

Abstract
Estrogen receptor alpha (ER/ESR1) mutations occur in 30–40% of endocrine resistant ER-positive (ER+) breast cancer. Forkhead Box A1 (FOXA1) is a key pioneer factor mediating ER-chromatin interactions and endocrine response in ER+ breast cancer, but its role in ESR1 mutant breast cancer remains unclear. Our previous FOXA1 ChIP-seq identified a large portion of redistributed binding sites in T47D genome-edited Y537S and D538G ESR1 mutant cells. Here, we further integrated FOXA1 genomic binding profile with the isogenic ER cistrome, accessible genome and transcriptome data of T47D cell model. FOXA1 redistribution was significantly associated with transcriptomic alterations caused by ESR1 mutations. Furthermore, in ESR1 mutant cells, FOXA1 binding sites less frequently overlapped with ER, and differential gene expression was less associated with the canonical FOXA1-ER axis. Motif analysis revealed a unique enrichment of retinoid X receptor (RXR) motifs in FOXA1 binding sites of ESR1 mutant cells. Consistently, ESR1 mutant cells were more sensitive to growth stimulation with the RXR agonist LG268. The mutant-specific response was dependent on two RXR isoforms, RXR-α and RXR-β, with a stronger dependency on the latter. In addition, T3, the agonist of thyroid receptor also showed a similar growth-promoting effect in ESR1 mutant cells. Importantly, RXR antagonist HX531 blocked growth of ESR1 mutant cells and a patient derived xenograft (PDX)-derived organoid with an ESR1 D538G mutation. Collectively, our data support evidence for a stronger RXR response associated with FOXA1 reprogramming in ESR1 mutant cells, suggesting development of therapeutic strategies targeting RXR pathways in breast tumors with ESR1 mutation.
Introduction
Estrogen receptor-α (ER/ESR1) is expressed in more than 70% of breast cancers and has an essential role in breast cancer tumorigenesis and progression (1–3). Endocrine therapy is the mainstay therapy to treat patients with ER+ breast cancer, including depletion of estradiol (E2) by aromatase inhibitors (AIs) or antagonizing ER activity by Selective Estrogen Receptor Modulators/Degraders (SERMs/SERDs) (4–6). The emergence of resistance to these therapies, however, is inevitable over time in a subset of patients, which remains a clinical and social challenge (7,8).
Previous studies have uncovered various pivotal mechanisms driving endocrine resistance, among which are hotspot ESR1 missense mutations that occur in approximately 30–40% of advanced breast cancers (9–12). ESR1 mutations cluster in the ligand binding domain, with Y537S and D538G being the most frequent variants, leading to a stabilized agonist state in the absence of E2 (13,14). Multiple studies have revealed a role for ESR1 mutations in causing constitutive ER activity and diminished sensitivity towards ER antagonists both in vitro and in vivo (15–20), as well as association with poor clinical outcomes in patients with advanced disease (21–23). Therefore, there is an urgent need to develop novel therapeutic strategies targeting tumors with ESR1 mutations. Recent studies have reported that the cyclin-dependent kinase 7 (cdk7) inhibitor THZ1 can block growth of ESR1 mutant cells (17), and cdk7 as a target was subsequently validated in a separate study using in vitro and in vivo approaches (24). Mao et al. described that the ER modulator BHP1 can hyperactivate the unfolded protein response and inhibit growth in ESR1 mutant cells (25). Gelsomino et al. uncovered enhanced IGF1 pathway activity in ESR1 mutant cells, which our group confirmed, suggesting co-targeting ER and IGF1R as a potential strategy (26,27). Yu et al. showed a key role of Myc pathway hyperactivation in the proliferation of ESR1 mutant cells (28). In addition, recent results by Dustin et al. and Gu et al. suggested the potential of a RON inhibitor (29) and androgen receptor (AR) agonist (30), respectively, to inhibit metastasis of mutant ESR1 PDX. Despite these exciting pre-clinical findings, no effective therapy targeting ESR1 mutations is approved for clinical application, and it remains imperative to identify novel therapeutic targets with clinical actionability.
Forkhead protein FOXA1 is a key pioneer factor mediating ER-chromatin interactions, and its impact on ER global binding, endocrine response and resistance has been broadly reported in ER+ breast cancer (31–33), but limited studies have examined a role in ESR1 mutant disease. Jeselsohn et al. revealed weaker FOXA1 binding in the Y537S mutant ER cistrome (24), while Arnesen et al. found FOXA1 bound to a significant fraction of accessible chromatin regions in both Y537S and D538G mutant cells (15). Recently our group has shown enhanced migration in T47D D538G cells dependent upon FOXA1-mediated Wnt pathway activation, and FOXA1 ChIP-seq indicated a distinct FOXA1 cistrome in T47D ESR1 mutant cells compared to ESR1 WT cells (34). Other than these limited data, the role of FOXA1 in ER mutant breast cancer has not been well characterized.
We set out to understand the impact of FOXA1 redistribution in ESR1 mutant cells. Integrative analysis with multi-omic datasets suggests that FOXA1 impacts transcriptional reprogramming, especially the expression of novel target genes in the context of an ESR1-mutant background. We show that FOXA1 interacts less with ER but confers potential RXR binding sites in mutant cells. Our in vitro experiments demonstrate that RXR agonist LG268 can promote growth and survival of ESR1 mutant cells, which can be inhibited with the RXR antagonist HX531. Our study provides novel mechanistic and therapeutic insights including blocking RXR activation in ESR1 mutant breast cancer.
Materials and Methods
Cell culture
Two genome-edited T47D ESR1 mutant cell models (Oesterreich and Gertz models) were established as previously described (15,18), and the parental T47D cells (RRID: CVCL_0553) were obtained from the American Type Culture Collection (ATCC). Cells were maintained in RPMI, supplemented with 10% FBS, 100μg/mL penicillin and 100mg/mL streptomycin, at 37°C in a humidified incubator with 5% CO2. Hormone deprivation was performed for all experiments, unless otherwise stated, by washing and maintaining cells in phenol-red-free IMEM (Gibco, A10488) with 10% charcoal-stripped serum (CSS, Gemini, #100-119), twice a day for three days. Clones with the same genotypes were equally pooled for subsequent experiments. All data were generated with Oesterreich T47D cells unless otherwise stated.
Organoid culture
All the organoid culture procedures and medium recipe followed the previous published protocol with the addition of E2 (35). Briefly, PDX tumor was digested in the dissociation medium, incubated on the rotator, sheared and filtered to collect organoids. For regular culture, organoids were embedded in Cultrex RGF Basement Membrane Extract (R&D Systems™ 353301002) in 24-well suspension culture plates (Greiner, M9312), passaged every 2 weeks.
