

Abstract
Background
MLH1 epimutation is characterised by constitutional monoallelic MLH1 promoter hypermethylation, which can cause colorectal cancer (CRC). Tumour molecular profiles of MLH1 epimutation CRCs were used to classify germline MLH1 promoter variants of uncertain significance and MLH1 methylated early-onset CRCs (EOCRCs). Genome-wide DNA methylation and somatic mutational profiles of tumours from two germline MLH1: c.-11C > T and one MLH1: c.-[28A > G; 7C > T] carriers and three MLH1 methylated EOCRCs (< 45 years) were compared with 38 reference CRCs. Methylation-sensitive droplet digital PCR (ddPCR) was used to detect mosaic MLH1 methylation in blood, normal mucosa and buccal DNA.
Results
Genome-wide methylation-based Consensus Clustering identified four clusters where the tumour methylation profiles of germline MLH1: c.-11C > T carriers and MLH1 methylated EOCRCs clustered with the constitutional MLH1 epimutation CRCs but not with the sporadic MLH1 methylated CRCs. Furthermore, monoallelic MLH1 methylation and APC promoter hypermethylation in tumour were observed in both MLH1 epimutation and germline MLH1: c.-11C > T carriers and MLH1 methylated EOCRCs. Mosaic constitutional MLH1 methylation in MLH1: c.-11C > T carriers and 1 of 3 MLH1 methylated EOCRCs was identified by methylation-sensitive ddPCR.
Conclusions
Mosaic MLH1 epimutation underlies the CRC aetiology in MLH1: c.-11C > T germline carriers and a subset of MLH1 methylated EOCRCs. Tumour profiling and ultra-sensitive ddPCR methylation testing can be used to identify mosaic MLH1 epimutation carriers.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13148-023-01511-y.
Keywords: MLH1 epimutation, Genome wide DNA methylation, MLH1 methylation, MMR deficiency, Colorectal cancer, Lynch syndrome
Background
Colorectal cancer (CRC) is the third most diagnosed cancer and the second leading cause of cancer-related death, responsible for ~ 10% of all cancer incidences and cancer-related deaths worldwide [1]. DNA methylation [2], together with inherited genetic predispositions, adverse environmental risk factors and ageing [3], plays an important role in CRC aetiology. Aberrant DNA methylation changes can be detected in virtually all CRC tumours [2], but it is the transcriptional silencing of MLH1 through promoter hypermethylation (referred to as MLH1 methylation) that is one of the most clinically important and well-characterised epigenetic events, seen in 10–20% of all CRCs [4].
Somatically acquired biallelic MLH1 methylation in CRC results in loss of immunohistochemical expression of the MLH1 and PMS2 DNA mismatch repair (MMR) proteins and microsatellite instability within the tumour (i.e. MMR-deficiency). Sporadic MLH1 methylation is associated with an older age of CRC diagnosis, females and features of the serrated pathway of neoplasia [5] namely the co-existence of somatic BRAF p.V600E mutations and genome-wide hypermethylation of tumour suppressor genes, commonly referred to as high levels of CIMP (CpG Island Methylator Phenotype) [6]. A second sporadic subtype of MMR-deficient CRC is caused by biallelic somatic mutations in one of the DNA MMR genes (often referred to as double somatic MMR mutations) [7]. In contrast, CRCs related to Lynch syndrome (LS) result from a germline pathogenic variant in one of the DNA MMR genes and a second somatic hit causing tumour MMR-deficiency. Rarely, constitutional mismatch deficiencies (CMMRD) occur when an individual inherits two germline pathogenic variants in the same MMR gene, leading to the loss of both alleles [8]. Lynch-related MLH1 deficiency occurs in the absence of both MLH1 methylation and features of the serrated neoplasia pathway (no BRAF p.V600E or CIMP-high) [9]. Therefore, tumour MLH1 methylation testing is used as the routine diagnostics testing to differentiate sporadic MLH1 methylated CRCs from inherited MLH1-deficient CRC caused by germline pathogenic variants (Lynch syndrome) [10, 11]. To further distinguish MLH1 epimutation CRCs from common sporadic MLH1 methylation CRCs, MLH1 methylation testing of non-tumour DNA sources (e.g. blood) is recommended [12].
A rarer subtype of MMR-deficient CRC is related to constitutional hypermethylation of the MLH1 gene promoter, referred to as MLH1 epimutation. MLH1 epimutations are characterised by monoallelic MLH1 promoter methylation [13], resulting from either idiopathic de novo methylation (“primary epimutation”) or from a cis-acting genetic variant (“secondary epimutation”), which determines the transgenerational transmissibility [14]. Primary and secondary MLH1 epimutations both present with tumour MLH1 methylation and resultant tumour MMR deficiency. In MLH1 epimutation carriers, soma-wide MLH1 methylation occurs in a monoallelic manner [15], although mosaic patterns have been described [16].
The prevalence of MLH1 epimutations is thought to be between 3 and 16% in Lynch-suspected cases with MLH1-deficient CRCs [10, 17–19]. There is currently a lack of consensus on the triaging approach to detect MLH1 epimutation carriers, largely due to highly variable inheritance and potentially mosaic constitutional methylation patterns [10]. Testing for MLH1 epimutation has been recommended in CRC cases diagnosed < 60 years with an MLH1 methylated tumour and those with a history of more than one Lynch-associated tumour [10], although it is unclear how routinely these criteria are applied, primarily due to their rarity.
Adding to the complexity, cases demonstrating mosaic patterns of constitutional MLH1 epimutation have been previously reported [16, 19]. Mosaic constitutional methylation has also been seen in other key cancer risk genes including BRCA1 [20] and RAD51C [21] in levels as low as 0.01% in non-neoplastic tissue and blood DNA samples from breast and ovarian cancer cases. Though primary MLH1 epimutations are largely thought to arise de novo, there has been a report of an early-onset colon cancer case who inherited a constitutional MLH1 epimutation from their asymptomatic mother who had low-level (3–5%) gonosomal mosaic MLH1 epimutation [22]. Therefore, identifying mosaic MLH1 epimutations poses a clinical challenge for assessing not only second primary cancer risks but also cancer risks in family members. The low MLH1 methylation levels present in mosaic cases are unlikely to be detectible by the Methylation-specific Multiplex Ligation-Dependent Probe Amplification (MS-MLPA) testing method commonly utilised in the clinical setting, highlighting the need for studies applying highly sensitive techniques such as methylation-sensitive droplet digital polymerase chain reaction (ddPCR) [23].
To date, the MLH1: c.-27C > A germline pathogenic variant is the only reported variant known to underlie secondary MLH1 epimutations [14]. Several other non-coding MLH1 promoter germline variants have been reported (e.g. c.-11C > T [19], c.-[28A > G; 7C > T] [24], c.-42C > T [25]), although their pathogenicity and effect on inducing MLH1 methylation are less defined and, as such, remain classified as variants of uncertain clinical significance (VUS). In CRCs associated with these VUS, a constitutional reduction of MLH1 expression was observed [19, 24, 25] but without a clear effect on MLH1 promoter methylation. Due to their rarity, studies of these variants are scarce and validation difficult, which impedes optimal clinical management in carriers.
Differentiating MLH1 epimutations from sporadic MLH1 methylated CRCs has important consequences for the clinical management of patients including prevention of second primary cancers and cancer prevention in relatives [10, 26]. This study investigated the genome-wide DNA methylation and somatic mutation profiles from clinically relevant subtypes of MMR-deficient CRCs, including those defined by sporadic MLH1 methylation or by constitutional MLH1 epimutation. The unique DNA methylation signatures demonstrated by the sporadic MLH1 methylated and constitutional MLH1 epimutation tumours were investigated in CRCs from carriers of a germline VUS in the MLH1 promoter or with tumour MLH1 methylation in an early-onset CRC (EOCRC) to support classification. Detection of low-level MLH1 methylation in blood and normal colonic tissue by ddPCR supported mosaic constitutional MLH1 epimutation for these clinically challenging cases.
Methods
Study participants and CRC tumour samples
We assessed genome-wide DNA methylation and somatic mutational profiles in 44 tumours and matched 14 normal colonic mucosa DNA samples from 43 participants with CRC (Fig. 1). All normal mucosa samples tested were from the surgical specimen from the furthest site of resection from the tumour (i.e. resection margin). Study participants were selected from the ANGELS study [27] or from the Australasian Colon Cancer Family Registry [28]. Immunohistochemical staining (IHC) for expression of the four MMR proteins (MLH1, MSH2, MSH6 and PMS2) was performed on each CRC using previously published protocols [29]. Tumour MLH1 gene promoter hypermethylation was tested using two locus-specific detection techniques, MethyLight [30] and methylation-sensitive high-resolution melting (MS-HRM) [31]. Tumours showing > 10% methylation by MethyLight and > 5% by MS-HRM were considered positive for MLH1 promoter methylation and further tested for MLH1 methylation in blood-derived DNA to identify MLH1 epimutation. For each of the 43 participants included in the study, the MMR genes, including the MLH1 gene promoter, were screened to identify germline pathogenic variants as previously described [29] or from multigene panel testing as part of the clinical management. Participants with MLH1 promoter hypermethylation (> 10%) in blood but without a germline pathogenic variant were classified as a primary MLH1 epimutation. Of 44 CRCs, 41 tumour DNA and matched blood-derived DNA were also sequenced using whole exome sequencing (WES; n = 27) [27] or by a custom designed 298 gene panel sequencing (Panel; n = 14) [32].
Fig. 1.

An overview of the study design including descriptions of CRC subgroups and key findings from three main analyses. Analysis 1—genome-wide DNA methylation-based Consensus clustering analysis identified four Consensus Clusters. Analysis 2—applying the Consensus Clustering to six diagnostically challenging CRCs and the classification of three MLH1 methylated EOCRCs and three MLH1 promoter germline VUS carriers into Consensus Cluster 4. Analysis 3—further assessment of DNA methylation and somatic mutational profiles associated with each Consensus Cluster
Thirty-eight CRCs from 37 participants that were classified into six confirmed CRC subtypes were used as reference groups (Fig. 1):
“LS-CRCs”—MMR-deficient CRCs from participants with LS including 2 × MLH1, 2 × MSH2, 3 × MSH6 and 2 × PMS2 germline pathogenic variant carriers with no MLH1 promoter hypermethylation in the tumour and blood-derived DNA (n = 9 CRCs from nine participants).
“Sporadic MLH1 methylated CRCs”—CRCs showing loss of MLH1/PMS2 by IHC with MLH1 promoter hypermethylation in the tumour but absent in the blood and/or normal mucosa-derived DNA and no germline MMR gene pathogenic variants identified (n = 9 CRCs from nine participants).
“Primary MLH1 epimutation CRCs”—CRCs showing loss of MLH1/PMS2 by IHC resulting from primary MLH1 epimutation in the absence of MLH1 promoter cis-variants with MLH1 promoter hypermethylation in the tumour and blood-derived DNA and no germline MMR gene pathogenic variants identified (n = 4 CRCs from three participants). One normal mucosa DNA was included to assess the constitutional nature of MLH1 methylation.
“Secondary MLH1 epimutation CRCs”—CRCs showing loss of MLH1/PMS2 by IHC resulting from a secondary MLH1 epimutation (MLH1: c.-27C > A), demonstrating MLH1 promoter hypermethylation in the tumour and blood-derived DNA (n = 2 CRCs from two participants). One normal mucosa DNA was included.
“Double MMR somatic CRCs”—MMR-deficient CRCs with two somatic mutations in the MMR gene indicated as defective by the pattern of protein loss by IHC and no MLH1 promoter hypermethylation in tumour and blood-derived DNA and no germline MMR gene pathogenic variants identified (n = 5 CRCs from five participants).
“MMR-proficient CRCs”—CRCs with retained/normal expression of all four MMR proteins by IHC and absence of MLH1 promoter hypermethylation in tumour and blood-derived DNA and no germline pathogenic variants (n = 9 CRCs from nine participants).
In addition to the six reference CRC subtypes, we tested two groups of six diagnostically challenging CRCs (Fig. 1):
“MLH1 promoter VUS CRCs”—Carriers of MLH1 promoter VUS including two carriers of germline MLH1: c.-11C > T and one carrier of germline MLH1: c.-[28A > G; 7C > T] in cis (n = 3 CRCs from three participants). Tumour MLH1 methylation was tested by two loci-specific techniques described above. No blood methylation was detected by the clinical testing methodology (i.e. MS-MLPA). All cases had no reported CRCs in the first-degree relatives.
“MLH1 methylated early-onset CRCs (EOCRCs)”— CRCs showing loss of MLH1/PMS2 by IHC with MLH1 promoter methylation in tumour and CRC diagnosis < 45 years and no germline MMR pathogenic variants or double somatic MMR gene mutations (n = 3 CRCs from three participants). No blood methylation was detected by the clinical testing methodology (i.e. MS-MLPA). All cases had no reported CRCs in the first- and second-degree relatives.
DNA methylation array processing
Tumour and normal mucosa DNA were isolated from macro-dissected formalin-fixed paraffin-embedded (FFPE) specimens using the QIAmp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany). Genomic DNA was bisulphite converted and restored as previously described [33, 34]. Tumour and normal mucosa DNA methylomes were profiled using the Infinium HumanMethylation EPIC platform (HMEPIC, Illumina, San Diego, United States) by the Australian Genome Research Facility (AGRF, Melbourne, Australia). Raw data were imported into the R programming software environment (v3.3.2) and processed using the minfi Bioconductor package (v1.38.0) [35]. The data underwent Functional normalisation [36] with noob background correction [37]. β-values were used for presenting the data and M-values were used for all statistical analyses [38]. Probes with detection P-values greater than 0.05 and probes on sex chromosomes were removed from all analyses. Methylation levels were measured from 771,234 probes in total.
