

Abstract
Zinc finger E-box binding homeobox 1 (ZEB1) is a transcription factor that can promote tumor invasion and metastasis by inducing epithelial to mesenchymal transition (EMT). To date, regulation of ZEB1 by RAS/RAF signaling remains unclear, and few studies have examined post translation modification of ZEB1 including its ubiquitination. In human colorectal cancer (CRC) cell lines with RAS/RAF/MEK/ERK activation, an interaction of ZEB1 with the deubiquitinase ubiquitin-specific protease 10 (USP10) was identified whereby USP10 modifies ZEB1 ubiquitination and promotes its proteasomal degradation. Regulation of the USP10-ZEB1 interaction by MEK-ERK signaling was shown whereby constitutive activation of ERK can phosphorylate USP10 at Ser236 to impair its interaction with ZEB1 and enable ZEB1 protein stabilization. Stabilized ZEB1 was shown to promote CRC metastatic colonization in a mouse tail vein injection model. Conversely, MEK-ERK inhibition blocked USP10 phosphorylation and enhanced the USP10-ZEB1 interaction shown to suppress ZEB1-mediated tumor cell migration and metastasis. In conclusion, we demonstrate a novel function of USP10 in the regulation of ZEB1 protein stability and its ability to mediate tumor metastasis in a preclinical model.
Introduction
Colorectal cancer (CRC) is the 3rd most common cancer in the U.S. and is 2nd only to lung cancer as a cause of cancer-related mortality (1). Of new cases of CRC, 20% of patients have metastatic disease and another 25% who present with localized disease will later develop metastases (2). Elucidating the molecular mechanisms underlying tumor metastasis is essential for the development of therapeutic strategies against CRC. Zinc finger E-box binding homeobox 1 (ZEB1) is a pleiotropic transcription factor that can regulate epithelial-to-mesenchymal transition (EMT) and promotes tumor metastasis in multiple tumor types including CRC (3–5). ZEB1 has been shown to repress the expression of epithelial genes such as E-cadherin at the transcriptional level (6–8). Tumor metastasis is mediated by EMT and this phenotype is associated with invasion, tumor progression and metastasis (9). With EMT, cells lose their epithelial characteristics and gain mesenchymal properties such as increased cell migration, tissue remodeling/wound repair and enhanced cancer stemness (10,11).
The ZEB1 protein is subject to ubiquitination and degradation, although the mechanism by which ZEB1 is stabilized in cancer cells is largely unknown (12,13). Ubiquitination is a posttranslational modification that regulates many cellular processes through protein stability, activity or localization (14). Ubiquitin has seven lysine residues (K6, K11, K27, K29, K33, K48, K63) and an N-terminus (M1) which can be conjugated by other ubiquitin moieties to form a polyubiquitination chain (15). Lysine 48-linked chains target substrate proteins for proteasomal degradation while the function of K27-linked ubiquitination is poorly understood. Ubiquitination can be reversed by deubiquitinase (DUB) enzymes that regulate protein stability of which only three DUBs have been shown to remove K27-linkage polyubiquitination chains from their substrates (16–18). Using the BioGrid database that includes manually curated protein and genetic interactions (19), we sought to identify a DUB(s) that interacts with ZEB1 and identified the ubiquitin-specific protease 10 (USP10) as a potential candidate. USP10 was shown to serve as a DUB for another EMT-related transcription factor in human cancer cells (20). USP10 is expressed in many human cancer cell types (21), and has been shown to stabilize p53 and SIRT6 proteins as well as the NOTCH1 intracellular domain (22,23,24). Previously, we found that USP10 can remove the K63-linkage polyubiquitination chain on AMP-activated protein kinase-alpha (AMPKα) and facilitate AMPKα phosphorylation by LKB1 (25). USP10 has also been reported to remove the K63 linkage chain from Phosphatase and tensin homolog (PTEN) which restored PTEN phosphatase activity (26). In this report, we determined whether the DUB USP10 could regulate ZEB1 stability in human CRC cells that may contribute to its role in tumor metastasis. ZEB1 has been shown to be regulated by MEK/ERK whereby the ERK consensus site at Thr-867 is phosphorylated on ZEB1 (27). We found that ZEB1 can be stabilized by MEK-ERK signaling and demonstrated for the first time that USP10 can modify ZEB1 ubiquitination and promote its proteasomal degradation. These events were shown to suppress the EMT-related processes of tumor cell migration and metastasis in a humanized mouse model.
Materials & Methods
Cell culture and reagents
The human CRC cell line RKO was purchased from the ATCC. Isogenic human parental RKO (BRAFV600E/V600E/wt; RRID:CVCL_HE16) and T29 (BRAFWT/−/−) CRC cell lines were obtained from Dr. B. Vogelstein [Genetic Resources Core Facility (GRCF), Johns Hopkins University, Baltimore, MD]. Colo320 (RRID:CVCL_1989) was a gift from Dr. S. Johnsen (Mayo Clinic). RPMI medium supplemented with 10% (v/v) fetal bovine serum (FBS) was used for culturing CRC cell lines. Dulbecco’s modified Eagle’s medium supplemented with 10% FBS was used for culturing HEK293T cells. All cell lines were authenticated by short tandem repeat analysis and routinely tested using MycoAlert Mycoplasma detection set (Lonza, OR). Cells were treated with the following drugs: cobimetinib (GDC-0973/XL-518; Selleckchem, TX), BRAF inhibitor vemurafenib (PLX-4032; Selleckchem), cetuximab (Eli Lilly, IN), encorafenib (Selleckchem) and trametinib (Selleckchem). The following antibodies were used in this study: anti-Snail (Cell Signaling Technology, MA.; Cat# 3879, RRID:AB_2255011), anti-Slug (Cell Signaling Technology Cat# 9585, RRID:AB_2239535), anti-β-tubulin (Cell Signaling Technology Cat# 2146, RRID:AB_2210545), and anti-Twist1 (Abcam, MA.; Cat#ab50887, RRID:AB_883294) and anti-Twist2 (Proteintech, IL., Cat# 11752-1-AP, RRID:AB_2877791). In other experiments, we utilized antibodies against phospho-p44/42 MAPK (ERK1/2) [Thr202/Tyr204] (Cell Signaling Technology Cat# 4370, RRID:AB_2315112), p44/42 MAPK (ERK1/2) (Cell Signaling Technology Cat# 4695, RRID:AB_390779), and E-cadherin (Cell Signaling Technology Cat# 3195, RRID:AB_2291471).
Antibodies against ZEB1 were used for immunoblotting (Bethyl Laboratories, TX., Cat# A301-922A, RRID:AB_1524126), and for immunoprecipitation (Santa Cruz Biotechnology, TX., Cat# sc-515797, RRID:AB_2934316). We also utilized an anti-ZEB2 antibody (E6U7Z)[Cell Signaling Technology Cat# 97885, RRID:AB_2934315]. Anti-USP10 antibodies were utilized for immunoprecipitation (Santa Cruz Biotechnology, Cat# sc-365828, RRID:AB_10846854), and antibodies to detect K48-linkage ubiquitination (Abcam Cat# ab140601, RRID:AB_2783797) and K27 linkage ubiquitination (Abcam Cat# ab181537, RRID:AB_2713902) were used, as was as anti-HA antibody (Proteintech Cat# 51064-2-AP, RRID:AB_11042321). For phospho-USP10 analysis, we utilized an antibody against phospho-MAPK Substrates Motif [PXpTP] MultiMab™ Rabbit mAb mix (Cell Signaling Technology Cat# 14378, RRID:AB_2798468).
Lentiviral shRNA mediated knockdown
Two Human USP10 shRNA were transfected with pMD2G (Addgene, MA.; plasmid #12259, RRID:Addgene_12259) and pSPAX2 (Addgene, plasmid #12260, RRID:Addgene_12260) into HEK293T (RRID:CVCL_0063) cells to generate lentivirus as previously described (25). CRC cells were incubated with lentiviruses and 8 μg/ml polybrene per the manufacturer’s instructions. When cells achieved 80% confluence after lentivirus infection, cells were incubated with medium containing puromycin (1:5000 v/v) with subsequent analysis of knockdown efficiency by immunoblotting. Production and transduction of lentivirus into target cells and elimination of non-transduced target cells were performed per standard procedures, as described previously (28).
Immunoblotting and immunoprecipitation
Immunoblot and immunoprecipitation assays were performed as described previously (29). Briefly, NETN buffer [20 mM Tris-HCl (pH 8.0), 1 mM EDTA, 0.5% Nonidet P-40, 100 mM NaCl], 10 mM NaF, 50 mM β-glycerophosphate, and 1 mg/ ml each of pepstatin A and aprotinin were used to lyze the cells. Cell lysates were centrifuged at 10,000 rpm for 15 min and the supernatant was removed and incubated with 2 μg of the indicated antibody and 20 μl protein A or protein G Sepharose beads (GE Healthcare, MA) overnight at 4 °C. Immunoprecipitates were centrifuged at 8000 rpm for 1 min, washed twice with cold NETN buffer and boiled with 1× Laemmli buffer for 10 min. The samples were then separated by SDS–PAGE and transferred to PVDF membranes using the semi-dry method (Trans-Blot® Turbo™ Transfer System, Bio-Rad, CA). PVDF membranes were incubated with 5% milk for 1 h, followed by incubation with the indicated primary antibody overnight at 4oC. On the following day, membranes were washed with PBST buffer x3, and then incubated with goat anti-rabbit HRP (Jackson Immunoresearch, AB_2313567, RRID: AB_2313567) or goat anti-mouse HRP (Jackson Immunoresearch, PA; AB_10015289, RRID: AB_10015289) secondary antibodies for 1 hr. After washing x3 in PBST buffer, the membranes were incubated with ECL and the signal was detected using an Azure imaging system (Dublin, CA) or by X-ray film.
