

Abstract
Oncogenic mutations in isocitrate dehydrogenase (IDH)-1 and -2 occur in a wide range of cancers, including acute myeloid leukemia (AML) and glioma. Mutant IDH enzymes convert 2-oxoglutarate (2OG) to (R)-2-hydroxyglutarate ((R)-2HG)), an oncometabolite that is hypothesized to promote cellular transformation by dysregulating 2OG-dependent enzymes. The only (R)-2HG target that has been convincingly shown to contribute to transformation by mutant IDH is the myeloid tumor suppressor TET2. However, there is ample evidence to suggest that (R)-2HG has other functionally relevant targets in IDH-mutant cancers. Here, we show that (R)-2HG inhibits KDM5 histone lysine demethylases and that this inhibition contributes to cellular transformation in IDH-mutant AML and IDH-mutant glioma. These studies provide the first evidence of a functional link between dysregulation of histone lysine methylation and transformation in IDH-mutant cancers.
Keywords: Mutant IDH, KDM5, H3K4 histone lysine demethylase, tumor suppressor, cancer
Introduction:
Gain-of-function mutations in isocitrate dehydrogenase enzymes IDH1 and IDH2 occur in ~10% of AMLs, are defining mutations in adult oligodendrogliomas and astrocytomas, and are highly prevalent in a number of other cancers [1]. Cancer-associated mutations at arginine-132 of IDH1 (IDH1R132X) and arginine-140 or arginine-172 of IDH2 alter the catalytic activity of IDH enzymes such that, instead of converting isocitrate to 2OG, the mutant enzymes convert 2OG to the structurally similar oncometabolite (R)-2HG [2]. (R)-2HG is hypothesized to promote leukemogenesis by inhibiting the 2OG-dependent dioxygenase (2OGDD) TET2, a myeloid tumor suppressor enzyme that catalyzes the conversion of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxycytosine (5caC) [3, 4]. The observations that TET2 loss-of-function mutations are largely mutually exclusive with IDH mutations in AML and that TET2 loss phenocopies mutant IDH expression in cell-based myeloid transformation assays have established TET2 as a bona fide functionally relevant target of (R)-2HG in IDH-mutant AML [5, 6].
Although attention has been largely focused on TET2 as a critical mediator of mutant IDH-mediated leukemogenesis, several lines of evidence suggest that the oncogenic mechanisms engaged by mutant IDH acquisition and TET2 loss are not identical. Firstly, the hematopoietic phenotypes of Idh1R132H knock-in mice and Tet2 knockout mice are quite different. Tet2 loss results in enhanced hematopoietic stem cell (HSC) self-renewal, bone marrow hypercellularity, and a skewing towards monocytic differentiation [6], whereas Idh1R132H acquisition results in normal HSC self-renewal, expansion of early hematopoietic progenitors (HPCs), bone marrow hypocellularity, and normal hematopoietic differentiation [7]. Secondly, IDH and TET2 mutations appear to have opposing prognostic significance in patients with intermediate-risk AML. IDH mutations are associated with improved overall survival whereas TET2 mutations are associated with reduced overall survival [8–10]. Finally, the frequencies of IDH and TET2 mutations in clonal myeloid disorders are notably different (Figure 1A, Supplementary Table S1). In the case of TET2, mutations occur at similar frequencies in clonal hematopoiesis of unknown significance (CHIP), and in lower- and higher-grade myeloproliferative (MPN) and myelodysplastic (MDS) disorders, suggesting that TET2 loss confers a clonal advantage to HSCs but does not define their ultimate disease state. IDH mutations, on the other hand, occur at significantly higher frequencies in high-grade and blast-phase MPN and MDS than in lower-grade clonal myeloid disorders and are very rare in CHIP, suggesting that mutant IDH promotes an aggressive hyperproliferative disease phenotype.
Figure 1: (R)-2HG likely has other functionally relevant targets besides TET2 in IDH-mutant AML.
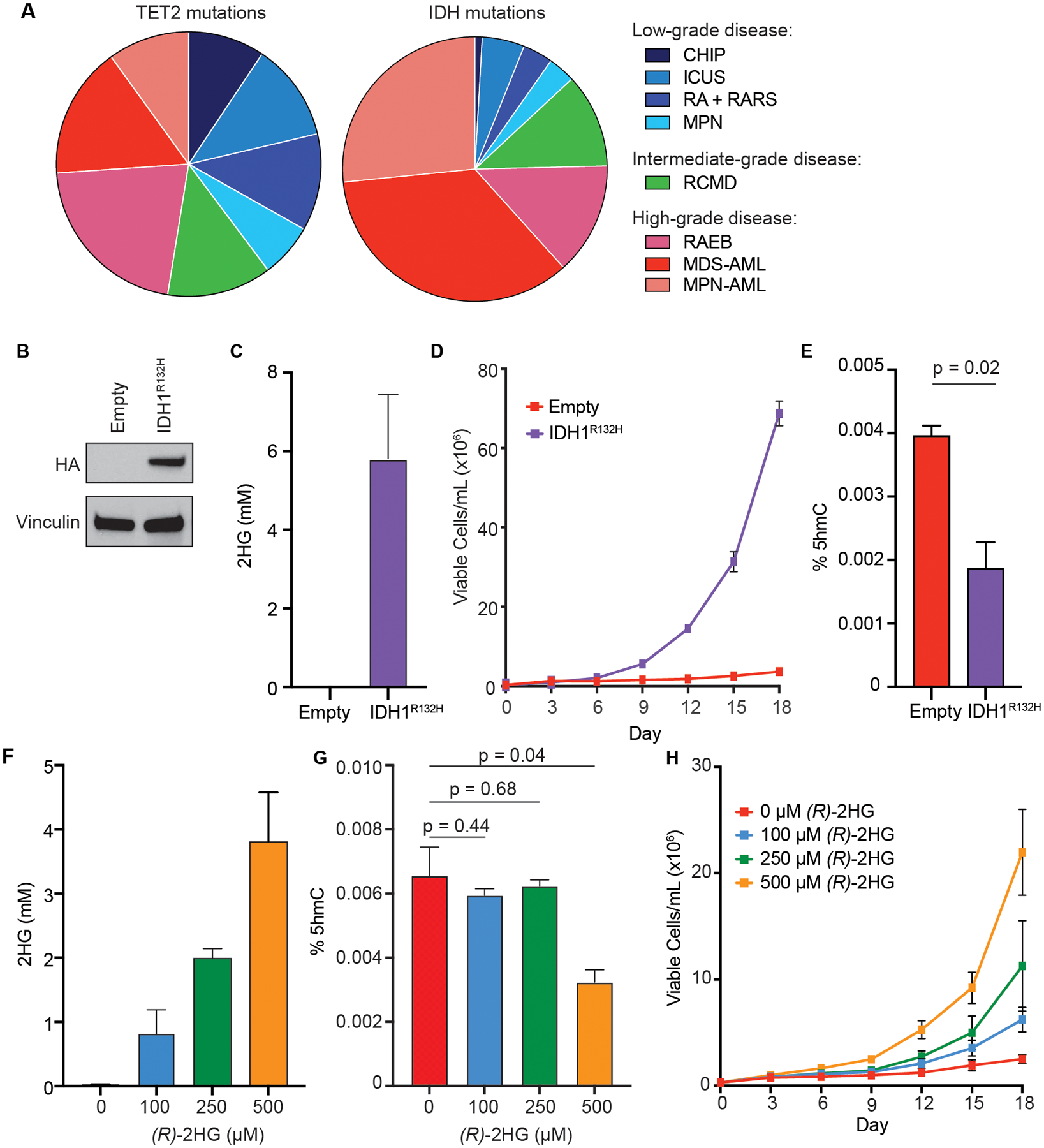
(A) Pie-charts illustrating the relative frequencies of TET2 (left) and IDH (right) mutations in clonal myeloid disorders (see Supplementary Table S1 for details). Low-grade clonal myeloid disorders: clonal hematopoiesis of indeterminate potential (CHIP), idiopathic cytopenia of undetermined significance (ICUS), refractory anemia (RA), refractory anemia with ring sideroblasts (RARS), and myeloproliferative neoplasms (MPN). Intermediate-grade clonal myeloid disorder: refractory cytopenia with multilineage dysplasia (RCMD). High-grade clonal myeloid disorders: refractory anemia with excess blasts (RAEB) and acute myeloid leukemias (AML) secondary to myelodysplastic disorder (MDS-AML) or myeloproliferative neoplasm (MPN-AML). (B) Immunoblot analysis of IDH1 expression in TF-1 cells expressing empty vector or HA-tagged R132H-mutant IDH1 (IDH1R132H). (C) GC-MS analysis of intracellular concentrations of 2HG in TF-1 cells expressing empty vector or IDH1R132H. Shown are mean values of duplicate experiments. (D) Proliferation under cytokine-poor conditions of TF-1 cells expressing empty vector or IDH1R132H. (E) Total 5hmC content in the TF-1 cells from (B-D) collected on day 0 of cytokine withdrawal, as measured by LC-MS. The average (± SD) of two independent biological replicates is shown. (F) GC-MS analysis of intracellular concentrations of 2HG under the following conditions: cultured in DMSO (0 μM R-2HG); cultured in 100 μM TFMB-(R)-2HG (100 μM R-2HG); cultured in 250 μM TFMB-(R)-2HG (250 μM R-2HG); and cultured in 500 μM TFMB-(R)-2HG (500 μM R-2HG). Shown are mean values of duplicate experiments. (G) Total 5hmC content in TF-1 cells following pre-treatment for 21 days with the indicated concentrations of TFMB-(R)-2-HG or DMSO-control (0 μM (R)-2HG), as measured by LC-MS. The average (± SD) of two independent biological replicates is shown. (H) Proliferation under cytokine-poor conditions of the TFMB-(R)-2-HG-treated TF-1 cells from (F-G). Representative results from at least three independent experiments are shown.
The mechanistic basis for the distinct hematologic effects of IDH and TET2 mutations is not well understood, although it has long been hypothesized that (R)-2HG-mediated disruption of other 2OGDD enzymes besides TET2 contributes to the oncogenic program induced by mutant IDH. In particular, the superfamily of 2OG-dependent Jumonji C domain-containing histone lysine demethylases (JmjC-KDMs) have been hypothesized to be important targets of (R)-2HG [11, 12]. A number of JmjC-KDM enzymes have been shown to be inhibited by (R)-2HG in vitro, and several of these same enzymes are recurrently mutated or otherwise downregulated in cancer [13]. This suggests that these enzymes can function as tumor suppressors and could plausibly contribute to mutant IDH-mediated transformation. However, histone lysine demethylase function is highly context dependent, and there are many examples of (R)-2HG-sensitive JmjC-KDM enzymes that can have either tumor suppressor or tumor-promoting functions, depending on the cellular context. For example, overexpression of KDM2A promotes stem-like phenotypes in hepatocellular carcinoma cells [14], whereas knockdown of KDM2A induces melanoma in zebrafish [15]; overexpression of KDM6B inhibits leukemogenesis in mouse models of AML1-ETO- and PML-RARα-translocated AML [16], but deletion of Kdm6b inhibits leukemogenesis in mouse models of MLL-AF9-translocated AML [17]; and inhibition of KDM4C blocks adipocyte differentiation of murine 3T3-L1 cells [11], but KDM4C is essential for normal HSC function and is a dependency in MLL-translocated AML [18, 19]. Given the complex functions of histone lysine demethylases in cancer, the ability of (R)-2HG to inhibit specific JmjC-KDM enzymes that can act as tumor suppressors does not necessarily indicate that those JmjC-KDM enzymes function as tumor suppressors in the context of IDH-mutant cancer.
One of the major challenges in studying mutant IDH-mediated transformation is the paucity of disease-relevant cell-based assays in which to study the functional consequences of mutant IDH expression. Cancer cell lines generated from IDH-mutant human tumors are not dependent on mutant IDH for growth in 2-dimensional culture, and ectopic expression of mutant IDH does not induce IL-3 independence in Ba/F3 or 32D hematopoietic transformation assays [20–22]. We previously reported that ectopic expression of mutant IDH induces growth factor independence and blocks the differentiation of TF-1 human AML cells and enhances the proliferation and soft agar colony formation of immortalized human astrocytes (IHA) [5, 23]. Herein, we used TF-1 cells to perform unbiased genetic screens to identify novel 2OG-dependent myeloid tumor suppressors that could plausibly be functionally relevant targets of (R)-2HG in AML. We identified three members of the KDM5 family of H3K4 histone lysine demethylases, KDM5A, KDM5C and KDM5D, as genes whose loss promotes TF-1 cytokine independence. All three KDM5 enzymes are inhibited by tumor-relevant concentrations of (R)-2HG and pan-KDM5 inhibition phenocopies mutant IDH expression in both the TF-1 myeloid transformation assay and the IHA glial transformation assay. These findings strongly suggest that KDM5A, KDM5C and KDM5D are functionally relevant targets of (R)-2HG in IDH-mutant AML and IDH-mutant glioma.
Results:
(R)-2HG promotes TF-1 cytokine independence at concentrations that do not affect global 5hmC levels.
5hmC is a biomarker of TET2 activity. TET2 mutations and IDH mutations are both associated with decreased 5hmC levels [24]. Likewise, loss of WT1, which disrupts TET function by a distinct mechanism, also reduces 5hmC levels. [25]. Consistent with these observations, TF-1 cells transformed to cytokine independence by ectopic expression of R132H-mutant IDH1 (TF1-IDH1R132H) have significantly lower 5hmC levels when compared to control TF-1 cells (Figure 1B–E).
Inhibition of TET2 is sufficient to phenocopy mutant IDH expression in cell-based transformation assays (Supplementary Figure 1A–B) [5, 23]. To determine if the transforming activity of mutant IDH correlates with (R)-2HG-mediated suppression of 5hmC, we treated TF-1 cells with a range of concentrations of cell-permeable α-trifluoromethyl benzyl-(R)-2HG (TFMB-(R)-2HG). We then measured (R)-2HG levels in the cells by gas chromatography-mass spectrometry (GC-MS), measured 5hmC levels in the cells by liquid chromatography-mass spectrometry (LC-MS), and evaluated transformation of the cells by assessing their ability to proliferate in the absence of cytokine. Treatment of TF-1 cells with increasing concentrations of TFMB-(R)-2HG resulted in dose-dependent accumulation of intracellular (R)-2HG (Figure 1F, Supplementary Table S2). Treatment with 100 μM and 250 μM TFMB-(R)-2HG, which resulted in intracellular (R)-2HG levels of 0.8 mM and 2.0 mM, respectively, did not suppress global 5hmC levels, whereas treatment with 500 μM TFMB-(R)-2HG resulted in intracellular (R)-2HG levels of ~4 mM and significant 5hmC suppression (Figure 1G). This is consistent with our previous finding that the (R)-2HG IC50 value for TET2 is ~5 mM [23]. Notably, although 500 μM TFMB-(R)-2HG more robustly transformed TF-1 cells, 100 μM and 250 μM TFMB-(R)-2HG were sufficient to induce TF-1 cytokine independence (Figure 1H).
