

Abstract
G protein-coupled receptors (GPCRs) selectively activate at least one of the four families of heterotrimeric G proteins, but the mechanism of coupling selectivity remains unclear. Structural studies emphasize structural complementarity of GPCRs and nucleotide-free G proteins, but selectivity is likely to be determined by transient intermediate state complexes that exist prior to nucleotide release. Here we study coupling to nucleotide-decoupled G protein variants that can adopt conformations similar to receptor-bound G proteins without releasing nucleotide, and are therefore able to bypass intermediate state complexes. We find that selectivity is degraded when nucleotide release is not required for GPCR-G protein complex formation, to the extent that most GPCRs interact with most nucleotide-decoupled G proteins. These findings demonstrate the absence of absolute structural incompatibility between noncognate receptor-G protein pairs, and are consistent with the hypothesis that transient intermediate states are partly responsible for coupling selectivity.
G protein-coupled receptors (GPCRs) sense extracellular cues and mediate many of their physiological responses by activating heterotrimeric G proteins1,2. Agonist-bound GPCRs adopt active conformations and associate with GDP-bound G proteins, which facilitates release of GDP from the Gα subunit. This is followed by binding of GTP and dissociation of the heterotrimer. Despite the vast number and diversity of external stimuli GPCRs share only four families of G proteins. Because members of each family interact with different intracellular effectors to produce distinct physiological responses, GPCRs must selectively activate specific subsets of G proteins.
The mechanism of coupling selectivity has been studied extensively using receptor and G protein chimeras and site-directed mutagenesis3,4. More recently, structural characterization of GPCR-G protein complexes has identified elements that are important for activation of specific G proteins5–8. However, structural studies require stable agonist-receptor-G protein complexes, and this almost always requires a nucleotide-free G protein. Therefore, such studies are largely limited to a state of the receptor-G protein complex that exists after nucleotide release has occurred, in other words after the critical step in G protein activation. Receptor-mediated nucleotide release involves extensive conformational rearrangement of the Gα subunit9, and it has been suggested that G protein selection occurs at an earlier intermediate state complex that is too energetically unstable to be captured by standard structural approaches10,11. A few studies have begun to characterize possible intermediate state complexes10,12, but evidence that such complexes are involved in coupling selectivity is lacking. Here we approach this question using G protein mutants where binding to receptors is decoupled from nucleotide release, and many of the structural changes associated with G protein activation are present prior to association with a receptor13. We find that receptor-G selectivity is substantially degraded with nucleotide-decoupled G proteins, suggesting that these mutants bypass intermediate steps that normally play a role in coupling selectivity. Moreover, most receptors spontaneously form complexes with cognate nucleotide-decoupled G proteins, implying that basal sampling of active conformations is a common feature of GPCRs. We exploited this finding to screen understudied orphan GPCRs and found many that failed to interact with nucleotide-decoupled G proteins, suggesting that these receptors may not activate G proteins at all.
Results
4A insertion decouples receptor association and nucleotide exchange
Biophysical and structural studies of GPCR-G protein complexes have revealed that Gα subunits undergo large scale conformational changes that lead to GDP release7,14. The C-terminal helix 5 (H5) of Gα interacts with the transmembrane core of active receptors, requiring translation of this helix towards the receptor7. This engagement, likely in concert with receptor interactions with the Gα N terminus, triggers several conformational changes that promote GDP release. These include: (1) disruption of a hydrophobic core comprised of H5, H1, and the S1-S3 strands; (2) increased disorder of the s6h5 loop (TCAT motif), which directly contacts the guanine ring of GDP; and (3) increased disorder of the P loop, which coordinates the β-phosphate of GDP9. In addition, separation of the Ras-homology and α-helical domains is necessary but not sufficient for GDP release14. These changes in Gα are energetically costly15, and presumably are compensated by formation of new contacts with the active receptor.
To test the idea that these changes might play a role in coupling selectivity we studied interactions between receptors and G protein mutants that mimicked the receptor-bound state. For this purpose, we chose a previously characterized design13 wherein the H5 helix is extended by inserting four alanine residues (4A) between H5.5 and H5.6 (according to the Common Gα Numbering (CGN) system)16. This disrupts the H5-H1-S1–3 hydrophobic core and adds one helical turn, lengthening H5 (Fig. 1a–c). The conserved residue FH5.8 is extracted from the hydrophobic core of the Gαi1 4A mutant, as is the case in nucleotide-free receptor-bound structures. Importantly, the Gi1 4A mutant remained bound to active rhodopsin even in the presence of guanine nucleotides13, suggesting that receptor association was decoupled from nucleotide binding.
Figure 1.
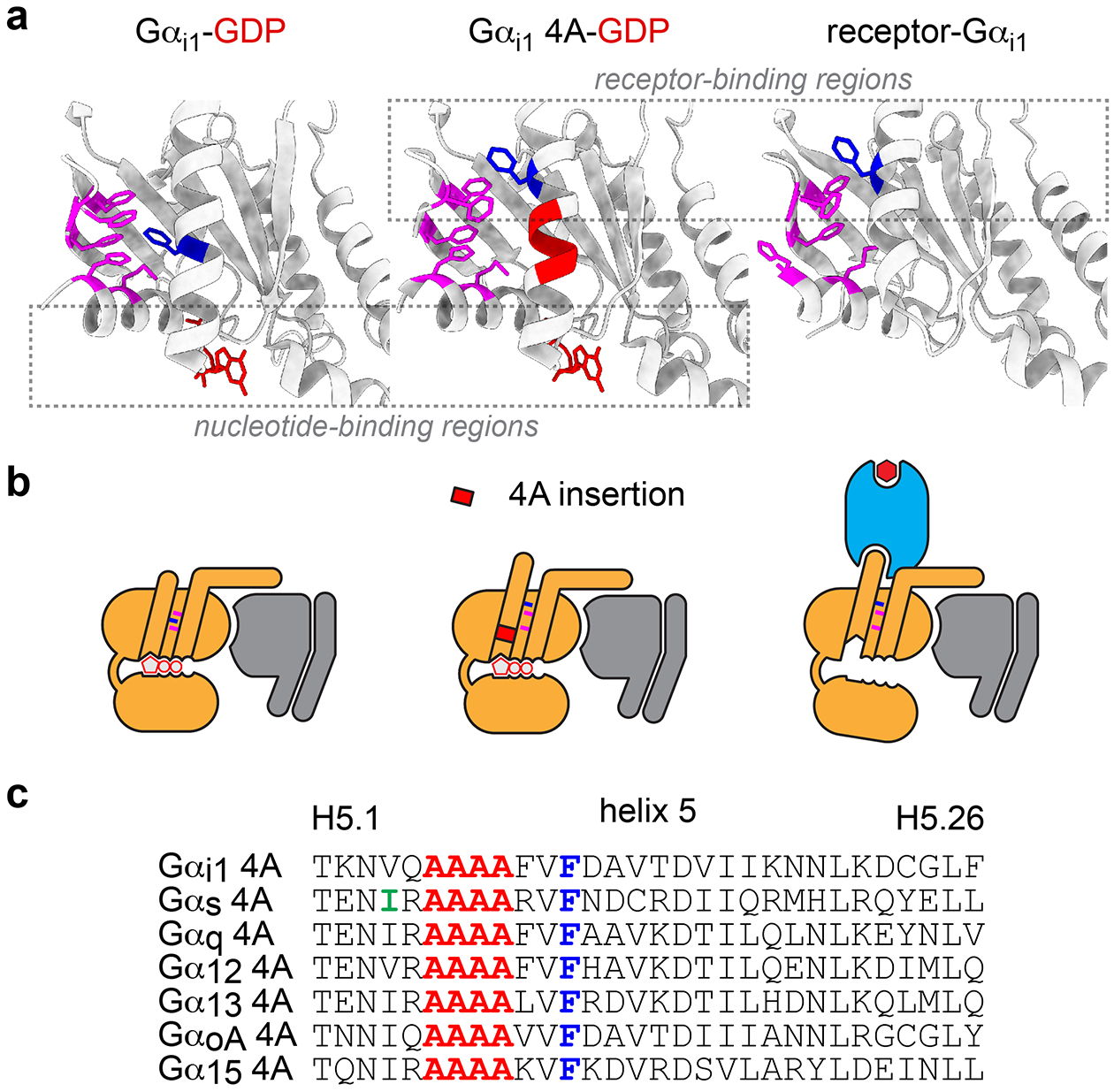
Structural features of nucleotide-decoupled G proteins. a, GDP-bound Gα subunits (left; PDB 1GP2) possess a hydrophobic core comprised of side chains of residues in alpha helix 1 (H1), and beta strands 1–3 (S1–3; magenta), as well as residues in helix 5 (H5), including FH5.8 (dark blue). In structures of nucleotide-free Gαi1 subunits bound to active receptors (right; PDB 7CMV) this hydrophobic core is disrupted, and FH5.8 is extracted. G protein 4A mutants (center; PDB 5KDO) include four alanine residues (red) between H5.5 and H5.6 which extends H5, disrupts the hydrophobic core, and extracts FH5.8. Receptor-binding regions of 4A mutants are similar to those of receptor-bound Gα subunits, whereas GDP-binding regions of 4A mutants are similar to those of GDP-bound wild-type Gα subunits (dashed rectangles). b, Cartoon representations of the three structures shown in panel a. c, Primary sequences of H5 in 4A mutants. Highlighted in green is residue IH5.4 in Gαs, which is mutated to alanine in mini Gs and midi Gs.