Reagents
17β-estradiol (E2, #E8875), LG100268 (#SML0279), HX531 (#SML2170), SR12813 (#S4194), 1α,25-Dihydroxyvitamin D3 (Vitamin D3, #D1530), Citco (#C6240), 3,3’,5-Triiodo-L-thyronine sodium salt (T3, #T6397), GW4064 (#G5172), GW3965 hydrochloride (#G6295), retinoic acid (#R2625), WY14643 (#C7081) were obtained from Sigma-Aldrich, and ICI 182,780 (Fulvestrant, #1047), GW0742 (#2229) was purchased from Tocris. For knockdown experiments, SMARTpool siRNAs against FOXA1 (#M-010319), RXRA (#L-003443) and RXRB (#L-003444) were obtained from Horizon Discovery. All siRNA sequences are indicated below:
FOXA1 siRNAs 1#: CCUCGGAGCAGCAGCAUAA; 2#: GCGCUGAGCCCGAGCGGCA; 3#: CGGGAAGACCGGCCAGCUA; 4#: GUGUAGACAUCCUCCGUAU.
RXRA siRNAs 1#: GCGCCAUCGUCCUCUUUAA; 2#: GCAAGGACCGGAACGAGAA; 3#: AGACCUACGUGGAGGCAAA; 4#: UCAAAUGCCUGGAACAUCU.
RXRB siRNAs 1#: GGGCAAUCAUUCUGUUUAA; 2#: GCAAACGGCUAUGUGCAAU; 3#: CGAAGAGGAUCCCACACUU; 4#: CCGCAAAGACCUUACAUAC.
Growth assay
4,000 T47D cells or 3,000 organoid cells were seeded into either regular 96-well plates (for 2D growth) (Corning, #353072), or round bottom 96-well ultra-low attachment plates (for 3D growth) (Corning, #353227). Cell numbers were quantified after the desired growth time course with either FluoReporter Blue Fluorometric dsDNA quantification kit (for 2D growth) (Invitrogen, F2962) or the Celltiter Glo luminescent cell viability kit (for 3D growth) (Promega, G7573). 100nM RXR agonist LG268 and 2μM RXR antagonist HX531 were used in the experiment. In the nuclear receptor (NR) screen part, the tested RXR partners and their agonist doses are indicated below: Retinoid Acid Receptor (RAR), 100nM retinoic acid; Thyroid Receptor (TR), 20nM T3; Vitamin D Receptor (VDR),10nM Vitamin D3; Liver X Receptor (LXR), 100nM GW3965; Peroxisome Proliferator-Activated Receptor Alpha (PPAR-α), 100nM WY14643; Peroxisome Proliferator-Activated Receptor Delta (PPAR-δ), 100nM GW0742; Farnesoid X receptor (FXR), 100nM GW4064; Constitutive Androstane Receptor (CAR), 100nM CITCO; Pregnane X Receptor (PXR), 100nM SR12813.
Clonogenic survival assay
5,000 T47D cells were seeded into 12-well plates, and formed colonies in 14 days under different treatment conditions. After that, cells were stained with 0.05 % crystal violet for 20min and scanned to get raw images. Then the crystal violet was dissolved in 10% acetic acid for 20min, absorbance was acquired using GloMax® Microplate Reader (Promega) at OD560. The raw absorbance OD560 was normalized to Veh group and presented as fold change.
qRT-PCR
T47D cells were seeded in triplicates into 6-well plates with 100,000 cells per well respectively. RNA was extracted after desired treatments, and then synthesized to cDNA using PrimeScript RT Master Mix kit (#RR036, Takara Bio). qRT-PCR reactions were performed with SybrGreen Supermix (#1726275, BioRad), and the ΔΔCt method was used to analyze relative mRNA fold changes with RPLP0 as the internal control. All primer sequences are indicated below:
RPLP0 (Forward): 5’-TAAACCCTGCGTGGCAATC-3’;
RPLP0 (Reverse): 5’-TTGTCTGCTCCCACAATGAAA-3’;
SERPINA1 (Forward): 5’- ACCTGGAAAATGAACTCACCC-3’;
SERPINA1 (Reverse): 5’- ACCTTAGTGATGCCCAGTTG-3’
RXRA (Forward): 5’- AGGACTGCCTGATTGACAAG-3’;
RXRA (Reverse): 5’- GACTCCACCTCATTCTCGTTC-3’
RXRB (Forward): 5’- GACCTTACATACTCTTGCCGG-3’;
RXRB (Reverse): 5’- CTTGTCCTTTCCCCGCTG-3’
RXRG (Forward): 5’- CAGATGGACAAGTCGGAACTG-3’;
RXRG (Reverse): 5’- GGATACTTCTGCTTGGTGTAGG-3’
GREB1 (Forward): 5’- GGTTCTTGCCAGATGACAATGG-3’;
GREB1 (Reverse): 5’- CTTGGGTTGAGTGGTCAGTTTC-3’
TFF1 (Forward): 5’- AGAACAAGGTGATCTGCGC-3’;
TFF1 (Reverse): 5’- TTATTTGCACACTGGGAGGG-3’
Chromatin Immunoprecipitation-sequencing (ChIP-seq) analysis
ChIP experimentation, peak calling and differentially binding analysis were performed as previously described (34). Additionally, Principal Component Analysis (PCA) plot, heatmap and genomic distribution were obtained using DiffBind (36) and ChIPseeker (37) package. Motif analysis including enrichment and scanning was performed on MEME suite (38). Specifically, Analysis for Motif Enrichment (AME) (39) used JASPAR CORE and UniPROBE Mouse database (2018) and identified significantly enriched motifs with E-value < 0.05, and Find Individual Motif Occurrences (FIMO) analysis (40) scanned the FOXA1 motif and output the significant matched ones with p-value < 0.0001. ERE motif numbers were quantified using EREFinder algorithm (41) with default parameters. Briefly, fasta files were converted from Bed format of each ChIP-seq profile using bedtools (RRID: SCR006646) (42). Fasta sequences were then used as input sources for EREFinder which scans ERE motifs under 10,000 bp of each slides window. ERE were defined as identified ERE motifs with 1/Kd>0.04. FOXA1 gained peaks annotated genes of mutant cells were input for Ingenuity Pathway Analysis (RRID: SCR008653) (43), and significantly enriched pathways (p-value<0.05) were obtained from Tox list.
Rapid immunoprecipitation mass spectrometry of endogenous proteins (RIME) analysis
FOXA1 RIME experiment was performed in the previous study (44). Briefly, proteins from MCF7 cells were immunoprecipitated, digested and measured by the mass spectrometer. Maximum allowed missed cleavage was 2, the peptide threshold was 95% and the protein false discovery rate (FDR) was set to 0.5%. Proteins with more than 2 unique peptides in the FOXA1 antibody group but no unique peptide in IgG group were classified as FOXA1-interacting proteins.
Binding and Expression Target Analysis (BETA)
BETA was performed as previously described (45). Briefly, differentially expressed (DE) genes were first computed using DESeq2 (RRID: 015687) and used as one of the inputs. DE genes between WT+E2 cells and WT cells were defined as FC>1.5, padj<0.01, and DE genes between mutant and WT cells were defined as FC>1.5, padj<0.005. In mutant cells, they were further divided into two groups: ligand-independent and novel target genes, the former were derived from intersection with E2-regulated genes in WT cells, while the latter not. In addition, gained or lost FOXA1 peaks were derived from DiffBind analysis with |FC|>2. BETA basic modules were used to compute the statistical associations between DE genes and DB peaks using 100kb as the ranges to link gene TSS to each peak. P values were derived using one-tailed Kolmogorov-Smirnov test for up-regulated and down-regulated genes respectively.
