

Abstract
BACKGROUND
Idiopathic CD4 lymphocytopenia (ICL) is a clinical syndrome that is defined by CD4 lymphopenia of less than 300 cells per cubic millimeter in the absence of any primary or acquired cause of immunodeficiency. Some 30 years after its original identification, ICL has remained a disease of obscure cause, with limited evidence with respect to its prognosis or management, despite diagnostic and therapeutic innovations.
METHODS
We evaluated the clinical, genetic, immunologic, and prognostic characteristics of 108 patients who were enrolled during an 11-year period. We performed whole-exome and targeted gene sequencing to identify genetic causes of lymphopenia. We also performed longitudinal linear mixed-model analyses of T-cell count trajectories and evaluated predictors of clinical events, the response to immunization against coronavirus disease 2019 (Covid-19), and mortality.
RESULTS
After the exclusion of patients with genetic and acquired causes of CD4 lymphopenia, the study population included 91 patients with ICL during 374 person-years of follow-up. The median CD4+ T-cell count among the patients was 80 cells per cubic millimeter. The most prevalent opportunistic infections were diseases related to human papillomavirus (in 29%), cryptococcosis (in 24%), molluscum contagiosum (in 9%), and nontuberculous mycobacterial diseases (in 5%). A reduced CD4 count (<100 cells per cubic millimeter), as compared with a CD4 count of 101 to 300 cells, was associated with a higher risk of opportunistic infection (odds ratio, 5.3; 95% confidence interval [CI], 2.8 to 10.7) and invasive cancer (odds ratio, 2.1; 95% CI, 1.1 to 4.3) and a lower risk of autoimmunity (odds ratio, 0.5; 95% CI, 0.2 to 0.9). The risk of death was similar to that in the age- and sex-adjusted general population, but the prevalence of cancer was higher.
CONCLUSIONS
Among the study patients, ICL continued to be associated with increased susceptibility to viral, encapsulated fungal, and mycobacterial diseases, as well as with a reduced response to novel antigens and an increased risk of cancer. (Funded by the National Institute of Allergy and Infectious Diseases and the National Cancer Institute; ClinicalTrials.gov number, NCT00867269.)
Idiopathic cd4 lymphocytopenia (icl) was initially described in 1992 after opportunistic infections that are known to be associated with human immmunodeficiency virus (HIV) infection were identified in patients with low CD4+ T-cell counts and negative HIV tests.1–5 In 1993, the ensuing investigation by the Centers for Disease Control and Prevention formulated the syndromic definition of ICL as a rare, idiopathic disease without evidence of familial linkage or a transmissible agent.6,7 Since then, the diagnosis of ICL has been defined as a CD4 count of less than 300 cells per cubic millimeter measured at least twice 6 weeks apart in the absence of any disease or therapy that may be associated with lymphopenia.
The discovery of ICL was contemporaneous to the identification of HIV and acquired immunodeficiency syndrome (AIDS) and was portrayed as “AIDS without HIV” on the basis of the presence of similar presenting opportunistic infections and a low CD4+ T-cell count but a negative HIV test. Today, 30 years later, HIV infection has become a chronic disease that is managed effectively with antiretroviral therapy with a nearly normal life expectancy.8 In sharp contrast, ICL has remained a disease of obscure cause, with limited evidence with respect to prognosis or management, despite diagnostic and therapeutic innovations.
The rarity and perceived heterogeneity of ICL cases have posed challenges to addressing the pathophysiological features, prognosis, and therapeutic strategies. Few studies have highlighted the main clinical and immunologic characteristics of this disease.9–11 More recently, the identification of ICL in infancy has added a new dimension to the syndrome by raising hopes of ascertaining potential genetic components.12,13 To evaluate the current knowledge regarding ICL and its clinical implications, we performed an observational study, called the Etiology, Pathogenesis, and Natural History of Idiopathic CD4 Lymphocytopenia, to describe the disease presentation, genetic workup, immunologic profile, and prognostic factors in a cohort of patients with ICL.
METHODS
PATIENTS
From January 2009 through March 2020, patients with ICL were enrolled in the study, which was approved by the internal review board at the National Institutes of Health (NIH). All the patients were seen at the NIH Clinical Center for all study visits at screening, baseline, follow-up, and ad hoc evaluations for incidental clinical needs.
Eligible patients were adults (≤18 years of age) with a CD4+ T-cell count of less than 300 cells per cubic millimeter or less than 20% of total T lymphocytes at the screening visit and on at least two previous occasions at least 6 weeks apart in the absence of any disease or therapy that may cause lymphopenia, as outlined in the protocol, available with the full text of this article at NEJM.org. Additional inclusion and exclusion criteria and all clinical and laboratory assessments that were performed at screening and at follow-up are described in the Supplementary Appendix, also available at NEJM.org. Ongoing care by a referring physician was required to coordinate care and access to clinical records for events that occurred before enrollment or outside of NIH study visits. All the patients provided written informed consent.
IMMUNOLOGIC STUDIES
We measured the number of CD4+ and CD8+ T cells, B cells, and natural killer (NK) cells on flow cytometry. Serologic analysis for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was performed with the use of the Elecsys SARS-CoV-2 Antigen assay (Roche Diagnostics).
STATISTICAL ANALYSIS
We performed two-tailed nonparametric tests to compare ranks between groups. We divided lymphocyte counts into thirds from the overall distribution of T cells and NK cells to evaluate the potential prognostic value of such numbers, according to measurements at study entry. To determine the association between the thirds of lymphocyte counts and a clinical condition associated with ICL, we performed Firth’s method of bias-reduced logistic-regression analysis14 using multiple outputation.15 To divide the distribution of lymphocyte counts into thirds, we used the cutoff points of 42 and 128 CD4+ T cells, 91 and 234 CD8+ T cells, and 120 and 167 NK cells per cubic millimeter; a predefined cutoff of 100 CD4+ T cells per cubic millimeter was used in a sensitivity analysis. Longitudinal linear mixed models were used to evaluate the trajectories of CD4+ and CD8+ T-cell counts.