Bioinformatic analysis
Forty-two CpGs overlapping the CpG island (hg19 chr3: 37033539–37036377) associated with the MLH1 promoter (NM_000249.3) were used for illustrating MLH1 promoter methylation. Of these, the mean methylation level was calculated across the four CpGs (cg23658326, cg11600697, cg21490561, cg00893636) overlapping the regulatory “C” region [39] and used to determine the MLH1 promoter methylation status. DNA samples with mean methylation (β-values) > 0.2 were considered MLH1 methylation positive. CIMP status was determined by assessing mean methylation levels across Infinium HMEPIC CpG probes overlapping or nearby five previously described gene promoter regions [40] (CACNA1G: cg18337803, cg20467136, cg23614229, cg11262815; RUNX3: cg06377278, cg27095256; SOCS1: cg06220235; NEUROG1: cg04620091; and IGF2: cg16977706). Samples with methylation (> 0.2) at 3 or more of these 5 gene regions were considered CIMP-high. Differentially methylated regions (DMRs) analysis was performed using “DMRcate” package (v2.6.0) [41].
The Consensus Cluster analysis was performed using “ConsensusClusterPlus” package (v1.56.0) [42] on the 77,113 most variably methylated CpGs between the 38 CRCs from the six reference groups ranked by standard deviation (SD). These probes constituted 10% of all CpG probes. The Consensus Cluster analysis was performed using the default setting and four total clusters (k’s) were selected after we found that testing for > 4 clusters provided no additional clusters from our 38 reference CRC samples. The “consensus class assignments” were used to define the sample classification. A principal component analysis (PCA) was performed to test the validity of the observed Consensus Clusters.
Tumour sequencing
Twenty-seven CRCs were sequenced by WES and 14 CRCs were sequenced by the targeted multigene panel. Three CRCs (1 LS-CRC, 1 sporadic MLH1 methylated, 1 primary MLH1 epimutation) were excluded from the methylation profiling due to insufficient tumour DNA material remaining for testing. Peripheral blood-derived DNA was extracted using the DNeasy blood and tissue kit (Qiagen) and sequenced as germline references. For WES capture, the Clinical Research Exome V2 kit (Agilent Technologies, Santa Clara, United States) was performed at AGRF as previously described [27].
Adaptor sequences were trimmed using trimmomatic v0.38 [43] and aligned to the GRCh37 human reference genome using the BWA (v.0.7.12). Germline variants were called using HaplotypeCaller (GATK library v.4.0.0, Broad Institute). Somatic single nucleotide variants (SNVs) and insertions/deletions (INDELs) were called using Strelka (v.2.9.2) [44] . For both WES and panel, variants were filtered for PASS called by Strelka with a minimum variant allele fraction (VAF) of 0.04 and a minimum coverage depth of 30× for tumour analyses. For consistency, non-overlapping regions between the WES and panel captures were removed, except for the MLH1 promoter region. WES and panel sequencing was used to identify germline variants across the MLH1 promoter region up to 1500 base pairs (bp) for the panel sequencing and 2125 bp for WES from the transcription start site. Tumour microsatellite instability (MSI) status was determined bioinformatically using MANTIS [45] with a cut-off for high levels of MSI (MSI-H) of ≥ 0.245 for WES and a cut-off of MSI-H of ≥ 0.252 for panel sequenced tumours [32]. Loss of heterozygosity (LOH) of MLH1 was determined using LOHdeTerminator v0.5 (https://github.com/supernifty/LOHdeTerminator) by assessing regions of the genome containing heterozygous germline variants that appear to be either homozygous reference or homozygous alternative in the somatic sample, based on an allele frequency range of 0.3 to 0.7 in the germline variant and a difference of greater than 0.3 in the somatic variant.
The maftools (v2.12.0) Bioconductor package was used for analysing and visualising somatic variants [46]. Unless described otherwise, P-values were derived from Fisher’s exact tests. The list of 32 genes that undergo frequent somatic mutations was retrieved from TCGA COAD samples [47]. Of those, 15 and 17 genes were identified from hypermutated (described as having a high TMB (10–100 mutations/megabase)) CRCs and non-hypermutated (TMB < 10 mutations/megabase) CRCs, respectively [47]. APC and TCFL2 overlapped in both lists and four genes (TTN, FAM123B, KIAA1804, EDNRB) were not captured by the panel sequencing used in this study. We also assessed somatic mutations in four commonly mutated genes (AXIN2, CCND1, ZNRF3, RNF43) associated with the Wnt pathway [48] as well as two DNA polymerase genes (POLE, POLD1) [49], and five genes (DNMT1, TET1, TET2, TET3, MBD4) related to DNA methylation machinery [50].
MLH1 promoter methylation detection using droplet digital PCR (ddPCR)
Twenty nanograms of bisulphite-modified blood, normal mucosa and buccal/saliva-derived DNA were tested using the Bio-Rad QX200 ddPCR system (Pleasanton, USA) with the ddPCR Supermix for Probes (no dUTP) (Bio-Rad) and the inclusion of 0.1X Q Solution (Qiagen), 800 nM of each primer and 400 nM of each probe (Bioneer Pacific, Daejeon, South Korea). Sequences for primers and probes are shown in Additional file 1: Table S1.
Detecting allelic MLH1 methylation using methylation-specific PCR and pyrosequencing
SMART-MSP (Sensitive Melting Analysis after Real-time Methylation-Specific PCR) reactions were performed in technical duplicates on a Mic qPCR Cycler (BMS, Sydney, Australia) as previously described [51] to specifically amplify only methylated epialleles. The primer sequences (Bioneer) can be found in Additional file 1: Table S1. The amplified methylated epialleles were then pyrosequenced on a Qseq instrument (BMS) using the Q48 Advanced CpG kit (Qiagen) and Streptavidin Mag Sepharose beads (Cytiva, MA, USA) to assess the genotypes of the SNPs on only the methylated epialleles. The pyrosequencing data were analysed with Qseq software 2.4.4 (BMS).
Results
The CRC subgroups tumour characteristics
The characteristics of the participants and their CRCs by subtype are described in Additional file 1: Tables S2 and S3. The HMEPIC-based DNA methylation levels (β-value) for each of the CRC and normal mucosa samples across the MLH1 promoter are shown in Additional file 2: Fig. S1 and Additional file 1: Table S3. CRC tumour samples from the primary (mean β = 0.77 ± 0.06 SD) and secondary (0.74 ± 0.16) MLH1 epimutation carriers showed MLH1 methylation levels (i.e. hypermethylation) consistent with tumour samples from people with sporadic MLH1 methylated CRCs (0.55 ± 0.09). For the normal mucosa samples, MLH1 promoter methylation was observed at high levels in only the primary (mean β-value = 0.40) and secondary (β = 0.39) MLH1 epimutation CRC groups. Each of the three MLH1 methylated EOCRCs from the diagnostically challenging group showed MLH1 promoter hypermethylation in their tumours (β = 0.34, 0.42 and 0.67) but not in the single normal mucosa sample tested (β = 0.07). Similarly, both the CRCs from the MLH1: c.-11C > T VUS carriers showed MLH1 promoter hypermethylation (β = 0.43 and 0.39, respectively), while the CRC from the MLH1: c.-[28A > G; 7C > T] VUS carrier was only moderately increased (β = 0.14), compared with their respective normal mucosa samples (β = 0.06, 0.1 and 0.09, respectively) but not meeting our threshold of 0.2 (Additional file 2: Fig. S1).
We assessed differential patterns of somatic mutations in key CRC genes between the six reference groups and the two groups of diagnostically challenging CRCs (Additional file 3: Fig. S2). MLH1 methylated CRCs were associated with less frequent mutations in APC, KRAS and TCF7L2, and frequent mutations in BRAF p.V600E and RNF43 (P < 0.05). Consistent with LS-CRCs, all MLH1 epimutation, MLH1 promoter VUS CRCs and MLH1 methylated EOCRCs carried at least APC or TCF7L2 somatic mutations and frequent KRAS codon 12&13 mutations (Additional file 1: Table S4). Unlike the sporadic MLH1 methylated CRCs, these groups’ CRCs also lacked the BRAF p.V600E somatic mutation (P < 0.01) and had less frequent RNF43 somatic mutations (P = 0.04). Of the five genes of the epigenetic machinery, only TET2 somatic mutations showed a significant enrichment in LS-CRC (P < 0.01; Additional file 1: Table S4).
Genome-wide DNA methylation consensus clusters distinguish MLH1 epimutation carriers CRCs from the other reference group CRCs
The genome-wide DNA methylation profiles were compared between 38 CRCs from the six reference tumour groups using a consensus clustering analysis based on the 77,113 most variably methylated CpGs. The consensus values are illustrated in a heatmap (Additional file 4: Fig. S3A), and the raw values are provided in Additional file 12: data. Six CRCs from two diagnostically challenging CRC groups (MLH promoter VUS CRCs and MLH1 methylated EOCRCs) were excluded in this training analysis. The analysis identified four Consensus Clusters (Table 1).
Consensus Cluster 1—comprised 20 CRCs (8/9 of LS-CRCs, 8/9 of the MMR-proficient CRCs and 4/5 of double MMR somatic CRCs). This cluster had the youngest mean age of CRC diagnosis (38.7 ± 10.6 interquartile range, IQR), the lowest level of MLH1 promoter methylation (mean β=0.08) and the lowest overall methylation levels across the variably methylated CpGs (mean β = 0.32).
Consensus Cluster 2—comprised three CRCs (1 LS-CRC, 1 MMR-proficient and 1 double MMR somatic CRCs) and was also characterised by low MLH1 promoter methylation (mean β=0.08). The double MMR somatic CRC (diagnosis age = 57 years) was also CIMP-high, though having low MLH1 promoter methylation. All samples from Consensus Cluster 2 had higher overall methylation (mean β = 0.42) across the variably methylated CpGs when compared with Consensus Cluster 1 group (P = 0.02).
Consensus Cluster 3—consisted of all nine sporadic MLH1 methylated CRCs and had the oldest mean age at CRC diagnosis (62.5 years ± 10.2 IQR). This cluster demonstrated the highest overall methylation across the variably methylated CpGs (mean β = 0.45).
Consensus Cluster 4—comprised all six primary and secondary MLH1 epimutation carrier CRCs. Tumours in this cluster demonstrated low overall methylation levels (mean β = 0.35) across the variably methylated CpGs similar to Consensus Cluster 1.
Table 1.
Overview of the sample composition and tumour characteristics within each of the four Consensus Clusters derived from genome-wide DNA methylation profiling of six CRC subtypes (reference groups)
| Consensus cluster 1 | Consensus cluster 2 | Consensus cluster 3 | Consensus cluster 4 | |
|---|---|---|---|---|
| Number of CRCs | 20 | 3 | 9 | 6 |
| Age at CRC diagnosis (mean ± s.d) | 38.7 ± 10.6 | 45.9 ± 12.8 | 65.5 ± 9.65 | 39.2 ± 11.3 |
| MLH1 promoter methylationa (mean ± s.d) | 0.076 ± 0.02 | 0.082 ± 0.02 | 0.548 ± 0.089 | 0.761 ± 0.045 |
| CIMPb (% positive) | 0 (0%) | 1 (33%) | 9 (100%) | 0 (0%) |
| Mean methylation across the VM-CpGsc | 0.32 | 0.42 | 0.45 | 0.35 |
| Lynch syndrome (n = 9) | 8 | 1 | 0 | 0 |
| MMR-proficient CRC (n = 9) | 8 | 1 | 0 | 0 |
| Double somatic MMR mutation CRC (n = 5) | 4 | 1 | 0 | 0 |
| Sporadic MLH1 methylated CRCs (n = 9) | 0 | 0 | 9 | 0 |
| MLH1 primary epimutation (n = 5) | 0 | 0 | 0 | 4 |
| MLH1 secondary epimutation (n = 1) | 0 | 0 | 0 | 2 |
aMean methylation (β-values) across the regulatory C region of MLH1
bCIMP was determined by the methylation levels of CpG probes overlapping five previously defined genes [40]
cMean methylation levels across the 77,113 most variably methylated CpGs (VM-CpGs), which was used for defining the Consensus Clusters. s.d.—standard deviation, VM-CpGs—(77,113) variably methylated CpGs defined by having high variation in the methylation patterns as ranked by standard deviation across 38 reference CRCs
The PCA analysis of the 77,113 most variably methylated CpGs, applied as an alternate approach to Consensus clustering, demonstrated three distinct groupings related to sporadic MLH1 methylated CRCs, primary and secondary MLH1 epimutation CRCs and a third group comprising LS-CRCs, double MMR somatic and MMR-proficient CRCs (Fig. 2) largely reflecting the groupings from the consensus cluster analysis. Additional file 5: Fig. S4 shows overall methylation patterns across the variably methylated CpGs.
Fig. 2.