Statistical analysis
All statistical analyses were performed by GraphPad Prism software version 7 (RRID: SCR002798) and R version 3.6.1. All experimental results included biological replicates and were indicated as mean ± standard deviation, unless otherwise stated. Specific statistical tests were highlighted in corresponding figure legends.
Data availability
The ER and FOXA1 ChIP-seq data has been deposited onto the Gene Expression Omnibus database (RRID: SCR005012) (GSE125117 and GSE165280). Other data resources used in this study are summarized in Supplementary Table S1. All raw data and scripts are available upon request from the corresponding author.
Results
Redistribution of FOXA1 in ESR1 mutant cells is associated with altered ER-FOXA1 interaction.
Given that FOXA1 is an important epigenetic regulator which determines ER-chromatin interactions (31–33), we sought to compare the global FOXA1-ER genomic occupancy in ESR1 WT and mutant cells. We previously performed FOXA1 ChIP-seq (34) and ER ChIP-seq analysis (46) in T47D Y537S and D538G genome-edited cells (Supplementary Table. S2). Intersecting ER and FOXA1 binding sites, we found that 1,628 ER binding sites (56%) overlapped with FOXA1 binding sites in WT cells after E2 treatment, consistent with previous studies (32,47). However, the ER/FOXA1 binding overlap was decreased to 403 (39%) and 325 (25%) in Y537S and D538G cells respectively despite a larger number of total FOXA1 binding sites (Fig. 1A). To build on this finding, we further performed FIMO (Find Individual Motif Occurrences) analysis (40) to quantify FOXA1 motifs enriched in ER binding sites and found that FOXA1 motifs were less frequently detected in mutant cells (Fig. 1B). We observed a similar pattern using FOXA1 and ER ChIP-seq data from other ESR1 mutant cell models (24,46) (Supplementary Fig. S1A–B). To further delineate the differences between FOXA1-dependent and independent ER action, we divided all ER binding sites into two groups based on intersection with FOXA1 binding sites. Unsupervised clustering revealed that ER binding sites overlapping with FOXA1 binding sites clustered together irrespective of ESR1 mutant status, but ER binding sites not overlapping with FOXA1 binding sites clearly segregated among WT and the two mutants (Fig. 1C and Supplementary Fig. S1C). Analysis of genomic feature distribution showed that ER binding sites that overlapped with FOXA1 binding sites were more enriched in promoter regions in both Y537S and D538G cells compared to WT cells, whereas ER binding sites that were distinct from FOXA1 sites were enriched in promoter regions in both WT and Y537S cells (Supplementary Fig. S1D–E). In ESR1 D538G cells, FOXA1 Non-overlapping ER binding sites were largely distinct with an enrichment in distal intergenic regions especially. There were fewer Estrogen Response Element (ERE) motifs in ER binding sites in the ER mutant compared to WT cells, independent of overlap with FOXA1 binding sites (Fig. 1D).
Figure 1. Redistribution of FOXA1 in ESR1 mutant cells is associated with altered ER-FOXA1 interaction.
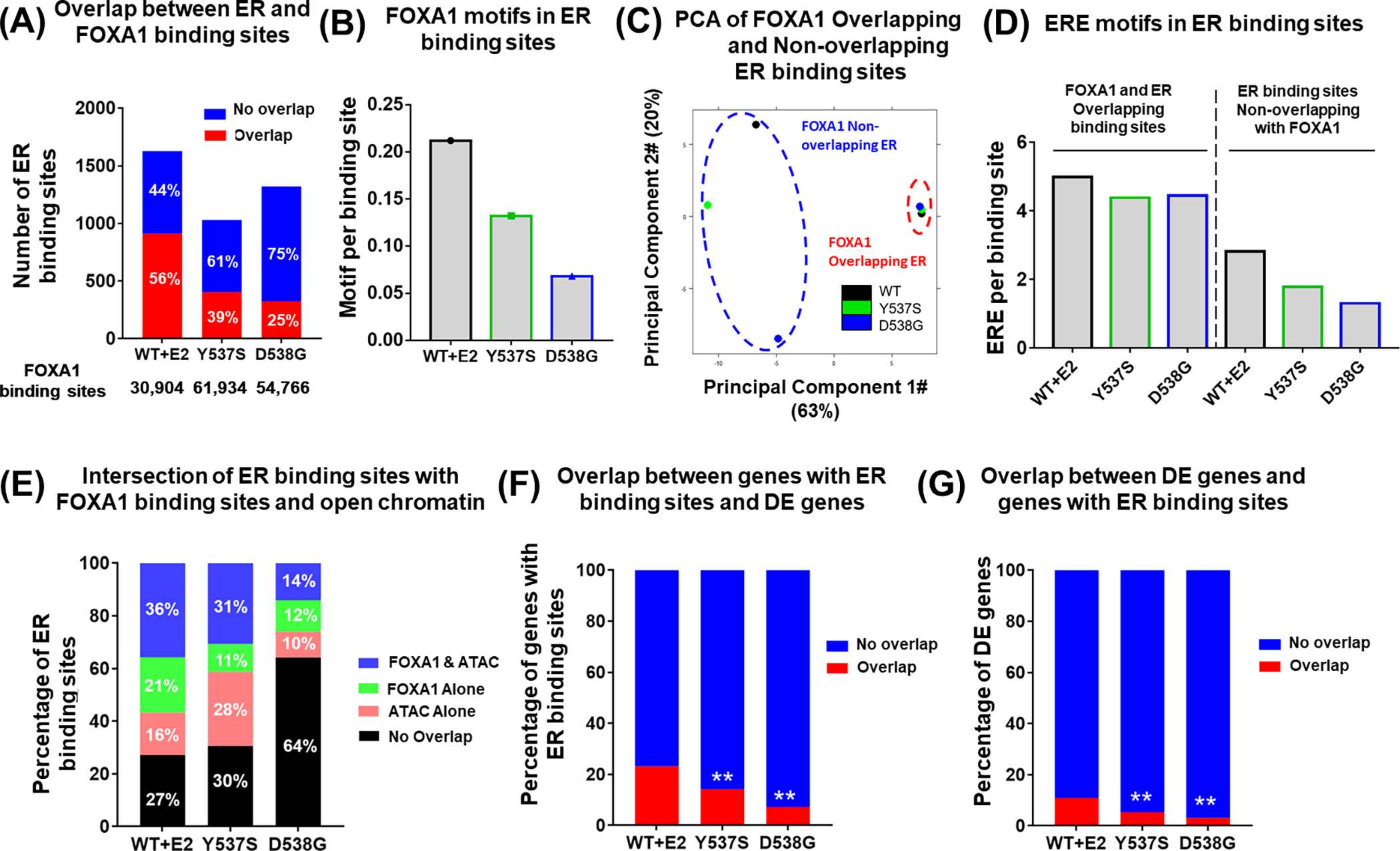
(A) Stacked bar plot showing the intersection of ER and FOXA1 binding sites in Oesterreich T47D cells. Percentages of each group of ER binding sites were indicated on each bar. Number of ER binding sites was shown on Y-axis, and number of total FOXA1 binding sites was shown below X-axis. Fisher’s exact test was performed to compare the overlapped part between WT and mutant cells.