We used a one-sided Poisson test to evaluate the difference in the prevalence and associated mortality between patients with ICL and the general population in the Surveillance, Epidemiology, and End Results (SEER) Program as matched according to age and sex. A P value of less than 0.05 was considered to indicate statistical significance.
RESULTS
PATIENTS
A total of 108 patients were enrolled in the study. Of these patients, 17 were excluded from our analysis: 5 who had a CD4+ T-cell count that was inconsistent with ICL, 5 who had an alternative diagnosis (common variable immunodeficiency in 2 patients, lymphodepleting chemotherapy for lymphoma in 1 patient, autoimmune disease in 1 patient, and Crohn’s disease with CD4+ T-cell recovery after treatment with a tumor necrosis factor α blocker in 1 patient), withdrawal of consent in 1 patient, and 6 cases of inborn errors of immunity16 with pathogenic variants in NFKB1, PI3KCD, FAS,17 IL2RG,18 DOCK8, and CD419 after genetic workup that was performed in 84 study patients (80 by whole-exome sequencing and 4 by targeted-gene-panel sequencing).20 Of the remaining 91 evaluable patients, 80 remained in the study through March 2020, whereas 11 patients who provided updated clinical information remotely were included in the analysis up to their last visit.
The baseline demographic and clinical characteristics of the patients are provided in Table 1. The median age at enrollment was 48 years (interquartile range, 36 to 59); 49% of the patients were women. Most of the patients were White (88%); 69% had an infectious complication at enrollment or during follow-up, and 36% had an autoimmune condition. The median cell counts per cubic millimeter at enrollment were as follows: CD4+ T cells, 80 (interquartile range, 25 to 168); CD8+ T cells, 130 (interquartile range, 58 to 317); NK cells, 142 (interquartile range, 100 to 378); and B cells, 144 (interquartile range, 92 to 218). Both CD4+ and CD8+ T-cell counts (and consequently, total CD3+ T-cell counts) were lower in the study patients than in healthy adults, but levels of NK and B cells were similar in the two groups. The median number of visits per patient was 7 (interquartile range, 7 to 14), with a median follow-up of 3 years (interquartile range, 0 to 6) and a total follow-up of 374 person-years.
Table 1.
Characteristics of the 91 Study Patients.*
| Characteristic | Value | Reference Range |
|---|---|---|
| Demographic features | ||
| Median age (IQR) — yr | ||
| At presentation | 48 (36–59) | — |
| At end of follow-up | 54 (38–62) | — |
| Sex — no. (%) | ||
| Male | 46 (51) | — |
| Female | 45 (49) | — |
| Race — no. (%)† | ||
| White | 86 (95) | — |
| Black | 5 (5) | — |
| Medical history | ||
| Median duration of follow-up (range) — yr | 3 (0–6) | — |
| General infectious complications — no. (%) | 63 (69) | — |
| Autoimmune clinical manifestations — no. (%)‡ | 33 (36) | — |
| Median white-cell count (IQR) — cells/mm3 | 4470 (3850–5480) | 4230–9070 |
| Median lymphocytes (IQR) | ||
| Absolute count — cells/mm3 | 650 (480–880) | 1320–3570 |
| Percentage of white-cell count | 15 (10–21) | 22–53 |
| Median T cells (IQR) — cells/mm3 | ||
| CD3 | 340 (154–565) | 714–2266 |
| CD4 | 80 (25–168) | 359–1565 |
| CD8 | 130 (58–317) | 178–853 |
| CD3+CD4−CD8− | 34 (15–64) | 24–223 |
| Median NK cells (IQR) — cells/mm3 | 142 (100–378) | 126–729 |
| Median B cells (IQR) — cells/mm3 | 144 (92–218) | 61–320 |
| Median immunoglobulin level (IQR) — mg/dl | ||
| IgG | 952 (713–1210) | 700–1600 |
| IgM | 89 (64–136) | 40–230 |
| IgA | 146 (63–196) | 80–350 |
The patients’ demographic characteristics were recorded at the time of enrollment unless otherwise specified (e.g., age at the end of follow-up). The medical history, cell counts, and immunoglobulin levels were recorded between enrollment and the end of follow-up or data censoring on March 2020. IQR denotes interquartile range, and NK natural killer.
Race was reported by the patients.
Autoimmune clinical manifestations were as follows: autoimmune thyroid diseases in 12 patients, sarcoidosis in 5, psoriatic rash in 4, psoriatic arthritis in 2, autoimmune hemolytic anemia in 3, immune thrombocytopenic purpura in 3, ulcerative colitis in 2, vitiligo in 2, discoid lupus in 2, alopecia in 2, systemic lupus erythematosus in 2, atopic dermatitis in 1, ankylosing spondylitis in 1, polyarticular rheumatoid arthritis in 1, autoimmune gastritis in 1, and antiphospholipid syndrome in 1. Patients could have more than one autoimmune condition.
INFECTIOUS COMPLICATIONS
Most infectious complications were registered during screening to describe the reason for the identification of CD4 lymphopenia and subsequent ICL diagnosis. To better delineate the clinical spectrum of the infections observed in the patients with ICL as compared with the general population, we categorized the patients as having “any infectious complication,” “an opportunistic infection of clinical significance,” or both. Of the 91 patients, 63 (69%) were categorized as having at least one infectious complication — 58 at screening and 5 during follow-up; 53 patients (58%) — 49 at screening and 4 during follow-up — were categorized as having at least one opportunistic infection of clinical significance (Table 2 and the Supplementary Appendix). We made this distinction because some infectious complications such as dermatomal shingles can occur in the general population and may not necessarily reflect an underlying severe immunologic defect, as compared with opportunistic infections of clinical significance such as cryptococcal meningoencephalitis and multidermatomal shingles.
