

Abstract
Crohn's disease (CD) is an inflammatory bowel disease (IBD) with complex clinical manifestations such as chronic diarrhea, weight loss and hematochezia. Despite the increasing incidence worldwide, cure of CD remains extremely difficult. The rapid development of high-throughput sequencing technology with integrated-omics analyses in recent years has provided a new means for exploring the pathogenesis, mining the biomarkers and designing targeted personalized therapeutics of CD. Host genomics and epigenomics unveil heredity-related mechanisms of susceptible individuals, while microbiome and metabolomics map host-microbe interactions in CD patients. Proteomics shows great potential in searching for promising biomarkers. Nonetheless, single omics technology cannot holistically connect the mechanisms with heterogeneity of pathological behavior in CD. The rise of multi-omics analysis integrates genetic/epigenetic profiles with protein/microbial metabolite functionality, providing new hope for comprehensive and in-depth exploration of CD. Herein, we emphasized the different omics features and applications of CD and discussed the current research and limitations of multi-omics in CD. This review will update and deepen our understanding of CD from integration of broad omics spectra and will provide new evidence for targeted individualized therapeutics.
Keywords: Crohn's disease, Multi-omics, Gut microbiota, Metagenomics, Metabolomics, Proteomics
Graphical Abstract
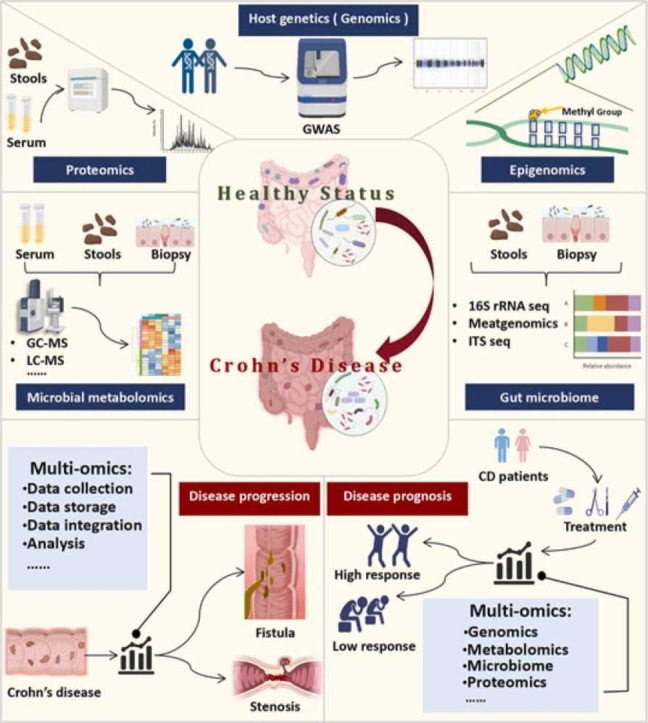
1. Introduction
Crohn's disease (CD) is a kind of chronic inflammatory gastrointestinal disease with a world significant increasing incidence in recent years [1], [2]. The main clinical manifestations include abdominal pain, diarrhea and weight loss. Unlike ulcerative colitis (UC), the lesions of CD can involve the whole digestive tract, characterized by transmural and segmental inflammation, most commonly in the terminal ileum and colon [3]. The clinical diagnosis and cure of CD remain extremely difficult due to the unknown pathogenesis and the complex and variable clinical manifestations, along with a high individual heterogeneity [4]. With the recurrence and progression of the disease, patients with CD are prone to develop many complications such as fistula, intestinal stenosis and perianal abscess. Up to 15 % patients suffer from inevitable surgical treatments with trauma and high postoperative recurrence rate [5]. Consequently, it is of vital importance to define the pathogenesis of CD, fully understand the relationship between possible molecular mechanisms and relevant disease characteristics, and identify biomarkers for prior diagnosis and prognosis as well as new effective therapeutics.
There exists evidence that the pathogenesis of CD is a gut inflammation caused by the combined effects of host genetic factors and gut microbial factors leading to abnormal host immune regulation [4], [6]. The emerging multi-omics analysis based on high-throughput sequencing technology gradually visualized the complex pathogenesis and corresponding disease characteristics of CD. Genome-wide association studies have shown that host genetics has a strong relationship with the pathogenesis of CD [7]. Only 13.1 % of the disease heritability is determined by genetic factors. Many susceptible genes have been identified and some of them are closely related to gut microbiota such as the intracellular pathogen molecular sensor nucleotide binding oligomerization domain 2 (NOD2) and the autophagy gene recombinant autophagy related protein 16 Like Protein 1 (ATG16L1) [8]. Epigenetic factors and intestinal microbial factors that can be affected by diet also play a crucial role in the pathogenesis of CD [9], [10], [11]. Microbial metagenomics and metabolomics suggest that compared with healthy controls, there are lower microbial diversity, different intestinal flora and derivative metabolites in CD patients, defined as dysbiosis such as reduced representation of Bacillota and Bacteroidota [12], [13], [14]. This results in impaired intestinal barrier and activation of inflammatory immune-related pathways, eventually leading to the occurrence of CD. Proteomic techniques help to discover protein biomarkers for phenotype identification and prognosis indication of CD [15], [16].
In this review, we systematically summarize the host and microbial characteristics of CD and their possible mechanisms from a new perspective of multi-omics in CD. Additionally, we enumerate and discuss the possibility and limitations of multi-omics analysis to specify the complex pathogenesis of CD with corresponding signaling pathways and disease phenotype. This review will provide new potential strategies on targeted and individualized treatments of CD through the integration of biomarker-targeted diagnosis, gene therapy and specific microbial or metabolite interventions.
2. Host genomics in CD
Previous studies have provided important information on CD genetic background [17], [18], [19], [20]. Since from the use of non-parametric linkage analysis of IBD patients with gene-positive scanning to find candidate loci in CD patients and accurately identified NOD2 as an important susceptibility gene [21], genome-wide association analysis (GWAS) has brought CD susceptibility genes research to a new era with the development of high-throughput sequencing technology. By detecting the genetic marker polymorphism of multiple individuals in the whole genome, the genotype and the phenotype of CD were statistically analyzed at the population level and the genetic variation most likely to affect the trait was screened according to the statistics or significant p value, thus to explore the susceptibility genes related to the pathogenesis, progression and treatment of CD. The pivotal methodological progress in achieving GWAS is powerfully driven by genome-wide single nucleotide polymorphism (SNP) chip which can classify a relatively limited number of selected polymorphisms to comprehensively analyze common genetic variations across the genome [22].
2.1. Host genomics indicate susceptible genes and pathogenesis of CD
The polymorphisms of substantial genes such as ATG16L1, IL-23R gene and fucosyltransferase 2 gene are identified to be relevant to the susceptibility to CD, attributing the success to the massive meta-analyses of CD genome-wide association studies [23], [24], [25], [26], [27], [28], [29], [30], [31]. Several GWAS studies have demonstrated that SNPs in the immunity-related GTPase family M (IRGM) and the deletion polymorphism of the IRGM promoter region are closely related to CD. Extensive researches have shown that SiRNA-mediated IRGM knockout significantly reduces the proportion of bacteria in autophagosome including CD-associated adhesive invasive Escherichia coli [32], [33], while over-expression of IRGM can lead to plasmid dose-dependent increase of autophagy response [34]. Using a mouse model of CD, Subhash Mehto et al. demonstrated that human IRGM and its mouse homologue Irgm1 controlled inflammation by inhibiting the activation of NLRP3 inflammasome, revealing the direct mechanism by which IRGM plays a protective role in CD [32], [35].
A GWAS study in the Korean population identified three new susceptibility loci suggesting that ATG16L2 and FCHSD2 may be new susceptibility genes for CD in addition to the previously reported four susceptibility loci including TNFSF15 and IL23R [36]. Cross-ancestor association study of IBD, including immunochip genotype data or genome-wide data from a cohort of European individuals and non-European individuals observed that previously reported European CD risk variation in ATG16L1 did not show evidence of association in East Asians [3], [37]. Beyond the leading susceptibility genes, a considerable amount of literature especially the GWAS meta-analysis has been published to expand our knowledge of new candidate genes such as the NCF4 locus [38], [39], [40], [41], [42]. GWAS meta-analysis of CD conducted by Franke A et al. found 30 new susceptibility loci (P < 5 ×10−8) in accordance with genome-wide significance including SMAD3, IL2RA, DNMT3A, ERAP2, TYK2, IL10, FUT2, BACH2, DENND1B, and TAGAP [43]. Based on the GWAS data, further attempts have been made to speculate that ATG16L1 and NOD2 risk variants may be related to autophagy pathways. By promoting imbalance of gut microbiota, these immune mechanisms lead to impaired mucosal barrier function and further participate in the pathogenesis of CD in a cell specific and functional specific manner [22], [44], [45], [46], [47]. With the deep look into genomics, investigators found that the mucosal inflammation observed in CD patients with NOD2 mutation might result from the increased transport of IgA-pathogen complex from lumen to PP immune cells through M cells, suggesting that the disease susceptibility of CD plays a pathogenic role through changing the signal interaction between intestinal microbiota and mucosal innate immunity [48].
2.2. Genomics indicate disease progression and prognosis of CD
Previous research has established that the variations of CLCA2, MAGI1, LY75 loci and 2q24.1 were associated with disease location, complex stenosis course, erythema nodosum, and mild course respectively [49]. Additionally, host genomics have also been applied to identifying individuals at higher risk of drug resistance to guide the clinical individualized therapeutic of CD. Retrospective studies have shown that drugs supported by GWAS evidence are more likely to receive clinical approval [50]. A large GWAS study included 1240 patients with CD monitored for anti-drug antibodies to reflect the patient 's immunogenicity found that HLA-DQA1 * 05 allele had a genome-wide significant correlation with the immunogenicity of anti-tumor necrosis factor therapy [51]. Lee JC et al. indicated that some SNPs in four risk genes XACT, MHC, FOXO3, IGFBP1/IGFBP3 were associated with the prognosis of CD, distinguished from those driving disease susceptibility, which provided new possibilities for CD treatment [52]. Retrospective study conducted by O 'Donnell S et al. focused on information of disease progression. It was reported that eight known IBD susceptibility SNPs were associated with abdominal surgery time, but none of the variations reached genome-wide significance. This is an attempt to apply GWAS to examining the relationship between inheritance and disease progression, suggesting that the impact of genetic factors in CD progression may be relatively small [53]. Moreover, cross-disease genome-wide association studies can also reveal pleiotropic genetic variations between CD and other immune-mediated diseases such as multiple sclerosis and systemic sclerosis [54], [55]. For instance, STAT3 has been confirmed as shared locus between systemic sclerosis and CD by relevant meta-analysis [54], [56], [57], which generates new insight into dealing with complicated case.
2.3. Limitations and future potential of genomics in CD
Most located in non-coding regions, GWAS trait-related SNPs can affect multiple genes over long distances, thus more potential candidate genes are usually only identified as low risk. Linkage disequilibrium (LD), meanwhile, leads to a complex and challenging identification of functional SNPs as well as specific pathways that mediate their action [58], [59]. As a consequence, host genomics alone cannot thoroughly explain the variation and phenotype of complicated genetic diseases leading that genetic assessment of CD hasn't been confirmed in clinical practice so far [60], [61], [62]. Computer biotech researchers still need to continually develop the complex and reliable algorithms to identify risk mutations and pathogenic genes [59], [63]. The latest research published on Nature Genetics developed a systematic and comprehensive analysis pipeline to prioritize causal genes for each published GWAS trait-related locus through fine mapping and co-localization analysis of systemic diseases and disease molecular characteristics [59]. Watanabe K et al. developed an interaction map to provide enrichment results based on gene, pathway and tissue, thereby promoting functional annotation, gene prioritization and interactive visualization of GWAS results [64]. Recently investigators from Massachusetts Institute of Technology and Harvard University demonstrated that large-scale exome sequencing can complement GWAS by pinpointing specific genes indirectly associated with GWAS, as well as genes that have not been observed in GWAS to fill in the low-frequency and rare variants of CD genetic profile [65]. These developments will provide a statistically convincing causal variation explanation for the results of GWAS which will contribute greater potential to the prediction, diagnosis and treatment of CD in the future. It is worth noting that the rise of genome-wide research joint with microbiome and metabolome also brings new insights into the pathogenesis and specific characterizations of CD.
3. Epigenomics in CD
Host genomics still have inevitable limitations while the rise of epigenomics in recent years seems to represent another dimension of host-environment interaction in the pathogenesis of CD. Epigenetic changes usually refer to heritable changes in gene structure such as histone modification, DNA methylation, and non-coding RNA, while the host gene sequence remains unchanged [66]. Previous research has established that epithelial DNA methylation is associated with inflammation and microbiota composition [67], [68] and microRNAs (miRNAs) can affect IBD by interfering with T cell differentiation, TH17 signaling pathway and autophagy-related pathways. Using transposase-accessible chromatin sequencing analysis with differentially expressed genes (DEGs) in CD-associated CD4+ lymphocytes, Gonzalez MM et al. demonstrated that loss of Treg identity disrupted the epigenome through genetic or transient BMI1 depletion and transformed Tregs into Th1/Th17-like pro-inflammatory cells, a transformation in CD4+ T cells associated with human CD [69]. Epigenome-wide association studies (EWAS) make it possible to identify differential epigenetic modification sites at the genomic level, which is conducive to revealing relevant disease characteristics. A latest meta-analysis summarized the epigenomics methylation studies of IBD peripheral blood up to February 2022, involving 177 CD patients and 243 healthy individuals. It was found that compared with healthy controls, FKBP5, BCL3, NLCR5, TRAF1, CDC42BPB, BAHCC1, LYN, TOLLIP and KCNAB2 were the most significantly different methylation sites in CD patients [70]. Other meta-analyses covering multiple epigenomic studies were also carried out to find possible IBD biomarkers such as blood miR-21 and miR-192 [71], [72], which might be promising in therapeutics. Serena C et al. used a methylation array (Illumina EPIC/850k array) to perform EWAS and gene expression analysis of peripheral blood mononuclear cells and human adipose-derived stem cells (hASCs) isolated from subcutaneous adipose tissue of CD patients and healthy controls. They found that hASCs in CD patients showed distinctive profiles of gene expression and DNA methylation, which helps to reveal the complex pathophysiological function of hASCs in CD [73]. These studies suggest that epigenomics may have important effects on the interaction between microbial and genetic factors involved in the development and progression of CD.
4. Gut microbiome in CD
Gut microbiota includes more than 10 billion microorganisms, whose genes are 100 times more than those in the host genome [74]. Symbiotic beneficial interactions between the microbiota and the host have received considerable critical attention, including nutrient metabolism, resistance to pathogens, and in particular, ensuring normal structure and function of the mucosal immune system [75]. The gut microbiome of IBD patients including CD differs significantly from that of healthy individuals, characterized by reduced abundance of Bifidobacterium, Bacillota and Fecalibacterium species, along with a decreased aggregate diversity [76]. Reduced Parabacteroides was reported in patients with familial CD [77]. It is previously well established that germ-free (GF) mice are less susceptible to colitis while fecal microbiota transplantation (FMT) from IBD patients to GF mice can reproduce the disease. A randomized controlled trial of FMT in ileocolic CD patients showed that patients receiving FMT had a decrease in disease severity index evaluated by endoscope after six weeks (P = 0.03), which strongly proves that gut microbiota is at the heart of our understanding of CD [74], [78], [79]. Intestinal inflammatory infiltration in CD includes Th1 and Th17 cells, whose response to bacteria or fungi is linked to the pathogenesis of the disease [3]. Moreover, creeping fat has been considered an extra-intestinal manifestation of CD, partially responsible for stenosis. The passage of viable bacteria or their products such as lipopolysaccharide and outer membrane vesicles from the intestine across the mucosa to mesenteric lymph nodes (MLN) is defined as bacterial translocation (BT) [80]. Recent studies have suggested that transplanting the microbiota from mesenteric fat of CD patients to IL10-/- mice can cause colitis, indicating that gut microbiota may play a fundamental role in the pathogenesis of fibrostenosis in CD [81]. Significant developments in DNA sequencing technology and analysis lay the foundation for CD microbiome research [82]. To date 16 S ribosomal RNA (16 S rRNA) sequencing and metagenomic sequencing are two of the most widely used high-throughput microbiome sequencing technologies. Polymerase chain reaction (PCR) amplification is performed on specific hypervariable regions of the DNA sequence encoding 16 S rRNA in prokaryotes to study the composition and the diversity of miacrobial community [83], [84]. Nevertheless, due to the inadequate coverage of non-full-length variable region sequences, species level cannot be identified while metagenomics can annotate at the species level and analyze microbial genes, functions, and metabolic pathways by performing high-throughput sequencing of whole-genome DNA from samples [85]. These new microbiome technologies can help reveal the microbial differences between healthy people and CD patients and clarify the links between the gut microbiome and complex pathological behaviors, providing new insights and evidence in pathogenesis, effective diagnosis, prediction of disease progression and prognosis, as well as individualized treatments of CD.