We first studied interactions of wild-type (wt) Gi1 and Gi1 4A with a prototypical ligand-activated GPCR, the M4 muscarinic acetylcholine receptor (M4R). We measured bioluminescence resonance energy transfer (BRET) between M4R fused to Renilla luciferase (Rluc8) and Gβ1 and Gγ2 subunits fused to complementary fragments of Venus (Fig. 2a). To minimize background from endogenous G proteins we used HEK 293 cells lacking endogenous Gs/olf, Gq/11 and G12/13 family proteins17. Cells were permeabilized and either treated with apyrase or supplemented with nucleotides. In the absence of nucleotides acetylcholine (Ach) elicited a large increase in BRET between M4R and wt Gi1 that was quickly reversed by addition of GDPβS (Fig. 2a). In contrast, Ach-induced BRET between M4R and Gi1 4A was largely resistant to GDPβS (Fig. 2a). Similar results were obtained with GTPγS, except that agonist-induced BRET was only partially resistant to GTPγS (Extended Data Fig. 1a–b). Resistance to GTPγS was enhanced if we introduced an additional mutation (RH2.04S) to prevent GTP-induced heterotrimer dissociation (Extended Data Fig. 1c)18. Using a protocol where GDPβS-GTPγS exchange is monitored indirectly with a membrane-anchored Gβγ sensor (memGRK3ct-Rluc8)19, we also found that spontaneous GDPβS-GTPγS exchange was more rapid for Gi1 4A than wt Gi1 (Fig. 2b), consistent with previous studies of isolated Gαi1 4A subunits13. Our results suggest that 4A mutants have structural (disrupted hydrophobic core) and functional (rapid GDP release) features of receptor-bound G proteins. These results are consistent with how Gi1 4A interacts with light-activated rhodopsin13, and show that the Gi1 4A interaction with M4R is decoupled from nucleotide binding.
Figure 2.

Functional properties of nucleotide-decoupled G proteins. a, BRET between M4R-Rluc8 and wild-type (wt) Gi1 heterotrimers induced by the agonist acetylcholine (100 μM) in the absence of nucleotides is largely reversed by addition of GDPβS (10 μM; 12 replicates from 3 experiments), whereas BRET between M4R-Rluc8 and Gi1 4A heterotrimers is largely resistant to GDPβS (16 replicates from 4 experiments). b, Wild-type Gi1 exchanges GDPβS for GTPγS slowly, whereas Gi1 4A exchanges nucleotides more rapidly. The rate of GTPγS binding to nucleotide-free heterotrimers is similar for wt and 4A mutant Gi1. Gαi1 subunits were ADP-ribosylated by pertussis toxin to prevent receptor-catalyzed nucleotide release; GTPγS binding was monitored indirectly using a membrane-bound free Gβγ sensor GRKct-Rluc8 after incubation in apyrase alone (for GTPγS binding) or apyrase plus GDBβS (for GDPβS-GTPγS exchange); ΔBRET is the change in BRET above or below the baseline, traces represent mean ± 95% C.I. for 7 replicates from 4 experiments (Gi1 wt) or 12 replicates from 6 experiments (Gi1 4A).
To test whether the properties of Gi1 4A are conserved across other G protein families, we constructed Gαs 4A, Gαq 4A, Gα12 4A subunits (Fig. 1c; Extended Data Fig. 1d) and examined interactions with cognate GPCRs. All of the 4A mutants expressed well enough to sequester free Gβγ dimers, although the extent of G protein sequestration was greatest for Gi1 4A and GoA 4A (Extended Data Fig. 2), and all of the 4A mutants supported interactions with GPCRs. Conventional allosteric coupling was observed for wt G proteins (Fig. 3a), in that receptor-G protein association was modest in the presence of GDP but much more robust in the absence of nucleotides, and inverse agonist responses were virtually undetectable (Fig. 3b). In contrast, 4A heterotrimers spontaneously formed complexes with cognate receptors, as indicated by high basal BRET that was increased only slightly by agonists (Fig. 3a). BRET between Gi1 4A and the α2 adrenoreceptor (α2AR) was only partially resistant to GDP when agonist was not present, whereas BRET between the other 4A mutants and their cognate receptors was completely resistant to GDP. Basal BRET between receptors and 4A G proteins was partially decreased by inverse agonists, consistent with disruption of active receptor-G protein 4A complexes (Fig. 3b).
Figure 3.

Nucleotide-decoupled G proteins bind spontaneously to cognate GPCRs. BRET between four representative Rluc8-tagged GPCRs and cognate wt and 4A G protein heterotrimers in response to agonists a and inverse agonists b. Compared to wt G proteins, 4A mutants display high basal BRET that is only modestly enhanced by agonists and only partially reversed by inverse agonists. Receptors are β2 and α2A adrenergic receptors (β2AR and α2AR), M3R acetylcholine receptors, and GPR35 (mean ± S.D of 5 independent experiments).
Further experiments indicated that complexes between receptors and 4A mutants were structurally analogous to complexes between active receptors and wild-type G proteins. Interactions between M4R and 4A mutants were completely blocked by pertussis toxin (PTX; for Gi1 4A) or deletion of 5 amino acids from the Gα subunit C terminus (Gi1 4A and Gs 4A), consistent with engagement of active receptors with the H5 helix (Extended Data Fig. 3a). We also found that a mutation that stabilizes the inactive state of the β2 adrenoreceptor (β2AR) decreased constitutive binding to Gs 4A (Extended Data Fig. 3b), and that expression of Gs 4A promoted high-affinity agonist binding to the β2AR in intact cells (Extended Data Fig. 3c). These results suggest that binding to 4A mutants requires receptors to adopt active states, and that the presence of 4A mutants in turn promotes receptor activation.
Nucleotide-decoupled G proteins bypass a selectivity barrier
The foregoing results indicate that association of receptors and 4A G proteins is decoupled from nucleotide exchange and that 4A G proteins associate spontaneously with cognate receptors. We reasoned that 4A mutants bypass the energetically-costly conformational changes associated with nucleotide release, and took advantage of this property to ask how such conformational changes contribute to receptor-G protein coupling selectivity. We hypothesized that if structural complementarity of nucleotide-free G protein-receptor complex was the sole driver of selectivity, then nucleotide-decoupled G protein mutants would maintain their selectivity. In contrast, if less stable intermediate precursors (nucleotide-bound G protein-receptor complexes) influenced selectivity, then selectivity would be degraded with G proteins that essentially bypass these states.
To test this idea, we measured BRET between 32 ligand-activated GPCRs and six nucleotide-decoupled G proteins (Gi1 4A, GoA 4A, Gs 4A, Gq 4A, G12 4A, and G13 4A) in intact cells. We found that basal net BRET was generally highest for cognate receptor-G protein 4A pairs. The G protein family with the highest basal net BRET matched the consensus primary coupling family in GPCRdb20 (defined by log(Emax/EC50)) in 27 out of 32 cases, demonstrating that selectivity between ligand-free receptors and G protein 4A families remained partially intact. However, agonist-mediated activation of receptors showed that selectivity was markedly degraded (Fig. 4a–d; Extended Data Fig. 4), such that 24 of the 32 receptors we studied met criteria (basal BRET>0.1 or agonist-induced BRET>20% over basal) for interaction with all six 4A mutants. Altogether 181 out of 192 possible pairings met these criteria. For example, M4R displayed robust agonist-induced BRET with 4A heterotrimers from all four families (Fig. 4a), even though these receptors cannot activate wt Gs/olf and G12/13 heterotrimers and only weakly activate Gq21. More stringent selectivity between 4A mutants was retained by a few receptors. For example, the Gs-coupled bile acid receptor GPBAR exhibited more stringent selectivity for Gs 4A heterotrimers, associating with other 4A mutants only in the presence of high concentrations of agonist (Fig. 4b). Notably, GPBAR is also highly selective for wt Gs/olf over other heterotrimers21. Likewise, 5-HT2A serotonin receptors (Fig. 4c) and GPR55 (Fig. 4d) retained fairly strict selectivity for Gq 4A and G13 4A, respectively. Interestingly, a few receptors selected one subtype yet rejected another within the same G protein family. For example, 5-HT5A associated with Gi1 4A and Gq 4A but largely rejected all other subtypes including GoA 4A, a subtype in the same family as Gi1. Similarly, GPR55 preferentially formed complexes with G13 4A over G12 4A. Reversal of receptor-G protein 4A association after addition of inverse agonist (Extended Data Fig. 5a) or activation of a competing receptor (Extended Data Fig. 5b) was more rapid and complete for noncognate complexes, suggesting that cognate complexes are more stable than noncognate complexes. These results indicate that 4A G protein heterotrimers interact promiscuously with active receptors, and this includes receptors that are unable to productively activate the corresponding wild-type G proteins (Extended Data Fig. 6). However, the extent to which selectivity is degraded varies widely between receptors, and a degree of selectivity is invariably retained.