Principal component analysis (PCA) showing genome-wide DNA methylation similarities between individual tumour, based on 77,113 variably methylated (VM)-CpG probes. Tumour samples of different CRC subgroups are shown in different colours. The MLH1-VUS group includes CRCs from 3 MLH1 germline VUS carriers (two MLH1: c.-11C > T and one MLH1: c.-[28A > G; 7C > T] carriers)
DNA methylation signatures associated with CRCs of MLH1 epimutation carriers
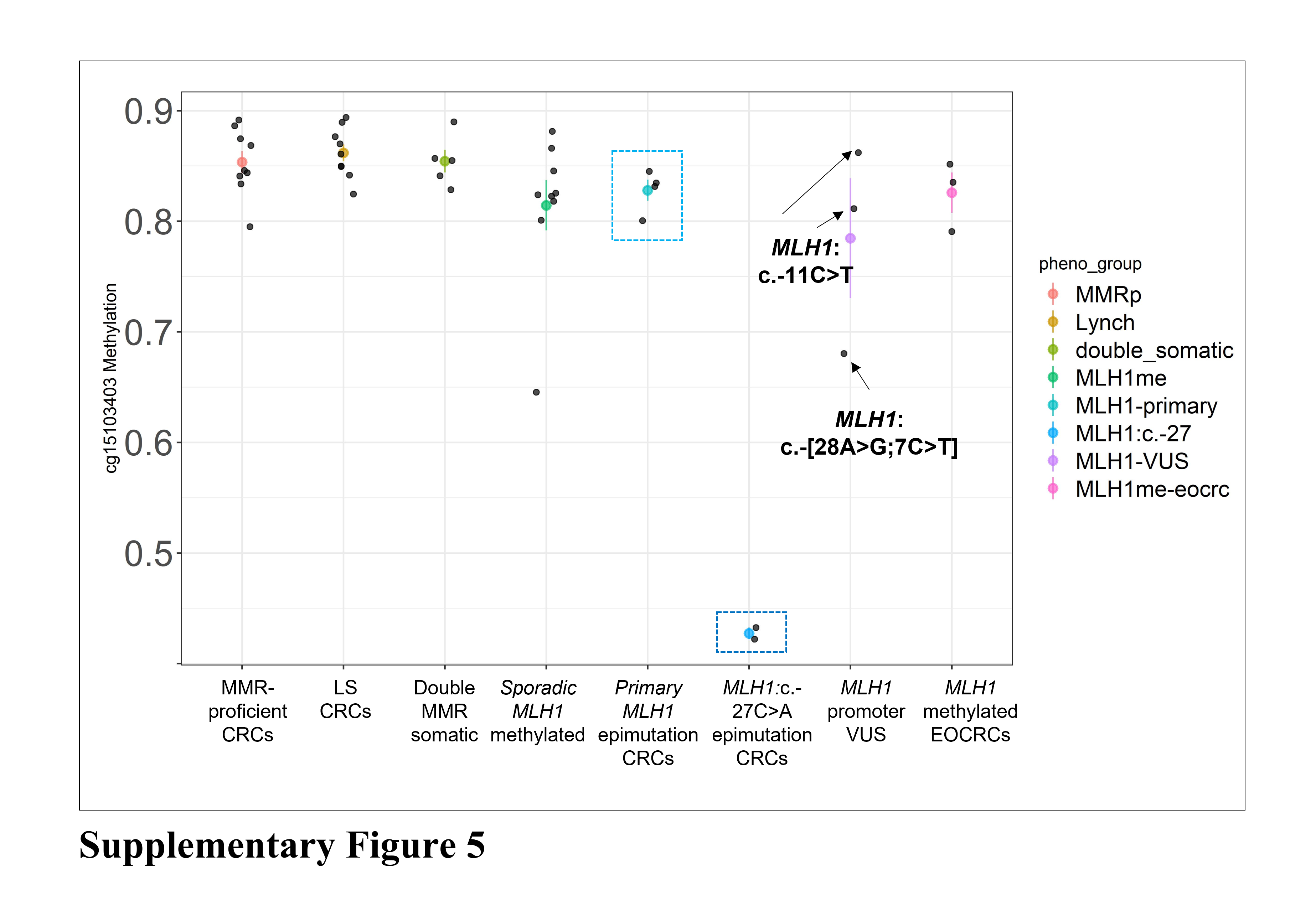
We performed differential methylation analysis between CRCs from primary and secondary (MLH1: c.-27C > A) epimutation carriers. A single differentially methylated (FDR or False Discovery Rate-adj P = 0.0004) CpG probe (cg15103403) was identified, located within the LRRFIP2 gene (chr3: 37110355). A clear hemi-methylation (~ 50%) pattern was observed in both CRCs from secondary epimutation carriers (Additional file 6: Fig. S5). There were no other CpG probes within the 4 kb flanking region and, therefore, regional methylation differences could not be assessed using the HMEPIC array data.
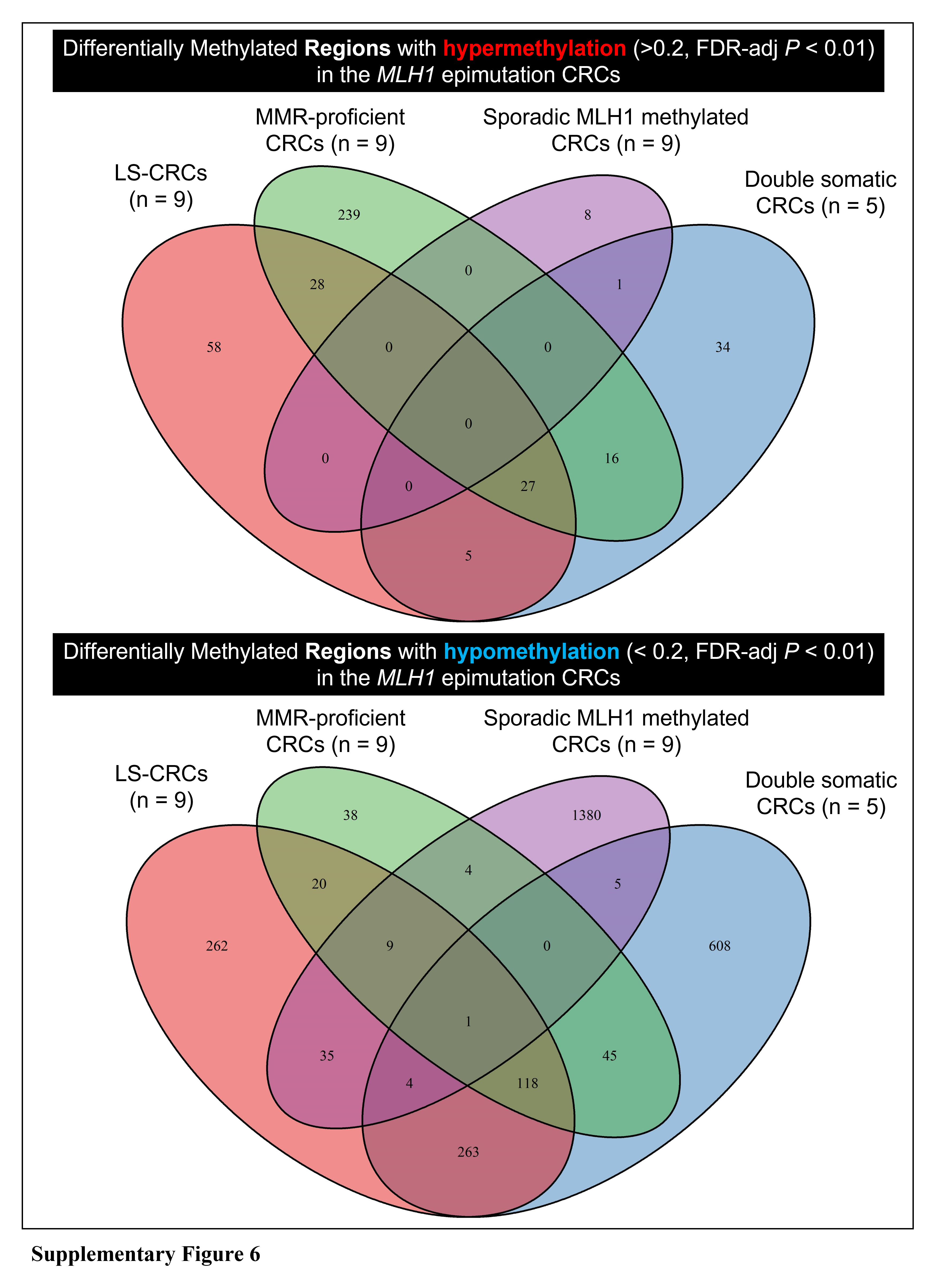
To further understand the tumour DNA methylation differences between those with sporadic (acquired) versus constitutional MLH1 promoter hypermethylation, we compared the genome-wide DNA methylation profiles of sporadic MLH1 methylated CRCs (n = 9) with those from MLH1 epimutation CRCs (n = 6) to identify DMRs. Given the paucity of differentially methylated CpGs sites between the primary and secondary MLH1 epimutation CRCs shown above, these two groups were combined as the MLH1 epimutation group. This identified 1447 DMRs (FDR-adj P < 0.01 & mean absolute β differences > 0.2) where 99% (1438/1447) of these DMRs were hypermethylated in the sporadic MLH1 methylated CRC group when compared with the epimutation group. In 9 (1%) of the DMRs, the mean methylation was greater by > 0.2 (β) in the MLH1 epimutation group and included the APC, MAD1L1, YPEL2, CRTC1, SSBP3, STARD13 genes and a non-coding RNA KLRK1-AS1 loci (Additional file 1: Table S5).
Of these seven genes, APC, a known driver of CRC tumourigenesis [52] , showed the most significant differences (Stouffer transformed P = 1.5 × 10–10) between the two groups. The APC promoter region (chr5: 112072926–112073958) showed higher methylation levels in the MLH1 epimutation group (mean β = 0.36 ± 0.14 SD) when compared with sporadic MLH1 promoter methylated CRCs (0.16 ± 0.08; P = 0.03) and when compared with the MMR-proficient CRCs (0.19 ± 0.18; P = 0.03), but were not different to the LS-CRCs (0.33 ± 0.11) or double MMR somatic CRCs (0.20 ± 0.15) (Fig. 3). The APC promoter hypermethylation (mean β > 0.2) was detected in only one of the five double MMR somatic CRCs. Both the unique and common DMRs to each reference group are shown in Additional file 7: Fig. S6.
Fig. 3.

DNA methylation patterns across the DMR (chr5: 112,072,926–112,073,958) overlapping the APC gene. A. The DMR (differentially methylated region) associated with the MLH1 epimutation CRCs. B. Bar plot showing mean DNA methylation levels by CRC subgroups. Error bars denote standard deviation. MLH1 epimutation group includes primary MLH1 epimutation and secondary MLH1 epimutation (c.-27C > A) CRCs. MLH1 promoter VUS group includes CRCs from two MLH1: c.-11C > T and one MLH1: c.-[28A > G; 7C > T] carriers
Genome-wide DNA methylation Consensus Clustering for categorising carriers of MLH1 promoter VUS and MLH1 methylated EOCRCs
We applied the Consensus Cluster approach using the same 77,113 variably methylated CpGs to MLH1 promoter VUS CRCs (n = 3) and MLH1 methylated EOCRCs (n = 3). The consensus values are illustrated in Additional file 4: Fig. S3B. The pedigrees for each case are shown in Additional file 8: Fig. S7. The two CRCs from MLH1: c.-11C > T and CRC from MLH1: c.-[28A > G; 7C > T] fitted to Consensus Cluster 4. Similarly, the three MLH1 methylated EOCRCs also fitted to Consensus Cluster 4, though no candidate germline cis variants or VUS were found in the MLH1 promoter region for these three participants.
CRCs from MLH1: c.-11C > T VUS and MLH1 methylated EOCRCs show tumour characteristics similar to known MLH1 epimutation CRCs and demonstrate mosaic monoallelic MLH1 epimutation patterns
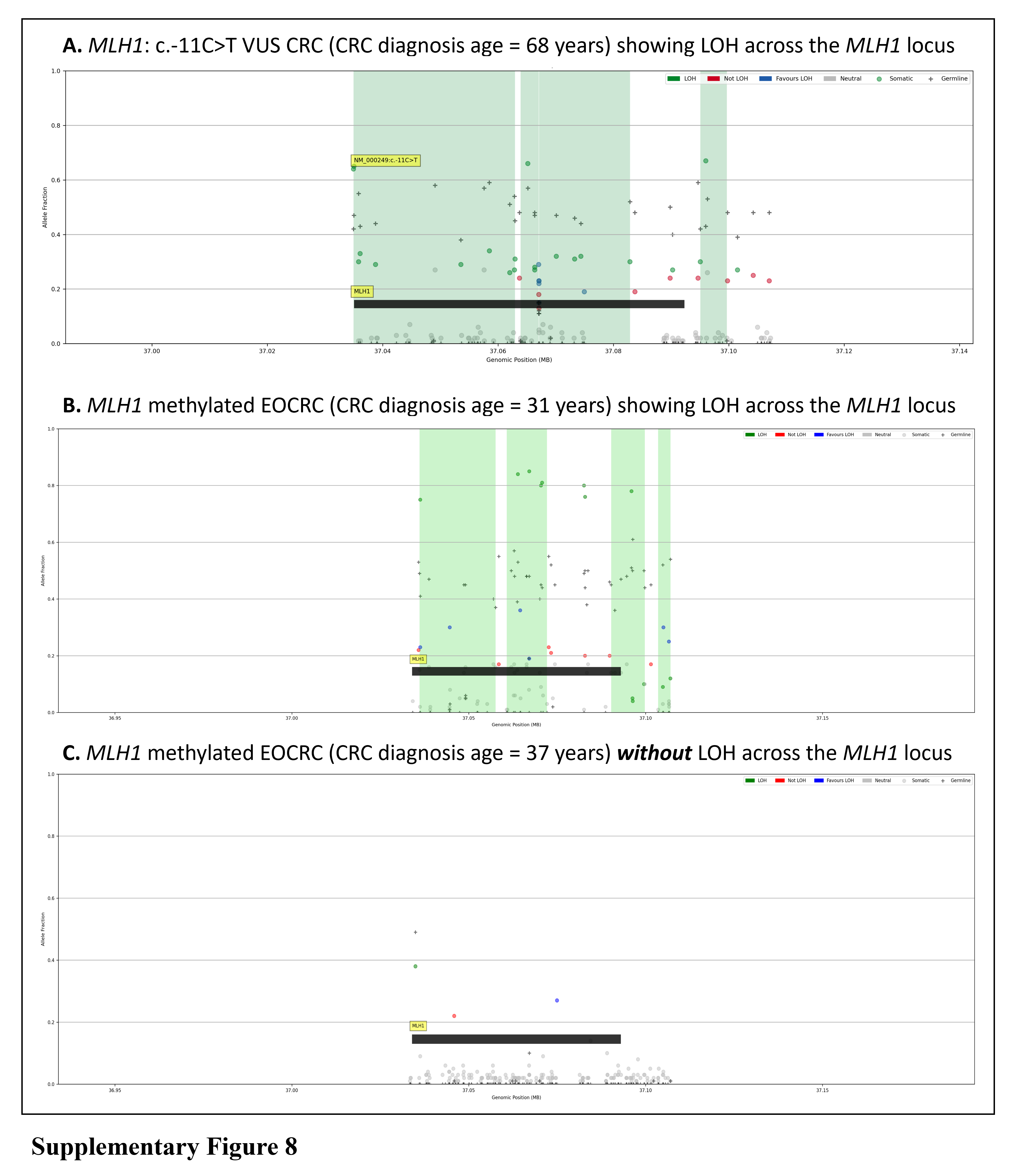
CRCs from MLH1: c.-11C > T VUS carriers had late diagnosis age, demonstrated loss of MLH1/PMS2 protein expression by IHC and showed MLH1 hypermethylation in the tumour concordantly by both loci-specific techniques and the HMEPIC data (Table 2). One of the MLH1: c.-11C > T VUS carriers demonstrated 29% methylation in the MLH1 gene promoter in a metachronous duodenal cancer and showed 1% MLH1 methylation in the blood-derived DNA as detected by MethyLight. The second MLH1: c.-11C > T VUS carrier demonstrated 0% MLH1 methylation in their blood-derived DNA by MethyLight. CRCs from both MLH1: c.-11C > T VUS carriers showed a “second somatic hit” in MLH1 as a single nucleotide variant or LOH (large deletion of wildtype allele, Additional file 9: Fig. S8A). Of note, a “second somatic hit” by LOH or single nucleotide variant in MLH1 was also observed in each of the primary and secondary MLH1 epimutation CRCs and in 6/8 (75%) of LS-CRCs but no second somatic hit in MLH1 was observed in the sporadic MLH1 methylated CRCs.