Table 2.
Infectious Complications in the 91 Study Patients.*
| Infectious Complication | Microorganism | Opportunistic Infection of Clinical Significance† | Any Infectious Complication‡ |
|---|---|---|---|
| number of patients (percent) | |||
| Human papillomavirus–related disease | HPV | 27 (29) | 35 (38) |
|
| |||
| Skin | 6 (7) | 8 (9) | |
|
| |||
| Mucosal disease | 9 (10)§ | 15 (16)¶ | |
|
| |||
| Skin and mucosal disease | 12 (13) | 12 (13) | |
|
| |||
| Cryptococcus disease | Cryptococcus neoformans and C. gattii | 22 (24) | 22 (24) |
|
| |||
| Meningoencephalitis | 16 (18)§ | 16 (18)¶ | |
|
| |||
| Pneumonia | 4 (4)§ | 4 (4)¶ | |
|
| |||
| Disseminated with skin or musculoskeletal involvement | 2 (2) | 2 (2) | |
|
| |||
| Varicella–zoster virus disease | VZV | 2 (2) | 13 (14) |
|
| |||
| Dermatomal zoster | 0 | 11 (12) | |
|
| |||
| Multidermatomal or complicated zoster | 2 (2) | 2 (2) | |
|
| |||
| Molluscum contagiosum | Poxvirus (molluscum contagiosum) | 8 (9) | 8 (9) |
|
| |||
| Histoplasmosis | Histoplasma capsulatum | 4 (4) | 4 (4) |
|
| |||
| Pulmonary | 1 (1) | 1 (1) | |
|
| |||
| Disseminated | 3 (3) | 3 (3) | |
|
| |||
| Nontuberculous mycobacterial diseases | Mycobacterium avium | 5 (5) | 5 (5) |
|
| |||
| Pulmonary | 2 (2) | 2 (2) | |
|
| |||
| Skin or musculoskeletal disease | 1 (1) | 1 (1) | |
|
| |||
| Disseminated | 2 (2)§ | 2 (2)¶ | |
|
| |||
| Progressive multifocal leukoencephalopathy | Human polyomavirus 2 (John Cunningham virus) | 3 (3) | 3 (3) |
|
| |||
| Cytomegalovirus disease | CMV | 2 (2) | 2 (2) |
|
| |||
| Colitis | 1 (1) | 1 (1) | |
|
| |||
| Retinitis | 1 (1) | 1 (1) | |
|
| |||
| Oral or anogenital ulcers | Herpes simplex virus | 0 | 2 (2) |
|
| |||
| Oral candidiasis | Candida albicans | 1 (1) | 1 (1) |
|
| |||
| Kaposi’s sarcoma | Kaposi’s sarcoma–associated herpesvirus | 1 (1) | 1 (1) |
|
| |||
| Pneumocystis pneumonia | Pneumocystis jirovecii | 1 (1) | 1 (1) |
Patients could have more than one type of infection.
Opportunistic infections of clinical significance include cryptococcosis, histoplasmosis, disseminated or pulmonary nontuberculous mycobacterial infections, severe diseases caused by human papillomavirus (HPV), complicated or disseminated varicella–zoster virus (VZV) infections, complicated or disseminated oral or anogenital herpetic infections, cancers associated with oncogenic viral infections (i.e., HPV, Kaposi’s sarcoma–associated herpesvirus, and Epstein–Barr virus), molluscum contagiosum, end-stage organ disease caused by cytomegalovrius (CMV), Pneumocystis jirovecii pneumonia, coccidioidomycosis, and progressive multifocal leukoencephalopathy.
This category includes all the opportunistic infections listed above as well as dermatomal and uncomplicated zoster, localized and uncomplicated oral or anogenital herpetic infections, HPV-related anogenital lesions without evidence of dysplasia, and skin warts (further details provided in the Statistical Analysis section in the Supplementary Appendix).
This opportunistic infection occurred during the follow-up period: a case each of disseminated nontuberculous mycobacterial disease (NTM), HPV-related cervical dysplasia, cryptococcal meningitis, and cryptococcal pneumonia.
This infectious complication occurred during the follow-up period: a case each of disseminated NTM, dermatomal shingles, HPV-related cervical dysplasia, cryptococcal meningitis, and cryptococcal pneumonia.
The most common opportunistic infections were severe skin or anogenital human papillomavirus (HPV)–related diseases (in 27 patients [29%]) and cryptococcosis (in 22 patients [24%]), which presented as meningoencephalitis (in 16 patients [18%]), pneumonia (in 4 patients), and disseminated skin or musculoskeletal diseases (in 2 patients). Eight patients had molluscum contagiosum; other opportunistic infections were disseminated nontuberculous mycobacterial infection (in 2 patients), pulmonary (in 2 patients) or skin or musculoskeletal diseases (in 1 patient), disseminated histoplasmosis (in 2 patients) and pulmonary histoplasmosis (in 2 patients), progressive multifocal leukoencephalopathy (in 3 patients), multidermatomal or complicated (Ramsay Hunt syndrome) shingles (in 2 patients), and cytomegalovirus diseases (colitis and retinitis in 1 patient each). Pneumocystis jirovecii pneumonia (PJP), oral candidiasis, pulmonary coccidioidomycosis, and pulmonary aspergillosis were observed in 1 patient each. A total of 21 of 53 patients (40%) had at least two different opportunistic infections.