There are two possible explanations for our observation that concentrations of (R)-2HG that have no discernable effect on global 5hmC can promote TF-1 cytokine independence. One possibility is that the level of TET2 inhibition that is required to promote TF-1 transformation is significantly less than that required to suppress global 5hmC levels. A recent study found that the level of TET2 activity at different methylated cytosines is highly dependent on their flanking sequence contexts [26]. It is possible that 5hmC-regulated loci that are critical for transformation of TF-1 cells are more sensitive to TET2 inhibition than are other sites, and that these specific loci are dysregulated at intracellular (R)-2HG levels that do not affect bulk TET activity. Another possible explanation is that (R)-2HG has other tumor suppressor targets that are more sensitive to inhibition by (R)-2HG than is TET2 and that that these other targets contribute to mutant IDH-mediated transformation. Consistent with this latter hypothesis, although partial TET2 knockdown is sufficient to promote the cytokine independent proliferation of TF-1 cells (Supplementary Figure S1A–B), this cytokine independence is significantly enhanced by pre-treatment with low-dose TFMB-(R)-2HG (Supplementary Figure S1C).
KDM5 histone lysine demethylases are myeloid tumor suppressors.
To identify novel myeloid tumor suppressors that could be (R)-2HG targets, we performed a pooled whole genome positive-selection CRISPR/Cas9 knockout screen in TF-1 cells (Figure 2A). We reasoned that single-guide RNAs (sgRNAs) that disrupt myeloid tumor suppressors would be enriched over time under cytokine-poor conditions by virtue of their ability to confer cytokine independence to TF-1 cells. Indeed, among the top-scoring sgRNAs in the screen were sgRNAs that target negative regulators of RAS signaling (RASA2, PTEN, RALGAPB and NF1) [27–29], and sgRNAs that target components of the PRC2 myeloid tumor suppressor complex (EZH2, EED and SUZ12) [30] (Figure 2B, Supplementary Table S3). Only two 2OGDD enzymes, the histone lysine demethylases KDM5A and KDM5C, scored with false discovery rates (FDR) <0.01. sgRNAs targeting other known tumor suppressors, including WT1 and KEAP1 [31, 32], were more modestly enriched.
Figure 2: Positive-selection CRISPR/Cas9 knockout screens identify KDM5 enzymes as 2OG-dependent myeloid tumor suppressors.
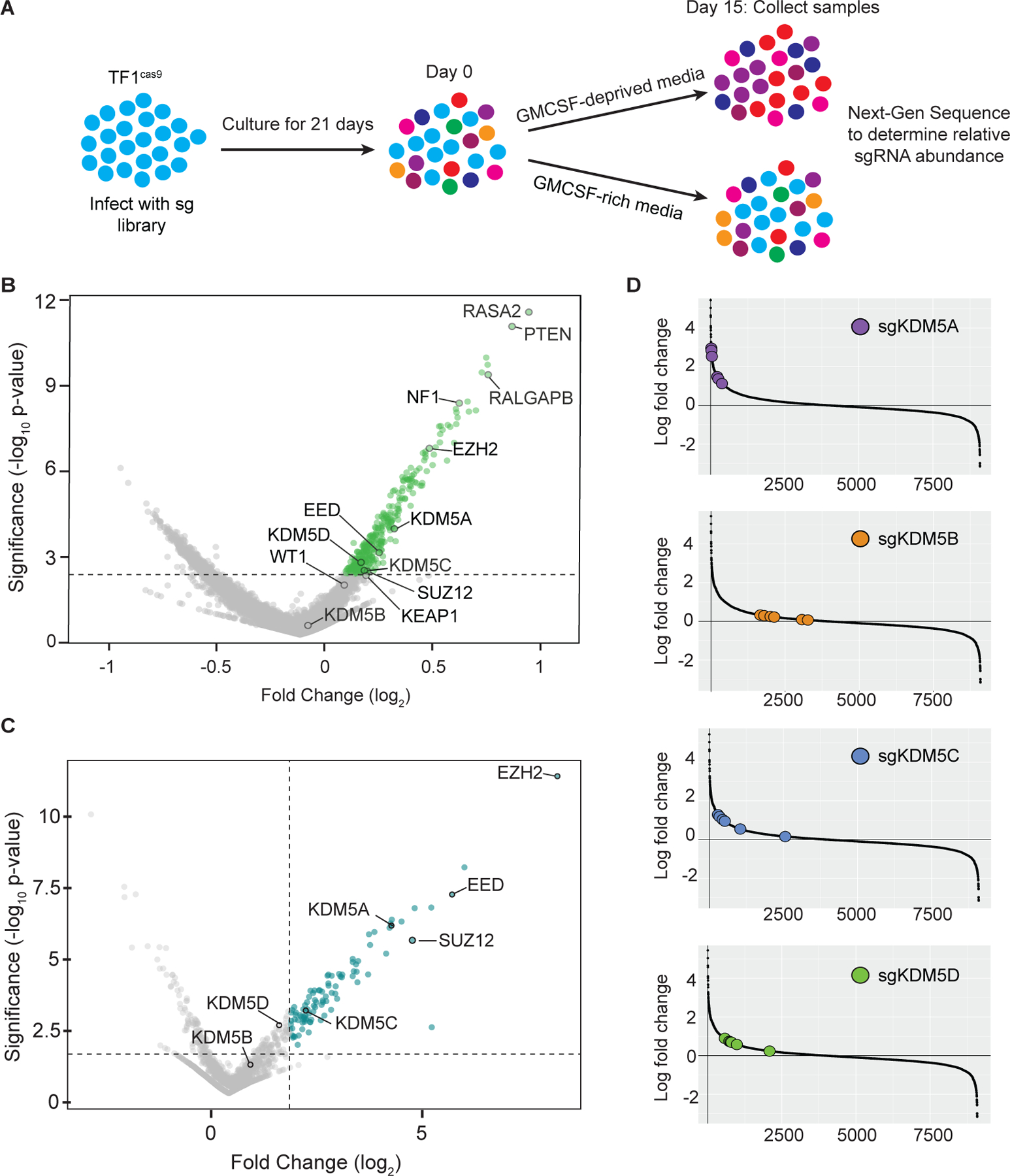
(A) Schematic of the CRISPR/Cas9 screens. TF-1 cells expressing Cas9 (TF1cas9) were infected with either the Brunello genome-wide CRISPR knockout sgRNA library or a custom library of 9,088 sgRNAs targeting 1355 genes (see Supplementary Table S4 for details), passaged for 21 days, split into GM-CSF-supplemented or GM-CSF-lacking media, and cultured for another 15 days. Cells were collected on day 0 and day 15 of differential cytokine treatment, gDNA was purified, and the abundance of each sgRNA in each sample was determined by next-generation sequencing. (B) Volcano plot illustrating the relative enrichment of sgRNAs targeting negative regulators of RAS signaling (RASA2, PTEN, RALGAP8 and NF1), known myeloid tumor suppressors (EZH2, EED, SUZ12, WT1 and KEAP1) and KDM5 family members in the genome-wide CRISPR/Cas9 knockout screen. Shown are the combined results of two biological replicates of the screen. (C) Volcano plot illustrating the relative enrichment of sgRNAs targeting EZH2, EED, SUZ12, and the KDM5 genes in the custom minipool CRISPR/Cas9 knockout screen. Shown are the combined results of two biological replicates of the screen. (D) Rank order and relative enrichment of the individual sgRNAs targeting KDM5A, KDM5B, KDM5C or KDM5D in the custom minipool CRISPR/Cas9 knockout screen.
In large-scale pooled screens, the precise ranking of sgRNAs can be ‘noisy,’ particularly when the observed phenotypic effect size is small. To identify 2OGDD-targeting sgRNAs that confer even a modest proliferative advantage to cytokine-starved TF-1 cells, we repeated the CRISPR/Cas9 knockout screen using a custom minipool library of sgRNAs that target all known 2OGDD enzymes as well as other chromatin remodelers and transcriptional regulators (Supplementary Table S4). Consistent with the results of the whole genome CRISPR/Cas9 screen, the PRC2 complex components EZH2, EED, and SUZ12 were among the top-scoring target genes, and KDM5A was the top-scoring 2OGDD (Figure 2C–D, Supplementary Table S5). sgRNAs targeting KDM5C and KDM5D were also significantly enriched, whereas sgRNAs targeting the KDM5 paralog KDM5B were not enriched. No other 2OGDD enzymes scored in the screen.
To validate the results of the CRISPR/Cas9 screens, we individually deleted KDM5A, KDM5B, KDM5C or KDM5D in TF-1 cells with one of two independent sgRNAs per gene and assessed the cytokine-independent proliferation of the cells. KDM5A, KDM5C and KDM5D knockout, but not KDM5B knockout, induced modest TF-1 cytokine independence (Figure 3A–B). Given that (R)-2HG could simultaneously inhibit multiple KDM5 enzymes, we generated TF-1 cells with combinatorial knockout of KDM5A, KDM5C, and KDM5D (TF1-sgACD) (Figure 3C). Deletion of all three KDM5 enzymes resulted in robust cytokine independent proliferation of TF-1 cells that phenocopied the effects of mutant IDH expression (Figure 3D).
Figure 3: Genetic or chemical inhibition of KDM5 enzymes phenocopies mutant IDH expression in TF-1 cells.
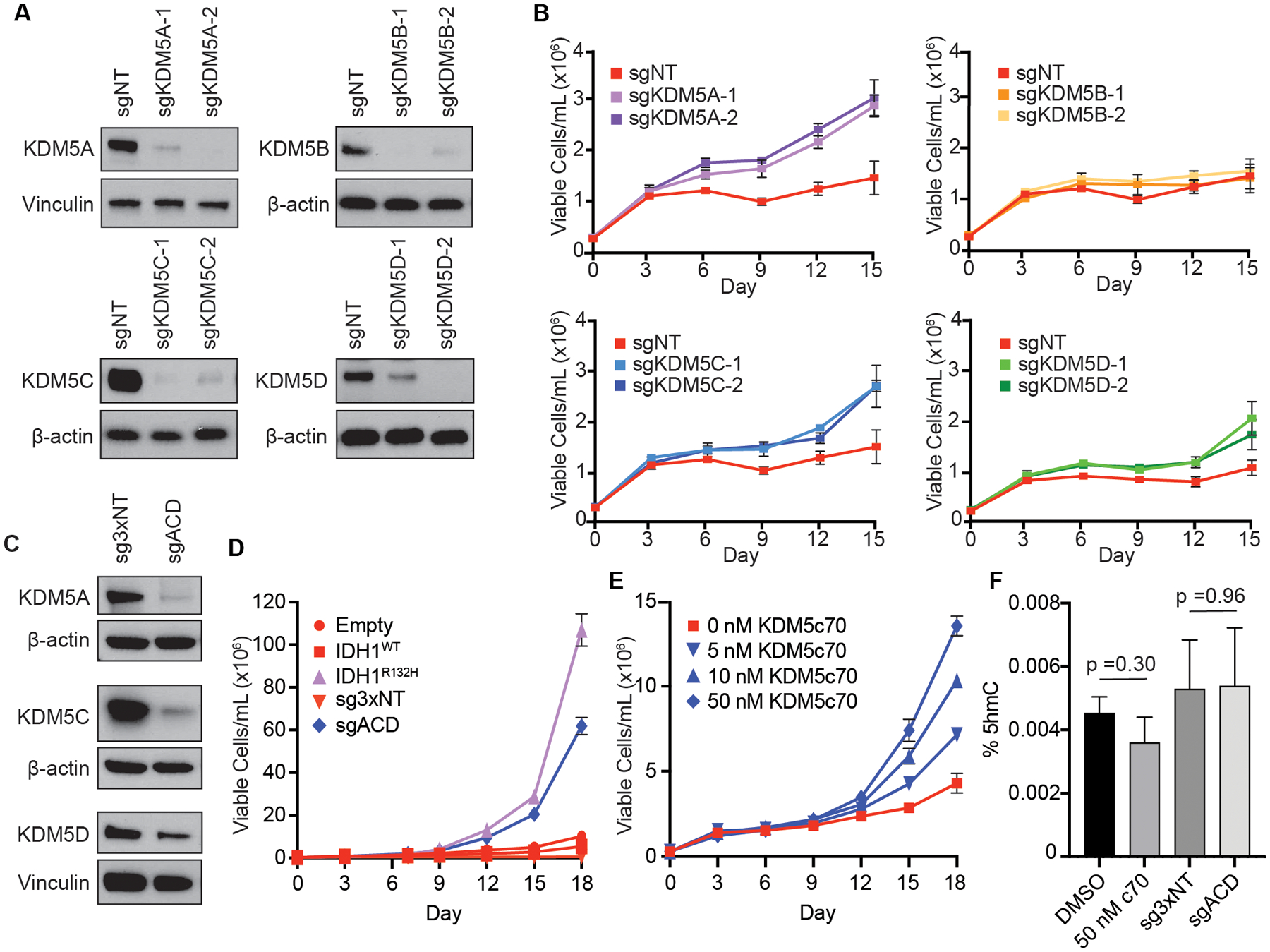
(A) Immunoblot analysis of KDM5 isoform expression in TF-1 cells infected with lentivirus co-expressing Cas9 and a non-targeting sgRNA (sgNT) or Cas9 and one of two sgRNAs targeting the indicated KDM5 paralogs: KDM5A (sgKDM5A), KDM5B (sgKDM5B), KDM5C (sgKDM5C), or KDM5D (sgKDM5D). (B) Proliferation under cytokine-poor conditions of the TF-1 cells from (A). (C) Immunoblot analysis of expression of the indicated KDM5 isoforms in TF-1 cells infected with lentiviruses co-expressing Cas9 and three non-targeting sgRNAs (sg3xNT) or Cas9 and sgRNAs targeting KDM5A, KDM5C and KDM5D (sgACD). (D) Proliferation under cytokine-poor conditions of the TF-1 cells from (C). Proliferation under cytokine-poor conditions of TF-1 cells expressing empty vector (Empty), HA-tagged wild-type IDH1 (IDH1WT) or HA-tagged R132H-mutant IDH1 (IDH1R132H) are shown for comparison. (E) Proliferation under cytokine-poor conditions of TF-1 cells treated with the indicated concentrations of the pan-KDM5 inhibitor KDM5c70 or DMSO-control (0 nM KDM5c70). (F) Total 5hmC content in DMSO- and KDM5c70-treated cells and sg3xNT and sgACD cells collected on day 0 of cytokine withdrawal, as measured by LC-MS. In all cases, cells were passaged for 21 days after lentiviral infection or for 21 days in the presence of DMSO or KDM5c70 prior to immunoblot analysis, cytokine withdrawal, and 5hmC quantification. Shown in (A-F) are representative results from three independent experiments.
