

Abstract
Alzheimer's disease (AD) is a neurodegenerative disorder characterized by dementia as the primary clinical symptom. The production and accumulation of aggregated β-amyloid (Aβ) in patient brain tissues is one of the hallmarks of AD pathogenesis. Microglia, brain-resident macrophages, produce inflammatory cytokines in response to Aβ oligomers or fibrils exacerbating Aβ pathology in AD. HMO6 cells were treated with Aβ42 in the presence or absence of 1,25-dihydroxy vitamin D3 (1,25(OH)2D3) to determine its potential immunomodulatory effects, and the expression of pro-/anti-inflammatory cytokines, M1/M2-associated markers, Toll-like receptors (TLRs), and triggering receptor expressed on myeloid cells 2 (TREM2) was examined. 1,25(OH)2D3 was found to suppress Aβ-induced expression of proinflammatory cytokines (TNF-α, IL-1β, and IL-6), M1 markers (CD86 and iNOS), and TLR2/4, whilst increasing the expression of anti-inflammatory cytokines (IL-4, IL-10, and CCL17) and M2 markers (CD206 and Arg-1). Furthermore, 1,25(OH)2D3 promoted TREM2 expression and Aβ uptake by HMO6 cells, and the enhancement of Aβ uptake and M2 polarization was revealed to be TREM2-dependent. The findings of this study suggest that 1,25(OH)2D3 facilitates M2 polarization and Aβ uptake in a TREM2-dependent manner.
1. Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder that accounts for 60-80% of dementia cases worldwide and is becoming an increasingly severe socioeconomic problem [1]. The major causes of the AD are the extracellular accumulation of beta-amyloid (Aβ) plaques and the intracellular accumulation of neurofibrillary tangles containing hyperphosphorylated tau proteins [2]. The accumulation of Aβ peptide is associated with proteolytic cleavage of the membrane Aβ precursor protein (APP), which is cleaved by β- and γ-secretases to yield a 37-49 amino acid residue peptide, Aβ [3]. Among the several APP cleavage products that contribute to AD, the 40- and 42-amino acid forms (Aβ40 and Aβ42, respectively) are the main final Aβ products [4]. Aβ42 is the more toxic form, which is attributable to the additional two amino acids that lead to misfolding and subsequent self-aggregation [5]. The accumulation of the more insoluble Aβ42 aggregates functions as a trigger for AD pathogenesis [6]. Aggregated extracellular Aβ may stimulate the activation of microglia, which are brain-resident macrophages that play a pivotal role in AD-associated neuroinflammation [7]. Activated microglia produce proinflammatory cytokines that are typically detected surrounding extracellular Aβ plaques in the brains of AD patients [8], and overactivation of these microglia promotes the development of inflammatory injuries and exacerbates the AD-associated pathology [9]. Thus, microglia-mediated inflammation is considered a promising therapeutic target for the treatment of AD.
TREM2 (triggering receptor expressed on myeloid cells 2) is a cell surface receptor expressed by microglia that plays an important role in the central nervous system (CNS) inflammation [10], and mutations in the TREM2 gene have been linked to an increased risk of developing AD [11, 12]. Ectodomain of TREM2 binds to a variety of ligands, including bacterial polysaccharides, lipoproteins, phospholipids, and DNA [13]. When such ligands bind to TREM2, intracellular signaling pathways are activated, which contribute to modulating cellular functions such as increased cell survival [14, 15], phagocytosis [14], cell adhesion and migration, and cytokine secretion [14]. Microglial TREM2 also detects extracellular Aβ, and its structure and function have been identified to influence the clearance and aggregation of Aβ [16–18].
The active form of vitamin D, 1,25-dihydroxy vitamin D3 (1,25(OH)2D3), is essential for calcium absorption and bone mineralization and is involved in a variety of biological processes, including cell growth and differentiation and immune response [19]. 1,25(OH)2D3 regulates target gene expression by binding to a nuclear hormone receptor, the vitamin D receptor (VDR) [20]. Emerging evidence suggests that vitamin D is involved in the early development of animal brains [21] and that maternal vitamin D deficiency during pregnancy is correlated with cognitive development disorders of offspring [22]. Importantly, patients with neurodegenerative diseases, including AD, have lower vitamin D serum levels [23, 24], and recent clinical studies have indicated the potentially beneficial effects of vitamin D supplements in the prevention of cognitive decline in AD patients [25, 26].
In this study, we treated human microglial cells (HMO6) with Aβ to mimic in vivo microglial activation and neuroinflammation in response to Aβ accumulation, which are the main pathological features of AD, and examined the potential immunomodulatory effects of 1,25(OH)2D3 in vitro.
2. Materials and Methods
2.1. Chemicals and Antibodies
Synthetic Aβ1−42 peptides (DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA) were purchased from LifeTein (Beijing, China); 1,25(OH)2D3 was purchased from Sigma-Aldrich (Sigma-Aldrich, Saint Louis, MO, USA); antibodies for TLR4, CD86, CD206, iNOS, and Arg-1 were obtained from Santa Cruz Biotechnology (Dallas, TX, USA); antibodies for TREM2 (B-3) and Aβ were purchased from BioLegend (San Diego, CA, USA); anti-TLR2 antibody was purchased from Novus Biologicals (Littleton, CO, USA); and TREM2 antibody (237920; R&D Systems, Minneapolis, MN, USA) was selected as blocking antibody according to a previous study [27].
2.2. Cell Culture and the Preparation of Aβ and 1,25(OH)2D3
An established human microglial cell line [28], HMO6 (accession number CVCL_5G94), was used for this study. Cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 1% antibiotic-antimycotic (Invitrogen Corp.), in a 5% CO2 at 37°C.
