

Abstract
Background:
Vascular Aβ accumulation is the hallmark of cerebral amyloid angiopathy (CAA). The composition of cerebrospinal fluid (CSF) of CAA patients may serve as diagnostic biomarker of CAA. We studied the diagnostic potential of the peptides Aβ38, Aβ40, Aβ42, and Aβ43 in patients with sporadic CAA (sCAA), hereditary Dutch-type CAA (D-CAA), and Alzheimer’s disease (AD).
Methods:
Aβ peptides were quantified by immunoassays in a discovery group (26 patients with sCAA and 40 controls), a validation group (40 patients with sCAA, 40 patients with AD and 37 controls) and a group of 22 patients with D-CAA and 54 controls. To determine the diagnostic accuracy, the area-under-the-curve (AUC) was calculated using a receiver operating characteristic curve with 95% confidence interval (CI)
Results:
We found decreased levels of all Aβ peptides in sCAA patients and D-CAA patients compared to controls. The difference was most prominent for Aβ42 (AUC of sCAA versus controls for discovery: 0.90 (95% CI: 0.82–0.99); for validation: 0.94 (95% CI: 0.89–0.99) and Aβ43 (AUC of sCAA versus controls for discovery: 0.95 (95% CI: 0.88–1.00); for validation: 0.91 (95% CI: 0.83–1.0). All Aβ peptides, except Aβ43, were also decreased in sCAA compared to AD (CSF Aβ38: AUC of 0.82 (95% CI: 0.71–0.93), CSF Aβ40: AUC of 0.88 (95% CI: 0.80–0.96), CSF Aβ42: AUC of 0.79 (95% CI: 0.66–0.92).
Conclusions:
A combined biomarker panel of CSF Aβ38, Aβ40, Aβ42, and Aβ43 has potential to differentiate sCAA from AD and controls, and D-CAA from controls.
Introduction
Cerebral amyloid angiopathy (CAA) is pathologically defined by the accumulation of amyloid β (Aβ) in the arterioles, leptomeningeal vessels and capillaries of the cerebral cortex (1). The clinical spectrum is characterized by intracerebral haemorrhage (ICH), cognitive impairment leading up to dementia and transient focal neurological episodes (2). Transient focal neurological episodes may mimic transient ischemic attacks, migraine auras, and focal seizures, but can be recognized by recurrent, stereotyped, transient episodes of spreading paraesthesias, numbness or weakness, lasting typically seconds to minutes, usually resolving over a similar period) (1, 3, 4). Histopathologically, moderate-to-severe CAA can be found in approximately one quarter of the elderly population upon examination of (post-mortem) brain tissue (5), although only a minority of them will develop symptoms. Dutch-type CAA (D-CAA) is a hereditary form of CAA, caused by a point mutation at codon 693 of the amyloid precursor protein (APP) gene on chromosome 21, with a young onset but with clinical symptoms similar to sCAA (6).
The diagnosis of sCAA during life relies on the modified Boston criteria, which include the presence of lobar haemorrhage(s), strictly lobar microbleeds and superficial siderosis on magnetic resonance imaging (MRI) (7). An update of these criteria, that additionally incorporates convexity subarachnoid haemorrhage and non-haemorrhagic markers, is recently published (8). Although these criteria are quite sensitive, they have some limitations. The imaging markers in these criteria are likely to reflect end-stage disease, and they are indirect reflections of CAA, since they represent the consequences of CAA instead of its aetiology, i.e. vascular amyloid deposition. In addition, they lack specificity, since the incorporated imaging markers can also be (partially) caused by arteriosclerotic small vessel disease. Moreover, they cannot provide a definitive diagnosis without a brain biopsy, which is rarely performed. Lastly, the updated criteria have a sensitivity of only 55% in patients without intracerebral haemorrhage (ICH).
Cerebrospinal fluid (CSF) biomarkers may provide an opportunity to identify CAA at an earlier stage and can be used to monitor disease progression. It has previously been demonstrated that CSF Aβ40 and Aβ42 levels are decreased in patients with sCAA compared to both controls and patients with AD (9–11). Furthermore, CSF Aβ40 and Aβ42 levels were decreased in patients with presymptomatic D-CAA before CAA-related imaging abnormalities were found, and they were further decreased in patients with symptomatic D-CAA (12), which indicates that these markers may serve as biomarkers of early stages of CAA.
In addition, reduced levels of CSF Aβ38 have been demonstrated in a single report on patients with sCAA versus controls (13). CSF levels of another common Aβ peptide, Aβ43, have not been studied in CSF from patients with sCAA, but reduced levels have been reported in patients with AD when compared to controls (14, 15).
The aim of our study was to examine if our previous findings of decreased CSF Aβ40 and Aβ42 can be extended to a larger panel of Aβ peptides in the CSF of patients with sCAA, and D-CAA as compared to patients with AD and controls. For this, we studied separate discovery and validation cohorts of patients with sCAA and controls, and additionally validated our findings in patients with (pre)symptomatic D-CAA and controls. Furthermore, we assessed CSF Aβ levels in patients with a CSF biomarker profile indicative of AD.
Methods
Participants
Patients with sCAA and controls; discovery
We included 26 patients with sCAA and 40 controls from the Radboud University Medical Center (RUMC), Nijmegen, the Netherlands (see figure S1). The inclusion criteria for the patients with sCAA were a diagnosis of probable (n=19) or possible (n=3) CAA according to the modified Boston criteria (7). Additionally, we included patients (n=4) with mixed lobar and deep hemorrhages/microbleeds. Availability of lumbar CSF was a prerequisite for inclusion. The CSF was collected in the context of either routine clinical workup (n=4) or two cross-sectional studies investigating new CSF biomarkers for CAA (Cererebral Amyloid Angiopathy: Vascular Imaging and fluid markers of Amyloid deposition (CAVIA; n=11) or “BIOmarkers for cogNitive Impairment due to Cerebral amyloid angiopathy (BIONIC; www.radboudumc.nl/BCS; n=11)). All but 2 CSF samples were taken at least 3 months after symptomatic intracerebral haemorrhage (ICH).
Part of the patients with sCAA (n=12) underwent a Montreal Cognitive Assessment (MoCA).
Part of the controls (n=30) underwent a lumbar puncture as part of a diagnostic workup in order to exclude central nervous system involvement of a systemic disease, a (central) neurological cause for their symptoms or a neurological infection or inflammation. Exclusion criteria were neurodegenerative disease, known cognitive impairment, sepsis, a recent stroke (<6 months) or a malignancy in the central nervous system. The other controls (n=10) were patients who underwent thoracoabdominal aortic aneurism repair, for which they had an external lumbar drain, of which CSF was sampled before the operation. They did not have known cognitive impairment or recent (<3 months) stroke or traumatic brain injury. The controls were sex-matched to the patients with sCAA (Table 1).
