

Abstract
Most solid tumors become stiff with progression of cancer. Cancer Associated Fibroblasts (CAFs), most abundant stromal cells in the tumor microenvironment (TME), are known to mediate such stiffening. While the biochemical crosstalk between CAFs and cancer cells have been widely investigated, it is not clear if and how CAFs in stiffer TME promote metastatic progression. To gather insights into the process, we controlled the mechanical stiffness of the substrates and collected gene expression data with human colorectal CAFs. We cultured human primary CAFs on 2D polyacrylamide hydrogels with increasing elastic modulus (E) of 1, 10 and 40 kPa, and performed genome-wide transcriptome analyses in these cells to identify expression levels of ~16000 genes. The high-quality RNAseq results can be an excellent data-source for bioinformatic analysis for identifying novel pathways and biomarkers in cancer development and metastatic progression. With thorough analysis and accurate interpretation, this data may help researchers understand the role of mechanical stiffness of the TME in CAF-cancer cell crosstalk.
Subject terms: Cancer microenvironment, Cancer genomics, Gene ontology, Gene expression, Computational biophysics
| Design Type(s) | solid tumor development model • biophysical progression in colorectal cancer • transcription profiling by high throughput sequencing design |
| Measurement Type(s) | transcription profiling assay |
| Technology Type(s) | RNA sequencing |
| Factor Type(s) | substrate mechanical stiffness • cancer progression stage |
| Sample Characteristic(s) | Human (Homo sapiens) • colorectal cancer (CRC)• cancer associated fibroblast |
Background & Summary
Cancer metastasis is a complex process that involves dynamic crosstalk between cancer and stromal cells. Only recently have we begun to understand that tumor microenvironment (TME) is a key mediator of such interactions. However, the precise mechanism by which TME influences metastasis is still not well understood. Both physical and chemical properties of TME play major roles in prompting the cancer cells to undergo epithelial to mesenchymal transition (EMT) that leads to metastasis1,2. Among many factors of the TME, the most prominent is increasing stiffness of solid tumors3–5. Extracellular matrix (ECM) stiffness and/or tumor rigidity are known to facilitate pro-metastatic progression in many types of cancer such as breast and colorectal cancer (CRC)2,6. Here we share transcriptome resources that researchers can utilize for understanding the role of TME stiffness in crosstalk between stromal fibroblasts and cancer cells.
Among the different types of stromal cells, CAFs are considered to be most important; because they are a key player in tumorigenesis and constitute the majority of the stromal population7,8. While genomic heterogeneity of cancer cells has been extensively studied, we know little about the heterogeneity of stromal cells. It is also not clear how the stromal population diversifies as the TME evolves with time. To address this gap, we focused on CAFs (CAF05, human colorectal primary CAFs from Neuromics) in this study to explore how increased substrate stiffness modulates their gene expressions that potentially facilitate metastatic progression. PolyA + paired-end RNA-seq was performed in biological duplicates from CAFs grown on polyacrylamide hydrogel (PA gel) substrates of 1 kPa, 10 kPa and 40 kPa elastic modulus. To represent typical in vitro culture condition, CAFs were also collected from polystyrene substrates (elastic modulus of 3.4 GPa). Figure 1 shows CAFs cultured on substrates with different mechanical stiffness. Currently, we do not have publicly available datasets for colorectal CAF transcriptome characterized with increasing substrate stiffness.
Fig. 1.

Morphologies/phenotypes of CAFs on substrates of (a) 1 kPa (b) 10 kPa, (c) 40 kPa and (d) 3.4 GPa (plastic) elastic modulus. Immunocytochemistry of α-Smooth Muscle Actin (red) and nuclei (blue) shows cell spread, stress fibers and nuclear shapes on different stiffness. Images are maximum intensity projections from a z-stack of confocal images. Scale bars: 50 um.
We have utilized high-precision sequencing technology (RNA-seq) to characterize gene expressions. Quality control and validation of the data is presented in Fig. 2. All relevant metrics indicate that the RNA-seq and analysis data is of high quality. This genome-wide transcriptome dataset may be mined by the research community for further studies. With detailed analysis and accurate interpretation, the data may help find novel pathways that get influenced by mechanical stiffness of the TME and understand CAF-cancer cell crosstalk. Analyses presented here were conducted using Illumina RNA-seq along with a bioinformatic algorithm described later to ensure sequence quality.