LYMPHOCYTE COUNTS AND CLINICAL IMPLICATIONS
We first evaluated whether the probability of having a specific clinical condition was associated with a particular third of CD4+ T-cell counts (Fig. 1). We found that the patients with a CD4+ T-cell count in the lowest third (0 to 42 cells per cubic millimeter) had a risk of having an opportunistic infection that was higher by a factor of 14.6 (95% confidence interval [CI], 6.2 to 47.4) than those in the highest third (128 to 300 cells per cubic millimeter); those in the middle third (42 to 128 cells per cubic millimeter) had a risk that was higher by a factor of 2.3 (95% CI, 1.1 to 5.0). In contrast, the odds of having an autoimmune disease were lower in the lowest and middle thirds of CD4+ T-cell counts — by a factor of 0.4 (95% CI, 0.2 to 0.8) in both thirds — than among those in the highest third. Moreover, the lowest third of CD4+ T-cell counts was associated with a higher risk of any cancer diagnosis (odds ratio, 2.5; 95% CI, 1.1 to 5.4).
Figure 1. Distribution of CD4, CD8, and NK Cell Counts in Patients with Opportunistic Infections, Cancers, and Autoimmune Diseases.
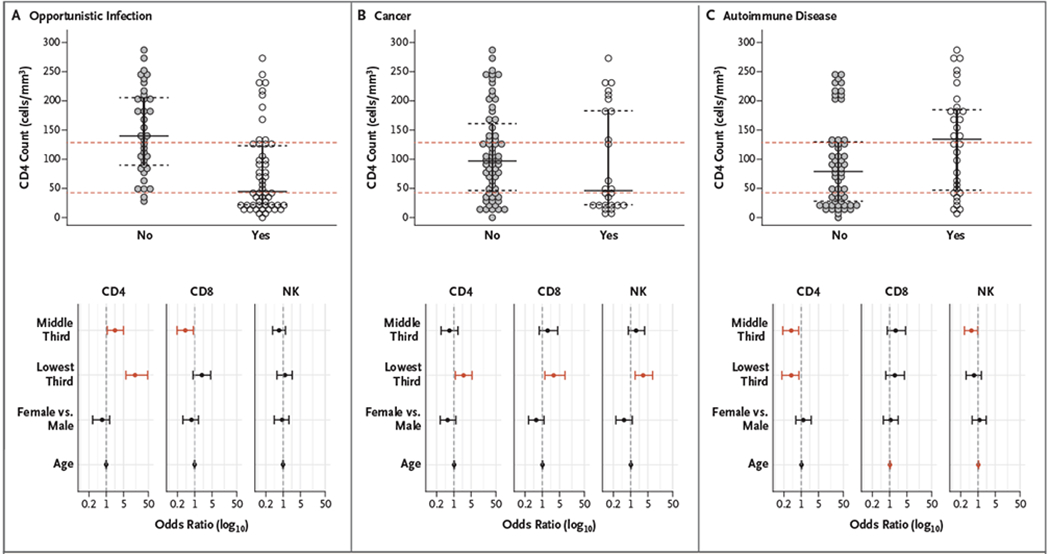
Shown are the median absolute numbers of CD4+ and CD8+ T cells and natural killer (NK) cells in the study patients, according to the presence or absence of opportunistic infection of clinical significance (Panel A), cancer (Panel B), and autoimmune disease (Panel C) (upper panels). The red dashed lines show the cutoffs for the lowest third, middle third, and highest third of the values. In each graph, the horizontal black bar indicates the median value, and the dashed black bars indicate the interquartile range. The lower panels show log-transformed odds ratios for opportunistic infection, cancer, or autoimmune disease among the study patients in the lowest third and middle third of values as compared with the highest third after adjustment for age and sex. Data points in red indicate the odds ratios in which the 95% confidence intervals (represented by error bars) exclude the null value of 1, indicating a significant increase or decrease in the risk of the specified outcome.
To validate our findings and their implications for risk stratification, we performed a sensitivity analysis by using a cutoff of 100 CD4+ T cells per cubic millimeter, a well-recognized clinical value in patients with HIV infection or AIDS. Such analysis showed results similar to those in the original analysis, with a CD4 count of less than 100 cells per cubic millimeter associated with a higher risk of invasive cancer (odds ratio, 2.1; 95% CI, 1.1 to 4.3) and opportunistic infection (odds ratio, 5.3; 95% CI, 2.8 to 10.7) and a lower risk of autoimmunity (odds ratio, 0.5; 95% CI, 0.2 to 0.9) (Fig. S2 in the Supplementary Appendix).
We found that patients with the middle third of CD8+ T-cell counts (91 to 234 cells per cubic millimeter) had lower risk of opportunistic infection than those in the highest third (odds ratio, 0.4; 95% CI, 0.2 to 0.9). The lowest third of CD8+ T-cell counts (0 to 91 cells per cubic millimeter) and NK cells (0 to 120 cells per cubic millimeter) were both associated with a higher risk of cancer than the highest third, with odds ratios of 2.9 (95% CI, 1.3 to 8.3) and 3.2 (95% CI, 1.5 to 7.7), respectively. CD8+ T-cell counts were not associated with autoimmunity, whereas the middle third of NK-cell counts (120 to 167 cells per cubic millimeter) was associated with a lower risk of autoimmunity than the highest third (odds ratio, 0.5; 95% CI, 0.3 to 0.98).
Because a progressive increase in CD4+ T-cell counts to 300 cells per cubic millimeter or more was observed in 2 patients, we assessed the trajectories of CD4+ and CD8+ T cells during the follow-up period (Fig. 2). Our analysis showed a significant, albeit minor, increase of CD4+ and CD8+ T-cell counts by a factor of 1.044 per year (95% CI, 1.030 to 1.059; P<0.001) and 1.031 per year (95% CI, 1.015 to 1.046; P<0.001), respectively. Therefore, if the CD4+ T-cell count was 100 cells per cubic millimeter, an increase to 104 cells per cubic millimeter would be expected in the following year. In contrast to T-cell lymphocytes, there were no significant changes in NK-cell numbers over time.