Several KDM enzymes, including KDM5A, have been found to have demethylase activity-independent functions [33]. To determine whether the cytokine independent proliferation induced by KDM5A/C/D triple knockout is due to loss of KDM5 catalytic activity, we treated parental TF-1 cells with KDM5c70, a small molecule inhibitor of KDM5 enzymes [34]. Direct chemical inhibition of KDM5 activity was sufficient to induce TF-1 cytokine independence (Figure 3E). Of note, neither KDM5A/C/D triple knockout nor treatment with KDM5c70 had a significant effect on global 5hmC levels (Figure 3F). This suggests that genetic and chemical inhibition of KDM5 enzymes does not transform TF-1 cells via off-target or secondary suppression of TET function.
KDM5 histone lysine demethylases are inhibited in vitro and in cells by tumor-relevant concentrations of (R)-2HG.
Concentrations of (R)-2HG that accumulate in primary IDH-mutant tumors range from 1 to 30 mM [2, 35]. However, the sensitivities of different 2OGDD enzymes to inhibition by (R)-2HG vary dramatically, and not all 2OGDD enzymes are susceptible to inhibition at tumor-relevant concentrations of (R)-2HG [13]. To ask if tumor-relevant concentrations of (R)-2HG can inhibit KDM5 enzymes, we expressed and purified full-length recombinant KDM5A, KDM5B, KDM5C, and KDM5D proteins and assessed their sensitivities to (R)-2HG. (R)-2HG inhibited KDM5A, KDM5C and KDM5D with IC50 values less than 1 mM (Table 1, Supplementary Figure S2A), which is significantly lower than the IC50 value of (R)-2HG for TET2 [23]. The (R)-2HG IC50 value for KDM5B was 3.6 mM, which is similar to that of TET2. KDM5A, KDM5C, and KDM5D were also all potently inhibited by the (S)-enantiomer of 2HG ((S)-2HG) (Table 1, Supplementary Figure S2B).
Table 1: 2OG Km values and (R)-2HG and (S)-2HG IC50 values for KDM5 enzymes and TET2.
The Km and IC50 values are the mean ± standard deviation of 3 to 7 independent assays (see also Supplementary Figure S2).
KDM5 enzymes are H3K4 histone lysine demethylases. To test whether KDM5 enzymes are inhibited in cells expressing mutant IDH, we compared the effects of IDH1R132H expression, KDM5A/C/D triple-knockout and KDM5c70 treatment on global H3K4 trimethylation in isogenic TF-1 cells by western blot. Each of the perturbations increased global levels of trimethyl-H3K4 when compared to IDH1WT-expressing (TF1-IDH1WT), Cas9 and non-targeting sgRNA-expressing (TF1-3xNT), and vehicle-control (DMSO)-treated TF-1 cells, respectively (Figure 4A). To test whether the same sites of H3K4 trimethylation are dysregulated by mutant IDH expression and KDM5 inhibition, we performed chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) on the isogenic TF-1 cell lines. We observed that genomic regions with increased trimethyl-H3K4 were significantly enriched in TF1-IDH1R132H cells when compared to TF1-IDH1WT cells (Figure 4B). These same trimethyl-H3K4-rich regions were enriched in TF1-sgACD and KDM5c70-treated TF-1 cells when compared to TF1-3xNT cells and TF-1 cells treated with DMSO, respectively (Figure 4C). To test whether 250 μM TFMB-(R)-2HG, a concentration of TFMB-(R)-2HG that does not discernably affect 5hmC levels but does promote cytokine independence, is sufficient to dysregulate KDM5 activity, we performed trimethyl-H3K4 ChIP-seq on TF-1 cells treated with 250 μM or 500 μM TFMB-(R)-2HG. Consistent with our observation that KDM5 enzymes are more sensitive to inhibition by (R)-2HG than is TET2, 250 μM TFMB-(R)-2HG was sufficient to induce increased trimethyl-H3K4 at the same sites as those differentially marked in TF1-IDH1R132H cells (Figure 4D). The intracellular (R)-2HG level achieved in the TF-1 cells treated with 250 μM TFMB-(R)-2HG was 2.0 mM (Supplementary Table S2), which is higher than the IC50 values of (R)-2HG for KDM5A, KDM5C and KDM5D and is well within the range of (R)-2HG levels that have been reported in primary IDH-mutant AML [35]. Of note, TET2 knockdown promoted cytokine independent proliferation of TF-1 cells (Supplementary Figure S1A) without causing an increase in global (Figure 4A) or locus-specific (Figure 4C and Supplementary Figure S3) H3K4 trimethylation when compared to non-targeting shRNA-expressing cells. This suggests that increased trimethyl-H3K4 is not a non-specific consequence of TF-1 cytokine independence. Finally, to test whether H3K4 trimethylation is increased in primary AML blast cells that harbor endogenous IDH mutations, we performed trimethyl-H3K4 ChIP-seq on two IDH-wild-type and two IDH-mutant AML patient samples (Supplementary Table S6). The two IDH-mutant samples clustered together (Supplementary Fig. S4A–B) and had higher levels of H3K4me3 when compared to the normal karyotype IDH-wild-type sample (Supplementary Fig. S4C). Of note, the IDH-wild-type sample of unknown karyotype did not cluster with either the IDH-mutant samples or with the normal karyotype IDH-wild-type sample and had even higher levels of H3K4me3 than the IDH-mutant AML samples. It is difficult to draw any definitive conclusions from this analysis given the small sample size. However, these results do suggest that mutant IDH is associated with increased H3K4 methylation in AML, and that H3K4 methylation can be dysregulated by other mechanisms in IDH-wild-type AML.
Figure 4: KDM5 histone lysine demethylases are inhibited by (R)-2HG in cells.
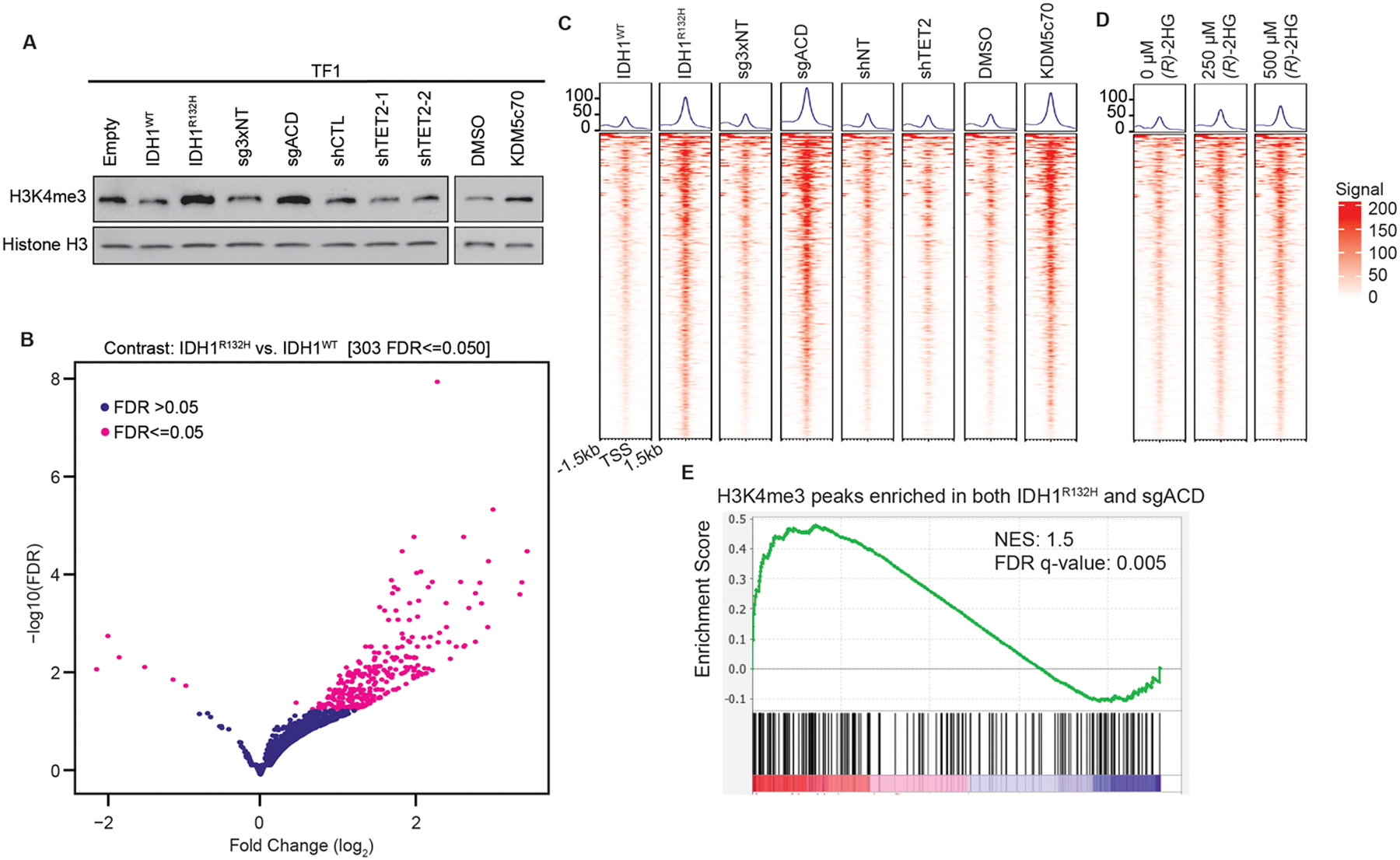
(A) Immunoblot analysis of trimethyl-H3K4 (H3K4me3) levels in isogenic TF-1 cell lines expressing empty vector (Empty), wild-type IDH1 (IDH1WT), or R132H-mutant IDH1 (IDH1R132H); Cas9 and three non-targeting sgRNAs (sg3xNT) or Cas9 and sgRNAs targeting KDM5A, KDM5C and KDM5D (sgACD); a non-targeting shRNA (shNT) or one of two shRNAs targeting TET2 mRNAs (shTET2-1 and shTET2-2); and TF-1 cells treated with vehicle-control (DMSO) or 50 nM KDM5c70. (B) Volcano plot illustrating the relative enrichment of trimethyl-H3K4 in IDH1R132H-expressing TF-1 cells when compared to IDH1WT-expressing TF-1 cells. (C) Tornado plots of the trimethyl-H3K4 ChIP-seq signals at the sites showing differential enrichment between IDH1WT and IDH1R132H identified in (B) in the TF-1 cells described in (A). (D) Tornado plots of the trimethyl-H3K4 ChIP-seq signals at sites showing differential enrichment between IDH1WT and IDH1R132H identified in (B) in TF-1 cells treated with the indicated concentrations of TFMB-(R)-2-HG [(R)-2HG] or DMSO-control (0 μM (R)-2HG). In all cases, cells were passaged for 21 days after lentiviral infection or 21 days in the presence of DMSO or KDM5c70 prior to harvesting for methyl-histone analysis. (E) GSEA of the transcripts detectable by RNA-seq in TF-1 cells that are associated with loci enriched for trimethyl-H3K4 in both IDH1R132H TF-1 cells and sgACD TF-1 cells when compared to Empty TF-1 cells and sg3xNT TF-1 cells (n=169 genes). Shown in (A) are representative results from at least two independent experiments. The ChIP-seq and RNA-seq experiments in (B-E) were done in duplicate, and the average signals from the two replicates is shown. All the experiments shown in (A-E) were done on cells cultured in the presence of GM-CSF. NES: normalized enrichment score; FDR: false discovery rate.
It is important to note that, although TET2 inhibition and KDM5A/C/D inhibition are each sufficient to recapitulate the effects of mutant IDH expression in TF-1 cells, it does not automatically follow that their inhibition is necessary for mutant IDH-mediated transformation. The only way to definitively ascertain whether inhibition of TET2, KDM5A, KDM5C, and/or KDM5D are necessary for mutant IDH-mediated transformation would be to ask if (R)-2HG-resistant variants of these enzymes are able to block transformation. To find such variants, we analyzed the structures of the TET and KDM5 enzymes to identify amino acid substitutions that would be predicted to impair (R)-2HG binding without affecting 2OG binding. In the case of the TET enzymes, we were not able to predict any such substitutions. For the KDM5 enzymes, we took advantage of the fact that KDM5B is significantly less sensitive to (R)-2HG than are KDM5A, KDM5C, and KDM5D (Table 1). When we compared the sequences of KDM5A, KDM5C and KDM5D to that of KDM5B, we identified two residues near the catalytic domain of the enzymes that are conserved in KDM5A, KDM5C and KDM5D but not KDM5B (Supplementary Figure S5A) that in silico modeling predicted would affect (R)-2HG binding. We generated KDM5A mutants in which these residues were replaced with their corresponding residues in KDM5B (KDM5AF68L and KDM5AR429I) and assessed the (R)-2HG sensitivity of the mutant enzymes. Wild-type KDM5A, KDM5AF68L and KDM5AR429I were all similarly sensitive to inhibition by (R)-2HG (Supplementary Figure S5B). As we were not able to generate (R)-2HG-resistant TET2 or KDM5 mutants, we cannot definitively prove that (R)-2HG-mediated inhibition of TET2, KDM5A, KDM5C and KDM5D are necessary for transformation by mutant IDH. However, the observation that low-dose TFMB-(R)-2HG significantly enhances the cytokine independent proliferation of TF-1 cells transformed by TET2 knockdown (Supplementary Figure S1C) does suggest that transformation by mutant IDH is mediated by combined inhibition of TET2, KDM5A, KDM5C and KDM5D.
IDH1R132H expression and co-deletion of KDM5A, KDM5C and KDM5D induce similar transcriptional changes.