The Aβ1−42 peptides and 1,25(OH)2D3 were dissolved in 5 mM dimethyl sulfoxide (DMSO) to prepare stock solutions and stored at -20°C. Before use, aliquots of these stock solutions were diluted to the requisite concentrations using DMEM containing 1% antibiotic-antimycotic.
2.3. RNA Preparation and Real-Time Quantitative PCR
Total RNA was extracted using an easy-BLUE™ Total RNA Extraction Kit (iNtRON Biotechnology, Inc., Seongnam, Korea) according to the manufacturer's instructions. The concentration of the extracted RNA was measured using a MaestroNano MicroVolume Spectrophotometer (Maestrogen, Las Vegas, NV, USA). Aliquots (2 μg) of the purified RNA were used for the synthesis of cDNA using a Hyperscript™ 2 × RT Master Mix (GeneAll Biotechnology, Seoul, Korea). Quantitative real-time PCR was performed on a Rotor-Gene system (Qiagen) using a QuantiSpeed SYBR NO-ROX kit (PhileKorea, Seoul, Korea). Amplifications were performed using the primer sets listed in Table 1. Sample normalization was performed using the human GAPDH gene as an endogenous control. For each sample, the relative abundance of the target mRNA was calculated from the -△cycle threshold (△Ct) values of the target and endogenous GAPDH reference genes using the 2−△△ Ct method.
Table 1.
qRT-PCR primers used in this study.
| Gene name | Forward primer (5′-3′) | Reverse primer (5′-3′) |
|---|---|---|
| GAPDH | ACAGCCTCAAGATCA TCAGCAAT | AGGAAATGAGCTTGACAAAGTGG |
| IL-1β | GGGATAACGAGGCTTA TGTGC | AGGTGGAGAGCTTTCAGTTCA |
| TNF-α | CAGAGGGCCTGTACCTCATC | GGAAGACCCCTCCCAGATAG |
| IL-6 | GACCCAACCACAAATGCCAG | GAGTTGTCATGTCCTGCAGC |
| IL-4 | CCGTAACAGACATCTTTGCTGCC | GAGTGTCCTTCTCATGGTGGCT |
| IL-10 | TCTCCGAGATGCGTTCAGCAGA | TCAGA CAAGGCTTGGCAACCCA |
| CCL17 | ACCCCAACAACAAGAGAGTGA | GAGGGCCCAGGTAGTCCC |
2.4. Flow Cytometry
To examine expression levels of the cell surface proteins TLR2, TLR4, CD86, CD206, and TREM2, cells were collected, washed with DPBS 2 times, incubated with primary antibody at 4°C for 30 min, and followed by phycoerythrin- (PE-) conjugated secondary antibodies at 4°C for 30 min. To determine intracellular levels of iNOS and Arg1, cells were treated with 4% formaldehyde and 1% Triton at room temperature for 20 min. Then, cells were stained with primary antibody at 4°C for 30 min, followed by staining with PE-conjugated secondary antibodies at 4°C for 30 min. Cells were resuspended in phosphate-buffered saline (PBS) and analyzed on a Cytomics FC500 MLP (Beckman Coulter Inc., Fullerton, CA, USA).
2.5. Enzyme-Linked Immunosorbent Assay (ELISA) for Aβ Measurements
The culture supernatant was collected and added to the 96-well culture plate overnight at 4°C. To prevent nonspecific binding of antigens and antibodies to the well, bovine serum albumin (BSA) 5% was used as a blocking agent. Quantification of soluble Aβ1−42 was performed using anti-Aβ primary antibody and followed by incubation with m-IgGκ BP-HRP secondary antibody at RT. Absorbance of ELISA test was detected at 450 nm using microplate reader.
2.6. Statistical Analysis
The experiments were conducted at least three times, and all data are independently showed as mean ± standard deviation (SD). Significant differences among groups were analyzed by one-way analysis of variance (ANOVA) followed by post hoc test using SPSS 12.0 for Windows. Differences were considered statistically significant at p value less than 0.05. T-test was applied to determine significance among the groups of Aβ42 uptake.
3. Results
3.1. 1,25(OH)2D3 Downregulates Aβ-Induced Proinflammatory Cytokines and Upregulates Anti-Inflammatory Cytokines
Given that Aβ is an aggregation-prone peptide and that monomeric Aβ can be converted to not only stable oligomeric forms but also short fibrillar Aβ under physiologic solution conditions at higher Aβ concentrations [29, 30], we used monomeric Aβ42 to stimulate microglia in this study. Initially, we examined the effects of Aβ42 on the expression of selected cytokines in the HMO6 human microglial cell line. qRT-PCR analysis revealed that mRNA expression of the proinflammatory cytokines TNF-α, IL-1β, and IL-6 was significantly increased at 6 h after treatment with 100 nM Aβ42, but thereafter, expression declined to pretreatment levels at 12 h. However, the expression of these cytokines was markedly inhibited when the HMO6 cells were treated with both 100 nM Aβ42 and 1 μM 1,25(OH)2D3 (Figures 1(a)–1(c)). Furthermore, we detected a significant reduction in the expression of the M2 cytokines IL-10 and CCL17 in Aβ42-treated cells, whereas that of IL-4 was increased. However, in response to the combined treatment with 100 nM Aβ42 and 1 μM 1,25(OH)2D3, there were significant increases in the expression of these cytokines (Figures 1(d)–1(f)). These findings accordingly indicate that 1,25(OH)2D3 can reverse the proinflammatory cytokine expression induced by Aβ42 and enhance the expression of anti-inflammatory cytokines.