Table 1:
Sporadic CAA patients and controls in the discovery experiments: characteristics and results of MRI and CSF analysis
| sCAA (n=26) | Probable CAA (n=19) | Controls (n=40) | p-value sCAA vs controls | p-value probable CAA vs controls | |
|---|---|---|---|---|---|
| Age (years) | 72±7 | 72±8 | 63±8 | p<0.001a | p<0.001a |
| Sex, M/F (n) | 18/8 | 7/12 | 28/12 | p=0.95b | p=0.60b |
| MoCA | 24 [20–28]d | 24 [20–28]e | N.A. | - | - |
| Hypertension | 48%f | 44%f | 43%g | p=0.72b | p=0.94b |
| CSF Aβ38 (pg/ml) | 1411±520 | 1400±396 | 1679±526 | p=0.015c | p=0.013c |
| CSF Aβ40 (ng/ml) | 7.55 [4.91–9.10] | 7.58 [6.07–9.04] | 9.08 [6.14–12.5] | p=0.015c | p=0.024c |
| CSF Aβ42 (pg/ml) | 356 [292–3447] | 353 [299–406] | 822 [572–1225] | p<0.001c | p<0.001c |
| CSF Aβ43 (pg/ml) | 11.5 [9.27–14.6] | 11.5 [10.6–12.8] | 40.2 [20.9–53.6] | p<0.001c | p<0.001c |
Table 1: Values are medians and [IQR] except for age, Aβ38 (mean ± SD), sex (n) and hypertension prevalence (%).
Student’s t-test,
chi-square test,
linear regression with age and sex as covariates,
n=12,
n=11,
hypertension status unknown for 1 patient,
hypertension status unknown for 10 controls. The sCAA patients consist of 19 patients with probable CAA, 4 patients with mixed microbleeds/ICH and 3 patients with possible CAA. The probable CAA patients are a subset of sCAA patients.
Abbreviations: Aβ= Amyloid beta, sCAA= sporadic cerebral amyloid angiopathy, F= Female, IQR= interquartile range, M= Male, MoCA: Montreal Cognitive Assessment, N.A.= not available, SD=standard deviation, SVD= small vessel disease.
Patients with sCAA and controls; validation
For validation purposes, we included a total of 40 patients with sCAA, 12 new patients from the RUMC, 19 patients from the Massachusetts General Hospital (MGH, Harvard Medical School, Boston, USA), and 9 patients from the Leiden University Medical Centre (LUMC, the Netherlands). Of the 40 patients, 1 patient had definite CAA, 5 patients had probable CAA with supporting pathology, and 34 patients had probable CAA (modified Boston criteria) (7). The CSF of the patients with sCAA from RUMC was collected in the context of a clinical workup (n=1) or the BIONIC study (n=11; see figure S1). The CSF of the patients with sCAA from LUMC was collected in the context of a clinical study investigating new CSF biomarkers (Following Sporadic CAA Study (FOCAS) (1)), and the CSF from the patients with sCAA from the MGH was also taken in the context of a research study. CSF samples were also taken at least 3 months after symptomatic intracerebral haemorrhage (ICH). Due to a limited amount of available CSF, Aβ38 was not measured in n=7 and Aβ43 was not measured in n=13 patients from MGH. Part of the patients with sCAA (n=21) underwent a MoCA.
We included 37 controls from the RUMC that met the same criteria as defined above for the discovery group. The controls were age- and sex-matched to the patients with sCAA (Table 2). Details about the final diagnosis of the controls are specified in the methods section of the supplementary material.
Table 2:
Sporadic CAA patients, AD patients and controls in the validation experiments: characteristics and results of MRI
| sCAA (n=40) | AD patients (n=40) | Controls (n=37) | p-value probable CAA vs controls | p-value probable CAA vs AD | p-value AD vs control | |
|---|---|---|---|---|---|---|
| Age (years) | 68±9 | 70±8 | 70±8 | 0.41a | 0.65a | 0.78a |
| Sex, M/F (n) | 19/21 | 17/23 | 15/22 | 0.54b | 0.27b | 0.86b |
| MoCA | 25 [22–26]d | N.A. | N.A. | - | - | - |
| Hypertension (%) | 50% | N.A. | 35%g | 0.20b | - | - |
| SVD burden score | 4 [3–6]d | N.A. | N.A. | - | - | - |
| CSF Aβ38 (pg/ml) | 2349 [1611–3153]e | 3355 [2893–4084] | 3947 [2567–5206] | <0.0001c | <0.0001c | 0.11c |
| CSF Aβ40 (ng/ml) | 4.86 [2.97–6.97] | 9.86 [7.73–12.34] | 11.1 [6.67–15.05] | <0.0001c | <0.0001c | 0.28c |
| CSF Aβ42 (pg/ml) | 240 [120–323] | 429 [312–486] | 755 [544–1068] | <0.0001c | 0.012c | <0.0001c |
| CSF Aβ43 (pg/ml) | 12.0 [7.49–16.5]f | 16.8 [12.3–20.7] | 42.6 [30.0–64.5] | <0.0001c | 0.86c | <0.0001c |
Table 2: Values are medians and [IQR] except for age (mean ± SD), sex (n) and hypertension prevalence (%).
Student’s t-test.
chi-square test,
linear regression with age and sex as covariates.
n=21.
n=33.
n=27.
hypertension status unknown for 3 controls.
Abbreviations: Aβ= Amyloid beta, sCAA= sporadic cerebral amyloid angiopathy, F= Female, IQR= interquartile range, M= Male, MoCA: Montreal Cognitive Assessment, SD=standard deviation, SVD=small vessel disease.
Patients with Alzheimer’s Disease (AD)
We included 40 patients with a CSF profile indicative of AD. These were patients who were referred to the RUMC for CSF diagnostics to investigate the etiology of their cognitive symptoms (Figure S1; Table 2). Patients were selected based on CSF biomarker evidence for amyloid deposition (A), tau accumulation (T), and neurodegeneration (N) (16). We used the following predefined local cutoff values: (CSF Aβ42 (A+): <659 pg/ml; phosphorylated tau (T+) :> 64 pg/ml; total tau (N+): > 400 pg/ml).
Patients with D-CAA
We included 10 patients with presymptomatic D-CAA from the LUMC and n=26 age-matched controls from RUMC, and 12 patients with symptomatic D-CAA patients from LUMC and n=28 age-matched controls from RUMC (Figure S1, Table 3). Patients with D-CAA were included if they had an available CSF sample. Presymptomatic D-CAA was defined as carriership of the c.2077G>C mutation in the amyloid-β precursor protein gene (resulting in p.Glu693Gln) and absence of symptomatic (haemorrhagic) strokes). Symptomatic D-CAA was defined by the presence of one or more symptomatic ICH(s) in combination with either confirmed carriership of the c.2077G>C mutation or one or more first-degree relative with D-CAA. The D-CAA CSF samples were collected in the context of a D-CAA natural history study of the LUMC (the AURORA study) (17).
Table 3:
Characteristics and results of CSF analysis in patients with D-CAA and controls.
| Presymp D-CAA (n=10) | Young controls (n=26) | Symptomatic D-CAA (n=12) | Old controls (n=28) | p-value Presymp vs Young | p-value Symp vs Old | p-value Presymp vs Symp | |
|---|---|---|---|---|---|---|---|
| Age (years) | 40±8 | 43±8 | 59±8 | 58±8 | 0.37a | 0.88a | <0.0001a |
| Sex, M/F (n) | 7/3 | 15/11 | 7/5 | 11/17 | 0.50b | 0.27b | 0.18b |
| Hypertension (%) | 20% | 8%c | 33% | 19%d | 0.31b | 0.34b | 0.48b |
| Aβ38 (pg/ml) | 1240 [1043–1701] | 3647 [3196–4833] | 747 [667–1087] | 3387 [2594–4318] | <0.0001e | <0.0001e | 0.14e |
| Aβ40 (ng/ml) | 2.34 [1.88–3.34] | 9.71 [7.12–11.2] | 1.55 [1.17–2.07] | 10.2 [6.52–12.2] | <0.0001e | <0.0001e | 0.047e |
| Aβ42 (pg/ml) | 108 [86.0–148] | 903 [643–1087] | 70.5 [53.3–89.8] | 782 [597–1300] | <0.0001e | <0.0001e | 0.026e |
| Aβ43 (pg/ml) | 9.77 [7.49–12.64] | 63.4 [48.9–63.4] | 6.53 [4.95–8.07] | 47.3 [35.4–68.5] | <0.0001e | <0.0001e | 0.14e |
Table 3: Values are medians and [IQR] except for age (mean ± SD), sex (n), and hypertension prevalence (%).