Deubiquitination assay
Deubiquitination assays were performed as previously described (25). Briefly, cells with the indicated treatments were incubated with a proteosome inhibitor MG132 at 10 μM for 6 hr. Cells were then lysed using 1% SDS, boiled at 100o C for 20 min, and diluted with NETN buffer containing a protease inhibitor cocktail. The cell lysate supernatant was immunoprecipitated with an anti-ZEB1 antibody and the immunocomplexes were analyzed by immunoblot assay.
Transwell assay
To assess cancer cell migration, RKO cells in serum free RPMI media were treated as indicated and then treatments were placed in the top chamber of transwell migration chambers (8.0 μm pore polycarbonate membrane insert; Corning, AZ). The lower wells were filled with RPMI supplemented with 10% FBS and the indicated treatment. Twenty-four hr. later, cells were removed from the top surface of the transwell membrane using a cotton swab, and migrated cells on the lower membrane surface were fixed, stained, photographed, and counted at light microscopy (20X magnification).
Animal studies
Female 4–6 weeks old Balb/c nude mice (strain #:002019, RRID:IMSR_JAX:002019) were purchased from Jackson Laboratory, ME. For analysis of lung metastasis, 1 × 106 RKO Luciferase-tagged parental cells or knockdown cells were resuspended in sterile PBS and injected into the lateral tail vein of nude mice using a 25-gauge needles and a model previously used for study of EMT-mediated CRC invasion and metastasis (30,31). At 7 days post injection, mice were randomized into control or treatment groups (n=5 each) and then treated with the indicated drugs either by gavage feeding or by i.p. injection until termination of the experiment. Mice were euthanized at 10 weeks after tail vein injection and examined for lung metastatic nodules using a dissecting microscope. Mouse lung tissues and tumor colonization were further examined in H&E-stainied tissue sections at light microscopy. Tumor quantification and related data analysis were performed blinded to treatment arms.
Statistical analysis
All cell culture experiments used to generate representative blots or figures were performed in triplicate. Data derived from the transwell assay and the tail vein injection assay were presented as mean ± standard deviation (SD) or standard error of the mean (SEM). Data were analyzed using the Student’s t test (two-sided) or presented as mean ± SEM using the one-way analysis of variance (ANOVA) followed by post hoc Tukey’s test. Results were considered statistically significant if *p < 0.05 or **p < 0.01, as indicated.
Study approval
All animal experiments were performed in accordance with a study protocol approved by the Institutional Animal Care and Use Committee of Mayo Clinic.
Data availability
All data generated or analyzed during this study are available within the article or available from the authors upon request.
Results
ZEB1 protein stability is regulated by MEK-ERK signaling
We examined protein products of EMT transcription factors in human CRC cell lines with constitutive MEK-ERK activation, including RKO cells that primarily exhibit mesenchymal features and carry one wild-type and two mutant BRAF alleles (V600E)(32). Of the EMT proteins examined, only ZEB1 and ZEB2 protein expression were increased in RKO isogenic BRAFV600E cells compared to BRAF wild-type (wt) [T29] cells (Fig.1A–B). Treatment of these cells with the MEK inhibitor cobimetinib suppressed expression of ZEB1 but not ZEB2 (Fig.1B). Similarly, treatment of RKO cells with the selective BRAFV600E inhibitors vemurafenib (Fig.1C) or encorafenib (Fig. 1D) were each shown to suppress ZEB1 expression. Another MEK inhibitor trametinib was also shown to decrease ZEB1 expression (Fig. 1D). To demonstrate induction of ZEB1 in cells with MEK-ERK activation, Colo320 cells were transfected with a mutant KRAS G12D plasmid wherein an increase in ZEB1 protein expression was observed (Fig.1E). Among EMT factors examined, preferential regulation of ZEB1 by MEK-ERK signaling was observed.
Fig.1.
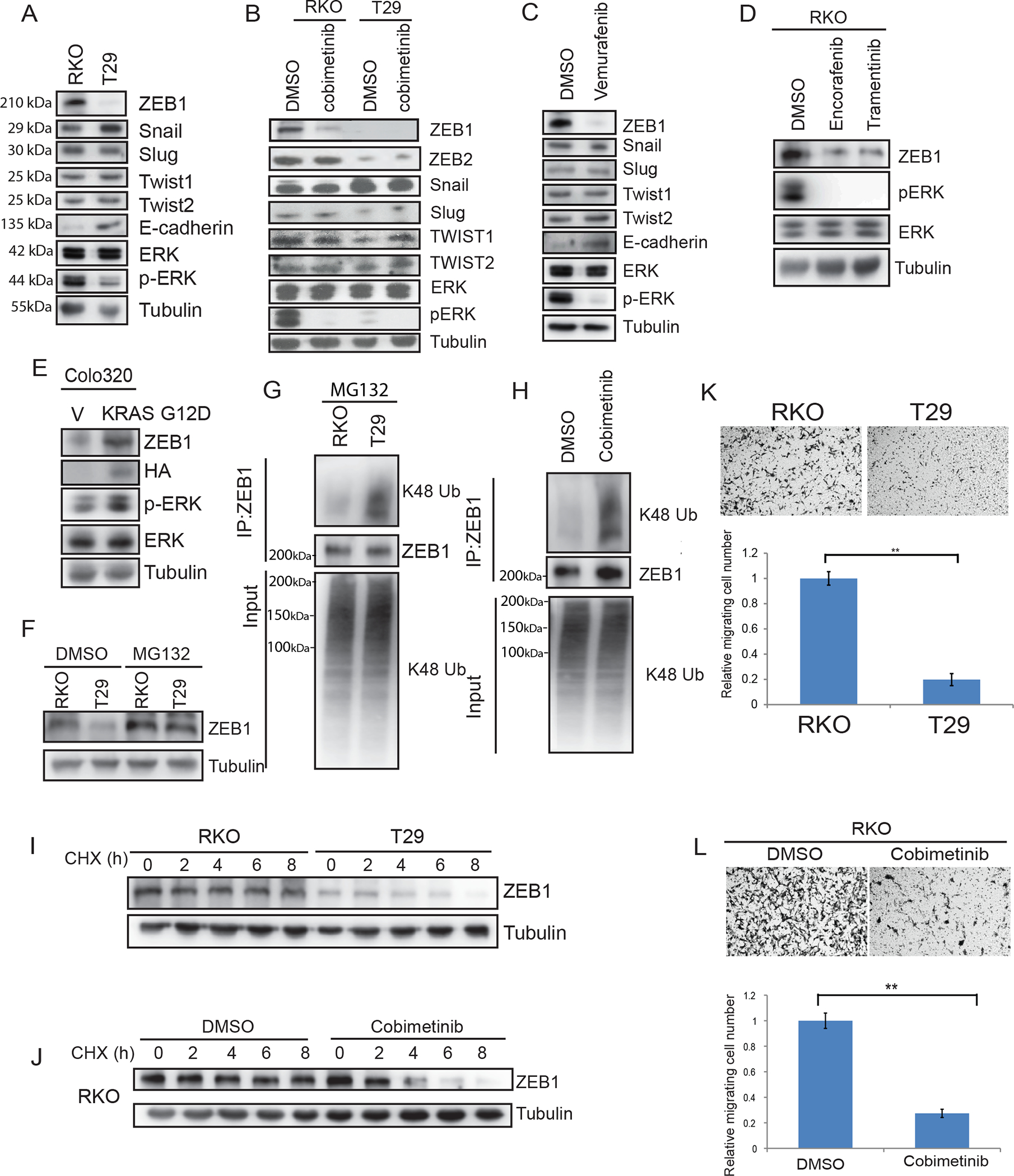
MEK-ERK signaling regulates ZEB1 protein stability and CRC cell migration.
A, Immunoblot analysis of expression of EMT transcription factors in isogenic CRC cell lines including parental RKO (BRAFV600E/V600E/wt) and T29 (BRAFwt/−/− ). B, Isogenic RKO cells treated with vehicle or MEK inhibitor cobimetinib (2 μM). C, EMT protein expression in RKO cells treated with BRAF inhibitor vemurafenib (5 μM) or vehicle. D, Detection of ZEB1 protein expression and p-ERK/ERK in RKO cells treated with BRAFV600E inhibitor encorafinib or MEK inhibitor trametinib (10 μM). (E), Colo320 cells transfected with mutant KRAS G12D. F, Analysis of ZEB1 expression in cell lines treated with the proteasome inhibitor, MG132. G-H, Cells were treated with MG132 and K48-linkage polyubiquitination of ZEB1 was analyzed in RKO and T29 cells (G), and in RKO cells treated with DMSO or cobimetinib (2 μM) [H]. I-J, Determination of ZEB1 protein half-life in RKO and T29 cells treated with cyclohexamide (I) and RKO cells treated with vehicle or cobimetinib (J). K, Analysis of isogenic RKO cell migration transwell assays and quantification of migrated cells. L, Analysis of RKO cell migration in presence of cobimetinib (2 μM) or vehicle by transwell assays and quantification of migrated cells in. Migrated cells were quantified from images (n=5) (K, L). Data are presented as mean ± SD. Student t test. **, p < 0.01.
Treatment of RKO isogenic cells with the proteasome inhibitor, MG132, enhanced ZEB1 expression in BRAFV600E and in BRAF wt T29 cell lines (Fig.1F) indicating that ZEB1 undergoes proteasomal degradation. Treatment of cells with MG132 showed decreased K48-linkage polyubiquitinated ZEB1 protein in RKO BRAFV600E in contrast to BRAF wt T29 cells (Fig.1G), and this ubiquitination could be reversed by cobimetinib (Fig.1H). Enhanced ZEB1 protein stabilization was also observed in BRAFV600E compared to BRAF wt cells by treatment of these isogenic cells with an inhibitor of protein synthesis, i.e., cycloheximide, that impaired protein degradation (Fig. 1I). Treatment with cobimetinib was shown to increase proteasomal degradation of ZEB1 (Fig.1J). Together, these results suggest that MEK-ERK activation can protect ZEB1 from proteasomal degradation to enhance its stability.
