate the stability of the protein–ligand complex and search for a more reliable binding position. The RMSD variation between the ligand conformation and the docked conformation was less than 1 Å throughout the MD simulation. After 38 ns of simulation, the ligand conformation converged, and the RMSD fluctuated by less than 0.5 Å (Fig. 12).
Fig. 12.
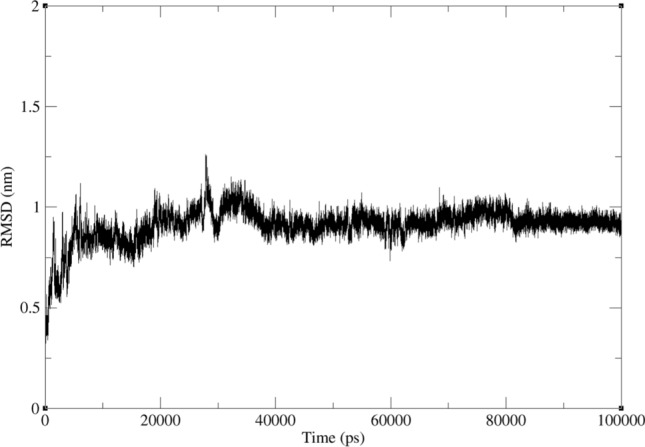
Results of the 100 ns molecular dynamics simulations for compound 3 with protein
Binding free energy was calculated using the last 10 ns of a complex trajectory based on the MMPBSA method. The average free energy of the binding of compound 3 with protein was -24.255 kcal/mol. This indicates that compound 3 effectively binds to protein.
We next assessed its binding ability to drug resistance mutant proteins. A published endonuclease inhibitor and Baloxavir were used as controls. The structure of this compound is 1-(4-(1H-tetrazol-5-yl)phenyl)-2-(((4-chlorophenyl)(methyl)amino)methyl)-5-hydroxypyridin-4(1H)-one, which we call the control group in this manuscript (Fig. 13) [17]. The control group was found to inhibit endonuclease activity with an IC50 of 14 nM in enzymatic assays. It is suitable for comparison with our compound, because it is not affected by the I38T mutation.
Fig. 13.
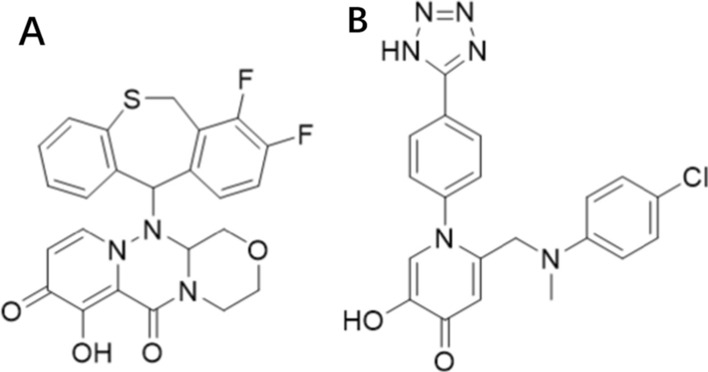
A The structure of Baloxavir. B The structure of control
The homologous model was subjected to an I38T mutation to create a mutant protein model that was stabilized through a 50 ns molecular dynamics simulation. Subsequently, molecular docking was performed to place three inhibitors into the binding pockets of the mutant protein, followed by 100 ns of molecular dynamics simulations to evaluate the binding free energy of each inhibitor. The control group and compound 3 remained embedded in its binding pocket throughout the simulations, while Baloxavir entered another binding conformation at 88 ns (Fig. 14).
Fig. 14.

A The RMSD of control. B The RMSD of Baloxavir. C The RMSD of compound 3
A MMPBSA analysis was performed to ensure the accuracy of the results. The control group and compound 3 had the lowest binding free energies of − 21.537 and − 27.802 kcal/mol, respectively. The binding free energy of Baloxavir was − 13.115 kcal/mol in the first conformation and − 10.273 kcal/mol in the second conformation. According to the results, compound 3 is suitable for further development as a lead compound.