Figure 2. Absolute Values and Trajectories of CD4 and CD8 Counts during Follow-up.
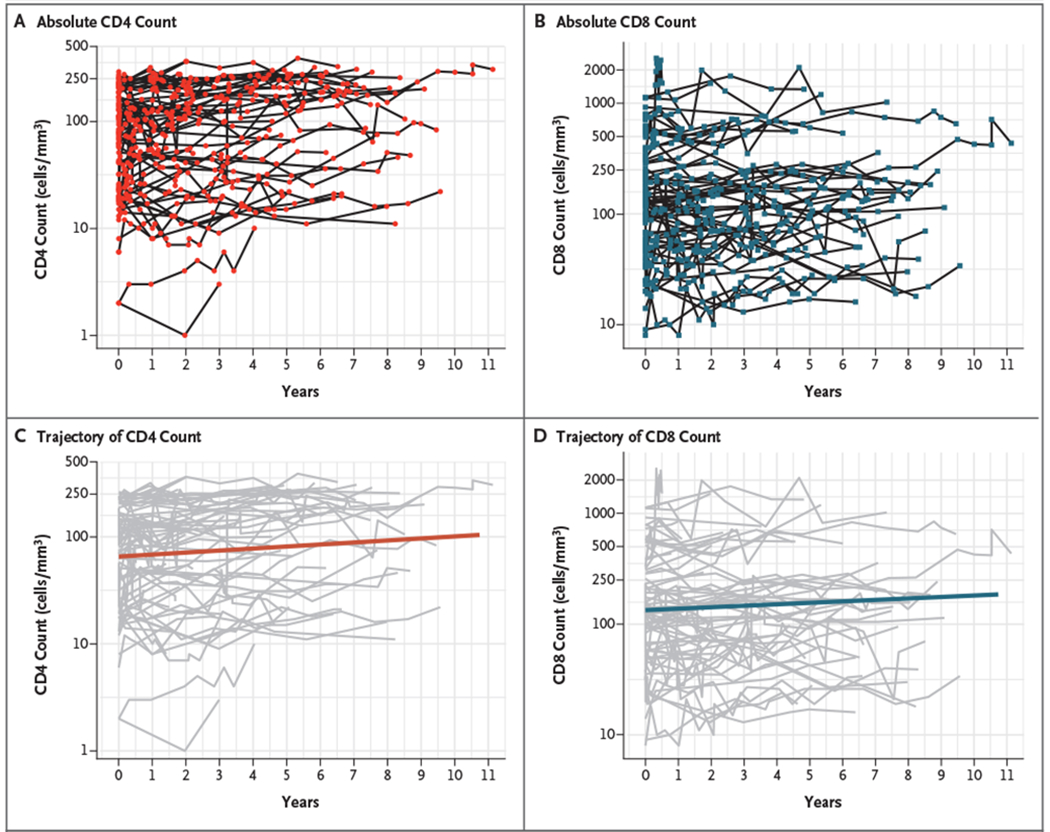
In the upper panels, shown are absolute numbers of CD4+ T cells (Panel A) and CD8+ T cells (Panel B) for all the study patients during 11 years of follow-up. In the lower panels, shown are the longitudinal trajectories of CD4+ T cells (Panel C) and CD8+ T cells (Panel D) over time as evaluated by linear mixed models for longitudinal data.
SEROLOGIC RESPONSE TO SARS-COV-2 IMMUNIZATION
As of October 2021, a total of 22 patients with ICL who had received two doses of SARS-CoV-2 messenger RNA vaccines had been evaluated. Of these patients, 8 (36%) did not have detectable antibodies against the SARS-CoV-2 spike protein. The lack of serologic response was associated with a lower median CD4+ T-cell count at the time of immunization (58 cells per cubic millimeter in those with no response vs. 165 cells per cubic millimeter among those with a response; P<0.001) (Fig. S1). The serologic response was not associated with age, sex, or B-cell count. As of May 2022, a total of 7 patients had received monoclonal-antibody therapy with tixagevimab–cilgavimab for preexposure prophylaxis. SARS-CoV-2 infection had developed in 10 patients, all of whom had mild disease that did not result in hospitalization, oxygen supplementation, or any clinical sequelae.
INCIDENTAL INFECTIOUS COMPLICATIONS, CANCERS OR AUTOIMMUNE DISEASES, AND MORTALITY DURING FOLLOW-UP
Incidental infectious complications were observed during follow-up in 5 patients (5%) for 1.47 events per 100 person-years. These complications were a case each of disseminated nontuberculous mycobacterial infection, dermatomal shingles, HPV-related cervical dysplasia, and cryptococcal meningitis, along with a case of cryptococcal pneumonia with cryptococcoma in a patient receiving fluconazole as primary prophylaxis (Table 2).
Fourteen invasive cancers of different histotypes were observed in 13 patients with ICL. These included anal and vulvovaginal cancers, tumors of the oral cavity or pharynx, lung squamous-cell carcinoma, papillary thyroid carcinoma, prostate or breast adenocarcinoma, non-Hodgkin’s lymphoma, and Kaposi’s sarcoma (Table 3). Eleven cancers were diagnosed after enrollment in our study, whereas three diagnoses preceded enrollment. The prevalence of any invasive cancer was higher among the study patients than in the general population after adjustment for age and sex for those with prevalent cancers (13 observed vs. 5.412 expected cases; P = 0.004) as well as for incident cancers (11 observed vs. 5.412 expected cases; P = 0.02). The study patients with ICL had a significantly higher prevalence of six cancers — anal, vulvovaginal, oral cavity or pharynx, papillary thyroid carcinoma, non-Hodgkin’s lymphoma, and Kaposi’s sarcoma — than expected cases in the general population. A total of 44 autoimmune diseases were diagnosed in 33 patients: 30 diagnoses that had been established at the time of enrollment and 14 during clinical follow-up.