Table 2.
The characteristics of the six CRCs in the diagnostically challenging group comprised of carriers of MLH1 promoter VUS (n = 3) and MLH1 methylated early-onset CRCs (n = 3)
| Inclusion Group | Dx age (yrs) | Sex | CRC anatomical site | MMR IHC | MSIe | Mean methylation in the VM-CpGsd | CIMP in CRC (Total positive)b | Mean MLH1 promoter methylation (Tumour) a | Blood MLH1 promoter methylation (MethyLight)c |
Non-tumour MLH1 promoter methylation (ddPCR)d | Additional tissue tested (MethyLight)c | MMR genes LOH/somatic mutation | APC promoter methylationf | KRAS somatic mutation |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
MLH1 VUS (MLH1: c.-11C > T) |
68 | F | Proximal | MLH1/PMS2 loss | MSI-H | 0.288 |
Negative (0/5) |
0.43 | 0% |
Blood—0% NMadj—2.3% NMdist—1.2% Buccal—6.5% |
– | MLH1 LOHg | 0.12 | None detected |
|
MLH1 VUS (MLH1: c.-11C > T) |
60 | M | Proximal | MLH1/PMS2 loss | MSI-H | 0.392 | Negative (2/5) | 0.39 | 1% |
Blood—4.5% NMadj—3.4% NMdist—nt Buccal—13.9% |
Duodenal cancer—29% | MLH1:p.S131Ter | 0.46 | p.G12D |
|
MLH1 VUS MLH1: c-[28A > G; 7C > T] |
35 | F | Proximal | MMRp (Incomplete loss of MLH1/PMS2) | MSS | 0.282 | Negative (1/5) | 0.14 | 0% |
Blood—0% NMadj—0% NMdist—0% Buccal—0% |
– | None detected | 0.14 | p.G12A |
| MLH1me EOCRC | 37 | F | Proximal | MLH1/PMS2 loss | MSI-H | 0.374 | Negative (2/5) | 0.34 | 0% |
Blood—0% NMadj—nt NMdist—0% Buccal—nt |
– | MLH1: p.S108AfsTer28 | 0.59 | p.G12D |
| MLH1me EOCRC | 43 | M | Proximal | MLH1/PMS2 loss | MSI-H | 0.338 | Negative (0/5) | 0.42 | 1% |
Blood—< 1% NMadj—nt NMdist—4.1% Buccal—2.4% |
TVA—49% | MLH1: p.X701_splice | 0.57 | None detected |
| MLH1me EOCRC | 31 | M | Proximal | MLH1/PMS2 loss | MSI-H | 0.353 | Negative (1/5) | 0.67 | 0% |
Blood—0% NMadj—nt NMdist—nt Buccal—0% |
– | MLH1 LOHg | 0.35 | None detected |
NMdist denotes distal normal mucosal samples derived from resection margin (i.e. non-adjacent), NMadj denotes normal mucosal samples derived from histologically normal mucosal samples adjacent to the tumour, TVA Tubulovillous Adenoma
aThe values indicate mean β-values estimated from 4 CpGs overlapping MLH1 promoter C region [39] derived from the HMEPIC data
bAs assessed from the HMEPIC
cAs assessed by MethyLight technique
dMean methylation levels across the 77,113 most variably methylated CpGs, which was used for defining the Consensus clusters
eEstimated using MANTIS
fMean methylation from 16 CpGs across the APC promoter c(hr3:112,072,926–11,207,395)
gLOH—loss of heterogygosity (across the MLH1 locus). The LOH plot is shown in Additional file 9: Fig. S8
The CRC from the carrier of in cis variants MLH1: c.-[28A > G; 7C > T] was diagnosed at 35 years of age, showed “patchy” loss of MLH1/PMS2 by IHC in both tumour and adjacent normal cells, with mean β of 0.14 in MLH1 promoter (Table 2 and Additional file 2: Fig. S1C). For this CRC, no “second somatic hit” in MLH1 was observed.
The three MLH1 methylated EOCRCs each showed loss of MLH1/PMS2 protein expression by IHC, showed high levels of MLH1 methylation in their tumours (β = 0.34, 0.42, 0.67) and did not have the BRAF p.V600E mutation or CIMP-high (Table 2 and Additional file 3: Fig. S2). One of the three demonstrated 1% MLH1 methylation in the blood-derived DNA by MethyLight and was additionally found to have MLH1 methylation (49%) in a conventional tubulovillous adenoma contiguous to the CRC. Consistent with constitutional MLH1 epimutation CRCs, all three EOCRC MLH1 methylated tumours showed a second somatic hit in MLH1 (Additional file 9: Fig. S8B). Tumour hypermethylation (~ 50%) of the APC promoter region was present in all three MLH1 methylated EOCRCs similar to MLH1 epimutation CRCs (mean β = 0.57) and LS-CRCs (0.33) but higher than the MMR-proficient (0.19) and sporadic MLH1 methylated CRCs (0.16).
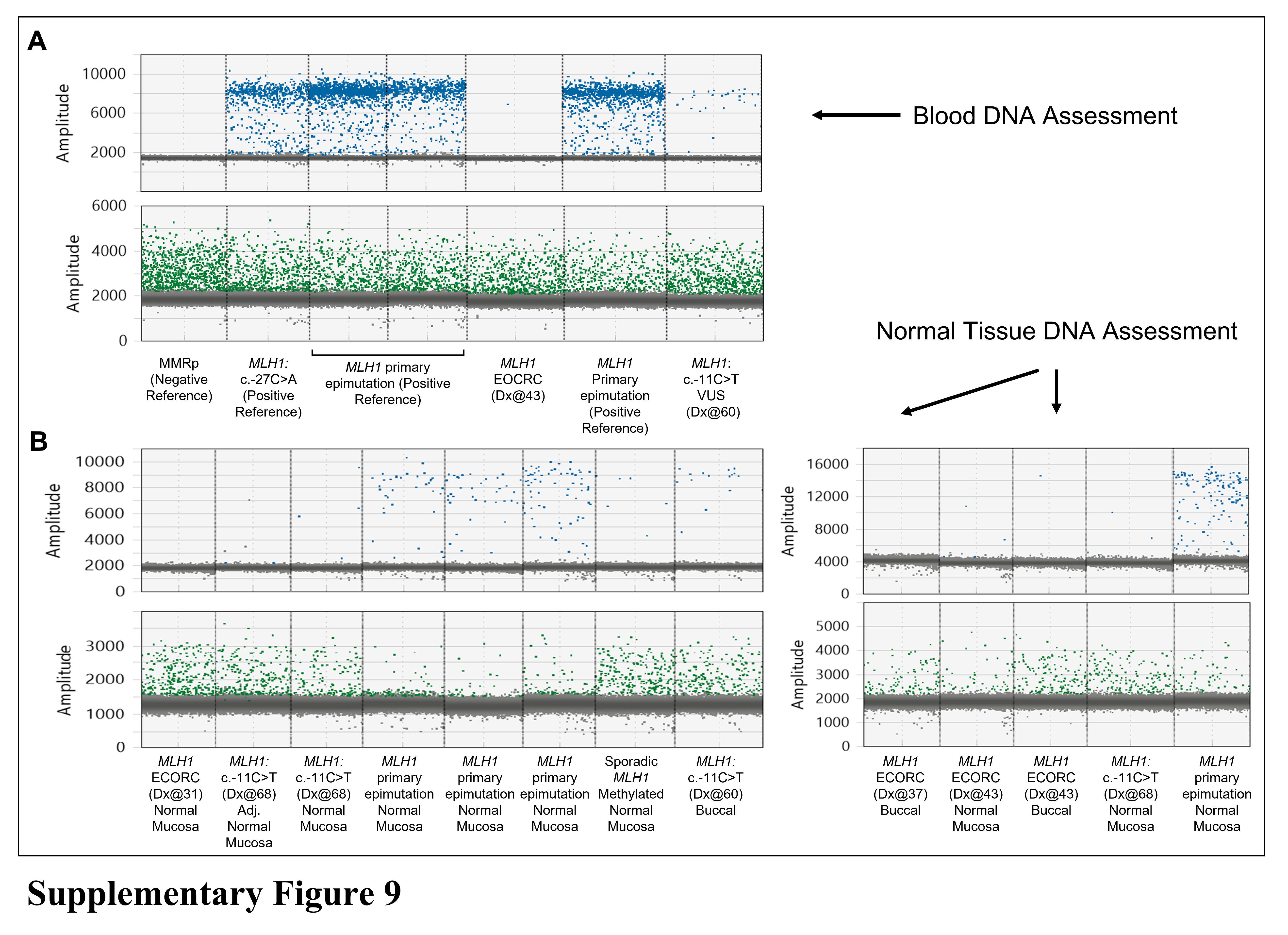
To further investigate potentially low-level mosaic constitutional MLH1 methylation, we employed methylation-sensitive ddPCR to measure MLH1 promoter methylation in blood, normal mucosa and buccal-derived DNA. Mosaic constitutional MLH1 methylation was confirmed in both MLH1: c.-11C > T VUS carriers present in low levels (1.6%-13.4%) across the three tissue types (Table 2 and Additional file 1: Table S6). Both carriers showed positive (> 1%) methylation in normal mucosa and buccal DNA samples, whilst one also showed MLH1 methylation in blood DNA samples. One of the three MLH1 methylated EOCRC cases showed a similar mosaic methylation pattern in all three tissue samples. In comparison, the three primary and two secondary MLH1 epimutation cases all showed MLH1 hypermethylation in blood and normal mucosa-derived DNA samples, whilst CRCs from other reference groups had no detectible methylation in blood or normal mucosa except for one sporadic MLH1 methylated CRC (age of CRC diagnosis = 78 years), which showed 3.8% methylation only in the distant normal mucosa but not in blood or adjacent normal mucosa. The MLH1: c.-[28A > G; 7C > T] VUS carrier did not show evidence of MLH1 methylation in blood, normal mucosa or buccal DNA samples. The ddPCR results are described in Additional file 1: Table S6 and also illustrated in Additional file 10: Fig. S9 for representative samples.
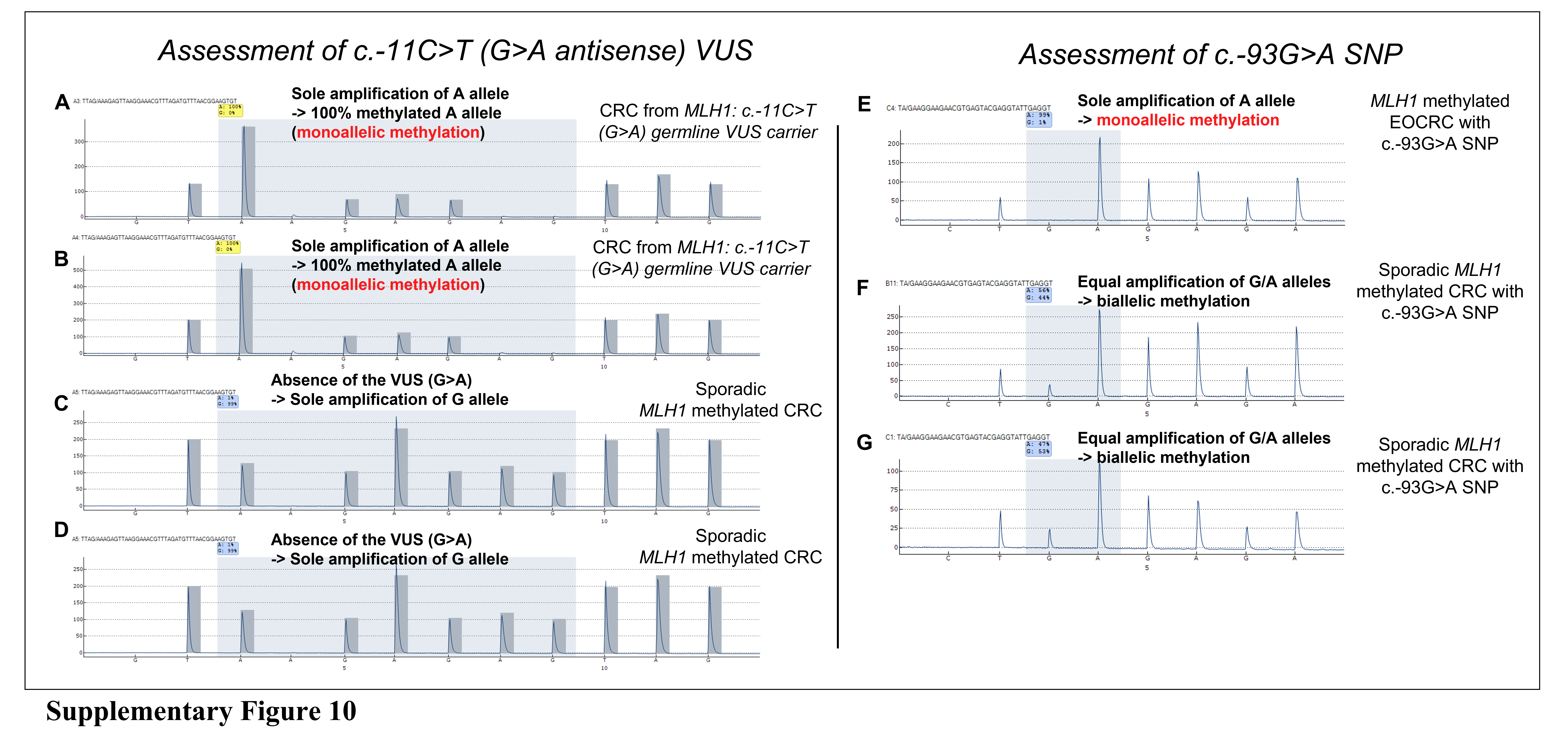
Using the SMART-PCR and pyrosequencing, we tested for allelic methylation levels in tumour DNA samples from two MLH1: c.-11C > T VUS carriers and one MLH1 methylated EOCRC case with a nearby single nucleotide polymorphism (SNP) (c.-93G > A/rs1800734). We found monoallelic MLH1 methylation in tumours from both MLH1: c.-11C > T VUS carriers where the allele carrying the c.-11C > T variant was methylated (Additional file 11: Fig. S10A & B). Here, as the MLH1: c.-11C reference residue is affected by the sodium bisulphite treatment, the methylation-specific PCR was targeted to the antisense strand (G > A). In two sporadic MLH1 methylated CRCs without the c.-11C > T (G > A antisense) variant (Additional file 11: Fig. S10C, D), the G (reference) allele showed sole amplification in absence of the A variant allele. In the MLH1 methylated EOCRC case with the heterozygous MLH1: c.-93G > A SNP, methylation was specifically associated with the SNP (A) allele showing sole amplification of the A allele (Additional file 11: Fig. S10E). In comparison, biallelic methylation was confirmed in all sporadic MLH1 methylated CRCs with methylation present in both alleles with MLH1: c.93A or G SNP showing equal amplification of both A and G alleles (Additional file 11: Fig. S10 F, G).