Figure 4.
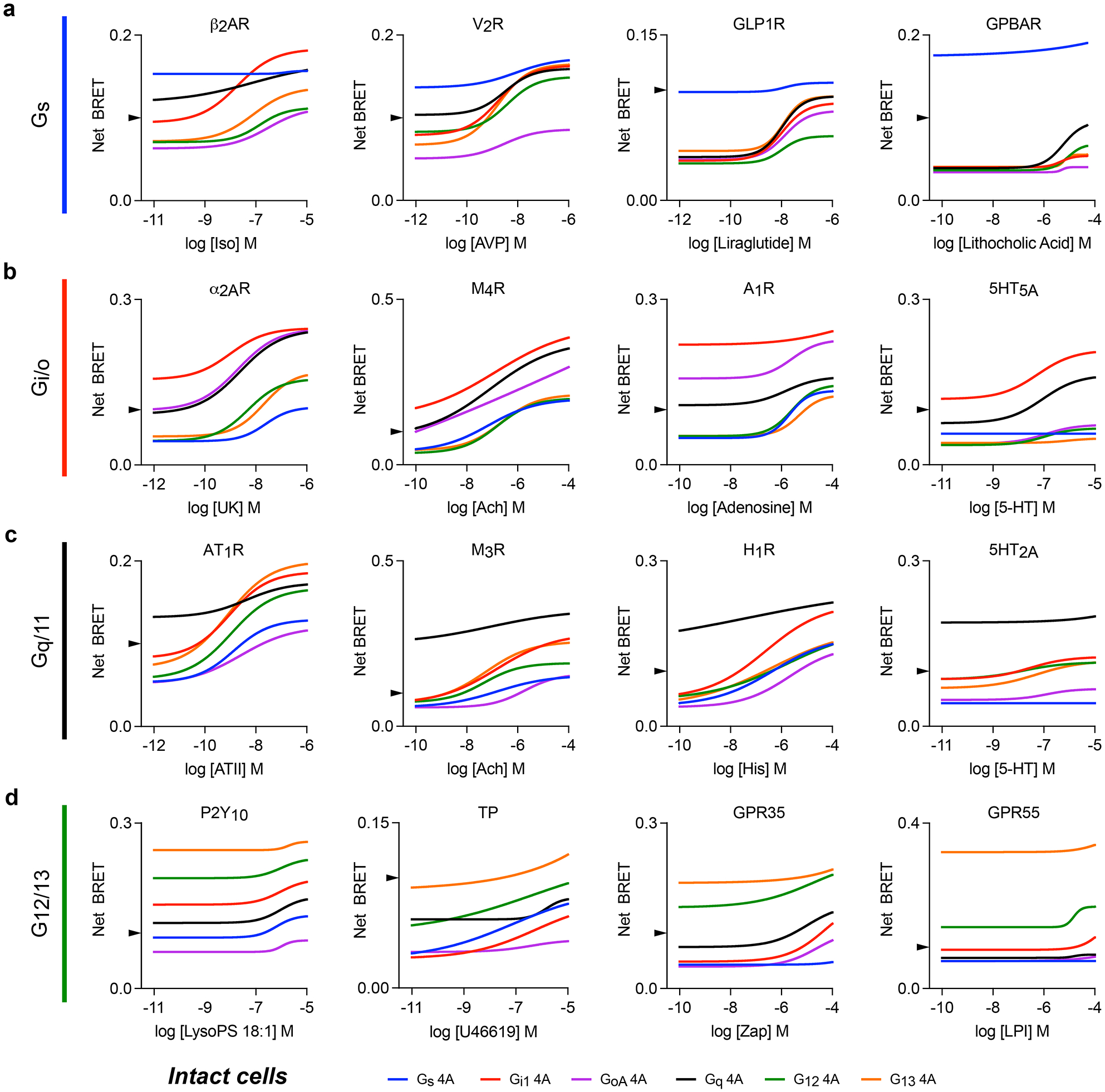
Nucleotide-decoupled G proteins bind promiscuously to noncognate GPCRs. BRET between sixteen representative Rluc8-tagged GPCRs and six different 4A G protein heterotrimers in response to agonists. Rows a, b, c and d show receptors that primarily are known to couple to Gs, Gi/o, Gq/11 and G12/13 heterotrimers respectively. Most receptors interact with all six 4A mutants in an agonist-dependent manner. Shown are least-square fits to a four-parameter concentration-response equation; data points have been removed for clarity. Concentration-response curves with data points from three independent experiments (± S.E.M.) are shown for all 32 receptors studied in Extended Data Figure 4. An arrowhead on each y axis marks Net BRET of 0.1, the threshold used for later analysis (Figure 6).
It was noteworthy that, except for 5-HT5A and Gs 4A, all of the Gi/o-coupled receptors that we studied formed agonist-dependent complexes with Gs 4A and Gq 4A heterotrimers (Extended Data Fig. 4). One mechanism that has been proposed to explain the selectivity of Gi/o-coupled receptors is a limited displacement of transmembrane helix 6 (TM6), which results in a binding pocket in the TM core that is too small to accommodate the relatively bulky C termini of Gαs and Gαq subunits22. This hypothesis is consistent with the limited displacement of TM6 observed in many receptor-Gi/o complex structures23–25. However, from structural studies alone it is unclear if receptors with small TM6 displacements are unable to adopt larger displacements, or if instead larger displacements are possible but not captured when receptors are in complexes with Gi or Go. Our results showing robust association of Gi/o-coupled receptors with Gs 4A and Gq 4A mutants are consistent with the latter explanation. To further support this conclusion, we verified that association of α2AR and M4R with Gs 4A required the Gαs C terminus (Extended Data Fig. 7a), and that neither receptor could detectably activate wt Gs heterotrimers (Extended Data Fig. 7b).
Coupling selectivity of engineered Gα subunits
Gα subunits engineered to form stable complexes with active receptors have been valuable for solving structures of GPCR-G protein complexes. For example, variants of mini G (mG) proteins have been used to solve several GPCR-G protein complex structures by x-ray crystallography and cryo-EM26. These engineered Gα subunits incorporate a mutation, IH5.4A (Fig. 1c), that was found to uncouple nucleotide binding and receptor association, such that GPCR-mG complexes would remain stable even in the presence of nucleotides27. Because this property is shared with 4A mutants we suspected that mG proteins might also have degraded selectivity. We tested this idea with mini Gs, because most other mG proteins used for structural studies are chimeras that include substantial sequence derived from Gαs. We constructed a variant of mini Gs (referred to here as midi Gs) with an intact αN helix, providing a membrane anchor as well as the capacity to bind Gβγ. We also constructed an analogous subunit (midi Gs wtct) in which the uncoupling mutation was reverted to the wt sequence (AH5.4I). As was the case with Gs 4A, midi Gs (expressed together with Venus-Gβγ) interacted spontaneously with β2AR. This interaction was only slightly enhanced by agonist, was weakly diminished by inverse agonist, and was completely insensitive to GDP (Fig. 5a). As was the case with Gs 4A, midi Gs clearly preferred the active state of the β2AR (Extended Data Fig. 3b), and in turn promoted the active, high-affinity agonist binding state in intact cells (Extended Data Fig. 3c). In contrast, midi Gs wtct interacted with β2AR in an agonist- and GDP-sensitive manner, supporting the conclusion that the IH5.4A mutation decoupled nucleotide binding from receptor association27. We then tested the ability of midi Gs and midi Gs wtct to interact with noncognate receptors in intact cells. Similar to what we observed with G protein 4A mutants, midi Gs interacted promiscuously with noncognate receptors in an agonist-dependent manner (Fig. 5b). In contrast, midi Gs wtct interacted poorly with noncognate receptors. That two different mutations (4A insertion and IH5.4A) that decouple nucleotide binding from receptor association also degrade coupling selectivity supports the notion that selectivity is partly determined by conformational changes that lead up to nucleotide release.
Figure 5.

Nucleotide-decoupled midi Gs also loses selectivity. a, In permeabilized cells midi Gs proteins bearing the nucleotide-decoupling mutation AH5.4 interact spontaneously with β2AR in a nucleotide-insensitive manner, whereas midi Gs bearing the wild-type residue in this position (wtct; IH5.4) interact with this receptor in a ligand- and nucleotide-sensitive manner (mean ± S.D. of 5 independent experiments. b, In intact cells midi Gs but not midi Gs wtct interacts promiscuously with Gi-, Gq- and G12/13-coupled receptors (α2AR, M3R and TP, respectively) in response to agonist stimulation (mean ± S.D. of 3 independent experiments).