Table 3.
Cancer Prevalence in the ICL Cohort as Compared with Expected Cases.*
| Variable | Histotype | Cases in ICL Cohort† | Expected Cases | P Value |
|---|---|---|---|---|
| number of patients | ||||
| Any prevalent or incidental cancer | 13 | 5.412 | 0.004 | |
|
| ||||
| Incidental cancer only | 11 | 5.412 | 0.02 | |
|
| ||||
| Type of cancer | ||||
|
| ||||
| Anus or anal canal | Squamous-cell carcinoma | 2 | 0.020 | |
|
| ||||
| Breast | Lobular or ductal carcinoma | 2 | 1.078 | |
|
| ||||
| Thyroid | Papillary thyroid carcinoma | 2 | 0.297 | |
|
| ||||
| Lung | Squamous-cell carcinoma | 1 | 0.186 | |
|
| ||||
| Prostate | Adenocarcinoma | 1 | 1.491 | |
|
| ||||
| Non-Hodgkin’s lymphoma | Marginal B-cell lymphoma, mycosis fungoides | 2 | 0.258 | |
|
| ||||
| Oral cavity or pharynx | Squamous-cell carcinoma | 2 | 0.154 | |
|
| ||||
| Vulvovagina | Squamous-cell carcinoma | 1 | 0.014 | |
|
| ||||
| Kaposi’s sarcoma | Cutaneous or visceral | 1 | 0.015 | |
Shown is the cancer prevalence in the cohort of patients with idiopathic CD4 lymphocytopenia (ICL) as compared with expected cases in the general population in the Surveillance, Epidemiology, and End Results Program as matched according to age and sex.
A total of 14 separate invasive cancers were diagnosed in 13 patients.
During 374 person-years of follow-up, the rate of death among the study patients was similar to that in the general population (1.337 deaths per 100 person-years [5 deaths in 91 patients] and 0.854 deaths per 100 person-years, respectively).
THERAPEUTIC INTERVENTIONS
In our cohort, 55% of the patients received prophylaxis for opportunistic infections. Of these patients, 88% continued prophylaxis for more than 1 year. A total of 67% of the patients received prophylaxis for PJP (48% with trimethoprim–sulfamethoxazole, 11% with atovaquone, and 8% with dapsone), 13% received azithromycin for nontuberculous mycobacterial infection prophylaxis, and 8% received fluconazole as antifungal prophylaxis. At the time of this report, one patient was receiving recombinant human interleukin-7 (efineptakin alfa) for disabling HPV-related skin disease and anogenital dysplasia. Two patients with progressive HPV-related cancers (skin and anogenital squamous-cell carcinoma) underwent allogeneic HLA-matched hematopoietic stem-cell transplantation and remain on follow-up with study data censored at the time of transplantation. Two patients were also enrolled in a pilot study of belimumab (ClinicalTrials.gov number, NCT04097561), but the data that were included in this analysis predated their belimumab infusions.
DISCUSSION
In this observational study, we sought to expand knowledge regarding the clinical, genetic, immunologic, and prognostic characteristics of patients with ICL. The rarity of ICL has constrained systematic clinical studies, with existing literature consisting of case reports or series and smaller retrospective cohort studies.9–11 The consensus had been that ICL represents a heterogeneous disorder and that widespread availability of genomic data would reveal novel mendelian genetic causes. However, the cause of ICL has remained elusive even in infants with lymphopenia that has been diagnosed by means of T-cell receptor excision circle screening,12,13 with only 10% currently receiving a specific genetic diagnosis. Similarly, in our cohort, despite extensive genetic workup, only 7% of the patients were found to have a monogenic cause of CD4 lymphopenia.
Nevertheless, our results support a prominent role of CD4+ T cells in disease presentation, which justifies the chosen name of the syndrome at its inception and expands the role of CD4+ T-cell counts in ICL as an important biomarker to inform management. We found that patients with reduced CD4+ T-cell counts were more likely to have opportunistic infections, a finding that supports a continuous relationship between CD4 counts and infection susceptibility. However, causality cannot be determined, and cutoffs of CD4 counts that originate from a single cohort may not be generalizable.
In addition, the spectrum of the opportunistic infections observed in patients with ICL is distinct from that in those with untreated HIV type 1, which suggests differences in pathogenesis. Although ICL has been consistently described as a heterogenous disorder, we found a rather unifying pattern of clinical presentations of HPV-related diseases, cryptococcosis, cancers, and autoimmunity, a compilation that should trigger an articulated clinical, immunologic, and genetic evaluation. We suspect that ICL may be more homogeneous than originally thought, because our study patients had an overall stable or modestly up-trending T-cell trajectory without a profound effect on mortality (very different from the trajectory in patients with HIV infection) and an apparent lack of a unifying monogenic cause. As compared with the first reported cases of ICL in the 1980s and 1990s, we have seen almost no cases of PJP, toxoplasmosis, mucosal candidiasis, and CMV retinitis as presenting opportunistic infections. Antimicrobial prophylaxis may only partially explain the lower frequency of PJP and toxoplasmosis in patients with ICL because most of the opportunistic infections that were identified in our study were recorded as the initial diagnosis before initiation of any prophylaxis. The distribution of opportunistic infections that we observed may also signal an overall shift in prevalent infections in the United States or a possible bias in ICL case identification at the height of the AIDS epidemic. Because long-term antibiotic prophylaxis poses risks and toxic effects, an individualized approach to PJP prophylaxis in patients with ICL may be preferred by weighing the presence of additional risk factors, such as underlying pulmonary disease, further immune suppression (e.g., with glucocorticoids), or logistic hurdles for prompt access to medical care. Prophylaxis or cryptococcal antigen screening for cryptococcosis species should be considered, given the severity and higher prevalence of these infections in our cohort.
