

Abstract
Objective
The idiopathic inflammatory myopathies (IIMs) are heterogeneous diseases thought to be initiated by immune activation in genetically predisposed individuals. We imputed variants from the ImmunoChip array using a large reference panel to fine‐map associations and identify novel associations in IIM.
Methods
We analyzed 2,565 Caucasian IIM patient samples collected through the Myositis Genetics Consortium (MYOGEN) and 10,260 ethnically matched control samples. We imputed 1,648,116 variants from the ImmunoChip array using the Haplotype Reference Consortium panel and conducted association analysis on IIM and clinical and serologic subgroups.
Results
The HLA locus was consistently the most significantly associated region. Four non‐HLA regions reached genome‐wide significance, SDK2 and LINC00924 (both novel) and STAT4 in the whole IIM cohort, with evidence of independent variants in STAT4, and NAB1 in the polymyositis (PM) subgroup. We also found suggestive evidence of association with loci previously associated with other autoimmune rheumatic diseases (TEC and LTBR). We identified more significant associations than those previously reported in IIM for STAT4 and DGKQ in the total cohort, for NAB1 and FAM167A‐BLK loci in PM, and for CCR5 in inclusion body myositis. We found enrichment of variants among DNase I hypersensitivity sites and histone marks associated with active transcription within blood cells.
Conclusion
We found novel and strong associations in IIM and PM and localized signals to single genes and immune cell types.
INTRODUCTION
The idiopathic inflammatory myopathies (IIMs) are a heterogeneous group of rare autoimmune diseases primarily characterized by muscle weakness with extramuscular manifestations. The strongest genetic risk for IIM resides in the HLA region, although non‐HLA associations have also been reported. To date, the largest genetic association studies have been conducted in Caucasian populations through the Myositis Genetics Consortium (MYOGEN) (1) and subsequent meta‐analyses (2). However, a genome‐wide association study (GWAS) in clinically amyopathic dermatomyositis in the Japanese population (3) and candidate gene studies in Japanese and Chinese populations have also identified significant genetic risk factors for IIM (4, 5, 6).
We previously published a genetic association study on 90,536 genetic variants from ImmunoChip, a targeted array containing coverage of 186 established autoimmune susceptibility loci (1). In this follow‐up study we re‐analyzed the IIM ImmunoChip data set after imputation of 1,648,116 variants to identify novel associations and to facilitate fine‐mapping of risk regions reported in IIM and clinical and serologic subgroups.
PATIENTS AND METHODS
Samples
Caucasian IIM patient samples were collected through MYOGEN (1). IIM samples were included if patients fulfilled probable or definite Bohan and Peter classification criteria (7) for polymyositis (PM), juvenile PM, dermatomyositis (DM), or juvenile DM, and Griggs, European Neuromuscular Centre, or Medical Research Council criteria for inclusion body myositis (IBM) (8). ImmunoChip control data from 12 countries were provided by 4 disease consortia. Anti–Jo‐1 (anti–histidyl–transfer RNA) autoantibodies were detected using either immunoprecipitation or line blot using methods described previously (9).
Genotyping and imputation
Analysis was conducted on the existing Illumina ImmunoChip array data consisting of 2,565 IIM patients (1). Clinical subgroup analysis was conducted on PM (n = 903), DM (n = 817), juvenile DM (n = 508), and IBM (n = 252) patients, and patients with anti–Jo‐1 autoantibodies (n = 311). Healthy control samples were collected from the same pool of controls but matched with patients 4:1 based on principal components analysis coordinates (9). Variants were imputed with the Michigan Imputation Server using the Haplotype Reference Consortium (HRC) panel version r1.1 2016. The HRC consists of 64,940 haplotypes from individuals of predominantly European ancestry. Poorly imputed genotypes (r2 < 0.5), single‐nucleotide polymorphisms (SNPs) deviating from Hardy‐Weinberg equilibrium in controls (P < 0.001), and variants with a low minor allele frequency of <0.01 were removed. After stringent SNP quality control, we analyzed 1,648,116 variants, of which 120,734 were directly genotyped SNPs. Population stratification was assessed by the genomic inflation factor (λ) scaled to 1,000 patients and 1,000 controls. Including the top 3 principal components as covariates was sufficient to control for population differences (λ for 1,000 samples = 1.04).