Dominant negative (DN) Gα subunits have also been valuable tools for structural determination of GPCR-G protein complexes28. DN G proteins have mutations that reduce nucleotide-binding affinity and enhance stability of ternary complexes. These functional properties prompted us to characterize their interaction with cognate and noncognate receptors, focusing on DN Gαs and Gαi1. Compared to G protein 4A mutants and midi Gs, association of DN Gαs and Gαi1 with cognate receptors (β2AR and α2AR) was more dependent upon agonist, and more sensitive to GDP, suggesting a milder nucleotide decoupling phenotype (Extended Data Fig. 8a). At the same time, DN Gαs and Gαi1 retained coupling selectivity to a greater degree than fully decoupled variants, as receptors interacted with noncognate DN G proteins only modestly, even in the absence of nucleotides (Extended Data Fig. 8b). These results suggest that degradation of coupling selectivity across G protein variants correlates with the extent to which receptor association is decoupled from nucleotide binding.
Constitutive conformational sampling of bona fide GPCRs
GPCRs are conformationally dynamic and can spontaneously transition between inactive and active states29,30. This property is likely to be responsible for constitutive G protein activation observed for some receptors. We found that 29 of the 32 receptors we studied had a basal net BRET value that exceeded an arbitrary threshold of 0.1 (Fig. 4, arrowheads) for their cognate G protein 4A family (Fig. 6a). Notably, even the three receptors where this threshold was not met displayed a clear preference for their cognate 4A family (Fig. 4 and Fig. 6a). This indicates that most receptors sample active states to some extent, and suggests that nucleotide-decoupled G proteins are sensitive probes for detecting this conformational sampling.
Figure 6.

Many orphan GPCRs may not activate G proteins. a, Heat maps showing basal BRET between 32 well-studied GPCRs and four different G protein 4A mutants. Only three of these receptors (GLP1R, ADCYAP1R1 and TBXA2, shown in red bold italic font) show basal BRET that fails to exceed 0.1 with their cognate 4A mutant. b, Three bona fide chemokine GPCRs show high basal BRET with Gi1 4A, whereas three known decoy receptors do not. c, Most orphan receptors that show constitutive coupling to wt G proteins (top) also interact spontaneously with at least one G protein 4A mutant, whereas a substantial fraction of orphan receptors that lack spontaneous coupling to wt G proteins also do not interact spontaneously with 4A mutants, suggesting that many of these receptors may not activate G proteins. Exceptions were GPR150, GPR151, GPR157 and GPR31, all of which showed basal BRET that exceeded 0.1 with Gi1 4A, although this was sensitive to pertussis toxin (PTX) only for the latter two receptors. Most adhesion receptors (expressed as C terminal fragments) also interact spontaneously with 4A mutants. Each cell represents the mean of at least 3 independent experiments.
We then asked if we could make use of this property to identify 7 transmembrane (7TM) proteins that are related to GPCRs but do not activate G proteins. Examples of this type of protein include so-called decoy receptors, which bind and sequester ligands yet fail to initiate G protein signaling. We predicted that such receptors would fail to interact with G protein 4A mutants. To test this prediction we measured basal BRET between 4A mutants, three bona fide Gi/o-coupled chemokine receptors (C5AR1, CCR2 and CXCR4), and three atypical chemokine receptors (C5AR2, ACKR2 and ACKR3) known to be decoys31. As expected, all three Gi/o-coupled receptors showed high basal BRET to Gi1 4A heterotrimers. In contrast, none of the known decoy receptors showed above-threshold basal BRET with any 4A mutant (Fig. 6b), and this could not be attributed to a failure of these receptors to traffic to the cell surface (Fig. 6 Source Data). We then applied this strategy to a panel of understudied orphan GPCRs (oGPCRs), many of which have not been shown to couple to any transducer. We divided this panel into two groups based on whether or not we previously detected conventional GDP-sensitive coupling to wt G proteins32. For oGPCRs shown previously to couple to wt G proteins 21 of 22 showed basal BRET signals greater than 0.1 for at least one 4A mutant, consistent with what we observed with liganded GPCRs. In contrast, only 4 of 16 receptors without detectable GDP-sensitive coupling to wt G proteins showed basal net BRET that exceeded 0.1 with any 4A mutant (Fig. 6c), even though all were expressed at the cell surface (Fig. 6 Source Data). We cannot rule out the possibility that some of these receptors failed to transfer energy due to a particularly unfavorable positioning of the C-terminal luciferase, although given the near universal competence of bona fide GPCRs we think this is highly unlikely. We also considered the possibility that some of these receptors might couple exclusively to G15 heterotrimers, but none of the oGPCRs that failed to interact with our original 4A mutants interacted well with G15 4A (Extended Data Fig. 9). The four receptors in this group that exceeded a basal BRET of 0.1 (GPR31, GPR150, GPR151 and GPR157) all did so with Gi1 4A. PTX nearly abolished basal BRET between Gi1 4A and GPR31 and GPR157, but did not significantly decrease energy transfer to the other two receptors (Extended Data Fig. 10), suggesting that GPR31 and GPR157 are both likely bona fide Gi-coupled GPCRs. Finally, we applied the same strategy to a sample of eight adhesion GPCRs (aGPCRs) expressed at the cell surface (Fig. 6 Source Data) as C terminal fragments (CTF), which lack the N-terminal GAIN domain and are often constitutively active33. Six of these aGPCRs interacted well with at least one 4A mutant, as indicated by a basal BRET value that exceeded 0.1. In contrast, neither ADGRA2 nor ADGRA3 CTFs exceeded this threshold with any 4A mutant (Fig. 6c). The fourteen oGPCRs and aGPCRs in our sample that did not transfer energy well to G protein 4A mutants may lack the ability to activate G proteins altogether, or may only be able to do so in the presence of an activating ligand or cofactor.
Discussion
The mechanism of selective GPCR-G protein coupling has been studied using a variety of experimental approaches, most of which have focused on the structural determinants of selectivity3. Recently, solved structures of receptors bound to nucleotide-free G proteins have been used to predict residues in the receptor-G protein interface that contribute to selectivity. These studies have provided valuable insights into how individual receptors discriminate G protein families, and suggest that the conformation of the receptor-G protein complexes that are critical for selectivity share many features with these empty-state complexes5–8. Nevertheless, structural studies are generally limited to stable complexes that exist only after GDP release, which may not resemble the key intermediates that immediately precede nucleotide release.
In the present study we considered the possibility that selectivity could be partly determined by the process whereby the receptor disrupts the hydrophobic core of the Gα subunit, concomitant with extension of the G protein C terminus. This change is predicted to be both energetically costly15 and critical for GDP release14. To test this idea we made use of G protein variants shown previously to bind to active GPCRs in the presence of guanine nucleotides13. In the case of G protein 4A mutants, the hydrophobic core of the Gα subunit is disrupted even though nucleotide is present, mimicking the receptor-bound empty state. We reasoned that such mutants would effectively bypass some of the steps leading up to disruption of the hydrophobic core and GDP release, and might therefore have less stringent coupling selectivity. Consistent with our hypothesis, we found that selectivity between GPCRs and nucleotide-decoupled G proteins was degraded to the point where most receptors could interact with G protein 4A mutants from all four families.
Our results are consistent with a model of selectivity wherein the conformational changes in Gα necessary to promote GDP release represent an energetically-insurmountable barrier for noncognate GPCR-G protein pairs. Bypassing this barrier reveals a latent capacity for most agonist-bound GPCRs and G proteins to interact. One straightforward conclusion that can be drawn from this observation is that there are no absolute structural barriers to complex formation for the majority of potential GPCR-G protein pairs. At the same time, our results reinforce the importance of structural complementarity in GPCR-G protein coupling, as the conformational plasticity presumably conferred on the G protein by nucleotide-decoupling did not abolish selectivity. Indeed, some receptors retained a high degree of selectivity between nucleotide-decoupled G proteins, consistent with highly variable selectivity mechanisms and stringency across GPCRs34,35. Our results argue against the specific hypothesis that Gi/o-coupled receptors generally reject Gs proteins because these receptors cannot accommodate the bulky C terminus of Gαs22. Most of the Gi/o-coupled receptors that we studied bound to Gs 4A in the presence of agonist. Once again there were exceptions to this trend (e.g. 5-HT5A), further reinforcing the notion that different receptors have different selectivity mechanisms.