The functional consequence of ZEB1 stabilization was examined in transwell assays. Isogenic BRAFV600E cells that overexpress ZEB1 were observed to migrate faster than did cells with BRAF wt (Fig.1K). Cobimetinib, shown to suppress ZEB1 (Fig. 1B), inhibited CRC cell migration in transwell assays (Fig.1 L). These data suggest that regulation of ZEB1 stability by MEK-ERK can modulate CRC cell migration, although cobimetinib-induced MEK suppression of migration could also be affected by other means.
USP10 can regulate ZEB1 protein stability
Deubiquitinases (DUBs) can regulate protein stability and we previously reported that ubiquitin-specific protease 10 (USP10) could deubiquitinate and thereby, activate AMPK (25). To identify potential DUBs that may interact with ZEB1, we examined the BioGrid database that includes manually curated protein and genetic interactions (19). We identified USP10 as a potential candidate in BioGrid (supplemental Table S1) and then demonstrated that USP10 co-immunoprecipitated with ZEB1 in RKO cells (Fig.2A–B). We confirmed the UP10-ZEB1 interaction in Colo320 cells where reciprocal immunoprecipitation using antibodies against USP10 or ZEB1 was shown to immunoprecipitate ZEB1 or USP10 proteins, respectively (Fig.2C–D). Since USP10 is a ubiquitin-specific protease, we determined if USP10 can stabilize ZEB1. Knockdown of USP10 in RKO cells was observed to significantly increase the level of endogenous ZEB1 protein (Fig.2E) that was also shown using a second USP10 shRNA (Fig.2E). Consistent results were observed in Colo320 cells (Fig.2F). To determine whether the increase in ZEB1 expression is due to protein stabilization, we treated cells expressing USP10 shRNA or control shRNA with cycloheximide and observed enhanced stability of ZEB1 in USP10 knockdown cells (Fig.2G). Furthermore, K48-linkage polyubiquitination of ZEB1 was decreased in cells expressing USP10 shRNA (Fig.2H), indicating impaired proteasomal degradation.
Fig.2.
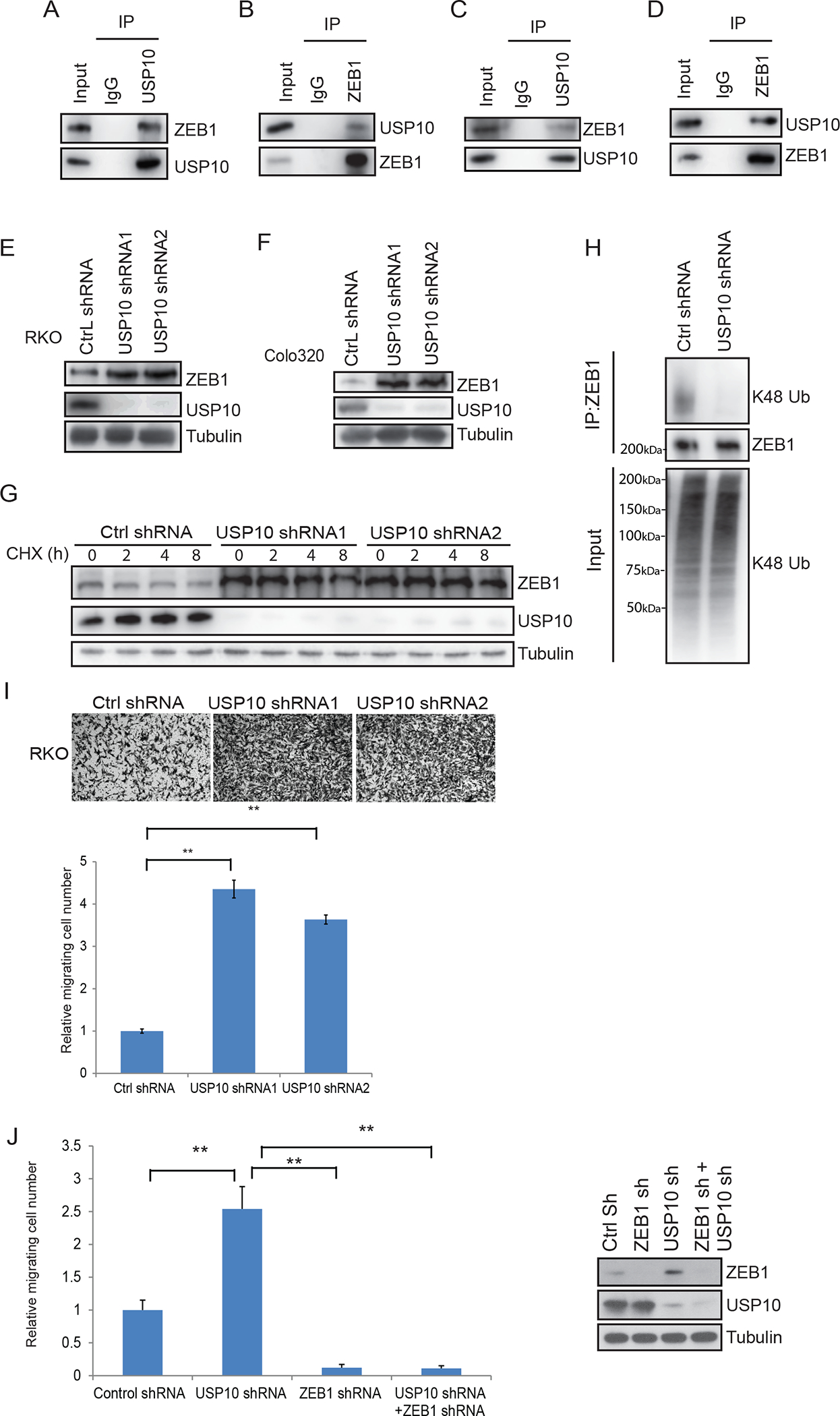
USP10 DUB interacts with and stabilizes ZEB1 leading to suppression of CRC cell migration. A-D, Analysis of interaction of ZEB1 and USP10 by immunoprecipitation in RKO (A-B) and Colo320 CRC cells (C-D). E-F, Immunoblot of ZEB1 and USP10 in RKO (E) and Colo320 (F) cells transfected with control shRNA or two different USP10 shRNA vectors. G, Determination of ZEB1 protein half-life in cycloheximide-treated RKO cells transfected with control shRNA or USP10 shRNA1 or 2. H, Analysis of K48-linkage polyubiquitination of ZEB1 in USP10 knockdown cells vs control shRNA cells. I, Analysis of cell migration by transwell assays in RKO cells expressing control shRNA or USP10 shRNA and quantification of migrated cells. J, Quantification of cell migration by transwell assays in RKO cells expressing control shRNA, USP10 shRNA, ZEB1 shRNA or USP10 shRNA/ ZEB1 shRNA. Migrated cells were quantified from the images (n=5) in I,J. Data are presented as mean ±SEM using the one-way ANOVA t-test followed by post hoc Tukey’s test. **, p < 0.01.
Since depletion of USP10 can induce ZEB1, we determined the effect of USP10 on the ability of ZEB1 to regulate CRC cell migration. Using transwell assays, we found that knockdown of USP10 and its associated induction of ZEB1 (Fig. 2E,F), can enhance cell migration in this assay (Fig.2I). To confirm that USP10 regulates cell migration through ZEB1, we generated cells with knockdown of USP10 or ZEB1 separately or combined and observed that CRC cells expressing USP10 shRNA showed faster migration whereas as cells with ZEB1 shRNA displayed slower migration (Fig. 2J). Knockdown of USP10 in cells with ZEB1 shRNA did not enhance cell migration (Fig.2J) indicating that USP10 regulates cell migration through ZEB1.
Activated MEK-ERK signaling promotes cell migration through USP10
We determined if MEK-ERK signaling can mediate ZEB1 stabilization in isogenic CRC cells through USP10. Knockdown of USP10 increased ZEB1 expression to a similar extent in BRAFV600E vs wild-type cells (Fig.3A) that was associated with decreased K48 ubiquitination of ZEB1 (Fig.3B). Analysis of cell migration in transwell assays revealed that RKO cells expressing BRAFV600E migrated faster than did T29 cells with control shRNA expression. However, USP10 knockdown in both cell lines was shown to increase cell migration to a similar extent (Fig. 3C). Depletion of USP10 was also shown to enhance migration of isogenic CRC cell lines in transwell assays (Fig.3 C). Since activation of MEK-ERK can stabilize ZEB1 and promote cell migration through USP10, we inhibited ERK signaling which was shown to suppress ZEB1 and its regulation by USP10. Treatment of RKO and Colo320 cells with cobimetinib decreased ZEB1 protein expression compared to control-treated cells (Fig.4 A,B). Knockdown of USP10 induced ZEB1 expression in the presence or absence of an inhibitor of cobimetinib (Fig.4 A,B) or vemurafenib that inhibits BRAF (Fig.4C). Enhanced K48 ubiquitination of ZEB1 was detected in both cobimetinib- and vemurafenib- treated cells, but knockdown of USP10 impaired these ubiquitination signals (Fig.4D–E).
Fig.3.