Statistical analysis
Association analysis was conducted on gene dosages using SNPTest version 2.5.2. Linkage disequilibrium between SNPs was calculated using Plink version 1.90. The first 3 principal components were included as covariates and used in a logistic regression analysis using an additive model. Forward stepwise logistic regression was used to test for independent effects conditional on the variant of interest. Genome‐wide significance was defined as a P value less than 5 × 10−8. Suggestive significance was defined as a P value less than 2.25 × 10−5, based on Bonferroni correction for the number of independent haplotype blocks on the original genotyping array. Regions were defined as novel if there was no genome‐wide or suggestive evidence of association in previous genetic analyses. Regional association plots were generated using LocusZoom.js (10).
Functional analysis
We reported the functional effect of the most strongly associated SNP in the locus from the dbSNP database's predicted functional effect. GARFIELD (GWAS Analysis of Regulatory and Functional Information Enrichment with Linkage Disequilibrium correction) analysis was used to characterize the cellular and regulatory contribution of the associated variants (11). The “gwas‐credible‐sets” package implemented in LocusZoom.js was used to calculate 95% credible SNP sets (10), which were annotated with functional information from public databases, including Genotype‐Tissue Expression (GTEx) and splicing quantitative trait locus (QTL), and regulatory information from the UCSC Genome Browser.
RESULTS
Regions of interest associated with IIM and clinical subgroups are presented in Table 1 and Figure 1. SNPs are reported if they reached genome‐wide significance, or reached suggestive significance and the locus has previous statistical evidence of association in an autoimmune rheumatic disease in the National Human Genome Research Institute–European Bioinformatics Institute GWAS Catalog. All associations reaching a suggestive significance threshold of P < 2.25 × 10−5 are included in the Supplementary Data, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42434. A Manhattan plot of the combined IIM analysis is shown in Figure 1. Manhattan plots for subgroup analyses and regional association plots for non‐HLA associations are included in Supplementary Figures 1–19, https://onlinelibrary.wiley.com/doi/10.1002/art.42434.
Table 1.
Loci associated with IIM reaching genome‐wide significance or suggestive significance compared to matched healthy controls*
| Gene region | Subgroup | SNP | Chromosome | Position | Minor Allele | MAF in Patients | MAF in Controls | P | OR (95% CI) | Function† | Overlap |
|---|---|---|---|---|---|---|---|---|---|---|---|
| HLA–DRA | IIM | rs9268813 | 6 | 32424594 | C | 0.24 | 0.11 | 6.69 × 10−120 | 2.49 (2.30–2.69) | – | Multiple |
| STAT4 ‡ | IIM | rs4853540 | 2 | 191917317 | T | 0.19 | 0.22 | 1.38 × 10−8 | 0.81 (0.75–0.87) | Intronic | RA (ref. 14), SLE (ref. 15), SSc (ref. 16) |
| SDK2 | IIM | rs7209879 | 17 | 71532097 | T | 0.37 | 0.34 | 1.46 × 10−8 | 1.15 (1.08–1.23) | Intronic | – |
| LINC00924 | IIM | rs8040452 | 15 | 96197257 | T | 0.37 | 0.42 | 1.9 × 10−8 | 0.84 (0.79–0.89) | Intergenic | – |
| PHTF1‐PTPN22 | PM | rs6679677 | 1 | 114303808 | A | 0.12 | 0.1 | 1.57 × 10−7 | 1.3 (1.19–1.43) | Exonic | RA (ref. 14), SLE (ref. 15) |