Student’s t-test.
Fisher’s exact test,
hypertenson status unknown of 1 control,
hypertension status unknown of 2 controls,
linear regression with age and sex as covariates.
Abbreviations: Aβ= Amyloid beta, CAA= cerebral amyloid angiopathy, F= Female, IQR= interquartile range, M= Male, MoCA: Montreal Cognitive Assessment, presymp D-CAA= patients with presymptomatic D-CAA patients, symp D-CAA=symptomatic D-CAA patients, SD=standard deviation.
The prevalence of hypertension in the patients and controls was assessed in all patients groups and controls, except in the patients with Alzheimer’s disease (Table 1,2 and 3).
CSF analysis
All participants in this study underwent a lumbar puncture according to state-of-the-art local protocols. At all participating hospitals, the CSF was collected in polypropylene tubes, centrifuged, aliquoted, and stored in polypropylene tubes at −80°C.
For all CSF analyses, patient and controls samples were randomly analysed to avoid bias. CSF Aβ40, Aβ42, tau phosphorylated at threonine 181 and total tau levels were quantified using the Lumipulse chemiluminescent immunoassay (Fujirebio, Gent, Belgium). The samples were analysed in different batches; however, we adhere to strict guidelines under the ISO15189 guidance to control that inter-assay variation is kept within predefined limits of variation for each assay.
CSF Aβ38 was quantified using ELISAs (Euroimmun, Lübeck, Germany for the discovery groups; due to a production stop that occurred between the execution of the discovery and validation stages of the study, we used an ELISA produced by IBL (Fujioka-Shi, Japan) for all other samples). CSF Aβ43 was quantified using an ELISA (IBL). The Aβ38 and Aβ43 ELISAs were done within a limited time window for both study cohorts separately (i.e. discovery and validation stage respectively).
For all ELISAs, five quality control samples were included on each plate to correct for any inconsistencies between plates. These controls consisted of pooled CSF samples that were stored in aliquots at −80 °C. For each analysis a fresh aliquot was used.
All CSF analysis were done at the RUMC.
MRI analysis
MRI acquisition
Patients with sCAA from the BIONIC study underwent a 3.0 Tesla MRI scan (Siemens Magnetom Prisma, Siemens Healthineers, Erlangen, Germany) using a 32-channel head coil. Participants were examined using a comprehensive protocol, and the for the current study, the 3D multi-echo gradient echo T2*-weighted sequence (voxel size 0.8 × 0.8 × 0.8 mm), the 3D T2-weighted sequence (voxel size 0.8 × 0.8 × 0.8 mm) and 3D fluid-attenuated inversion recovery (FLAIR) sequence (voxel size 0.8 × 0.8 × 0.8 mm) were analyzed. Magnitude and phase data from the multi-echo gradient sequence was processed to a SWI using the “Contrast-weighted, Laplace-unwrapped, bipolar multi-Echo, ASPIRE-combined, homogeneous, improved Resolution SWI” (CLEAR-SWI) method (18). Patients with sCAA and D-CAA patients from the FOCAS and AURORA studies were scanned on a 3.0 Tesla MRI scanner (Philips Healthcare, Best, the Netherlands) with a 32-channel head coil. This protocol included SWI, T2 and FLAIR sequences. Further details are described in (17).
MRIs from the BIONIC study were rated independently by AK and HBS and MRIs from the AURORA and FOCAS study by EK. In case of disagreement between AK and HBS, FS (senior vascular neurologist) was consulted before final consensus was reached.
The following markers were assessed following the STRIVE criteria: cerebral microbleeds (CMBs) (19), cortical superficial siderosis (CSS)(7), enlarged perivascular spaces (EPVS) in the centrum semi-ovale (CSO; using a dichotomized classification: high (≥21 EPVS) or low (≤20 EPVS)) and white matter hyperintensities according to the Fazekas Scale (WMH)(20, 21). In addition, we assessed the total burden score of small vessel disease (SVD) in CAA (22), henceforth referred to as SVD burden score. This is an ordinal composite score, ranging from 0 to 6 points, which represents the small vessel disease burden in CAA and incorporates lobar CMBs, CSS, CSO-PVS and WMH. In case of 2–4 lobar CMBs, one point is awarded; in case of 5 of more CMBs, two points are awarded. Focal CSS is awarded with 1 point, and disseminated CSS with 2 points. Periventricular WMH Fazekas score of 3 and/or deep WMH Fazekas score of ≥2 (23) was awarded 1 point. Lastly, CSO-PVS ≥21 was also awarded with 1 point (22).
Data analysis
The Shapiro-Wilk test was used to analyze the normality of the data. If parameters were normally distributed, they were depicted as mean ± standard deviation and group differences were analyzed with a Student’s t-test or an ANOVA. Otherwise, they were stated as medians with interquartile ranges (IQR) and differences were analyzed with a Mann-Whitney U test or a Kruskal-Wallis test. Depending on group size, sex frequency was analyzed by a chi-square test or Fisher’s exact test.
In order to measure the magnitude of difference between patients and controls, we compared the mean biomarker levels of the different groups, expressed as a fold change (FC; patients/controls). When comparing group differences of CSF Aβ levels, we adjusted for age and sex by performing multiple regression analysis with patient group, age and sex as independent variables. In the patients with sCAA and controls used for discovery, we performed a sensitivity analysis comparing probable CAA patients with controls. To determine the diagnostic accuracy of CSF Aβ38, 40, 42, 43 for the distinction between groups, we determined the area under the curve (AUC) using a receiver operating characteristic curve (ROC) with 95% confidence interval (CI). The Youden index was determined (sensitivity + specificity − 1.0) to find the optimal cutoff value, and the sensitivity and specificity for that cutoff was calculated. We also evaluated whether ratio’s or combinations of the various Aβ peptides yielded a higher AUC than a single Aβ peptide.
Spearman rank correlation (rSP) was used to evaluate correlations between the CSF Aβ peptides with each other, age and MoCA scores. Using partial correlation, the correlation between the CSF Aβ peptides with small vessel disease (SVD) burden score was adjusted for age.
Ethical statement
Lumbar punctures were performed after informed consent from the patients themselves. CAA patients underwent a lumbar puncture in the context of cross-sectional studies on biomarkers for CAA, which were approved by the local medical ethics committees (CAVIA; no. 2014–1401, BIONIC no. 2017–3810, AURORA; NL62670.058.17, FOCAS: NL63256.058.17, patients from MGH: no. 2006P000664). For AD patients and controls, CSF was used that remained after a clinical diagnostic work-up (no. 2016–3011).
Results
CSF Aβ levels in patients with sCAA and controls; discovery
Patients with sCAA (age 72±7 years) were older than the controls (age 63±8 years; p<0.0001). The sex distribution was similar (Table 1).