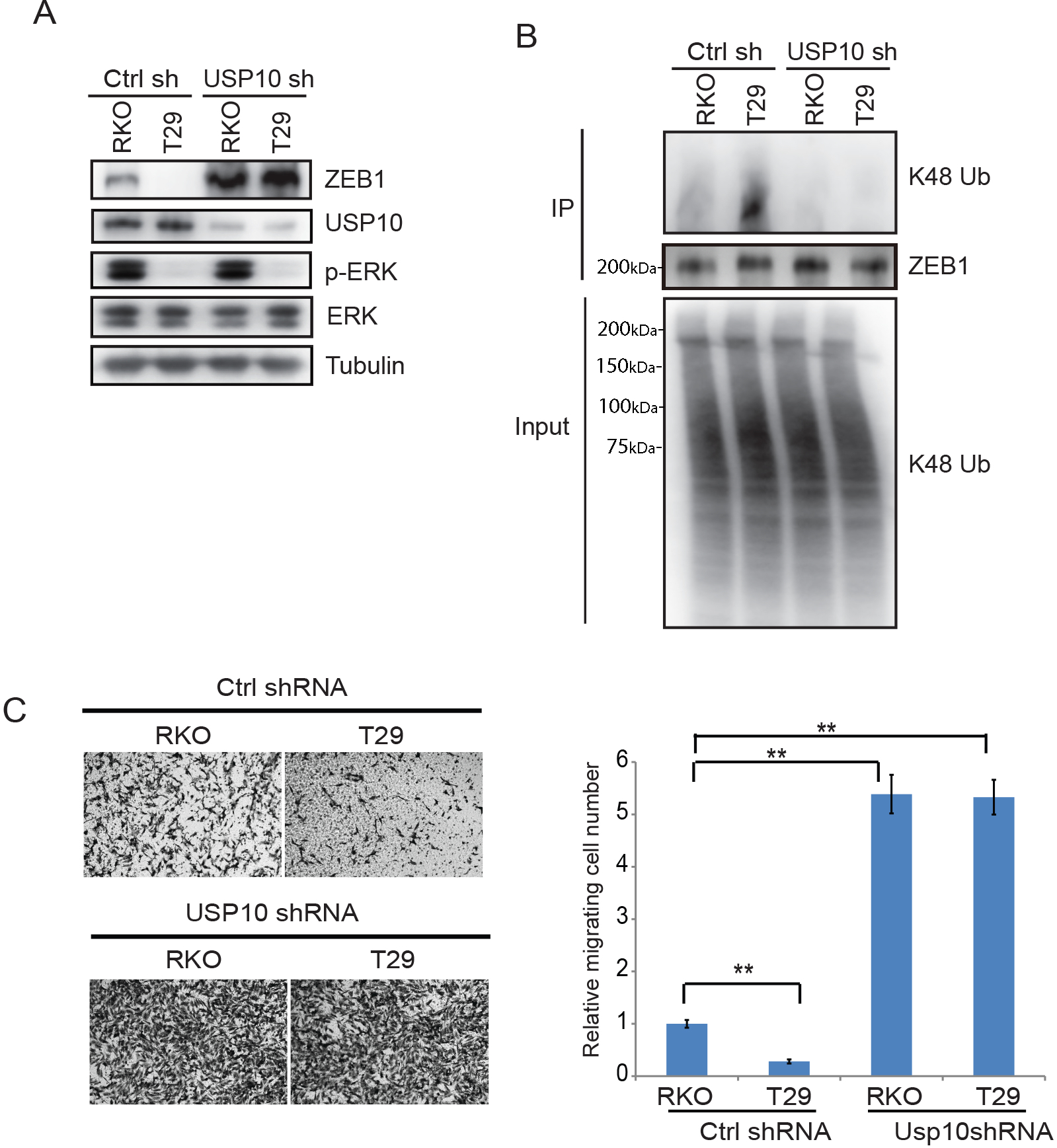
Constitutive activation of MEK-ERK signaling phosphorylates USP10 to impair its interaction with ZEB1 and thereby, stabilize ZEB1 protein. A, Analysis of ZEB1 protein expression in isogenic RKO cells expressing control shRNA or USP10 shRNA. B, Immunoblotting of K48-linkage polyubiquitination of ZEB1 in isogenic RKO cells expressing control shRNA or USP10 shRNA. C, Assessment of cell migration in isogenic RKO cells expressing control shRNA or USP10 shRNA in transwell assays and quantification of migrated cells. Migrated cells were quantified from the images (n=5) (C). Data are presented as mean ±SEM using the one-way ANOVA followed by post hoc Tukey’s test. **, p < 0.01.
Fig.4.
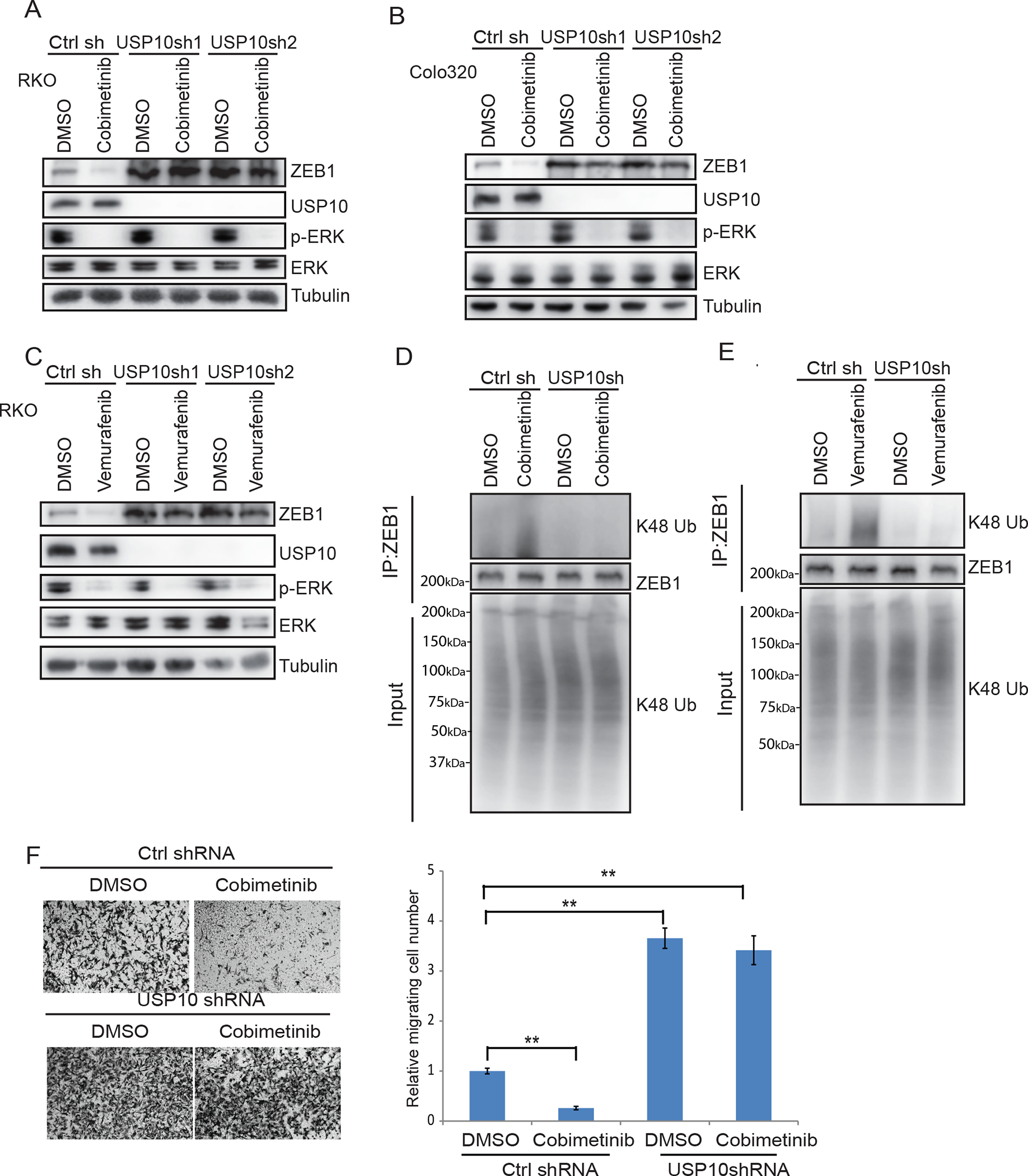
USP10 can edit ZEB1 ubiquitination and modify cell migration.
A-B, Immunoblot analysis of ZEB1 and p-ERK/ERK protein expression in RKO cells (A) or Colo320 cells (B) transfected with control shRNA or two different USP10 shRNA vectors in the presence or absence of treatment with cobimetinib or the BRAF inhibitor, vemurafenib (C). D-E, Analysis of K48-linkage polyubiquitination of ZEB1 in RKO cells expressing control shRNA or USP10 shRNA with or without cobimetinib (D) or vemurafenib (E). F, Assessment of cell migration in RKO cells expressing control shRNA or USP10 shRNA treated with vehicle or cobimetinib in transwell assays and quantification of migrated cells. Migrated cells were quantified from the images (n=5) (F). Data are presented as mean ±SEM using the one-way ANOVA followed by post hoc Tukey’s test. **, p < 0.01.
Treatment of CRC cells with cobimetinib suppressed cell migration that could be reversed or enhanced with depletion of USP10 even in the presence of MEK inhibition (Fig.4F).
USP10 can edit ZEB1 ubiquitination
USP10 can stabilize its substrate or regulate substrate activity. To elucidate the mechanism by which USP10 regulates ZEB1 protein stability, we transfected control and USP10 depleted RKO cells with a panel of ubiquitin mutants where only one of seven lysine residues was inactivated. USP10 knockdown in CRC cells was shown to increase K27 ubiquitination and to inhibit K48 ubiquitination of ZEB1 (Fig. 5A). This finding suggests that USP10 can selectively remove K27-linkage that may allow for subsequent K48-linked ubiquitination of ZEB1. To confirm this result, we re-introduced wild-type USP10 or a catalytically dead mutant of USP10 into USP10 knockdown cells. Reconstitution of wild-type USP10 was shown to reverse the ubiquitination pattern of ZEB1, but not in cells with the catalytic dead mutant (Fig. 5B).
Fig 5.