The somatic mutation profiles differ between the four methylation-derived consensus clusters
We investigated differences in the somatic mutational profiles between the four Consensus Clusters. An enrichment analysis identified 20 genes in which somatic mutations were significantly (P < 0.05) associated with each of the Consensus Clusters (Fig. 4). Specifically, six genes (ACVR2A, DCC, RNF43, TCF7, B2M and BRAF) were associated (P < 0.05) with Consensus Cluster 3 CRCs. Additionally, Consensus Cluster 3 CRCs (P < 0.05) were also associated with less frequent mutations in four genes (APC, KMT2C, KRAS and TCFL2) when compared to the rest. Consensus Cluster 1 CRCs were associated (P < 0.05) with infrequent mutations in three genes (BRCA2, SETD2 and BMPR2) but no frequently mutated genes were identified. Consensus Cluster 4 CRCs were associated with frequent somatic mutations in seven genes (LMO7, MYH9, UTP20, FAN1, LARP7, MEN1 and TERT) (P < 0.05). MLH1 promoter VUS CRCs and MLH1 methylated EOCRCs clustered synonymously with MLH1 epimutation CRCs, showing infrequent mutations in DCC, RNF43, TCF7, B2M and BRAF but showing frequent mutations in APC, KMT2C, KRAS and TCF7L2.
Fig. 4.

Somatic mutational “oncoplot” showing differential mutational patterns by Consensus Clusters as defined by genome-wide DNA methylation. Somatic mutational profiles of 20 novel genes, including 3 genes associated with Consensus Cluster 3, 10 genes associated with Consensus Cluster 3 and 7 genes associated with Consensus Cluster 4 are shown. The samples are shown in vertical lines and ordered by CRC subgroup and their Consensus Clusters. The total mutational burden (TMB) shows the accumulative numbers of somatic mutations identified in each tumour sample. Individual tumour samples are further annotated by CRC subgroup, anatomical location, gender and CRC diagnosis age (years). The different types of somatic mutations are shown in different colours, and the compositional barplots illustrate the total loads of somatic mutations separated by the nucleotide changes
Discussion
This integrative analysis of genome-wide DNA methylation and somatic mutational profiles of 38 CRCs of six clinically relevant subgroups of sporadic and inherited CRCs provides insight into these tumours’ molecular heterogeneity. This study identified unique genome-wide DNA methylation aberrations and somatic mutations associated with rare MLH1 epimutation CRCs and assessed CRC aetiologies in three germline carriers of MLH1 promoter VUS and three EOCRCs with MLH1 methylation (Fig. 1). Unique tumour features were identified that may augment the detection of MLH1 epimutation carriers, including genome-wide DNA methylation as depicted by Consensus Clusters, frequent somatic mutations in APC, KRAS codons 12&13, KMT2C and TCF7L2, a second somatic hit in MLH1 with monoallelic methylation [12], and APC promoter methylation.
This study identified mosaic constitutional MLH1 epimutation associated with the MLH1: c.-11C > T germline VUS in two CRCs, detected in non-tumour DNA samples. Both CRCs showed tumour features concordant with CRCs from known constitutional MLH1 epimutation cases, suggesting the same CRC aetiology. Using methylation-sensitive ddPCR, we identified low-level methylation in blood, normal mucosa and buccal DNA from MLH1: c.-11C > T germline VUS carriers. The absence of both the BRAF p.V600E mutation and the widespread hypermethylation across the variably methylated CpGs suggests that these CRCs and the MLH1 promoter hypermethylation have not arisen through the serrated pathway [53], despite their later age at CRC diagnosis. A previous study showed that this variant induced a significant reduction of the MLH1 transcription [19], though was unable to detect methylation in the blood DNA. These authors described variable CRC diagnosis age with no remarkable family history associated with the c.-11C > T variant [19], suggesting a lower penetrance of this variant.
No evidence of constitutional MLH1 methylation was identified in normal mucosa, blood or buccal DNA for the MLH1: c.-[28A > G; 7C > T] in cis germline variant, despite observing weak (β = 0.14) methylation in the tumour DNA, heterogenous loss of MLH1/PMS2 protein expression and some of the tumour features associated with known MLH1 epimutation carriers. In previous studies, these germline variants have been shown to induce a partial and constitutional reduction of MLH1 expression, however, without causing significant MLH1 promoter methylation in tumour or normal tissue [24, 54]. Therefore, the findings from the present study remain inconclusive for determining the mechanism of pathogenesis of these germline variants.
This study showed that constitutional MLH1 epimutation underlie a subset of EOCRCs with MLH1 methylation by identifying tumour features associated with MLH1 epimutation and low-level mosaic MLH1 methylation in non-tumour DNA. Since none were found to carry germline pathogenic variants or VUS in the promoter region, it suggests a de novo origin of mosaic MLH1 epimutation. These EOCRCs were BRAF wildtype with no presentation of CIMP or widespread genome-wide DNA methylation aberrations, suggesting these CRCs did not develop via the serrated pathway [55, 56]. Whilst no evidence that this was related to secondary epimutation or methylation quantitative trait locus was found, we cannot exclude the possibility that the constitutional MLH1 methylation was caused by germline and probable de novo, structural variants including insertion of repetitive elements [57], or a large inversion or duplication involving the MLH1 gene region [58], for which our sequencing platform did not provide the resolution to detect. Furthermore, the three EOCRCs did not show a family history of Lynch syndrome spectrum cancers, further supporting the absence of a highly penetrant, inherited cis-acting genetic variant in these participants (see Additional file 8: Fig. S7 for pedigrees).
Although triaging CRC cases for MLH1 epimutation testing varies between clinics, young cases with ostensibly sporadic CRCs with MLH1 methylation have been recommended to be screened for possible MLH1 epimutation [10, 26] and our findings support the importance of this. Although the transgenerational heritability of primary MLH1 epimutation is yet to be completely understood [10], a transmission of mosaic MLH1 epimutation from an asymptomatic carrier into a full-blown MLH1 epimutation in the offspring has been reported [22], highlighting the clinical importance of early detection of such cases. This will also help alleviate the subsequent cancer risk by recommending appropriate surveillance.
A significant association between APC promoter hypermethylation in the CRCs from constitutional MLH1 epimutation carriers and the MLH1 methylated EOCRCs was observed. Though not extensively reported, APC methylation has been shown to be inversely correlated with CIMP in BRAF wildtype CRCs [59], that is consistent with the findings of the current study. Given its primary role as the main Wnt regulator, our finding warrants further investigation into the functional importance of APC hypermethylation in MLH1 epimutation and as an additional feature to distinguish MLH1 epimutation carriers from sporadic MLH1 methylated CRCs.
This study had several limitations including the small number of MLH1 epimutation CRCs, germline MLH1 promoter VUS and MLH1 methylated EOCRCs. Despite these being rare subtypes of CRCs, further validation in additional CRCs from these groups is needed to confirm our findings and the likely prevalence of MLH1 methylation mosaicism in EOCRCs. Further studies will also be needed to identify the mechanism underlying mosaicism of MLH1 methylated EOCRCs. The ability to differentiate MLH1 epimutations arising de novo (primary MLH1 epimutation) from those with a genetic basis (secondary) has implications for the relative testing and clinical management. Although no difference was observed in this study, the identification of genome-wide DNA methylation profiles that differentiate primary and secondary MLH1 epimutation CRCs may require larger sample sizes and, if present, would indicate the need for further genetic testing such as long-read or RNA-sequencing to identify a causative germline variant.
In our study, transcriptional loss associated with monoallelic MLH1 methylation in MLH1: c.-11C > T VUS CRCs and MLH1 methylated EOCRCs within the blood was not confirmed by expression studies. However, determining reduced monoallelic expression is not feasible due to the low proportion of MLH1 methylated alleles (mosaicism) in blood DNA. Detecting small changes in expression resulting from only a few methylated alleles in the background of many non-methylated alleles is not feasible. Transcriptional loss of both alleles within the tumour is confirmed by the loss of MLH1 protein expression determined by IHC where one allele is defective due to hypermethylation and the other allele through a second somatic hit.
Conclusions
MLH1 epimutations may account for up to 10% of all CRCs with MLH1 protein loss without germline MLH1 mutation [26] suggesting that currently, MLH1 epimutations might be underdiagnosed and consequently the true disease burden caused by MLH1 epimutation is unknown [15, 60]. Currently, no consensus guidelines for triaging potential MLH1 epimutation carriers exist. Further, unpredictable transgenerational inheritance patterns and the presence of mosaic patterns seen in the carriers, as well as the lack of sensitive testing tools such as genome-wide methylation or ddPCR as demonstrated in this study, contribute towards the current impediment in identifying MLH1 epimutation carriers and providing personalised clinical management [10]. Here, our study has provided additional molecular features based on genome-wide DNA methylation and somatic mutational landscapes that may be useful for triaging MLH1 epimutation carriers and provide supporting evidence for resolving VUS associated with MLH1 epimutation and identifying potential epimutation carriers among young cancer cases with mosaic constitutional MLH1 epimutation.
Supplementary Information
Additional file 1. Table S1: Sequences of primers included in this study. Table S2: The characteristics of 38 CRCs from six reference CRC groups. Table S3: The characteristics of 44 tumour and 14 normal colonic mucosal samples from the 43 people included in the study. Table S4: Summary of somatic mutations occurrent in key CRC genes and CRC subgroups associated with frequent or less frequent somatic mutation in the described gene. Table S5: Differentially Methylated Regions between tumours of MLH1 epimutation carriers and sporadic MLH1 methylated CRCs. Table S6: MLH1 promoter methylation levels in non-tumour DNA samples measured by methylation-sensitive digital droplet PCR.
{kind=link}
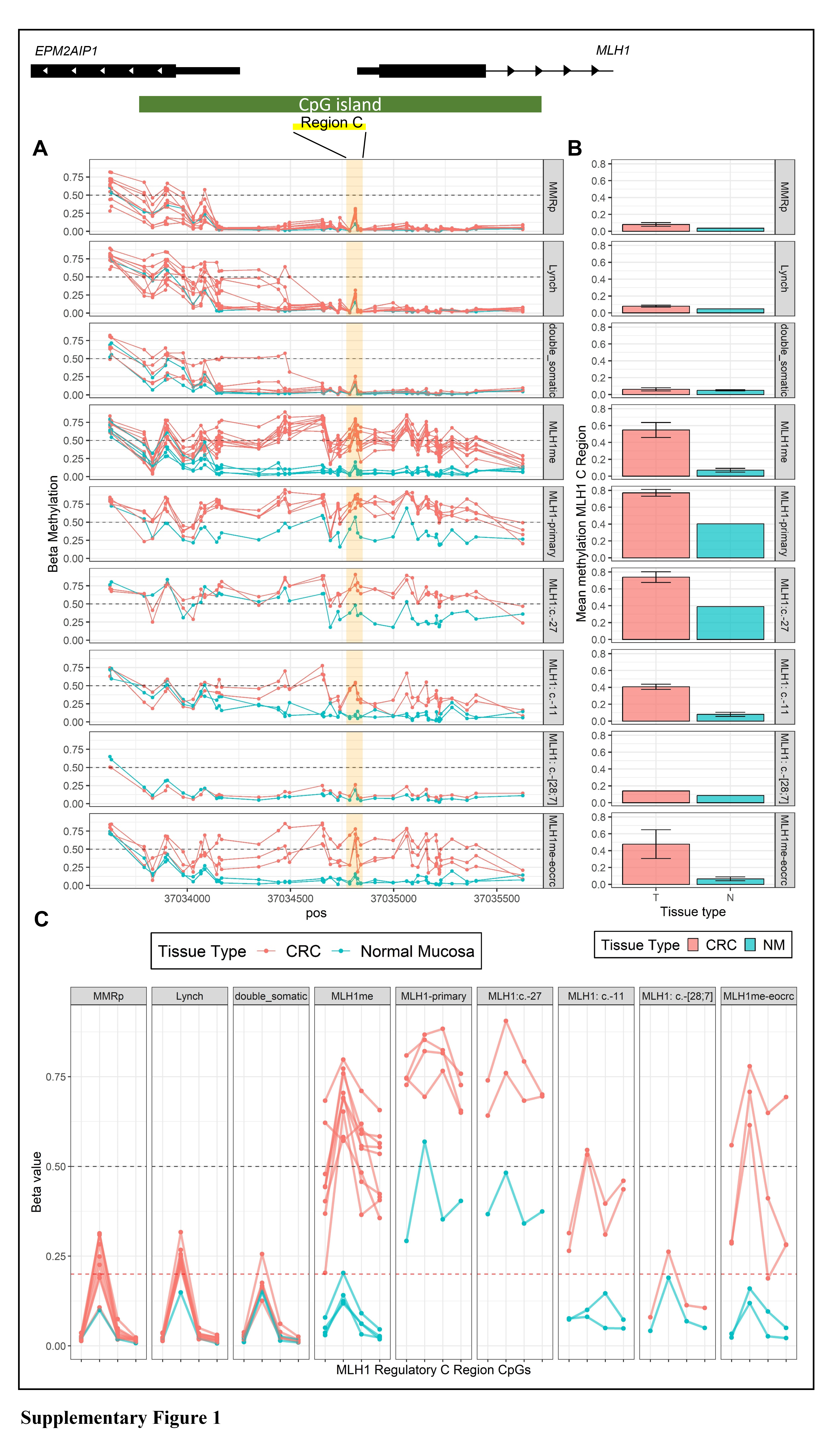
Additional file 2. Figure S1:DNA methylation levels across the CpG island associated with the MLH1 promoter by CRC subgroups: 1) MMR-proficient CRCs, 2) Lynch-syndrome associated CRCs, 3) double MMR somatic mutation CRCs, 4) sporadic MLH1 methylated CRCs, 5) constitutional primary MLH1 epimutation CRCs and 6) constitutional secondary MLH1 epimutation CRCs, as well as two diagnostically challenging CRC groups: 1) MLH1 promoter VUS carriers with MLH1: c.-11C>T and MLH1: c.-[28A>G; 7C>T] and 2) MLH1 methylated EOCRCs. DNA methylation of tumour-derived DNA are shown in red and normal mucosal DNA is shown in blue. All normal mucosa samples were from resection margin, not adjacent to the tumour. Yellow boxes denote the regulatory C region as described in Deng et al. Bar plots showing mean methylation within the regulatory C region of MLH1 gene promoter across all samples for each CRC group. Tumour and normal mucosal samples are separately shown. Error bars denote standard deviation. Close-up view of four CpGs overlapping the regulatory C region.