CSF Aβ38 (FC:0.84), Aβ40 (FC: 0.80), Aβ42 (FC: 0.48) and Aβ43 (FC: 0.37) levels, adjusted for age and sex, were all significantly lower in patients with sCAA than in controls (Table 1; Figure 1A–D; results of unadjusted comparisons are presented in Table S1). The differences were most pronounced for Aβ42 and Aβ43. The comparison between the subset of patients with probable CAA and controls yielded similar results (Table 1). ROC analysis (probable CAA versus controls) yielded area under the curve (AUC) values between 0.65 (Aβ38) and 0.95 (Aβ43; for details see Figure 1E and Table S3). Any ratio or any combination of Aβ biomarkers did not yield a higher AUC than calculated for any of the individual peptides (data not shown).
Figure 1:

CSF Aβ38, Aβ40, Aβ42 and Aβ43 levels in sCAA patients and controls for discovery. (A) Scatterplots in all panels (depicting median and interquartile range). P-values adjusted for age and sex are depicted. Panel A: CSF Aβ38 levels were decreased in sCAA patients (p=0.015). Panel B: CSF Aβ40 levels were decreased in sCAA patients (p=0.015). Panel C: CSF Aβ42 levels were decreased in sCAA patients (p<0.0001). Panel D: CSF Aβ43 levels were decreased in sCAA patients (p<0.0001). Panel E: ROC analysis showed moderately high to high accuracy levels for discrimination of probable CAA from controls in the discovery group (CSF Aβ38: AUC of 0.65 (95% confidence interval (CI): 0.51–0.79); CSF Aβ40: AUC of 0.63 (95% CI: 0.49–0.75); CSF Aβ42: AUC of 0.90 (95% CI: 0.82–0.99); CSF Aβ43: AUC of 0.95 (95% CI: 0.88–1.00). Abbreviations: sCAA= sporadic cerebral amyloid angiopathy, CSF= cerebrospinal fluid, ROC: receiver operator curve. * = p<0.05, *** = p<0.001.
CSF Aβ levels in patients with sCAA and controls; validation
The age and sex distribution of the patients with sCAA was similar (Table 2).
CSF Aβ38 (FC: 0.61), Aβ40 (FC:0.48), Aβ42 (FC: 0.30) and Aβ43 (FC: 0.36) levels, adjusted for age and sex, were all significantly decreased in patients with sCAA compared to controls (Table 2; Figure 2A–D; results of unadjusted comparisons are presented in Table S2). The differences were most pronounced for Aβ42 and Aβ43. ROC analysis (probable CAA versus controls) yielded AUC values between 0.81 (Aβ38) and 0.94 (Aβ42; for details see Figure 2E and Table S3). Any ratio or any combination of Aβ biomarkers did not yield a higher AUC than calculated for any individual peptides (data not shown).
Figure 2:

CSF Aβ38, Aβ40, Aβ42 and Aβ43 levels in sCAA patients, AD patients and controls for validation (A). Scatterplots in all panels (depicting median and interquartile range). p-values adjusted for age and sex are depicted. Panel A: CSF Aβ38 levels were decreased in sCAA patients compared to controls (p<0.0001), and compared to AD patients (p<0.0001). Panel B: CSF Aβ40 levels were decreased in sCAA patients compared to controls (p<0.001), and compared to AD patients (p<0.0001). Panel C: CSF Aβ42 levels were decreased in sCAA patients compared to controls (p<0.0001) and compared to AD patients (p=0.012), and decreased in AD patients compared to controls (p<0.0001). Panel D: CSF Aβ43 levels were significantly decreased in sCAA patients compared to controls (p<0.001) and in AD patients compared to controls (p< 0.001), but similar between AD and sCAA patients (p=0.86). Panel E: ROC analysis showed moderately high to high accuracy levels for discrimination of probable sCAA from controls in the validation group (CSF Aβ38: AUC of 0.81 (95% CI: 0.71–0.91); CSF Aβ40: AUC of 0.61 (95% CI: 0.46–0.75); CSF Aβ42: AUC of 0.94 (95% CI: 0.89–0.99) CSF Aβ43: AUC of 0.91 (95% CI: 0.83–1.0). Panel F: ROC analysis showed moderately high to high accuracy levels for discrimination of probable sCAA from AD patients (CSF Aβ38: 0.82 (95% CI: 0.71–0.93), CSF Aβ40: AUC of 0.88 (95% CI: 0.80–0.96), CSF Aβ42: AUC of 0.79 (95% CI: 0.66–0.92), CSF Aβ43: AUC of 0.68 (95% CI: 0.53–0.82). Abbreviations: Abbreviations: AD= Alzheimer patients with a CSF biomarker profile indicative of Alzheimer’s disease. AUC= area under the curve, sCAA= sporadic cerebral amyloid angiopathy, CSF= cerebrospinal fluid, *p<0.05, *** = p<0.001.
Comparison of Aβ levels of patients with sCAA of different centers
CSF Aβ38, CSF Aβ40 and CSF Aβ42, adjusted for age and sex, were significantly decreased in the patients with sCAA from LUMC compared to the patients with sCAA from RUMC (Figure 3). CSF Aβ43, however, was similar in patients with sCAA from the LUMC and RUMC. There were no significant differences in any CSF Aβ peptide between the patients with sCAA from MGH and patients with sCAA from RUMC or LUMC (Figure 3).
Figure 3:

Effect of center on CSF Aβ38, Aβ40, Aβ42 and Aβ43 levels. CSF Aβ38, Aβ40, Aβ42 and Aβ43 levels in sCAA patients from the validation cohort: CAA patients from RUMC, LUMC and MGH. In all panels scatterplots are shown, depicting median and interquartile range. The mean age of the sCAA patients from RUMC (n=12) was 71±7 years and 33% was male, the mean age from the sCAA patients from LUMC (n=9) was 74±9 years and 30% was male, and the mean age from the sCAA patients from MGH (n=19) was 63±8 years and 63% was male. Panel A: CSF Aβ38 levels in patients with sCAA from LUMC were decreased compared to the patients with sCAA from RUMC (p=0.046). Panel B: CSF Aβ40 levels were decreased in patients with sCAA from LUMC compared to patients with sCAA from RUMC (p<0.0001). Panel C: CSF Aβ42 levels were decreased in LUMC patients compared to the RUMC patients (p=0.013). Panel D: CSF Aβ43 levels were similar in patients with sCAA from LUMC, RUMC and MGH. p-values are adjusted for age and sex. Abbreviations: CAA= cerebral amyloid angiopathy, CSF= cerebrospinal fluid, LUMC= Leiden University Medical Centre, Leiden, the Netherlands, MGH= Harvard Medical School, Boston, USA, RUMC= Radboud University Medical Center, Nijmegen. *=p<0.05, *** = p<0.001.
Correlations of CSF Aβ isoforms with each other, age, MoCa and MRI parameters in sCAA and controls.
In the patients with sCAA and controls for discovery, both CSF Aβ42 (rSP=-0.30; p=0.02) and CSF Aβ43 (rSP=-0.30; p=0.02) correlated with age, whereas CSF Aβ38 and CSF Aβ40 did not (Figure 4A). In the patients with sCAA and controls for validation, none of the CSF Aβ peptides correlated with age (Figure 4A). Furthermore, all CSF Aβ peptides correlated with each other, with the strongest correlations between CSF Aβ38 and Aβ40 (discovery: rSP=0.86; p< 0.001, validation: rSP=0.93; p< 0.001; Figure 4A and 4B) and CSF Aβ42 and Aβ43 (discovery: rSP=0.93; p< 0.001, validation: rSP=0.97; p< 0.001; Figure 4A and 4B). There were no significant correlations between MoCA scores and CSF Aβ levels in patients with sCAA of both groups (Figure 4A and 4B). In the validation cohort, there were significant age-adjusted correlations between all Aβ peptides and SVD burden (rSP= (-0.57) – (-0.67); p=0.001–0.009; Figure 4B). Lastly, we found no significant correlations between the time (in days) between most recent ICH and lumbar puncture and CSF Aβ isoforms (data not shown). The median time between lumbar puncture and most recent ICH in the patients with sCAA in the discovery cohort was 476 days [189–1320] and in the validation cohort 425 days [249–1228].