Certain caveats should be considered when interpreting these results. First, we used mutant G proteins with features that are predicted to mimic receptor-bound G proteins to infer what likely occurs during the process of GPCR-G protein coupling. An obvious limitation of this approach is that receptor-bound wild-type G proteins may differ from nucleotide-decoupled G proteins in unpredictable ways. A second limitation is that this approach does not directly assess the intermediate-state complexes that we suggest play a role in coupling selectivity, but instead indirectly assesses the importance of such complexes by bypassing them. We believe these are acceptable tradeoffs given that the entire process of receptor-G protein coupling is not readily visualized by more direct methods. It is also worth considering why, in previous studies35, we found that wild-type G proteins maintain more stringent coupling selectivity under nucleotide-free conditions that what we observed with nucleotide-decoupled G proteins. We suspect that one reason for this difference is that nucleotide-free wild-type G proteins will sample states where the hydrophobic core is intact, whereas the mutants we studied have the hydrophobic core permanently disrupted. In other words, formation of receptor-G protein complexes with already-empty G proteins will still need to surmount an energy barrier associated with disruption of the G protein hydrophobic core, albeit a lowered barrier due to the absence of GDP14. A second possible reason for the greater selectivity observed with wild-type G proteins in nucleotide-free conditions is the likelihood that some G proteins will still be bound to GDP even in the presence of apyrase, as spontaneous GDP release can be quite slow when Gβγ dimers are present36,37. Supporting the latter hypothesis is the finding that the more modest promiscuity that was observed with wild-type G proteins in the absence of nucleotides was greatest for Gi1 heterotrimers, which have the fastest rate of spontaneous GDP release among the heterotrimers tested38. Indeed, every receptor sampled in a previous study was able to interact with Gi1 in an agonist-dependent manner under nucleotide-free conditions, including receptors that cannot productively activate Gi135. In this respect these prior results are also consistent with a model wherein coupling selectivity is partly due to the conformational changes associated with nucleotide release. Finally, our sample of GPCRs was not comprehensive and focused largely on family A receptors. It is possible that individual receptors or groups of receptors will be found that interact with nucleotide-decoupled G proteins with a high degree of selectivity, which would imply that a different selectivity mechanism is utilized by these receptors.
One somewhat surprising observation was that nucleotide-decoupled G proteins spontaneously formed complexes with cognate GPCRs. Moreover, agonist activation was only modestly effective at increasing BRET between some receptors and 4A mutants, and inverse agonists were only partially effective at decreasing this signal. These results imply that nucleotide-decoupled G proteins can be highly effective at stabilizing the active state of cognate GPCRs. Almost all of the bona fide GPCRs that we studied showed robust constitutive signals with nucleotide-decoupled G proteins. This finding suggests that most ligand-activated GPCRs frequently sample active states, even when no ligands are present. This is not unexpected for receptors known to constitutively activate G proteins, but our results suggest this property extends even to receptors with low constitutive signaling activity. We made use of this finding to probe a collection of orphan GPCRs and found that basal interaction with 4A mutants correlated well with GDP-sensitive coupling to wild-type G proteins. Most of the oGPCRs that did not couple detectably with wild-type G proteins also did not interact with 4A mutants. Two exceptions to this were GPR31 and GPR157, both of which showed PTX-sensitive interactions with Gi1 4A. These results agree with previous studies suggesting that GPR31 couples to Gi/o proteins39, but contrast with a report suggesting that GPR157 may activate Gq/11 proteins40. Most of the oGPCRs that failed to interact well with 4A mutants have not been shown to activate G proteins. One exception is GPR18, which preferred Gi1 4A but did not meet our established threshold (Fig. 6). This receptor is similar to cannabinoid receptors and has been shown to couple to Gi/o heterotrimers41. Likewise, neither of the aGPCRs in our sample that failed to interact with 4A mutants (ADGRA2 and ADGRA3) have been shown to activate G proteins. Instead, both of these receptors can recruit disheveled (DVL) to the plasma membrane42,43, suggesting that these adhesion receptors may function in a manner similar to Frizzled receptors. Our results imply that many of the proteins that did not interact with 4A mutants may function in a G protein-independent fashion. However, it is possible that such receptors will ultimately be shown to activate G proteins in response to ligands or in the presence of other cofactors.
In summary, nucleotide-decoupled G proteins bind promiscuously to agonist-activated GPCRs, but retain a preference for cognate receptors. To our knowledge this represents the first perturbation that degrades GPCR-G protein selectivity across receptors and G protein families, likely because these mutations enhance the conformational flexibility of the G protein instead of reshaping the receptor-G protein interface. Our findings are consistent with a model where coupling selectivity is determined at several steps during receptor-catalyzed nucleotide exchange, at several sites across an evolving receptor-G protein interface, and by mechanisms that differ widely between receptors.
Methods
Materials
Trypsin, culture media, PBS, DPBS, penicillin/streptomycin and L-glutamine were from GIBCO (ThermoFisher Scientific, Waltham, MA, USA). PEI MAX was purchased from Polysciences Inc. (Warrington, PA, USA). Digitonin, apyrase and GDP were purchased from MilliporeSigma (St. Louis, MO, USA). Coelenterazine h was purchased from Nanolight Technologies (Pinetop, AZ, USA) and furimazine (NanoGlo) was purchased from Promega (Madison, WI, USA). [3H]-CGP12177 was from Perkin-Elmer (Waltham, MA, USA).
Plasmid DNA constructs
GPCR coding sequences were provided by Bryan Roth (University of North Carolina, Chapel Hill, NC; PRESTO-Tango Kit—#1000000068, Addgene, Watertown), MA, USA)44, except for GPR139, which was a gift from Kirill Martemyanov. For each class A receptor the coding sequence was amplified with a common forward primer (corresponding to a cleavable signal sequence) and custom reverse primer (corresponding to the receptor C terminus) and ligated into a pRluc8-N1 cloning vector. For each aGPCR a custom forward primer corresponding to the Stachel peptide preceded by an initiating methionine was used with a custom reverse primer corresponding to the receptor C terminus. Venus-1–155-Gγ2, Venus-155–239-Gβ1, and memGRK3ct-Rluc8 were described previously19. Gα subunit plasmids were purchased from cdna.org (Bloomsburg University, Bloomsburg, PA. 4A insertion and R208S (H2.04) mutations were introduced to Gα subunits using the QuikChange Mutagenesis Kit (Agilent Technologies) and oligonucleotides (Integrated DNA Technologies) as primers. Gαi1 4A was kindly provided by Heidi Hamm (Vanderbilt University, Nashville, TN). Midi Gs was created by replacing NES-Venus in NES-Venus-mGs45 with the αN helix of Gαs (MGCTLSSKTEDQRNEEKAQREANKK) bearing mutations (LGN>TLS) that direct N-myristoylation. Midi Gs wtct was created by reverting AH5.4 in midi Gs to isoleucine using QuikChange Mutagenesis. Dominant negative Gαs and Gαi subunits were kindly provided by Cheng Zhang (University of Pittsburgh, Pittsburgh, PA), and included mutations S54N (H1.02), G226A (s3h2.02), E268A (H3.04), N271K (H3.07), K274D (H3.10), R280K (H3.16), T284D (h3s5.02), I285T (h3s5.03) for DN Gαs and S47N (H1.02), G203A (s3h2.02), E245A (H3.04), A326S (s6h5.03) for DN Gαi. A plasmid encoding the Nluc-EPAC-VV cAMP sensor was kindly provided by Kirill Martemyanov (Scripps Research Institute, Jupiter, FL). Venus-kras and Venus-PTP1bct were described previously46. All plasmid constructs were verified by Sanger sequencing. A plasmid encoding the S1 subunit of pertussis toxin (PTX-S1) was kindly provided by Stephen R. Ikeda (NIAAA, Rockville, MD, USA).
Cell culture and transfection
HEK 293 cells (ATCC) were propagated in plastic flasks and on 6-well plates according to the supplier’s protocol. HEK 293 cells with targeted deletion of GNAS, GNAL, GNAQ, GNA11, GNA12, and GNA13 (G protein three family knockouts; 3GKO) were derived, authenticated and propagated as previously described17. Cells were transfected in growth medium using linear polyethyleneimine MAX (PEI MAX; MW 40,000) at an nitrogen/phosphate ratio of 20 and were used for experiments 24–48 hours later. Up to 3.0 μg of plasmid DNA was transfected in each well of a 6-well plate.
Measurement of coupling between receptors and G proteins
For G protein coupling in intact cells, HEK cells were transfected with a GPCR-Rluc8, Gα WT or 4A subunit, Venus-1–155-Gγ2, Venus-155–239-Gβ1, and pcDNA3.1(+) or PTX-S1 in a (1:10:5:5:5) ratio for a total of 2.6 μg of plasmid DNA in each well of a 6-well plate. After a 24–48-h incubation, cells were washed twice in PBS and harvested by trituration in DPBS. Measurements were made from intact cells with either agonist, inverse agonist or DPBS control.
For G protein coupling in nucleotide-depleted cells, 3GKO cells were transfected with the same plasmids and ratio for the intact cell G protein coupling experiments. After a 48-h incubation, cells were washed twice with permeabilization buffer (KPS) containing 140 mM KCl, 10 mM NaCl, 1 mM MgCl2, 0.1 mM Potassium EGTA, 20 mM NaHEPES (pH 7.2); harvested by trituration; permeabilized in KPS buffer containing 10 μg mL−1 high-purity digitonin; and transferred to opaque black 96-well plate. Measurements were made from permeabilized cells supplemented either with 100 μM GDP or 2U mL−1 apyrase, in both cases with either agonist, inverse agonist or KPS control.