| DGKQ | IIM | rs6599390 | 4 | 956047 | A | 0.3 | 0.34 | 1.64 × 10−7 | 0.84 (0.78–0.89) | Intronic | Seropositive RD (ref. 2), SSc (ref. 16) |
| UBE2L3‐YDJC | IIM | rs11089637 | 22 | 21979096 | C | 0.19 | 0.16 | 1.23 × 10−6 | 1.23 (1.13–1.33) | Intergenic | RA (ref. 14), JIA (ref. 13), SLE (ref. 15) |
| TEC | IIM | rs80105690 | 4 | 48155618 | T | 0.09 | 0.07 | 6 × 10−6 | 1.27 (1.14–1.42) | Intronic | RA (ref. 14) |
| PLCL1 | IIM | rs1518359 | 2 | 198847383 | T | 0.52 | 0.49 | 1.45 × 10−5 | 1.14 (1.07–1.21) | Intronic | SLE (ref. 17) |
| LTBR | IIM | rs11064180 | 12 | 6523249 | T | 0.38 | 0.41 | 1.58 × 10−5 | 0.87 (0.81–0.92) | Intergenic | JIA (ref. 13), AS (ref. 12) |
| HLA–DQB1 | PM | rs3129716 | 6 | 32657436 | C | 0.29 | 0.13 | 1.54 × 10−54 | 2.65 (2.35–3.00) | – | Multiple |
| NAB1 | PM | rs6733720 | 2 | 191516020 | G | 0.22 | 0.17 | 1.96 × 10−8 | 1.41 (1.24–1.60) | Intronic | Seropositive RD (ref. 2), SSc (ref. 16) |
| PTPN22 | PM | rs2476601 | 1 | 114377568 | A | 0.15 | 0.1 | 1.25 × 10−6 | 1.46 (1.26–1.70) | Exonic | RA (ref. 14), SLE (ref. 15) |
| FAM167A‐BLK | PM | rs17799348 | 8 | 11333521 | T | 0.33 | 0.39 | 1.72 × 10−6 | 0.77 (0.69–0.85) | Intergenic | Seropositive RD (ref. 2), SSc (ref. 16), RA (ref. 14), SLE (ref. 15) |
| HLA–B | DM | rs7748141 | 6 | 31288877 | C | 0.23 | 0.12 | 2.70 × 10−28 | 2.17 (1.89–2.49) | – | Multiple |
| HLA–DRA | JDM | rs12204922 | 6 | 32451613 | C | 0.3 | 0.18 | 7.99 × 10−16 | 1.91 (1.63–2.23) | – | Multiple |
| HLA–DQB1 | IBM | rs4713570 | 6 | 32626040 | T | 0.57 | 0.24 | 3.9 × 10−45 | 4.15 (3.39–5.09) | – | Multiple |
| CCR5 | IBM | rs41490645 | 3 | 46410137 | C | 0.08 | 0.16 | 5.96 × 10−7 | 0.45 (0.32–0.63) | Upstream Variant | JIA (ref. 18) |
| HLA–DRA | Jo‐1 | rs9268813 | 6 | 32424594 | C | 0.39 | 0.11 | 5.01 × 10−65 | 5.15 (4.21–6.32) | – | Multiple |
Genome‐wide significance was defined as P < 5 × 10−8; suggestive significance was defined as P < 2.25 × 10−5 with previous statistical evidence of association in an autoimmune rheumatic disease (RD) in the National Human Genome Research Institute–European Bioinformatics Institute Genome‐Wide Association Study Catalog. IIM = idiopathic inflammatory myopathy; SNP = single‐nucleotide polymorphisms; MAF = minor allele frequency; OR = odds ratio; 95% CI = 95% confidence interval; RA = rheumatoid arthritis; SLE = systemic lupus erythematosus; SSc = systemic sclerosis; PM = polymyositis; JIA = juvenile idiopathic arthritis; AS = ankylosing spondylitis; DM = dermatomyositis; JDM = juvenile dermatomyositis; IBM = inclusion body myositis.
Function obtained from the dbSNP database's predicted functional effect.
Locus with evidence of independent effects.
Figure 1.
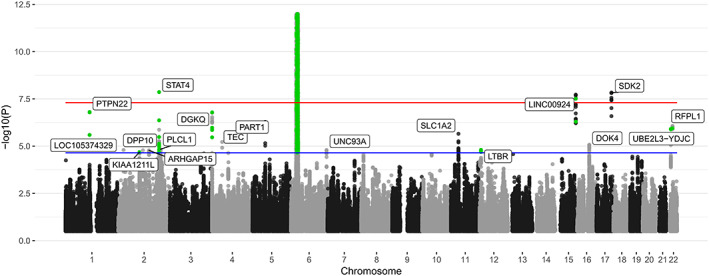
Manhattan plot of the total idiopathic inflammatory myopathy (n = 2,565) association analysis. The red line represents genome‐wide level of significance (P < 5 × 10−8); the blue line represents suggestive significance calculated from prior coverage from the ImmunoChip array (P < 2.25 × 10−5). Single‐nucleotide polymorphisms reaching P < 2.25 × 10−5 that were directly genotyped are colored green to differentiate from imputed variants. For visualization purposes, the y‐axis has a cutoff in the HLA region (chromosome 6, 25–35 Mb) of P < 1 × 10−1.