Figure 4:

(A) Correlation of CSF Aβ38, Aβ40, Aβ42 and Aβ43 with age, MoCA score, and amongst the Aβ peptides (patients with sCAA and controls for discovery (n=67)). MoCa score (n=12) was only available for a subset of sCAA patients. Spearman correlation coefficients are stated. (B): Correlation of CSF Aβ38, Aβ40, Aβ42 and Aβ43 with age, MoCa score, SVD burden score, and amongst the Aβ peptides (patients with sCAA and controls for validation (n=78). Spearman correlation coefficients are stated. MoCA and SVD burden scores were only available for a subset of sCAA patients (both n=21). (C) Correlation of CSF Aβ38, Aβ40, Aβ42 and Aβ43 with age, MoCA score, SVD burden score, and amongst the Aβ peptides in patients with D-CAA and controls. Spearman correlation coefficients are stated. MoCA (n=21) and SVD burden scores (n=19) were only available for (a subset of) D-CAA patients. The correlation of SVD burden score with the Aβ peptides was adjusted for age. The correlation between CSF Aβ40 and SVD burden score showed a trend (p=0.075). * indicates a significant correlation (p<0.05). Abbreviations: CSF= cerebrospinal fluid; Aβ= amyloid beta, MoCA= Montreal Cognitive Assessment, SVD = small vessel disease.
CSF Aβ levels in patients with AD compared to patients with sCAA and controls; validation group
The age of the patients with AD was similar to the age of controls and of patients with sCAA and sex distribution was similar as well (Table 2).
CSF Aβ38 and CSF Aβ40 levels, adjusted for age and sex, were significantly higher in patients with AD compared to patients with sCAA, but similar to controls (Table 2; Figure 2A–B; results of unadjusted comparisons are presented in in Table S2). Furthermore, CSF Aβ42 levels were significantly higher in patients with AD compared to patients with sCAA, and were significantly decreased in both sCAA and AD compared to controls (Table 2; Figure 2C). Lastly, CSF Aβ43 levels were similar in patients with AD and patients with sCAA, but were decreased compared to controls (Table 2, Figure 2D).
ROC analysis (AD versus probable CAA) yielded AUC values between 0.68 (Aβ43) and 0.88 (Aβ40; for details see Figure 2F and Table S3). The ratio of CSF Aβ42/Aβ43 yielded an AUC of 0.92 (95% CI: 0.85–0.99). The combination of CSF Aβ40 and Aβ43 in combination with either Aβ38 or Aβ42 both yielded the highest AUC (0.96, 95% CI: 0.92–1.0).
CSF Aβ levels in D-CAA
Age and sex were statistically similar in patients with presymptomatic D-CAA (8 of these patients did not have microbleeds or cortical superficial siderosis) versus the young controls, and in the patients with symptomatic D-CAA patients versus the old controls (Table 3). CSF Aβ38, Aβ40, Aβ42 and Aβ43 levels, adjusted for age and sex, were all significantly decreased in both patients with presymptomatic and symptomatic versus controls. CSF Aβ40 and Aβ42, adjusted for age and sex, were decreased in patients with symptomatic D-CAA compared to patients with asymptomatic CAA, and unadjusted analysis showed a decrease in all Aβ isoforms (Table 3; Figure 5; Table S4).
Figure 5:
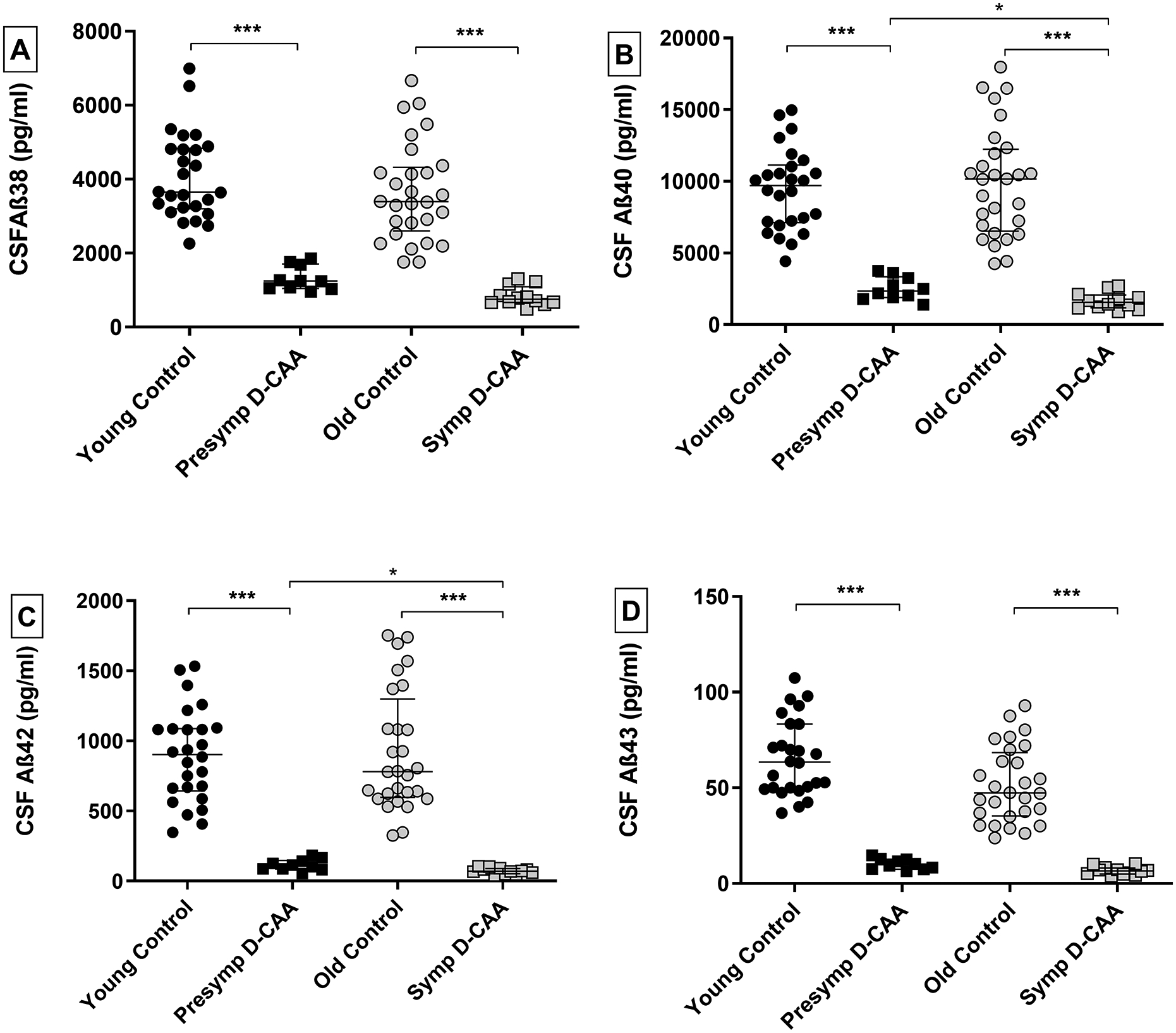
CSF Aβ38, Aβ40, Aβ42 and Aβ43 levels in D-CAA patients and controls. (A) Scatterplots in all panels (depicting median and interquartile range). p-values adjusted for age and sex are depicted. Panel A: CSF Aβ38 levels were significantly decreased in presymptomatic and symptomatic D-CAA patients versus their respective controls (both p<0.0001). Panel B: CSF Aβ40 levels were significantly decreased in presymptomatic and presymptomatic D-CAA patients versus their respective controls (both p<0.001), and in symptomatic versus presymptomatic patients (p=0.047). Panel C: CSF Aβ42 levels were significantly decreased in presymptomatic and symptomatic D-CAA patients versus their respective controls (both<0.001), in symptomatic D-CAA patients versus controls (p<0.001), and in symptomatic versus presymptomatic patients (p=0.026). Panel D were significantly decreased in presymptomatic and presymptomatic D-CAA patients versus their respective controls (both p<0.0001). Abbreviations: D-CAA= Dutch-type cerebral amyloid angiopathy, CSF= cerebrospinal fluid. Presymp D-CAA= presymptomatic D-CAA patients, Symp D-CAA=symptomatic D-CAA patients. *=p<0.05, *** = p<0.001.