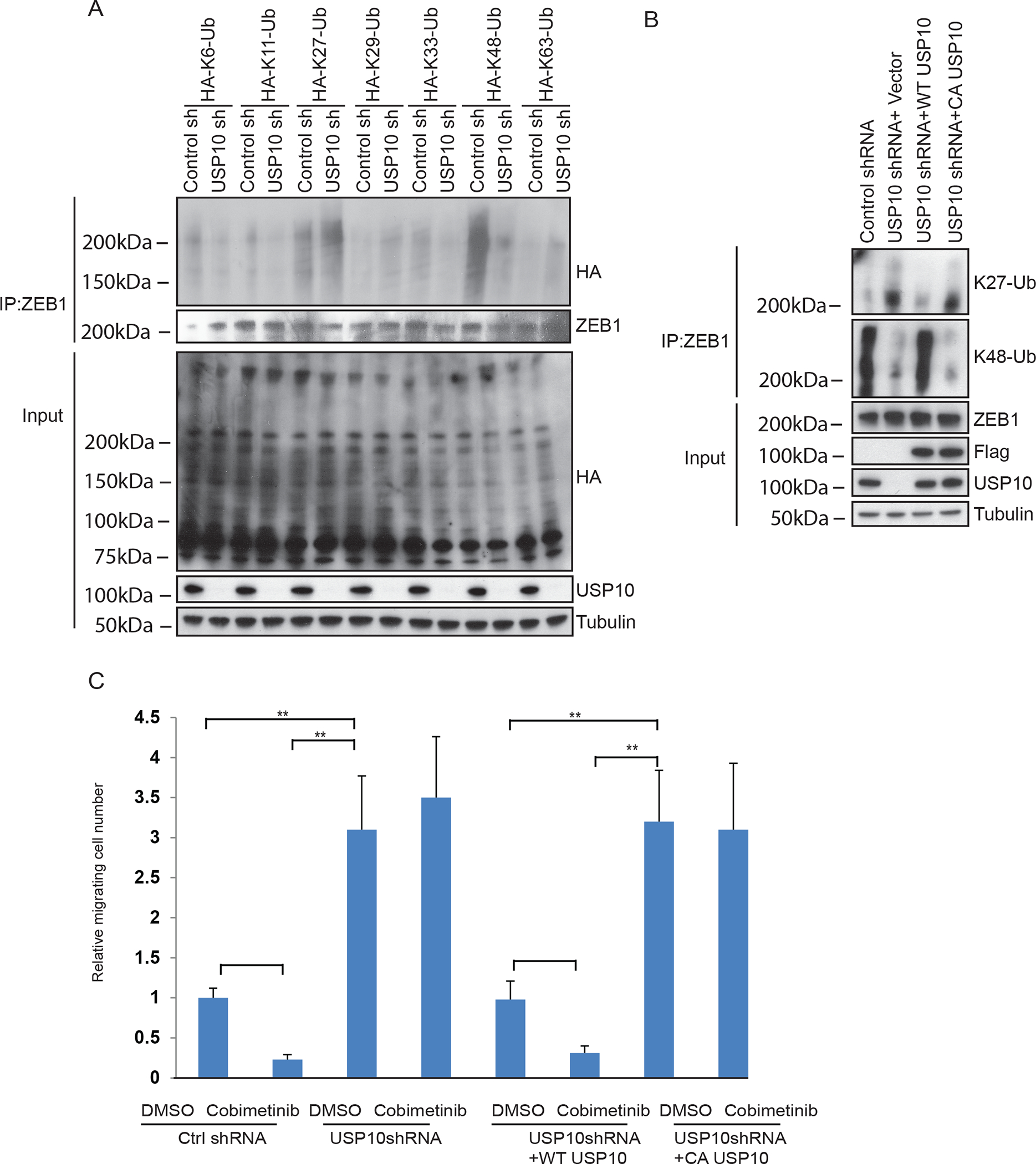
USP10 edits ZEB1 ubiquitination.
To elucidate the mechanism by which USP10 regulates ZEB1 protein stability, control and USP10 depleted RKO cells were generated with a panel of ubiquitin mutants with inactivation of specific lysine residues. A, Analysis of effect of USP10 on ZEB1 ubiquitination using a panel of ubiquitin mutants. B, Analysis of ZEB1 K27 linkage and K48-linkage chain polyubiquitination in RKO cells expressing control shRNA, USP10 shRNA, wild-type USP10 reconstituted cells, or in USP10 catalytically dead mutant cells. C, Quantification of cell migration by the transwell assays in RKO cells expressing control shRNA, USP10 shRNA, USP10 shRNA+ wt USP10, and USP10 shRNA+ CA USP10 in the presence or absence of cobimetinib. Migrated cells were quantified from the images (n=5) (C). Data are presented as mean ±SEM using the one-way ANOVA followed by post hoc Tukey’s test. **, p < 0.01.
To assess the functional consequence of regulation of ZEB1 by USP10, CRC cell migration was again analyzed. We observed increased cell migration in USP10 knockdown cells in presence or absence of cobimetinib (Fig.5C). Next, we assessed the effect of a USP10 catalytically dead mutant on cell migration. While re-introduction of wt USP10 into USP10 depleted cells was shown to suppress cell migration, this was not observed for the catalytically dead USP10 mutant. These results suggest that USP10 can edit ZEB1 ubiquitination and thereby, influence CRC cell migration through its DUB activity.
ERK phosphorylates USP10 at S236 and promotes its disassociation from ZEB1
We examined the interaction of USP10 with ZEB1 in CRC cells with constitutive activation of MEK-ERK signaling. The USP10-ZEB1 interaction was found to be attenuated in cells with BRAFV600E compared to BRAF wt cells (Fig.6A), and this interaction was enhanced by treatment with cobimetinib (Fig.6B). Since ERK kinases have various cytosolic and nuclear substrates in contrast to RAF and MEK1/2 kinases that have narrow substrate specificity (33), we hypothesized that ERK can phosphorylate USP10 which may alter its interaction with ZEB1. ERK-activated BRAFV600E cells showed enhanced phosphorylation of USP10 compared to wt cells, and binding between USP10 and ERK is demonstrated (Fig. 6C). Suppression of MEK-ERK by cobimetinib attenuated USP10 phosphorylation and enhanced the USP10-ZEB1 interaction (Fig.6D). Analysis of the USP10 protein sequence identified two potential ERK phosphorylation sites, T74 and S236, based on the ERK substrate motif. Whereas mutation of Thr to Ala at 74 had no effect on phosphorylation of USP10 and its interaction with ZEB1, mutation of Ser to Ala at 236 suppressed ERK-mediated USP10 phosphorylation and enhanced the USP10-ZEB1 interaction (Fig. 6E). Treatment of cells expressing this phosphorylation mutant (S236A) with cobimetinib did not alter the phosphorylation event nor the interaction between USP10 and ZEB1 (Fig.6E). Furthermore, a double mutant behaved similarly to the S236A mutant (Fig.6E). Importantly, expression of the USP10 S236A mutant was shown to suppress K27 ubiquitination and to increase K48 ubiquitination of ZEB1 (Fig.6F) that resulted in its degradation (Fig. 6G) and its suppression of cell migration (Fig. 6H). Lastly, inhibition of MEK/ERK failed to alter ZEB1 ubiquitination pattern, protein stability and cell migration in cells expressing the S236A mutant (Fig.6F–H).
Fig.6.
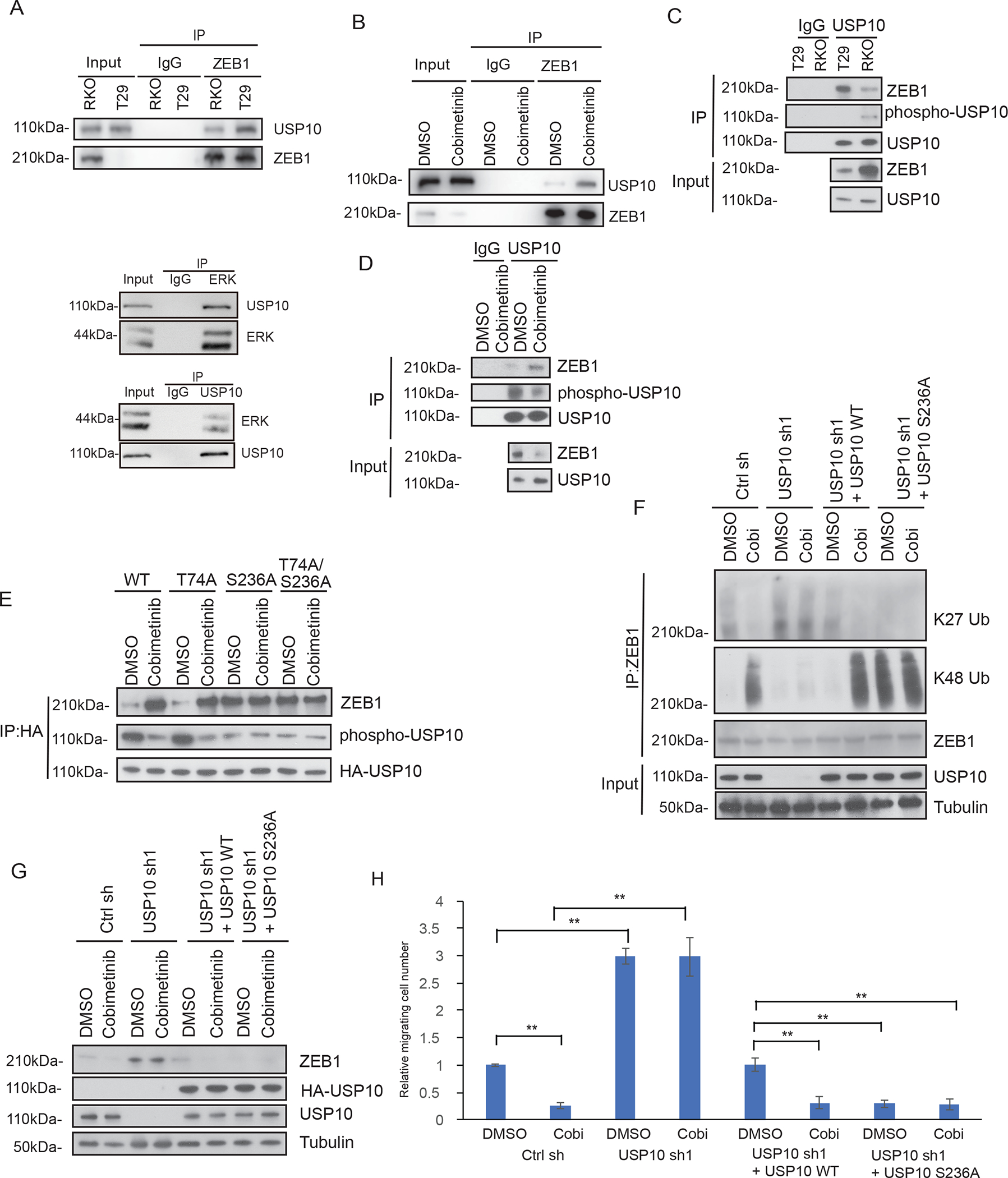
Phosphorylation of USP10 by ERK impairs its interaction with ZEB1 and results in ZEB1 stabilization. A-D, Analysis of interaction between ZEB1 and USP10 by co-immunoprecipitation in isogenic RKO CRC cells including binding between ERK and USP10 (A,C), or in RKO cells treated with vehicle or cobimetinib (B,D). E, Identification of the ERK-mediated phosphorylation site on the USP10 protein using a phospho-MAPK substrate antibody. Potential ERK phosphorylation sites, T74 and S236, were identified based on the ERK substrate motif and mutants were generated. Effect of treatment of cells vehicle (DMSO) versus the MEK inhibitor cobimetinib on USP10 phosphorylation events and the interaction of USP10 and ZEB1. F-H, Analysis of ZEB1 K27 linkage and K48-linkage chain polyubiquitination in RKO cells expressing control shRNA, USP10 shRNA, wildtype USP10 reconstituted cells or USP10 S236A mutant expressing cells in the presence or absence of vehicle (DMSO) or cobimetinib treatment (F). Analysis of ZEB1 protein expression (G) and assessment of cell migration (H) were also performed in the same RKO expressing cell lines. Migrated cells were quantified from the images (n=5) (H). Data are presented as mean ±SEM using the one-way ANOVA followed by post hoc Tukey’s test. **, p < 0.01.