For IIM as a whole and all clinical subgroups, the HLA region was the most significant genetic risk factor. Three non‐HLA regions, STAT4 (P = 1.38 × 10−8, odds ratio [OR] 0.81 [95% confidence interval (95% CI) 0.75–0.87]), SDK2 (P = 1.46 × 10−8, OR 1.15 [95% CI 1.08–1.23]), and LINC00924 (P = 1.9 × 10−8, OR 0.84 [95% CI 0.79–0.89]), reached genome‐wide significance in the total IIM cohort, and 1 non‐HLA region, NAB1 (P = 1.96 × 10−8, OR 1.41 [95% CI 1.24–1.60]), reached genome‐wide significance in the PM subgroup. This is the first time these loci have been found to reach genome‐wide significance in IIM. After conditioning on the SNP with the lowest P value in these regions, there were no additional independent effects reaching genome‐wide significance. However, an independent intronic variant in STAT4 remained suggestively significant in IIM (rs6752770, P = 6.1 × 10−6, OR 1.11 [95% CI 1.04–1.19]) (Supplementary Figure 7, https://onlinelibrary.wiley.com/doi/10.1002/art.42434).
The 95% credible SNP sets for the 4 regions reaching genome‐wide significance are included in the Supplementary Data (https://onlinelibrary.wiley.com/doi/10.1002/art.42434), along with predicted deleteriousness and functionality using Combined Annotation Dependent Depletion (CADD) and RegulomeDB, respectively. For the STAT4 and NAB1 regions, a substantial proportion of the 95% posterior probability for credible SNPs could be attributed to a single SNP; rs4853540 (STAT4) maps to an enhancer for STAT1, and rs6733720 (NAB1) is a significant splicing QTL for NAB1 in 26 tissues (P < 1.6 × 10−6) and a significant expression QTL for several genes, including NAB1 and GLS (glutaminase), in multiple tissues (see Supplementary Data).
The LINC00924 locus is a novel association in autoimmune disease, reaching genome‐wide significance. Most variants in these regions were imputed, explaining the lack of association in our prior IIM ImmunoChip study, where the original SNP may have been removed during quality control (1). Notably, we also found suggestive evidence of association with TEC and LTBR, which have previously been associated with autoimmune rheumatic diseases (12, 13, 14). Associations with the STAT4 and DGKQ loci were more significant in the current study than in the prior IIM ImmunoChip study, as were associations previously reported in IIM clinical subgroups, such as NAB1 and FAM167A‐BLK loci in PM and CCR5 in IBM (Table 1).
We used GARFIELD to assess whether IIM‐associated variants are enriched in regulatory elements of specific cell types (11). We found enrichment of variants among DNase I hypersensitivity sites and histone marks associated with active transcription within blood cells (Supplementary Figure 20 and Supplementary Data, https://onlinelibrary.wiley.com/doi/10.1002/art.42434). Specifically, IIM variants were most enriched within DNase I hypersensitivity hotspots of primary CD19+ B cells (P = 1.1 × 10−16) and CD3+ T cells (P = 3 × 10−15).
DISCUSSION
By using imputation to identify and fine‐map genetic associations in IIM, we found 3 new genome‐wide associations in the combined IIM cohort, STAT4, SDK2, and LINC00924. Conditional analysis revealed evidence of independent associations in the STAT4 region. For the whole IIM group and clinical subgroups, the HLA region was the most significant genetic risk factor. The strongest genetic risk in this region was for the anti–Jo‐1 subgroup despite a sample size of only 331 patients. In the PM subgroup, we found 1 novel genome‐wide association with NAB1. This is the first time this data set has been used for genome‐wide imputation, and the first time this data set has been stratified by adult‐ and juvenile‐onset myositis subgroups and by individuals with anti–Jo‐1 autoantibodies.