ADMET prediction
The screened molecule was a compound close to the edge of Lipinski's rule. The prediction of physicochemical properties showed that the compound had moderate solubility (LogS = 40.901 μg/mL). It was mainly distributed by passive diffusion (LogD7.4 = 0.386). The predicted oil–water partition coefficient was 1.881, which had good permeability in both phases.
The absorption prediction indicated that this compound was mainly absorbed in the human intestinal tract. Its Papp (caco-2 permeability) was − 6.148 cm/s because the compound was not particularly absorbed well by the human body due to its excessive molecular weight.
The distribution prediction showed that this compound could effectively bind to protein (PPB = 88.28%).
The metabolism prediction indicated that the compound had 74, 50, and 55% inhibition potential for P450 CYP3A4, P450 CYP3A4, and P450 CYP2C9, respectively, with less than 50% inhibition of other P450 enzymes.
The elimination prediction showed that the compound could efficiently remain in the human body with a T1/2 of 1.242 h and a clearance rate of 1.017 mL/min/kg.
Toxicity predictions indicated that this compound had low toxicity, with an LD50 of 1013.301 mg/kg. The probabilities that the compound caused this to occur were 89, 93, 29, and 31% for human hepatotoxicity, drug-induced liver injury, skin sensitization, and Ames mutagenicity, respectively.
The ADMET prediction showed that the compound had good properties and was suitable for the development as a lead compound.
Material and methods
Sequence alignment of influenza virus PA protein
Influenza virus PA protein sequences were retrieved from the UniProt database [18], and we retained only the reviewed sequences and saved the sequences in FASTA format. A total of 103 influenza virus PA sequences were obtained, including 95 influenza A viruses, 5 influenza B viruses, and 3 influenza C viruses. Each sequence was truncated to leave only residues 1–198, which were responsible for endonuclease activity. In order to compare the variability of amino acid sequences in parallel, we performed data processing on the sequences. Specifically, we standardized the data quantity of the three subtypes of ABC to 95, 95, 96 (Only copy, not expand).
The protein sequences were submitted to multiple sequence alignment by CLUSTALX [19]. The online protein variability server (PVS) was used for protein variability analysis [20], with the select consensus sequence as the reference sequence and Shannon entropy analysis as the scoring function [21]. The visualization of sequence alignment was done by ENDscript [13].
Homologous modeling and mutation modeling
The consensus sequence of influenza A virus PA sequence was selected, and the inhibitor-bound (Baloxavir) influenza A virus PA protein (PDB: 6FS6) was used as the template for homology modeling by Discovery Studio 2020 (Accelrys Software Inc., Santiago, USA). For the modeling, we used the parameters as default parameters. We selected the model with the lowest PDF total energy as the result of the homogenous modeling. We first generated the models of the I38T mutant protein by Discovery Studio 2020 (Accelrys Software Inc., Santiago, USA).
The models of the I38T mutant protein were subjected to 50 ns of free molecular dynamics simulation. Resulting from the MD simulation analyses, we performed the molecular dynamics simulation to examine whether the I38T mutation alters the structure of protein and ensures the rationality of amino acids in mutant protein. Models showed stable conformation with an RMSD until the end of the MD production run (Fig. 15).
Fig. 15.