Fig. 2.

Quality assessment by FastQC shows high quality sequences for all samples. (a) General quality statistics of each RNA-seq sample from FastQC. (b) Principal Component Analysis (PCA) showing the variations between the RNA-seq samples. (c) Sequence quality score per base from FastQC. y-axis shows the mean Phred score, wherein Phred score >28 was defined as “very good quality”. x-axis shows the base position in the read. (d) Quality score per sequence from FastQC. y-axis shows the number of reads and x-axis shows the Phred score. (e) GC content per sequence.
Materials and Methods
Substrate preparation
Substrates with different elastic moduli (1, 10 and 40 kPa) were prepared with Polyacrylamide (PA) gel layered on top of glass-bottom petri dishes (Cellvis). First, the glass surfaces were silanized with (3-Aminopropyl)trimethoxysilane (APTES) solution (Sigma-Aldrich) for 5 mins. Next, the silanized surfaces were functionalized with 0.5% glutaraldehyde solution (Polysciences) for 30 mins. PA gel precursor solutions were prepared by mixing Acrylamide (40% sol., Sigma-Aldrich), N,N′-Methylenebis- acrylamide (2% sol., Sigma-Aldrich) and Phosphate Buffered Saline (PBS, Corning) at specific ratios according to the protocol9,10. Addition of 1% (v/v) of ammonium persulfate (APS) solution (10% w/v APS, Bio-Rad) and 0.1% (v/v) of Tetramethylethylenediamine (TEMED) solution (Bio-Rad) starts the polymerization of the gel. For a 30 mm diameter surface, 95 ul PA gel solution is added to bottom glasses, and then sandwiched with clean coverslips. After polymerization, the top coverslips were removed and 0.2 mg/ml sufosuccinimidyl-6- (4′-azido- 2′-nitrophenylamino)- hexanoate (Sulfo-SANPAH, ThermoFisher Scientific) solution in HEPES buffer (50 mM HEPES at pH 8.5, ThermoFisher Scientific) was applied. Next, activation with 365 nm UV light (8 Watt, UVP UVL-28, Analytik Jena, US) for 20 mins binds the Sulfo-SANPAH cross-linkers to the PA gel. For ECM functionalization, 25 µg/ml Fibronectin (Human, Corning) solution in HEPES buffer was prepared and the substrates were soaked in the ECM solution overnight. After ECM coating, 3 times rinsing with PBS prepared the substrates for cell plating.
Cell culture
Human primary colorectal tumor CAFs, CAF05 (Neuromics), were maintained in VitroPlus III Low Serum, Complete medium (Cat. # PC00B1, Neuromics) supplemented with 1% penicillin-streptomycin (Lonza). Cells were grown at 37 °C in a humidified incubator with 5% CO2. We used the CAFs with low passage numbers (passage 3–9), and the CAFs are alpha-SMA positive. This indicates that this population does not have any epithelial cancer cells. The culture media was not supplemented with exogenous TGF-beta or Matrigel. However, the substrates were coated with fibronectin for cell attachment. Other relevant metadata is presented in Table 1.
Table 1.
Clinical information on CAF05.
| Cell | Tissue Type | Tumor Stage | Grade | T/N/M | Donor ID | Sex | Age | Race | Treatment |
|---|---|---|---|---|---|---|---|---|---|
| CAF05 | Colo- rectal tumor | N/A | N/A | N/A | BTC1000-E1110035885100614HS | Male | 55 | Caucasian | N/A |
RNA extraction and sequencing
After 48 hours of culture on substrates of specified stiffness, RNA was extracted using Trizol reagent (Invitrogen) as per manufacturer instructions. Samples for RNA-seq were further cleaned up by RNeasy Mini Kit (QIAGEN). The RNA-seq libraries were prepared with Illumina TruSeq Stranded mRNAseq Sample Prep kit (Illumina). Paired-end, polyA + RNA-sequencing was performed on Illumina platform at the Roy J. Carver Biotechnology Center at UIUC.