4.1. Alteration of microbiome characteristics in CD
A large cohort study covering sequence data from four European countries established by Pascal, V. et al. performed 16 S rRNA sequencing on more than 2000 fecal samples to analyze their microbiome characteristics. It was found that the gut dysbiosis of CD patients was significantly more severe with decreased diversity and unstable microbial community comparing to that of UC patients. Decreased abundance of Anaerostipes, Faecalibacterium, Methanobrevibacter, an unknown Peptostreptococcaceae, Collinsella and an unknown Christensenellaceae with increased abundance of Escherichia and Fusobacterium were identified to be unique to CD [12]. Other investigators carried out similar consequence that despite decreased abundance of most gut bacteria in CD patients, the abundance of Ruminococcus gnavus, Shigella spp. and Echerichia spp. increased conversely [86]. Existing research shows that children with CD are significantly different from adults in disease leisions [75]. Gevers D et al. conducted a large cohort study to explore the gut microbiome characteristics of children with newly diagnosed CD. Based on the 16 S rRNA sequencing of gut microbiome, MaAsLin pipeline was used to identify microbes that were statistically significantly associated with disease phenotype. Results turned out that CD was positively correlated with the abundance of Neisseriaceae, Pasteurellaceae, Veillonellaceae, and Fusobacteriaceae. Compared with the control group, several genera such as Ruminococcus, Bacteroides, Roseburia, Faecalibacterium, Blautia, Coprococcus and a number of taxa within the families of Lachnospiraceae and Oscillospiraceae were demonstrated to be negatively correlated with CD [87]. Due to the wide heterogeneity of microbiome analysis methods or experimental designs, meta-analysis of changes in gut microbiome helps to master comprehensive and critical information of CD [88]. Recent meta-analyses indicate that the abundance of beneficial microorganisms such as Lachnospiraceae, Ruminococcacae and F. prausnitzii are reduced, whereas some pathogens such as Fusobacterium, Streptococcus, Enterococcus, Blautia, Flavonifractor and Veilonella are increased in CD [88], [89], [90]. As the most consistent and particularly related genus in CD, Fusobacterium is identified as one of the most valuable biological diagnostic marker for CD [90]. Most studies to date have tended to focus on fecal microbiome rather than the microbiota that attach to the intestinal mucosa, but there is evidence that the mucosal microbiome is closer to immune cells which may cause a more robust immune response in gut [75]. A noteworthy phenomenon was reported through microbiome analysis that only the content of Bacteriaceae, Fusobacteriaceae and Neisseriaceae in the fecal microbiome decreased, while most of the microbial inflammatory markers significantly related to the disease suggested by the intestinal mucosal microbiome were lost in the fecal microbiome, indicating that the intestinal mucosal microbiome plays a key role in the pathogenesis and diagnosis of CD, distinguished from the fecal microbiome [87]. A systematic review indicated that for CD, stool samples might be more consistent than biopsy samples for associating taxa with disease [91].
Characterization of gut microbial flora is no longer content with changes at phylum level and genus level with the continuous development of metagenomics technology and a growing number of specific strains are identified. Furthermore, researches on intestinal flora tend to focus not only on the composition but also on functional changes, providing more in-depth insights into the pathogenesis and treatment of Crohn's disease. The strain abundance of Roseburia intestinalis, an acetate butyrate transformant present in the intestinal mucus layer, has been reported to decrease in stool samples from patients with Crohn's disease [92]. It is reported that Enterobacteriaceae and in particular certain strains of adherent-invasive E. coli (AIEC) have been associated with the ileal mucosa of patients with Crohn's disease, which can enter the lamina propria and be engulfed by macrophages. These functions make it possible to replicate continuously with secretion of high levels of TNF, leading to intestinal inflammation [93], [94]. On genetic levels, biosynthetic gene clusters (BGCs) are getting more and more attention. BGCs are the genes encoding secondary metabolites (SMs), a cluster of bacterium-originated chemical compounds with bioactivities and diverse structures. As the bridge between microbiome and host, BGCs have proven to be associated with diseases [95], [96]. Jaeyun Sung et al. utilized TaxiBGC on gut microbiome samples from three published case-control studies and found 5 BGCs elevated in CD patients, which encoded for gassericin E, gassericin S, gassericin T, acticin Q and ruminococcin A, respectively [95].
Role of intestinal fungal disorders in the pathogenesis of CD is increasingly concerned apart from bacteria. Several studies have confirmed that there are iconic anti- Saccharomyces cerevisiae antibodies (ASCA) in the serum of patients with CD. The depletion of CX3XR1 gene in mouse macrophages, which is related to the significant reduction of antifungal antibodies in CD patients, can aggravate intestinal diseases after fungal colonization [97]. A study of Japanese IBD patients analyzed the fecal microbiome using 16 S rRNA sequencing and ITS sequencing and found a higher abundance of Candida in CD patients than that in UC patients or healthy individuals [14]. Five fungal communities were found to be positively associated with CD including Cyberlindnera jadinii, Saccharomyces cerevisiae, Kluyveromyces marxianus, Clavispora lusitaniae, and Candida albicans. Among them, Clavispora lusitaniae was the most important fungi contributing to the disease prediction model [98].
4.2. Microbiome identifies distinctive status of CD
The clinical phenotype of CD is complex with an extremely high heterogeneity of disease progression as well as terrible complications. Microbiome provides new basis for predicting disease progression and distinguishing complex phenotype. Gut microbiome analysis of patients with active CD revealed a significant increase in Enterobacteriaceae, Klebsiella, Pseudomonadota and Fusobacterium, etc. together with a decrease in Clostridia cluster IV of anaerobic bacteria compared to patients with inactive CD [99]. A systematic review focusing on the application of microbiome in IBD also confirmed a significant decrease in Bifidobacterium species in fecal samples during active CD compared to remission [91]. A prospective cohort study of 913 children with CD who had no complications within 90 days of diagnosis, published on Lancet, collected ileal and rectal stool samples for 16 S rRNA sequencing to analyze their gut microbiome. In addition to the aforementioned CD-related genera, Campylobacter, Akkermansia, Collinsella and Desulfovibrio species were also significantly associated with CD in children. Meanwhile, the study found that Ruminococcus and Rothia were associated with stenosis complications of CD. In penetrating CD, the abundance of Collinsella increased significantly whereas the abundance of Veillonella increased specifically in the ileum [100]. Microbiome technologies also set up new concern about the microbial mechanisms of creeping fat expansion in CD. Ha CWY et al. collected mesenteric creeping fat from affected sites in CD patients with complications requiring surgical treatment and performed deep shotgun metagenomic sequencing. It was found that compared with the microbiome in mesenteric fat of the uninvolved intestinal segment and mucosa, the relative abundance of Erysipelotrichaceae in the creeping fat of the affected intestinal segment increased with a lower total diversity. By comparing the viable bacteria isolated from the mesenteric fat of UC patients and healthy controls, a subset of five bacteria unique to creeping fat in CD was identified including Clostridium symbiosum, Parabacteroides distasonis, Erysipelocridium ramosum, Clostridium innocuum, and Bifidobacterium pseudolongum [101]. Consistent with the results, Clostridium innocuum prefers lipid-derived metabolic substrates and has been shown to be a potentially invasive pathogen with cytotoxicity that can cause severe colitis [102].
4.3. Microbiome influences therapeutic outcomes and prognosis of CD
Gut microbiome characterization of CD correlated with response to multiple therapeutics [103]. A latest systematic review has shown a sustained increased abundance of short-chain fatty acid producing bacteria Blautia in fecal samples from CD patients treated with infliximab [89]. A comparison of gut microbiome analysis between responsive and unresponsive CD patients to anti-TNF-α therapy found increase in Bifidobacterium, Roseburia, Collinsella, Lachnospiraceae, Eggerthella taxa and decrease in Phascolarctobacterium [77]. Bifidobacterium is a well-studied probiotic that can metabolize oligosaccharides to resist IBD. Lachnospiraceae, Oscillospiraceae and Roseburia can produce short-chain fatty acids (SCFAs), a class of metabolites that have been widely shown to regulate intestinal immunity to produce anti-inflammatory effects. For example, butyric acid can inhibit lipopolysaccharide-induced nuclear factor kappa B activity and increase mucin and antimicrobial peptide production [75]. Disorders in the association network involving Lachnospiraceae and Oscillospiraceae have been reported to be associated with frequent recurrence of Crohn's disease and poor response to against TNF-α treatment [77]. Owing to complications such as stenosis, obstruction and fistula accompanied with CD, more than 50 % of cases require surgery within 20 years after diagnosis. Surgical treatment has become a common method to alleviate CD [75]. Decrease in Parabacteroides and Clostridiales along with increase in Enterobacteriaceae in the gut microbiome of post-operative CD patients have been reported, which almost inevitably lead to a relapse if left untreated [77]. 16 S rRNA sequencing revealed increased abundance of Alistipes and decreased abundance of Actinomycetota and Bifidobacterium spp. in the gut microbiome of CD patients with endoscopic disease remission 1 year after surgery compared to patients with disease relapse [86].
Additionally, dietary regulation is considered to be a relatively safe and noninvasive treatment for CD without causing immunosuppression, primarily by altering the gut microbiota and mucosal integrity of CD patients. The application of high-throughput sequencing revealed the characteristics of gut microbiome changes in different dietary therapies and provides a reference for its effectiveness. Shotgun metagenomic sequencing in a single-blind clinical controlled trial of dietary intervention in IBD patients found that compared with Sham diet, CD patients received diet low in fermentable oligosaccharides, disaccharides, monosaccharides, and polyols (FODMAP) showed significant differences in gut microbiome including increased abundance of Bacteroides xylanisolvens, Enterocloster citroniae and decreased abundance of Anaerobutyricum hallii and Faecalibacterium prausnitzii. The study demonstrated that a low FODMAP 4-week diet was safe and effective for the treatment of persistent intestinal symptoms in patients with stationary IBD by comprehensive disease assessment and other data analysis [104]. These are effective attempts to provide possible evidence for appropriate dietary treatment of CD through microbiome.
4.4. Limitations and future potential of microbiome in CD
Although microbiome analysis in CD has become a very active field with the development of high-throughput sequencing technologies, the results have not yet been translated into clinical practice, as most strategies to control the microbiome (probiotics or antibiotics) have failed [3]. Microbiome technology still needs to be improved in many aspects, such as the improvement of analysis pipeline and the development of analysis software [85], [105], [106]. Recent studies have reported that metagenomic next-generation-sequencing (mNGS) can improve the ratio of microbial readings and the ability to target and identify sample microorganisms [107]. Furthermore, this realistic result is more likely to be attributed to the fact that microbiome is influenced by many factors such as host genetics and diet, etc., among which there are indivisible interaction networks that affect host immunity and cause specific pathological behaviors and disease characteristics. Just as host genetics alone cannot explain CD, microbiome only provides the superficial microbiological characteristics of CD.
5. Microbial metabolomics in CD
As the intermediates of host-microbiota interplay, metabolites reflect the bacterial function directly, hence the combination of microbiome and metabolomics allows for a deeper understanding of disease process and the function of a specific metabolite, aiming in the detecting of new therapeutic targets [108]. Numerous metabolites like amino acids and SCFAs influence the gut wellbeing by various ways like affecting the gut barrier and regulating the immune system [109], [110], [111], [112], [113]. Up-down-regulation of metabolite levels can directly reflect the effects of various pathophysiological stimuli or even genetic modifications on human body under specific environments [114]. Metabolomics refers to the quantitatively and qualitatively analysis of small molecular metabolites in biological samples [115]. The commonly used analytical techniques for metabolomics include liquid chromatography-mass spectrometry (LC-MS), gas chromatography-mass spectrometry (GC-MS) and nuclear magnetic resonance (NMR) spectroscopy [109]. With regard to their application, GC-MS is frequently used for the analysis of the fatty acids, amino acids and other small molecular weight, volatile non-polar substances, whereas LC-MS is widely applicable and sensitive, which is mostly used in clinical examination and non-targeted metabolomics. Although NMR is less sensitive than MS, the relatively simple preparation and rapid sample detection as well as non-destructive property [116] make NMR a popular platform for metabolomics. According to diverse research purpose and methods, metabolomics can be divided into targeted or untargeted metabolomics. Targeted metabolomics focus on specific metabolites while untargeted metabolomics detect the dynamic changes of small molecule metabolites in samples without bias and screen differential metabolites through bio-informatics analysis. Several studies have been conducted to investigate the changes and fluctuation of multiple metabolites from urine, feces and serum samples of CD patients compared with the control group. In this part, we will briefly summary present metabonomics research of fatty acids, amino acids and secondary bile acids to trace and assess the significance and application potential of microbial metabolism in CD.
5.1. Fatty acids
Fatty acids make up a great part of gut microbial metabolites and consist of SCFAs (<6 carbons), medium-chain (7–12 carbons, MCFAs) and long-chain fatty acids (more than 12 carbons, LCFAs) [108]. Among them, SCFAs mainly including acetate, butyrate and propionate play an important part in gut hemostasis through various ways, positively correlated with the gut wellbeing [113]. As a kind of typical intermediate of host-microbiota interplay, SCFAs protect the gut barrier integrity through accelerating mucin and IgA production and facilitate to the expression of tight junction proteins [112], [113]. Apart from the effects on the gut barrier, SCFAs also participate in regulating immune system through alleviating the pro-inflammatory factors production, regulating the differentiation and function of Treg cells and promoting the chemotaxis of neutrophils [113].
Gut inflammation leads to the disturbance of microbiota-host interplay, manifested as the alteration of the microbiome and metabolome, hence it is possible that the level of metabolites may reflect the disease state in turn. Efforts into the confirmation of this concept promoted the rapid advancement of metabolomics. Researches have already been carried out to validate the diagnostic function of fatty acids using numerous metabolomics methods, where different levels of fatty acids were found in people with different status. For instance, De Preter V et al. identified MCFAs as one of differentiating metabolites in CD patients by detecting decreased levels of butyrate, pentanoate, hexanoate and heptanoate in CD, which indicated the potential diagnostic function of MCFAs [117]. Likewise, LCFAs including arachidic and oleic acid, etc and MCFAs such as sebacic acid and isocaproic acid were found to be decreased in IBD status [118], [119]. A recent meta-analysis indicated a decrease in acetate, butyrate, and valerate levels in patients with CD compared to healthy controls (HC). However, there was no significant difference in propionic acid between CD and HC. Due to the limited data of all included studies, subgroup analysis cannot be performed to explore the SCFA changes in different disease stages of CD [120]. Distinction of metabolites helps to diagnosis between diseases with similar phenotype. Butyrate, decreased in IBD patients, changed more significantly in CD compared to UC patients, serving as a candidate marker to distinguish between CD and UC [111], [121]. Treatments may alter the level of metabolites through making a difference to gut microbiota, providing a potential to evaluate the prognosis in a noninvasive way. Increased level of butyrate after probiotic oligofructose-enriched inulin intervention pointed out the potential role of butyrate in evaluating disease prognosis [117]. Combining the curative effect with levels of metabolites at baseline proposed assumption about selecting markers predictive of treatment effects. Based on this, fatty acids were found in association with CD exclusion diet plus partial enteral nutrition (CDED+PEN) and exclusive enteral nutrition (EEN) induced remission [122].