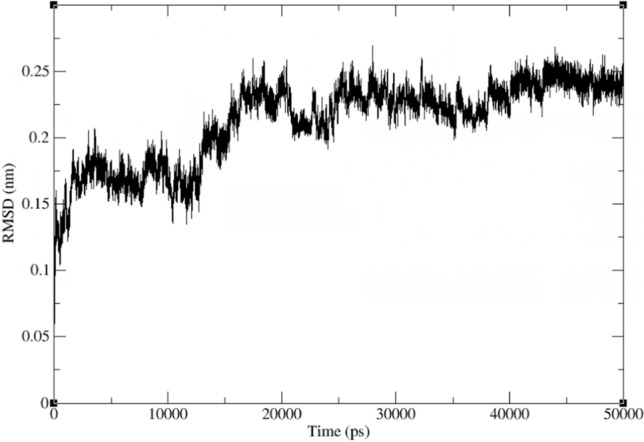
Results of the 50 ns molecular dynamic simulations for the I38T mutant protein
Molecular docking
OpenBabel software was used to convert molecules into pdbqt format, and the energy was minimized using a high-precision MMFF94 force field [22]. The 3D structures that were ultimately generated were used in the molecular docking studies.
Molecular docking was performed using AutoDock Vina 1.2 [23, 24]. The dock pocket was set to a square with x = 19.226, y = 94.398, and z = 44.662 as the center and a side length of 20. The exhaustiveness was set to 32. We selected the other default parameters.
The molecules used for verification were selected from the 10 molecules with different structures reported in the existing single crystals (PDB: 4AVG, 4AWM, 4KIL, 4LN7, 4M4Q, 4YYL, 5W3I, 5W44, 6FS8, and 6YEM) [25–29]. The molecular docking results were analyzed using Discovery Studio software.
Fragment growth
The outward growth of ligands using DeepFrag constituted the screening database [30]. Fragment replacement was conducted using FragRep [31]. Deepfrag is a software developed based on the local receptor environment and the existing ligand scaffold for fragment growth at specific sites. It was used to search for excellent compound structures in the pocket. Fragrep was used to check whether the small molecules generated by Deepfrag had better structures.
According to the reported PA inhibitors, three structures were selected as the parent ring structure for fragment growth (Fig. 16), and the upper limit of growth iteration was 600 molecular weights. The parent ring A was used primarily to establish those accessible amino acids in the C region. The parent rings B and C retained the most active structure of the inhibitor, and we eliminated the heteroatoms on the ring to ensure that the whole binding pocket could be accessed.
Fig. 16.
Parent structure of fragment growth. Growth site: red
The growth point of the parent ring A was designed to be outside the binding pocket and the molecular segments parallel to the metal chelating structure. Because of this, there could be distortion during the growth process. To ensure accurate sampling, molecules with a growth score greater than 0.35 were selected as the molecular library A.
As the most rigid structure, the parent ring B obtained the molecules that interacted with different regions through derivations from five growth points.
The parent ring C only retained the toothed structure of small molecules and metals to determine whether a more appropriate binding posture existed near the metal ions.
Each growth point of parent rings A, B, and C has generated 3816, 498, and 4216 compounds.
Simulation of molecular dynamics
All of the molecular dynamic simulations described in this article are based on a set of unified standards, as outlined below. All MD simulations were performed using the GROMACS package, version 2020.3 [32–34]. The AMBER99SB-ILDN force field was used to optimize proteins [35], and water molecules were modeled with TIP3P [36, 37]. We used ACPYPE to calculate ligand charges and generate topology files for GAFF force [38]. The RESP charge of Mn2+ ions was determined by conducting a single point energy calculation of neighboring amino acids and small molecules using Gaussian 16 and Multiwfn software at the B3LYP-D3 (BJ)/def2-TZVP level [39, 40]. The MD box was set up as a cube, with the distance between each atom and the box exceeding 0.8 nm. SOL water was added to the box at a density of 1000 g/L. Chloride ions were used to replace water molecules in the system to achieve an electrically neutral simulation system. The relaxation system was then simulated using the Verlet algorithm for energy minimization to eliminate the overlap between atoms and avoid problems such as overly close proximity of homogeneous charges [41]. The system was subjected to a 100 ps restricted kinetic simulation at 298.15 K. Lastly, the regular dynamics of the system were simulated using the Verlet algorithm [41]. The integration step was set at 0.002 ps, and simulations were conducted for 100 ns in total. The simulations were performed in an isothermal isobaric regime at 298.15 K and under 1 bar of pressure, with temperature and pressure, respectively, controlled with the V-rescale and Parrinello-Rahman methods [42], and PBC (Periodic Boundary Condition) was enabled. The RMSD (Root Mean Squared Deviation) was calculated for protein–protein and protein–molecule interactions. MD trajectories were viewed using VMD software [43].