(B) Bar plot representing the percentage of FOXA1 motifs present in the ER binding sites in Oesterreich T47D cells.
(C) PCA plot showing the similarity among FOXA1 Overlappping ER and FOXA1 Non- Overlapping ER binding sites in T47D ESR1 WT and mutant models.
(D) Bar plot comparing ERE motif occurrence per binding site among FOXA1 Overlappping ER and FOXA1 Non-overlapping ER binding sites in T47D ESR1 WT and mutant models.
(E) Stacked bar plot showing the intersection of ER peaks with FOXA1 and ATAC peaks, Percentages of each group of ER peaks are indicated on each bar.
(F, G) Stacked bar plot representing the intersection between ER binding sites annotated genes and differentially expressed genes. F) shows the percentage of ER binding sites annotated genes which have overlap with differentially expressed genes, G) shows the percentage of differentially expressed genes which have overlap with ER binding sites annotated genes. Fisher’s exact test was performed to compare the overlapped part between WT and mutant cells. (** p<0.01)
Since the regulation of ER-FOXA1 interaction is mainly reflected through the nexus of ER, FOXA1, and chromatin accessibility, in which FOXA1 increases accessibility of certain chromatin regions and then ER binds to the open chromatin (31–33), we next intersected ER and FOXA1 binding sites with accessible chromatin regions previously identified through ATAC-seq analysis in ESR1 WT and mutant T47D cells (34). We observed distinct binding patterns comparing WT and each mutant ER: ER bound to FOXA1-associated open chromatin at a rate of ~30% in WT and Y537S cells, but this rate was decreased to only 14% in D538G cells (Fig. 1E, Supplementary Fig. S1F). Notably, Y537S cells showed 28% of ER binding localized to open chromatin without FOXA1 binding sites suggesting that this part of ER-chromatin interactions is independent of FOXA1 and may rely upon other factors. D538G cells had 64% of ER binding sites mapped to closed chromatin without FOXA1 binding, suggesting that a large portion of mutant ER binds to chromatin with low accessibility. Given the more prevalent noncanonical ER binding pattern (i.e. exclusive from FOXA1 and open chromatin) observed in ER mutant cells, we next questioned whether the transcriptomic alterations are also less associated with mutant ER binding. We defined differentially expressed (DE) genes between WT+E2 cells and WT cells as |FC|>1.5, padj<0.01, and DE genes between mutant and WT cells as |FC|>1.5, padj<0.005 from previous T47D cell RNA-seq data (Supplementary Table. S3) (18), intersected the genes associated with ER binding sites with those DE genes and checked the proportion of the overlapped part in either of them respectively. Both analyses showed that mutant ER binding sites were less associated with DE genes (Fig. 1F–G), suggesting mutant ER likely hijacks other factors to reshape transcriptomes in additional to the known ligand-independent binding. Collectively, our findings suggest that in the context of ESR1 mutation, FOXA1 plays a less dominant role in facilitating the binding of ER on accessible chromatin and further regulating canonical downstream genes.
ESR1 mutant-specific de novo transcriptomic alterations are associated with FOXA1-RXR nexus.
Since the actual number of FOXA1 binding sites was significantly higher in ESR1 mutant compared to WT cells, these data suggested that FOXA1 may have gained novel functions in the mutant cells which are independent of ER-FOXA1 interaction on chromatin. To test this hypothesis, we first directly examined the role of FOXA1 in regulation of gene expression. Our recent FOXA1 binding analysis identified 2,447, 15,870, and 10,451 binding sites with increased intensity (FC>2) and 3,732, 4,064, and 2,687 binding sites with decreased intensity (FC<−2) in WT+E2, Y537S and D538G cells, respectively, compared to WT+veh (34). To test the association between the differential FOXA1 binding sites and DE genes in mutant cells, we applied the Binding and Expression Target Analysis (BETA) algorithm (45). In this analysis, DE genes in mutant cells were further divided into two groups: ligand-independent and novel target genes, the former were derived from intersection with E2-regulated genes in WT cells, while the latter are genes not regulated by E2 in WT cells. First in WT+E2, Y537S, and D538G cells, gained FOXA1 binding sites (FC>2 versus WT+Veh cells) were preferentially associated with upregulated genes and lost FOXA1 binding sites (FC<−2 versus WT+Veh cells) were associated with downregulated genes, confirming that FOXA1 binding allows transcriptional activators to induce gene expression (Fig. 2A, and Supplementary Fig. S2A). Notably, lost FOXA1 binding sites were associated with upregulated genes in WT+E2 cells but not mutant cells, indicating ESR1 mutation might change the impact of FOXA1 binding on transcription regulatory directionality. In addition, gained FOXA1 binding sites were significantly associated with both ligand-independent and novel target genes, in Y537S but not D538G mutant cells, suggesting that FOXA1 might play a more pivotal role in Y537S mutant specific transcriptomic reprogramming.
Figure 2. ESR1 mutant-specific de novo transcriptomic alterations are associated with FOXA1-RXR nexus.
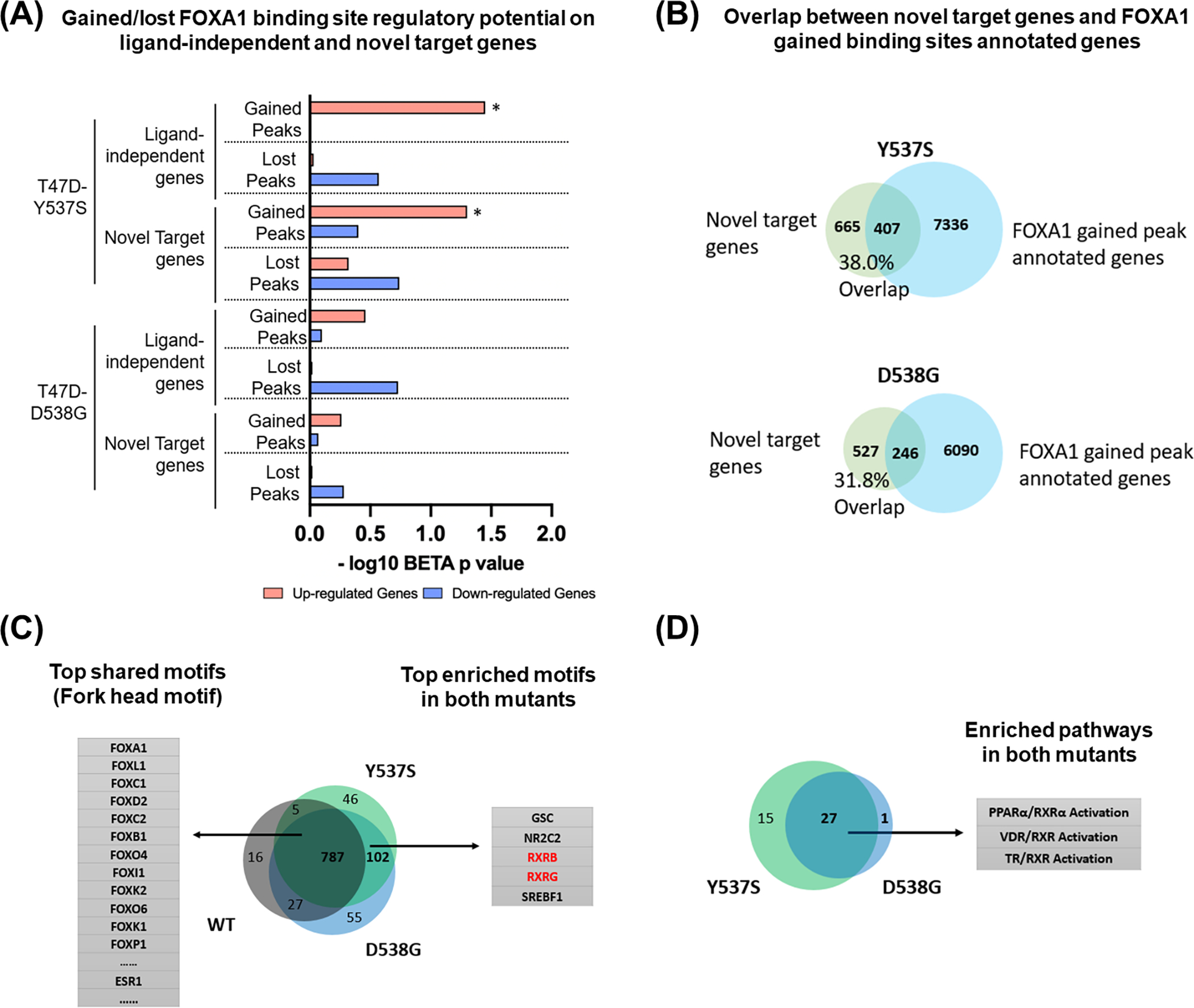
(A) Bar plot showing the regulatory potential of gained/lost FOXA1 peaks on ligand independent and novel target genes in T47D ESR1 Y537S and D538G cells. p values were obtained by one-tailed Kolmogorov-Smirnov test from BETA. (* p<0.05)
(B) Venn diagram showing the overlap between novel target genes and FOXA1 gained binding sites annotated genes in Oesterreich T47D ESR1 Y537S can D538G cells.