Targeting the MEK-ERK-USP10-ZEB1 axis inhibits metastasis
ZEB1 mRNA expression, but not USP10, was significantly increased in human colon cancers that display a transcriptomically-determined mesenchymal and EMT-associated subtype known as consensus molecular subtype 4 (CMS4)](34) in pooled data from 10 human colon cancer cohorts (stage I-IV; N=1483)[Fig. 7A]. To study the role of the MEK-ERK USP10-ZEB1 axis in tumor metastasis, we utilized a humanized mouse model. RKO cells with knockdown of USP10 were injected via the tail vein into mice and shown to promote lung tumor colonization to a significantly greater extent compared to injected control shRNA cells (Fig. 7B,C). In contrast, knockdown of ZEB1 dramatically suppressed lung tumor colonization and in these cells, combined knockdown of USP10 failed to overcome the effect of ZEB1 suppression and enhance lung colonization (Fig.7B,C). These results suggest that USP10 regulates CRC cell metastasis through the EMT transcription factor ZEB1. We then determined whether MEK-ERK inhibition can suppress tumor metastasis through the USP10-ZEB1 axis in our CRC murine model. EGFR-expressing RKO cells (35) were treated with encorafenib (BRAF inhibitor) plus cetuximab (EGFR inhibitor) since this combination is needed to suppress rebound EGFR activation when RAS/RAF MEK-ERK signaling is inhibited (36). Treatment with encorafenib plus cetuximab, in contrast to either drug alone, significantly suppressed lung tumor colonization resulting from tumor cell tail vein injection (Fig.7D,E). In cells with knockdown of USP10, however, a marked increase lung colony number was observed in the presence or absence of treatment with encorafenib, cetuximab or their combination (Fig.7D,E).
Fig.7.
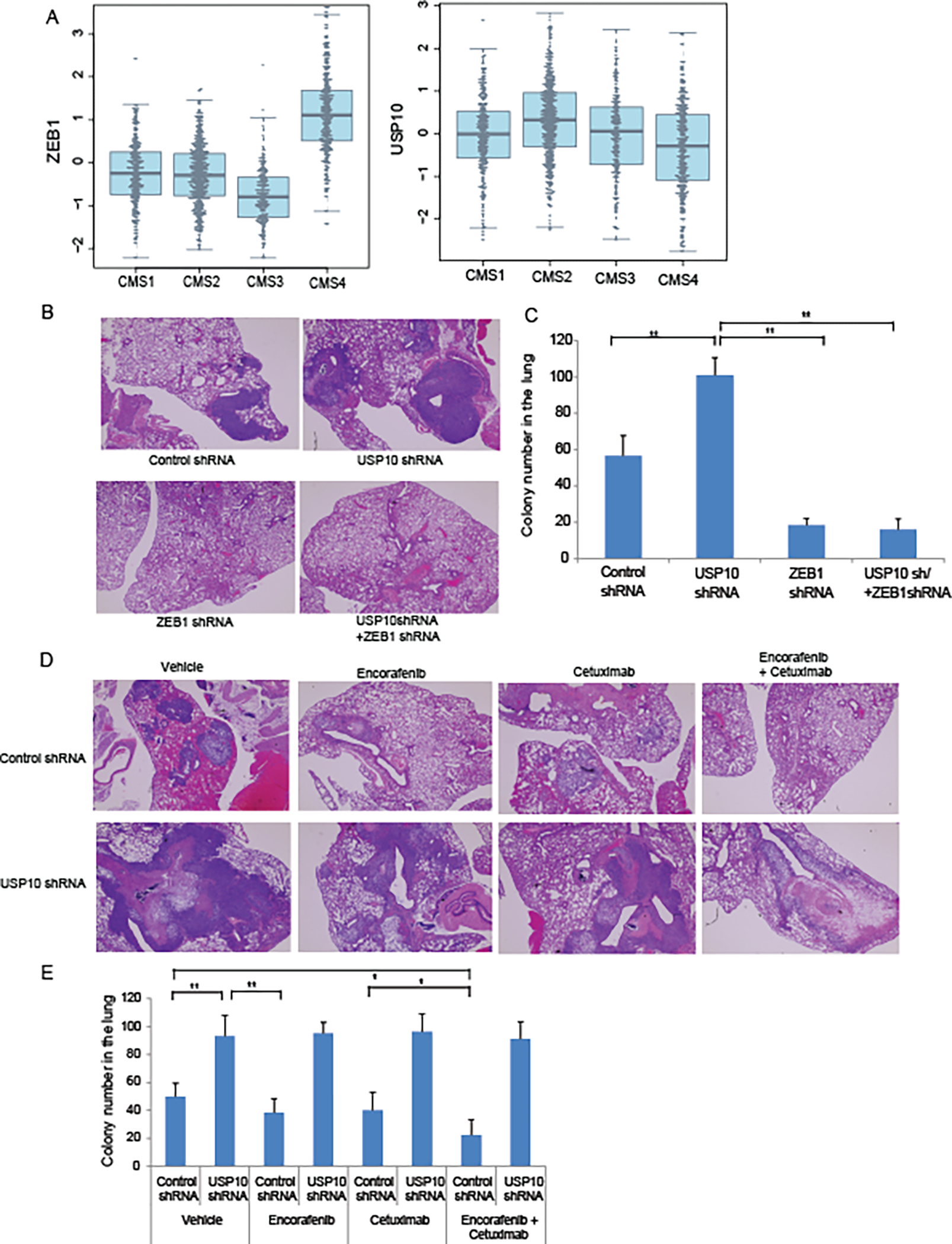
Inhibition of MEK-ERK-USP10-ZEB1 signaling suppresses CRC metastatic colonization.A, Association of ZEB 1 and USP10 mRNA expression in human colon cancers with transcriptomically-determined consensus molecular subtypes (CMS1-4)(34). Data are from 10 cohorts of patients with stage I-IV colon cancers (n=1483; see supplemental Table S2). P values for ZEB1 (3.57 × 10−84) and USP10 (7.78 ×10−11) by CMS were determined using Stouffer’s test. B, Representative images (H&E sections) of metastatic lung nodules generated by tail vein injection of RKO cells into nude mice. Injected RKO cells included those generated to express control shRNA, USP10 shRNA, ZEB1 shRNA, and combined shRNA of USP10 plus ZEB1. C, Quantification of metastatic lung nodules from mice injected with cell lines described in B. D, Representative image (H&E) of metastatic lung nodules induced by injection of RKO cells expressing control shRNA or USP10 shRNA in presence of vehicle, BRAF inhibitor encorafenib, EGFR inhibitor cetuximab, or their combination. E, Quantification of metastatic lung nodules from experiments outlined in D. Metastatic lung colonies were quantified from lung tissue (n=5) (C,E). Data are presented as mean ±SEM using the one-way ANOVA followed by post hoc Tukey’s test. *, p < 0.05,**, p < 0.01.
Discussion
ZEB1 is a transcription factor that promotes tumor invasion and metastasis via EMT, and its expression increases during neoplastic progression (37). We found that ZEB1 was associated with a mesenchymal phenotype in pooled data from human colon cancers whereby ZEB1 expression was significantly increased in the transcriptomically-determined mesenchymal and EMT-associated consensus molecular subtype 4 (CMS4). CMS4 colon cancers have been shown to be more aggressive, metastatic and to have significantly worse survival compared to other CMS groups (34). Among the EMT master transcription factors, ZEB1 was found to be regulated by the RAS/RAF/MEK/ERK signaling pathway. Constitutive activation of MEK-ERK stabilized ZEB1 protein, and inhibitors of this signaling pathway suppressed ZEB1 due to its degradation. Posttranslational modifications (PTMs) regulate protein half-life, sub-cellular localization, and DNA/protein binding ability (38). To date, few studies have focused on PTMs of ZEB1 which may be critical to its ability to promote tumor metastasis. In this report, we made the novel observation that ERK kinase can phosphorylate USP10 (at serine 236) that was potently suppressed by MEK-ERK inhibition resulting in an enhanced USP10-ZEB1 interaction. These events lead to enhanced CRC cell migration and tumor metastasis consistent with the function of ZEB1 as a regulator of EMT and CRC metastasis.