The binding free energy between the protein–ligand complexes was estimated using the MM/PBSA equation [44, 45].
ADMET prediction and synthetic accessibility score
We used ADMETlab to perform the ADMET prediction, which is a model of 30 different datasets consisting of 280,000 molecules, and the R2 of each model was greater than 0.95, so the prediction result was statistically significant [46]. The synthetic accessibility scores (SA) were calculated using the Ambit SA module of MolAICal [47, 48], which scores compounds on a scale of 0–100. A higher score indicates that a compound is easier to synthesize, while a lower score indicates that it is more difficult to synthesize.
Conclusion
All the influenza outbreaks in recent years did not consist of neuraminidase inhibitor-resistant strains, but there have been many times in history when there have been neuraminidase inhibitor resistant strains. Although we can treat the current strain with Xofluza, the clinical experience showed that the drug developed resistance after one course of use. This suggests that Xofluza is not suitable for treating seasonal influenza on a large scale over an extended period of time. In this study, we started with the Xofluza resistance mutation and selected amino acid residues with high stability for binding while retaining the core structure of this type of inhibitor. The three-dimensional structure of the protein with the characteristics of a broad-spectrum influenza virus was constructed by consensus sequences, and a fragment structure suitable for development was obtained by combining fragment growth and molecular docking technology based on the existing active metal skeleton and molecular fragment library. After optimization of the combination of fragments and the linking structure, we obtained a leader compound with a similar binding capacity as baloxavir. This compound completely bypassed the I38 mutation, which is the main reason for Xofluza resistance. However, this compound is only computationally comparable to existing active compounds, it demonstrated a lower SA score compared to the active compounds, indicating a potential difficulty in synthesizing the molecule. To improve synthetic accessibility, the introduction of easily synthesized link structures is recommended. Furthermore, additional studies are required to assess the compound's activity through testing.
Supplementary Information
Below is the link to the electronic supplementary material.
Author contributions
YR and LW wrote the main manuscript text. SC completed the article conception and proofreading.
Funding
This research received no external funding.
Data availability
The data presented in this study are available on request from the corresponding author.
Declarations
Conflicts of interest
The authors declare no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Yixin Ren and Li Wan have contributed equally to this work.
References
- 1.Flannery B, Chung JR, Thaker SN, Monto AS, Martin ET, Belongia EA, McLean HQ, Gaglani M, Murthy K, Zimmerman RK, Nowalk MP, Jackson ML, Jackson LA, Foust A, Sessions W, Berman L, Spencer S, Fry AM. Interim estimates of 2016–17 seasonal influenza vaccine effectiveness—United States. MMWR Morb Mortal Wkly Rep. 2017;66(6):167–171. doi: 10.1558/mmwr.mm6606a3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baranovich T, Saito R, Suzuki Y, Zaraket H, Dapat C, Caperig-Dapat I, Oguma T, Shabana II, Saito T, Suzuki H. Emergence of H274Y oseltamivir-resistant A(H1N1) influenza viruses in Japan during the 2008–2009 season. J Clin Virol. 2010;47(1):23–28. doi: 10.1016/j.jcv.