All ROC analyses, for both presymptomatic and symptomatic D-CAA patients compared to age-matched controls, and for all Aβ peptides, yielded an AUC of 1 (95% CI: 1–1).
CSF Aβ38 (rSP=-0.53; p=0.01) and Aβ43 correlated with age (rSP=-0.49; p<0.001).
Furthermore, all CSF Aβ peptides correlated with each other, with the strongest correlations between CSF Aβ38 and Aβ40 (rSP=0.94; p< 0.001, Figure 4C) and CSF Aβ40 and Aβ42 (rSP=0.95; p< 0.001). There were no correlations between CSF Aβ isoforms and MoCA scores (Figure 4C). Lastly, there was a significant age-adjusted correlation of SVD score with Aβ42 (rSP=-0.52; p=0.026; Figure 4C). Lastly, we found no significant correlations between the time (in days) between most recent ICH and lumbar puncture (data not shown) CSF Aβ isoforms in the patients with symptomatic D-CAA. The median time was between most recent ICH and lumbar puncture was 392 days [264–969].
Discussion
The main findings of our study are as follows: (1) In patients with sCAA, levels of CSF Aβ38, Aβ40, Aβ42, and Aβ43 are significantly decreased compared to controls, and the decreases in CSF levels of Aβ42 and Aβ43 (AUC value of up to 0.94) appeared stronger than in the other CSF Aβ species, as established in two independent sample sets (2). All four Aβ peptide levels are also decreased in presymptomatic and symptomatic D-CAA patients, discriminating patients from controls with 100% specificity and 100% sensitivity; (3) Levels of CSF Aβ38, Aβ40, and Aβ42, but not Aβ43, are lower in sCAA as compared to AD; (4) The combination of Aβ40, Aβ43, and either Aβ38 or Aβ42 yielded an AUC of 0.96 to discriminate sCAA from AD patients.
With our results, we confirm previous findings of decreased CSF Aβ40 and Aβ42 levels in patients with sCAA compared to patients with AD and controls (9, 24). In addition, we demonstrate reduced CSF levels of Aβ38 and Aβ43 in patients with sCAA compared to controls. In our study, Aβ42 and Aβ43 yield a better AUC compared to Aβ38 and Aβ40, and also compared to earlier reported AUC’s of Aβ40 (0.74) and Aβ42 (0.68) (9). Thus, Aβ42 and Aβ43 seem superior in their discrimination of sCAA and controls. These markers may be used in clinical practice when the diagnosis of CAA is uncertain, for instance in patients with possible CAA, or at an usual age of onset, or in patients with a contra-indication for MRI. In addition, since the Boston criteria v2.0 only have 55% sensitivity in patients without ICH, the CSF Aβ panel may add more diagnostic certainty in this patient group. Furthermore, this panel of four Aβ peptides could aid in the discrimination of sCAA from AD when there is uncertainty about the predominant pathology in patients presenting with cognitive symptoms: a decrease in all four peptides would be more indicative of CAA, while only a decrease in CSF Aβ42 and Aβ43, but not of Aβ38 and Aβ40 would be more indicative of AD. Another possible application of our results could be the detection of CAA in healthy subjects or patients with (mild) AD, although this should be validated first in a study specifically designed for this aim. In case CSF Aβ peptides are indeed capable to detect CAA in patients with AD, this would be highly relevant in the light of the emergence of anti-Aβ immunotherapy, especially with regard to the (controversial) approvals in 2022/2023 of aducanumab and lecanumab by the federal drug administration (25). These therapies are accompanied by a risk of side effects, the so called “anti-amyloid therapy-related imaging abnormalities”(ARIA): brain edema and hemorrhages, which occurred likely more frequent in AD patients with concomitant CAA (26). Thus, CSF biomarkers that detect CAA in patients with AD may potentially help with patient selection and exclusion of patients with (a high load of) CAA from these and other anti Aβ immunotherapy trails.
In patients with D-CAA, we also found decreased levels of Aβ38, Aβ40, Aβ42, and Aβ43 peptides, with no overlap between both presymptomatic and symptomatic patients and their respective controls. This confirms earlier findings of decreased Aβ40 and Aβ42 levels in D-CAA patients (12) and extends these findings to decreased levels of Aβ38 and Aβ43. Our results also show that CAA pathogenesis and the consequent alterations in all four CSF Aβ isoforms occur before symptomatic ICH, and even before the occurrence of neuroimaging features of CAA such as microbleeds or superficial siderosis.
We observe for D-CAA the strongest decreases in Aβ42 and Aβ43, similar to the pattern in patients with sCAA. This finding supports the hypothesis that in the process of vascular Aβ accumulation, Aβ42 deposits early and may serve as a “seed” in cerebral vessels, followed by growth of these deposits by Aβ40 deposition (27–31). There is also some evidence that Aβ43 can act as a seed in the parenchyma (32), although it has not been established if this occurs in vessels as well. Our findings indicate that CSF Aβ42 and Aβ43 in patients with D-CAA (and sCAA) may be used for monitoring of the efficacy of disease-modifying drugs in clinical trials.
It is known that a proportion of patients with AD has concurrent CAA-pathology: based on MRI a prevalence of 14% has been described for probable CAA, 22% for strictly lobar microbleeds, and 5% of cortical superficial siderosis, and based on neuropathology, the prevalence is even higher: almost 50% (5). Despite the fact that AD and CAA are not two entirely separate entities and often co-occur, we show that levels of CSF Aβ38, Aβ40, and Aβ42, but not Aβ43, are lower in sCAA as compared to AD. The use of CSF Aβ40 and Aβ42 to discriminate sCAA from AD has been described earlier (9). However, we show that the combination of Aβ40, Aβ43, and either Aβ38 or Aβ42, yielded an AUC of 0.96 to discriminate sCAA from AD. We unfortunately lack information on imaging markers associated with CAA in the patients with AD, but interestingly, despite that a proportion of our AD patients may have CAA, still, the aforementioned distinct CSF Aβ pattern (normal levels of Aβ38, Aβ40, decreased levels of Aβ42 and Aβ43) was found at the group level in AD patients.