Quality validation and RNA-seq analysis
The quality of the RNA-seq libraries were evaluated using FastQC (http://www.bioinformatics.babraham. ac.uk/projects/fastqc/). The reads were aligned to the human genome assembly GRCh38.p13 using HISAT211. The gene counts were first quantified using HTseq-Count12, then the gene expression was analyzed using edgeR13. Normalization of library size was performed by calcNormFactors in edgeR with the default TMM method. Heatmaps were plotted using the coolmap function from the limma package14 with row centering and scaling. Hierarchical clustering of genes was performed with average-linkage method. Differentially expressed genes were defined by |log2(fold change)| >1 and FDR <0.05. Gene ontology analyses, GSEA (gene set enrichment analysis), and disease ontology analysis were performed using clusterProfiler of Bioconductor15,16.
Data Records
Data from the RNA-seq were deposited to the NCBI Gene Expression Omnibus (GEO) under accession number GSE22974217.
Technical validation
Library quality
RNA quality was assessed using Agilent Fragment Analyzer in the Roy J. Carver Biotechnology Center, and all samples were determined to be suitable for poly (A) selection and sequencing. 49 to 87 million poly (A)-selected paired-end sequence reads were obtained per sample from Illumina NovaSeq 6000 in the Roy J. Carver Biotechnology Center (Fig. 2A). The quality of the RNA-seq libraries were evaluated using FastQC (http://www.bioinformatics.babraham. ac.uk/projects/fastqc/).
Quality validation
The mean Phred quality score across the reads were evaluated and plotted by FastQC, which shows that all samples have a mean per base Phred score >34 (Fig. 2C). The Phred quality score per sequence also shows that the sequencing reads have an overall high quality, Phred score >28 (Fig. 2D). Principal Component Analysis (PCA) shown in Fig. 2B confirms the similarity between most biological replicates PC1 explains 42.62% of the variance and PC2 explains 23.65% of the variance. The GC contents of the samples are also within 40–50% (Fig. 2E).
Immunostaining
Cells were fixed, permeabilized and blocked with 4% Paraformaldehyde (PFA) in PBS (30 mins), 0.2% Triton X-100 in PBS, and 2.5% bovine serum albumin (BSA) with 2% normal goat serum (NGS) in PBS. Following overnight incubation in αSMA antibody (1:500) (Sigma, cat. no. A5228), the samples were incubated with Alexa Fluor 568 conjugated secondary antibody (1:1000) (Abcam Inc., cat. no. ab175695) at 4 °C for 12 hrs. Next, the samples were incubated in 4′,6-diamidino-2-phenylindole (DAPI) (1:1000) (Invitrogen, cat. no. D1306) for 10 minutes and imaged with LSM710 (Zeiss) confocal microscope using an EC Plan-Neofluar 20X/0.5 NA objective lens (Zeiss).
Usage Notes
The RNA-seq data shared in this article can be processed using a collection of open access tools. For instance, the raw fastq data can be aligned to human reference genome assembly (e.g. GRCh38.p13) using aligners such as STAR11 and HISAT218. For this study, we used the HISAT2 aligner. Other compatible aligners can also be used for this purpose and alignment can be accessed by various genome browsers such as ZENBU19, Integrative Genome Viewer (IGV)20 or UCSC Genome Browser21. Differential gene expression analysis can be performed using publicly available packages such as edgeR13, DESeq 222 and CuffDiff223. We have used edgeR with TMM normalization to analyze the differentially expressed genes from various mechanical stiffness of the substrates and performed hierarchical clustering of genes with average-linkage method. Gene ontology (GO) analyses, GSEA (gene set enrichment analysis), and disease ontology analysis can be performed using clusterProfiler of Bioconductor15,16. It should be noted that there are methods available for alignment-free differential gene expression analysis. For such applications, transcript quantification can be performed using Sailfish24 or Kallisto25. Gene-level abundance estimates, and statistical inference can be made using packages such as tximport26 and then differential expression can be determined with DESeq 2 or edgeR.
Data shared here provides insights into the biological processes that take place in CAFs while they adapt to increasing rigidity in the tumor microenvironment. Although our cells are taken from colorectal cancer, these results should also be relevant to other solid form of tumors such as breast, prostate and lung cancers. Also, the culture conditions do not allow cross-talk between CAFs and cancer cells. Hence, these results strictly represent the effects of mechanical stimulation of the CAFs. Hierarchical clustered heatmaps show that stiffness alone can result in differential expression of many genes (Fig. 3). Gene ontology (GO) analyses and gene set enrichment analyses (GSEA) show that substrate stiffness has profound influence on many signaling pathways such as chromatin assembly, nuclear organization, cell membrane function and cytoskeleton organization in CAFs. Further analysis of the data is required to identify novel pathways and biomarkers in cancer development and metastatic progression. In addition, this data can be compared with RNAseq data from primary cancer cells to identify pathways that may be regulated by stiffness-dependent cross-talk between CAFs and cancer cells. This data may also provide guidance for choosing biomechanics based therapeutic targets. As a result, we are publicly sharing this data for researchers in relevant fields to encourage further reuse of the transcriptomics.