Associations between ICL and the risk of cancer (especially lymphomas) and primary immunodeficiencies (particularly common variable immunodeficiency)21,22 have been identified previously. following similar methods, we found that the overall prevalence of cancers was increased in our ICL cohort, which was driven by HPV-related squamous-cell carcinoma, non-Hodgkin’s lymphoma, papillary thyroid carcinoma, and Kaposi’s sarcoma. The increased risk of cancer in patients with ICL with concomitantly reduced numbers of CD4, CD8, and NK cells further emphasizes the clinical implication of severe lymphopenia on cancer immunosurveillance. Deficient immune responses against viral or somatic neoantigens may underlie the increased susceptibility to virus-associated cancers (i.e., HPV, Kaposi’s sarcoma–associated herpesvirus, and Epstein–Barr virus) and non-Hodgkin’s lymphoma, whereas impaired CD4+ T-cell regulatory functions causing autoimmune thyroiditis may promote papillary thyroid carcinoma, as suggested by accumulating epidemiologic evidence in the general population,23 in patients with common variable immunodeficiency,20 and in patients undergoing organ transplantation.24
Understanding the pathogenesis of ICL remains an important obstacle to designing clinical interventions. Immune-based therapies and hematopoietic stem-cell transplantation25–27 have been attempted,28 including at our institution, with mixed results. We previously reported on the high prevalence of autoantibodies29,30 in patients with ICL, which may cause or exacerbate lymphopenia, and a pilot study of belimumab is currently under way.29 The late clinical onset of ICL with prominent loss of naive T cells9,10 may result in a premature progressive erosion of adaptive immunosurveillance against some opportunistic pathogens and somatic antigens, a hypothesis that provides an additional argument for a distinct pathogenesis as compared with other primary or acquired immunodeficiencies.
Our data did not support a short-term higher risk of death among patients with ICL than that in the general population, but our sample was small and follow-up time was relatively short to ascertain such an outcome. Regardless, if untreated patients with HIV infection with similar CD4 counts were followed for 3 years, the mortality would have been higher than what we observed in our study cohort, which further supports the distinct clinical implications of CD4 lymphopenia in patients with ICL.31–35 Another limitation of our study is that we recruited patients from the entire country, and we cannot exclude the possibility that the presence of mild cases, disabling opportunistic infections (i.e., progressive multifocal leukoencephalopathy), or coexisting conditions may have affected the decision for NIH referral. We did not identify clear differences between our study patients and those in other smaller cohorts,11 but we cannot rule out the possibility that the clinical spectrum may have been skewed by other referral biases based on NIH clinical expertise or disparities in the ability to diagnose opportunistic infections or to acquire clinical and microbiologic records. Regardless, a national registry would greatly enhance the study of the clinical and epidemiologic features of ICL, including important morbidity and mortality data.
We present clinical and immunologic characteristics and outcomes of the patients with ICL who participated in our study. The lower CD4+ T-cell counts that we encountered in our study patients were associated with both opportunistic infections and cancers, which led to clinical monitoring and individualized approaches to antimicrobial prophylaxis. Although our study patients did not have a higher short-term risk of death than that in the general population, the increased cancer prevalence supports primary and secondary cancer preventive strategies in ICL management.
Supplementary Material
Acknowledgments
Supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (NIH) and by federal funds from the National Cancer Institute (contract number, 75N91019D00024).
We thank the staff members of the Center for Information Technology of the NIH for providing access to the Biowulf cluster computer system and its high-performance computational capabilities.
Footnotes
The views expressed in this article are those of the authors and do not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
A data sharing statement provided by the authors is available with the full text of this article at NEJM.org.
REFERENCES
- 1.Centers for Disease Control. Unexplained CD4+ T-lymphocyte depletion in persons without evident HIV infection — United States. MMWR Morb Mortal Wkly Rep 1992;41:541–5. [PubMed] [Google Scholar]
- 2.Duncan RA, von Reyn CF, Alliegro GM, Toossi Z, Sugar AM, Levitz SM. Idiopathic CD4+ T-lymphocytopenia — four patients with opportunistic infections and no evidence of HIV infection. N Engl J Med 1993;328:393–8. [DOI] [PubMed] [Google Scholar]
- 3.Ho DD, Cao Y, Zhu T, et al. Idiopathic CD4+ T-lymphocytopenia — immunodeficiency without evidence of HIV infection. N Engl J Med 1993;328:380–5. [DOI] [PubMed] [Google Scholar]
- 4.Laurence J, Mitra D, Steiner M, Lynch DH, Siegal FP, Staiano-Coico L. Apoptotic depletion of CD4+ T cells in idiopathic CD4+ T lymphocytopenia. J Clin Invest 1996;97:672–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spira TJ, Jones BM, Nicholson JK, et al. Idiopathic CD4+ T-lymphocytopenia — an analysis of five patients with unexplained opportunistic infections. N Engl J Med 1993;328:386–92. [DOI] [PubMed] [Google Scholar]
- 6.Smith DK, Neal JJ, Holmberg SD. Centers for Disease Control Idiopathic CD4+ T-lymphocytopenia Task Force. Unexplained opportunistic infections and CD4+ T-lymphocytopenia without HIV infection — an investigation of cases in the United States. N Engl J Med 1993;328:373–9. [DOI] [PubMed] [Google Scholar]
- 7.Fauci AS. CD4+ T-lymphocytopenia without HIV infection — no lights, no camera, just facts. N Engl J Med 1993;328:429–31. [DOI] [PubMed] [Google Scholar]