GPCR competition assays
Cells were transfected with an untagged GPCR, GPCR-Rluc8, Gα 4A, Venus-1–155-Gγ2, and Venus-155–239-Gβ1 in a (3:3:8:4:4) ratio for a total of 2.2 μg of plasmid DNA in each well of a 6-well plate. After a 48-h incubation, cells were washed twice with DPBS, harvested by trituration, and transferred to opaque white 96-well plates.
Nluc-EPAC-VV cAMP assay
For testing Gs activation by Gi-coupled receptors (Extended Data Fig. 7), 3GKO cells were transfected with a GPCR, Gαs subunit, Gβ1, Gγ2, Nluc-EPAC-VV, pcDNA3.1(+), and PTX-S1 in a (15:5:10:10:1:49:10) ratio. After a 24-h incubation, cells were washed twice with DPBS, harvested by trituration, and transferred to opaque black 96-well plates.
Nucleotide exchange rate assay
Cells were transfected with a memGRK3ct-Rluc8, Gα 4A, Venus-1–155-Gγ2, Venus-155–239-Gβ1, and PTX-S1 in a (3:10:5:5:3) ratio for a total of 2.6 μg of plasmid DNA in each well of a 6-well plate. After a 48-h incubation, cells were washed twice with KPS and harvested by trituration in KPS containing digitonin. Permeabilized cells were supplemented with 2U mL−1 apyrase with or without 10 μM GDPβS for at least 30 minutes. The rate at which free Gβγ-Venus is released upon addition of 10 μM GTPγS in the absence of GDPβS reflects GTPγS incorporation into nucleotide-free Gα, whereas in the presence of GDPβS the rate of GTPγS incorporation and Gβγ-Venus release is limited by GDPβS dissociation (nucleotide exchange).
Bystander BRET trafficking assay
Cells were transfected with GPCR-Rluc8 and either Venus-kras or Venus-PTP1bct in a (1:5) ratio for a total of 1.2 μg of DNA in each well of a 6-well plate. After 24 hours cells were washed twice in PBS and harvested by trituration in DPBS. Bystander BRET signals with the plasma membrane marker Venus-kras indicated trafficking to the plasma membrane (PM), whereas bystander BRET signals with the endoplasmic reticulum (ER) marker Venus-PTP1bct indicated retention in the endoplasmic reticulum. The decoy receptors and oGPCRs with subthreshold basal BRET to 4A mutants (Fig. 6) all trafficked well to the plasma membrane, as indicated by PM/ER BRET ratios that exceeded 1 (Extended Data Fig. 6 Source Data).
BRET measurements
Steady-state BRET and luminescence measurements were made using a Mithras LB940 photon-counting plate reader (Berthold Technologies GmbH) running MicroWin2000 software. Kinetic BRET and luminescence time course measurements were made using a Polarstar Optima plate reader (BMG Labtech) running BMG Optima version 2.20R2 software. Coelenterazine h (5 μM) or furimazine (1:1,000) were added to all wells immediately prior to making measurements with Rluc8 and Nluc, respectively. Raw BRET signals were calculated as the emission intensity at 520–545 nm divided by the emission intensity at 475–495 nm. Net BRET signals were calculated as the raw BRET signal minus the raw BRET signal measured from cells expressing only the Rluc8 donor. Concentration-response curves were fitted to a four-parameter binding logistic model using GraphPad Prism Version 9.2.0 (GraphPad, San Diego, CA, USA).
Radioligand binding
HEK293 cells were seeded into 10 cm dishes for 24 h and then transfected with 5 μg of β2AR-Rluc8, 4 μg of Venus-1–155-Gγ2, 4 μg of Venus-155–239-Gβ1 and 8 μg of Gαs (Gs long), Gs 4A or midi Gs + myr. After 6 h, the cells were split into 24-well plates and cultured for additional 24 h. The cells were incubated with 250 μl of 5 nM [3H]-CGP12177, together with different concentrations of isoproterenol (from 1 nM to 1 mM) for 90 min at 4°C. After washing twice with 500 μl of PBS, cells were treated with 250 μl of 1M NaOH for 1 h at room temperature. Radioactivity was measured by liquid scintillation counting in 2 ml of Ecoscint A (National Diagnostics, Atlanta, GA, USA). Non-specific binding to β2AR-Rluc8 was determined in the presence of 100 μM alprenolol. Data points were fitted to a two-site binding model using GraphPad Prism Version 9.2.0 (GraphPad, San Diego, CA, USA).
Extended Data
Extended Data Fig. 1.

G protein 4A interaction with the ligand-activated M4R is decoupled from nucleotide binding. A) BRET between M4R-Rluc8 and wild-type (wt) Gi1 heterotrimers induced by the agonist acetylcholine (100 μM) in the absence of nucleotides was largely reversed by the addition of GTPγS or GDPβS (mean ± 95% C.I.; n=12). B) The response for Gi1 4A was insensitive to GDPβS and partially sensitive to GTPγS (mean ± 95% C.I.; n=16–20). C) βγ release-deficient Gi1 4A R208S nearly abolished GTPγS sensitivity of the receptor-G protein complex, suggesting that the nucleotide sensitivity of Gi1 4A reflects heterotrimer dissociation (mean ± 95% C.I.; n=12). D) BRET between M4R-Rluc8 and Gs 4A, Gq 4A and G13 4A is largely resistant to GDPβS (mean ± 95% C.I.; n=16); ΔBRET is the change in BRET above or below the baseline.
Extended Data Fig. 2.
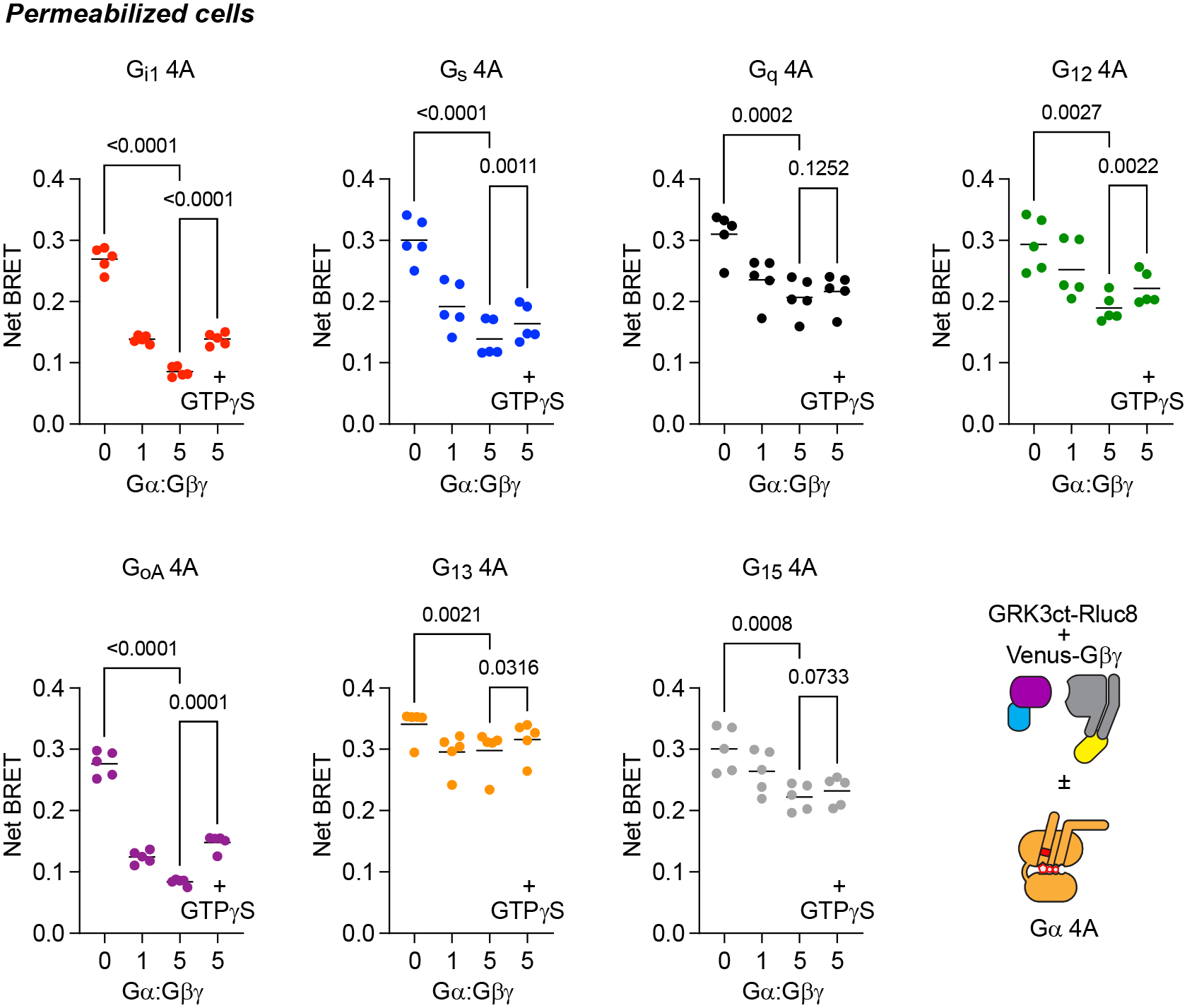
4A mutant Gα subunits sequester Gβγ dimers. BRET between the βγ sensor memGRKct-Rluc8 and Venus-βγ dimers decreased when 4A mutant Gα subunits were expressed, indicating formation of heterotrimers and sequestration of free Gβγ dimers; Gα:Gβγ refers to the ratio of plasmid DNA used for transfection. In most cases Gβγ sequestration was partially reversed by the addition of GTPγS (100 μM). P values represent one-way ANOVA, Dunnett’s multiple comparisons; n=5.