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 3.Meijer A, Lackenby A, Hungnes O, Lina B, van der Werf S, Schweiger B, Opp M, Paget J, van de Kassteele J, Hay A, Zambon M, European Influenza Surveillance Scheme Oseltamivir-resistant influenza virus A (H1N1), Europe, 2007–08 season. Emerg Infect Dis. 2009;15(4):552–560. doi: 10.3201/eid1504.181280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Webster D, Li Y, Bastien N, Garceau R, Hatchette TF. Oseltamivir-resistant pandemic H1N1 influenza. CMAJ: Can Med Assoc J. 2011;183(7):E420–422. doi: 10.1503/cmaj.100313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kati WM, Saldivar AS, Mohamadi F, Sham HL, Laver WG, Kohlbrenner WE. GS4071 is a slow-binding inhibitor of influenza neuraminidase from both A and B strains. Biochem Biophys Res Commun. 1998;244(2):408–413. doi: 10.1006/bbrc.1998.8282. [DOI] [PubMed] [Google Scholar]
- 6.Babu YS, Chand P, Bantia S, Kotian P, Dehghani A, El-Kattan Y, Lin TH, Hutchison TL, Elliott AJ, Parker CD, Ananth SL, Horn LL, Laver GW, Montgomery JA. BCX-1812 (RWJ-270201): discovery of a novel, highly potent, orally active, and selective influenza neuraminidase inhibitor through structure-based drug design. J Med Chem. 2000;43(19):3482–3486. doi: 10.1021/jm0002679. [DOI] [PubMed] [Google Scholar]
- 7.Zhou Z, Liu T, Zhang J, Zhan P, Liu X. Influenza A virus polymerase: an attractive target for next-generation anti-influenza therapeutics. Drug Discov Today. 2018;23(3):503–518. doi: 10.1016/j.drudis.2018.01.028. [DOI] [PubMed] [Google Scholar]
- 8.Takashita E, Kawakami C, Morita H, Ogawa R, Fujisaki S, Shirakura M, Miura H, Nakamura K, Kishida N, Kuwahara T, Mitamura K, Abe T, Ichikawa M, Yamazaki M, Watanabe S, Odagiri T, Influenza Virus Surveillance Group of Japan Detection of influenza A(H3N2) viruses exhibiting reduced susceptibility to the novel cap-dependent endonuclease inhibitor baloxavir in Japan, December 2018. Euro Surveill. 2019;24(3):1800698. doi: 10.2807/1560-7917.ES.2019.24.3.1800698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Checkmahomed L, M'Hamdi Z, Carbonneau J, Venable MC, Baz M, Abed Y, Boivin G. Impact of the Baloxavir-resistant polymerase acid I38T substitution on the fitness of contemporary influenza A(H1N1)pdm09 and A(H3N2) strains. J Infect Dis. 2020;221(1):63–70. doi: 10.1093/infdis/jiz418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones JC, Kumar G, Barman S, Najera I, White SW, Webby RJ, Govorkova EA. Identification of the I38T PA substitution as a resistance marker for next-generation influenza virus endonuclease inhibitors. mbio. 2018 doi: 10.1128/mBio.00430-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hayden FG, Sugaya N, Hirotsu N, Lee N, de Jong MD, Hurt AC, Ishida T, Sekino H, Yamada K, Portsmouth S, Kawaguchi K, Shishido T, Arai M, Tsuchiya K, Uehara T, Watanabe A. Baloxavir marboxil for uncomplicated influenza in adults and adolescents. N Engl J Med. 2018;379(10):913–923. doi: 10.1056/NEJMoa1716197. [DOI] [PubMed] [Google Scholar]
- 12.Tomassini J, Selnick H, Davies ME, Armstrong ME, Baldwin J, Bourgeois M, Hastings J, Hazuda D, Lewis J, McClements W, et al. Inhibition of cap (m7G pppXm)-dependent endonuclease of influenza virus by 4-substituted 2,4-dioxobutanoic acid compounds. Antimicrob Agents Chemother. 1994;38(12):2827–2837. doi: 10.1128/aac.38.12.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cianci C, Chung TDY, Meanwell N, Putz H, Hagen M, Colonno RJ, Krystal M. Identification of N-hydroxamic acid and N-hydroxyimide compounds that inhibit the influenza virus polymerase. Antiviral Chem Chemother. 1996;7(6):353–360. doi: 10.1177/095632029600700609. [DOI] [Google Scholar]