(C) Motif enrichment analysis of FOXA1 binding sites in Oesterreich T47D ESR1 WT and mutant cells. Venn diagram shows the overlap of significantly enriched motifs (E-value<0.05, obtained by Fisher’s Exact Test from MEME-Suite) in WT, Y537S and D538G cells. The top shared motifs and mutant-enriched motifs were shown on each side, motifs of RXR subtypes were highlighted.
(D) Venn diagram showing the overlap of significantly enriched pathways (p-value<0.05, obtained by Fisher’s Exact Test from IPA) in genes with gained FOXA1 binding sites between T47D ESR1 Y537S and D538G cells. Among the 27 overlapped pathways, 3 RXR-related pathways were highlighted on the right side.
Given the reduced functional interaction between FOXA1 and ER in ESR1 mutant cells, we next focused on novel target genes with gained FOXA1 binding sites. We found >30% of novel target genes are associated with gained FOXA1 binding sites in ESR1 mutant cells, suggesting a de novo FOXA1-driven transcriptional program (Fig. 2B). For example, we identified a Y537S-unqiue novel target gene SERPINA1 with increased FOXA1 but not ER binding intensity at SERPINA1 proximity (30 kb from TSS) in Y537S cells (Supplementary Fig. S2B). Consistent with this, SERPINA1 was upregulated in Y537S cells, but showed weak, if any, response to E2 stimulation in WT cells from two independent ESR1 mutant cell models (Supplementary Fig. S2C). In addition, depletion of FOXA1 but not ER significantly dampened SERPINA1 expression in Y537S cells (Supplementary Fig. S2D–E), suggesting that the expression is regulated by a FOXA1-dependent but ER-independent mechanism. Importantly, SERPINA1 was also consistently upregulated in ESR1 mutant metastatic tumors in four publicly available cohorts (24,48–52) (Supplementary Fig. S2F). Taken together, FOXA1 redistribution in ESR1 mutant cells causes alterations in the transcriptome, especially effecting expression of novel target genes (i.e. genes that are not regulated by E2 in WT cells but show differential expression in ESR1 mutant cells).
To further delineate alternative FOXA1 downstream pathways uniquely involved in ESR1 mutant tumors, we performed motif enrichment analysis in FOXA1 binding sites of WT and mutant cells (Fig. 2C and Supplementary Table. S4). The intersection of the significantly enriched motifs (E<0.05) confirmed Fork Head motifs as the top shared targets among all groups. In addition, there were 102 motifs uniquely enriched in both mutants, and among those, motifs corresponding to retinoid X receptors (RXR) were among the top five. In addition, RXR motifs were also identified as being enriched in ESR1 mutant T47D cells when compared to WT+E2 (Supplementary Fig. S3A and Supplementary Table. S4), although this was not seen analyzing data from FOXA1 binding sites in an independent MCF7 Y537S cell model, possibly due to model differences as previously described (24). Analysis of previous FOXA1 rapid immunoprecipitation mass spectrometry of endogenous proteins (RIME) in MCF7 cells to screen for FOXA1-interacting proteins confirmed FOXA1-RXR interaction (44). The results showed that both RXR-α and RXR-β had 6 unique peptides in the FOXA1 antibody group which was similar to the ER-α-FOXA1 interaction (Supplementary Fig. S3B), confirming the specific FOXA1-RXR-α/β interactions in ER+ breast cancer cell line.
RNA expression analysis showed that RXRA (RXR-α) and RXRB (RXR-β) but not RXRG (RXR-γ) were robustly expressed in ESR1 WT and mutant cell models and tumor specimens (Supplementary Fig. S3C–D). In support of the results from the motif enrichment analysis, Ingenuity Pathway Analysis (IPA) identified three RXR-related pathways, PPARα/RXRα activation, VDR/RXR activation and TR/RXR activation, which are significantly associated with genes linked to gained FOXA1 binding sites in both ESR1 mutant cells (Fig. 2D and Supplementary Table. S5). Furthermore, a gene set enrichment screen of all 187 KEGG pathways between ESR1 WT and mutant metastatic tumors from four clinical cohorts (24,48–52) revealed RETINOL_METABOLISM pathway as the 2nd consistently increased pathway in ESR1 mutant tumors (Supplementary Fig. S3E), suggesting the hyperactivated metabolism of multiple precursors of RXR ligands. Collectively, these findings encouraged us to further study RXR related phenotypes in the setting of ESR1 mutations.
ESR1 mutant cells show stronger response to RXR agonist LG268-mediated survival and 2D growth.