While ZEB1 protein is subject to ubiquitination and degradation, the mechanism by which cellular ZEB1 is stabilized remains unclear. Mutation of the ERK-regulated phosphorylation site on USP10 changed serine to alanine at residue Ser 236, and impaired the dissociation of ZEB1 from its novel interaction with the DUB USP10. Our finding of deubiquitination of ZEB1 by USP10 resulting in its proteasomal degradation is mechanistically distinct from the reported interaction of ZEB1 with USP51 resulting in its deubiquitination and stabilization (39). Ubiquitination is achieved through ubiquitin-activating (E1) and -conjugating enzymes (E2) as well as ubiquitin-protein ligase (E3) resulting in ubiquitin being conjugated to a substrate protein. The E3 ligases SIAH1, TRIM26 and FBXO45 are responsible for K48 polyubiquitinated ZEB1 which can promote ZEB1 degradation. Lysine 48- and lysine 63-linked chains are the best characterized linkages while atypical lysine 27-linked chains remain poorly understood. Our data suggest that K27-linkage polyubiquitination chains can modulate ZEB1 protein stability. Specifically, the DUB activity of USP10 can remove the K27-linked ubiquitin chain from ZEB1 with a potential increase in K48-linked ubiquitinated ZEB1 that led to its proteasomal degradation. To date, only three DUBs (USP19, OTUD6A, USP38) have been shown to remove K27-linkage polyubiquitination chains from their substrates (16–18). Other examples exist for fine tuning of protein stability by competition with different polyubiquitinated chains. USP38 cleaves K33-linkage chains on TBK1 allowing for K48-linked ubiquitination mediated by DTX4 and TRIP(40). Elucidating the mechanisms that regulate ZEB1 ubiquitination levels may provide a promising strategy for targeting E3 ubiquitin ligase and DUBs in cancer treatment. Two known E3 ligases responsible for K27-linked ubiquitination have been reported, including NEDD4 and HACE1 (41,42). Planned studies will explore whether these or other E3 ligases utilized by USP10 contribute to its ability to modify the ubiquitination of ZEB1.
We studied the role of the MEK-ERK-USP10-ZEB1 axis in a tumor metastasis model that involves cancer cell adherence, transendothelial migration and colonization of a secondary organ site (43). Knockdown of ZEB1 was shown to dramatically suppress lung tumor colony colonization. While knockdown of USP10 promoted lung metastasis, cells with combined knockdown of USP10 and ZEB1 failed to promote lung colonization, suggesting that USP10 regulates CRC metastatic colonization through ZEB1. Consistent results were shown in a prostate cancer model where knockdown of ZEB1 partially restored an epithelial phenotype and reduced transendothelial migration and metastatic colonization of prostate cancer cells (44). Mutations in RAS/RAF are key drivers of MEK-ERK signaling and in human CRCs that harbor a BRAFV600E mutation, treatment with encorafenib plus cetuximab was shown to extend patient survival and is a standard treatment regimen for this molecularly-defined tumor subset (45). Treatment with encorafenib plus cetuximab, but not either drug alone, suppressed the number of lung metastatic colonies when USP10 was expressed, but not in cells with USP10 knockdown. These data suggest that inhibition of MEK-ERK signaling can suppress tumor colonization through the USP10-ZEB1 axis.
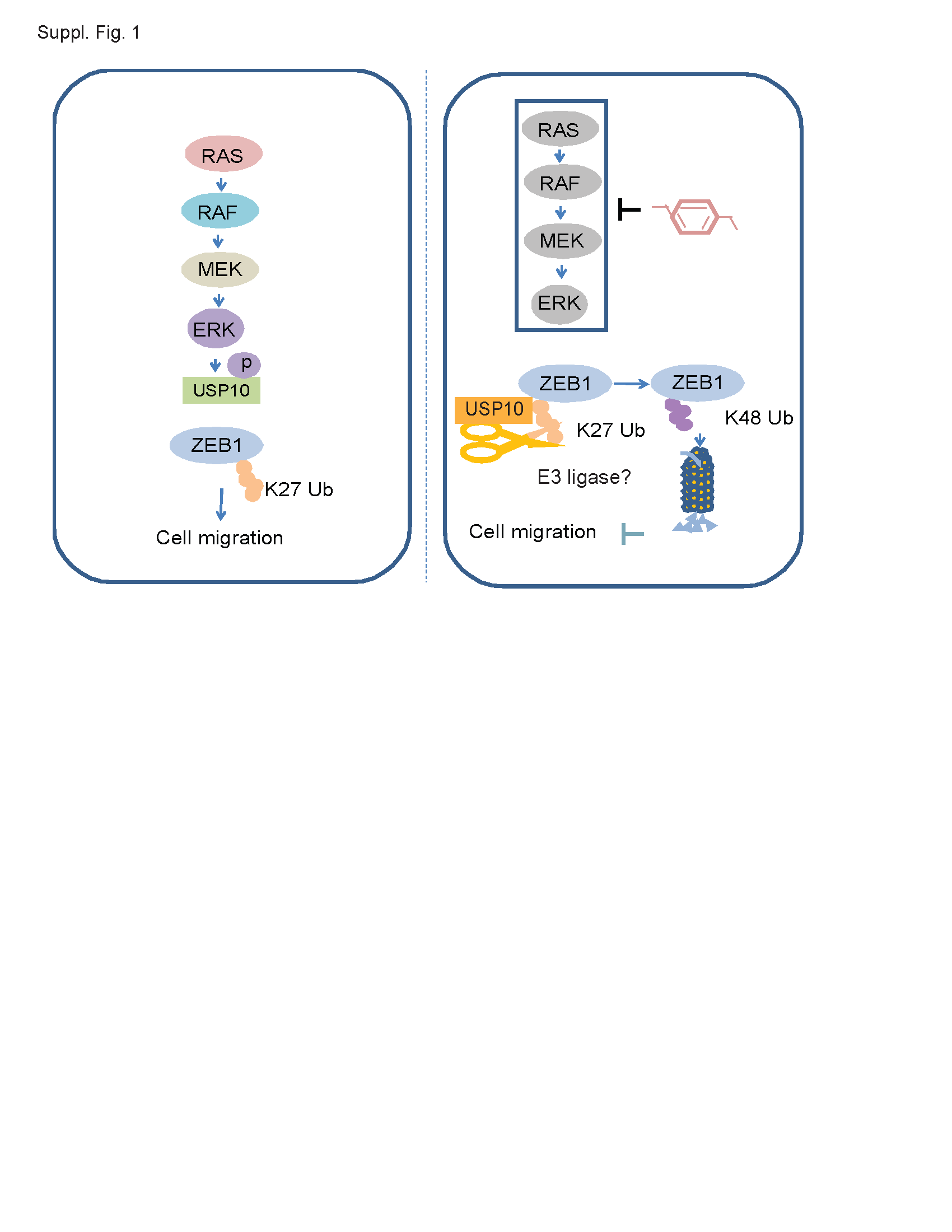
In summary, constitutive activation of MEK-ERK can phosphorylate USP10 at a novel site (S326) shown to disassociate USP10 from ZEB1 resulting in ZEB1 protein stabilization, enhanced cell migration, and tumor metastasis. Inhibition of MEK-ERK signaling suppressed USP10 phosphorylation, enhanced its interaction with ZEB1, and promoted its proteasomal degradation (shown schematically in Supplemental Fig. 1). These events served to suppress ZEB1-regulated EMT and tumor colonization in vivo. While our study demonstrates the deubiquitination of ZEB1 by USP10, identifying the specific E3 ligase responsible for adding polyubiquitinated chains to ZEB1 is under investigation. Taken together, these findings demonstrate a novel function of USP10 in the regulation of ZEB1 ubiquitination and protein stability that can inhibit ZEB1-mediated tumor colonization.
Supplementary Material
{kind=link}
Implications.
The MEK-ERK regulated interaction of USP10 with ZEB1 can promote the proteasomal degradation of ZEB1 and thereby suppress its demonstrated ability to mediate tumor metastasis.
Acknowledgements
This study was supported, in part, by NCI R01 CA210509 and Mayo Clinic Center for Biomedical Discovery Pilot Grant Program (both to FAS). LS is supported by the Second Affiliated Hospital of Guangzhou Medical University, Guangzhou, China. The authors wish to express their appreciation to Bahar Saberzadeh-Ardestani for her critical review of the manuscript.
Footnotes
Conflict of interest statement
The authors declare no potential conflicts of interest.