H3K4 trimethylation is associated with activation of gene transcription [36, 37]. To ask if (R)-2HG-mediated inhibition of KDM5 activity results in increased expression of KDM5-regulated genes in IDH-mutant cells, we performed RNA sequencing (RNA-seq) on TF1-IDH1R132H cells and empty vector control-expressing TF-1 cells. We then performed Gene Set Enrichment Analysis (GSEA) using the set of genes marked by trimethyl-H3K4 peaks that were enriched in both TF1-IDH1R132H and TF1-sgACD cells when compared to their respective control cells. Genes and pathways associated with loci that were enriched for trimethyl-H3K4 in both TF1-IDH1R132H and TF1-sgACD cells were significantly upregulated in TF1-IDH1R132H cells when compared to empty vector-expressing TF-1 cells (Normalized Enrichment Score: 1.5; FDR: 0.005) (Figure 4E, Supplementary Table S7–S8). This suggests that a subset of the transcriptional changes induced by mutant IDH expression in TF-1 cells are the result of (R)-2HG-mediated inhibition of KDM5 activity.
To ask if the transcriptional effects of IDH1R132H expression in TF-1 cells recapitulate the transcriptional effects of mutant IDH expression in primary IDH-mutant human AML cells, we used publicly available expression data (GSE24505) to perform GSEA on IDH-mutant (n=49) and IDH wild-type (n=306) AML patient samples. Although a subset of genes upregulated in TF1-IDH1R132H cells when compared to TF1-IDH1WT cells were similarly upregulated in IDH-mutant AML patient samples when compared to IDH-wild-type AML patient samples, this correlation did not reach statistical significance (Normalized Enrichment Score: 1.08; FDR: 0.331) (Supplementary Figure S6A, Supplementary Table S9). We also identified a subset of genes with increased trimethyl-H3K4 in both TF1-IDH1R132H and TF1-sgACD cells that were expressed at higher levels in IDH-mutant AML patient samples when compared to IDH-wild-type AML patient samples, but this enrichment likewise did not reach statistical significance (Normalized Enrichment Score: 1.03, FDR: 0.457) (Supplementary Figure S6B, Supplementary Table S10).
IDH-mutant and IDH-wild-type gliomas have similarly low levels of 5hmC.
In AML, IDH-mutant disease is associated with significantly lower levels of 5hmC than IDH-wild-type disease [24]. In contrast, in glioma, several studies have found no clear correlation between IDH mutation status and 5hmC levels [38, 39]. To determine whether the relationship between mutant IDH-positivity and 5hmC loss is, indeed, discordant in AML and glioma, we measured 5hmC levels in a panel of primary IDH-wild-type and IDH-mutant glioma samples (Supplementary Table S11) and a panel of primary IDH-wild-type and IDH-mutant AML samples using the identical LC-MS methodology. Although 5hmC levels varied across the samples, there was no correlation between IDH mutation status and loss of 5hmC in the primary glioma samples (IDH-wild-type: median = 0.032%, range = 0.157%; IDH1-mutant: median = 0.049%, range = 0.083%; IDH2-mutant: median = 0.039%, range = 0.019%) (Figure 5A). In contrast, in the primary AML samples, mutant IDH positivity was associated with significantly lower 5hmC levels compared to IDH-wild-type AML (IDH-wild-type: median = 0.078%, range = 0.096%; IDH-mutant: median = 0.029%, range = 0.066%) (Figure 5B). To rule out the possibility that the lack of difference in 5hmC levels between the IDH-mutant and IDH-wild-type gliomas was due to infiltration of IDH-wild-type non-tumor cells in the IDH-mutant glioma samples, we measured 5hmC levels by LC-MS and (R)-2HG levels by GC-MS in a panel of patient-derived multicellular tumor spheroid (MCTS) lines from IDH1 wild-type and IDH1-mutant gliomas. We saw no correlation between global 5hmC levels and either IDH mutation status or intracellular (R)-2HG levels in the glioma MCTS lines (Figure 5C–D).
Figure 5: Inhibition of KDM5 enzymes, but not TET enzymes, is specifically associated with mutant IDH positivity in gliomas.
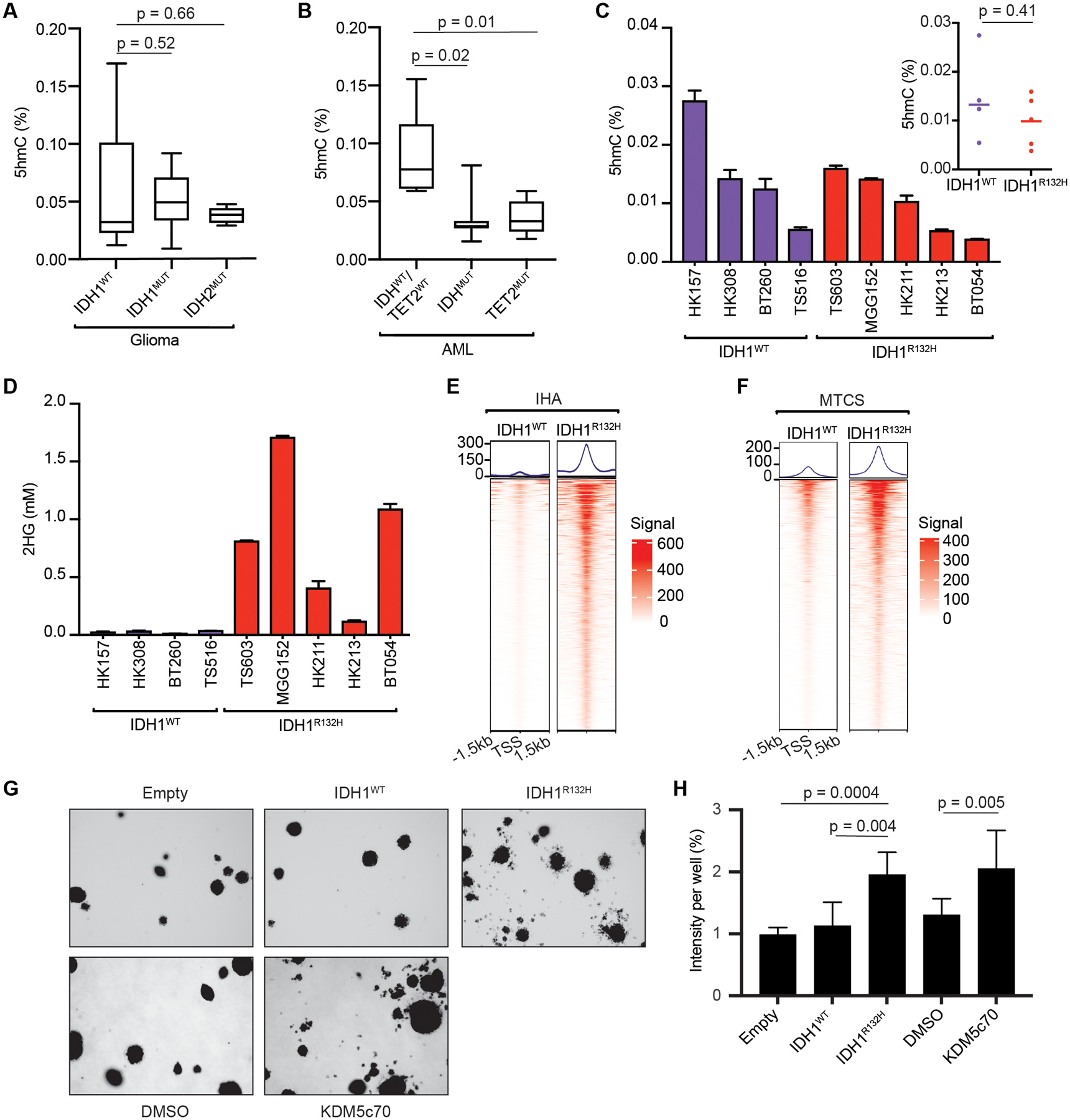
(A-C) Total and median 5hmC content in primary IDH-wild-type (IDH1/2WT) (N=6), IDH1-mutant (IDH1MUT) (N=11) and IDH2-mutant (IDH2MUT) (N=5) glioma patient samples (A); in primary IDH-wild-type and TET2-wild-type (IDHWT/TET2WT) (N=5), IDH-mutant (IDHMUT) (N=8) and TET2-mutant (TET2MUT) (N=5) AML patient samples (B); and in IDH1 wild-type (IDH1WT) and R132H-mutant IDH1 (IDH1R132H) patient-derived glioma multicellular tumor spheroid (MCTS) lines (C), as measured by LC-MS. (D) 2HG levels in the glioma MCTS lines from (C), as measured by GC-MS. (E) Tornado plots of the trimethyl-H3K4 ChIP-seq signals at sites showing differential enrichment in isogenic IHA cells stably expressing IDH1WT or IDH1R132H. (F) Tornado plots of the composite trimethyl-H3K4 ChIP-seq signals at sites showing differential enrichment in IDH1WT (n=4) and IDH1R132H (n=5) glioma MCTS lines (see also Supplementary Figure S9). (G) Soft agar colony formation by IHA cells stably expressing empty vector (Empty), IDH1WT or IDH1R132H, or treated with vehicle (DMSO) or 50 nM KDM5c70, as indicted. (H) Quantification of colony formation by the IHA cells from (G). Shown are mean values for duplicate (for IDH expression) or triplicate (for KDM5c70 treatment) experiments ± SD. The experiments in IHA cells were performed with cells that had been passaged ~35 times after lentiviral infection or passaged 7 times in the presence of DMSO or KDM5c70. The ChIP-seq experiments were performed in duplicate and the average signals from the two replicates is shown.
Although TET enzymes have been shown to be involved in DNA demethylation [40], there is conflicting data regarding whether IDH mutations are associated with global DNA hypermethylation in AML and glioma, with some studies showing a strong association [6, 41], and other studies showing no association [24, 38]. To ask if mutant IDH-positivity is associated with global DNA hypermethylation in our study samples, we measured total 5mC levels by LC-MS in the primary IDH-wild-type and IDH-mutant glioma and AML samples, and the IDH1-wild-type and IDH1-mutant glioma MCTS lines. We found a modest, but not statistically significant, increase in total 5mC levels in the IDH-mutant AML samples when compared to the IDH-wild-type AML samples (Supplementary Figure S7A) and no difference in total 5mC levels in the IDH-mutant primary glioma and MCTS samples when compared to their IDH-wild-type counterparts (Supplementary Figure S7B–C).
IDH-mutant glioma cells have increased trimethyl-H3K4 levels when compared to IDH-wild-type glioma cells.
To test whether KDM5 enzymes are inhibited by mutant IDH in glial cells, we generated isogenic IHA cells ectopically expressing either IDH1WT (IHA-IDH1WT) or IDH1R132H (IHA-IDH1R132H) (Supplementary Figure S8A–B) and assessed global H3K4 trimethylation by western blot and global 5hmC levels by LC-MS. Consistent with previous reports that ectopic expression of mutant IDH in IHA cells results in histone hypermethylation and suppression of 5hmC [42], we found that global trimethyl-H3K4 levels were significantly increased (Supplementary Figure S8C) and global 5hmC levels were significantly decreased (Supplementary Figure S8D) in the IHA-IDH1R132H cells when compared to the IHA-IDH1WT cells. To test whether locus-specific H3K4 trimethylation is similarly increased in glial cells that ectopically express mutant IDH and glial cells that harbor endogenous IDH mutations, we performed trimethyl-H3K4 ChIP-seq on the isogenic IHA-IDH1WT and IHA-IDH1R132H cells and the IDH1-wild-type and IDH1-mutant glioma MCTS lines. Genomic regions marked by increased trimethyl-H3K4 were similarly enriched in the IHA-IDH1R132H cells (Figure 5E) and IDH1-mutant glioma MCTS lines (Figure 5F and Supplementary Figure S9) when compared to the IHA-IDH1WT cells and IDH1-wild-type glioma MCTS lines, respectively.
Inhibition of KDM5 histone lysine demethylases phenocopies mutant IDH expression in an in vitro glial transformation assay.
To test whether small-molecule inhibition of KDM5 enzymes is sufficient to phenocopy mutant IDH expression in the in vitro glial transformation assay [23], we assessed the effect of KDM5c70 treatment on soft agar colony formation by IHA cells. We found that KDM5c70 treatment is sufficient to induce soft agar colony formation and recapitulate the phenotype induced by mutant IDH expression in IHA cells Figure 5G–H).
Inhibition of H3K4 methyltransferase activity reverses TF-1 cytokine independence induced by mutant IDH expression and TET2 loss.
To ask if inhibition of H3K4 methyltransferase activity reverses mutant IDH-mediated transformation, we treated TF-1 cells with the MEN1-MLL1 inhibitor VTP50469 [43]. First, we assessed the effect of VTP50469 on TF-1 cells grown in cytokine-rich media, reasoning that any cytotoxic effects of VTP50469 under these conditions would be unlikely to be due to reversal of mutant IDH-induced H3K4 hypermethylation since TF-1 cells do not require increased H3K4 methylation for growth in cytokine-rich media. Indeed, at doses up to 100 nM, VTP50469 had no effect on the cytokine-dependent proliferation of TF1-IDH1R132H cells or control TF-1 cells (Figure 6A and Supplementary Figure S10). We then assessed the effects of VTP50469 on the cytokine-independent proliferation of TF1-IDH1R132H cells under two conditions, when drug treatment was started on day 0 of cytokine withdrawal and when drug treatment was started two weeks prior to cytokine withdrawal. We reasoned that any immediate effects of VTP50469 would be unlikely to be on-target as reversal of transformation of TF1-IDH1R132H cells is time-dependent [5]. VTP50469 had no effect on TF1-IDH1R132H cytokine independence when treatment was started on day 0 of cytokine withdrawal (Figure 6B). However, pretreatment with VTP50469 did inhibit TF1-IDH1R132H cytokine independence in a dose-dependent manner (Figure 6C–D). Finally, to ask if the transformation-reversing effects of VTP50469 are specific to TF-1 cells transformed by mutant IDH, we assessed the effect of VTP50469 on TF-1 cells transformed by knockdown of TET2 (TF1-shTET2 cells). To our surprise, pre-treatment with VTP50469 inhibited the cytokine independent proliferation of TF1-shTET2 cells to a similar extent as TF1-IDH1R132H cells (Figure 6E–F). Although this could be the result of an off-target toxic effect of VTP50469, it is also possible that VTP50469 inhibits the accumulation of H3K4 methyl marks that are generally required for TF-1 cytokine independence.
Figure 6: MLL inhibition reverses TF-1 transformation induced by mutant IDH expression and TET2 loss.