Figure 1.
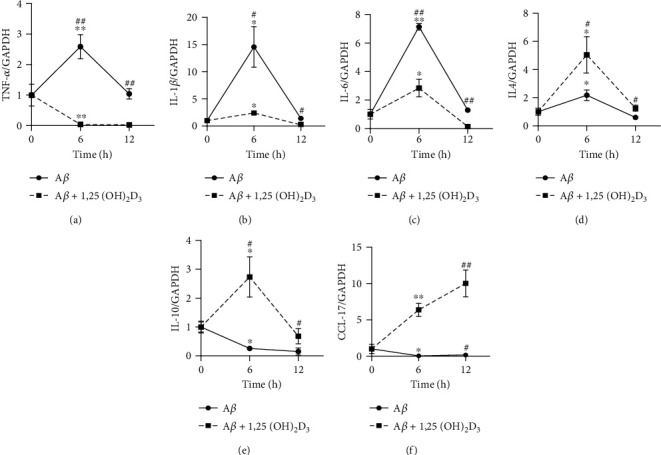
1,25(OH)2D3 downregulates Aβ-induced M1 cytokine expression, whilst upregulating M2 cytokine expression in HMO6 cells. Cells were incubated in DMEM exposed to 100 nM Aβ1-42 and treated with or without 1 μM of 1,25(OH)2D3 for 6 h or 12 h. mRNA was extracted, and expression of proinflammatory cytokines TNF-α (a), IL-1β (b), and IL-6 (c) or anti-inflammatory cytokines IL-4 (d), IL-10 (e), and CCL17 (f) was measured by quantitative real-time PCR. ∗p < 0.05 and ∗∗p < 0.005 vs. 0 h control group; #p < 0.05 and ##p < 0.005 vs. Aβ1-42-treated group.
3.2. 1,25(OH)2D3 Reverses the Aβ-Induced Upregulation of TLR4 and TLR2 Expressions in HMO6 Cells
Toll-like receptors (TLRs) are implicated in a range of neurological disorders, including AD, and are accordingly emerging as promising therapeutic targets for AD treatment [31]. Among the 10 human TLRs identified, TLR4 and TLR2 are reported to function as microglial Aβ receptors [32], which can induce proinflammatory cytokine production via the transcription factor activation of nuclear factor- (NF-) κB in response to Aβ treatment [33]. We thus examined the surface expression of TLR4 and TLR2 on Aβ42-treated HMO6 cells. As shown in Figure 2, there was a significant enhancement of in the expression of these two receptors in Aβ42-treated cells, which was markedly reversed following treatment with 1 μM 1,25(OH)2D3.
Figure 2.

1,25(OH)2D3 downregulates the Aβ42-induced expression of TLR4 and TLR2 in HMO6 cells. Cells were treated with or without 1 μM 1,25(OH)2D3 in the presence of 100 nM Aβ42 at 37°C for 24 h. Cell surface levels of TLR4 (a) and TLR2 (b) were measured by flow cytometry. Bar graphs indicate the relative expression ± SD (left panels). Right panels designate representative histograms. ∗p < 0.05 and ∗∗p < 0.0005 vs. the DMSO control. #p < 0.05 and ##p < 0.0005.
3.3. 1,25(OH)2D3 Downregulates Aβ-Induced M1 Marker Expression in HMO6 Cells, Whilst Upregulating M2 Marker Expression
Similar to macrophages, microglia can be classified into two distinct phenotypes based on their activation state, namely, M1 microglia, which produce proinflammatory cytokines, such as TNF-α, IL-1ß, and IL-6 [34], and contribute to inducing neuronal damage [35], and M2 microglia, which release anti-inflammatory cytokines, such as IL-4 and IL-10, and the Th2 chemokine CCL17 [36] and play role in neuroprotection [37]. In the present study, we examined the effects of Aβ42 treatment on the expression of M1 and M2 markers in HMO6 cells. In Aβ-treated cells, we detected a significant increase in the expression of the M1 markers CD86 and iNOS, which was reversed following treatment with 1,25(OH)2D3 (Figures 3(a) and 3(b)). Expression of the M2 marker arginase I (Arg-1), which metabolically dampens T-cell responses by causing arginine deprivation [38], in Aβ42-treated cells was significantly reduced, whereas that of CD206 remained unchanged. However, in cells cotreated with Aβ42 and 1,25(OH)2D3, we detected the markedly enhanced expression of both these M2 markers (Figures 3(c) and 3(d)). These findings thus provide evidence to indicate that Aβ42 treatment leads to M1 polarization in HMO6 cells and that 1,25(OH)2D3 skews the Aβ42-induced M1 phenotype toward an M2 phenotype in these cells.
Figure 3.

1,25(OH)2D3 downregulates Aβ42-induced M1 marker expression in HMO6 cells, whilst upregulating M2 marker expression. Cells were treated with or without 1 μM 1,25(OH)2D3 in the presence of 100 nM Aβ42 for 12 h, and protein expressions of M1 markers CD86 (a) and iNOS (b) and M2 markers CD206 (c) and Arg-1 (d) were measured by flow cytometry. Bar graphs indicate the relative expression ± SD (left panels). Right panels designate representative histograms. ∗p < 0.05 vs. the DMSO control; #p < 0.05 vs. the Aβ42-treated group.