References:
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin 2021;71(1):7–33 doi 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 2.Biller LH, Schrag D. Diagnosis and Treatment of Metastatic Colorectal Cancer: A Review. JAMA 2021;325(7):669–85 doi 10.1001/jama.2021.0106. [DOI] [PubMed] [Google Scholar]
- 3.Lindner P, Paul S, Eckstein M, Hampel C, Muenzner JK, Erlenbach-Wuensch K, et al. EMT transcription factor ZEB1 alters the epigenetic landscape of colorectal cancer cells. Cell Death Dis 2020;11(2):147 doi 10.1038/s41419-020-2340-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caramel J, Ligier M, Puisieux A. Pleiotropic Roles for ZEB1 in Cancer. Cancer Res 2018;78(1):30–5 doi 10.1158/0008-5472.CAN-17-2476. [DOI] [PubMed] [Google Scholar]
- 5.Liu Y, Lu X, Huang L, Wang W, Jiang G, Dean KC, et al. Different thresholds of ZEB1 are required for Ras-mediated tumour initiation and metastasis. Nat Commun 2014;5:5660 doi 10.1038/ncomms6660. [DOI] [PubMed] [Google Scholar]
- 6.Manshouri R, Coyaud E, Kundu ST, Peng DH, Stratton SA, Alton K, et al. ZEB1/NuRD complex suppresses TBC1D2b to stimulate E-cadherin internalization and promote metastasis in lung cancer. Nat Commun 2019;10(1):5125 doi 10.1038/s41467-019-12832-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanchez-Tillo E, Lazaro A, Torrent R, Cuatrecasas M, Vaquero EC, Castells A, et al. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene 2010;29(24):3490–500 doi 10.1038/onc.2010.102. [DOI] [PubMed] [Google Scholar]
- 8.Na TY, Schecterson L, Mendonsa AM, Gumbiner BM. The functional activity of E-cadherin controls tumor cell metastasis at multiple steps. Proc Natl Acad Sci U S A 2020;117(11):5931–7 doi 10.1073/pnas.1918167117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mittal V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu Rev Pathol 2018;13:395–412 doi 10.1146/annurev-pathol-020117-043854. [DOI] [PubMed] [Google Scholar]
- 10.De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer 2013;13(2):97–110 doi 10.1038/nrc3447. [DOI] [PubMed] [Google Scholar]
- 11.Ribatti D, Tamma R, Annese T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl Oncol 2020;13(6):100773 doi 10.1016/j.tranon.2020.100773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen A, Wong CS, Liu MC, House CM, Sceneay J, Bowtell DD, et al. The ubiquitin ligase Siah is a novel regulator of Zeb1 in breast cancer. Oncotarget 2015;6(2):862–73 doi 10.18632/oncotarget.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu M, Zhu C, Zhao X, Chen C, Zhang H, Yuan H, et al. Atypical ubiquitin E3 ligase complex Skp1-Pam-Fbxo45 controls the core epithelial-to-mesenchymal transition-inducing transcription factors. Oncotarget 2015;6(2):979–94 doi 10.18632/oncotarget.2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem 1998;67:425–79 doi 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 15.Akutsu M, Dikic I, Bremm A. Ubiquitin chain diversity at a glance. J Cell Sci 2016;129(5):875–80 doi 10.1242/jcs.183954. [DOI] [PubMed] [Google Scholar]
- 16.Wu X, Lei C, Xia T, Zhong X, Yang Q, Shu HB. Regulation of TRIF-mediated innate immune response by K27-linked polyubiquitination and deubiquitination. Nat Commun 2019;10(1):4115 doi 10.1038/s41467-019-12145-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu X, Zhao J, Yu G, Zhang X, Sun J, Li L, et al. OTUD6A promotes prostate tumorigenesis via deubiquitinating Brg1 and AR. Commun Biol 2022;5(1):182 doi 10.1038/s42003-022-03133-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yi XM, Li M, Chen YD, Shu HB, Li S. Reciprocal regulation of IL-33 receptor-mediated inflammatory response and pulmonary fibrosis by TRAF6 and USP38. Proc Natl Acad Sci U S A 2022;119(10):e2116279119 doi 10.1073/pnas.2116279119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oughtred R, Rust J, Chang C, Breitkreutz BJ, Stark C, Willems A, et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci 2021;30(1):187–200 doi 10.1002/pro.3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ouchida AT, Kacal M, Zheng A, Ambroise G, Zhang B, Norberg E, et al. USP10 regulates the stability of the EMT-transcription factor Slug/SNAI2. Biochem Biophys Res Commun 2018;502(4):429–34 doi 10.1016/j.bbrc.2018.05.156. [DOI] [PubMed] [Google Scholar]
- 21.Gao D, Zhang Z, Xu R, He Z, Li F, Hu Y, et al. The Prognostic Value and Immune Infiltration of USP10 in Pan-Cancer: A Potential Therapeutic Target. Front Oncol 2022;12:829705 doi 10.3389/fonc.2022.829705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan J, Luo K, Zhang L, Cheville JC, Lou Z. USP10 regulates p53 localization and stability by deubiquitinating p53. Cell 2010;140(3):384–96 doi 10.1016/j.cell.2009.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim R, Sugino T, Nolte H, Andrade J, Zimmermann B, Shi C, et al. Deubiquitinase USP10 regulates Notch signaling in the endothelium. Science 2019;364(6436):188–93 doi 10.1126/science.aat0778. [DOI] [PubMed] [Google Scholar]
- 24.Lin Z, Yang H, Tan C, Li J, Liu Z, Quan Q, et al. USP10 antagonizes c-Myc transcriptional activation through SIRT6 stabilization to suppress tumor formation. Cell Rep 2013;5(6):1639–49 doi 10.1016/j.celrep.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deng M, Yang X, Qin B, Liu T, Zhang H, Guo W, et al. Deubiquitination and Activation of AMPK by USP10. Mol Cell 2016;61(4):614–24 doi 10.1016/j.molcel.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun J, Li T, Zhao Y, Huang L, Sun H, Wu H, et al. USP10 inhibits lung cancer cell growth and invasion by upregulating PTEN. Mol Cell Biochem 2018;441(1–2):1–7 doi 10.1007/s11010-017-3170-2. [DOI] [PubMed] [Google Scholar]
- 27.Llorens MC, Lorenzatti G, Cavallo NL, Vaglienti MV, Perrone AP, Carenbauer AL, et al. Phosphorylation Regulates Functions of ZEB1 Transcription Factor. J Cell Physiol 2016;231(10):2205–17 doi 10.1002/jcp.25338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu J, Qin B, Moyer AM, Nowsheen S, Tu X, Dong H, et al. Regulation of sister chromatid cohesion by nuclear PD-L1. Cell Res 2020;30(7):590–601 doi 10.1038/s41422-020-0315-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun L, Patai AV, Hogenson TL, Fernandez-Zapico ME, Qin B, Sinicrope FA. Irreversible JNK blockade overcomes PD-L1-mediated resistance to chemotherapy in colorectal cancer. Oncogene 2021;40(32):5105–15 doi 10.1038/s41388-021-01910-6. [DOI] [PubMed] [Google Scholar]
- 30.Hu JL, Wang W, Lan XL, Zeng ZC, Liang YS, Yan YR, et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol Cancer 2019;18(1):91 doi 10.1186/s12943-019-1019-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rokavec M, Oner MG, Li H, Jackstadt R, Jiang L, Lodygin D, et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J Clin Invest 2014;124(4):1853–67 doi 10.1172/JCI73531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feng B, Dong TT, Wang LL, Zhou HM, Zhao HC, Dong F, et al. Colorectal cancer migration and invasion initiated by microRNA-106a. PLoS One 2012;7(8):e43452 doi 10.1371/journal.pone.0043452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roskoski R Jr. ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res 2012;66(2):105–43 doi 10.1016/j.phrs.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 34.Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nature Medicine 2015;21(11):1350–6 doi 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang G, Huang L, Jia H, Aikemu B, Zhang S, Shao Y, et al. NDRG1 enhances the sensitivity of cetuximab by modulating EGFR trafficking in colorectal cancer. Oncogene 2021;40(41):5993–6006 doi 10.1038/s41388-021-01962-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012;483(7387):100–3 doi 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 37.Drapela S, Bouchal J, Jolly MK, Culig Z, Soucek K. ZEB1: A Critical Regulator of Cell Plasticity, DNA Damage Response, and Therapy Resistance. Front Mol Biosci 2020;7:36 doi 10.3389/fmolb.2020.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perez-Oquendo M, Gibbons DL. Regulation of ZEB1 Function and Molecular Associations in Tumor Progression and Metastasis. Cancers (Basel) 2022;14(8) doi 10.3390/cancers14081864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou Z, Zhang P, Hu X, Kim J, Yao F, Xiao Z, et al. USP51 promotes deubiquitination and stabilization of ZEB1. Am J Cancer Res 2017;7(10):2020–31. [PMC free article] [PubMed] [Google Scholar]
- 40.Lin M, Zhao Z, Yang Z, Meng Q, Tan P, Xie W, et al. USP38 Inhibits Type I Interferon Signaling by Editing TBK1 Ubiquitination through NLRP4 Signalosome. Mol Cell 2016;64(2):267–81 doi 10.1016/j.molcel.2016.08.029. [DOI] [PubMed] [Google Scholar]
- 41.Pei G, Buijze H, Liu H, Moura-Alves P, Goosmann C, Brinkmann V, et al. The E3 ubiquitin ligase NEDD4 enhances killing of membrane-perturbing intracellular bacteria by promoting autophagy. Autophagy 2017;13(12):2041–55 doi 10.1080/15548627.2017.1376160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Palicharla VR, Maddika S. HACE1 mediated K27 ubiquitin linkage leads to YB-1 protein secretion. Cell Signal 2015;27(12):2355–62 doi 10.1016/j.cellsig.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 43.Zhang D, Bi J, Liang Q, Wang S, Zhang L, Han F, et al. VCAM1 Promotes Tumor Cell Invasion and Metastasis by Inducing EMT and Transendothelial Migration in Colorectal Cancer. Front Oncol 2020;10:1066 doi 10.3389/fonc.2020.01066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Drake JM, Strohbehn G, Bair TB, Moreland JG, Henry MD. ZEB1 enhances transendothelial migration and represses the epithelial phenotype of prostate cancer cells. Mol Biol Cell 2009;20(8):2207–17 doi 10.1091/mbc.E08-10-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tabernero J, Grothey A, Van Cutsem E, Yaeger R, Wasan H, Yoshino T, et al. Encorafenib Plus Cetuximab as a New Standard of Care for Previously Treated BRAF V600E-Mutant Metastatic Colorectal Cancer: Updated Survival Results and Subgroup Analyses from the BEACON Study. J Clin Oncol 2021;39(4):273–84 doi 10.1200/JCO.20.02088. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are available within the article or available from the authors upon request.