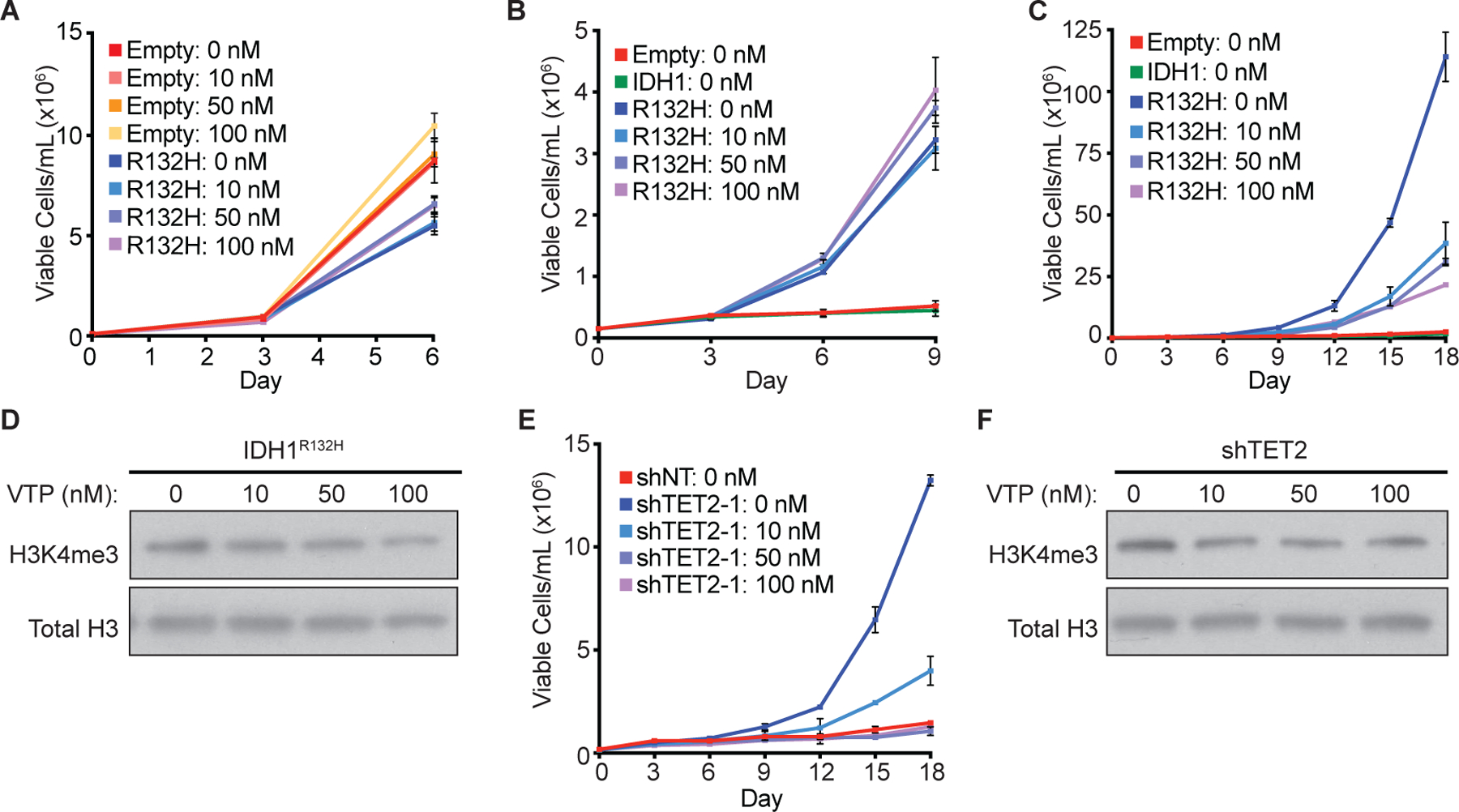
(A) Proliferation under cytokine-rich conditions of TF-1 cells expressing empty vector (Empty) or HA-tagged R132H-mutant IDH1 (R132H) treated with the indicated concentrations of the MEN1-MLL1 inhibitor VTP50469 or DMSO-control (0 nM). (B) Proliferation under cytokine-poor conditions of TF-1 cells expressing empty vector (Empty), HA-tagged wild-type IDH1 (IDH1), or HA-tagged R132H-mutant IDH1 (R132H) treated with the indicated concentrations of VTP50469 or DMSO-control (0 nM) starting on day 0 of cytokine withdrawal. (C) Proliferation under cytokine-poor conditions of the TF-1 cells from (B) pre-treated with the indicated concentrations of VTP50469 or DMSO-control (0 nM) for two weeks prior to cytokine withdrawal. (D) Immunoblot analysis of trimethyl-H3K4 (H3K4me3) levels in the R132H-mutant expressing TF-1 cells from (C). (E) Proliferation under cytokine-poor conditions of TF-1 cells expressing a non-targeting shRNA (shNT) or a TET2-targeting shRNA (shTET2-1) pre-treated with the indicated concentrations of VTP50469 or DMSO-control (0 nM) for two weeks prior to cytokine withdrawal. (F) Immunoblot analysis of H3K4me3 levels in the TET2-targeting shRNA-expressing TF-1 cells from (E).
Discussion:
A number of 2OG-dependent cellular pathways other than TET2 have been suggested as tumor suppressor targets that contribute to (R)-2HG-mediated transformation, most notably the JmjC-KDM superfamily of histone lysine demethylases. Indeed, many JmjC-KDM enzymes have been found to be recurrently mutated, deleted, or silenced in cancer [13]. Moreover, several of these same JmjC-KDM enzymes are inhibited by tumor-relevant concentrations of (R)-2HG in vitro, and ectopic expression of mutant IDH in cells results in the accumulation of many histone methyl marks [7, 11]. However, it is not known to what extent inhibition of JmjC-KDM enzymes functionally contributes to mutant IDH-mediated transformation as opposed to being a ‘bystander’ effect of (R)-2HG accumulation, and there is scant evidence to directly implicate dysregulation of specific JmjC-KDM enzymes in mutant IDH-mediated transformation.
To identify 2OGDD enzymes that could be functionally relevant tumor suppressor targets of (R)-2HG, we performed positive-selection CRISPR/Cas9 screens in TF-1 cells, an IDH-wild-type cytokine-dependent AML cell line that is transformed to cytokine independence by expression of mutant IDH and by treatment with cell-permeable esterified (R)-2HG [5]. The screens identified three members of the KDM5 family of 2OG-dependent H3K4 histone lysine demethylases, KDM5A, KDM5C and KDM5D, as genes whose loss induces modest TF-1 cytokine independence. Combined genetic knockout of KDM5A, KDM5C and KDM5D and pan-inhibition of KDM5 activity by a KDM5-selective small molecule both induce robust TF-1 cytokine independence.
The role of KDM5 enzymes in AML is somewhat controversial. RNAi-mediated knockdown of KDM5A and KDM5B have been shown to induce apoptosis of HL60 and NB4 cells [44, 45]. Conversely, loss-of-function mutations in KDM5A and KDM5C have been reported in pediatric and adult AML [46, 47]; Kdm5b depletion has been shown to promote the proliferation and survival of MLL-rearranged AML cells [47]; and Kdm5d deletion has been shown to promote leukemogenesis in a mouse model of AML1-ETO-positive AML [48]. Our observation that combined loss of KDM5A, KDM5C and KDM5D phenocopies the effects of mutant IDH expression and (R)-2HG accumulation in a mutant IDH myeloid transformation assay suggests that KDM5A, KDM5C and KDM5D function as tumor suppressors in a hematologic context in which mutant IDH has oncogenic activity. Moreover, the observation that direct chemical inhibition of KDM5 activity recapitulates the effects of genetic deletion of KDM5A, KDM5C and KDM5D indicates that the tumor suppressive functions of KDM5A, KDM5C and KDM5D are mediated by their catalytic activities, which is a prerequisite for a 2OGDD enzyme to qualify as a tumor suppressor target of (R)-2HG. Finally, we show that KDM5A, KDM5C and KDM5D are all inhibited by tumor-relevant concentrations (R)-2HG. Taken together, these results strongly suggest that (R)-2HG-mediated suppression of KDM5 activity contributes to mutant IDH-mediated transformation and that inhibition of these additional tumor suppressor targets cooperates with inhibition of TET2 to induce a more leukemogenic program than that induced by TET2 loss alone. This could explain, at least in part, why IDH mutations are predominantly associated with high-grade MDS/MPN and AML whereas TET2 mutations are equally associated with low-grade and high-grade clonal myeloid disorders.
To date, efforts to define a specific transcriptional program associated with mutant IDH-positivity in primary human AML have been unsuccessful. IDH-mutant AML cases do not segregate from IDH wild-type cases in unsupervised clustering analysis of gene expression data [6] and, in a focused analysis of primary IDH-mutant and IDH-wild-type acute erythroleukemias, the transcriptional profiles of the leukemias were found to reflect not their IDH mutation status but, rather, their specific morphologic subtype [49]. Consistent with these previous reports, we found that the transcriptional effects of IDH1R132H expression in TF-1 cells are not fully recapitulated in primary IDH-mutant AML. It is likely that many of the same genes that are dysregulated by (R)-2HG accumulation in primary IDH-mutant AML are dysregulated by other mechanisms in other AML subtypes. It is also likely that some of the gene expression changes induced by mutant IDH are further modulated by other co-mutated genes. Indeed, there is evidence to suggest that growth-antagonizing expression changes induced by mutant IDH in leukemia cells are specifically reversed by other co-occurring disease alleles [50].
In glioma, IDH mutations define a clinically distinct subset of tumors that are more indolent, more responsive to chemoradiation therapy, and have significantly prolonged survival compared to IDH-wild-type tumors [51]. In trying to understand the molecular basis for these phenotypic differences, studies have largely focused on the role of altered DNA methylation in mutant IDH-mediated transformation. The CpG island methylator phenotype (CIMP) characterizes a distinct molecular subclass of oligodendrogliomas and astrocytomas that are defined by locus-specific DNA hypermethylation at CpG-rich promoter regions [42, 52]. Given that TET enzymes play a critical role in DNA demethylation [40], TET inhibition by (R)-2HG has been proposed as a mechanism to explain the finding that IDH-mutant gliomas are near-universally CIMP-positive. However, it is not clear whether the association between CIMP-positivity and mutant IDH-positivity in glioma is causal or merely correlative. CIMP-positivity is seen in a number of tumor types in which IDH mutations are not found, including colorectal and ovarian cancer [53, 54], and mutant IDH-independent CIMP-positivity has been described in AML [55]. It is also not clear whether CIMP-positivity in IDH-mutant glioma is the result of (R)-2HG-mediated inhibition of TET enzymes. Recent studies have found that 5hmC levels are significantly lower in brain tumors than in normal brain tissue irrespective of IDH mutation status [38, 39]. This is likely because TET enzymes are critical glial tumor suppressors that can be inhibited by multiple mechanisms besides (R)-2HG-mediated chemical inhibition, including genomic deletion [56], transcriptional silencing [57, 58], and nuclear exclusion [59]. Given that loss of TET function appears to play an important role in both IDH-wild-type and IDH-mutant gliomagenesis, TET inhibition by (R)-2HG cannot explain the unique clinical and biological features of IDH-mutant glioma. We found that H3K4 trimethylation is specifically dysregulated in IDH-mutant gliomas when compared to IDH-wild-type gliomas and that direct inhibition of KDM5 activity promotes glial cell transformation. These results suggest that (R)-2HG-mediated inhibition of KDM5 enzymes contributes to mutant IDH-mediated transformation in glioma and plays a role in inducing the unique biology of IDH-mutant brain tumors.
The positive-selection CRISPR/Cas9 screens were highly sensitive. They were able to identify individual KDM5 genes as 2OG-dependent myeloid tumor suppressors even though loss or inhibition of multiple KDM5 family members is required to fully recapitulate the transforming effects of mutant IDH and (R)-2HG. This apparent functional redundancy between KDM5 enzymes is likely why KDM5 mutations are rare in AML [46, 47], as multiple family members would have to be biallelically inactivated to recapitulate the effects of acquisition of a single oncogenic IDH mutation. Although no other 2OGDD enzymes scored in our CRISPR/Cas9 screens, we cannot exclude the possibility that dysregulation of other 2OGDD enzymes contributes to the leukemogenic activity of mutant IDH and (R)-2HG. The screens were designed such that only a single target gene is disrupted per cell. It is possible that other myeloid tumor suppressors did not score in the CRISPR/Cas9 screens because they are completely functionally redundant with other closely related enzymes and knockout of multiple paralogs is required to induce TF-1 cytokine independence. It is also important to note that, although shRNA-mediated knockdown of TET2 induces TF-1 cytokine independence [5], TET2 did not score in the CRISPR/Cas9 screen. Whether this is the result of a technical limitation of CRISPR or is due to biological differences between complete genetic deletion of TET2 and partial loss of TET2 expression is unclear. However, it does reinforce the point that failure of a gene to score in the TF-1 CRISPR/Cas9 screen does not rule out that the gene has myeloid tumor suppressor activity.
In summary, we have identified KDM5 enzymes as tumor suppressor targets of (R)-2HG that play a role in the pathogenesis of IDH-mutant AML and IDH-mutant glioma. Further studies will be needed to determine whether targeting H3K4 hypermethylation is an effective therapeutic strategy in IDH-mutant cancers. It will also be important to determine whether (R)-2HG-independent dysregulation of KDM5 tumor suppressor activity plays a role in cancers that do not harbor IDH mutations. Our study, by demonstrating that inhibition of multiple KDM5 family members by (R)-2HG functionally contributes to mutant IDH-mediated transformation, provides an important rationale for further investigation of the role of dysregulated H3K4 methylation in cancer.
Materials and Methods:
Cell lines:
HEK-293T cells (ATCC, RRID:CVCL_0063) were maintained at low passage number in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin and split every 2–3 days before reaching confluence. After being thawed, HEK-293T cells were passaged a maximum of 5 times (2 weeks) prior to use in experiments. TF-1 cells (ATCC, RRID:CVCL_0559) were derived from a XY-male with erythroleukemia and were maintained in RPMI containing 10% FBS, 1% penicillin/streptomycin and 2 ng/mL recombinant human GM-CSF (R&D Systems, 215-GM). TF-1 cells infected with IDH1-, sgRNA- and/or Cas9-expressing lentiviruses were selected with 1 μg/mL puromycin, 200 μg/mL hygromycin and/or 1 mg/mL neomycin. After being thawed, TF-1 cells were passaged a maximum of 3 times (10 days) prior to use in experiments. Immortalized Human Astrocyte (IHA) cells, which were derived from an XX-female and immortalized with E6/E7/hTERT, were a gift from Russell O. Pieper (University of California, San Francisco). IHA cells were maintained in DMEM containing 10% FBS and 1% penicillin/streptomycin. IHA cells infected with empty vector or IDH1-expressing lentiviruses were selected with 500 μg/mL hygromycin. After being thawed, IHA cells were passaged a maximum of 2 times (1 week) prior to use in experiments. The cell lines were all authenticated by Short Tandem Repeat profiling (ATCC) and were periodically tested for mycoplasma throughout the experimental period (most recent negative testing was November 5th, 2018).