3.4. 1,25(OH)2D3 Enhances Surface TREM2 Expression and Aβ42 Uptake by HMO6 Cells
As a cell surface receptor, TREM2 has been established to interact with Aβ and plays a central role in AD pathogenesis, and thus, we examined the effects of different concentrations of 1,25(OH)2D3 on the expression of TREM2 in HMO6 cells treated with 100 nM Aβ42. We accordingly found that when applied at concentrations of 100 and 1000 nM, 1,25(OH)2D3 promoted the enhanced expression of cell surface TREM2 in HMO6 cells treated with 100 nM Aβ42 (Figure 4(a)). To determine the capacity of human microglial cells to take up exogenous Aβ42 peptides, HMO6 cells were treated with 100 nM Aβ42 in the presence or absence of 1 μM 1,25(OH)2D3 for different periods (6-24 h). Uptake of Aβ42 by HMO6 cells was determined by measuring the intracellular levels of Aβ42 by flow cytometry, and nonuptake of Aβ42 was accessed by measuring levels of Aβ42 in culture supernatants based on ELISA. The results revealed that at all assessed time points, the levels of Aβ42 uptake by HMO6 cells cultured in the presence of 1,25(OH)2D3 were higher than those in the absence of 1,25(OH)2D3 (Figure 4(b)). In line with expectation, nonuptake levels of Aβ42 in the culture supernatants of 1,25(OH)2D3-treated cells were significantly lower than those in the supernatants of nontreated cells (Figure 4(c)). These findings thus indicate that 1,25(OH)2D3 facilitates the uptake of exogenous Aβ42 by HMO6 cells. The phagocytosis of Aβ by microglial cells has been reported to be dependent on TREM2 [39], and thus, to determine the potential involvement of cell surface TREM2 in the 1,25(OH)2D3-enhanced uptake of exogenous Aβ42 by HMO6 cells, we used a TREM2-blocking antibody to block these receptors. In HMO6 cells pretreated with anti-TREM2 antibodies before treatment with Aβ42, we detected significantly lower levels of intracellular Aβ42 than in nontreated cells and even in DMSO-treated cells (Figure 4(d)). Correspondingly, thus nonuptake levels of Aβ42 in the culture supernatants of anti-TREM2 antibody-treated cells were significantly higher than those of nontreated cells and DMSO-treated cells (Figure 4(e)). These observations accordingly provide convincing evidence to indicate that cell surface TREM2 is involved in the 1,25(OH)2D3-enhanced uptake of exogenous Aβ42 by HMO6 cells.
Figure 4.

1,25(OH)2D3 enhances surface TREM2 expression and Aβ42 uptake by HMO6 cells. (a) Cells were treated with different concentrations of 1,25(OH)2D3 in the presence of 100 nM Aβ42 for 12 h, and cell surface expression of TREM2 was measured by flow cytometry. (b) Cells were treated with 100 nM Aβ42 in the presence or absence of 1 μM 1,25(OH)2D3 at 37°C for different periods, and the levels of intracellular Aβ were measured by flow cytometry using anti-Aβ antibodies. (c) The culture supernatants were collected, and levels of Aβ were measured by ELISA. (d) Cells were treated with or without TREM2-blocking antibodies for 2 h and then treated with 100 nM Aβ42 in the presence or absence of 1 μM 1,25(OH)2D3 at 37°C for 24 h, after which, the levels of intracellular Aβ were measured by flow cytometry. Bar graphs indicate the relative expression ± SD (left panels). Right panels designate representative histograms. (e) Culture supernatants were collected, and levels of Aβ were measured by ELISA. ∗p < 0.05 and ∗∗p < 0.005 vs. the DMSO control. ##p < 0.005.
3.5. 1,25(OH)2D3-Induced M2 Polarization Is Dependent on Cell Surface TREM2
Given that TREM2 has been proposed to play a vital role in the anti-inflammatory responses of microglia in AD [40], we examined the expression of M1/M2 markers and pro-/anti-inflammatory cytokines in HMO6 cells following stimulation with Aβ42 in the presence or absence of either 1,25(OH)2D3 or anti-TREM2-blocking antibody. Upon exposure to Aβ42, we detected a significant enhancement of M1 marker CD86 expression in HMO6 cells, whereas that of the M2 marker CD206 was reduced, and we found that both these responses could be reversed in the presence of 1,25(OH)2D3 (Figures 5(a) and 5(b)). However, the reduced expression of CD86 and enhanced expression of CD206 in 1,25(OH)2D3-treated cells were reversed in cells exposed to anti-TREM2-blocking antibodies. Similar to the expression patterns of M1/M2 markers detected upon challenge with Aβ42, the mRNA expression of IL-1β was significantly enhanced, whereas IL-4 was reduced in HMO6 cells, and these responses were reversed in the presence of 1,25(OH)2D3 (Figures 5(c) and 5(d)). Again, these 1,25(OH)2D3-mediated effects were reversed in the presence of anti-TREM2-blocking antibodies. These findings thus indicate that the cell surface receptor TREM2 may play a role in the anti-inflammatory responses of human microglial cells by facilitating M2 polarization.
Figure 5.

1,25(OH)2D3-induced M2 polarization of microglia is dependent on cell surface TREM2. HMO6 cells were pretreated with or without TREM2-blocking antibody for 2 h and then treated with or without 1 μM 1,25(OH)2D3 in the presence of 100 nM Aβ42 at 37°C for 6 h (for mRNA expression) or 12 h (surface protein expression). Levels of cell surface CD86 (a) and CD206 (b) were measured by flow cytometry. Bar graphs indicate the relative expression ± SD (left panels). Right panels designate representative histograms. Levels of IL-1β (c) and IL-4 (d) mRNA expression were analyzed based on quantitative real-time PCR. ∗p < 0.05 and ∗∗p < 0.005 vs. the DMSO control; #p < 0.05 and ##p < 0.005 vs. the Aβ42-treated group.