5.2. Secondary bile acids
Bile acids are important components of bile that contribute to lipid digestion and absorption [110]. Previous studies have shown evidence that primary bile acids are converted to secondary bile acids including deoxycholic acid, lithocholic acid, and compounds they bind to glycine or taurine in intestine lumen, suggesting gut microbial metabolism plays a crucial role in determining bile salt intestinal pools [110]. Several attempts have been made to suggest a critical role for secondary bile acids in intestinal inflammation. Metabolomics research indicated that lithocholic acid salt,deoxycholate glycolate, taurocholate and chenodeoxycholate were significantly reduced in feces of CD patients compared to healthy individuals [111], [123]. It's well established that conversion of primary bile acids to secondary bile acids is processed via deconjugation catalyzed by bile salt hydrolase (BSH) [124]. BSH producers such as Bifidobacterium and Clostridium are less abundant in CD patients, and infliximab can inhibit inflammation by enriching BSH producers in the treatment of CD [111]. Similarly, a prospective cohort study showed that the concentrations of fecal secondary bile acids were significantly increased in CD patients who received anti-TNF treatment [125]. Metabolomics suggested higher deoxycholic acid levels in CD patients who were more likely to respond to anti-TNF therapy while CD patients who showed low response had higher circulating unconjugated bile acid levels consistent with the higher levels of conjugated bile acids in urine and feces [126]. Overall, secondary bile acids may serve as efficacy biomarkers in CD patients treated with infliximab and anti-TNF. In accordance with those results, investigators have found that specific derivatives of lithocholic acid (LCA) such as 3-oxoLCA and isoalloLCA can respectively inhibit the differentiation of Th17 cells along with promotion of RORγ-expressing T regs to play an anti-inflammatory role [127], [128]. This concept has recently been challenged by studies demonstrating that increased deoxycholic acid causes Paneth cell dysfunction by over-activating the farnesoid X receptor (FXR) and type I interferon (IFN) signals in intestinal epithelial cells, which contributes to the pathogenesis of CD [129]. Investigators have already drawn attention to the paradox in bile acids and proved the variable role in the regulation of intestinal barrier homeostasis [130]. These controversial researches may rise from intricate physicochemical properties, different immune mechanisms and unclear interactions with gut microbes.
5.3. Amino acids
Gut homeostasis is correlated with amino acids from two opposite aspects. Amino acids can protect the gut barrier through promoting mucosal healing and serving as energy substrates and precursors of multifarious bio-active metabolites, thus leading to the alleviation of IBD. Nonetheless, some specific amino acids have negative correlation with gut homeostasis through the biosynthesis of some potentially harmful gut metabolites, contributing to the development of IBD [126], [131], [132]. Offering the snapshot of the metabolic states, metabolomics technologies are extensively used to assess the morbid state. Previous works shed light on the potential function of amino acids as instructors for clinical applications such as clinical assessment and treatment, the development of which relays largely on metabolomics.
The level of amino acids may relate to IBD states to some extent, as Balasubramanian K et al. demonstrated previously [133]. Combined with the assessment of CD treatment using clinical and objective markers, patterns of metabolome profiles at different times from the baseline were detected to find out the specific amino acids in association with the post-treatment response of CD. This paved the way for the search for metabolic biomarkers predictive of the curative effect. Consistent with the finding that amino acid profiles were different between pediatric CD patients with or without remission after EEN [134], numerous amino acid biomarkers in variety of CD therapies such as biotherapy, surgery and nutritional therapy were reported in previous works. These include serum histidine and urine cysteine (for anti-TNF response) [126], serum tryptophan (for biotherapy or surgery) [135], serum L-promote acid (for pediatric IFX therapy) [111] and fecal kynurenine (for CDED+PEN and EEN) [122]. IBD influences the metabolism of gut microbes, leading to levels of specific amino acids captured by metabolomics change with IBD progression, which makes it possible to find metabolic biomarkers for disease status identification. Various findings coincident with this idea already validated numerous biomarkers for diagnosis, such as valine along with isoleucine for early CD diagnosis, a panel of amino acids including leucine and histidine for IBD diagnosis and glycine for the active IBD & IBD in remission differentiation [136], [137].
As stated above, amino acids have dual effects on the gut homeostasis, thus showing contradictory effects on CD. Although the likelihood of amino acids supplement to treat IBD has been confirmed in previous work [131], [135], there exists controversial discovery showing that dietary amino acids supplementation exacerbated colitis in DSS induced mice colitis model [134]. These conflicting discoveries enlightened us on the research for amino acids function of alleviation or exacerbation, for the sake of accurate treatment. More efforts need to be put in finding more targeted amino acids for nutritional therapy, following the discovery of Nikolaus S et al. which showed the beneficial effect of tryptophan to alleviate colitis [135].
As the bridge between microbes and the host, metabolites are strongly related with the function of bacteria. It is of great significance to analyze the microbiota and metabolites comprehensively to understand the ongoing of diseases [111], [134], [135].
5.4. Proteomics in CD
Proteomics, picturing the landscape of entire proteins of the whole organism [138], provides the insight into their interplay, abundance, amounts and modifications on a comprehensive and dynamic level [139], [140], thus facilitating a deep understanding of the progression of a specific disease. As the result of interactions between genes and environmental factors, proteome can imply changes of proteins under disturbance, offering the direct information of life status. Proteomic analysis favours the discovery of candidate biomarkers, exploration of disease pathogenesis and development of targeted medicine [141].
A large quantity of proteins alters in CD [142], [143], making it possible to apply proteomics to seek out biomarkers of this disease. Fields in demand of comprehensive use of biomarkers encompass disease prediction, differential diagnosis, therapeutic evaluation and forecast of prognosis [144]. The combination of multiple biomarkers, especially the anti-Saccharomyces cerevisiae antibodies (ASCA)-IgA combination, can be used to sort out patients in preclinical phase of disease in IBD, contributing to the prediction and prevention of CD [145]. Furthermore, biomarkers identified through proteomics can also be utilized ulteriorly to predict the relapse risk for confirmed patients, other than predicting the risk of CD occurrence for undiagnosed subpopulation [146], [147]. Changing in proteome with states makes sense to conjecture the course of disease using proteomics [148]. In line with this view, Lehmann, T et al. regarded human sucrose-isomaltase as CD specific biomarker and transcriptional regulatory protein RprY from Bacteroides fragilis as microbial candidate biomarkers for some gastrointestinal diseases [149]. Likewise, a set of serum proteome was validated for distinguishing CD from primary intestinal lymphoma and intestinal tuberculosis [150], [151]; AKR1C3 was traced for potential differentiation of UC and CD [152]. Panels of blood protein markers were identified as biomarkers indicating the complications risk in pediatric CD patients, among which ECM1 acted as the indicator of fibrostenotic [153], [154]. Šimurina M et al. collected 3441 plasma samples from two independent cohorts (1265 CD patients, 1309 UC patients, and 867 control subjects) to detect IgG Fc-glycosylation (tryptic glycopeptides) levels by LC-MS. Subsequently, a meta-analysis of the obtained results was conducted and found that IgG galactosylation levels in patients with IBD were lower than those in the control group. Decreased galactosylation was associated with more severe IBD, indicating the potential role of IgGFc-glycosylation as a diagnostic and prognostic tool. In addition, the fucosylation of IgG increased in CD patients while decreased in UC patients compared to HC, indicating that the fucosylation level of IgG could be used to distinguish CD and UC155]. Coupled with other methods, proteomics isextensively involved in the illustrations of mechanisms hidden in a biological function. Proteomics is employed to find the differentially expressed proteins (DEPs) for the seeking of the upstream or downstream molecule, so as to find associated pathways. In extracellular vesicles (EVs), host defense proteins especially the reactive oxidant-producing enzymes were found present and elevated, indicating the mechanism of mucosal inflammation may lie in acceleration intestinal oxidative stress of proteins [156]. Sun, Xue-Liang et al. confirmed fibrinogen-like protein 1 could induce gut inflammation in CD by activating NF-κB signalling pathway [139]. Other investigators identified the role that AGR2 and epithelia ER stress may play in the process of CD fibrotic strictures [157]. In terms of practical application, the illustration of mechanism paves path for the selection of medicine target. Proteomics is often applied to identify the DEPs during treatment, providing potential targets such as anemoside B4 and elafin [158], [159].
Despite all these strengths above, limitations remain. Acting in networks makes proteins vast in scale and complicated in function, adding difficulty in data processing. Proteome profiles are not always consistent when established through different methods or in different laboratories. Metaproteome can change with sampling position, with disparities existing among metaproteome of serum, colon tissues, fecal materials and even formalin-fixed and paraffin-embedded tissue specimens [143].
6. Multi-omics integration launches a new era of CD research
Multi-omics integration that advanced the identification as well as linkage of key features in CD has taken CD research by storm. Through integrating and analyzing the collected data of different omics molecular characteristics, multi-omics provides information from the original etiological mechanism to the functional outcome of the disease, thus to build an important basis for finding targeted therapeutic sites for diseases [160], [161]. Some significant findings have been achieved in the systematic study of Crohn 's disease with the exploratory application of multi-omics techniques. Ling-Jie Huang et al. integrated single cell sequencing technology, transcriptome sequencing analysis, and metabolomics sequencing to reveal that selenium can mediate cell reactive oxygen species clearance through selenoprotein W, and inhibit the differentiation of Th1 cells, thus to inhibit the pathogenesis of CD [162]. This systematic discovery of biological mechanism depends on the efficient and reliable immune-metabolites pathway research model formed under multi-omics technology. Compared with single analysis, the integrated analysis of microbiome and metabolite profiles in CD patients who received autologous hematopoietic stem cell transplantation showed a more significant separation effect on disease activity. By integrating and summarizing the interactions between OTU and metabolites, the original key role of sulfur metabolism in linking disease activity to human microbiome was revealed [163]. In addition to the disease mechanism, large-scale multi-omics studies of Crohn 's disease have enabled researchers to more accurately describe the characteristics of the location variation. By combining metaproteomics, shotgun metagenomics, 16 S rRNA sequencing, metabolomic profiling, and host genetics, great molecular and microbial features associated with CD locations were revealed, which helped to generate disease severity biomarkers at specific locations. The performance of these biomarkers even exceed Calprotectin [164], which means multi-omics have great prospects in the clinical classification and diagnosis of Crohn 's disease.
It is well established that almost all human cells have the same genome but their expression profiles are tissue specific. Development of epigenomics, metatranscriptomics and metaproteomics combined to provide critical supplement and extension for genomics and visualize the links between pathogenesis and disease features from gene post-transcriptional regulation to functional proteins [165], [166]. On the basis of transcript abundance, transcriptional Risk Scores (TRS) integrate GWAS and expression quantitative trait locus (eQTL) results to measure individual risk of CD suggested 29 genes with the strongest co-evidence of association signals and demonstrating high confidence in distinguishing disease status and progression [167]. Intestinal epithelial cells (IECs) in children with CD were analyzed for genome-wide DNA methylation patterns and transcriptomes. Results turned out that compared with control group, the DNA methylation and transcription patterns of ileum and colon epithelium in CD patients significantly changed, which proposed that IECs represented promising potential in CD [168]. Due to the pivotal role of gut microbiota in the pathogenesis of CD, microbiome has been widely used in multi-omics studies of CD. Through comprehensive analysis of microbiome from CD patients with epigenomic data, Ryan FJ et al. identified a large number of CpG sites that are differentially methylated in clusters defined by the microbiome and demonstrated in vitro that pro-inflammatory bacteria are associated with immune-related epigenetic markers [68]. The Integrative Human Microbiome Project (HMP2 or iHMP) has greatly deepened our understanding of host-microbiome interactions. A multi-omics IHMP study published in Nature in 2019 collected blood, colonic biopsy, and fecal specimens from 132 subjects for one year to generate a comprehensive longitudinal molecular map of host and microbiome during IBD [169]. This study constructed a large-scale cross-measurement association network, including significant correlation analysis of 10 aspects of host-microbiome interactions and found interesting associations of multi-level host-microbiome interactions. By analyzing the ranscripts covaried with the microbiome, the authors found that indirect microbial modulators DUOX2 and its maturation factor DUOXA2 were negatively correlated with the abundance of Oscillospiraceae UCG 005 (OTU 89) in the ileum. The abundance of Agathobacter rectalis (OTU 120) in ileum was negatively correlated with the expression of antimicrobial CXCL6 and chemokine CCL20, so was the abundance of Streptococcus (OTU 37) and Eikenella (OTU 39) in rectum. A recent host-centered study has identified microbiome-derived protein clusters with high sequence identity to human protein-protein interactions (PPIs) by collecting existing interspecies PPIs data [170]. By combining PPIs with metagenome analysis, host functional annotations were added to microbiome-related diseases such as IBD, so as to provide a more reliable reference for the study of disease mechanism and promising therapeutic molecular targets. Undoubtedly, these high-throughput host-microbiome interaction networks still need to be continuously improved in the future. The main limitation of single metanolomics analysis lies in distinguishing whether the metabolites are host- or microbe-derived,which is compensated by the integration of microbiome and metabolome. With the development of multi-omics technology, growing metabolomics and microbiome research are closely integrated, providing a qualitative leap for revealing the mechanism of disease. Common variations between species and metabolites are captured, leading fresh insight on specific metabolism pathway such as sulfur metabolism. Multi-omics in microbiome and metabolome showed operational taxonomic units (OTUs) corresponding to Desulfovibeio and Escherichia/Shigella were enriched in active CD, while OTUs corresponding to Bacteroides, Parabacteroides, Bilophila, Acidamicococcus and Odoribacter were enriched in inactive CD. Targeted and non-targeted metabolomics analysis of mice receiving fecal transplantation from patients was consistent with what was observed in CD patients. Sulfur metabolism disorders, aurine binding bile acids and glycocholic acids were associated with the development of inflammation in humanized mice, manifested as a relatively high number of sulfated compounds, including bile acids, polyphenols and biogenic amines [163]. Recently attention has been paid to mesenteric adipose tissue (mAT) in CD from the prospective of multi-omics. Four families of Alcaligenaceae, Brucellaceae, Pseudomonadaceae and Hyphomicrobiaceae in mAT showed significant covariation with the host transcriptome and metabolome, abundance of which had positive correlation with endoscopic recurrence rate [81]. This provides help for people to deeply understand the pathological mechanism of mesenteric crawling fat in CD. Despite the proliferation of microbiome and metabolome studies, methods for integrating microbiome and metabolome data remain imperfect. Most studies usually focused on the superficial statistical association of two types of data rather than the complex interaction between the microbial genome and its corresponding metabolic functions, thus could not really systematically integrate these data [171], [172]. In 2016, researchers have tried to construct an analytical framework that can link species abundance to metabolite changes [173]. In short, this framework infers the metagenomic quantity of each sample based on microbial composition and available or inferred genome information. Metagenomic Universal Single-Copy Correction (MUSiCC) was then used to normalize the inferred metagenomes [174], followed by translating the enzymatic gene abundance estimates into community-based metabolite potential (CMP) scores through Predicted Reactive Metabolic Turnover (PRMT) approach. Finally, the consistency between the predicted changes in CMP score and the measured changes in metabolite was evaluated. This kind of computational framework can predict the changes of metabolites to a certain extent through the changes of microbial community composition and metabolic capacity of various member species, which provides precondition for the emergence of 'predictive metabolomics'. Predictive metabolomics can break through the obstacles that metabolomics data are expensive and difficult to obtain on a large scale. Using microbial sequencing data to quickly and easily predict microbial metabolite markers, predictive metabolomics provide deeper insights into the pathogenesis of diseases and the possibility for microbial intervention treatment [175]. However, this model is hard to apply or verify in a data-driven manner or cannot be well extended to complex communities with partially referenced taxa or metabolites [176]. Due to the complexity and instability of the intestinal microbial environment, the prediction accuracy of similar models applied to IBD data is greatly reduced [173], [177], [178]. In order to improve the accuracy of metabolite prediction in complex environments, researchers developed Model-based Genomically Informed High-dimensional Predictor of Community Metabolic Profiles (MelonnPan), an optimized computational framework to predict metabolomics from microbial community profiles. This model was validated by applying melonpan to two independent intestinal metagenomic datasets, including 200 participants with CD, UC and healthy controls. The results showed that up to 50 % of the trend of intestinal metabolites can be successfully predicted by this framework [176], thus laying a foundation for the multi-omics comprehensive study of CD based on model. In order to address the poorly understood pathogenesis that how isolated CD phenotype influences molecular profiles, Gonzalez CG et al. integrated host genetics, microbiome, metabolomics and metaproteomics to analyze the location characteristics of CD patients [164]. They demonstrated that primary and secondary bile acid levels in ileal CD increased with Faecalibacterium prausnitzii and other species preferred to acid-rich environment compared to colonic CD. Meanwhile, metaproteomics indicated enrichments of neutrophil-related protein in colonic CD, whereas host genetics didn't contribute to the distinction [164]. Last but not least, multi-omics generated unique profile for therapeutic including diet therapy and anti- cytokine treatment in CD associated with remission [179], [180]. These profiles recognized by multi-omics may facilitate a prior determination of optimal and targeted therapeutic for patients with CD. Table 1, Table 2.