Mammalian expression constructs:
For constructs that co-express Cas9 and an individual sgRNA, the forward and reverse sgRNA sequences were annealed and the sgRNA was cloned into lentiCRISPR-v2 (RRID:Addgene_52961). For constructs that express an individual sgRNA alone, the individual sgRNA was cloned into lentiGuide-Puro (RRID:Addgene_52963); lentiGuide-Hygro (RRID:Addgene_139462) or lentiGuide-Neo (RRID:Addgene_139449). To express Cas9 alone, lentiCas9-Blast (RRID:Addgene_52962) was used, and to assess Cas9 expression, the Cas9-GFP reporter (RRID:Addgene_59702) was used. The lentiviral pLKO.1 human TET2 shRNA vectors have been previously described [5]. For expression of IDH1 variant cDNAs in TF-1 cells, LeGO-iG2 (RRID:Addgene_27341) expression vectors containing no cDNA insert, or human wild-type (Accession# NM_005896) or R132H-mutant HA-tagged IDH1 cDNAs [5] were digested with NotI and BsrGI restriction enzymes to remove their IRES-iGFP cassettes, and a gblock (IDT) encoding the sequence for IRES-puro was subcloned into the NotI-BsrGI sites to produce the vectors LeGO-IRES-Puro, LeGO-IDH1WT-Puro, and LeGO-IDH1R132H-Puro. For expression of IDH1 variant cDNAs in IHA cells, plenti-EF1α-IRES-hygro (RRID:Addgene_85134) expression vectors encoding human wild-type and R132H-mutant HA-tagged IDH1 cDNAs were used [60].
KDM5-targeting sgRNA oligonucleotide sequences:
sgA1: CACCGCACTCTGGATTTAACCA and AAACTGGTTAAATCCAGAGTGC
sgA2: CACCGTCAAAGACGGGGCACTCTGG and AAACCCAGAGTGCCCCGTCTTTGAC
sgB1: CACCGTGGGCTCACATATCAG and AAACCTGATATGTGAGCCCAC
sgB2: CACCGCATTTCCCCAAAAGTACGGA and AAACTCCGTACTTTTGGGGAAATGC
sgC1: CACCGAGGCTACAACTTTGCCG and AAACCGGCAAAGTTGTAGCCTC
sgC2: CACCGGCTAGACCTGAACCTGG and AAACCCAGGTTCAGGTCTAGCC
sgD1: CACCGCTTATCATCTTCATCCCCA and AAACTGGGGATGAAGATGATAAGC
sgD2: CACCGCTATTGTGTCTTCTCCCACG and AAACCGTGGGAGAAGACACAATAGC
Chemical treatments:
TFMB-(R)-2HG was synthesized as previously described [5] and was resuspended in dimethyl sulfoxide (DMSO) at a stock concentration of 1M. KDM5c70 (Xcessbio, M60192–2S) was resuspended in DMSO at a stock concentration of 10mM. VTP50469 (Selleck Chemicals, S8934) was resuspended in DMSO at a stock concentration of 5mM.
Metabolite extraction and (R)-2HG quantification:
Metabolites were extracted from exponentially growing TF-1 cells, IHA cells and glioma MCTS cultures using 80% aqueous methanol (−80 °C) and were profiled by gas chromatography-electrospray ionization-mass spectrometry (GC–MS), as previously described [60].
5hmC and 5mC quantification:
To measure total 5hmC and total 5mC levels as a fraction of total cytosines, DNA was purified using phenol-chloroform extraction. 2 μg of purified DNA was then hydrolyzed to single nucleosides by nuclease P1 (Sigma-Aldrich), phosphodiesterase I (Sigma-Aldrich), and calf intestine alkaline phosphatase (Thermo Fisher Scientific). Hydrolyzed nucleosides were separated by an Acquility UPLC Oligonucleotide BEH C18 Column (Waters) in an Agilent 1290 liquid chromatography system, before being measured by Agilent 6460 Triple-Quadrupole tandem mass spectrometry. The mass spectrometry protocol was optimized to detect deoxyadenosine (dA), thymidine (T), deoxyguanosine (dG), deoxycytidine (dC), 5-methyl-deoxycytidine (5mdC), and 5-hydroxymethyl-deoxycytidine (5hmdC). Standards containing mixtures of all detection targets in different known ratios were run alongside to correct for detection bias. Total cytosine levels were estimated by the total dG signals since cytosine and guanine bases exist in equal amounts in the genome. 5hmC and 5mC levels were calculated as a percentage of total cytosine from the output signals, which were then normalized to the measured standards. Statistical analysis was carried out using GraphPad Prism (RRID:SCR_002798) software. Significance was calculated by unpaired two-tailed t-test using a cutoff of p <0.05. For comparisons of two groups with significantly different variances, Welch’s t-test was used. For comparisons of two groups without significant differences in variances, Student’s t-test was used.
Whole genome sgRNA library construction:
The Brunello human CRISPR knockout pooled sgRNA library (RRID:Addgene_73179) has been previously described [61].
Custom minipool sgRNA library construction:
Gene-targeting sgRNAs and appropriate controls were designed using established algorithms to optimize on-target and minimize off-target genome editing, as described at the Genetic Perturbation Platform (GPP) portal of the Broad Institute (http://portals.broadinstitute.org/gpp/public/). Oligonucleotides were flanked by PCR primer sites, and PCR was used to amplify DNA using NEBNext kits (NEB). The PCR products were purified using Qiagen PCR cleanup kits and cloned into lentiGuide-Puro (RRID:Addgene_52963) using Golden Gate cloning reactions (NEB). Pooled libraries were amplified using electrocompetent Stbl4 cells (Thermo Fisher Scientific). Viruses were generated as outlined at the GPP portal. The gene list for the sgRNA library targeting epigenetics regulators and all known 2OG-dependent processes was compiled in collaboration with the laboratories of Dr. Bradley Bernstein (Massachusetts General Hospital, MGH) and Dr. William G. Kaelin, Jr. (DFCI). The sgRNA library consisted of 9,088 sgRNAs targeting 1,355 genes (5–6 sgRNAs targeting each gene) and 1,000 nontargeting sgRNAs.
CRISPR/Cas9 screens:
To perform large-scale infections for the Brunello genome-wide CRISPR/Cas9 screens, 1.5 × 108 Cas9-expressing TF-1 cells per replicate (~2,000 cell/sgRNA) were resuspended in 75 ml of growth media containing 8 μg/ml polybrene. To perform large-scale infections for the custom minipool screens, 5.4 × 107 Cas9-expressing TF-1 cells per replicate (~5,000 cells/sgRNA) were resuspended in 25 ml of growth media containing 8 μg/ml polybrene. In each case, the cells were split across 12-well plates at 2 ml per well and an appropriate volume of pooled high-titer lentivirus was added to achieve a multiplicity of infection (MOI) of 0.3. Spin infections were performed by centrifugation at 2000 rpm for 2 hours at RT. After spin infection, cells were incubated for 4 hours and then the cells from each well were combined and fresh growth media was added. Puromycin was added 24 hours after infection. Each replicate was passaged for 21 days, and the cells were then washed four times with plain RPMI and plated at a density of 0.2 × 106 cells/mL in media either containing or lacking GM-CSF. The cells were then passaged for another 15 days. A minimum of 1.5 × 108 cells per condition for the genome-wide screens and 5 × 107 cells per condition for the custom minipool screens were maintained throughout the experiment. Aliquots of cells, 1.5 × 108 cells for the genome-wide screens and 2.5 × 107 cells for the custom minipool screens, were collected on day 0 and day 15 of growth factor deprivation. Following completion of the screens, genomic DNA (gDNA) was isolated using a Qiagen Genomic DNA midi prep kit according to the manufacturer’s protocol. 60μg of gDNA was submitted for sequencing per sample. PCR of the gDNA was performed as previously described [61] and sgRNA abundance was determined by sequencing on an Illumina HiSeq.
CRISPR/Cas9 screen data analysis:
Reads mapping to each sgRNA were counted, and the log2-normalized-reads per-million was calculated for each sgRNA. To rank genes and calculate FDRs, a STARS analysis was performed using negative binomial distribution, as previously described [61]. To determine average log-fold change per gene target, the log2-normalized-reads per-million per sgRNA were averaged across the 5–6 sgRNAs targeting each gene and the day 15 GM-CSF-rich condition was subtracted from the day 15 GM-CSF-deprived condition using the Hypergeometric Distribution Tool available online (https://portals.broadinstitute.org/gpp/public/analysis-tools/crispr-gene-scoring). Volcano plots were generated with VolcaNoseR [62], using p-values calculated with the Hypergeometric Distribution Tool. The data from both biological replicates of each screen were used for all analyses.
Individual lentiviral infections:
Lentiviral particles were generated by Lipofectamine 2000 (Invitrogen) co-transfection of HEK-293T cells with cDNA, sgRNA or shRNA expression vectors and the lentiviral packaging constructs psPAX2 (RRID:Addgene_14887) and pMD2.G (RRID:Addgene_14888) in a 2:2:1 ratio. To perform lentiviral infections in TF-1 cells, the cells were spin-infected in 12-well plates as follows: 2 × 106 TF-1 cells were plated in growth media with lentiviral supernatant and 8 μg/mL polybrene, and the cells were centrifuged at 2000 rpm for 2 hours at RT. To create KDM5A/C/D triple knockout cells, TF-1 cells were infected with lentiCrispr-v2-Puro with the first sgRNA on day 1, lentiGuide-neo with the second sgRNA on day 2, and a lentiGuide-hygro with the third sgRNA on day 3. Antibiotics were added on day 4. To perform lentiviral infections in IHA cells, the cells were spin-infected in 6-well plates as follows: 3 × 106 IHA cells were plated in growth media with lentiviral supernatant and 8 μg/mL polybrene, and the cells were centrifuged at 2000 rpm for 2 hours at RT. The infected cells were cultured overnight and expanded the following morning. Antibiotic selection was started 24 hours after infection. Lentiviral expression and shRNA- and sgRNA-mediated inhibition of gene expression were assessed by immunoblot analysis.
Immunoblot analysis:
Whole cells extracts were prepared using lysis buffer (50mM Tris-HCl pH 7.9, 400mM NaCl and 0.5% NP40) supplemented with a protease inhibitor cocktail (Roche), resolved on 4–20% SDS-PAGE gels (BioRad), and transferred to 0.45 μm PVDF membranes (Millipore). Histone extracts were prepared using a histone extraction kit (Abcam), as per manufacturer’s instructions, resolved on 4–20% SDS-PAGE gels (BioRad), and transferred to 0.2 μm PVDF membranes. Membranes were blocked in TBST with 5% non-fat milk, probed with primary antibodies, and detected with horseradish-peroxidase-conjugated anti-rabbit (Jackson ImmunoResearch Labs Cat# 211–032-171, RRID:AB_2339149) or anti-mouse (Cell Signaling Technology Cat# 7076, RRID:AB_330924) antibodies. Primary antibodies used: rabbit monoclonal HA-tag antibody (Cell Signaling Technology Cat# 3724, RRID:AB_1549585), rabbit monoclonal anti-KDM5A antibody (Cell Signaling Technology Cat# 3876, RRID:AB_2129055), rabbit polyclonal anti-KDM5B antibody (Cell Signaling Technology Cat# 3273, RRID:AB_1264191), rabbit polyclonal anti-KDM5C antibodies (Abcam Cat# ab34718, RRID:AB_881090 and Abcam Cat# ab190180, RRID:AB_2927800), rabbit polyclonal anti-KDM5D antibody (Millipore Cat# ABE203, RRID:AB_11205069), TET2 antibody (Cell Signaling Technology Cat# 18950, RRID:AB_2798809), rabbit monoclonal anti-trimethyl-histone H3 lysine 4 antibody (Cell Signaling Technology Cat# 9751, RRID:AB_2616028), rabbit monoclonal anti-histone H3 antibody (Cell Signaling Technology Cat# 4499, RRID:AB_10544537), rabbit monoclonal anti-β-actin antibody (Cell Signaling Technology Cat# 4970, RRID:AB_2223172), and mouse monoclonal anti-vinculin antibody (Sigma-Aldrich Cat# V9131, RRID:AB_477629).
TF-1 cytokine-withdrawal assays:
TF-1 stable cell lines were washed four times with plain RPMI. Cells were then counted, and 1 × 106 cells were plated in duplicate or triplicate in 5 mL RPMI containing 10% FBS and 1% penicillin/streptomycin. Cell proliferation and viability were assessed by counting the number of viable cells/mL every 3 days using a Vi-Cell Cell Viability Analyzer (Beckman Coulter), and the cultures were periodically split and fed with fresh growth media lacking GM-CSF.
Baculoviral protein expression and purification:
The baculovirus for the C-terminal FLAG-tagged human KDM5B was a kind gift from Dr. Qin Yan (Yale University). The C-terminal FLAG-tagged human full length KDM5A (Accession# NG_046993.1) was generated by PCR and was subcloned into the pVL1393 backbone (Sigma-Aldrich, E8772). The corresponding baculovirus was generated using the BacMagic-3 DNA kit (Novagen). The FLAG-tagged full-length human KDM5C (Accession# NG_008085.2) and KDM5D (Accession# NG_032920.1) were generated by PCR and subcloned into pFastBac Dual plasmid (Thermo Fisher Scientific, #10712024). KDM5C and KDM5D bacmids were generated using DH10Bac cells (Invitrogen) and the standard Bac-to-Bac protocol, and the corresponding baculoviruses were generated by transfecting the bacmid DNA into Sf9 cells. Recombinant proteins were produced by transducing Sf9 insect cells with the corresponding baculoviruses for 72 h. The cells were then washed with cold 1 X PBS and homogenized in a buffer containing 10 mM Tris-HCl pH 7.8, 150 mM NaCl, 100 mM glycine, 5 μM FeSO4, 0.1% Triton X-100 and protease inhibitor cocktail (PIC). The soluble fractions of the FLAG-tagged enzymes were affinity purified using anti-FLAG M2 affinity gel (Sigma-Aldrich Cat# A2220, RRID:AB_10063035), washed with TBS containing 5 μM FeSO4 and PIC, and eluted with wash buffer containing 150 μg/ml FLAG-peptide. The fractions collected were analyzed using 10% SDS-PAGE under reducing conditions followed by Coomassie Blue staining.