We defined an associated region by proximity to the nearest gene. For example, in the LINC00924 region, the strongest association was intergenic, lying approximately 150 kb from this long intergenic noncoding RNA, which has been associated with a number of traits such as coronary heart disease and ischemic stroke. However, it may be that these associations are influencing a different gene or regulatory element lying further away from the most associated SNP. It is worth noting that the lead variant in the LINC00924 locus was removed during quality control in our prior ImmunoChip analysis. In other instances, such as in the SDK2 region, the most associated variants lie intronic of the gene. SDK2 is a member of the immunoglobulin superfamily, although the specific function is not yet known. In both instances, there is no previous evidence of genetic association with rheumatic diseases. A limitation of this study is the lack of a replication cohort due to the rarity of IIM, although patient recruitment is ongoing within the MYOGEN consortium.
For some regions, we found suggestive novel associations that have prior evidence of association with autoimmune disease, such as TEC, a tyrosine kinase involved in T cell signalling and activation, and LTBR, a signalling receptor expressed on myeloid cells. In other instances, we strengthened previously observed associations with IIM (STAT4 and DGKQ). In addition, it is interesting to note that we observed a suggestive association with PLCL1 (P = 1.45 × 10−5, OR 1.14 [95% CI 1.07–1.21]) in the combined IIM analysis. Variants in PLCL1, a gene encoding the intracellular signalling molecule phospholipase C–like 1, were previously reported in the MYOGEN DM GWAS, although it was not identified in the subsequent ImmunoChip analysis. We have identified more significant associations than were previously reported in IIM clinical subgroups, such as NAB1 and FAM167A‐BLK loci in PM and CCR5 in IBM, which also have prior evidence of association with rheumatic disease. Although this study comprised the same cohort as previous studies, this analysis can be viewed as a fine‐mapping experiment; imputation from a large reference panel allows better coverage to localize further signals. Indeed, we found that many associations could be localized to single genes or credible SNPs with high posterior probability, likely due to the coverage of coding genes on the ImmunoChip array resulting in high‐quality imputation. We note that the ImmunoChip is a targeted array and therefore does not have genome‐wide imputation coverage.
Functional analysis of variants reaching suggestive significance in IIM was conducted using GARFIELD. This method uses functional annotation from primary tissues and cell lines from the ENCODE and Roadmap Epigenomics projects. As expected from data generated using the ImmunoChip array, associated variants in IIM were enriched in regulatory elements of blood cells. In particular, the strongest relative enrichment was seen in regions of open chromatin in CD19+ B cells and CD3+ T cells. The role of T and B cells is well known in IIM through muscle immunohistopathology and the presence of autoantibodies, and our findings suggest their contribution to disease pathology may be genetically encoded.
Increasing the number of patient samples in an analysis should statistically strengthen suggestive associations from previous studies. A recent genome‐wide meta‐analysis of 4 seropositive rheumatic diseases revealed several novel loci, for example, NAB1, DGKQ, and YDJC, where IIM contributed to the association. Using additional IIM patients to those included in the meta‐analysis, we were able to identify these associations in IIM only, and in some cases, such as NAB1 in PM, attribute these associations to specific clinical subgroups of IIM. The lead variant rs6733720 in NAB1 is a significant splicing QTL for NAB1 in 26 tissues and an expression QTL for several genes, including NAB1 and GLS, in different tissues. These NAB1 variants are also in moderate linkage disequilibrium (r2 = 0.66), with a variant previously identified in systemic sclerosis and rheumatoid arthritis, which acts as an expression QTL for NAB1 expression in lymphoblastoid cell lines (2).