Table 1.
Multi-omics researches on patients with Crohn's disease.
| Publications | Involved omics | Samples & Subjects | Methods | Major valuable omics signatures |
|---|---|---|---|---|
| Jacobs, Jonathan P et al. 2022 [184] | Microbiome | MLI samples from 176 CD and 219 non-IBD | 16 S rRNA Seq |
|
| Metabolomics | MLI samples from 176 CD and 219 non-IBD | Ultra-performance LC-MS untargeted metabolomics | ||
| Genomics | Blood samples from 75 CD and 97 non-IBD | GRS | ||
| Gonzalez, Carlos G et al. 2022 [164] | Proteomics | 10 patients with any strictures or penetrating wounds and 9 without | Offline basic pH reverse-phase liquid chromatography and LC-MS2/MS3 analysis |
|
| 103 CD, 60 UC and 19 healthy controls | Offline basic pH reverse-phase liquid chromatography and LC-MS2/MS3 analysis | |||
| Microbiome | 103 CD, 60 UC and 19 healthy controls | 16 S rRNA Seq and shotgun metagenomics | ||
| Metabolomics | 103 CD, 60 UC and 19 healthy controls | Untargeted MS data, Shotgun LC-MS/MS and in-line HPLC fractionation | ||
| Genomics | 126 IBD | SNP arrays | ||
| He, Zhen et al. 2021 [81] | Microbiome | Mesenteric fat samples from 48 CD and 16 non-CD | 16 S rRNA Seq |
|
| Metabolomics | Mesenteric fat samples from 48 CD and 16 non-CD | Metabolic analysis | ||
| Transcriptomics | Mesenteric tissue from 46 CD and 15 non-CD | Host RNA-Seq | ||
| Lee, Jonathan Wei Jie et al. 2021 [180] | Microbiome | 114 baseline fecal samples from 108 CD and 77 UC | Metagenomic sequencing |
|
| Metabolomics | Baseline blood samples | LC-MS | ||
| Proteomics | Baseline blood samples | Multiplex PEA technology | ||
| Wang, Yizhong et al. 2021 [111] | Microbiome | Fecal samples from 20 healthy children and 29 pediatric CD patients | 16 S rRNA Seq |
|
| Fecal samples from baseline and post-treatment CD patients | 16 S rRNA Seq | |||
| Fecal samples from 20 healthy children and 29 pediatric CD patients | ITS2 gene sequencing | |||
| Fecal samples from baseline and post-treatment CD patients | ITS2 gene sequencing | |||
| Metabolomics | Fecal samples from 20 healthy children and 29 pediatric CD patients | Targeted metabolomics analysis | ||
| Fecal samples from baseline and post-treatment CD patients | Targeted metabolomics analysis | |||
| Berlinberg, Adam J et al. 2021 [185] | Microbiome | Featal samples from 21 axSpA, 27 CD, 12 CD-axSpA and 24 HC | Shotgun metagenomics sequencing |
|
| Metabolomics | 4 non-inflamed pinch biopsies of the distal colon | LC-MS | ||
| Kakuta, Yoichi et al. 2020 [186] | Genomics | Genotyping data of 713 Japanese CD patients and 2063 controls | GWAS |
|
| TEM cells located in the LPMCs from 20 IBD patients (15 CD and 5 UC) | eQTL analyses | |||
| Transcriptomics | TEM cells located in the LPMCs from 20 IBD patients (15 CD and 5 UC) | RNA-Seq | ||
| eQTL from Genotype Tissue Expression project | TWAS | |||
| Ryan, F J et al. 2020 [68] | Microbiome | Matched inflamed and non-inflamed colonic mucosa from 50 CD, 80 UC and 31 healthy people | 16 S rRNA Seq |
|
| Epigenomics | Biopsies from 72 paired CD. Matching host transcriptome data from 71 samples |
Genome-wide DNA methylation analysis | ||
| Transcriptomics | Matching host transcriptome data from 71 samples | Polyadenylated capture and RNA-Seq approach | ||
| Metwaly, Amira et al. 2020 [163] | Microbiome | Post-HSCT fecal samples from 35 active CD and 83 inactive disease | 16 S rRNA Seq |
|
| Mice colonized with microbiota from active or inactive CD patients | Shotgun metagenomics sequencing | |||
| Metabolomics | Post-HSCT fecal samples from 18 active CD and 36 inactive disease | RP-UHPLC and HILIC-UHPLC | ||
| CD patients and respective humanized mice | Untargeted UPLC/TOF-MS metabolomics | |||
| CD patients and respective humanized mice | Targeted metabolomic approach | |||
| Suskind, David L et al. 2020 [179] | Microbiome | Fecal samples taken at enrollment (baseline) and at the end of the treatment course (week 12) from 5 patients | 16 S rRNA and metagenomic shotgun sequencing |
|
| Proteomics | Fecal samples from 5 patients (3 whole food diets and 2 modified SCD) were obtained prior to adopting the diet (baseline) and at 2 and 12 weeks afterwards | HPLC-MS | ||
| Metabolomics | Fecal samples from 5 patients (3 whole food diets and 2 modified SCD) were obtained prior to adopting the diet (baseline) and at 2 and 12 weeks afterwards | GC-MS | ||
| Connors, Jessica et al. 2020 [187] | Microbiome | 11 priBA-dominant and 44 secBA-dominant samples | 16 S rRNA and metagenomic shotgun sequencing |
|
| Metabolomics | 57 fecal (13 Remissio, 4 NR, 7 SR and 6 NSR) samples from 17 pediatric CD | LC-MS,MS | ||
| Brückner, Annecarin et al. 2020 [188] | Metabolomics | Plasma samples from pediatric patients (22 PEN and 19 non-PEN) | LC-MS |
|
| Microbiome | Fecal samples from pediatric patients (22 PEN and 19 non-PEN) | 16 S rRNA Seq | ||
| Borren, Nienke Z et al. 2020 [189] | Proteomics | Serum samples | Multiplex proximity extension assay technologies |
|
| Metabolomics | Serum samples | LC-MS | ||
| Microbiome | 85 Fecal samples (12 relapse and 73 remission) | Shotgun metagenomic sequencing | ||
| Franzosa, Eric A et al. 2019 [190] | Metabolomics | Cross-sectional fecal samples from discovery (88 CD, 53 UC and 34 controls) and validation (20 CD, 23 UC and 22 control) | Untargeted LC-MS metabolomic profiling |
|
| Microbiome | Cross-sectional fecal samples from discovery (88 CD, 53 UC and 34 controls) and validation (20 CD, 23 UC and 22 control) | Taxonomic profiling of subjects’ gut microbiomes | ||
| Lloyd-Price, Jason et al. 2019 [123] | Metabolomics | Fecal samples from 132 IBD | LC–MS |
|
| Proteomics | Fecal samples from 132 IBD | LC–MS/MS | ||
| Transcriptomics | 252 biopsies (baseline) of 132 IBD | Host RNA-seq | ||
| Microbiome | 178 biopsies (baseline) of 132 IBD | 16 S rRNA Seq | ||
| Howell, Kate Joanne et al. 2018 [168] | Epigenomics | 7 healthy children and 5 pediatric CD patients | Genome-wide DNA methylation patterns analysis |
|
| Ileal and colonic biopsies from IBD patients both at diagnosis and at a later stage in their disease (14 CD and 9 UC) | Genome-wide DNA methylation patterns analysis | |||
| Control, CD-, and UC-derived datasets for each gut segment | Genome-wide DNA methylation patterns analysis | |||
| 170 samples of mucosal biopsies collected from different gut segments of IBD and control | Genome-wide DNA methylation patterns analysis | |||
| Transcriptomics | 170 samples of mucosal biopsies collected from different gut segments of IBD and control | Gene expression profile analysis | ||
| Control, CD-, and UC-derived datasets for each gut segment | Gene expression profile analysis | |||
| Genomics | Disease-specific DMRs, DEGs, and rDMRs within genomic IBD risk loci | Epithelial cell-derived molecular signatures | ||
| Microbiome | Adjacent microbiota isolated from biopsies | 16 S rRNA Seq |
AC, ascending colon; axSpA, axial spondyloarthritis; BAs, bile acids; CCD, colonic CD; CD, Crohn 's disease; CD8A, CD8 alpha chain; DEGs, differentially expressed genes; DMPs, differentially methylated positions; DMRs, differentially methylated regions; DNAm, DNA methylation; ECM, extracellular matrix; eQTL, expression quantitative trait locus; GC-MS, gas chromatography–mass spectrometry; GDNF, glial cell line–derived neurotrophic factor; GRS, genetic risk score; GWAS, genome-wide association study; HC, healthy controls; HILIC-UHPLC, hydrophilic interaction liquid chromatography UHPLC; HPLC, high performance liquid chromatography; IAA, indole-3-acetate; IBD, inflammatory bowel disease; ICD, ileal CD; IECs, intestinal epithelial cells; IFX, infliximab; ITS2, internal transcribed spacer 2; I3Ald, indole-3-acetaldehyde; LC-MS, liquid chromatography tandem mass spectrometry; LC-MS-MS, liquid chromatography tandem mass spectrometry; LPMCs, lamina propria mononuclear cells; mAT, mesenteric adipose tissue; MDS, multidimensional scaling; MLI, mucosal luminal interface; MS, mass spectrometry; MSCD, modified SCD; NR, non-remission; NSR, non-sustained remission; PC, phosphatidylcholines; PCA, principal component analysis; PCaa, diacyl phosphatidylcholines; PCDAI, pediatric Crohn’s disease activity index; PEA, proximity extension assays; PEN, partial enteral nutrition; PL, phospholipids; PLC, propionyl-L-carnitine; post-HSCT, post hematopoietic stem cell transplantation; priBA-dominant, primary bile acid dominant; RARRES3, retinoic acid receptor responder 3; rDMRs, regulatory DMRs; RNA-Seq, RNA sequencing; RP-UHPLC, reversed phase UHPLC; RSVs, Ribosomal sequence variants; SC, sigmoid colon; SCD, specific carbohydrate diet; SCFAs, short-chain fatty acids; secBA-dominant, secondary bile acid dominant; SNP, single-nucleotide polymorphism; SR, sustained remission; TEM, effector memory T cells; TOF-MS, time of flight MS; TWAS, transcriptome wide association study; UC, ulcerative colitis; UHPLC, ultra high performance liquid chromatography; UPLC, ultra performance liquid chromatography; WF, whole food; 16 S rRNA Seq, 16 S ribosomal RNA sequencing.
Table 2.
Brief summary of the most common pipelines in omics.
| Omics | Common pipelines | Advantages | Disadvantages |
|---|---|---|---|
| Host-genomics | Genome-wide association analysis (GWAS) |
|
|
| Epigenomics | Epigenome-wide association studies (EWAS) | ||
| Microbiome | 16 S rRNA gene sequence (16 S rDNA sequence) | ||
| Shotgun metagenomics | |||
| Long-read metagenomics | |||
| Metabolomics | Nuclear magnetic resonance (NMR) | ||
| Gas chromatography-mass spectrometry (GC-MS) |
|
||
| Liquid chromatography-mass spectrometry (LC-MS) | |||
| Proteomics | Data-independent acquisition (DIA)/ Sequential windowed acquisition of all theoretical fragment ions (SWATH) |
|
|
| Label-free quantification (LFQ) |
CD, Crohn’s disease; DIA, data-independent acquisition; EWAS, epigenome-wide association studies; GWAS, genome-wide association analysis; GC-MS, gas chromatography-mass spectrometry; LC-MS, liquid chromatography-mass spectrometry; LD, linkage disequilibrium; LFQ, label-free quantification; NMR, nuclear magnetic resonance; PCR, polymerase chain reaction; SWATH, sequential windowed acquisition of all theoretical fragment ions; TMT, tandem mass tag.
Summary and outlook
Host genomics, epigenomics, microbiome, metabolomics and metaproteomics reveal the disease characteristics of CD from different dimensions, which helps people further understand the various molecular mechanisms and predict disease progression. Nevertheless, there exists a long path to identify applicable markers with clinical therapeutic value only based on the restricted features in complicated pathogenesis demonstrated by single omics. Multi-omics experimental design can better capture the dynamic changes and functional activities of microbes and their metabolites along with the causal relationship between host gene expressions and microbial interactions during disease progression. Rise of these studies accumulates database for the establishment of a multi-omics joint platform for CD. In reality, emerged multi-omics research have limitations of high cost and great technology challenges. In addition to the technical barriers of different omics, multi-omics integration requires higher accuracy and quality of clinical sample information [181]. Major technical challenge in integration of multi-omics data lies in high heterogeneity of clinical information and a large number of dimensional variables generated to satisfy unchanged biological sample size [182]. Due to the complexity and diversity of clinical manifestations, more multi-omics research models of CD specific to different characteristics demand to be established in the future. Only based on these accurate models can we generate better non-intuitive design predictions, promote more in depth multi-omics CD research, and establish sophisticated database [183]. With great vision, the perfection of complete CD multi-omics networks and bio-informatics analysis tools are expected to provide the pivotal basis for comprehensive analysis of complex mechanisms of CD and targeted therapeutics.
CRediT authorship contribution statement
Chenlu Mu, Qianjing Zhao, Qing Zhao were the major contributors. They prepared and revised the draft. Chenlu Mu: Writing-prepare most of original draft, Designing the graphical abstract, Writing - review & editing. Qianjing Zhao: Writing-Original draft preparation, Writing - review & editing. Qing Zhao: Writing-Original draft preparation, Writing - review & editing. Lijiao Yang: Designing and drawing the graphical abstract, reviewing the draft. Xiaoqi Pang, Tianyu Liu, Xiaomeng Li: Revising the draft critically. Bangmao Wang: Reviewing the draft and put forward valuable suggestions for the manuscript. Shan-Yu Fung: Co-corresponding author; Involved in the study design and the critical revision of the manuscript. Hailong Cao: Corresponding author; Involved in the study design and the critical revision of the manuscript.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by grants (82070545, 82100574, 82270574 and 81971549) from the National Natural Science Foundation of China.
Contributor Information
Shan-Yu Fung, Email: shanefung@tmu.edu.cn.
Hailong Cao, Email: caohailong@tmu.edu.cn.