{kind=link}
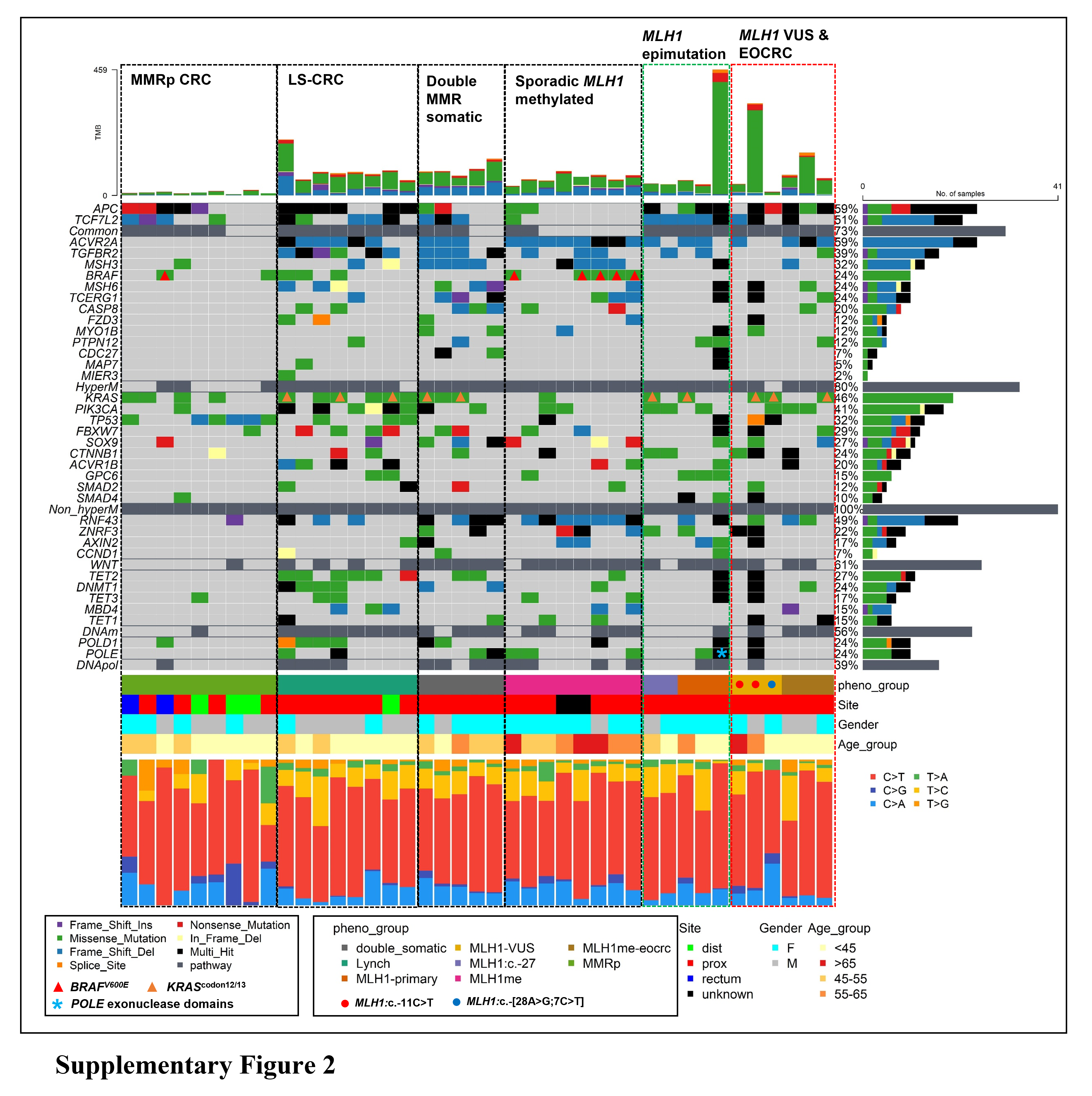
Additional file 3. Figure S2: Oncoplot showing somatic mutational profiles at the 26 recurrently mutated CRC genes identified in CRC tumours from TCGA, 4 additional Wnt pathway associated genes and 5 genes related to the DNA methylation machinery. The total mutational burden shows the accumulative numbers of somatic mutations identified in each tumour sample. Individual tumour samples are further annotated by CRC subgroup, anatomical location, gender and CRC diagnosis age. The different types of somatic mutations are shown in different colours and the compositional barplots illustrate the total loads of somatic mutations separated by the nucleotide changes. One primary MLH1 epimutation CRC had a POLE somatic mutation in the exonuclease domain and also had the highest TMB with 459 total somatic mutations.
{kind=link}
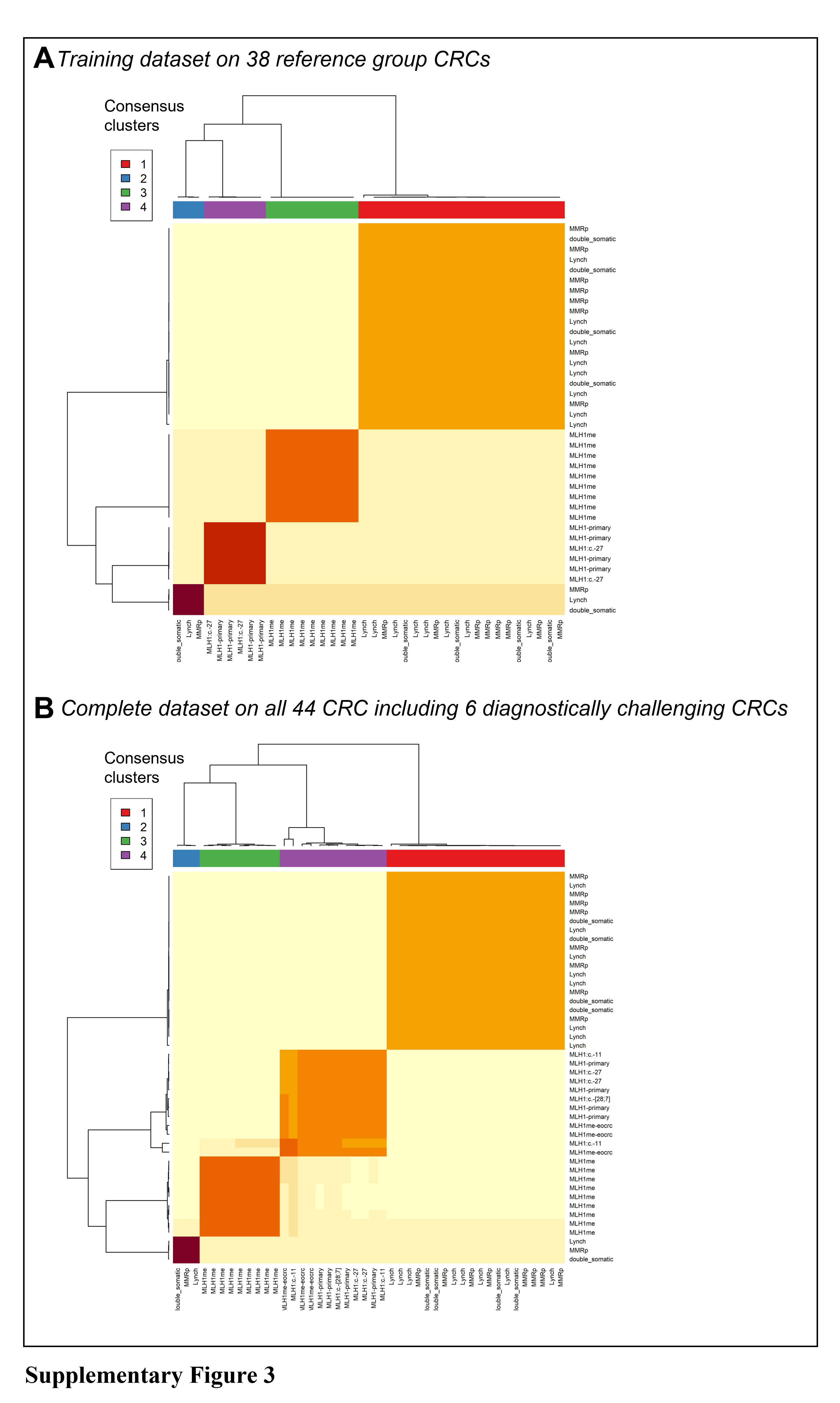
Additional file 4. Figure S3: Heatmaps illustrating the consensus matrix identified by the ConsensuClusterPlus analysis. A illustrates the consensus matrix estimated on 38 reference group CRCs. B illustrates the same analysis but performed on the complete dataset of 44 CRCs including six diagnostically challenging CRCs. The final consensus cluster classification for individual samples are shown in four different colours. The darker heatmaps indicate the stability evidence for classifying individual samples into each cluster. The raw consensus values are provided in Supplementary data.
{kind=link}
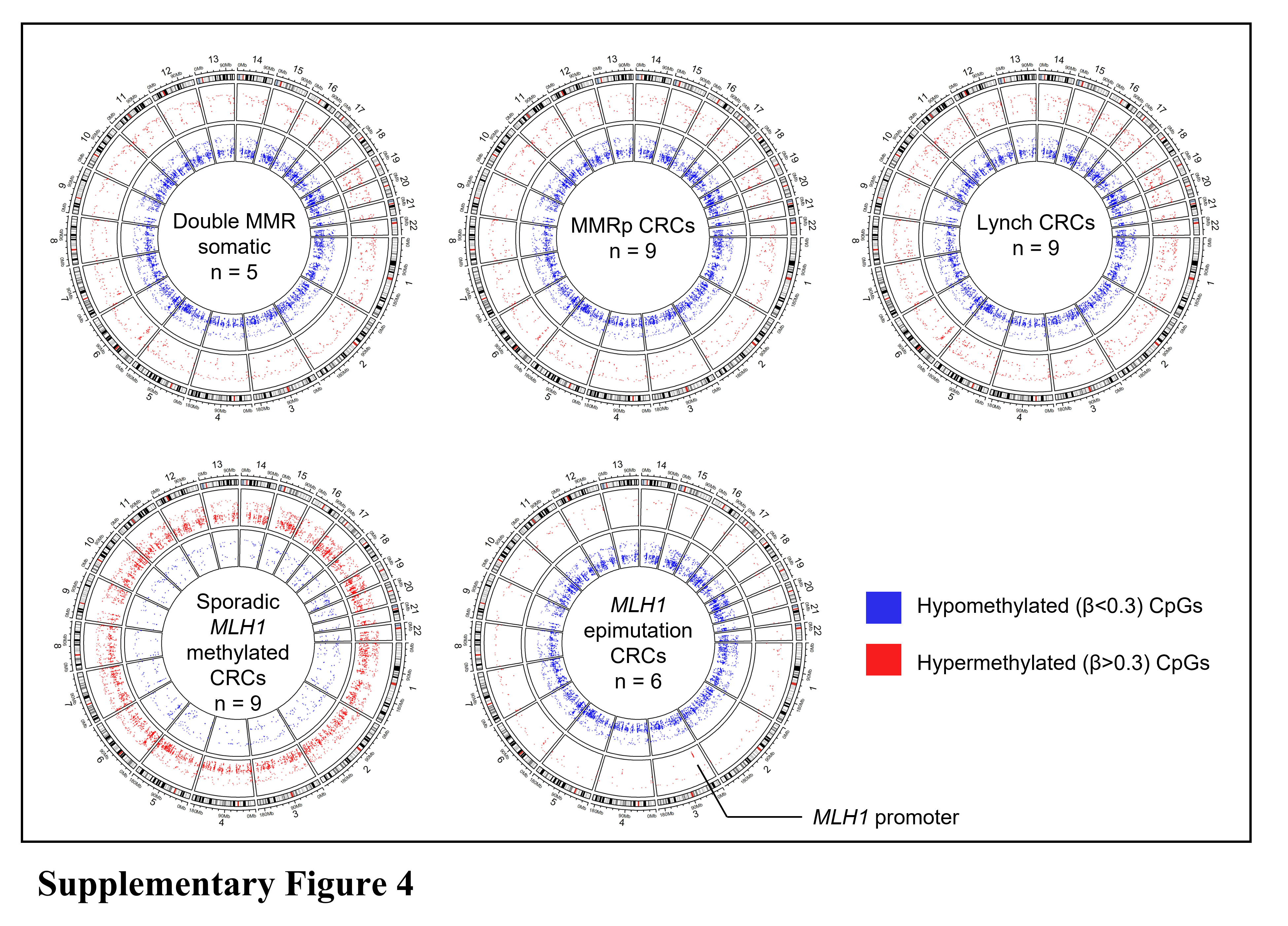
Additional file 5. Figure S4: “Circos” plots showing mean genomic hypomethylation and hypermethylation patterns in tumours of five reference CRC groups. For the MLH1 epimutation group, five primary and two secondary epimutation CRCs were combined in this analysis. Mean methylation values are shown per each CRC group.