Because RXR is a subclass of ligand-dependent nuclear receptors which regulate a variety of cellular processes including proliferation and apoptosis (53,54), we tested whether ESR1 WT and mutant cells show different response in survival and growth under RXR agonist stimulation. After treatment with pan-RXR agonist LG268 at 100nM in hormone deprived condition for 14 days, both T47D ESR1 WT and mutant cells showed significantly stronger clonogenic survival compared to Vehicle (Veh) treatment, and this effect was significantly more pronounced in Y537S cells (Fig. 3A–B and Supplementary Fig. S4A). Consistent with this, dose response studies showed that Y537S cells exhibited greater sensitivity and maximal response towards LG268 (Supplementary Fig. S4B–C). In addition, we compared the clonogenic stimulation effect between LG268 and E2, and we observed both of them could promote the clonogenic survival in WT and mutant cells (Supplementary Fig. S4D–E). Interestingly, the response towards E2 and LG286 was negatively correlated across the three groups, and Y537S cells showed the strongest effect of LG268 normalized to E2 (Supplementary Fig. S4F). To test the reproducibility of the phenotype in an independent model, we performed the same experiments using Gertz T47D cell model (15), and the results showed that ESR1 mutant cells uniquely developed more colonies after LG268 treatment (Fig. 3C–D). 2D growth of both mutant cells in hormone deprived condition was promoted by LG268 with a more pronounced enhancement in Y537S cells, while it had no effect on WT cells (Fig. 3E and Supplementary Fig. S4G), and again the same phenotype was reproduced in an independent T47D model (Fig. 3F). Furthermore, dose response assays showed that both Oesterreich and Gertz T47D ESR1 mutant cells were more sensitive towards LG268 stimulated growth (Fig. 3G–H). We also performed the same assays in Park and Gertz MCF7 cell models, but only observed the similar phenotype in Gertz MCF7 D538G cells (Supplementary Fig. S5A–F). Together, these results suggest that T47D ESR1 mutant cells have stronger response to RXR agonist-mediated survival and 2D growth.
Figure 3. ESR1 mutant cells show stronger response to RXR agonist LG268 promoted survival.
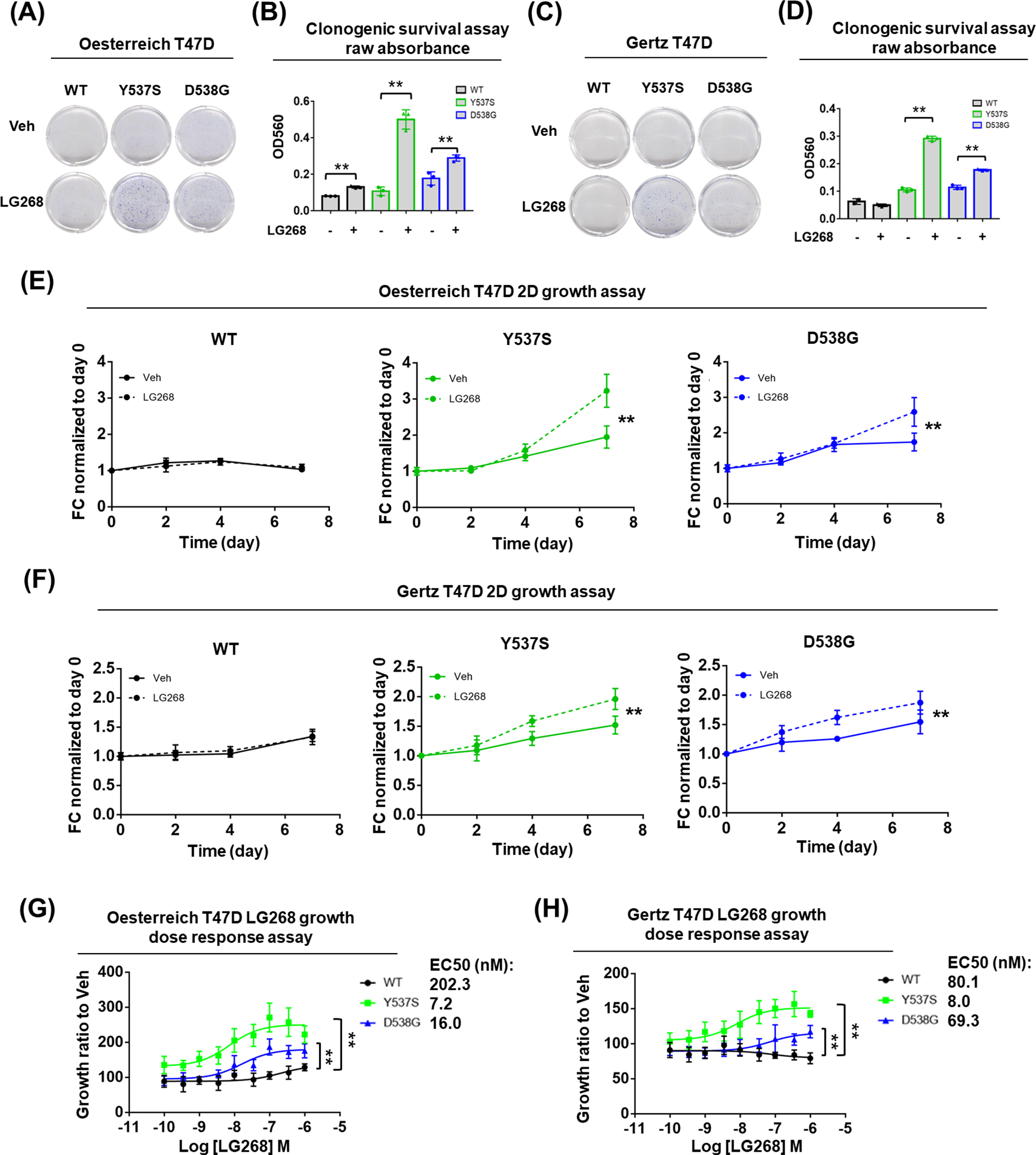
(A, C) Representative images of colonies formed after Veh or 100nM LG268 treatment in hormone deprived condition with Oesterreich (A) and Gertz (C) models for 14 days.
(B, D) Bar plot showing the quantification of A and C with raw absorbance OD560 of each group after crystal violet staining. Each bar represents mean ± SD with three biological replicates. Student’s t test was used to examine the effects of treatment between each group’s OD560. (** p<0.01)
(E, F) Growth curve of Oesterreich (E) and Gertz (F) T47D cells under Veh or 100nM LG268 treatment in hormone deprived condition. Cell amount quantified by FluoReporter kit at day 7 was normalized to day 0 and presented as fold change (FC). Each bar represents mean ± SD with five biological replicates. Two-way ANOVA was performed to compare the 100nM LG268 group and Veh group. (** p<0.01)
(G, H) LG268 dose response curve of Oesterreich (G) and Gertz (H) T47D cells. Cell amount quantified by FluoReporter kit at day 7 of all dose groups was normalized to Veh and presented as fold change (FC). Each bar represents mean ± SD with five biological replicates. EC50s were shown on the right side. Two-way ANOVA test was performed to compare the dose response between mutant and WT cells. (** p<0.01)
The response to RXR agonist in ESR1 mutant cells depends on RXR-α and RXR-β, RXR partner TR can also promote ESR1 mutant cell growth.
To examine which receptor(s) was causing the observed RXR agonist response, we knocked down RXRA, RXRB, or the combination using siRNA. RXR subtype-specific knockdown was validated using qRT-PCR (Fig. 4A). Knockdown of RXRA alone slightly but significantly reduced clonogenic survival induced by LG268, whereas it was completed abolished by knockdown of RXRB alone or both RXRA and RXRB (Fig. 4B–C). Comparison of fold change further validated the significant reduction of cell colonization in all RXR siRNA knockdown groups (Fig. 4C). Similar results were obtained with a growth assay in hormone deprived condition (Fig. 4D). Taken together, these data demonstrate that the responses to RXR agonist LG268 are dependent upon both RXR-α and RXR-β, with a relatively stronger dependency on RXR-β.