References
- 1.Ng S.C., Shi H.Y., Hamidi N., Underwood F.E., Tang W., Benchimol E.I., et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2017;390(10114):2769–2778. doi: 10.1016/S0140-6736(17)32448-0. [published Online First: 2017/10/21] [DOI] [PubMed] [Google Scholar]
- 2.Shouval D.S., Rufo P.A. The role of environmental factors in the pathogenesis of inflammatory bowel diseases: a review. JAMA Pediatr. 2017;171(10):999–1005. doi: 10.1001/jamapediatrics.2017.2571. [published Online First: 2017/08/29] [DOI] [PubMed] [Google Scholar]
- 3.Torres J., Mehandru S., Colombel J.F., Peyrin-Biroulet L. Crohn's disease. Lancet. 2017;389(10080):1741–1755. doi: 10.1016/s0140-6736(16)31711-1. [published Online First: 2016/12/05] [DOI] [PubMed] [Google Scholar]
- 4.Roda G., Chien Ng S., Kotze P.G., Argollo M., Panaccione R., Spinelli A., et al. Crohn's disease. Nat Rev Dis Primers. 2020;6(1):22. doi: 10.1038/s41572-020-0156-2. published Online First: 2020/04/04] [DOI] [PubMed] [Google Scholar]
- 5.Fiorino G., Bonifacio C., Peyrin-Biroulet L., Danese S. Preventing collateral damage in Crohn's Disease: the Lemann index. J Crohns Colitis. 2016;10(4):495–500. doi: 10.1093/ecco-jcc/jjv240. [published Online First: 2016/01/09] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramos G.P., Papadakis K.A. Mechanisms of disease: inflammatory bowel diseases. Mayo Clin Proc. 2019;94(1):155–165. doi: 10.1016/j.mayocp.2018.09.013. [published Online First: 2019/01/07] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang H., Fang M., Jostins L., Umicevic Mirkov M., Boucher G., Anderson C.A., et al. Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature. 2017;547(7662):173–178. doi: 10.1038/nature22969. [published Online First: 2017/06/29] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ellinghaus D., Jostins L., Spain S.L., Cortes A., Bethune J., Han B., et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat Genet. 2016;48(5):510–518. doi: 10.1038/ng.3528. [published Online First: 2016/03/15] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang C., Haritunians T., Okou D.T., Cutler D.J., Zwick M.E., Taylor K.D., et al. Characterization of genetic loci that affect susceptibility to inflammatory bowel diseases in African Americans. Gastroenterology. 2015;149(6):1575–1586. doi: 10.1053/j.gastro.2015.07.065. [published Online First: 2015/08/19] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levine A., Sigall Boneh R., Wine E. Evolving role of diet in the pathogenesis and treatment of inflammatory bowel diseases. Gut. 2018;67(9):1726–1738. doi: 10.1136/gutjnl-2017-315866. [published Online First: 2018/05/20] [DOI] [PubMed] [Google Scholar]
- 11.Uniken Venema W.T., Voskuil M.D., Dijkstra G., Weersma R.K., Festen E.A. The genetic background of inflammatory bowel disease: from correlation to causality. J Pathol. 2017;241(2):146–158. doi: 10.1002/path.4817. [published Online First: 2016/10/28] [DOI] [PubMed] [Google Scholar]
- 12.Pascal V., Pozuelo M., Borruel N., Casellas F., Campos D., Santiago A., et al. A microbial signature for Crohn's disease. Gut. 2017;66(5):813–822. doi: 10.1136/gutjnl-2016-313235. [published Online First: 2017/02/10] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Palmela C., Chevarin C., Xu Z., Torres J., Sevrin G., Hirten R., et al. Adherent-invasive Escherichia coli in inflammatory bowel disease. Gut. 2018;67(3):574–587. doi: 10.1136/gutjnl-2017-314903. [published Online First: 2017/11/17] [DOI] [PubMed] [Google Scholar]
- 14.Imai T., Inoue R., Kawada Y., Morita Y., Inatomi O., Nishida A., et al. Characterization of fungal dysbiosis in Japanese patients with inflammatory bowel disease. J Gastroenterol. 2019;54(2):149–159. doi: 10.1007/s00535-018-1530-7. [published Online First: 2018/11/28] [DOI] [PubMed] [Google Scholar]
- 15.Sands B.E. Biomarkers of inflammation in inflammatory bowel disease. Gastroenterology. 2015;149(5):1275–1285. doi: 10.1053/j.gastro.2015.07.003. [published Online First: 2015/07/15] [DOI] [PubMed] [Google Scholar]
- 16.Pierre N., Salee C., Massot C., Bletard N., Mazzucchelli G., Smargiasso N., et al. Proteomics highlights common and distinct pathophysiological processes associated with lleal and colonic ulcers in Crohn's disease. J Crohns Colitis. 2020;14(2):205–215. doi: 10.1093/ecco-jcc/jjz130. [published Online First: 2019/07/10] [DOI] [PubMed] [Google Scholar]
- 17.Moller F.T., Andersen V., Wohlfahrt J., Jess T. Familial risk of inflammatory bowel disease: a population-based cohort study 1977-2011. Am J Gastroenterol. 2015;110(4):564–571. doi: 10.1038/ajg.2015.50. [published Online First: 2015/03/25] [DOI] [PubMed] [Google Scholar]
- 18.Halfvarson J., Bodin L., Tysk C., Lindberg E., Jarnerot G. Inflammatory bowel disease in a Swedish twin cohort: a long-term follow-up of concordance and clinical characteristics. Gastroenterology. 2003;124(7):1767–1773. doi: 10.1016/s0016-5085(03)00385-8. [published Online First: 2003/06/14] [DOI] [PubMed] [Google Scholar]
- 19.Spehlmann M.E., Begun A.Z., Burghardt J., Lepage P., Raedler A., Schreiber S. Epidemiology of inflammatory bowel disease in a German twin cohort: results of a nationwide study. Inflamm Bowel Dis. 2008;14(7):968–976. doi: 10.1002/ibd.20380. [published Online First: 2008/02/07] [DOI] [PubMed] [Google Scholar]
- 20.Khor B., Gardet A., Xavier R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474(7351):307–317. doi: 10.1038/nature10209. [published Online First: 2011/06/17] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ogura Y., Bonen D.K., Inohara N., Nicolae D.L., Chen F.F., Ramos R., et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411(6837):603–606. doi: 10.1038/35079114. [published Online First: 2001/06/01] [DOI] [PubMed] [Google Scholar]
- 22.McGovern D.P., Kugathasan S., Cho J.H. Genetics of inflammatory bowel diseases. Gastroenterology. 2015;149(5):1163–1176. doi: 10.1053/j.gastro.2015.08.001. [published Online First: 2015/08/11] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cotterill L., Payne D., Levinson S., McLaughlin J., Wesley E., Feeney M., et al. Replication and meta-analysis of 13,000 cases defines the risk for interleukin-23 receptor and autophagy-related 16-like 1 variants in Crohn's disease. Can J Gastroenterol. 2010;24(5):297–302. doi: 10.1155/2010/480458. [published Online First: 2010/05/21] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mao Y.Q., Dong S.Q., Gao M. Association between TNF-α rs1799724 and rs1800629 polymorphisms and the risk of Crohn's disease. Genet Mol Res. 2015;14(4):15811–15821. doi: 10.4238/2015.December.1.33. [published Online First: 2015/12/05] [DOI] [PubMed] [Google Scholar]
- 25.Petryszyn P., Dudkowiak R., Gruca A., Jaźwińska-Tarnawska E., Ekk-Cierniakowski P., Poniewierka E., et al. C3435T polymorphism of the ABCB1 gene in polish patients with inflammatory bowel disease: a case-control and meta-analysis study. Genes (Basel) 2021;12(9) doi: 10.3390/genes12091419. [published Online First: 2021/09/29] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cho Y.A., Lee J., Oh J.H., Chang H.J., Sohn D.K., Shin A., et al. Vitamin D receptor fokI polymorphism and the risks of colorectal cancer, inflammatory bowel disease, and colorectal adenoma. Sci Rep. 2018;8(1) doi: 10.1038/s41598-018-31244-5. [published Online First: 2018/08/29] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou F., Zhu Q., Zheng P.F., Feng Y.L. Association of fucosyltransferase 2 gene variant with inflammatory bowel diseases: a meta-analysis. Med Sci Monit. 2019;25:184–192. doi: 10.12659/msm.911857. [published Online First: 2019/01/08] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu W.D., Xie Q.B., Zhao Y., Liu Y. Association of Interleukin-23 receptor gene polymorphisms with susceptibility to Crohn's disease: A meta-analysis. Sci Rep. 2015;5 doi: 10.1038/srep18584. [published Online First: 2015/12/19] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang L., Lu Y., Ge Y., Shi Y., Wu X., Xu Q., et al. Interleukin-23R rs7517847 T/G polymorphism contributes to the risk of Crohn's disease in Caucasians: a meta-analysis. J Immunol Res. 2015;2015 doi: 10.1155/2015/279849. [published Online First: 2015/06/20] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang B.B., Liang Y., Yang B., Tan Y.J. Association between ATG16L1 gene polymorphism and the risk of Crohn's disease. J Int Med Res. 2017;45(6):1636–1650. doi: 10.1177/0300060516662404. [published Online First: 2016/10/05] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shafieyoun A., Moraveji S., Bashashati M., Rezaei N. Discs large homolog 5 (DLG5) gene polymorphism and Crohn's disease: a meta-analysis of the published studies. Acta Med Iran. 2016;54(5):289–295. [published Online First: 2016/06/17] [PubMed] [Google Scholar]
- 32.Mehto S., Jena K.K., Nath P., Chauhan S., Kolapalli S.P., Das S.K., et al. The Crohn's disease risk factor IRGM limits NLRP3 inflammasome activation by impeding its assembly and by mediating its selective autophagy. Mol Cell. 2019;73(3):429–445. doi: 10.1016/j.molcel.2018.11.018. [published Online First: 2019/01/08] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Larabi A., Barnich N., Nguyen H.T.T. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy. 2020;16(1):38–51. doi: 10.1080/15548627.2019.1635384. [published Online First: 2019/07/10] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen H.T., Lapaquette P., Bringer M.A., Darfeuille-Michaud A. Autophagy and Crohn's disease. J Innate Immun. 2013;5(5):434–443. doi: 10.1159/000345129. [published Online First: 2013/01/19] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brest P., Lapaquette P., Souidi M., Lebrigand K., Cesaro A., Vouret-Craviari V., et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn's disease. Nat Genet. 2011;43(3):242–245. doi: 10.1038/ng.762. [published Online First: 2011/02/01] [DOI] [PubMed] [Google Scholar]
- 36.Yang S.K., Hong M., Zhao W., Jung Y., Baek J., Tayebi N., et al. Genome-wide association study of Crohn's disease in Koreans revealed three new susceptibility loci and common attributes of genetic susceptibility across ethnic populations. Gut. 2014;63(1):80–87. doi: 10.1136/gutjnl-2013-305193. [published Online First: 2013/07/16] [DOI] [PubMed] [Google Scholar]
- 37.Liu J.Z., van Sommeren S., Huang H., Ng S.C., Alberts R., Takahashi A., et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47(9):979–986. doi: 10.1038/ng.3359. [published Online First: 2015/07/21] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jostins L., Ripke S., Weersma R.K., Duerr R.H., McGovern D.P., Hui K.Y., et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491(7422):119–124. doi: 10.1038/nature11582. [published Online First: 2012/11/07] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu J.Z., van Sommeren S., Huang H.L., Ng S.C., Alberts R., Takahashi A., et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations Nature Genetics. 2015;47(9):979. doi: 10.1038/ng.3359. -+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hong M., Ye B.D., Yang S.K., Jung S., Lee H.S., Kim B.M., et al. Immunochip meta-analysis of inflammatory bowel disease identifies three novel loci and four novel associations in previously reported loci. J Crohns Colitis. 2018;12(6):730–741. doi: 10.1093/ecco-jcc/jjy002. [published Online First: 2018/03/28] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arimura Y., Isshiki H., Onodera K., Nagaishi K., Yamashita K., Sonoda T., et al. Characteristics of Japanese inflammatory bowel disease susceptibility loci. J Gastroenterol. 2014;49(8):1217–1230. doi: 10.1007/s00535-013-0866-2. [published Online First: 2013/08/15] [DOI] [PubMed] [Google Scholar]
- 42.Jung S., Ye B.D., Lee H.S., Baek J., Kim G., Park D., et al. Identification of three novel susceptibility loci for inflammatory bowel disease in koreans in an extended genome-wide association study. J Crohns Colitis. 2021;15(11):1898–1907. doi: 10.1093/ecco-jcc/jjab060. [published Online First: 2021/04/15] [DOI] [PubMed] [Google Scholar]
- 43.Franke A., McGovern D.P., Barrett J.C., Wang K., Radford-Smith G.L., Ahmad T., et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet. 2010;42(12):1118–1125. doi: 10.1038/ng.717. [published Online First: 2010/11/26] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shawki A., McCole D.F. Mechanisms of intestinal epithelial barrier dysfunction by adherent-invasive escherichia coli. Cell Mol Gastroenterol Hepatol. 2017;3(1):41–50. doi: 10.1016/j.jcmgh.2016.10.004. [published Online First: 2017/02/09] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kennedy N.A., Lamb C.A., Berry S.H., Walker A.W., Mansfield J., Parkes M., et al. The impact of NOD2 variants on fecal microbiota in Crohn's disease and controls without gastrointestinal disease. Inflamm Bowel Dis. 2018;24(3):583–592. doi: 10.1093/ibd/izx061. [published Online First: 2018/02/21] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fritz T., Niederreiter L., Adolph T., Blumberg R.S., Kaser A. Crohn's disease: NOD2, autophagy and ER stress converge. Gut. 2011;60(11):1580–1588. doi: 10.1136/gut.2009.206466. [published Online First: 2011/01/22] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lauro M.L., Burch J.M., Grimes C.L. The effect of NOD2 on the microbiota in Crohn's disease. Curr Opin Biotechnol. 2016;40:97–102. doi: 10.1016/j.copbio.2016.02.028. [published Online First: 2016/04/02] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rochereau N., Roblin X., Michaud E., Gayet R., Chanut B., Jospin F., et al. NOD2 deficiency increases retrograde transport of secretory IgA complexes in Crohn's disease. Nat Commun. 2021;12(1) doi: 10.1038/s41467-020-20348-0. [published Online First: 2021/01/13] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alonso A., Domènech E., Julià A., Panés J., García-Sánchez V., Mateu P.N., et al. Identification of risk loci for Crohn's disease phenotypes using a genome-wide association study. Gastroenterology. 2015;148(4):794–805. doi: 10.1053/j.gastro.2014.12.030. [published Online First: 2015/01/06] [DOI] [PubMed] [Google Scholar]
- 50.Nelson M.R., Tipney H., Painter J.L., Shen J., Nicoletti P., Shen Y., et al. The support of human genetic evidence for approved drug indications. Nat Genet. 2015;47(8):856–860. doi: 10.1038/ng.3314. [published Online First: 2015/06/30] [DOI] [PubMed] [Google Scholar]
- 51.Sazonovs A., Kennedy N.A., Moutsianas L., Heap G.A., Rice D.L., Reppell M., et al. HLA-DQA1*05 carriage associated with development of anti-drug antibodies to infliximab and adalimumab in patients with Crohn's disease. Gastroenterology. 2020;158(1):189–199. doi: 10.1053/j.gastro.2019.09.041. [published Online First: 2019/10/11] [DOI] [PubMed] [Google Scholar]
- 52.Lee J.C., Biasci D., Roberts R., Gearry R.B., Mansfield J.C., Ahmad T., et al. Genome-wide association study identifies distinct genetic contributions to prognosis and susceptibility in Crohn's disease. Nat Genet. 2017;49(2):262–268. doi: 10.1038/ng.3755. [published Online First: 2017/01/10] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.O'Donnell S., Borowski K., Espin-Garcia O., Milgrom R., Kabakchiev B., Stempak J., et al. The unsolved link of genetic markers and Crohn's disease progression: a North American Cohort experience. Inflamm Bowel Dis. 2019;25(9):1541–1549. doi: 10.1093/ibd/izz016. [published Online First: 2019/02/26] [DOI] [PubMed] [Google Scholar]