Although we found a number of interesting associations in the PM subgroup, one limitation of these findings is the potential heterogeneity within this subgroup. The Bohan and Peter criteria do not differentiate between PM and immune‐mediated necrotizing myopathy. In addition, we included patients classified as having PM that have autoantibodies against transfer RNA synthetases, although antisynthetase syndrome is increasingly recognised as a separate entity. Some previous genetic studies in IIM combined adult‐ and juvenile‐onset DM for analysis. This study stratified DM by age of onset to investigate non‐HLA associations. However, there do not seem to be strong signals that differentiate these clinical subgroups. Our previous work has shown that stratifying IIM cohorts by autoantibody status may increase power to detect genetic associations within the HLA region (9). Therefore, we also analyzed a subgroup of patients with anti–Jo‐1 autoantibodies; however, we did not find any significant associations outside the HLA region. Although the anti–Jo‐1 subgroup is thought to be more clinically homogeneous, there may have been a lack of power to detect associations with only 331 patients in the analysis. For this reason, we did not investigate genetic associations with other less common autoantibodies.
To our knowledge, this is the largest genetic association study investigating non‐HLA genes in patients with anti–Jo‐1 autoantibodies. A recent study targeted a number of SNPs in the IL1B locus in a Mexican cohort of 154 antisynthetase‐positive IIM patients and found an association with a synonymous SNP in IL1B (19). We could not replicate this association in our Caucasian cohort (rs1143634, P = 0.1). In the prior ImmunoChip analysis, PTPN22 was the only non‐HLA region to reach genome‐wide significance; however, in this analysis, PTPN22 did not reach genome‐wide significance. This disparity may be due to the more accurate matching of patients to controls, as it is known that there is a wide variation of allele frequency among different European populations of the PTPN22 R620W risk polymorphism (20).
In summary, we used imputation to identify and fine‐map genetic associations in IIM. We found 4 new genome‐wide associations in IIM and PM, and we found associations reaching suggestive significance that have previously been associated with autoimmune rheumatic disease. These risk variants are functionally enriched in relevant immune cells, expanding our knowledge of the genetic architecture of IIM.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Rothwell had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Rothwell, Amos, Miller, Rider, Lundberg, Wedderburn, Chinoy, Lamb.
Acquisition of data
Rothwell, Miller, Rider, Lundberg, Gregersen, Vencovsky, McHugh, Limaye, Selva‐O'Callaghan, Hanna, Machado, Pachman, Reed, Molberg, Benveniste, Mathiesen, Radstake, Doria, De Bleecker, De Paepe, Maurer, Ollier, Padyukov, O'Hanlon, Lee, Wedderburn, Chinoy, Lamb.
Analysis and interpretation of data
Rothwell, Amos, Miller, Rider, Lundberg, McHugh, Ollier, Padyukov, Wedderburn, Chinoy, Lamb.
Supporting information
Disclosure Form
Appendix S1: Supplementary Material
Data S1: Supplementary Data
Figure S1: Supplementary Figure
ACKNOWLEDGMENTS
We would like to thank Drs. Elaine Remmers and Michael Ombrello for their outstanding advice. Additionally, we would like to thank all the patients and healthy individuals for their essential collaboration. A full list of the MYOGEN collaborators is included in the Supplementary Appendix, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42434.
The views expressed in this publication are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health.
Supported by the Intramural Research Program, National Institute of Environmental Health Sciences, NIH. Dr. Lundberg's work was supported by grants from the Swedish Research Council and Stockholm Regional Council (ALF). Dr. Vencovsky's work was supported by the Czech Ministry of Health–Conceptual Development of Research Organization (award 00023728) (Institute of Rheumatology). Drs. Hanna and Machado's work were supported by the NIHR University College London Hospitals Biomedical Research Centre. Drs. De Bleecker and De Paepe's work were supported by the European Reference Network for Rare Neuromuscular Diseases (ERN EURO‐NMD). Dr. Wedderburn's work was supported by Versus Arthritis (awards 21593 and 21552), the Wellcome trust (award 085860), Myositis UK, the Cure JM Foundation, the Remission Charity, and the NIHR Biomedical research Centre at GOSH. Dr. Chinoy's work was supported by the Medical Research Council UK (award MR/N003322/1), Myositis UK, and the NIHR Manchester Biomedical Research Centre Funding Scheme. Dr. Lamb's work was supported by the Medical Research Council UK (award MR/N003322/1) and Myositis UK.
Author disclosures are available online at https://onlinelibrary.wiley.com/doi/10.1002/art.42434.