Fig. 3.
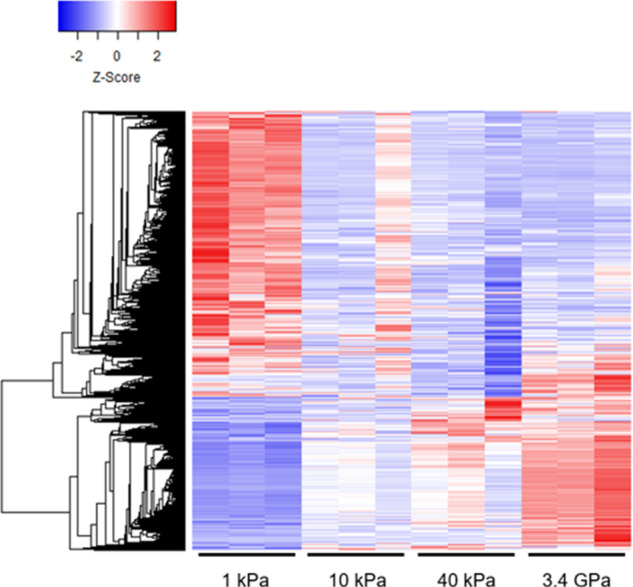
CAFs undergo significant gene expression changes when grown on substrates of increasing stiffness. Hierarchical clustered heatmaps showing the relative expression levels of differentially expressed genes based on RNA-seq. Rows represent genes which are hierarchically clustered using the average-linkage clustering method. Columns represent the samples, showing three biological replicates of CAFs grown at 1 kPa, 10 ka, 40 kPa, and 3.4 GPa of stiffness.
Acknowledgements
We thank Minxue Liu for preparing RNA samples for RNA-seq analyses. Research reported in this publication was partially supported by the National Institute of Biomedical Imaging and Bioengineering of the NIH under award number T32EB019944 to B.E and Beckman Institute Graduate Fellowship program. Funding was also provided by NSF grant NSF ECCS 1934991, Mayo Clinic grant PO 66236006, and Cancer Center at Illinois seed grants to M.T.A.S. Work conducted in K.V.P. laboratory is supported by grants from National Institute of Health (R21-AG065748 & R01-GM132458), Cancer Center at Illinois seed grants, Prairie Dragon Paddlers and NSF EAGER 1723008. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Author contributions
B.E., K.V.P. and M.T.A.S. conceived and designed the experiments. B.E. prepared the substrates, cultured cells and harvested RNAs. Y.J.S. performed the RNA-seq analyses. M.V.K. and Y.J.S. performed the validation of the RNA-seq targets. B.E., Y.J.S., K.V.P. and M.T.A.S. wrote the manuscript. All authors have read and approved the final manuscript.