Extended Data Fig. 3:

Nucleotide-decoupled G proteins bind to and promote the active state of GPCRs. A) BRET between M4R and Gi1 4A is blocked by expression of the S1 subunit of pertussis toxin (PTX), and BRET between M4R Gi1 4A and Gs 4A is blocked by deletion of the last five amino acids of each Gα subunit (Δ5); mean ± S.D.; n=3–5. B) BRET between β2AR wild-type (wt) or N322K, which stabilizes the inactive state of the receptor, and Gs wt, Gs 4A, and midi Gs heterotrimers. The inactivating mutation abolished agonist-dependent association with Gs wt and converted spontaneous association with Gs 4A and midi Gs into agonist-dependent association; mean ± S.D.; n=4. C) Agonist displacement of antagonist (CGP 12177) binding in intact cells expressing β2AR-Rluc8 and heterotrimers containing Gs wt, Gs 4A, or midi Gs. Expression of either nucleotide-decoupled G protein increases agonist binding affinity, although midi Gs appears to convert a higher fraction of the binding sites to a high affinity (active) state. Curves were fit to a two-state competition binding model; mean ± S.D.; n=3. All experiments were performed using intact cells, and A and B monitored BRET between Rluc8-tagged receptors and Venus-βγ.
Extended Data Fig. 4.

Concentration-response curves of BRET between six different 4A G protein heterotrimers and 32 Rluc8-tagged GPCRs in response to agonists; mean ± S.E.M.; n=3. Data points are fitted to a 4-parameter variable-slope model. Receptors are arranged according to their putative primary G protein coupling families; when available, IUPHAR primary and secondary G protein coupling (primary, secondary) is shown in parentheses beside each receptor name. All experiments were performed using intact cells.
Extended Data Fig. 5.

Noncognate receptor-G protein 4A complexes are less stable than cognate complexes. A) Time course of complex dissociation in intact cells expressing M4R-Rluc8 and various 4A heterotrimers; receptors were preactivated with 100 μM acetylcholine (Ach) and complex dissociation was triggered by rapid addition of 10 μM atropine. Traces represent BRET normalized to baseline prior to addition of atropine and are the average of 12 replicates from 3 independent experiments. B) Time course of complex dissociation in intact cells in response to activation of unlabeled α2AR by rapid addition UK 14,304 (UK; 1 μM) in the continuous presence of 100 μM acetylcholine. Traces represent BRET normalized to baseline prior to addition of UK 14,303 and are the average of 5–9 replicates from 3 independent experiments. In both cases cognate (Gi1 and Go 4A) complexes dissociate more slowly than noncognate complexes. All experiments were performed using intact cells.
Extended Data Fig. 6.

Receptor interaction with nucleotide-decoupled 4A mutants is promiscuous compared to G protein activation. Comparison of normalized maximum BRET (Emax) between receptors and 4A heterotrimers and G protein activation assessed using effector membrane translocation assays (EMTA) as reported by Avet and colleagues1, in both cases in the presence of saturating concentrations of agonists.
Extended Data Fig. 7.

Gi-coupled receptors accommodate the C terminus of nucleotide-decoupled Gs heterotrimers yet cannot activate wild-type Gs. A) Agonist-dependent BRET between Rluc8-tagged Gi-coupled receptors α2AR (top) and M4R (bottom) and Gs 4A heterotrimers is nearly abolished when Gαs 4A is truncated by removing 5 amino acids from the C terminus (Δ5); mean ± S.D. n=3–5. B) Neither of these receptors is capable of stimulating adenylyl cyclase by activating wild-type Gs. Cyclic AMP (cAMP) was monitored using an EPAC-based BRET sensor; α2AR was stimulated with 1 μM UK 14,304 (UK) and M4R was stimulated with 10 μM oxotremorine M (OXO) agonist-induced activation; responses to 10 μM forskolin (FSK) are shown for comparison; *****P < 0.05, one-way ANOVA with Dunnett’s multiple comparisons; ns, not significant.
Extended Data Fig. 8.

Dominant negative (DN) G proteins have a milder nucleotide decoupling phenotype. A) In permeabilized cells association of heterotrimers containing DN Gαs and Gαi1 with their cognate receptors (β2AR and α2AR) are both agonist- and nucleotide-dependent; mean ± S.D. n=5. B) DN Gs and Gi1 heterotrimers associated with noncognate receptors only modestly in the absence of nucleotides; mean ± S.D. n=4.
Extended Data Fig. 9.

Screening of orphan GPCRs with G15 4A. A) Agonist-dependent BRET between M3R-Rluc8 and G15 4A heterotrimers is insensitive to GDPβS (100 μM) at steady-state (left; mean ± S.D.; n=3) or after rapid addition (right; mean ± 95% C.I.; n=12). B) Heat map of basal BRET between the indicated oGPCRs and G protein 4A heterotrimers. Receptors shown in red bold italic font failed to reach the arbitrary net BRET threshold of 0.1 with any 4A heterotrimer; the promiscuous receptor MRGPRD is shown as a positive control. Each cell represents the mean of at least three independent experiments.
Extended Data Fig. 10.
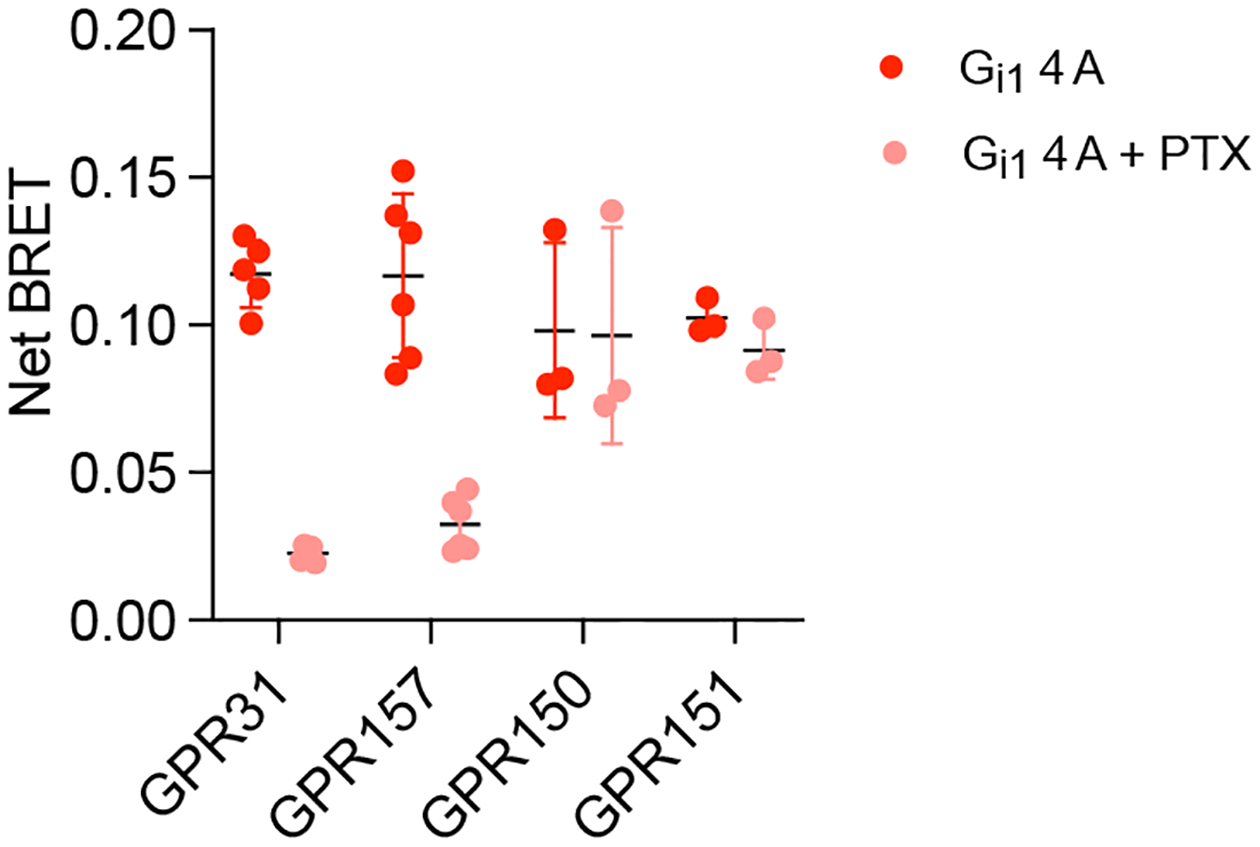
Pertussis toxin (PTX) sensitivity of basal BRET between oGPCRs and Gi1 4A. Shown is basal BRET (mean ± S.D.) between oGPCR-Rluc8 receptors (also indicated on Figure 6) and Gi1 4A in the absence and presence of coexpressed PTX.