REFERENCES
- 1. Rothwell S, Cooper RG, Lundberg IE, et al. Dense genotyping of immune‐related loci in idiopathic inflammatory myopathies confirms HLA alleles as the strongest genetic risk factor and suggests different genetic background for major clinical subgroups. Ann Rheum Dis 2016;75:1558–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Acosta‐Herrera M, Kerick M, González‐Serna D, et al. Genome‐wide meta‐analysis reveals shared new loci in systemic seropositive rheumatic diseases. Ann Rheum Dis 2019;78:311–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kochi Y, Kamatani Y, Kondo Y, et al. Splicing variant of WDFY4 augments MDA5 signalling and the risk of clinically amyopathic dermatomyositis. Ann Rheum Dis 2018;77:602–11. [DOI] [PubMed] [Google Scholar]
- 4. Sugiura T, Kawaguchi Y, Goto K, et al. Positive association between STAT4 polymorphisms and polymyositis/dermatomyositis in a Japanese population. Ann Rheum Dis 2012;71:1646–50. [DOI] [PubMed] [Google Scholar]
- 5. Chen S, Wang Q, Wu Z, et al. Genetic association study of TNFAIP3, IFIH1, IRF5 polymorphisms with polymyositis/dermatomyositis in Chinese Han population. PLoS One 2014;9:e110044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen S, Wu W, Li J, et al. Single nucleotide polymorphisms in the FAM167A‐BLK gene are associated with polymyositis/dermatomyositis in the Han Chinese population. Immunol Res 2015;62:153–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med 1975;292:344–7. [DOI] [PubMed] [Google Scholar]
- 8. Rothwell S, Cooper RG, Lundberg IE, et al. Immune‐array analysis in sporadic inclusion body myositis reveals HLA–DRB1 amino acid heterogeneity across the myositis spectrum. Arthritis Rheumatol 2017;69:1090–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rothwell S, Chinoy H, Lamb JA, et al. Focused HLA analysis in Caucasians with myositis identifies significant associations with autoantibody subgroups. Ann Rheum Dis 2019;78:996–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boughton AP, Welch RP, Flickinger M, et al. LocusZoom.js: interactive and embeddable visualization of genetic association study results. Bioinformatics 2021;37:3017–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Iotchkova V, Ritchie GRS, Geihs M, et al. GARFIELD classifies disease‐relevant genomic features through integration of functional annotations with association signals. Nat Genet 2019;51:343–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Evans DM, Spencer CCA, Pointon JJ, et al. Interaction between ERAP1 and HLA‐B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA‐B27 in disease susceptibility [published correction appears in Nat Genet 2011;43:919]. Nat Genet 2011;43:761–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hinks A, Cobb J, Marion MC, et al. Dense genotyping of immune‐related disease regions identifies 14 new susceptibility loci for juvenile idiopathic arthritis. Nat Genet 2013;45:664–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Okada Y, Wu D, Trynka G, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014;506:376–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bentham J, Morris DL, Cunninghame Graham DS, et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat Genet 2015;47:1457–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. López‐Isac E, Acosta‐Herrera M, Kerick M, et al. GWAS for systemic sclerosis identifies multiple risk loci and highlights fibrotic and vasculopathy pathways. Nat Commun 2019;10:4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang YF, Zhang Y, Lin Z, et al. Identification of 38 novel loci for systemic lupus erythematosus and genetic heterogeneity between ancestral groups. Nat Commun 2021;12:772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. López‐Isac E, Smith SL, Marion MC, et al. Combined genetic analysis of juvenile idiopathic arthritis clinical subtypes identifies novel risk loci, target genes and key regulatory mechanisms. Ann Rheum Dis 2021;80:321–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ponce‐Gallegos MA, Ramos‐Martínez E, García‐Carmona A, et al. Genetic susceptibility to antisynthetase syndrome associated with single‐nucleotide variants in the IL1B gene that lead variation in IL‐1β serum levels. Front Med (Lusanne) 2020;7:547186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Burn GL, Svensson L, Sanchez‐Blanco C, et al. Why is PTPN22 a good candidate susceptibility gene for autoimmune disease? [review]. FEBS Lett 2011;585:3689–98. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclosure Form
Appendix S1: Supplementary Material
Data S1: Supplementary Data
Figure S1: Supplementary Figure