Figure 4. The response to RXR agonist in ESR1 mutant cells depends on RXR-α and RXR-β, RXR partner TR can also promote ESR1 mutant cell growth.
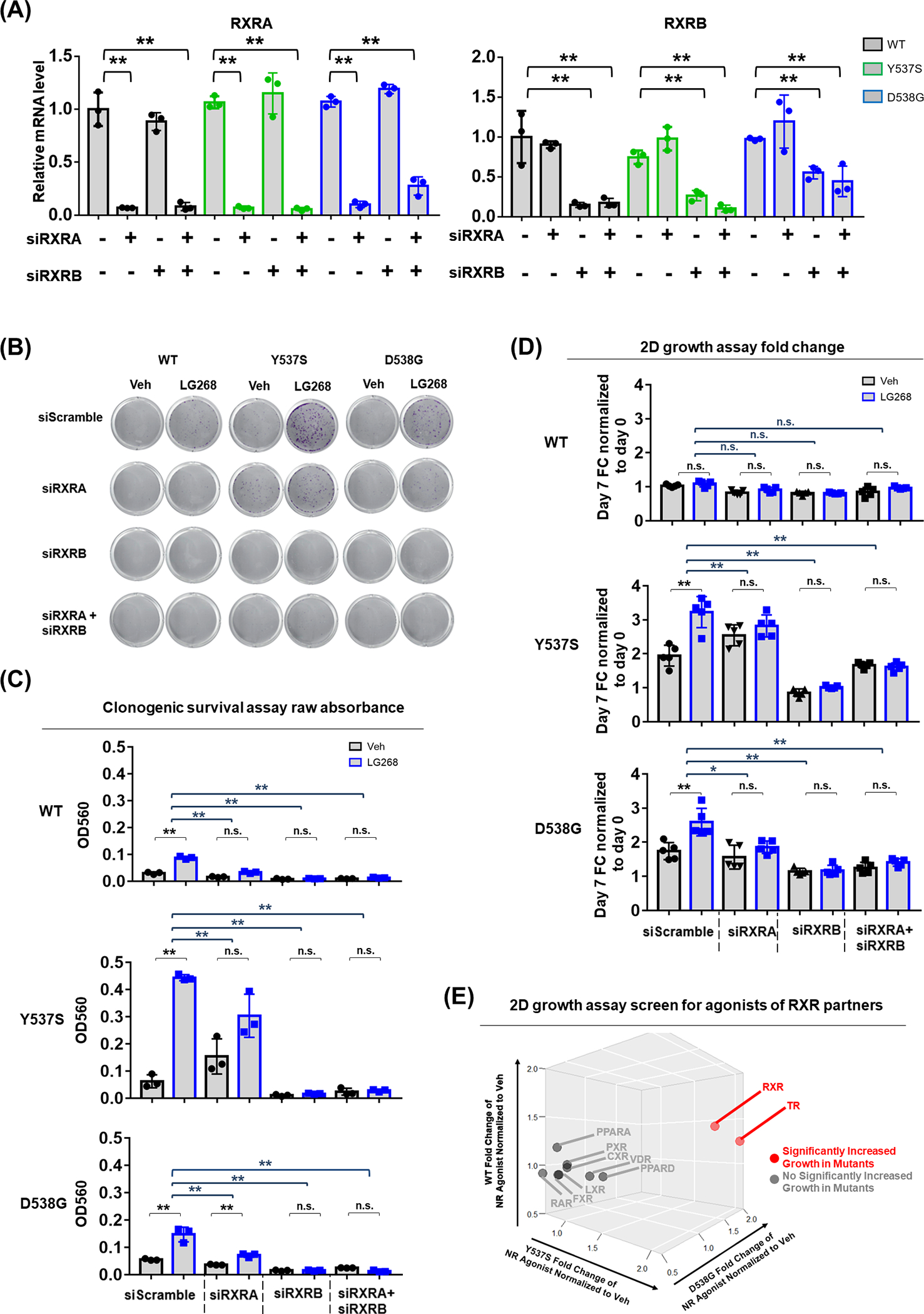
(A) qRT-PCR validation of RXRA and RXRB mRNA levels in T47D WT and mutant cells with RXRA and RXRB siRNA knockdown. mRNA fold changes were normalized to RPLP0 and further normalized to WT scramble group. Each bar represents mean ± SD with three biological replicates. Dunnett’s test was used to compare the gene expression between scramble and knockdown groups. (** p<0.01)
(B - C) Colony formation assay result after RXRA and RXRB siRNA knockdown in hormone deprived condition. (B) Representative images of colonies formed after Veh or 100nM LG268 treatment; (C) Bar plot showing the raw absorbance OD560 of each group after crystal violet staining and acetic acid dissolving. Each bar represents mean ± SD with three biological replicates. Student’s t test was used to examine the effects of treatment between LG268 group and Veh group. Dunnett’s test was used to compare the fold change between scramble and knockdown groups. (* p<0.05, ** p<0.01)
(D) Growth assay result after RXRA and RXRB siRNA knockdown in hormone deprived condition. Cell amount quantified by FluoReporter kit at day 7 was normalized to day 0 and presented as fold change (FC). Bar plot showing the fold change of each group. Each bar represents mean ± SD with five biological replicates. Student’s t test was used to examine the effects of treatment between LG268 group and Veh group. Dunnett’s test was used to compare the fold change between scramble and knockdown groups. (* p<0.05, ** p<0.01);
(E) Growth assay screen for agonists of RXR partners in hormone deprived condition. Cell amount of nuclear receptor (NR) agonist group quantified by FluoReporter kit at day 7 was normalized to day 0 and further normalized to Veh group as fold change (FC), represented by three-dimensional plot for T47D WT, Y537S and D538G cells respectively. The agonists of following nuclear receptors have been used for the screen: Retinoid Acid Receptor (RAR), Thyroid Receptor (TR), Vitamin D Receptor (VDR), Liver X Receptor (LXR), Peroxisome Proliferator-Activated Receptor Alpha (PPAR-α), Peroxisome Proliferator-Activated Receptor Delta (PPAR-δ), Farnesoid X receptor (FXR), Constitutive Androstane Receptor (CAR), Pregnane X Receptor (PXR). Student’s t test was used to examine the effects of treatment between agonist group and Veh group (p<0.05). Agonists significantly increased growth in mutant cells were highlighted in red and labeled with its receptor name. Agonists not significantly increased growth in mutant cells were highlighted in grey and labeled with its receptor name.
We also tried to test whether the RXR agonist stimulated growth was dependent on FOXA1, but FOXA1 knockdown drastically attenuated the base line growth of both ESR1 WT and mutant cells, with stronger effects observed in mutant cells (Supplementary Fig. S6). Given FOXA1’s essential role in ER+ breast cancer cells growth through its regulation of multiple cell proliferation-related factors (55–57), it is challenging to specifically examine the specific impact of FOXA1 knockdown on RXR agonist-induced cell growth phenotype.