4. Discussion
In this study, we demonstrated that in Aβ42-treated human microglial cells, the active form of vitamin D, 1,25(OH)2D3, promotes the downregulated expression of the proinflammatory cytokines TNF-α, IL-1β, and IL-6 and induces the upregulated expression of the anti-inflammatory cytokines IL-4, IL-10, and CCL17. In the CNS, microglia can recognize extracellular Aβ oligomers and fibrils and are thereby activated with the associated release of proinflammatory cytokines [41]. The sustained production of these cytokines from activated microglia can exacerbate the AD process [42]. In addition to their direct adverse effect on neighboring neurons [43, 44], the proinflammatory cytokines secreted by activated microglia can activate astrocytes that contribute to promoting neuronal loss in AD [45]. In this regard, the downregulation of proinflammatory cytokines and upregulation of anti-inflammatory cytokines induced by 1,25(OH)2D3 in Aβ42-treated HMO6 cells indicate that 1,25(OH)2D3 may have applicability in the treatment of AD. Interestingly, IL-4 was increased at 6 h after Aβ treatment in the absence of 1,25(OH)2D3. Mechanisms underlying the induction of IL-4 expression in microglia after Aβ treatment are currently unknown. Possible explanation for this induction is that IL-4 may be involved in counteracting the inflammatory response of microglia to Aβ or enhancing the phagocytic activity of microglia to promote the uptake and clearance of Aβ [46]. The application of 1,25(OH)2D3 has also been demonstrated to reduce the expression of TLR2 and TLR4, which are cell surface receptors for Aβ, the activation of which in turn induces microglial activation and neuroinflammation in AD [32]. The Aβ-induced activation of TLR2 and TLR4 promotes intracellular signaling leading to the production of proinflammatory cytokines. Consequently, the observed reduction in the expression of such proinflammatory cytokines in Aβ42-treated HMO6 cells in the presence of 1,25(OH)2D3 can probably be ascribed to the 1,25(OH)2D3-induced downregulation of TLR2 and TLR4 expressions.
Given that Aβ aggregates induce M1 microglial polarization [45] and that, in turn, the M1 inflammatory response led to the formation of Aβ aggregates [45, 47], it is important to facilitate either M2 polarization or Aβ clearance to suppress the progression of AD. In this regard, it is noteworthy that in Aβ42-treated HMO6 cells, 1,25(OH)2D3 downregulates M1 marker expression, whilst upregulating M2 marker expression, thereby indicating that 1,25(OH)2D3 has the effect of inducing M2 polarization. In this context, it has recently been suggested that promoting a phenotypic shift from M1 to M2 microglia may have therapeutic potential for the treatment of AD [48, 49], and accordingly, the 1,25(OH)2D3-induced polarization of a microglial phenotype from M1 to M2 could be considered as a promising therapeutic option.
Microglia play a central role in the clearance of Aβ aggregates via phagocytosis or receptor-mediated endocytosis, which are mediated by several types of cell surface receptors, including scavenger receptors, receptors for advanced glycation end products (RAGE), lipoprotein receptor-related proteins (LRPs), and TREM2 [50], among which TREM2 has been the most extensively studied. On the surface of microglial cells, TREM2 binds directly to Aβ oligomers with high affinity, and either the loss [51] or alteration [17] of TREM2 has been found to impair Aβ clearance by microglia. Consequently, it would appear that the maintenance of sufficient levels of TREM2 expression and function is important for retarding the progression of AD [52]. Our findings in this study revealed that 1,25(OH)2D3 enhances surface TREM2 expression and extracellular Aβ42 uptake by microglia and that the 1,25(OH)2D3-induced increase in microglial Aβ42 uptake is almost completely reversed in the presence of a TREM2-blocking antibody. These findings indicate that 1,25(OH)2D3 facilitates microglial Aβ42 uptake by upregulating the expression of cell surface TREM2. Consistent with this interpretation, the findings of a recent study have indicated that an agonistic TREM2 antibody, which can induce intracellular signaling, enhances the phagocytosis of oligomeric Aβ by microglia in vitro and improves cognitive function by attenuating chronic inflammatory responses in a murine model of AD [53, 54].
It is generally accepted that vitamin D plays an essential role in maintaining cognitive function in old age [55] and patients receiving hemodialysis [56], and the findings of several recent meta-analyses have indicated that lower serum levels of vitamin D are associated with a heightened risk of AD [57–60]. Moreover, human and animal studies have indicated that vitamin D contributes a reduction in Aβ deposition in the brain [61, 62]. Consequently, our observations in the present study, indicating that 1,25(OH)2D3 enhances the M2 polarization and Aβ uptake of human microglial cells in a TREM2-dependent manner, suggest a potential therapeutic option for the treatment of AD. However, more elaborate studies will be necessary to elucidate the precise mechanisms whereby vitamin D (1,25(OH)2D3)-enhanced TREM2 expression is involved in facilitating the M2 polarization and Aβ uptake of HMO6 cells. In addition, in vitro studies using primary microglial cells and animal AD model studies are needed to evaluate the potential clinical applications of 1,25(OH)2D3.
In conclusion, the findings of this study provide convincing evidence to indicate the potential protective effect of 1,25(OH)2D3 on Aβ42-treated human microglial cells by suppressing the expression levels of proinflammatory mediators, whilst enhancing the expression of anti-inflammatory mediators, which we suspect is associated with M2 polarization and Aβ uptake, plausibly mediated via an increase in cell surface TREM2 expression.