Enzyme kinetic assays:
2OG Km values for the KDM5 paralogues were measured using the stoichiometric coupling of lysine demethylation to 2OG decarboxylation as previously described [63]. Each 50 μl reaction volume consisted of 50 mM MES [pH 6.5] for KDM5A and 50 mM Tris-HCl [pH 7.5] for KDM5C and KDM5D, 2 mg/ml bovine serum albumin (Roche), 60 μg/ml catalase (Sigma-Aldrich), 0.1 mM dithiothreitol, 2 mM sodium ascorbate, 50 μM iron(II) sulfate, and 10% (v/v) DMSO. The 2OG mixture contained 11% 2-oxo[1–14C]glutarate (Perkin-Elmer). The peptide histone H3(1–21)K4me3 (Innovagen) was used as a substrate at saturating concentrations; 30 μM for KDM5A and 15 μM for KDM5C and KDM5D. The peptide substrate contained an additional glycine and a biotinylated lysine residue at their C-termini. Enzyme concentrations used were 0.2–1.5 μM. Enzymatic reactions were simultaneously performed in 8–9 different 2OG concentration at 37 °C under ambient oxygen concentration for 3 min. Reactions were stopped by adding 100 μl of 1M potassium phosphate pH 5 and the amount of 14C-labeled CO2 generated was scintillated in a Tri-Carb 2900TR (Perkin Elmer). Km values were determined from the Michaelis–Menten saturation curves and Lineweaver–Burk plots using Excel (Microsoft). IC50 values of (R)- and (S)-2HG were determined in the presence of fourfold 2OG concentration relative to the 2OG Km value of the enzyme by increasing the concentrations of (R)- and (S)-2HG in the reaction.
ChIP-seq sample preparation:
2 × 107 cells (TF-1 or IHA cells), 5 × 106 cells (AML primary samples), or 1 × 106 cells (glioma MCTS lines) were collected per replicate, cross-linked with 1% formaldehyde for 10 minutes at room temperature on a rocker, and quenched with 140mM glycine for 5 minutes at room temperature. Cells were washed 3 times with PBS and snap frozen. Cell pellets were resuspended in ChIP lysis buffer (1% SDS, 10mM EDTA, 50mM Tris-HCl pH 8.1) containing protease inhibitor cocktail (Roche). Chromatin was sonicated using a Sonic Dismembrator Model 100 (Thermo Fisher Scientific) with rounds of 10 seconds on, 30 seconds off at 30% power (TF-1 cells: 11 rounds; primary AML samples: 9 rounds; IHA cells: 9 rounds; glioma MCTS lines: 7 rounds). Shearing efficacy was confirmed by agarose gel electrophoresis. Immunoprecipitations were performed with the equivalent of 1 × 106 cells for each sample using the Magna ChIP A/G kit (Millipore) and rabbit monoclonal H3K4me3 antibody (Millipore Cat# 05–745R, RRID:AB_1587134) or normal rabbit IgG (Millipore Cat# 12–370, RRID:AB_145841). Drosophila spike-in chromatin (Active Motif) was added along with Drosophila anti-H2Av (Active Motif Cat# 61686, RRID:AB_2737370) to samples prior to immunoprecipitation. Chromatin was incubated with primary antibodies overnight at 4 °C with constant agitation. Inputs were prepared from sonicated controls for each cell line. QC, library preparation and sequencing were performed by the Molecular Biology Core Facilities at DFCI. Sequencing libraries were generated from the purified immunoprecipitated DNA samples using Swift 2S ligation chemistry and were sequenced using 50-base paired-end reads on an Illumina platform (Novogene).
ChIP-seq analysis:
Sequencing quality control was performed using FastQC [64]. Reads were aligned to the human genome (hg38) or drosophila genome (dm3) using Bowtie2 v2.2.9 (RRID:SCR_016368) [65]. Samtools v1.3.1 (RRID:SCR_002105) was used to convert SAM files to BAM format, reads were sorted and filtered for duplicates using Sambamba, and were indexed with Samtools [66, 67]. Peaks were called using MACS2 v 2.1.1.20160309 with the ‘--BAMPE’ option [68]. Sample quality was assessed using CHIPQC [69]. Blacklist removal, drosophila spike-in normalization (for all samples except the primary AML samples), differential binding analysis, and tornado plot generation were performed using Diffbind v3.2.5 (RRID:SCR_012918) [70, 71]. For the AML primary samples, spike-in drosophila DNA read counts were insufficient, and normalization by sequencing depth using the full library sizes was performed. For the MTCS lines, an additional normalization step was required due to the chromosomal instability and genome duplication that is associated with IDH mutations in glioma [72, 73]. To account for the increased DNA content of the IDH1-mutant glioma MCTS cells, the DNA concentrations of each of the MTCS input samples was assessed using a Quant-it dsDNA Assay Kit (Invitrogen), and a DNA concentration correction (DCC) for each individual sample was derived by dividing the DNA concentration of each sample by the average concentration of all the glioma MCTS samples. The Drosophila spike-in normalization factor calculated using Diffbind was multiplied by this DCC to determine final normalization factors. Due to variation in background signal across samples, greylist removal was also performed for MCTS samples. The data from both biological replicates of each ChIP-seq sample were used for all analyses.
Principal Component Analysis (PCA):
PCA analysis was performed using Diffbind v3.2.5 (RRID:SCR_012918). Calculations were based on normalized read counts across all binding sites.
RNA-seq sample preparation:
Cell pellets were snap-frozen and RNA was extracted using RNeasy mini kit (Qiagen #74106). QC, library preparation, sequencing, and mapping were performed by Novogene using an Illumina Novaseq 6000 with 150bp paired-end reads.
RNA-seq analysis:
Reads were mapped using STAR v2.5, mismatch=2 (RRID:SCR_004463). Reads mapping to each gene were counted using HTSeq v0.6.1 (RRID:SCR_005514). DESeq2 v2_1.6.3 (RRID:SCR_015687) was used to normalize the data and calculate log2(fold-change) and p-values. Genes with an adjusted P-value <0.05 and fold-change of ≥1.5 were considered upregulated. For GSEA (RRID:SCR_003199), software was downloaded from the GSEA website (http://www.broad.mit.edu.ezp-prod1.hul.harvard.edu/gsea/downloads.jsp) [74]. Gene sets with an FDR <0.25 were considered significant. For the TF-1 RNA-seq data set, GSEAPreranked was used. Ranking metrics for each transcript were calculated by multiplying the -log10(p-value) by the sign of the log2(fold-change). The data set was then collapsed to gene symbols and default settings were used (enrichment statistic=weighted p=1). For the primary patient data set from GSE24505, data were downloaded from the GEO database [6, 75]. Default settings were used (the data set was collapsed to gene symbol, permutation type=phenotype, enrichment statistic=weighted p=1, ranking metric=Signal2Noise).
Soft agar colony-forming assays and quantification:
12,000 cells per condition were resuspended in a top layer of 0.4% soft agar (Lonza SeaPlaque Agarose, Thermo Fisher Scientific) and plated on a bottom layer of 1% soft agar containing complete DMEM supplemented with 10% FBS in 6-well plates. DMSO or KDM5c70 were added to the top layer where indicated. 2 to 3 wells per condition were plated per experiment. Cells were fed with 2 mL of media, which was exchanged every 3 to 4 days. After 3 to 4 weeks, colonies were stained with 0.1% iodonitrotetrazolium chloride (Sigma-Aldrich) and were imaged using a Nikon inverted live-cell imaging system at 4X magnification. To quantify colony formation, the intensity-weighted colony area percentages were calculated on images that had been cropped to individual wells and converted to grayscale, and a background threshold was applied using the java-based plugin ColonyArea for ImageJ (RRID:SCR_003070) [76]. When a user-defined region was used to exclude artifacts, the analysis was repeated, and the mean value was used.
Patient-derived glioma multicellular tumor spheroid (MCTS) lines:
Glioma MCTS lines were generated as 3-dimensional spheroid/organoid non-adherent cultures in defined neural stem cell media containing EGF/bFGF supplementation. BT260 was derived from an IDH-wild-type anaplastic oligodendroglioma and is available from the DFCI Center for Patient Derived Models (http://dana-farber.org/cpdm) [23]. MGG152 was derived from an IDH1 R132H-mutant astrocytoma and was provided by Drs. Daniel Cahill and Hiroaki Wakimoto (MGH) [77]. TS603 was derived from an IDH1 R132H-mutant oligodendroglioma [78] and TS516 was derived from a primary IDH-wild-type glioblastoma. Both lines were provided by Dr. Ingo Mellinghoff (Memorial Sloan Kettering Cancer Center, MSKCC). HK157 was derived from a primary IDH-wild-type glioblastoma; HK308 was derived from an IDH-wild-type, EGFR VIII+ recurrent primary glioblastoma; and HK211 and HK213 were derived from IDH1 R132H-mutant recurrent secondary glioblastomas [79]. BT054 was derived from an IDH1 R132H-mutant oligodendroglioma and was provided by Dr. Samuel Weiss (University of Calgary) [80].
Primary patient AML samples:
Primary AML patient samples for 5hmC quantification were obtained from Dr. Ross Levine (MSKCC) and have been previously described [25]. Primary AML patient samples for ChIP-seq have been previously described [81].
Culturing of primary AML samples:
Primary AML samples for ChIP-seq were expanded ex vivo as previously described [81].
Primary patient glioma samples:
Patient glioma tissues were obtained with written informed consent from the patients, and the studies were conducted in accordance with recognized ethical guidelines (e.g., Declaration of Helsinki, CIOMS, Belmont Report, U.S. Common Rule) and were approved by the Institutional Review Board of the Preston Robert Tisch Brain Tumor Center BioRepository at Duke University Medical Center. Tissue sections were reviewed by board-certified neuropathologists to confirm histopathological diagnosis in accordance with WHO guidelines, and samples with ≥70% tumor cellularity by hematoxylin and eosin (H&E) staining were selected for 5hmC analysis. DNA isolation, PCR amplification and Sanger sequencing of IDH1 and IDH2 mutation hotspots were performed as previously described [82].
Supplementary Material
Statement of Significance:
Mutant IDH is known to induce histone hypermethylation. However, it is not known if this hypermethylation is functionally significant or is a bystander effect of (R)-2HG accumulation in IDH-mutant cells. Here, we provide evidence that KDM5 inhibition by (R)-2HG contributes to mutant IDH-mediated transformation in AML and glioma.
Acknowledgements:
We thank Dr. Ingo Mellinghoff (MSKCC), Dr. Samuel Weiss (University of Calgary), Dr. Hiroaki Wakimoto (MGH), and Dr. Daniel P. Cahill (MGH) for kindly providing glioma MCTS models. We thank Dr. Russell O. Pieper for kindly providing the IHA cells. We thank Dr. Ross Levine (MSKCC) for use of the leukemia 5hmC data. We thank Dr. William G. Kaelin, Jr. (DFCI) and Dr. Bradley E. Bernstein (MGH) for assistance with designing and generating the sgRNA minipool library. We thank Dr. Qin Yan (Yale) for use of the KDM5B expression construct. We thank Dr. Rikkert Wierenga (University of Oulu) and Dr. Kristian Koski (University of Oulu) for assistance with the structural analysis of the KDM5 enzymes and Dr. Stephen Blacklow (Harvard University) for assistance with the structural analysis of the TET enzymes. We thank the staff of the Broad GPP for help with the CRISPR/Cas9 screens. We thank members of the Losman, Kaelin and Tothova laboratories for technical help and valuable discussions. We thank Eeva Lehtimäki for expert technical assistance. We thank Dr. Richard Koche (MSKCC), Dr. Rameen Beroukhim (DFCI), Dr. Ramesh Shivdasani (DFCI) and Dr. Miles Brown (DFCI) for invaluable discussions and assistance with bioinformatic analysis of the CHIP-seq and RNA-seq data.
Conflict of Interest Disclosure Statement:
J.-A. Losman has received funding support unrelated to this project via the DFCI from Lilly and via the Broad Institute from Bayer. C.D. receives research funding from Janssen. H.Y. is the chief scientific officer and has ownership interest in Genetron Holdings and receives royalties from Agios, Genetron and Personal Genome Diagnostics. K. L. Ligon is a founder and equity holder in Travera Inc., is a consultant for Bristol Myers Squibb (BMS) and Integragen and receives funding support via DFCI from BMS and Lilly. J.G.D. consults for Microsoft Research, Agios, Phenomic AI, Maze Therapeutics, BioNTech, and Pfizer; and consults for and has equity in Tango Therapeutics. J.G.D.’s interests were reviewed and are managed by the Broad Institute in accordance with its conflict-of-interest policies. The remainder of the authors report that they have nothing to disclose.
Financial support:
Investigators who conducted this research report individual research support from the National Institutes of Health-National Cancer Institute (R01CA227640 to J.-A. Losman; R01CA188228 and 50CA165962 to K. L. Ligon; P50CA211015 to H. I. Kornblum) and the National Institutes of Health-National Institute of Allergy and Infectious Diseases (R01AI127724 to R. Looper), the Cancer Prevention and Research Institute of Texas (RR190034 to S.K. McBrayer), the Sidney Kimmel Foundation for Cancer Research (J.-A. Losman), the Forbeck Foundation (J.-A. Losman), the Wong Family (J.-A. Losman), the Academy of Finland Project (#308009 to P. Koivunen), the Sigrid Jusélius Foundation (P. Koivunen), the Jane and Aatos Erkko Foundation (P. Koivunen), Cancer Foundation Finland (P. Koivunen), the V Foundation for Cancer Research (C. Duy), Leukemia Research Foundation (C. Duy), the W.W. Smith Charitable Trust (C. Duy), the Alternatives Research and Development Foundation (C. Duy), the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (H. I. Kornblum), OligoNation (K. L. Ligon), and the Dana-Farber Cancer Institute (J.-A. Losman and K. L. Ligon).
Data availability statement:
Further information, resources and reagents are available upon request. Inquiries should be directed to and will be fulfilled by Julie-Aurore Losman (julieaurore_losman@dfci.harvard.edu).