{kind=link}
Additional file 6. Figure S5: DNA methylation patterns at CpG site cg15103403 within LRRFIP2 gene for each of the six reference CRC subgroups and the two diagnostically challenging CRC subgroups.
{kind=link}
Additional file 7. Figure S6: Venn diagrams showing numbers of Differentially Methylated Regions of LS-CRCs, MMR-proficient CRCs, sporadic MLH1 methylated CRCs and double somatic MMR CRCs when compared with the primary and secondary MLH1 epimutation CRCs. Venn diagram shows the number of hypermethylated DMRs and Venn diagram showing the number of hypomethylated DMRs in common with the MLH1 epimutation group.
{kind=link}
Additional file 8. Figure S7: Pedigrees for each the six people in the diagnostically challenging group comprised of the MLH1: c.-11C>T, MLH1: c.-[28A>G; 7C>T] VUS carriers, and the three people with MLH1 methylated EOCRCs.
{kind=link}
Additional file 9. Figure S8: Loss of heterozygosity plots in the tumours of the MLH1: c.-11C>T VUS carrier and the MLH1 methylated EOCRC across the MLH1 locus. Points represent somatic and germline variants plotted by genomic position and variant allele fraction, where green circles/shading support LOH of the region. C showing the variants across the MLH1 locus in tumour samples from one of the MLH1 methylated EOCRCs without LOH.
{kind=link}
Additional file 10. Figure S9: Droplet digital PCR results in representative samples illustrated as “1D Amplitude” plots. Methylation positive and negative droplets are shown in blue and green dots, respectively. A illustrates MLH1 methylation results in samples demonstrating negative MLH1 methylation, hypermethylation of MLH1, and low mosaic methylation in MLH1 methylated EOCRC and MLH1: c.-11C>T VUS carrier. Similarly, B shows MLH1 methylation patterns in normal and buccal-derived DNA samples.
{kind=link}
Additional file 11. Figure S10: Pyrosequencing profiles of tumour DNA samples underwent for SMART-PCR to assess monoallelic methylation pattern of the MLH1 promoter. A-B, CRCs from two heterozygous MLH1: c.-11C>T VUS carriers showing occurrent MLH1 promoter methylation specifically in the variant allele. C-D, two reference sporadic MLH1 methylated CRCs without the c.-11C>T variant and hence showing sole amplification of G reference allele. E, one MLH1 methylated EOCRC that was heterozygous for the c.-93G>A promoter SNP showing sole amplification of the A SNP allele indicating monoallelic methylation associated only with this SNP allele. F-G, two sporadic MLH1 methylated CRCs that both were heterozygous for the c.-93G>A SNP showing the amplification of both G and A alleles indicating biallelic methylation of both A and G alleles at MLH1: c.-93 locus.
Additional file 12. Supplementary data: Raw consensus matrix values derived from the Consensus clustering analysis on 77,113 most variably methylated CpGs, which was performed on 44 CRCs including the 6 diagnostically challenging CRCs.
Acknowledgements
We thank the members of the Colorectal Oncogenomics Group for their support of this manuscript. We thank the participants and staff from the Australasian Colorectal Cancer Family Registries (ACCFR) and the ANGELS study. We especially thank Maggie Angelakos, Samantha Fox and Allyson Templeton for supporting this study. We thank the Australian Genome Research Facility for their collaboration on this project and Melbourne Bioinformatics for their support of this work. “The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the Colon Cancer Family Registry (Colon-CFR), nor does mention of trade names, commercial products, or organisations imply endorsement by the US Government or the Colon-CFR.”
Abbreviations
- ANGELS
Applying Novel Genomics approaches to Early-onset and suspected Lynch Syndrome colorectal and endometrial cancers
- ACCFR
Australasian Colorectal Cancer Family Registry
- CRC
Colorectal cancer
- ddPCR
Droplet digital PCR
- DMR
Differentially methylated region
- EOCRC
Early-onset colorectal cancer
- FFPE
Formalin-fixed paraffin-embedded
- HMEPIC
HumanMethylationEPIC
- IHC
Immunohistochemical staining
- LS
Lynch syndrome
- MSI
Microsatellite instability
- MMR
DNA mismatch repair
- MMRd
MMR-deficient
- MMRp
MMR-proficient
- PCR
Polymerase chain reaction
- SBS
Single base substitution
- TMB
Tumour mutational burden
- TMS
Tumour mutational signatures
- VUS
Variants of uncertain clinical significance
- WES
Whole exome sequencing
Author contributions
DDB and JEJ conceived the original study concept and design and designed the analysis. LG, AB, CR, FAM, IMW, AKW, JLH, MAJ and DDB contributed to the acquisition of study data. MW, MF, ML, JW, HM, RS, JI, EE, MB, JK, EI, LM and YA recruited and referred CRC patients to the study. The sample curation and laboratory testing were performed by JEJ, RW, IC, MC, JC, SJ, SP and AD. JEJ, KM and PG performed the bioinformatics analysis. JEJ and DDB prepared the manuscript. All authors provided critical revisions to the manuscript for important intellectual content and have read and approved of the final manuscript.
Funding
The design, analysis and interpretation of data for this study was supported by a National Health and Medical Research Council of Australia (NHMRC) project grant GNT1125269 (PI- Daniel Buchanan). In addition, this study was supported by the Australian Genome Research Facility’s 2020 Summer Oncology Mini Grant Competition. DDB is supported by an NHMRC Investigator grant (GNT1194896) and University of Melbourne Dame Kate Campbell Fellowship. RW is supported by the Margaret and Irene Stewardson Fund Scholarship and by the Melbourne Research Scholarship. PG is supported by the University of Melbourne Research Scholarship. MAJ is supported by an NHMRC Investigator grant (GNT1195099). JLH is supported by the University of Melbourne Dame Kate Campbell Fellowship. Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number U01CA167551 and through a cooperative agreement with the Australasian Colorectal Cancer Family Registry (NCI/NIH U01 CA074778 and U01/U24 CA097735) and by the Victorian Cancer Registry, Australia. This research was performed under CCFR approved projects C-AU-0818-01, C-AU-1014-01, C-AU-0312-01.
Availability of data and materials
Genome-wide DNA methylation data (HumanMethylationEPIC array) has been deposited to GEO and accessible through Accession No. GSE233854.
Declarations
Ethics approval and consent to participate
All study participants provided written informed consent. The study was conducted to the guidelines of the Declaration of Helsinki and approved by the University of Melbourne Human Research Ethics Committee (HREC#1750748).
Completing interests
The authors have no conflicts of interest to declare.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Baylin SB, Jones PA. Epigenetic determinants of cancer. Cold Spring Harbor Persp Biol. 2016;8(9):a019505. doi: 10.1101/cshperspect.a019505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC, et al. Colorectal cancer statistics, 2020. CA Cancer J Clin. 2020;70(3):145–164. doi: 10.3322/caac.21601. [DOI] [PubMed] [Google Scholar]
- 4.Dekker E, Tanis PJ, Vleugels JLA, Kasi PM, Wallace MB. Colorectal cancer. Lancet. 2019;394(10207):1467–1480. doi: 10.1016/S0140-6736(19)32319-0. [DOI] [PubMed] [Google Scholar]
- 5.Weisenberger DJ, Liang G, Lenz HJ. DNA methylation aberrancies delineate clinically distinct subsets of colorectal cancer and provide novel targets for epigenetic therapies. Oncogene. 2018;37(5):566–577. doi: 10.1038/onc.2017.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toyota M, Ohe-Toyota M, Ahuja N, Issa JP. Distinct genetic profiles in colorectal tumors with or without the CpG island methylator phenotype. Proc Natl Acad Sci USA. 2000;97(2):710–715. doi: 10.1073/pnas.97.2.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pearlman R, Haraldsdottir S, de la Chapelle A, Jonasson JG, Liyanarachchi S, Frankel WL, et al. Clinical characteristics of patients with colorectal cancer with double somatic mismatch repair mutations compared with Lynch syndrome. J Med Genet. 2019;56(7):462–470. doi: 10.1136/jmedgenet-2018-105698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aronson M, Colas C, Shuen A, Hampel H, Foulkes WD, Baris Feldman H, et al. Diagnostic criteria for constitutional mismatch repair deficiency (CMMRD): recommendations from the international consensus working group. J Med Genet. 2022;59(4):318–327. doi: 10.1136/jmedgenet-2020-107627. [DOI] [PubMed] [Google Scholar]
- 9.Toon CW, Walsh MD, Chou A, Capper D, Clarkson A, Sioson L, et al. BRAFV600E immunohistochemistry facilitates universal screening of colorectal cancers for Lynch syndrome. Am J Surg Pathol. 2013;37(10):1592–1602. doi: 10.1097/PAS.0b013e31828f233d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hitchins MP. Finding the needle in a haystack: identification of cases of Lynch syndrome with MLH1 epimutation. Fam Cancer. 2016;15(3):413–422. doi: 10.1007/s10689-016-9887-3. [DOI] [PubMed] [Google Scholar]
- 11.Parsons MT, Buchanan DD, Thompson B, Young JP, Spurdle AB. Correlation of tumour BRAF mutations and MLH1 methylation with germline mismatch repair (MMR) gene mutation status: a literature review assessing utility of tumour features for MMR variant classification. J Med Genet. 2012;49(3):151–157. doi: 10.1136/jmedgenet-2011-100714. [DOI] [PubMed] [Google Scholar]
- 12.Hitchins MP. The role of epigenetics in Lynch syndrome. Fam Cancer. 2013;12(2):189–205. doi: 10.1007/s10689-013-9613-3. [DOI] [PubMed] [Google Scholar]
- 13.Hitchins MP, Ward RL. Constitutional (germline) MLH1 epimutation as an aetiological mechanism for hereditary non-polyposis colorectal cancer. J Med Genet. 2009;46(12):793–802. doi: 10.1136/jmg.2009.068122. [DOI] [PubMed] [Google Scholar]
- 14.Hitchins MP, Rapkins RW, Kwok CT, Srivastava S, Wong JJ, Khachigian LM, et al. Dominantly inherited constitutional epigenetic silencing of MLH1 in a cancer-affected family is linked to a single nucleotide variant within the 5'UTR. Cancer Cell. 2011;20(2):200–213. doi: 10.1016/j.ccr.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 15.Hitchins MP. Constitutional epimutation as a mechanism for cancer causality and heritability? Nat Rev Cancer. 2015;15(10):625–634. doi: 10.1038/nrc4001. [DOI] [PubMed] [Google Scholar]
- 16.Damaso E, Canet-Hermida J, Vargas-Parra G, Velasco A, Marin F, Darder E, et al. Highly sensitive MLH1 methylation analysis in blood identifies a cancer patient with low-level mosaic MLH1 epimutation. Clin Epigenetics. 2019;11(1):171. doi: 10.1186/s13148-019-0762-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Niessen RC, Hofstra RM, Westers H, Ligtenberg MJ, Kooi K, Jager PO, et al. Germline hypermethylation of MLH1 and EPCAM deletions are a frequent cause of Lynch syndrome. Genes Chromosomes Cancer. 2009;48(8):737–744. doi: 10.1002/gcc.20678. [DOI] [PubMed] [Google Scholar]
- 18.Pinto D, Pinto C, Guerra J, Pinheiro M, Santos R, Vedeld HM, et al. Contribution of MLH1 constitutional methylation for Lynch syndrome diagnosis in patients with tumor MLH1 downregulation. Cancer Med. 2018;7(2):433–444. doi: 10.1002/cam4.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ward RL, Dobbins T, Lindor NM, Rapkins RW, Hitchins MP. Identification of constitutional MLH1 epimutations and promoter variants in colorectal cancer patients from the Colon Cancer Family Registry. Genet Med. 2013;15(1):25–35. doi: 10.1038/gim.2012.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Snell C, Krypuy M, Wong EM, Loughrey MB, Dobrovic A. BRCA1 promoter methylation in peripheral blood DNA of mutation negative familial breast cancer patients with a BRCA1 tumour phenotype. Breast Cancer Res. 2008;10(1):12. doi: 10.1186/bcr1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hansmann T, Pliushch G, Leubner M, Kroll P, Endt D, Gehrig A, et al. Constitutive promoter methylation of BRCA1 and RAD51C in patients with familial ovarian cancer and early-onset sporadic breast cancer. Hum Mol Genet. 2012;21(21):4669–4679. doi: 10.1093/hmg/dds308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sloane MA, Nunez AC, Packham D, Kwok CT, Suthers G, Hesson LB, et al. Mosaic epigenetic inheritance as a cause of early-onset colorectal cancer. JAMA Oncol. 2015;1(7):953–957. doi: 10.1001/jamaoncol.2015.1484. [DOI] [PubMed] [Google Scholar]
- 23.Yu M, Heinzerling TJ, Grady WM. DNA methylation analysis using droplet digital PCR. Methods Mol Biol. 2018;1768:363–383. doi: 10.1007/978-1-4939-7778-9_21. [DOI] [PubMed] [Google Scholar]
- 24.Hesson LB, Packham D, Kwok CT, Nunez AC, Ng B, Schmidt C, et al. Lynch syndrome associated with two MLH1 promoter variants and allelic imbalance of MLH1 expression. Hum Mutat. 2015;36(6):622–630. doi: 10.1002/humu.22785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Q, Thompson BA, Ward RL, Hesson LB, Sloane MA. Understanding the Pathogenicity of noncoding mismatch repair gene promoter variants in lynch syndrome. Hum Mutat. 2016;37(5):417–426. doi: 10.1002/humu.22971. [DOI] [PubMed] [Google Scholar]
- 26.Hitchins MP, Lynch HT. Dawning of the epigenetic era in hereditary cancer. Clin Genet. 2014;85(5):413–416. doi: 10.1111/cge.12369. [DOI] [PubMed] [Google Scholar]