- 14.Bauman JD, Patel D, Baker SF, Vijayan RSK, Xiang A, Parhi AK, Martínez-Sobrido L, LaVoie EJ, Das K, Arnold E. Crystallographic fragment screening and structure-based optimization yields a new class of influenza endonuclease inhibitors. ACS Chem Biol. 2013;8(11):2501–2508. doi: 10.1021/cb400400j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Omoto S, Speranzini V, Hashimoto T, Noshi T, Yamaguchi H, Kawai M, Kawaguchi K, Uehara T, Shishido T, Naito A, Cusack S. Characterization of influenza virus variants induced by treatment with the endonuclease inhibitor Baloxavir marboxil. Sci Rep. 2018;8(1):9633. doi: 10.1038/s41598-018-27890-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Credille CV, Dick BL, Morrison CN, Stokes RW, Adamek RN, Wu NC, Wilson IA, Cohen SM. Structure activity relationships in metal-binding pharmacophores for influenza endonuclease. J Med Chem. 2018;61(22):10206–10217. doi: 10.1021/acs.jmedchem.8b01363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Credille CV, Chen Y, Cohen SM. Fragment-based identification of influenza endonuclease inhibitors. J Med Chem. 2016;59(13):6444–6454. doi: 10.1021/acs.jmedchem.6b00628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.The UniProt C. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021;49(D1):D480–D489. doi: 10.1093/nar/gkaa1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics (Oxford, England) 2007;23(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 20.Garcia-Boronat M, Diez-Rivero CM, Reinherz EL, Reche PA. PVS: a web server for protein sequence variability analysis tuned to facilitate conserved epitope discovery. Nucleic Acids Res. 2008;36:W35–W41. doi: 10.1093/nar/gkn211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shannon CE. A mathematical theory of communication. Bell Syst Tech J. 1948;27(3):379–423. doi: 10.1002/j.1538-7305.1948.tb01338.x. [DOI] [Google Scholar]
- 22.O'Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: an open chemical toolbox. J Cheminform. 2011;3(1):33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eberhardt J, Santos-Martins D, Tillack AF, Forli S. AutoDock Vina 1.2.0: new docking methods, expanded force field, and python bindings. J Chem Inform Model. 2021;61(8):3891–3898. doi: 10.1021/acs.jcim.1c00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kowalinski E, Zubieta C, Wolkerstorfer A, Szolar OH, Ruigrok RW, Cusack S. Structural analysis of specific metal chelating inhibitor binding to the endonuclease domain of influenza pH1N1 (2009) polymerase. PLoS pathogens. 2012;8(8):e1002831. doi: 10.1371/journal.ppat.1002831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sagong HY, Parhi A, Bauman JD, Patel D, Vijayan RS, Das K, Arnold E, LaVoie EJ. 3-Hydroxyquinolin-2(1H)-ones as inhibitors of influenza A endonuclease. ACS Med Chem Lett. 2013;4(6):547–550. doi: 10.1021/ml4001112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fudo S, Yamamoto N, Nukaga M, Odagiri T, Tashiro M, Neya S, Hoshino T. Structural and computational study on inhibitory compounds for endonuclease activity of influenza virus polymerase. Bioorg Med Chem. 2015;23(17):5466–5475. doi: 10.1016/j.bmc.2015.07.046. [DOI] [PubMed] [Google Scholar]
- 28.Beylkin D, Kumar G, Zhou W, Park J, Jeevan T, Lagisetti C, Harfoot R, Webby RJ, White SW, Webb TR. Protein-structure assisted optimization of 4,5-dihydroxypyrimidine-6-carboxamide inhibitors of influenza virus endonuclease. Sci Rep. 2017;7(1):17139. doi: 10.1038/s41598-017-17419-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zima V, Radilová K, Kožíšek M, Albiñana CB, Karlukova E, Brynda J, Fanfrlík J, Flieger M, Hodek J, Weber J, Majer P, Konvalinka J, Machara A. Unraveling the anti-influenza effect of flavonoids: Experimental validation of luteolin and its congeners as potent influenza endonuclease inhibitors. Eur J Med Chem. 2020;208:112754. doi: 10.1016/j.ejmech.2020.112754. [DOI] [PubMed] [Google Scholar]