Our observations of aberrant CSF Aβ levels may also provide insights into the underlying pathophysiology of CAA. Alterations in the dynamic equilibrium of Aβ production, clearance, and accumulation may lead to changes in CSF Aβ concentrations (9). Decreased Aβ levels in the CSF are thought to be the result of a net reduction of Aβ peptides eluted with interstitial fluid towards the CSF. This may be partly explained by trapping of the Aβ peptides in the cerebral vasculature or parenchyma. Longer Aβ peptides such as Aβ42 and Aβ43 are less soluble, more prone to aggregate, and mostly found in amyloid plaques in the brain parenchyma (33–35), whereas shorter peptides such as Aβ38 and Aβ40 are more soluble, can diffuse along perivascular drainage pathways, and accumulate predominantly in CAA (36–38). This corresponds to our findings that Aβ38 and Aβ40 are decreased only in patients with sCAA, and not in AD patients. Furthermore, we show that Aβ42 and Aβ43 are decreased in AD patients, which may be a result of the predominant accumulation of these peptides in amyloid plaques.
We found inconsistent results regarding the correlation between SVD burden score and CSF Aβ levels in the patients with sCAA and D-CAA. The SVD burden score is an ordinal score, based on the presence of four small vessel disease imaging markers and has been associated with the severity of post-mortem CAA-associated vasculopathic changes (22). In patients with sCAA, we found strong correlations with all Aβ peptides, which indicates that CSF Aβ may be a reflection of CAA-related pathologic changes. In the D-CAA group, we only found a weak correlation between SVD score and CSF Aβ42, and a trend for Aβ40. This may be explained by a the relatively small sample size of this group. Another explanation may be that the CSF Aβ levels in symptomatic D-CAA reach a plateau level earlier than the SVD burden score.
It is notable that the absolute values of Aβ38, Aβ40, Aβ42 and Aβ43 differed between the discovery and the validation cohort. Furthermore, levels of the Aβ peptides were lower in patients with sCAA from the LUMC compared to patients with sCAA from the RUMC. It is well-known that pre-analytical factors may affect the results of CSF Aβ analysis (39). Although we harmonized CSF protocols in terms of, e.g., centrifugation, storage temperature and tube use across the centres, it cannot be ruled out that unknown pre-analytical factors may have affected our results. Furthermore, it cannot be excluded that the observed differences were partly caused by a difference in patient characteristics in the various cohorts. Nevertheless, a center effect on Aβ measurements cannot be ruled out, which complicates the interpretation of the results in the validation cohort. Future studies that combine cohorts from different centers should also take this possible center effect into account.
Strengths of our study are that we could validate our findings in two independent, extensively characterized, patients with sCAA from various centers, and in the biggest group of patients with D-CAA to date.
A limitation of this study is that we did not have controls from centers other than the RUMC, which complicates interpretation of results, since the origin of the CSF samples may affect the concentration of CSF Aβ peptides. In addition, although neuroimaging was not the primary type of investigation of our study, we did not have standardized MRI available for the patients with AD and controls. Another limitation is that we did not have information on the final clinical diagnosis in our patients with AD, although the ATN classification has been accepted as a proxy for AD pathology (16). Furthermore, we lacked information on the presence of strictly lobar microbleeds or cortical superficial siderosis in our controls, although the reported prevalence of these markers in the general population controls is quite low and likely did not affect our results (7% for lobar microbleeds and 0.5% for cortical superficial siderosis (5)). Another limitation is that we had to use a different Aβ38 assay for the validation experiment, since the Aβ38 assay that we used in the discovery experiment went out of production during our study.
Conclusion:
A biomarker panel consisting of CSF Aβ38, Aβ40, Aβ42, and Aβ43 has great potential to distinguish sCAA from controls and from AD patients: Aβ38 and Aβ40 are only decreased in CAA, but not in AD. On the other hand, all four peptides are decreased in CAA compared to controls. Furthermore, Aβ38 and Aβ40 are most specific for sCAA in the comparison with AD, while Aβ42 and Aβ43 are superior for the distinction between sCAA and controls.
Supplementary Material
Acknowledgements:
This study was supported by the BIONIC project (no. 733050822, which has been made possible by ZonMW as part of ‘Memorabel’, the research and innovation program for dementia, as part of the Dutch national ‘Deltaplan for Dementia’: zonmw.nl/dementiaresearch), the SCALA project, funded by “The Galen and Hilary Weston Foundation” (NR170024) and the CAFÉ project (the National Institutes of Health, USA, grant number 5R01NS104147-02). We would additionally like to thank Mengfei Cai for his assistance with MRI processing and Nina Hilkens for her advice on statistical methods.
Footnotes
Potential conflicts of interest
Nothing to report.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
- 1.Charidimou A, Boulouis G, Gurol ME, Ayata C, Bacskai BJ, Frosch MP, et al. Emerging concepts in sporadic cerebral amyloid angiopathy. Brain. 2017;140(7):1829–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wermer MJH, Greenberg SM. The growing clinical spectrum of cerebral amyloid angiopathy. Current Opinion in Neurology. 2018;31(1):28–35. [DOI] [PubMed] [Google Scholar]
- 3.Charidimou A, Perosa V, Frosch MP, Scherlek AA, Greenberg SM, van Veluw SJ. Neuropathological correlates of cortical superficial siderosis in cerebral amyloid angiopathy. Brain. 2020;143(11):3343–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vales-Montero M, García-Pastor A, Iglesias-Mohedano AM, Esteban-de Antonio E, Salgado-Cámara P, García-Domínguez JM, et al. Cerebral amyloid angiopathy-related transient focal neurological episodes: A transient ischemic attack mimic with an increased risk of intracranial hemorrhage. Journal of the Neurological Sciences. 2019;406:116452. [DOI] [PubMed] [Google Scholar]
- 5.Jäkel L, De Kort AM, Klijn CJM, Schreuder FHBM, Verbeek MM. Prevalence of cerebral amyloid angiopathy: A systematic review and meta-analysis. Alzheimer’s & Dementia. 2022;18(1):10–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bornebroek M, Haan J, Maat-Schieman ML, Van Duinen SG, Roos RA. Hereditary cerebral hemorrhage with amyloidosis-Dutch type (HCHWA-D): I--A review of clinical, radiologic and genetic aspects. Brain Pathology 1996;6(2):111–4. [DOI] [PubMed] [Google Scholar]