- 54.González-Serna D., Ochoa E., López-Isac E., Julià A., Degenhardt F., Ortego-Centeno N., et al. A cross-disease meta-GWAS identifies four new susceptibility loci shared between systemic sclerosis and Crohn's disease. Sci Rep. 2020;10(1) doi: 10.1038/s41598-020-58741-w. [published Online First: 2020/02/07] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang Y., Musco H., Simpson-Yap S., Zhu Z., Wang Y., Lin X., et al. Investigating the shared genetic architecture between multiple sclerosis and inflammatory bowel diseases. Nat Commun. 2021;12(1) doi: 10.1038/s41467-021-25768-0. [published Online First: 2021/09/26] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Festen E.A., Goyette P., Green T., Boucher G., Beauchamp C., Trynka G., et al. A meta-analysis of genome-wide association scans identifies IL18RAP, PTPN2, TAGAP, and PUS10 as shared risk loci for Crohn's disease and celiac disease. PLoS Genet. 2011;7(1) doi: 10.1371/journal.pgen.1001283. [published Online First: 2011/02/08] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li P., Yang X.K., Wang X., Zhao M.Q., Zhang C., Tao S.S., et al. A meta-analysis of the relationship between MYO9B gene polymorphisms and susceptibility to Crohn's disease and ulcerative colitis. Hum Immunol. 2016;77(10):990–996. doi: 10.1016/j.humimm.2016.07.008. [published Online First: 2016/07/21] [DOI] [PubMed] [Google Scholar]
- 58.Zhu Z., Zhang F., Hu H., Bakshi A., Robinson M.R., Powell J.E., et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48(5):481–487. doi: 10.1038/ng.3538. [published Online First: 2016/03/29] [DOI] [PubMed] [Google Scholar]
- 59.Mountjoy E., Schmidt E.M., Carmona M., Schwartzentruber J., Peat G., Miranda A., et al. An open approach to systematically prioritize causal variants and genes at all published human GWAS trait-associated loci. Nat Genet. 2021;53(11):1527–1533. doi: 10.1038/s41588-021-00945-5. [published Online First: 2021/10/30] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee H.S., Cleynen I. Molecular profiling of inflammatory bowel disease: Is it ready for use in clinical decision-making? Cells. 2019;8(6) doi: 10.3390/cells8060535. [published Online First: 2019/06/07] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Torres J., Colombel J.F. Genetics and phenotypes in inflammatory bowel disease. Lancet. 2016;387(10014):98–100. doi: 10.1016/s0140-6736(15)00464-x. [published Online First: 2015/10/23] [DOI] [PubMed] [Google Scholar]
- 62.Paternoster L., Tilling K., Davey Smith G. Genetic epidemiology and Mendelian randomization for informing disease therapeutics: Conceptual and methodological challenges. PLoS Genet. 2017;13(10) doi: 10.1371/journal.pgen.1006944. [published Online First: 2017/10/06] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.GWAS to the people. Nat Med. 2018;24(10):1483. doi: 10.1038/s41591-018-0231-3. [published Online First: 2018/10/10] [DOI] [PubMed] [Google Scholar]
- 64.Watanabe K., Taskesen E., van Bochoven A., Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8(1) doi: 10.1038/s41467-017-01261-5. [published Online First: 2017/12/01] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sazonovs A., Stevens C.R., Venkataraman G.R., Yuan K., Avila B., Abreu M.T., et al. Large-scale sequencing identifies multiple genes and rare variants associated with Crohn's disease susceptibility. Nat Genet. 2022;54(9):1275–1283. doi: 10.1038/s41588-022-01156-2. [published Online First: 2022/08/30] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zeng Z., Mukherjee A., Zhang H. From genetics to epigenetics, roles of epigenetics in inflammatory bowel disease. Front Genet. 2019;10 doi: 10.3389/fgene.2019.01017. [published Online First: 2019/11/19] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Annese V. Genetics and epigenetics of IBD. Pharmacol Res. 2020;159 doi: 10.1016/j.phrs.2020.104892. [published Online First: 2020/05/29] [DOI] [PubMed] [Google Scholar]
- 68.Ryan F.J., Ahern A.M., Fitzgerald R.S., Laserna-Mendieta E.J., Power E.M., Clooney A.G., et al. Colonic microbiota is associated with inflammation and host epigenomic alterations in inflammatory bowel disease. Nat Commun. 2020;11(1) doi: 10.1038/s41467-020-15342-5. [published Online First: 2020/04/07] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gonzalez M.M., Bamidele A.O., Svingen P.A., Sagstetter M.R., Smyrk T.C., Gaballa J.M., et al. BMI1 maintains the Treg epigenomic landscape to prevent inflammatory bowel disease. J Clin Invest. 2021;131(12) doi: 10.1172/JCI140755. [published Online First: 2021/06/16] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Joustra V., Hageman I.L., Satsangi J., Adams A., Ventham N.T., de Jonge W.J., et al. Systematic review and meta-analysis of peripheral blood DNA methylation studies in inflammatory bowel disease. J Crohns Colitis. 2022 doi: 10.1093/ecco-jcc/jjac119. [published Online First: 2022/08/24] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yarani R., Shojaeian A., Palasca O., Doncheva N.T., Jensen L.J., Gorodkin J., et al. Differentially expressed miRNAs in ulcerative colitis and Crohn's disease. Front Immunol. 2022;13 doi: 10.3389/fimmu.2022.865777. [published Online First: 2022/06/24] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sun L., Han Y., Wang H., Liu H., Liu S., Yang H., et al. MicroRNAs as potential biomarkers for the diagnosis of inflammatory bowel disease: a systematic review and meta-analysis. J Int Med Res. 2022;50(4) doi: 10.1177/03000605221089503. [published Online First: 2022/04/22] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Serena C., Millan M., Ejarque M., Saera-Vila A., Maymó-Masip E., Núñez-Roa C., et al. Adipose stem cells from patients with Crohn's disease show a distinctive DNA methylation pattern. Clin Epigenetics. 2020;12(1) doi: 10.1186/s13148-020-00843-3. [published Online First: 2020/04/08] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Glassner K.L., Abraham B.P., Quigley E.M.M. The microbiome and inflammatory bowel disease. J Allergy Clin Immunol. 2020;145(1):16–27. doi: 10.1016/j.jaci.2019.11.003. [published Online First: 2020/01/09] [DOI] [PubMed] [Google Scholar]
- 75.Brusaferro A., Cavalli E., Farinelli E., Cozzali R., Principi N., Esposito S. Gut dysbiosis and paediatric Crohn's disease. J Infect. 2019;78(1):1–7. doi: 10.1016/j.jinf.2018.10.005. [published Online First: 2018/10/20] [DOI] [PubMed] [Google Scholar]
- 76.Starz E., Wzorek K., Folwarski M., Kaźmierczak-Siedlecka K., Stachowska L., Przewłócka K., et al. The modification of the gut microbiota via selected specific diets in patients with Crohn's disease. Nutrients. 2021;13(7) doi: 10.3390/nu13072125. [published Online First: 2021/07/03] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yilmaz B., Juillerat P., Øyås O., Ramon C., Bravo F.D., Franc Y., et al. Microbial network disturbances in relapsing refractory Crohn's disease. Nat Med. 2019;25(2):323–336. doi: 10.1038/s41591-018-0308-z. [published Online First: 2019/01/22] [DOI] [PubMed] [Google Scholar]
- 78.Sokol H., Landman C., Seksik P., Berard L., Montil M., Nion-Larmurier I., et al. Fecal microbiota transplantation to maintain remission in Crohn's disease: a pilot randomized controlled study. Microbiome. 2020;8(1) doi: 10.1186/s40168-020-0792-5. [published Online First: 2020/02/06] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Paramsothy S., Paramsothy R., Rubin D.T., Kamm M.A., Kaakoush N.O., Mitchell H.M., et al. Faecal microbiota transplantation for inflammatory bowel disease: a systematic review and meta-analysis. J Crohns Colitis. 2017;11(10):1180–1199. doi: 10.1093/ecco-jcc/jjx063. [published Online First: 2017/05/10] [DOI] [PubMed] [Google Scholar]
- 80.Caparros E., Wiest R., Scharl M., Rogler G., Gutierrez Casbas A., Yilmaz B., et al. Dysbiotic microbiota interactions in Crohn's disease. Gut Microbes. 2021;13(1) doi: 10.1080/19490976.2021.1949096. [published Online First: 2021/07/28] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.He Z., Wu J., Gong J., Ke J., Ding T., Zhao W., et al. Microbiota in mesenteric adipose tissue from Crohn's disease promote colitis in mice. Microbiome. 2021;9(1) doi: 10.1186/s40168-021-01178-8. [published Online First: 2021/11/25] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kostic A.D., Xavier R.J., Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology. 2014;146(6):1489–1499. doi: 10.1053/j.gastro.2014.02.009. [published Online First: 2014/02/25] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Smith B.A., Gupta N., Denny K., Culver G.M. Characterization of 16S rRNA processing with Pre-30S subunit assembly intermediates from E. coli. J Mol Biol. 2018;430(12):1745–1759. doi: 10.1016/j.jmb.2018.04.009. [published Online First: 2018/04/17] [DOI] [PubMed] [Google Scholar]
- 84.Clarridge J.E., 3rd Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin Microbiol Rev. 2004;17(4):840–862. doi: 10.1128/CMR.17.4.840-862.2004. [published Online First: 2004/10/19] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ye S.H., Siddle K.J., Park D.J., Sabeti P.C. Benchmarking metagenomics tools for taxonomic classification. Cell. 2019;178(4):779–794. doi: 10.1016/j.cell.2019.07.010. [published Online First: 2019/08/10] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Strömbeck A., Lasson A., Strid H., Sundin J., Stotzer P.O., Simrén M., et al. Fecal microbiota composition is linked to the postoperative disease course in patients with Crohn's disease. BMC Gastroenterol. 2020;20(1) doi: 10.1186/s12876-020-01281-4. [published Online First: 2020/05/06] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gevers D., Kugathasan S., Denson L.A., Vazquez-Baeza Y., Van Treuren W., Ren B., et al. The treatment-naive microbiome in new-onset Crohn's disease. Cell Host Microbe. 2014;15(3):382–392. doi: 10.1016/j.chom.2014.02.005. [published Online First: 2014/03/19] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Aldars-Garcia L., Chaparro M., Gisbert J.P. Systematic review: the gut microbiome and its potential clinical application in inflammatory bowel disease. Microorganisms. 2021;9(5) doi: 10.3390/microorganisms9050977. [published Online First: 2021/05/06] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mah C., Jayawardana T., Leong G., Koentgen S., Lemberg D., Connor S.J., et al. Assessing the relationship between the gut microbiota and inflammatory bowel disease therapeutics: a systematic review. Pathogens. 2023;12(2) doi: 10.3390/pathogens12020262. [published Online First: 2023/02/26] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Abdel-Rahman L.I.H., Morgan X.C. Searching for a consensus among inflammatory bowel disease studies: a systematic meta-analysis. Inflamm Bowel Dis. 2023;29(1):125–139. doi: 10.1093/ibd/izac194. [published Online First: 2022/09/17] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Aldars-García L., Chaparro M., Gisbert J.P. Systematic review: the gut microbiome and its potential clinical application in inflammatory bowel disease. Microorganisms. 2021;9(5) doi: 10.3390/microorganisms9050977. [published Online First: 2021/05/06] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liu S., Zhao W., Lan P., Mou X. The microbiome in inflammatory bowel diseases: from pathogenesis to therapy. Protein Cell. 2021;12(5):331–345. doi: 10.1007/s13238-020-00745-3. [published Online First: 2020/07/01] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schirmer M., Garner A., Vlamakis H., Xavier R.J. Microbial genes and pathways in inflammatory bowel disease. Nat Rev Microbiol. 2019;17(8):497–511. doi: 10.1038/s41579-019-0213-6. [published Online First: 2019/06/30] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ni J., Wu G.D., Albenberg L., Tomov V.T. Gut microbiota and IBD: causation or correlation? Nat Rev Gastroenterol Hepatol. 2017;14(10):573–584. doi: 10.1038/nrgastro.2017.88. [published Online First: 2017/07/27] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sanchez-Navarro R., Nuhamunada M., Mohite O.S., Wasmund K., Albertsen M., Gram L., et al. Long-read metagenome-assembled genomes improve identification of novel complete biosynthetic gene clusters in a complex microbial activated sludge ecosystem. mSystems. 2022;7(6) doi: 10.1128/msystems.00632-22. [published Online First: 2022/11/30] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gupta V.K., Bakshi U., Chang D., Lee A.R., Davis J.M., 3rd, Chandrasekaran S., et al. TaxiBGC: a taxonomy-guided approach for profiling experimentally characterized microbial biosynthetic gene clusters and secondary metabolite production potential in metagenomes. mSystems. 2022;7(6) doi: 10.1128/msystems.00925-22. [published Online First: 2022/11/16] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Doron I., Mesko M., Li X.V., Kusakabe T., Leonardi I., Shaw D.G., et al. Mycobiota-induced IgA antibodies regulate fungal commensalism in the gut and are dysregulated in Crohn's disease. Nat Microbiol. 2021;6(12):1493–1504. doi: 10.1038/s41564-021-00983-z. [published Online First: 2021/11/24] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lewis J.D., Chen E.Z., Baldassano R.N., Otley A.R., Griffiths A.M., Lee D., et al. Inflammation, antibiotics, and diet as environmental stressors of the gut microbiome in pediatric Crohn's disease. Cell Host Microbe. 2015;18(4):489–500. doi: 10.1016/j.chom.2015.09.008. [published Online First: 2015/10/16] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yilmaz B., Juillerat P., Oyas O., Ramon C., Bravo F.D., Franc Y., et al. Microbial network disturbances in relapsing refractory Crohn's disease. Nat Med. 2019;25(2):323–336. doi: 10.1038/s41591-018-0308-z. [published Online First: 2019/01/22] [DOI] [PubMed] [Google Scholar]
- 100.Kugathasan S., Denson L.A., Walters T.D., Kim M.O., Marigorta U.M., Schirmer M., et al. Prediction of complicated disease course for children newly diagnosed with Crohn's disease: a multicentre inception cohort study. Lancet. 2017;389(10080):1710–1718. doi: 10.1016/S0140-6736(17)30317-3. [published Online First: 2017/03/06] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Miquel S., Martín R., Rossi O., Bermúdez-Humarán L.G., Chatel J.M., Sokol H., et al. Faecalibacterium prausnitzii and human intestinal health. Curr Opin Microbiol. 2013;16(3):255–261. doi: 10.1016/j.mib.2013.06.003. [published Online First: 2013/07/09] [DOI] [PubMed] [Google Scholar]
- 102.Chen Y.C., Kuo Y.C., Chen M.C., Zhang Y.D., Chen C.L., Le P.H., et al. Case-Control Study of Clostridium innocuum Infection, Taiwan. Emerg Infect Dis. 2022;28(3):599–607. doi: 10.3201/eid2803.204421. [published Online First: 2022/02/24] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Caparrós E., Wiest R., Scharl M., Rogler G., Gutiérrez Casbas A., Yilmaz B., et al. Dysbiotic microbiota interactions in Crohn's disease. Gut Microbes. 2021;13(1) doi: 10.1080/19490976.2021.1949096. [published Online First: 2021/07/28] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cox S.R., Lindsay J.O., Fromentin S., Stagg A.J., McCarthy N.E., Galleron N., et al. Effects of low FODMAP diet on symptoms, fecal Microbiome, and markers of inflammation in patients with quiescent inflammatory bowel disease in a randomized trial. Gastroenterology. 2020;158(1):176–188. doi: 10.1053/j.gastro.2019.09.024. [published Online First: 2019/10/06] [DOI] [PubMed] [Google Scholar]
- 105.Kang X., Deng D.M., Crielaard W., Brandt B.W. Reprocessing 16S rRNA gene amplicon sequencing studies: (Meta)data issues, robustness, and reproducibility. Front Cell Infect Microbiol. 2021;11 doi: 10.3389/fcimb.2021.720637. [published Online First: 2021/11/09] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Saa P., Urrutia A., Silva-Andrade C., Martín A.J., Garrido D. Modeling approaches for probing cross-feeding interactions in the human gut microbiome. Comput Struct Biotechnol J. 2022;20:79–89. doi: 10.1016/j.csbj.2021.12.006. [published Online First: 2022/01/04] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhao N., Cao J., Xu J., Liu B., Liu B., Chen D., et al. Targeting RNA with Next- and Third-Generation Sequencing Improves Pathogen Identification in Clinical Samples. Adv Sci (Weinh) 2021;8(23) doi: 10.1002/advs.202102593. [published Online First: 2021/10/24] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lavelle A., Sokol H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2020;17(4):223–237. doi: 10.1038/s41575-019-0258-z. [published Online First: 2020/02/23] [DOI] [PubMed] [Google Scholar]