Code availability
The following software and versions were used for quality control and data analysis:
1. FastQC, version 0.11.8 and MultiQC, version 1.7 were used for quality analysis of raw FASTQ sequencing data: http://www.bioinformatics.babraham. ac.uk/projects/fastqc/
2. HISAT2 was used for mapping of sequence reads to the human GRCh38.13 genome assembly: http://www.ccb.jhu.edu/software/hisat/index.shtml
3. HTSeq, version 0.9.1 was used for calculating the gene counts: http://bioinf.wehi.edu.au/featureCounts/
4. edgeR, version 3.34.1 was used for normalization and visualization of differential gene expression analysis output: https://bioconductor.org/packages/release/bioc/html/edgeR.html
Software and codes are open source and readily available.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Bashar Emon, You Jin Song
References
- 1.Emon, B., Bauer, J., Jain, Y., Jung, B. & Saif, T. Biophysics of Tumor Microenvironment and Cancer Metastasis - A Mini Review. Computational and Structural Biotechnology Journal16, 279–287, 10.1016/j.csbj.2018.07.003 (2018). [DOI] [PMC free article] [PubMed]
- 2.Bauer J, et al. Increased stiffness of the tumor microenvironment in colon cancer stimulates cancer associated fibroblast-mediated prometastatic activin A signaling. Sci Rep. 2020;10:1–11. doi: 10.1038/s41598-019-55687-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levental KR, et al. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Handorf, A. M., Zhou, Y., Halanski, M. A. & Li, W. J. Tissue stiffness dictates development, homeostasis, and disease progression. Organogenesis11, 1–15, 10.1080/15476278.2015.1019687 (2015). [DOI] [PMC free article] [PubMed]
- 5.Malik, R., Lelkes, P. I. & Cukierman, E. Biomechanical and biochemical remodeling of stromal extracellular matrix in cancer. Trends in Biotechnology33, 230–236, 10.1016/j.tibtech.2015.01.004 (2015). [DOI] [PMC free article] [PubMed]
- 6.Cox TR, Erler JT. Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis Model Mech. 2011;4:165–178. doi: 10.1242/dmm.004077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bussard KM, Mutkus L, Stumpf K, Gomez-Manzano C, Marini FC. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Research 2016 18:1. 2016;18:1–11. doi: 10.1186/s13058-016-0740-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Öhlund D, Elyada E, Tuveson D. Fibroblast heterogeneity in the cancer wound. Journal of Experimental Medicine. 2014;211:1503–1523. doi: 10.1084/jem.20140692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tse JR, Engler AJ. Preparation of Hydrogel Substrates with Tunable Mechanical Properties. Curr Protoc Cell Biol. 2010;47:10.16.1–10.16.16. doi: 10.1002/0471143030.cb1016s47. [DOI] [PubMed] [Google Scholar]
- 10.Knoll, S. G., Ali, M. Y. & Saif, M. T. A. A Novel Method for Localizing Reporter Fluorescent Beads Near the Cell Culture Surface for Traction Force Microscopy. Journal of Visualized Experiments, 10.3791/51873 (2014). [DOI] [PMC free article] [PubMed]
- 11.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ritchie ME, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu G, Wang LG, Han Y, He QY. ClusterProfiler: An R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Piñero, J. et al. DisGeNET: a discovery platform for the dynamical exploration of human diseases and their genes. Database (Oxford)2015 (2015). [DOI] [PMC free article] [PubMed]
- 17.Emon B, Song YJ, Prasanth KV, Saif MTA. 2023. RNA-seq of CAF05s grown on different substrates. Gene Expression Omnibus. //identifiers.org/geo/GSE229742
- 18.HISAT. http://www.ccb.jhu.edu/software/hisat/index.shtml.
- 19.Severin J, et al. Interactive visualization and analysis of large-scale sequencing datasets using ZENBU. Nat Biotechnol. 2014;32:217–219. doi: 10.1038/nbt.2840. [DOI] [PubMed] [Google Scholar]
- 20.Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kent WJ, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq 2. Genome Biol. 2014;15:1–21. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trapnell C, et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nature Biotechnology 2012 31:1. 2012;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patro R, Mount SM, Kingsford C. Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nature Biotechnology 2014 32:5. 2014;32:462–464. doi: 10.1038/nbt.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nature Biotechnology 2016 34:5. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 26.Soneson, C., Love, M. I. & Robinson, M. D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Res4 (2016). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Emon B, Song YJ, Prasanth KV, Saif MTA. 2023. RNA-seq of CAF05s grown on different substrates. Gene Expression Omnibus. //identifiers.org/geo/GSE229742
Data Availability Statement
The following software and versions were used for quality control and data analysis:
1. FastQC, version 0.11.8 and MultiQC, version 1.7 were used for quality analysis of raw FASTQ sequencing data: http://www.bioinformatics.babraham. ac.uk/projects/fastqc/
2. HISAT2 was used for mapping of sequence reads to the human GRCh38.13 genome assembly: http://www.ccb.jhu.edu/software/hisat/index.shtml
3. HTSeq, version 0.9.1 was used for calculating the gene counts: http://bioinf.wehi.edu.au/featureCounts/
4. edgeR, version 3.34.1 was used for normalization and visualization of differential gene expression analysis output: https://bioconductor.org/packages/release/bioc/html/edgeR.html
Software and codes are open source and readily available.