- 7.Linn J, Halpin A, Demaerel P, Ruhland J, Giese AD, Dichgans M, et al. Prevalence of superficial siderosis in patients with cerebral amyloid angiopathy. Neurology. 2010;74(17):1346–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Charidimou A, Boulouis G, Frosch MP, Baron J-C, Pasi M, Albucher JF, et al. The Boston criteria version 2.0 for cerebral amyloid angiopathy: a multicentre, retrospective, MRI–neuropathology diagnostic accuracy study. The Lancet Neurology. 2022;21(8):714–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verbeek MM, Kremer BPH, Rikkert MO, Van Domburg PHMF, Skehan ME, Greenberg SM. Cerebrospinal fluid amyloid β40 is decreased in cerebral amyloid angiopathy. Annals of Neurology. 2009;66(2):245–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martínez-Lizana E, Carmona-Iragui M, Alcolea D, Gómez-Choco M, Vilaplana E, Sánchez-Saudinós MB, et al. Cerebral amyloid angiopathy-related atraumatic convexal subarachnoid hemorrhage: an ARIA before the tsunami. Journal of Cerebral Blood Flow & Metabolism. 2015;35(5):710–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Renard D, Gabelle A, Hirtz C, Demattei C, Thouvenot E, Lehmann S. Cerebrospinal Fluid Alzheimer’s Disease Biomarkers in Isolated Supratentorial Cortical Superficial Siderosis. Journal of Alzheimer’s Disease. 2016;54(4):1291–5. [DOI] [PubMed] [Google Scholar]
- 12.van Etten ES, Verbeek MM, van der Grond J, Zielman R, van Rooden S, van Zwet EW, et al. β-Amyloid in CSF: Biomarker for preclinical cerebral amyloid angiopathy. Neurology. 2017;88(2):169–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Banerjee G, Ambler G, Keshavan A, Paterson RW, Foiani MS, Toombs J, et al. Cerebrospinal Fluid Biomarkers in Cerebral Amyloid Angiopathy. Journal of Alzheimer’s Disease. 2020;74:1189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Almdahl IS, Lauridsen C, Selnes P, Kalheim LF, Coello C, Gajdzik B, et al. Cerebrospinal Fluid Levels of Amyloid Beta 1–43 Mirror 1–42 in Relation to Imaging Biomarkers of Alzheimer’s Disease. Frontiers in Aging Neuroscience. 2017;9(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu L, Lauro BM, He A, Lee H, Bhattarai S, Wolfe MS, et al. Identification of the Aβ37/42 peptide ratio in CSF as an improved Aβ biomarker for Alzheimer’s disease. Alzheimers & Dementia. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers & Dementia. 2018;14(4):535–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koemans EA, Voigt S, Rasing I, van Harten TW, Jolink WM, Schreuder FH, et al. Cerebellar Superficial Siderosis in Cerebral Amyloid Angiopathy. Stroke. 2021:552–7. [DOI] [PubMed] [Google Scholar]
- 18.Eckstein K, Bachrata B, Hangel G, Widhalm G, Enzinger C, Barth M, et al. Improved susceptibility weighted imaging at ultra-high field using bipolar multi-echo acquisition and optimized image processing: CLEAR-SWI. Neuroimage. 2021;237:118175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gregoire S, Chaudhary U, Brown M, Yousry T, Kallis C, Jäger H, et al. The Microbleed Anatomical Rating Scale (MARS): reliability of a tool to map brain microbleeds. Neurology. 2009;73(21):1759–66. [DOI] [PubMed] [Google Scholar]
- 20.Wardlaw JM, Smith EE, Biessels GJ, Cordonnier C, Fazekas F, Frayne R, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurology. 2013;12(8):822–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fazekas F, Chawluk JB, Alavi A, Hurtig HI, Zimmerman RA. MR signal abnormalities at 1.5 T in Alzheimer’s dementia and normal aging. American Journal of Roentgenology. 1987;149(2):351–6. [DOI] [PubMed] [Google Scholar]
- 22.Charidimou A, Martinez-Ramirez S, Reijmer YD, Oliveira-Filho J, Lauer A, Roongpiboonsopit D, et al. Total Magnetic Resonance Imaging Burden of Small Vessel Disease in Cerebral Amyloid Angiopathy: An Imaging-Pathologic Study of Concept Validation. JAMA Neurology. 2016;73(8):994–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Staals J, Makin SD, Doubal FN, Dennis MS, Wardlaw JM. Stroke subtype, vascular risk factors, and total MRI brain small-vessel disease burden. Neurology. 2014;83(14):1228–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Charidimou A, Friedrich JO, Greenberg SM, Viswanathan A. Core cerebrospinal fluid biomarker profile in cerebral amyloid angiopathy: a meta analysis. Neurology. 2018;90(9):e754–e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nature 613, 227–228 (2023). [DOI] [PubMed] [Google Scholar]
- 26.Greenberg SM, Bacskai BJ, Hernandez-Guillamon M, Pruzin J, Sperling R, van Veluw SJ. Cerebral amyloid angiopathy and Alzheimer disease — one peptide, two pathways. Nature Reviews Neurology. 2020;16(1):30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alonzo NC, Hyman BT, Rebeck GW, Greenberg SM. Progression of cerebral amyloid angiopathy: accumulation of amyloid-beta40 in affected vessels. Journal of Neuropathology & Experimental Neurology. 1998;57(4):353–9. [DOI] [PubMed] [Google Scholar]
- 28.Maat-Schieman ML, van Duinen SG, Rozemuller AJ, Haan J, Roos RA. Association of vascular amyloid beta and cells of the mononuclear phagocyte system in hereditary cerebral hemorrhage with amyloidosis (Dutch) and Alzheimer disease. Journal of Neuropathology & Experimental Neurology. 1997;56(3):273–84. [DOI] [PubMed] [Google Scholar]
- 29.Natté R, Yamaguchi H, Maat-Schieman MLC, Prins FA, Neeskens P, Roos RAC, et al. Ultrastructural evidence of early non-fibrillar Aβ42 in the capillary basement membrane of patients with hereditary cerebral hemorrhage with amyloidosis, Dutch type. Acta Neuropathologica. 1999;98(6):577–82. [DOI] [PubMed] [Google Scholar]
- 30.Shinkai Y, Yoshimura M, Ito Y, Odaka A, Suzuki N, Yanagisawa K, et al. Amyloid beta-proteins 1–40 and 1–42(43) in the soluble fraction of extra- and intracranial blood vessels. Annals of Neurology. 1995;38(3):421–8. [DOI] [PubMed] [Google Scholar]
- 31.Vinters HV, Secor DL, Read SL, Frazee JG, Tomiyasu U, Stanley TM, et al. Microvasculature in Brain Biopsy Specimens from Patients with Alzheimer’s Disease: An Immunohistochemical and Ultrastructural Study. Ultrastructural Pathology. 1994;18(3):333–48. [DOI] [PubMed] [Google Scholar]
- 32.Ruiz-Riquelme A, Mao A, Barghash MM, Lau HHC, Stuart E, Kovacs GG, et al. Aβ43 aggregates exhibit enhanced prion-like seeding activity in mice. Acta Neuropathologica Communications. 2021;9(1):83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Veluw SJ, Frosch MP, Scherlek AA, Lee D, Greenberg SM, Bacskai BJ. In vivo characterization of spontaneous microhemorrhage formation in mice with cerebral amyloid angiopathy. Journal of Cerebral Blood Flow & Metabolism. 2021;41(1):82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saito T, Suemoto T, Brouwers N, Sleegers K, Funamoto S, Mihira N, et al. Potent amyloidogenicity and pathogenicity of Aβ43. Nature Neuroscience. 2011;14(8):1023–32. [DOI] [PubMed] [Google Scholar]
- 35.Jarrett JT, Berger EP, Lansbury PT, Jr. The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer’s disease. Biochemistry. 1993;32(18):4693–7. [DOI] [PubMed] [Google Scholar]
- 36.Jäkel L, Boche D, Nicoll JAR, Verbeek MM. Aβ43 in human Alzheimer’s disease: effects of active Aβ42 immunization. Acta Neuropathologica Communications. 2019;7(1):141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duyckaerts C, Delatour B, Potier MC. Classification and basic pathology of Alzheimer disease. Acta Neuropathologica. 2009;118(1):5–36. [DOI] [PubMed] [Google Scholar]
- 38.Moro ML, Giaccone G, Lombardi R, Indaco A, Uggetti A, Morbin M, et al. APP mutations in the Aβ coding region are associated with abundant cerebral deposition of Aβ38. Acta Neuropathologica. 2012;124(6):809–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fourier A, Portelius E, Zetterberg H, Blennow K, Quadrio I, Perret-Liaudet A. Pre-analytical and analytical factors influencing Alzheimer’s disease cerebrospinal fluid biomarker variability. Clinica Chimica Acta. 2015;449:9–15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.