- 27.Georgeson P, Pope BJ, Rosty C, Clendenning M, Mahmood K, Joo JE, et al. Evaluating the utility of tumour mutational signatures for identifying hereditary colorectal cancer and polyposis syndrome carriers. Gut. 2021;70:2138. doi: 10.1136/gutjnl-2019-320462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jenkins MA, Win AK, Templeton AS, Angelakos MS, Buchanan DD, Cotterchio M, et al. Cohort profile: the colon cancer family registry cohort (CCFRC) Int J Epidemiol. 2018;47:387. doi: 10.1093/ije/dyy006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buchanan DD, Clendenning M, Rosty C, Eriksen SV, Walsh MD, Walters RJ, et al. Tumor testing to identify lynch syndrome in two Australian colorectal cancer cohorts. J Gastroenterol Hepatol. 2017;32(2):427–438. doi: 10.1111/jgh.13468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, et al. MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000;28(8):E32. doi: 10.1093/nar/28.8.e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wojdacz TK, Dobrovic A. Methylation-sensitive high resolution melting (MS-HRM): a new approach for sensitive and high-throughput assessment of methylation. Nucleic Acids Res. 2007;35(6):e41. doi: 10.1093/nar/gkm013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walker R, Georgeson P, Mahmood K, Joo JE, Makalic E, Clendenning M, et al. Evaluating multiple next-generation sequencing derived tumor features to accurately predict DNA mismatch repair status. medRxiv. 2022. [DOI] [PMC free article] [PubMed]
- 33.Joo JE, Clendenning M, Wong EM, Rosty C, Mahmood K, Georgeson P, et al. DNA methylation signatures and the contribution of age-associated methylomic drift to carcinogenesis in early-onset colorectal cancer. Cancers. 2021;13(11):2589. doi: 10.3390/cancers13112589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joo JE, Jayasekara H, Wong EM, Clendenning M, Rosty C, Winship IM, et al. Assessing the ProMCol classifier as a prognostic marker for non-metastatic colorectal cancer within the Melbourne Collaborative Cohort Study. Gut. 2019;68(4):761–762. doi: 10.1136/gutjnl-2018-316122. [DOI] [PubMed] [Google Scholar]
- 35.Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fortin JP, Labbe A, Lemire M, Zanke BW, Hudson TJ, Fertig EJ, et al. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014;15(12):503. doi: 10.1186/s13059-014-0503-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Triche TJ, Jr, Weisenberger DJ, Van Den Berg D, Laird PW, Siegmund KD. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res. 2013;41(7):e90. doi: 10.1093/nar/gkt090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010;11:587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deng G, Chen A, Hong J, Chae HS, Kim YS. Methylation of CpG in a small region of the hMLH1 promoter invariably correlates with the absence of gene expression. Can Res. 1999;59(9):2029–2033. [PubMed] [Google Scholar]
- 40.Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38(7):787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 41.Peters TJ, Buckley MJ, Statham AL, Pidsley R, et al. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin. 2015;8:6. doi: 10.1186/1756-8935-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26(12):1572–1573. doi: 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28(14):1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 45.Kautto EA, Bonneville R, Miya J, Yu L, Krook MA, Reeser JW, et al. Performance evaluation for rapid detection of pan-cancer microsatellite instability with MANTIS. Oncotarget. 2017;8(5):7452–7463. doi: 10.18632/oncotarget.13918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mayakonda A, Lin DC, Assenov Y, Plass C, Koeffler HP. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018;28(11):1747–1756. doi: 10.1101/gr.239244.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cancer Genome Atlas N Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lannagan TRM, Lee YK, Wang T, Roper J, Bettington ML, Fennell L, et al. Genetic editing of colonic organoids provides a molecularly distinct and orthotopic preclinical model of serrated carcinogenesis. Gut. 2019;68(4):684–692. doi: 10.1136/gutjnl-2017-315920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Briggs S, Tomlinson I. Germline and somatic polymerase epsilon and delta mutations define a new class of hypermutated colorectal and endometrial cancers. J Pathol. 2013;230(2):148–153. doi: 10.1002/path.4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38(1):23–38. doi: 10.1038/npp.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Candiloro IL, Dobrovic A. Detection of MGMT promoter methylation in normal individuals is strongly associated with the T allele of the rs16906252 MGMT promoter single nucleotide polymorphism. Cancer Prev Res. 2009;2(10):862–867. doi: 10.1158/1940-6207.CAPR-09-0056. [DOI] [PubMed] [Google Scholar]
- 52.Zhang L, Shay JW. Multiple roles of apc and its therapeutic implications in colorectal cancer. J Natl Cancer Inst. 2017 doi: 10.1093/jnci/djw332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rosty C, Hewett DG, Brown IS, Leggett BA, Whitehall VL. Serrated polyps of the large intestine: current understanding of diagnosis, pathogenesis, and clinical management. J Gastroenterol. 2013;48(3):287–302. doi: 10.1007/s00535-012-0720-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morak M, Ibisler A, Keller G, Jessen E, Laner A, Gonzales-Fassrainer D, et al. Comprehensive analysis of the MLH1 promoter region in 480 patients with colorectal cancer and 1150 controls reveals new variants including one with a heritable constitutional MLH1 epimutation. J Med Genet. 2018;55(4):240–248. doi: 10.1136/jmedgenet-2017-104744. [DOI] [PubMed] [Google Scholar]
- 55.Crockett SD, Nagtegaal ID. Terminology, molecular features, epidemiology, and management of serrated colorectal neoplasia. Gastroenterology. 2019;157(4):949–66 e4. doi: 10.1053/j.gastro.2019.06.041. [DOI] [PubMed] [Google Scholar]
- 56.Tao Y, Kang B, Petkovich DA, Bhandari YR, In J, Stein-O'Brien G, et al. Aging-like spontaneous epigenetic silencing facilitates Wnt Activation, stemness, and Braf (V600E)-induced tumorigenesis. Cancer Cell. 2019;35(2):315–28 e6. doi: 10.1016/j.ccell.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leclerc J, Flament C, Lovecchio T, Delattre L, Ait Yahya E, Baert-Desurmont S, et al. Diversity of genetic events associated with MLH1 promoter methylation in Lynch syndrome families with heritable constitutional epimutation. Genet Med. 2018;20(12):1589–1599. doi: 10.1038/gim.2018.47. [DOI] [PubMed] [Google Scholar]
- 58.Morak M, Koehler U, Schackert HK, Steinke V, Royer-Pokora B, Schulmann K, et al. Biallelic MLH1 SNP cDNA expression or constitutional promoter methylation can hide genomic rearrangements causing Lynch syndrome. J Med Genet. 2011;48(8):513–519. doi: 10.1136/jmedgenet-2011-100050. [DOI] [PubMed] [Google Scholar]
- 59.Iacopetta B, Grieu F, Li W, Ruszkiewicz A, Caruso M, Moore J, et al. APC gene methylation is inversely correlated with features of the CpG island methylator phenotype in colorectal cancer. Int J Cancer. 2006;119(10):2272–2278. doi: 10.1002/ijc.22237. [DOI] [PubMed] [Google Scholar]
- 60.Pineda M, Mur P, Iniesta MD, Borras E, Campos O, Vargas G, et al. MLH1 methylation screening is effective in identifying epimutation carriers. Eur J Hum Genet. 2012;20(12):1256–1264. doi: 10.1038/ejhg.2012.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Table S1: Sequences of primers included in this study. Table S2: The characteristics of 38 CRCs from six reference CRC groups. Table S3: The characteristics of 44 tumour and 14 normal colonic mucosal samples from the 43 people included in the study. Table S4: Summary of somatic mutations occurrent in key CRC genes and CRC subgroups associated with frequent or less frequent somatic mutation in the described gene. Table S5: Differentially Methylated Regions between tumours of MLH1 epimutation carriers and sporadic MLH1 methylated CRCs. Table S6: MLH1 promoter methylation levels in non-tumour DNA samples measured by methylation-sensitive digital droplet PCR.
Additional file 2. Figure S1:DNA methylation levels across the CpG island associated with the MLH1 promoter by CRC subgroups: 1) MMR-proficient CRCs, 2) Lynch-syndrome associated CRCs, 3) double MMR somatic mutation CRCs, 4) sporadic MLH1 methylated CRCs, 5) constitutional primary MLH1 epimutation CRCs and 6) constitutional secondary MLH1 epimutation CRCs, as well as two diagnostically challenging CRC groups: 1) MLH1 promoter VUS carriers with MLH1: c.-11C>T and MLH1: c.-[28A>G; 7C>T] and 2) MLH1 methylated EOCRCs. DNA methylation of tumour-derived DNA are shown in red and normal mucosal DNA is shown in blue. All normal mucosa samples were from resection margin, not adjacent to the tumour. Yellow boxes denote the regulatory C region as described in Deng et al. Bar plots showing mean methylation within the regulatory C region of MLH1 gene promoter across all samples for each CRC group. Tumour and normal mucosal samples are separately shown. Error bars denote standard deviation. Close-up view of four CpGs overlapping the regulatory C region.
Additional file 3. Figure S2: Oncoplot showing somatic mutational profiles at the 26 recurrently mutated CRC genes identified in CRC tumours from TCGA, 4 additional Wnt pathway associated genes and 5 genes related to the DNA methylation machinery. The total mutational burden shows the accumulative numbers of somatic mutations identified in each tumour sample. Individual tumour samples are further annotated by CRC subgroup, anatomical location, gender and CRC diagnosis age. The different types of somatic mutations are shown in different colours and the compositional barplots illustrate the total loads of somatic mutations separated by the nucleotide changes. One primary MLH1 epimutation CRC had a POLE somatic mutation in the exonuclease domain and also had the highest TMB with 459 total somatic mutations.
Additional file 4. Figure S3: Heatmaps illustrating the consensus matrix identified by the ConsensuClusterPlus analysis. A illustrates the consensus matrix estimated on 38 reference group CRCs. B illustrates the same analysis but performed on the complete dataset of 44 CRCs including six diagnostically challenging CRCs. The final consensus cluster classification for individual samples are shown in four different colours. The darker heatmaps indicate the stability evidence for classifying individual samples into each cluster. The raw consensus values are provided in Supplementary data.
Additional file 5. Figure S4: “Circos” plots showing mean genomic hypomethylation and hypermethylation patterns in tumours of five reference CRC groups. For the MLH1 epimutation group, five primary and two secondary epimutation CRCs were combined in this analysis. Mean methylation values are shown per each CRC group.
Additional file 6. Figure S5: DNA methylation patterns at CpG site cg15103403 within LRRFIP2 gene for each of the six reference CRC subgroups and the two diagnostically challenging CRC subgroups.
Additional file 7. Figure S6: Venn diagrams showing numbers of Differentially Methylated Regions of LS-CRCs, MMR-proficient CRCs, sporadic MLH1 methylated CRCs and double somatic MMR CRCs when compared with the primary and secondary MLH1 epimutation CRCs. Venn diagram shows the number of hypermethylated DMRs and Venn diagram showing the number of hypomethylated DMRs in common with the MLH1 epimutation group.
Additional file 8. Figure S7: Pedigrees for each the six people in the diagnostically challenging group comprised of the MLH1: c.-11C>T, MLH1: c.-[28A>G; 7C>T] VUS carriers, and the three people with MLH1 methylated EOCRCs.
Additional file 9. Figure S8: Loss of heterozygosity plots in the tumours of the MLH1: c.-11C>T VUS carrier and the MLH1 methylated EOCRC across the MLH1 locus. Points represent somatic and germline variants plotted by genomic position and variant allele fraction, where green circles/shading support LOH of the region. C showing the variants across the MLH1 locus in tumour samples from one of the MLH1 methylated EOCRCs without LOH.
Additional file 10. Figure S9: Droplet digital PCR results in representative samples illustrated as “1D Amplitude” plots. Methylation positive and negative droplets are shown in blue and green dots, respectively. A illustrates MLH1 methylation results in samples demonstrating negative MLH1 methylation, hypermethylation of MLH1, and low mosaic methylation in MLH1 methylated EOCRC and MLH1: c.-11C>T VUS carrier. Similarly, B shows MLH1 methylation patterns in normal and buccal-derived DNA samples.
Additional file 11. Figure S10: Pyrosequencing profiles of tumour DNA samples underwent for SMART-PCR to assess monoallelic methylation pattern of the MLH1 promoter. A-B, CRCs from two heterozygous MLH1: c.-11C>T VUS carriers showing occurrent MLH1 promoter methylation specifically in the variant allele. C-D, two reference sporadic MLH1 methylated CRCs without the c.-11C>T variant and hence showing sole amplification of G reference allele. E, one MLH1 methylated EOCRC that was heterozygous for the c.-93G>A promoter SNP showing sole amplification of the A SNP allele indicating monoallelic methylation associated only with this SNP allele. F-G, two sporadic MLH1 methylated CRCs that both were heterozygous for the c.-93G>A SNP showing the amplification of both G and A alleles indicating biallelic methylation of both A and G alleles at MLH1: c.-93 locus.
Additional file 12. Supplementary data: Raw consensus matrix values derived from the Consensus clustering analysis on 77,113 most variably methylated CpGs, which was performed on 44 CRCs including the 6 diagnostically challenging CRCs.
Data Availability Statement
Genome-wide DNA methylation data (HumanMethylationEPIC array) has been deposited to GEO and accessible through Accession No. GSE233854.