- 30.Green H, Koes DR, Durrant JD. DeepFrag: a deep convolutional neural network for fragment-based lead optimization. Chem Sci. 2021;12(23):8036–8047. doi: 10.1039/d1sc00163a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shan J, Pan X, Wang X, Xiao X, Ji C. FragRep: a web server for structure-based drug design by fragment replacement. J Chem Inf Model. 2020;60(12):5900–5906. doi: 10.1021/acs.jcim.0c00767. [DOI] [PubMed] [Google Scholar]
- 32.Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19–25. doi: 10.1016/J.SOFTX.2015.06.001. [DOI] [Google Scholar]
- 33.Berendsen HJ, van der Spoel D, van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput Phys Commun. 1995;91(1–3):43–56. doi: 10.1016/0010-4655(95)00042-E. [DOI] [Google Scholar]
- 34.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26(16):1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 35.Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78(8):1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu JB, Qiu YQ, Baron R, Molinero V. Coarse-graining of TIP4P/2005, TIP4P-Ew, SPC/E, and TIP3P to monatomic anisotropic water models using relative entropy minimization. J Chem Theory Comput. 2014;10(9):4104–4120. doi: 10.1021/ct500487h. [DOI] [PubMed] [Google Scholar]
- 37.Price DJ, Brooks CL. A modified TIP3P water potential for simulation with Ewald summation. J Chem Phys. 2004;121(20):10096–10103. doi: 10.1063/1.1808117. [DOI] [PubMed] [Google Scholar]
- 38.Sousa da Silva AW, Vranken WF. ACPYPE—AnteChamber PYthon parser interface. BMC Res Notes. 2012;5:367. doi: 10.1186/1756-0500-5-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery Jr. JA, Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ (2016) Gaussian 16 Rev. C.01
- 40.Lu T, Chen F. Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem. 2012;33(5):580–592. doi: 10.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
- 41.Grubmüller H, Heller H, Windemuth A, Schulten K. Generalized verlet algorithm for efficient molecular dynamics simulations with long-range interactions. Mol Simul. 1991;6(1–3):121–142. doi: 10.1080/08927029108022142. [DOI] [Google Scholar]
- 42.Parrinello M, Rahman A. Crystal structure and pair potentials: a molecular-dynamics study. Phys Rev Lett. 1980;45(14):1196. doi: 10.1103/physrevlett.45.1196. [DOI] [Google Scholar]
- 43.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Grapzh. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 44.Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov. 2015;10(5):449–461. doi: 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Valdés-Tresanco MS, Valdés-Tresanco ME, Valiente PA, Moreno E. gmx_MMPBSA: a new tool to perform end-state free energy calculations with GROMACS. J Chem Theory Comput. 2021;17(10):6281–6291. doi: 10.1021/acs.jctc.1c00645. [DOI] [PubMed] [Google Scholar]
- 46.Dong J, Wang NN, Yao ZJ, Zhang L, Cheng Y, Ouyang DF, Lu AP, Cao DS. ADMETlab: a platform for systematic ADMET evaluation based on a comprehensively collected ADMET database. J Cheminform. 2018 doi: 10.1186/s13321-018-0283-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bai Q, Tan S, Xu T, Liu H, Huang J, Yao X. MolAICal: a soft tool for 3D drug design of protein targets by artificial intelligence and classical algorithm. Brief Bioinform. 2021;22(3):161. doi: 10.1093/bib/bbaa161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ertl P, Schuffenhauer A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J Cheminform. 2009;1(1):8. doi: 10.1186/1758-2946-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data presented in this study are available on request from the corresponding author.