Acknowledgments
This work was supported by the Gachon University research fund of 2019 (GCU-2019-0820).
Abbreviations
- 1,25(OH)2D3:
1,25-Dihydroxy vitamin D3
- AD:
Alzheimer's disease
- TREM2:
Triggering receptor expressed on myeloid cells 2
- TLR:
Toll-like receptor
- VDR:
Vitamin D receptor.
Data Availability
The data used to support the findings of this study are available from the corresponding author upon request.
Disclosure
This study was partly based on the first author's master thesis submitted in 2021 to the Gachon University in Korea.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- 1.Huat T. J., Camats-Perna J., Newcombe E. A., Valmas N., Kitazawa M., Medeiros R. Metal toxicity links to Alzheimer's disease and neuroinflammation. Journal of Molecular Biology . 2019;431(9):1843–1868. doi: 10.1016/j.jmb.2019.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zheng H., Cheng B., Li Y., Li X., Chen X., Zhang Y. W. TREM2 in Alzheimer’s disease: microglial survival and energy metabolism. Frontiers in Aging Neuroscience . 2018;10:p. 395. doi: 10.3389/fnagi.2018.00395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nunan J., Small D. H. Regulation of APP cleavage by α-, β-and γ-secretases. FEBS Letters . 2000;483(1):6–10. doi: 10.1016/S0014-5793(00)02076-7. [DOI] [PubMed] [Google Scholar]
- 4.Olsson F., Schmidt S., Althoff V., et al. Mechanism of APP and Aβ processing and modulation. Journal of Biological Chemistry . 2014;289(3):1540–1550. doi: 10.1074/jbc.M113.498246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahmed M., Davis J., Aucoin D., et al. Structural conversion of neurotoxic amyloid-β1-42 oligomers to fibrils. Nature Structural & Molecular Biology . 2010;17(5):561–567. doi: 10.1038/nsmb.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balducci C., Forloni G. Novel targets in Alzheimer’s disease: a special focus on microglia. Pharmacological Research . 2018;130:402–413. doi: 10.1016/j.phrs.2018.01.017. [DOI] [PubMed] [Google Scholar]
- 7.Heneka M. T., Golenbock D. T., Latz E. Innate immunity in Alzheimer’s disease. Nature Immunology . 2015;16(3):229–236. doi: 10.1038/ni.3102. [DOI] [PubMed] [Google Scholar]
- 8.McGeer P. L., Itagaki S., Tago H., McGeer E. G. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neuroscience Letters . 1987;79(1-2):195–200. doi: 10.1016/0304-3940(87)90696-3. [DOI] [PubMed] [Google Scholar]
- 9.Lue L. F., Kuo Y. M., Beach T., Walker D. G. Microglia activation and anti-inflammatory regulation in Alzheimer’s disease. Molecular Neurobiology . 2010;41(2-3):115–128. doi: 10.1007/s12035-010-8106-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolfe C. M., Fitz N. F., Nam K. N., Lefterov I., Koldamova R. The role of APOE and TREM2 in Alzheimer′s disease—current understanding and perspectives. International Journal of Molecular Sciences . 2019;20(1):p. 81. doi: 10.3390/ijms20010081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guerreiro R., Wojtas A., Bras J., et al. TREM2 variants in Alzheimer’s disease. New England Journal of Medicine . 2013;368(2):117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jonsson T., Stefansson H., Steinberg S., et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. New England Journal of Medicine . 2013;368(2):107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kober D. L., Brett T. J. TREM2-ligand interactions in health and disease. Journal of Molecular Biology . 2017;429(11):1607–1629. doi: 10.1016/j.jmb.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Colonna M., Wang Y. TREM2 variants: new keys to decipher Alzheimer disease pathogenesis. Nature Reviews Neuroscience . 2016;17(4):201–207. doi: 10.1038/nrn.2016.7. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y., Cella M., Mallinson K., et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell . 2015;160(6):1061–1071. doi: 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lessard C. B., Malnik S. L., Zhou Y., et al. High-affinity interactions and signal transduction between Aβ oligomers and TREM 2. EMBO Molecular Medicine . 2018;10(11, article e9027) doi: 10.15252/emmm.201809027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao Y., Wu X., Li X., et al. TREM2 is a receptor for β-amyloid that mediates microglial function. Neuron . 2018;97(5):1023–1031.e7. doi: 10.1016/j.neuron.2018.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhong L., Wang Z., Wang D., et al. Amyloid-beta modulates microglial responses by binding to the triggering receptor expressed on myeloid cells 2 (TREM2) Molecular Neurodegeneration . 2018;13(1):p. 15. doi: 10.1186/s13024-018-0247-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pike J. W., Christakos S. Biology and mechanisms of action of the vitamin D hormone. Endocrinology and Metabolism Clinics . 2017;46(4):815–843. doi: 10.1016/j.ecl.2017.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faye P. A., Poumeaud F., Miressi F., et al. Focus on 1, 25-dihydroxyvitamin D3 in the peripheral nervous system. Frontiers in Neuroscience . 2019;13:p. 348. doi: 10.3389/fnins.2019.00348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bivona G., Gambino C. M., Iacolino G., Ciaccio M. Vitamin D and the nervous system. Neurological Research . 2019;41(9):827–835. doi: 10.1080/01616412.2019.1622872. [DOI] [PubMed] [Google Scholar]