- 109.Gallagher K., Catesson A., Griffin J.L., Holmes E., Williams H.R.T. Metabolomic Analysis in Inflammatory Bowel Disease: A Systematic Review. J Crohns Colitis. 2021;15(5):813–826. doi: 10.1093/ecco-jcc/jjaa227. [published Online First: 2020/11/12] [DOI] [PubMed] [Google Scholar]
- 110.Wahlström A., Sayin S.I., Marschall H.U., Bäckhed F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016;24(1):41–50. doi: 10.1016/j.cmet.2016.05.005. [published Online First: 2016/06/21] [DOI] [PubMed] [Google Scholar]
- 111.Wang Y., Gao X., Zhang X., Xiao F., Hu H., Li X., et al. Microbial and metabolic features associated with outcome of infliximab therapy in pediatric Crohn's disease. Gut Microbes. 2021;13(1):1–18. doi: 10.1080/19490976.2020.1865708. [published Online First: 2021/01/13] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Russo E., Giudici F., Fiorindi C., Ficari F., Scaringi S., Amedei A. Immunomodulating activity and therapeutic effects of short chain fatty acids and tryptophan post-biotics in inflammatory bowel disease. Front Immunol. 2019;10 doi: 10.3389/fimmu.2019.02754. [published Online First: 2019/12/12] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li M., Yang L., Mu C., Sun Y., Gu Y., Chen D., et al. Gut microbial metabolome in inflammatory bowel disease: From association to therapeutic perspectives. Comput Struct Biotechnol J. 2022;20:2402–2414. doi: 10.1016/j.csbj.2022.03.038. [published Online First: 2022/06/07] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chen R., Zheng J., Li L., Li C., Chao K., Zeng Z., et al. Metabolomics facilitate the personalized management in inflammatory bowel disease. Therap Adv Gastroenterol. 2021;14 doi: 10.1177/17562848211064489. published Online First: 2022/01/07] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kaddurah-Daouk R., Kristal B.S., Weinshilboum R.M. Metabolomics: a global biochemical approach to drug response and disease. Annu Rev Pharmacol Toxicol. 2008;48:653–683. doi: 10.1146/annurev.pharmtox.48.113006.094715. [published Online First: 2008/01/11] [DOI] [PubMed] [Google Scholar]
- 116.Nicholson J.K., Lindon J.C. Systems biology: metabonomics. Nature. 2008;455(7216):1054–1056. doi: 10.1038/4551054a. [published Online First: 2008/10/25] [DOI] [PubMed] [Google Scholar]
- 117.De Preter V., Joossens M., Ballet V., Shkedy Z., Rutgeerts P., Vermeire S., et al. Metabolic profiling of the impact of oligofructose-enriched inulin in Crohn's disease patients: a double-blinded randomized controlled trial. Clin Transl Gastroenterol. 2013;4 doi: 10.1038/ctg.2012.24. [published Online First: 2013/01/11] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Weng Y.J., Gan H.Y., Li X., Huang Y., Li Z.C., Deng H.M., et al. Correlation of diet, microbiota and metabolite networks in inflammatory bowel disease. J Dig Dis. 2019;20(9):447–459. doi: 10.1111/1751-2980.12795. [published Online First: 2019/06/27] [DOI] [PubMed] [Google Scholar]
- 119.De Preter V., Machiels K., Joossens M., Arijs I., Matthys C., Vermeire S., et al. Faecal metabolite profiling identifies medium-chain fatty acids as discriminating compounds in IBD. Gut. 2015;64(3):447–458. doi: 10.1136/gutjnl-2013-306423. [published Online First: 2014/05/09] [DOI] [PubMed] [Google Scholar]
- 120.Zhuang X., Li T., Li M., Huang S., Qiu Y., Feng R., et al. Systematic review and meta-analysis: short-chain fatty acid characterization in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2019;25(11):1751–1763. doi: 10.1093/ibd/izz188. [published Online First: 2019/09/10] [DOI] [PubMed] [Google Scholar]
- 121.Marchesi J.R., Holmes E., Khan F., Kochhar S., Scanlan P., Shanahan F., et al. Rapid and noninvasive metabonomic characterization of inflammatory bowel disease. J Proteome Res. 2007;6(2):546–551. doi: 10.1021/pr060470d. [published Online First: 2007/02/03] [DOI] [PubMed] [Google Scholar]
- 122.Ghiboub M., Penny S., Verburgt C.M., Boneh R.S., Wine E., Cohen A., et al. Metabolome changes with diet-induced remission in pediatric Crohn's disease. Gastroenterology. 2022;163(4):922–936. doi: 10.1053/j.gastro.2022.05.050. [published Online First: 2022/06/10] [DOI] [PubMed] [Google Scholar]
- 123.Lloyd-Price J., Arze C., Ananthakrishnan A.N., Schirmer M., Avila-Pacheco J., Poon T.W., et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019;569(7758):655–662. doi: 10.1038/s41586-019-1237-9. [published Online First: 2019/05/31] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ridlon J.M., Kang D.J., Hylemon P.B. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47(2):241–259. doi: 10.1194/jlr.R500013-JLR200. [published Online First: 2005/11/22] [DOI] [PubMed] [Google Scholar]
- 125.Roda G., Porru E., Katsanos K., Skamnelos A., Kyriakidi K., Fiorino G., et al. Serum bile acids profiling in inflammatory bowel disease patients treated with anti-TNFs. Cells. 2019;8(8) doi: 10.3390/cells8080817. [published Online First: 2019/08/07] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ding N.S., McDonald J.A.K., Perdones-Montero A., Rees D.N., Adegbola S.O., Misra R., et al. Metabonomics and the gut microbiome associated with primary response to anti-TNF therapy in Crohn's disease. J Crohns Colitis. 2020;14(8):1090–1102. doi: 10.1093/ecco-jcc/jjaa039. [published Online First: 2020/03/03] [DOI] [PubMed] [Google Scholar]
- 127.Hang S., Paik D., Yao L., Kim E., Trinath J., Lu J., et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature. 2019;576(7785):143–148. doi: 10.1038/s41586-019-1785-z. [published Online First: 2019/11/30] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Song X., Sun X., Oh S.F., Wu M., Zhang Y., Zheng W., et al. Microbial bile acid metabolites modulate gut RORgamma(+) regulatory T cell homeostasis. Nature. 2020;577(7790):410–415. doi: 10.1038/s41586-019-1865-0. [published Online First: 2019/12/27] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Liu T.C., Kern J.T., Jain U., Sonnek N.M., Xiong S., Simpson K.F., et al. Western diet induces Paneth cell defects through microbiome alterations and farnesoid X receptor and type I interferon activation. Cell Host Microbe. 2021;29(6):988–1001.e6. doi: 10.1016/j.chom.2021.04.004. [published Online First: 2021/05/20] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pavlidis P., Powell N., Vincent R.P., Ehrlich D., Bjarnason I., Hayee B. Systematic review: bile acids and intestinal inflammation-luminal aggressors or regulators of mucosal defence? Aliment Pharmacol Ther. 2015;42(7):802–817. doi: 10.1111/apt.13333. [published Online First: 2015/08/01] [DOI] [PubMed] [Google Scholar]
- 131.Vidal-Lletjós S., Beaumont M., Tomé D., Benamouzig R., Blachier F., Lan A. Dietary protein and amino acid supplementation in inflammatory bowel disease course: what impact on the colonic mucosa? Nutrients. 2017;9(3) doi: 10.3390/nu9030310. [published Online First: 2017/03/25] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Liu Y., Wang X., Hu C.A. Therapeutic potential of amino acids in inflammatory bowel disease. Nutrients. 2017;9(9) doi: 10.3390/nu9090920. [published Online First: 2017/08/24] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Balasubramanian K., Kumar S., Singh R.R., Sharma U., Ahuja V., Makharia G.K., et al. Metabolism of the colonic mucosa in patients with inflammatory bowel diseases: an in vitro proton magnetic resonance spectroscopy study. Magn Reson Imaging. 2009;27(1):79–86. doi: 10.1016/j.mri.2008.05.014. [published Online First: 2008/07/05] [DOI] [PubMed] [Google Scholar]
- 134.Diederen K., Li J.V., Donachie G.E., de Meij T.G., de Waart D.R., Hakvoort T.B.M., et al. Exclusive enteral nutrition mediates gut microbial and metabolic changes that are associated with remission in children with Crohn's disease. Sci Rep. 2020;10(1) doi: 10.1038/s41598-020-75306-z. [published Online First: 2020/11/05] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Nikolaus S., Schulte B., Al-Massad N., Thieme F., Schulte D.M., Bethge J., et al. Increased tryptophan metabolism is associated with activity of inflammatory bowel diseases. Gastroenterology. 2017;153(6):1504–1516. doi: 10.1053/j.gastro.2017.08.028. [published Online First: 2017/08/23] [DOI] [PubMed] [Google Scholar]
- 136.Dawiskiba T., Deja S., Mulak A., Zabek A., Jawien E., Pawelka D., et al. Serum and urine metabolomic fingerprinting in diagnostics of inflammatory bowel diseases. World J Gastroenterol. 2014;20(1):163–174. doi: 10.3748/wjg.v20.i1.163. [published Online First: 2014/01/15] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Fathi F., Majari-Kasmaee L., Mani-Varnosfaderani A., Kyani A., Rostami-Nejad M., Sohrabzadeh K., et al. 1H NMR based metabolic profiling in Crohn's disease by random forest methodology. Magn Reson Chem. 2014;52(7):370–376. doi: 10.1002/mrc.4074. [published Online First: 2014/04/24] [DOI] [PubMed] [Google Scholar]
- 138.Saleh S., Staes A., Deborggraeve S., Gevaert K. Targeted proteomics for studying pathogenic bacteria. Proteomics. 2019;19(16) doi: 10.1002/pmic.201800435. [published Online First: 2019/06/27] [DOI] [PubMed] [Google Scholar]
- 139.Sun X.L., Qiao L.C., Gong J., Wen K., Xu Z.Z., Yang B.L. Proteomics identifies a novel role of fibrinogen-like protein 1 in Crohn's disease. World J Gastroenterol. 2021;27(35):5946–5957. doi: 10.3748/wjg.v27.i35.5946. [published Online First: 2021/10/12] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Baldan-Martin M., Chaparro M., Gisbert J.P. Tissue proteomic approaches to understand the pathogenesis of inflammatory bowel disease. Inflamm Bowel Dis. 2021;27(8):1184–1200. doi: 10.1093/ibd/izaa352. [published Online First: 2021/02/03] [DOI] [PubMed] [Google Scholar]
- 141.Moriggi M., Pastorelli L., Torretta E., Tontini G.E., Capitanio D., Bogetto S.F., et al. Contribution of extracellular matrix and signal mechanotransduction to epithelial cell damage in inflammatory bowel disease patients: a proteomic study. Proteomics. 2017;17(23-24) doi: 10.1002/pmic.201700164. [published Online First: 2017/10/14] [DOI] [PubMed] [Google Scholar]
- 142.Di Narzo A.F., Brodmerkel C., Telesco S.E., Argmann C., Peters L.A., Li K., et al. High-Throughput Identification of the Plasma Proteomic Signature of Inflammatory Bowel Disease. J Crohns Colitis. 2019;13(4):462–471. doi: 10.1093/ecco-jcc/jjy190. [published Online First: 2018/11/18] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Segal J.P., Mullish B.H., Quraishi M.N., Acharjee A., Williams H.R.T., Iqbal T., et al. The application of omics techniques to understand the role of the gut microbiota in inflammatory bowel disease. Therap Adv Gastroenterol. 2019;12 doi: 10.1177/1756284818822250. [published Online First: 2019/02/06] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lopez R.N., Leach S.T., Lemberg D.A., Duvoisin G., Gearry R.B., Day A.S. Fecal biomarkers in inflammatory bowel disease. J Gastroenterol Hepatol. 2017;32(3):577–582. doi: 10.1111/jgh.13611. [published Online First: 2016/10/11] [DOI] [PubMed] [Google Scholar]
- 145.Torres J., Petralia F., Sato T., Wang P., Telesco S.E., Choung R.S., et al. Serum biomarkers identify patients who will develop inflammatory bowel diseases up to 5 years before diagnosis. Gastroenterology. 2020;159(1):96–104. doi: 10.1053/j.gastro.2020.03.007. [published Online First: 2020/03/14] [DOI] [PubMed] [Google Scholar]
- 146.Pierre N., Baiwir D., Huynh-Thu V.A., Mazzucchelli G., Smargiasso N., De Pauw E., et al. Discovery of biomarker candidates associated with the risk of short-term and mid/long-term relapse after infliximab withdrawal in Crohn's patients: a proteomics-based study. Gut. 2020 doi: 10.1136/gutjnl-2020-322100. [published Online First: 2020/10/28] [DOI] [PubMed] [Google Scholar]
- 147.Pierre N., Huynh-Thu V.A., Marichal T., Allez M., Bouhnik Y., Laharie D., et al. Distinct blood protein profiles associated with the risk of short-term and mid/long-term clinical relapse in patients with Crohn's disease stopping infliximab: when the remission state hides different types of residual disease activity. Gut. 2022 doi: 10.1136/gutjnl-2022-327321. [published Online First: 2022/08/26] [DOI] [PubMed] [Google Scholar]
- 148.Vitali R., Palone F., Armuzzi A., Fulci V., Negroni A., Carissimi C., et al. Proteomic analysis identifies three reliable biomarkers of intestinal inflammation in the stools of patients with Inflammatory Bowel Disease. J Crohns Colitis. 2022 doi: 10.1093/ecco-jcc/jjac110. [published Online First: 2022/08/31] [DOI] [PubMed] [Google Scholar]
- 149.Lehmann T., Schallert K., Vilchez-Vargas R., Benndorf D., Puttker S., Sydor S., et al. Metaproteomics of fecal samples of Crohn's disease and Ulcerative Colitis. J Proteomics. 2019;201:93–103. doi: 10.1016/j.jprot.2019.04.009. [published Online First: 2019/04/23] [DOI] [PubMed] [Google Scholar]
- 150.Ning L., Shan G., Sun Z., Lou X., Zhang F., Li S., et al. Serum proteome profiles to differentiate Crohn disease from intestinal tuberculosis and primary intestinal lymphoma: A pilot study. Medicine (Baltimore) 2019;98(50) doi: 10.1097/MD.0000000000018304. [published Online First: 2019/12/20] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sharma V. Differentiating intestinal tuberculosis and Crohn disease: Quo Vadis. Expert Rev Gastroenterol Hepatol. 2020;14(8):647–650. doi: 10.1080/17474124.2020.1785870. published Online First: 2020/06/20] [DOI] [PubMed] [Google Scholar]
- 152.Arafah K., Kriegsmann M., Renner M., Lasitschka F., Fresnais M., Kriegsmann K., et al. Microproteomics and Immunohistochemistry Reveal Differences in Aldo-Keto Reductase Family 1 Member C3 in Tissue Specimens of Ulcerative Colitis and Crohn's Disease. Proteomics Clin Appl. 2020;14(4) doi: 10.1002/prca.201900110. [published Online First: 2020/02/01] [DOI] [PubMed] [Google Scholar]
- 153.Wu J., Lubman D.M., Kugathasan S., Denson L.A., Hyams J.S., Dubinsky M.C., et al. Serum Protein Biomarkers of Fibrosis Aid in Risk Stratification of Future Stricturing Complications in Pediatric Crohn's Disease. Am J Gastroenterol. 2019;114(5):777–785. doi: 10.14309/ajg.0000000000000237. [published Online First: 2019/05/07] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ungaro R.C., Hu L., Ji J., Nayar S., Kugathasan S., Denson L.A., et al. Machine learning identifies novel blood protein predictors of penetrating and stricturing complications in newly diagnosed paediatric Crohn's disease. Aliment Pharmacol Ther. 2021;53(2):281–290. doi: 10.1111/apt.16136. [published Online First: 2020/11/02] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Šimurina M., de Haan N., Vučković F., Kennedy N.A., Štambuk J., Falck D., et al. Glycosylation of Immunoglobulin G Associates With Clinical Features of Inflammatory Bowel Diseases. Gastroenterology. 2018;154(5):1320–1333. doi: 10.1053/j.gastro.2018.01.002. [published Online First: 2018/01/09] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zhang X., Deeke S.A., Ning Z., Starr A.E., Butcher J., Li J., et al. Metaproteomics reveals associations between microbiome and intestinal extracellular vesicle proteins in pediatric inflammatory bowel disease. Nat Commun. 2018;9(1) doi: 10.1038/s41467-018-05357-4. [published Online First: 2018/07/22] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Vieujean S., Hu S., Bequet E., Salee C., Massot C., Bletard N., et al. Potential Role of Epithelial Endoplasmic Reticulum Stress and Anterior Gradient Protein 2 Homologue in Crohn's Disease Fibrosis. J Crohns Colitis. 2021;15(10):1737–1750. doi: 10.1093/ecco-jcc/jjab061. [published Online First: 2021/04/07] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zhang Y., Zha Z., Shen W., Li D., Kang N., Chen Z., et al. Anemoside B4 ameliorates TNBS-induced colitis through S100A9/MAPK/NF-κB signaling pathway. Chin Med. 2021;16(1) doi: 10.1186/s13020-020-00410-1. [published Online First: 2021/01/20] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Xie Y., Fontenot L., Chupina Estrada A., Nelson B., Wang J., Shih D.Q., et al. Elafin Reverses Intestinal Fibrosis by Inhibiting Cathepsin S-Mediated Protease-Activated Receptor 2. Cell Mol Gastroenterol Hepatol. 2022;14(4):841–876. doi: 10.1016/j.jcmgh.2022.06.011. [published Online First: 2022/07/16] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Misra B.B., Langefeld C.D., Olivier M., Cox L.A. Integrated Omics: Tools, Advances, and Future Approaches. J Mol Endocrinol. 2018 doi: 10.1530/jme-18-0055. [published Online First: 2018/07/15] [DOI] [PubMed] [Google Scholar]