- 8.Edwards JK, Cole SR, Breger TL, et al. Mortality among persons entering HIV care compared with the general U.S. population: an observational study. Ann Intern Med 2021;174:1197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zonios DI, Falloon J, Bennett JE, et al. Idiopathic CD4+ lymphocytopenia: natural history and prognostic factors. Blood 2008;112:287–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Régent A, Autran B, Carcelain G, et al. Idiopathic CD4 lymphocytopenia: clinical and immunologic characteristics and follow-up of 40 patients. Medicine (Baltimore) 2014;93:61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yarmohammadi H, Cunningham-Rundles C. Idiopathic CD4 lymphocytopenia: pathogenesis, etiologies, clinical presentations and treatment strategies. Ann Allergy Asthma Immunol 2017;119:374–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buchbinder D, Walter JE, Butte MJ, et al. When screening for severe combined immunodeficiency (SCID) with T cell receptor excision circles is not SCID: a case-based review. J Clin Immunol 2021;41:294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jongco AM III, Sporter R, Hon E, et al. Characterization of infants with idiopathic transient and persistent T cell lymphopenia identified by newborn screening-a single-center experience in New York State. J Clin Immunol 2021;41:610–20. [DOI] [PubMed] [Google Scholar]
- 14.Firth D. Bias reduction of maximum likelihood estimates. Biometrika 1993;80:27–38. [Google Scholar]
- 15.Follmann D, Proschan M, Leifer E. Multiple outputation: inference for complex clustered data by averaging analyses from independent data. Biometrics 2003;59:420–9. [DOI] [PubMed] [Google Scholar]
- 16.Tangye SG, Al-Herz W, Bousfiha A, et al. Human inborn errors of immunity: 2022 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol 2022;42:1473–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lisco A, Wong CS, Price S, et al. Paradoxical CD4 lymphopenia in autoimmune lymphoproliferative syndrome (ALPS). Front Immunol 2019;10:1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lisco A, Hsu AP, Dimitrova D, et al. Treatment of relapsing HPV diseases by restored function of natural killer cells. N Engl J Med 2021;385:921–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lisco A, Ye P, Wong C-S, et al. Lost in translation: lack of CD4 expression due to a novel genetic defect. J Infect Dis 2021;223:645–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Similuk MN, Yan J, Ghosh R, et al. Clinical exome sequencing of 1000 families with complex immune phenotypes: toward comprehensive genomic evaluations. J Allergy Clin Immunol 2022;150:947–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mayor PC, Eng KH, Singel KL, et al. Cancer in primary immunodeficiency diseases: cancer incidence in the United States Immune Deficiency Network Registry. J Allergy Clin Immunol 2018;141:1028–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shavit R, Maoz-Segal R, Frizinsky S, et al. Combined immunodeficiency (CVID and CD4 lymphopenia) is associated with a high risk of malignancy among adults with primary immune deficiency. Clin Exp Immunol 2021;204:251–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JH, Kim Y, Choi J-W, Kim YS. The association between papillary thyroid carcinoma and histologically proven Hashimoto’s thyroiditis: a meta-analysis. Eur J Endocrinol 2013;168:343–9. [DOI] [PubMed] [Google Scholar]
- 24.Kitahara CM, Yanik EL, Ladenson PW, et al. Risk of thyroid cancer among solid organ transplant recipients. Am J Transplant 2017;17:2911–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petersen EJ, Rozenberg-Arska M, Dekker AW, Clevers HC, Verdonck LF. Allogeneic bone marrow transplantation can restore CD4+ T-lymphocyte count and immune function in idiopathic CD4+ T-lymphocytopenia. Bone Marrow Transplant 1996;18:813–5. [PubMed] [Google Scholar]
- 26.Cervera C, Fernádez-Avilés F, de la Calle-Martin O, et al. Non-myeloablative hematopoietic stem cell transplantation in the treatment of severe idiopathic CD4+ lymphocytopenia. Eur J Haematol 2011;87:87–91. [DOI] [PubMed] [Google Scholar]
- 27.Hamidieh AA, Pourpak Z, Hamdi A, Nabavi M, Ghavamzadeh A. Successful fludarabine-based hematopoietic stem cell transplantation in a pediatric patient with idiopathic CD4+ lymphocytopenia. Pediatr Transplant 2013;17(4):E109–E111. [DOI] [PubMed] [Google Scholar]
- 28.Sheikh V, Porter BO, DerSimonian R, et al. Administration of interleukin-7 increases CD4 T cells in idiopathic CD4 lymphocytopenia. Blood 2016;127:977–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perez-Diez A, Wong CS, Liu X, et al. Prevalence and pathogenicity of autoantibodies in patients with idiopathic CD4 lymphopenia. J Clin Invest 2020;130:5326–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cudrici CD, Boulougoura A, Sheikh V, et al. Characterization of autoantibodies, immunophenotype and autoimmune disease in a prospective cohort of patients with idiopathic CD4 lymphocytopenia. Clin Immunol 2021;224:108664. [DOI] [PubMed] [Google Scholar]
- 31.Boulle A, Schomaker M, May MT, et al. Mortality in patients with HIV-1 infection starting antiretroviral therapy in South Africa, Europe, or North America: a collaborative analysis of prospective studies. PLoS Med 2014;11(9):e1001718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Young J, Psichogiou M, Meyer L, et al. CD4 cell count and the risk of AIDS or death in HIV-Infected adults on combination antiretroviral therapy with a suppressed viral load: a longitudinal cohort study from COHERE. PLoS Med 2012;9(3):e1001194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mills GD, Jones PD. Relationship between CD4 lymphocyte count and AIDS mortality, 1986–1991. AIDS 1993;7:1383–6. [DOI] [PubMed] [Google Scholar]
- 34.National Cancer Institute. Surveillance, epidemiology, and end results program (https://seer.cancer.gov/). [Google Scholar]
- 35.Phillips AN, Elford J, Sabin C, Bofill M, Janossy G, Lee CA. Immunodeficiency and the risk of death in HIV infection. JAMA 1992;268:2662–6. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.