CRISPR/Cas9 screen data, ChIP-seq data, and RNA-seq data are publicly available in Gene Expression Omnibus (GEO) at GSE184611. All additional data reported in this paper will be shared by the lead contact upon request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References:
- 1.Pirozzi CJ, Yan H. The implications of IDH mutations for cancer development and therapy. Nat Rev Clin Oncol 2021;18:645–61. [DOI] [PubMed] [Google Scholar]
- 2.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009;462:739–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009;324:930–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011;333:1300–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 2013;339:1621–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010;18:553–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sasaki M, Knobbe CB, Munger JC, Lind EF, Brenner D, Brüstle A, et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature 2012;488:656–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012;366:1079–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chou WC, Lei WC, Ko BS, Hou HA, Chen CY, Tang JL, et al. The prognostic impact and stability of Isocitrate dehydrogenase 2 mutation in adult patients with acute myeloid leukemia. Leukemia 2011;25:246–53. [DOI] [PubMed] [Google Scholar]
- 10.Chou WC, Chou SC, Liu CY, Chen CY, Hou HA, Kuo YY, et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood 2011;118:3803–10. [DOI] [PubMed] [Google Scholar]
- 11.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012;483:474–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep 2011;12:463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Losman JA, Koivunen P, Kaelin WG Jr. 2-Oxoglutarate-dependent dioxygenases in cancer. Nat Rev Cancer 2020;20:710–26. [DOI] [PubMed] [Google Scholar]
- 14.Lin Q, Wu Z, Yue X, Yu X, Wang Z, Song X, et al. ZHX2 restricts hepatocellular carcinoma by suppressing stem cell-like traits through KDM2A-mediated H3K36 demethylation. EBioMedicine 2020;53:102676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scahill CM, Digby Z, Sealy IM, Wojciechowska S, White RJ, Collins JE, et al. Loss of the chromatin modifier Kdm2aa causes BrafV600E-independent spontaneous melanoma in zebrafish. PLoS Genet 2017;13:e1006959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu SH, Zhu KY, Chen J, Liu XZ, Xu PF, Zhang W, et al. JMJD3 facilitates C/EBPβ-centered transcriptional program to exert oncorepressor activity in AML. Nat Commun 2018;9:3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mallaney C, Ostrander EL, Celik H, Kramer AC, Martens A, Kothari A, et al. Kdm6b regulates context-dependent hematopoietic stem cell self-renewal and leukemogenesis. Leukemia 2019;33:2506–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheung N, Fung TK, Zeisig BB, Holmes K, Rane JK, Mowen KA, et al. Targeting Aberrant Epigenetic Networks Mediated by PRMT1 and KDM4C in Acute Myeloid Leukemia. Cancer Cell 2016;29:32–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Agger K, Nishimura K, Miyagi S, Messling JE, Rasmussen KD, Helin K. The KDM4/JMJD2 histone demethylases are required for hematopoietic stem cell maintenance. Blood 2019;134:1154–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li L, Paz AC, Wilky BA, Johnson B, Galoian K, Rosenberg A, et al. Treatment with a Small Molecule Mutant IDH1 Inhibitor Suppresses Tumorigenic Activity and Decreases Production of the Oncometabolite 2-Hydroxyglutarate in Human Chondrosarcoma Cells. PLoS One 2015;10:e0133813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis MI, Gross S, Shen M, Straley KS, Pragani R, Lea WA, et al. Biochemical, cellular, and biophysical characterization of a potent inhibitor of mutant isocitrate dehydrogenase IDH1. J Biol Chem 2014;289:13717–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saha SK, Gordan JD, Kleinstiver BP, Vu P, Najem MS, Yeo JC, et al. Isocitrate Dehydrogenase Mutations Confer Dasatinib Hypersensitivity and SRC Dependence in Intrahepatic Cholangiocarcinoma. Cancer Discov 2016;6(7):727–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012;483:484–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kroeze LI, Aslanyan MG, van Rooij A, Koorenhof-Scheele TN, Massop M, Carell T, et al. Characterization of acute myeloid leukemia based on levels of global hydroxymethylation. Blood 2014;124:1110–8. [DOI] [PubMed] [Google Scholar]
- 25.Rampal R, Alkalin A, Madzo J, Vasanthakumar A, Pronier E, Patel J, et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep 2014;9:1841–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adam S, Bräcker J, Klingel V, Osteresch B, Radde NE, Brockmeyer J, et al. Flanking sequences influence the activity of TET1 and TET2 methylcytosine dioxygenases and affect genomic 5hmC patterns. Commun Biol 2022;5:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sever R, Brugge JS. Signal transduction in cancer. Cold Spring Harb Perspect Med 2015;5:a006098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arafeh R, Di Pizio A, Elkahloun AG, Dym O, Niv MY, Samuels Y. RASA2 and NF1; two-negative regulators of Ras with complementary functions in melanoma. Oncogene 2019;38:2432–4. [DOI] [PubMed] [Google Scholar]
- 29.Yoshimachi S, Shirakawa R, Cao M, Trinh DA, Gao P, Sakata N, et al. GTPase-activating protein regulates the malignancy of pancreatic ductal adenocarcinoma. Cancer Sci 2021;112:3064–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaito S, Iwama A. Pathogenic Impacts of Dysregulated Polycomb Repressive Complex Function in Hematological Malignancies. Int J Mol Sci 2020;22:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Owen C, Fitzgibbon J, Paschka P. The clinical relevance of Wilms Tumour 1 (WT1) gene mutations in acute leukaemia. Hematol Oncol 2010;28:13–9. [DOI] [PubMed] [Google Scholar]
- 32.Gañán-Gómez I, Wei Y, Yang H, Boyano-Adánez MC, García-Manero G. Oncogenic functions of the transcription factor Nrf2. Free Radic Biol Med 2013;65:750–64. [DOI] [PubMed] [Google Scholar]
- 33.DiTacchio L, Le HD, Vollmers C, Hatori M, Witcher M, Secombe J, et al. Histone lysine demethylase JARID1a activates CLOCK-BMAL1 and influences the circadian clock. Science 2011;333:1881–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johansson C, Velupillai S, Tumber A, Szykowska A, Hookway ES, Nowak RP, et al. Structural analysis of human KDM5B guides histone demethylase inhibitor development. Nat Chem Biol 2016;12:539–45. [DOI] [PubMed] [Google Scholar]
- 35.Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG, et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med 2010;207:339–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, et al. Active genes are tri-methylated at K4 of histone H3. Nature 2002;419:407–11. [DOI] [PubMed] [Google Scholar]
- 37.Schneider R, Bannister AJ, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat Cell Biol 2004;6:73–7. [DOI] [PubMed] [Google Scholar]
- 38.Kraus TF, Globisch D, Wagner M, Eigenbrod S, Widmann D, Münzel M, et al. Low values of 5-hydroxymethylcytosine (5hmC), the “sixth base,” are associated with anaplasia in human brain tumors. Int J Cancer 2012;131:1577–90. [DOI] [PubMed] [Google Scholar]
- 39.Orr BA, Haffner MC, Nelson WG, Yegnasubramanian S, Eberhart CG. Decreased 5-hydroxymethylcytosine is associated with neural progenitor phenotype in normal brain and shorter survival in malignant glioma. PLoS One 2012;7:e41036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pastor WA, Aravind L, Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol 2013;14:341–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012;22:425–37. [DOI] [PubMed] [Google Scholar]
- 42.Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012;483:479–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krivtsov AV, Evans K, Gadrey JY, Eschle BK, Hatton C, Uckelmann HJ, et al. A Menin-MLL Inhibitor Induces Specific Chromatin Changes and Eradicates Disease in Models of MLL-Rearranged Leukemia. Cancer Cell 2019;36:660–673.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shokri G, Doudi S, Fathi-Roudsari M, Kouhkan F, Sanati MH. Targeting histone demethylases KDM5A and KDM5B in AML cancer cells: A comparative view. Leuk Res 2018;68:105–11. [DOI] [PubMed] [Google Scholar]
- 45.Xu S, Wang S, Xing S, Yu D, Rong B, Gao H, et al. KDM5A suppresses PML-RARα target gene expression and APL differentiation through repressing H3K4me2. Blood Adv 2021;5:3241–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med 2016;374:2209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wong SH, Goode DL, Iwasaki M, Wei MC, Kuo HP, Zhu L, et al. The H3K4-Methyl Epigenome Regulates Leukemia Stem Cell Oncogenic Potential. Cancer Cell 2015;28:198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Q, Zhao L, Yang Y, Li S, Liu Y, Chen C. Mosaic loss of chromosome Y promotes leukemogenesis and clonal hematopoiesis. JCI Insight. 2022;7:e153768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fagnan A, Bagger FO, Piqué-Borràs MR, Ignacimouttou C, Caulier A, Lopez CK, et al. Human erythroleukemia genetics and transcriptomes identify master transcription factors as functional disease drivers. Blood 2020;136:698–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marshall A, Kasturiarachchi J, Datta P, Guo Y, Deltcheva E, James C, et al. Mir142 loss unlocks IDH2R140-dependent leukemogenesis through antagonistic regulation of HOX genes. Sci Rep 2020;10:19390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mellinghoff IK, Chang SM, Jaeckle KA, van den Bent M. Isocitrate Dehydrogenase Mutant Grade II and III Glial Neoplasms. Hematol Oncol Clin North Am 2022;36:95–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016;529:110–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A 1999;96:8681–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Strathdee G, Appleton K, Illand M, Millan DW, Sargent J, Paul J, et al. Primary ovarian carcinomas display multiple methylator phenotypes involving known tumor suppressor genes. Am J Pathol 2001;158:1121–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kelly AD, Kroeger H, Yamazaki J, Taby R, Neumann F, Yu S, et al. A CpG island methylator phenotype in acute myeloid leukemia independent of IDH mutations and associated with a favorable outcome. Leukemia 2017;31:2011–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stasik S, Juratli TA, Petzold A, Richter S, Zolal A, Schackert G, et al. Exome sequencing identifies frequent genomic loss of TET1 in IDH-wild-type glioblastoma. Neoplasia 2020;22:800–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Forloni M, Gupta R, Nagarajan A, Sun LS, Dong Y, Pirazzoli V, et al. Oncogenic EGFR Represses the TET1 DNA Demethylase to Induce Silencing of Tumor Suppressors in Cancer Cells. Cell Rep 2016;16:457–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim YH, Pierscianek D, Mittelbronn M, Vital A, Mariani L, Hasselblatt M, et al. TET2 promoter methylation in low-grade diffuse gliomas lacking IDH1/2 mutations. J Clin Pathol 2011;64:850–2. [DOI] [PubMed] [Google Scholar]
- 59.Müller T, Gessi M, Waha A, Isselstein LJ, Luxen D, Freihoff D, et al. Nuclear exclusion of TET1 is associated with loss of 5-hydroxymethylcytosine in IDH1 wild-type gliomas. Am J Pathol 2012;181:675–83. [DOI] [PubMed] [Google Scholar]
- 60.McBrayer SK, Mayers JR, DiNatale GJ, Shi DD, Khanal J, Chakraborty AA, et al. Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 2018;175:101–16.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 2016;34:184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goedhart J, Luijsterburg MS. VolcaNoseR is a web app for creating, exploring, labeling and sharing volcano plots. Sci Rep 2020;10:20560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Laukka T, Myllykoski M, Looper RE, Koivunen P. Cancer-associated 2-oxoglutarate analogues modify histone methylation by inhibiting histone lysine demethylases. J Mol Biol 2018;430:3081–92. [DOI] [PubMed] [Google Scholar]
- 64.Wingett SW, Andrews S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Res. 2018;7:1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics 2015;31:2032–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carroll TS, Liang Z, Salama R, Stark R, de Santiago I. Impact of artifact removal on ChIP quality metrics in ChIP-seq and ChIP-exo data. Front Genet 2014;5:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012;481:389–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stark R, Brown G. DiffBind: differential binding analysis of ChIP-Seq peak data. Bioconductor.org [Internet]. 2011. [edited 2022 October 4]. Available from: http://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf. 2011. [Google Scholar]
- 72.Mirchia K, Sathe AA, Walker JM, Fudym Y, Galbraith K, Viapiano MS, et al. Total copy number variation as a prognostic factor in adult astrocytoma subtypes. Acta Neuropathol Commun 2019;7:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cohen A, Sato M, Aldape K, Mason CC, Alfaro-Munoz K, Heathcock L, et al. DNA copy number analysis of Grade II-III and Grade IV gliomas reveals differences in molecular ontogeny including chromothripsis associated with IDH mutation status. Acta Neuropathol Commun 2015;3:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 2002;30:207–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guzmán C, Bagga M, Kaur A, Westermarck J, Abankwa D. ColonyArea: an ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS One 2014;9:e92444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wakimoto H, Tanaka S, Curry WT, Loebel F, Zhao D, Tateishi K, et al. Targetable signaling pathway mutations are associated with malignant phenotype in IDH-mutant gliomas. Clin Cancer Res 2014;20:2898–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013;340:626–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Laks DR, Crisman TJ, Shih MY, Mottahedeh J, Gao F, Sperry J, et al. Large-scale assessment of the gliomasphere model system. Neuro Oncol 2016;18:1367–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kelly JJ, Blough MD, Stechishin OD, Chan JA, Beauchamp D, Perizzolo M, et al. Oligodendroglioma cell lines containing t(1;19)(q10;p10). Neuro Oncol 2010;12:745–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Duy C, Teater M, Garrett-Bakelman FE, Lee TC, Meydan C, Glass JL, et al. Rational Targeting of Cooperating Layers of the Epigenome Yields Enhanced Therapeutic Efficacy against AML. Cancer Discov. 2019;9:872–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Killela PJ, Pirozzi CJ, Healy P, Reitman ZJ, Lipp E, Rasheed BA, et al. Mutations in IDH1, IDH2, and in the TERT promoter define clinically distinct subgroups of adult malignant gliomas. Oncotarget 2014;5:1515–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Laukka T, Mariani CJ, Ihantola T, Cao JZ, Hokkanen J, Kaelin WG Jr, et al. Fumarate and Succinate Regulate Expression of Hypoxia-inducible Genes via TET Enzymes. J Biol Chem 2016;291:4256–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Further information, resources and reagents are available upon request. Inquiries should be directed to and will be fulfilled by Julie-Aurore Losman (julieaurore_losman@dfci.harvard.edu).
CRISPR/Cas9 screen data, ChIP-seq data, and RNA-seq data are publicly available in Gene Expression Omnibus (GEO) at GSE184611. All additional data reported in this paper will be shared by the lead contact upon request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.