- 22.Garcia-Serna A. M., Morales E. Neurodevelopmental effects of prenatal vitamin D in humans: systematic review and meta-analysis. Molecular Psychiatry . 2020;25(10):2468–2481. doi: 10.1038/s41380-019-0357-9. [DOI] [PubMed] [Google Scholar]
- 23.Łukaszyk E., Bień-Barkowska K., Bień B. Cognitive functioning of geriatric patients: is hypovitaminosis D the next marker of cognitive dysfunction and dementia? Nutrients . 2018;10(8):p. 1104. doi: 10.3390/nu10081104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jayedi A., Rashidy-Pour A., Shab-Bidar S. Vitamin D status and risk of dementia and Alzheimer’s disease: a meta-analysis of dose-response. Nutritional Neuroscience . 2019;22(11):750–759. doi: 10.1080/1028415X.2018.1436639. [DOI] [PubMed] [Google Scholar]
- 25.Jia J., Hu J., Huo X., Miao R., Zhang Y., Ma F. Effects of vitamin D supplementation on cognitive function and blood Aβ-related biomarkers in older adults with Alzheimer’s disease: a randomised, double-blind, placebo-controlled trial. Journal of Neurology, Neurosurgery & Psychiatry . 2019;90(12):1347–1352. doi: 10.1136/jnnp-2018-320199. [DOI] [PubMed] [Google Scholar]
- 26.Yang T., Wang H., Xiong Y., et al. Vitamin D supplementation improves cognitive function through reducing oxidative stress regulated by telomere length in older adults with mild cognitive impairment: a 12-month randomized controlled trial. Journal of Alzheimer’s Disease . 2020;78(4):1509–1518. doi: 10.3233/JAD-200926. PMID: 33164936. [DOI] [PubMed] [Google Scholar]
- 27.Shirotani K., Hori Y., Yoshizaki R., et al. Aminophospholipids are signal-transducing TREM2 ligands on apoptotic cells. Scientific Reports . 2019;9(1):p. 7508. doi: 10.1038/s41598-019-43535-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagai A., Nakagawa E., Hatori K., et al. Generation and characterization of immortalized human microglial cell lines: expression of cytokines and chemokines. Neurobiology of Disease . 2001;8(6):1057–1068. doi: 10.1006/nbdi.2001.0437. [DOI] [PubMed] [Google Scholar]
- 29.Stine W. B., Dahlgren K. N., Krafft G. A., LaDu M. J. In vitro characterization of conditions for amyloid-β peptide oligomerization and fibrillogenesis. Journal of Biological Chemistry . 2003;278(13):11612–11622. doi: 10.1074/jbc.M210207200. [DOI] [PubMed] [Google Scholar]
- 30.Morel B., Carrasco-Jiménez M. P., Jurado S., Conejero-Lara F. Rapid conversion of amyloid-beta 1-40 oligomers to mature fibrils through a self-catalytic bimolecular process. International Journal of Molecular Sciences . 2021;22(12):p. 6370. doi: 10.3390/ijms22126370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gambuzza E. M., Sofo V., Salmeri M. F., Soraci L., Marino S., Bramanti P. Toll-like receptors in Alzheimer’s disease: a therapeutic perspective. CNS & Neurological Disorders-Drug Targets (Formerly Current Drug Targets-CNS & Neurological Disorders) . 2014;13(9):1542–1558. doi: 10.2174/1871527313666140806124850. [DOI] [PubMed] [Google Scholar]
- 32.Reed-Geaghan E. G., Savage J. C., Hise A. G., Landreth G. E. CD14 and toll-like receptors 2 and 4 are required for fibrillar Aβ-stimulated microglial activation. Journal of Neuroscience . 2009;29(38):11982–11992. doi: 10.1523/JNEUROSCI.3158-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Momtazmanesh S., Perry G., Rezaei N. Toll-like receptors in Alzheimer's disease. Journal of Neuroimmunology . 2020;348:p. 577362. doi: 10.1016/j.jneuroim.2020.577362. [DOI] [PubMed] [Google Scholar]
- 34.Perry V. H., Nicoll J. A., Holmes C. Microglia in neurodegenerative disease. Revista de Neurologia . 2010;6(4):193–201. doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]
- 35.Li Z., Ma L., Kulesskaya N., Võikar V., Tian L. Microglia are polarized to M1 type in high-anxiety inbred mice in response to lipopolysaccharide challenge. Brain, Behavior, and Immunity . 2014;38:237–248. doi: 10.1016/j.bbi.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 36.Kalkman H. O., Feuerbach D. Antidepressant therapies inhibit inflammation and microglial M1-polarization. Pharmacology & Therapeutics . 2016;163:82–93. doi: 10.1016/j.pharmthera.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 37.Almolda B., de Labra C., Barrera I., et al. Alterations in microglial phenotype and hippocampal neuronal function in transgenic mice with astrocyte-targeted production of interleukin-10. Brain, Behavior, and Immunity . 2015;45:80–97. doi: 10.1016/j.bbi.2014.10.015. [DOI] [PubMed] [Google Scholar]
- 38.Rath M., Müller I., Kropf P., Closs E. I., Munder M. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Frontiers in Immunology . 2014;5:p. 532. doi: 10.3389/fimmu.2014.00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hemonnot A. L., Hua J., Ulmann L., Hirbec H. Microglia in Alzheimer disease: well-known targets and new opportunities. Frontiers in Aging Neuroscience . 2019;11:p. 233. doi: 10.3389/fnagi.2019.00233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xue F., Du H. TREM2 mediates microglial anti-inflammatory activations in Alzheimer’s disease: lessons learned from transcriptomics. Cell . 2021;10(2):p. 321. doi: 10.3390/cells10020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Selkoe D. J. Alzheimer's disease: genes, proteins, and therapy. Physiological Reviews . 