- 161.Karczewski K.J., Snyder M.P. Integrative omics for health and disease. Nat Rev Genet. 2018;19(5):299–310. doi: 10.1038/nrg.2018.4. [published Online First: 2018/02/27] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Huang L.J., Mao X.T., Li Y.Y., Liu D.D., Fan K.Q., Liu R.B., et al. Multiomics analyses reveal a critical role of selenium in controlling T cell differentiation in Crohn's disease. Immunity. 2021;54(8):1728–1744. doi: 10.1016/j.immuni.2021.07.004. [published Online First: 2021/08/04] [DOI] [PubMed] [Google Scholar]
- 163.Metwaly A., Dunkel A., Waldschmitt N., Raj A.C.D., Lagkouvardos I., Corraliza A.M., et al. Integrated microbiota and metabolite profiles link Crohn's disease to sulfur metabolism. Nat Commun. 2020;11(1) doi: 10.1038/s41467-020-17956-1. [published Online First: 2020/08/30] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Gonzalez C.G., Mills R.H., Zhu Q., Sauceda C., Knight R., Dulai P.S., et al. Location-specific signatures of Crohn's disease at a multi-omics scale. Microbiome. 2022;10(1) doi: 10.1186/s40168-022-01331-x. [published Online First: 2022/08/24] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Zhang X., Li L., Butcher J., Stintzi A., Figeys D. Advancing functional and translational microbiome research using meta-omics approaches. Microbiome. 2019;7(1) doi: 10.1186/s40168-019-0767-6. [published Online First: 2019/12/08] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zhou M., Varol A., Efferth T. Multi-omics approaches to improve malaria therapy. Pharmacol Res. 2021;167 doi: 10.1016/j.phrs.2021.105570. [published Online First: 2021/03/27] [DOI] [PubMed] [Google Scholar]
- 167.Marigorta U.M., Denson L.A., Hyams J.S., Mondal K., Prince J., Walters T.D., et al. Transcriptional risk scores link GWAS to eQTLs and predict complications in Crohn's disease. Nat Genet. 2017;49(10):1517–1521. doi: 10.1038/ng.3936. published Online First: 2017/08/15] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Howell K.J., Kraiczy J., Nayak K.M., Gasparetto M., Ross A., Lee C., et al. DNA methylation and transcription patterns in intestinal epithelial cells from pediatric patients with inflammatory bowel diseases differentiate disease subtypes and associate with outcome. Gastroenterology. 2018;154(3):585–598. doi: 10.1053/j.gastro.2017.10.007. [published Online First: 2017/10/17] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lloyd-Price J., Arze C., Ananthakrishnan A.N., Schirmer M., Avila-Pacheco J., Poon T.W., et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019;569(7758):655–662. doi: 10.1038/s41586-019-1237-9. [published Online First: 2019/05/31] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zhou H., Beltrán J.F., Brito I.L. Host-microbiome protein-protein interactions capture disease-relevant pathways. Genome Biol. 2022;23(1) doi: 10.1186/s13059-022-02643-9. [published Online First: 2022/03/06] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.McHardy I.H., Goudarzi M., Tong M., Ruegger P.M., Schwager E., Weger J.R., et al. Integrative analysis of the microbiome and metabolome of the human intestinal mucosal surface reveals exquisite inter-relationships. Microbiome. 2013;1(1) doi: 10.1186/2049-2618-1-17. [published Online First: 2014/01/24] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Heinken A., Thiele I. Systematic prediction of health-relevant human-microbial co-metabolism through a computational framework. Gut Microbes. 2015;6(2):120–130. doi: 10.1080/19490976.2015.1023494. [published Online First: 2015/04/23] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Noecker C., Eng A., Srinivasan S., Theriot C.M., Young V.B., Jansson J.K., et al. Metabolic Model-Based Integration of Microbiome Taxonomic and Metabolomic Profiles Elucidates Mechanistic Links between Ecological and Metabolic Variation. mSystems. 2016;1(1) doi: 10.1128/mSystems.00013-15. [published Online First: 2016/05/31] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Manor O., Borenstein E. MUSiCC: a marker genes based framework for metagenomic normalization and accurate profiling of gene abundances in the microbiome. Genome Biol. 2015;16(1) doi: 10.1186/s13059-015-0610-8. [published Online First: 2015/04/18] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Dorrestein P.C., Mazmanian S.K., Knight R. Finding the missing links among metabolites, microbes, and the host. Immunity. 2014;40(6):824–832. doi: 10.1016/j.immuni.2014.05.015. [published Online First: 2014/06/21] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Mallick H., Franzosa E.A., McLver L.J., Banerjee S., Sirota-Madi A., Kostic A.D., et al. Predictive metabolomic profiling of microbial communities using amplicon or metagenomic sequences. Nat Commun. 2019;10(1) doi: 10.1038/s41467-019-10927-1. [published Online First: 2019/07/19] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Erickson A.R., Cantarel B.L., Lamendella R., Darzi Y., Mongodin E.F., Pan C., et al. Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of Crohn's disease. PLoS One. 2012;7(11) doi: 10.1371/journal.pone.0049138. [published Online First: 2012/12/05] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Jansson J., Willing B., Lucio M., Fekete A., Dicksved J., Halfvarson J., et al. Metabolomics reveals metabolic biomarkers of Crohn's disease. PLoS One. 2009;4(7) doi: 10.1371/journal.pone.0006386. [published Online First: 2009/07/29] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Suskind D.L., Lee D., Kim Y.M., Wahbeh G., Singh N., Braly K., et al. The specific carbohydrate diet and diet modification as induction therapy for pediatric Crohn's disease: a randomized diet controlled trial. Nutrients. 2020;12(12) doi: 10.3390/nu12123749. [published Online First: 2020/12/10] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lee J.W.J., Plichta D., Hogstrom L., Borren N.Z., Lau H., Gregory S.M., et al. Multi-omics reveal microbial determinants impacting responses to biologic therapies in inflammatory bowel disease. Cell Host Microbe. 2021;29(8):1294–1304. doi: 10.1016/j.chom.2021.06.019. [published Online First: 2021/07/24] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kim M., Tagkopoulos I. Data integration and predictive modeling methods for multi-omics datasets. Mol Omics. 2018;14(1):8–25. doi: 10.1039/c7mo00051k. [published Online First: 2018/05/05] [DOI] [PubMed] [Google Scholar]
- 182.Wani N., Raza K. Integrative approaches to reconstruct regulatory networks from multi-omics data: A review of state-of-the-art methods. Comput Biol Chem. 2019;83 doi: 10.1016/j.compbiolchem.2019.107120. [published Online First: 2019/09/10] [DOI] [PubMed] [Google Scholar]
- 183.St John P.C., Bomble Y.J. Approaches to computational strain design in the multiomics era. Front Microbiol. 2019;10 doi: 10.3389/fmicb.2019.00597. published Online First: 2019/04/27] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Jacobs J.P., Goudarzi M., Lagishetty V., Li D., Mak T., Tong M., et al. Crohn's disease in endoscopic remission, obesity, and cases of high genetic risk demonstrates overlapping shifts in the colonic mucosal-luminal interface microbiome. Genome Med. 2022;14(1) doi: 10.1186/s13073-022-01099-7. [published Online First: 2022/08/16] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Berlinberg A.J., Regner E.H., Stahly A., Brar A., Reisz J.A., Gerich M.E., et al. Multi 'omics analysis of intestinal tissue in ankylosing Spondylitis identifies alterations in the tryptophan metabolism pathway. Front Immunol. 2021;12 doi: 10.3389/fimmu.2021.587119. [published Online First: 2021/03/23] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kakuta Y., Ichikawa R., Fuyuno Y., Hirano A., Umeno J., Torisu T., et al. An integrated genomic and transcriptomic analysis reveals candidates of susceptibility genes for Crohn's disease in japanese populations. Sci Rep. 2020;10(1) doi: 10.1038/s41598-020-66951-5. [published Online First: 2020/06/26] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Connors J., Dunn K.A., Allott J., Bandsma R., Rashid M., Otley A.R., et al. The relationship between fecal bile acids and microbiome community structure in pediatric Crohn's disease. Isme j. 2020;14(3):702–713. doi: 10.1038/s41396-019-0560-3. [published Online First: 2019/12/05] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Brückner A., Werkstetter K.J., Frivolt K., Shokry E., Ahmed M., Metwaly A., et al. Partial enteral nutrition has no benefit on bone health but improves growth in paediatric patients with quiescent or mild Crohn's disease. Clin Nutr. 2020;39(12):3786–3796. doi: 10.1016/j.clnu.2020.04.012. [published Online First: 2020/05/08] [DOI] [PubMed] [Google Scholar]
- 189.Borren N.Z., Plichta D., Joshi A.D., Bonilla G., Sadreyev R., Vlamakis H., et al. Multi-"-omics" profiling in patients with quiescent inflammatory bowel disease identifies biomarkers predicting relapse. Inflamm Bowel Dis. 2020;26(10):1524–1532. doi: 10.1093/ibd/izaa183. [published Online First: 2020/08/09] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Franzosa E.A., Sirota-Madi A., Avila-Pacheco J., Fornelos N., Haiser H.J., Reinker S., et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol. 2019;4(2):293–305. doi: 10.1038/s41564-018-0306-4. [published Online First: 2018/12/12] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Parkes M. The genetics universe of Crohn's disease and ulcerative colitis. Dig Dis. 2012;30(Suppl 1):78–81. doi: 10.1159/000341130. published Online First: 2012/10/25] [DOI] [PubMed] [Google Scholar]
- 192.Tam V., Patel N., Turcotte M., Bossé Y., Paré G., Meyre D. Benefits and limitations of genome-wide association studies. Nat Rev Genet. 2019;20(8):467–484. doi: 10.1038/s41576-019-0127-1. [published Online First: 2019/05/10] [DOI] [PubMed] [Google Scholar]
- 193.Lee J.C., Parkes M. Genome-wide association studies and Crohn's disease. Brief Funct Genomics. 2011;10(2):71–76. doi: 10.1093/bfgp/elr009. [published Online First: 2011/03/26] [DOI] [PubMed] [Google Scholar]
- 194.Xie T., Gorenjak V., GS M., Dadé S., Marouli E., Masson C., et al. Epigenome-wide association study (EWAS) of blood lipids in healthy population from STANISLAS family study (SFS) Int J Mol Sci. 2019;20(5) doi: 10.3390/ijms20051014. [published Online First: 2019/03/01] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Langevin S.M., Kelsey K.T. The fate is not always written in the genes: epigenomics in epidemiologic studies. Environ Mol Mutagen. 2013;54(7):533–541. doi: 10.1002/em.21762. [published Online First: 2013/02/28] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Breeze C.E., Wong J.Y.Y., Beck S., Berndt S.I., Franceschini N. Diversity in EWAS: current state, challenges, and solutions. Genome Med. 2022;14(1) doi: 10.1186/s13073-022-01065-3. [published Online First: 2022/07/07] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Paul D.S., Beck S. Advances in epigenome-wide association studies for common diseases. Trends Mol Med. 2014;20(10):541–543. doi: 10.1016/j.molmed.2014.07.002. [published Online First: 2014/08/06] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Fuks G., Elgart M., Amir A., Zeisel A., Turnbaugh P.J., Soen Y., et al. Combining 16S rRNA gene variable regions enables high-resolution microbial community profiling. Microbiome. 2018;6(1) doi: 10.1186/s40168-017-0396-x. [published Online First: 2018/01/28] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Quince C., Walker A.W., Simpson J.T., Loman N.J., Segata N. Shotgun metagenomics, from sampling to analysis. Nat Biotechnol. 2017;35(9):833–844. doi: 10.1038/nbt.3935. [published Online First: 2017/09/13] [DOI] [PubMed] [Google Scholar]
- 200.Laudadio I., Fulci V., Palone F., Stronati L., Cucchiara S., Carissimi C. Quantitative Assessment of Shotgun Metagenomics and 16S rDNA Amplicon Sequencing in the Study of Human Gut Microbiome. Omics. 2018;22(4):248–254. doi: 10.1089/omi.2018.0013. [published Online First: 2018/04/14] [DOI] [PubMed] [Google Scholar]
- 201.Albertsen M. Long-read metagenomics paves the way toward a complete microbial tree of life. Nat Methods. 2023;20(1):30–31. doi: 10.1038/s41592-022-01726-6. [published Online First: 2023/01/13] [DOI] [PubMed] [Google Scholar]
- 202.Azad R.K., Shulaev V. Metabolomics technology and bioinformatics for precision medicine. Brief Bioinform. 2019;20(6):1957–1971. doi: 10.1093/bib/bbx170. [published Online First: 2018/01/06] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Wishart D.S. Metabolomics for investigating physiological and pathophysiological processes. Physiol Rev. 2019;99(4):1819–1875. doi: 10.1152/physrev.00035.2018. [published Online First: 2019/08/23] [DOI] [PubMed] [Google Scholar]
- 204.Sas K.M., Karnovsky A., Michailidis G., Pennathur S. Metabolomics and diabetes: analytical and computational approaches. Diabetes. 2015;64(3):718–732. doi: 10.2337/db14-0509. [published Online First: 2015/02/26] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Collins B.C., Hunter C.L., Liu Y., Schilling B., Rosenberger G., Bader S.L., et al. Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry. Nat Commun. 2017;8(1) doi: 10.1038/s41467-017-00249-5. [published Online First: 2017/08/23] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Anjo S.I., Santa C., Manadas B. SWATH-MS as a tool for biomarker discovery: From basic research to clinical applications. Proteomics. 2017;17(3-4) doi: 10.1002/pmic.201600278. [published Online First: 2017/01/28] [DOI] [PubMed] [Google Scholar]
- 207.Gillet L.C., Navarro P., Tate S., Rost H., Selevsek N., Reiter L., et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics. 2012;11(6) doi: 10.1074/mcp.O111.016717. [published Online First: 2012/01/21] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Sidoli S., Fujiwara R., Garcia B.A. Multiplexed data independent acquisition (MSX-DIA) applied by high resolution mass spectrometry improves quantification quality for the analysis of histone peptides. Proteomics. 2016;16(15-16):2095–2105. doi: 10.1002/pmic.201500527. [published Online First: 2016/05/20] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Beer L.A., Liu P., Ky B., Barnhart K.T., Speicher D.W. Efficient quantitative comparisons of plasma proteomes using label-free analysis with MaxQuant. Methods Mol Biol. 2017;1619:339–352. doi: 10.1007/978-1-4939-7057-5_23. [published Online First: 2017/07/05] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Neilson K.A., Ali N.A., Muralidharan S., Mirzaei M., Mariani M., Assadourian G., et al. Less label, more free: approaches in label-free quantitative mass spectrometry. Proteomics. 2011;11(4):535–553. doi: 10.1002/pmic.201000553. [published Online First: 2011/01/19] [DOI] [PubMed] [Google Scholar]