Acknowledgments
We thank Aska Inoue for providing CRISPR-modified cells lacking Gα subunits, and Heidi Hamm, Steve Ikeda, Kirill Martemyanov, Bryan Roth and Cheng Zhang for providing plasmid DNA. This study was supported by NIH grants GM130142 and GM145284 (N.A.L.), GM136397 (G.W.) and a PhRMA Foundation Predoctoral Fellowship in Drug Discovery (W.J.).
Footnotes
Competing interests
The authors declare no competing interests.
Data availability
All data generated or analyzed during this study are included in this published article and the accompanying Source Data files. Plasmids encoding GPCR-Rluc8 and nucleotide-decoupled G protein constructs used in this study are freely available upon request. Structures used to construct Figure 1 are deposited in the Protein Database (PDB) at https://www.rcsb.org/.
References
- 1.Pierce KL, Premont RT & Lefkowitz RJ Seven-transmembrane receptors. Nat Rev Mol Cell Biol 3, 639–650 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Gilman AG G proteins: transducers of receptor-generated signals. Annual Review of Biochemistry 56, 615–649 (1987). [DOI] [PubMed] [Google Scholar]
- 3.Wess J Molecular basis of receptor/G-protein-coupling selectivity. Pharmacology & Therapeutics 80, 231–264 (1998). [DOI] [PubMed] [Google Scholar]
- 4.Conklin BR, Farfel Z, Lustig KD, Julius D & Bourne HR Substitution of three amino acids switches receptor specificity of Gqα to that of Giα. Nature 363, 274–276 (1993). [DOI] [PubMed] [Google Scholar]
- 5.Qiao A et al. Structural basis of G(s) and G(i) recognition by the human glucagon receptor. Science 367, 1346–1352 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Mobbs JI et al. Structures of the human cholecystokinin 1 (CCK1) receptor bound to Gs and Gq mimetic proteins provide insight into mechanisms of G protein selectivity. PLOS Biology 19, e3001295–e3001295 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rasmussen SGF et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harris JA et al. Selective G protein signaling driven by substance P-neurokinin receptor dynamics. Nat Chem Biol 18, 109–115 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mahoney JP & Sunahara RK Mechanistic insights into GPCR–G protein interactions. in Current Opinion in Structural Biology Vol. 41 247–254 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu X et al. Structural Insights into the Process of GPCR-G Protein Complex Formation. Cell 177, 1243–1251.e12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Du Y et al. Assembly of a GPCR-G Protein Complex. Cell 177, 1232–1242.e11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kato HE et al. Conformational transitions of a neurotensin receptor 1–Gi1 complex. Nature 572, 80–85 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaya AI et al. A Conserved Hydrophobic Core in Gα subi1/sub Regulates G Protein Activation and Release from Activated Receptor. Journal of Biological Chemistry 291, 19674–19686 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dror RO et al. SIGNAL TRANSDUCTION. Structural basis for nucleotide exchange in heterotrimeric G proteins. Science (New York, N.Y.) 348, 1361–1365 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alexander NS et al. Energetic analysis of the rhodopsin-G-protein complex links the α5 helix to GDP release. Nat Struct Mol Biol 21, 56–63 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flock T et al. Universal allosteric mechanism for Gα activation by GPCRs. Nature 524, 173–179 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grundmann M et al. Lack of beta-arrestin signaling in the absence of active G proteins. Nature Communications 9, 341–341 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knight KM et al. A universal allosteric mechanism for G protein activation. Mol Cell 81, 1384–1396.e6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hollins B, Kuravi S, Digby GJ & Lambert NA The c-terminus of GRK3 indicates rapid dissociation of G protein heterotrimers. Cellular Signalling 21(2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kooistra AJ et al. GPCRdb in 2021: Integrating GPCR sequence, structure and function. Nucleic Acids Research 49, D335–D343 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Avet C et al. Effector membrane translocation biosensors reveal G protein and βarrestin coupling profiles of 100 therapeutically relevant GPCRs. Elife 11, e74101 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rose AS et al. Position of Transmembrane Helix 6 Determines Receptor G Protein Coupling Specificity. Journal of the American Chemical Society 136, 11244–11247 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Kang Y et al. Cryo-EM structure of human rhodopsin bound to an inhibitory G protein. Nature 558, 553–558 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.García-Nafría J, Nehmé R, Edwards PC & Tate CG Cryo-EM structure of the serotonin 5-HT1B receptor coupled to heterotrimeric Go. Nature 558, 620–623 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koehl A et al. Structure of the μ-opioid receptor–Gi protein complex. Nature 558, 547–552 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nehmé R et al. Mini-G proteins: Novel tools for studying GPCRs in their active conformation. PLOS ONE 12, e0175642–e0175642 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carpenter B & Tate CG Engineering a minimal G protein to facilitate crystallisation of G protein-coupled receptors in their active conformation. Protein Engineering Design and Selection 29, 583–594 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang YL et al. Dominant Negative G Proteins Enhance Formation and Purification of Agonist-GPCR-G Protein Complexes for Structure Determination. ACS Pharmacol Transl Sci 1, 12–20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yao XJ et al. The effect of ligand efficacy on the formation and stability of a GPCR-G protein complex. Proceedings of the National Academy of Sciences of the United States of America 106, 9501–9506 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gregorio GG et al. Single-molecule analysis of ligand efficacy in β2AR-G-protein activation. Nature 547, 68–73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Graham GJ D6 and the atypical chemokine receptor family: novel regulators of immune and inflammatory processes. Eur J Immunol 39, 342–51 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Lu S, Jang W, Inoue A & Lambert NA Constitutive G protein coupling profiles of understudied orphan GPCRs. PloS one 16, e0247743–e0247743 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vizurraga A, Adhikari R, Yeung J, Yu M & Tall GG Mechanisms of adhesion G protein-coupled receptor activation. Journal of Biological Chemistry 295, 14065–14083 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flock T et al. Selectivity determinants of GPCR-G-protein binding. Nature 545, 317–322 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okashah N et al. Variable G protein determinants of GPCR coupling selectivity. Proceedings of the National Academy of Sciences of the United States of America 116, 12054–12059 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brandt DR & Ross EM GTPase activity of the stimulatory GTP-binding regulatory protein of adenylate cyclase, Gs. Accumulation and turnover of enzyme-nucleotide intermediates. Journal of Biological Chemistry 260, 266–72 (1985). [PubMed] [Google Scholar]
- 37.Higashijima T, Ferguson KM, Sternweis PC, Smigel MD & Gilman AG Effects of Mg2+ and the beta gamma-subunit complex on the interactions of guanine nucleotides with G proteins. Journal of Biological Chemistry 262, 762–6 (1987). [PubMed] [Google Scholar]
- 38.Stoveken HM, Hajduczok AG, Xu L & Tall GG Adhesion G protein-coupled receptors are activated by exposure of a cryptic tethered agonist. Proc Natl Acad Sci U S A 112, 6194–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin AL, Steurer MA & Aronstam RS Constitutive Activity among Orphan Class-A G Protein Coupled Receptors. PLoS One 10, e0138463 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takeo Y, Kurabayashi N, Nguyen MD & Sanada K The G protein-coupled receptor GPR157 regulates neuronal differentiation of radial glial progenitors through the Gq-IP3 pathway. Sci Rep 6, 25180 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kohno M et al. Identification of N-arachidonylglycine as the endogenous ligand for orphan G-protein-coupled receptor GPR18. Biochem Biophys Res Commun 347, 827–32 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Li X et al. Gpr125 modulates Dishevelled distribution and planar cell polarity signaling. Development 140, 3028–39 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eubelen M et al. A molecular mechanism for Wnt ligand-specific signaling. Science 361, eaat1178 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Kroeze WK et al. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nature Structural & Molecular Biology 22, 362–369 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wan Q et al. Mini G protein probes for active G protein–coupled receptors (GPCRs) in live cells. Journal of Biological Chemistry 293, 7466–7473 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lan T-H, Liu Q, Li C, Wu G & Lambert NA Sensitive and high resolution localization and tracking of membrane proteins in live cells with BRET. Traffic (Copenhagen, Denmark) 13, 1450–1456 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article and the accompanying Source Data files. Plasmids encoding GPCR-Rluc8 and nucleotide-decoupled G protein constructs used in this study are freely available upon request. Structures used to construct Figure 1 are deposited in the Protein Database (PDB) at https://www.rcsb.org/.