2001;81(2):741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 42.Rubio-Perez J. M., Morillas-Ruiz J. M. A review: inflammatory process in Alzheimer's disease, role of cytokines. The Scientific World Journal . 2012;2012:15. doi: 10.1100/2012/756357.756357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park K. M., Bowers W. J. Tumor necrosis factor-alpha mediated signaling in neuronal homeostasis and dysfunction. Cellular Signalling . 2010;22(7):977–983. doi: 10.1016/j.cellsig.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Conroy S. M., Nguyen V., Quina L. A., et al. Interleukin-6 produces neuronal loss in developing cerebellar granule neuron cultures. Journal of Neuroimmunology . 2004;155(1-2):43–54. doi: 10.1016/j.jneuroim.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 45.Glass C. K., Saijo K., Winner B., Marchetto M. C., Gage F. H. Mechanisms underlying inflammation in neurodegeneration. Cell . 2010;140(6):918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang R. H., Qi R. Q., Liu H. Y. Interleukin-4 affects microglial autophagic flux. Neural Regeneration Research . 2019;14(9):1594–1602. doi: 10.4103/1673-5374.255975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang X., He Q., Huang T., et al. Treadmill exercise decreases Aβ deposition and counteracts cognitive decline in APP/PS1 mice, possibly via hippocampal microglia modifications. Frontiers in Aging Neuroscience . 2019;11:p. 78. doi: 10.3389/fnagi.2019.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Q., Yao H., Liu W., et al. Microglia polarization in Alzheimer’s disease: mechanisms and a potential therapeutic target. Frontiers in Aging Neuroscience . 2021;13 doi: 10.3389/fnagi.2021.772717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sanjay, Kim J. Y. Anti-inflammatory effects of 9-cis-retinoic acid on β-amyloid treated human microglial cells. European Journal of Inflammation . 2022;20, article 1721727X2211436 doi: 10.1177/1721727X221143651. [DOI] [Google Scholar]
- 50.Ries M., Sastre M. Mechanisms of Aβ clearance and degradation by glial cells. Frontiers in Aging Neuroscience . 2016;8:p. 160. doi: 10.3389/fnagi.2016.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kleinberger G., Yamanishi Y., Suárez-Calvet M., et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Science Translational Medicine . 2014;6(243):p. 243ra86. doi: 10.1126/scitranslmed.3009093. [DOI] [PubMed] [Google Scholar]
- 52.Wang Q., Xu Y., Qi C., Liu A., Zhao Y. Association study of serum soluble TREM2 with vascular dementia in Chinese Han population. International Journal of Neuroscience . 2020;130(7):708–712. doi: 10.1080/00207454.2019.1702548. [DOI] [PubMed] [Google Scholar]
- 53.Fassler M., Rappaport M. S., Cuño C. B., George J. Engagement of TREM2 by a novel monoclonal antibody induces activation of microglia and improves cognitive function in Alzheimer’s disease models. Journal of Neuroinflammation . 2021;18(1):p. 19. doi: 10.1186/s12974-020-01980-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Q., Yang W., Zhang J., Zhao Y., Xu Y. TREM2 overexpression attenuates cognitive deficits in experimental models of vascular dementia. Neural Plasticity . 2020;2020:10. doi: 10.1155/2020/8834275.8834275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pérez-López F. R., Chedraui P., Fernández-Alonso A. M. Vitamin D and aging: beyond calcium and bone metabolism. Maturitas . 2011;69(1):27–36. doi: 10.1016/j.maturitas.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 56.Zhang J., Hu J., Zhou R., Xu Y. Cognitive function and vitamin D status in the Chinese hemodialysis patients. Computational & Mathematical Methods in Medicine . 2022;2022, article 2175020:6. doi: 10.1155/2022/2175020. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 57.Balion C., Griffith L. E., Strifler L., et al. Vitamin D, cognition, and dementia: a systematic review and meta-analysis. Neurology . 2012;79(13):1397–1405. doi: 10.1212/WNL.0b013e31826c197f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Annweiler C., Montero-Odasso M., Llewellyn D. J., Richard-Devantoy S., Duque G., Beauchet O. Meta-analysis of memory and executive dysfunctions in relation to vitamin D. Journal of Alzheimer’s Disease . 2013;37(1):147–171. doi: 10.3233/JAD-130452. [DOI] [PubMed] [Google Scholar]
- 59.Zhao Y., Sun Y., Ji H. F., Shen L. Vitamin D levels in Alzheimer’s and Parkinson’s diseases: a meta-analysis. Nutrition . 2013;29(6):828–832. doi: 10.1016/j.nut.2012.11.018. [DOI] [PubMed] [Google Scholar]
- 60.Chai B., Gao F., Wu R., et al. Vitamin D deficiency as a risk factor for dementia and Alzheimer’s disease: an updated meta-analysis. BMC Neurology . 2019;19(1):p. 284. doi: 10.1186/s12883-019-1500-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miller B. J., Whisner C. M., Johnston C. S. Vitamin D supplementation appears to increase plasma Aβ 40 in vitamin D insufficient older adults: a pilot randomized controlled trial. Journal of Alzheimer's Disease . 2016;52(3):843–847. doi: 10.3233/JAD-150901. [DOI] [PubMed] [Google Scholar]
- 62.Durk M. R., Han K., Chow E. C., et al. 1α, 25-Dihydroxyvitamin D3 reduces cerebral amyloid-β accumulation and improves cognition in mouse models of Alzheimer’s disease. Journal of Neuroscience . 2014;34(21):7091–7101. doi: 10.1523/JNEUROSCI.2711-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.