Since RXR is known to form heterodimers with other nuclear receptors (NR) in a ligand-dependent manner (53,54,58), we treated our T47D cells with agonists of nine NRs widely reported to form heterodimers with RXR (58,59), and tested their impact on cell growth. The screen revealed that the addition of 20nM of thyroid hormone T3, the agonist for thyroid receptor (TR), could significantly promote growth of ESR1 mutant cells to an equivalent level as the RXR agonist (Fig. 4E), suggesting a potential role of TR as a binding partner with RXR in this setting, warranting further investigation.
RXR antagonist HX531 can inhibit the response of ESR1 mutant cells and a patient-derived xenograft organoid (PDXO) to RXR agonist.
Given that multiple endogenous RXR agonists have been identified (60), we speculated that ESR1 mutant breast cancer cells may utilize these physiological RXR agonists to gain an additional survival and proliferation advantage in the absence of estrogen. Thus, blocking the endogenous RXR pathway might be a novel therapeutic strategy to treat patients with ESR1 mutant breast cancer, and thus we tested the effect of a highly-selective RXR antagonist HX531 (61). As shown in Fig. 5A–B, 2μM HX531 diminished LG268-promoted colonization. In addition, combinations of varying doses of agonist/antagonist showed that the response to 10nM LG268 could be completely blocked by 2μM HX531. 5μM HX531 blocked the response to 10nM or 100nM LG268 but also decreased baseline levels of growth (Supplementary Fig. S7). In addition, in the presence of 100nM LG268, ESR1 mutant cells were significantly more sensitive to antagonist inhibition compared to WT cells (Fig. 5C).
Figure 5. RXR antagonist HX531 can inhibit the response of ESR1 mutant cells and organoids to RXR agonist.
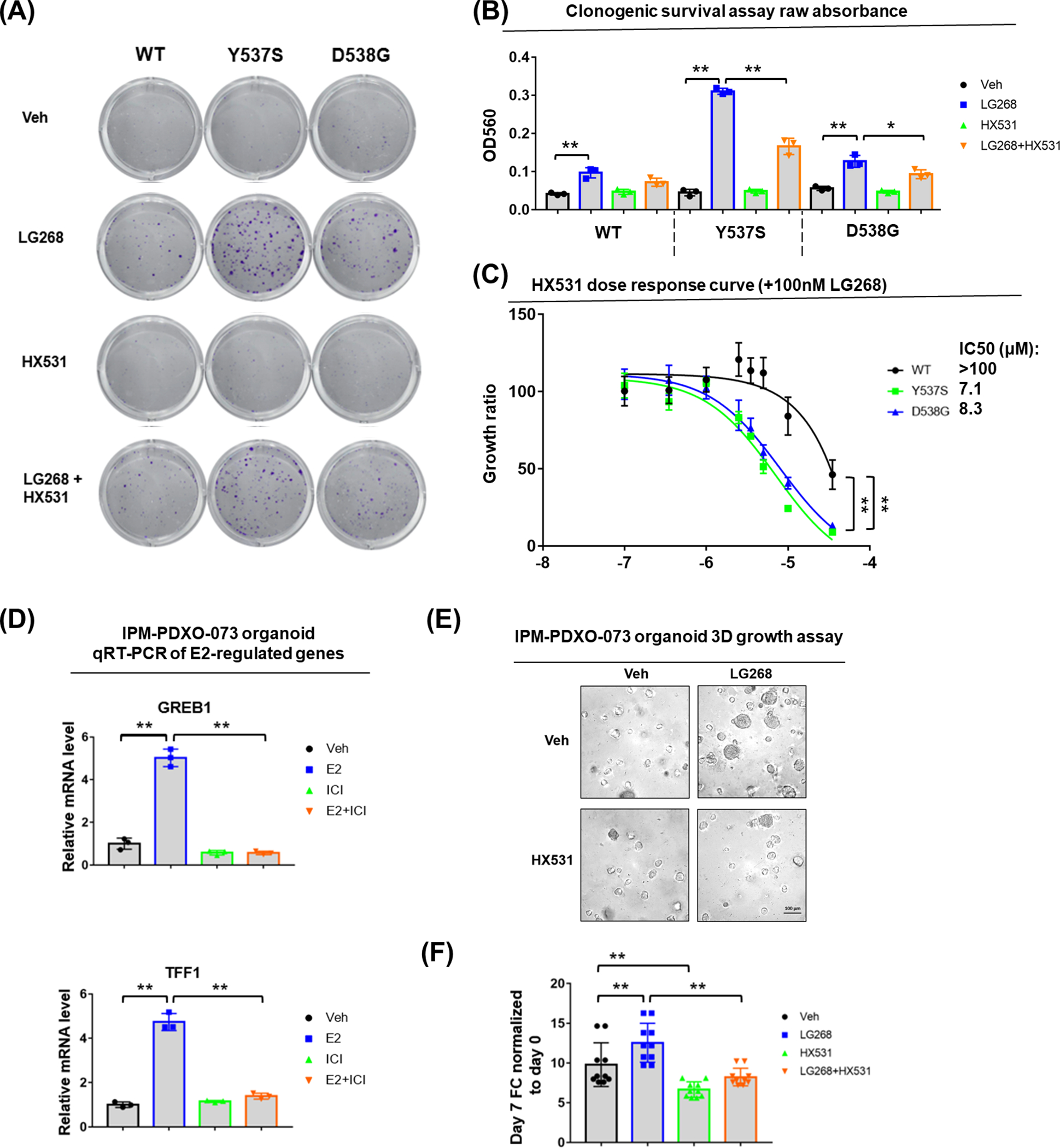
(A, B) Colony formation assay result after RXR agonist or antagonist treatment. (A) Representative images of colonies formed after Veh or 100nM LG268 treatment or 2μM HX531 or 100nM LG268 + 2μM HX531 treatment; (B) Bar plot showing the raw absorbance OD560 of each group after crystal violet staining and acetic acid dissolving. Each bar represents mean ± SD with three biological replicates. Student’s t test was used to examine the effects of treatment between LG268 group and Veh group, and between LG268+HX531 and LG268 group. (* p<0.05, ** p<0.01).
(C) HX531 dose response curve of T47D cells under fixed 100nM LG268 treatment. Cell amount quantified by FluoReporter kit at day 7 of all dose groups was normalized to 100nM LG268 group and presented as fold change (FC). Each bar represents mean ± SD with five biological replicates. IC50s were shown on the right side. Two-way ANOVA were performed to compare the HX531 dose response between mutant and WT cells. (** p<0.01)
(D) qRT-PCR measurement of GREB1 and TFF1 mRNA level in IPM-PDXO-073 organoid with Veh or 1nM E2 or 1μM ICI or 1nM E2 + 1μM ICI treatment. mRNA fold changes were normalized to RPLP0 and further normalized to Veh group. Each bar represents mean ± SD with three biological replicates. Dunnett’s test was used to compare the expression between Veh and E2, E2 and E2+ICI groups. (** p<0.01)
(E-F) Growth assay result on IPM-PDXO-073 organoid with Veh or 100nM LG268 treatment or 2μM HX531 or 100nM LG268 + 2μM HX531 treatment. (E) Representative images of IPM-PDXO-073 organoid in round bottom 96-well ultra-low attachment plate after 7 days’ treatment. The organoids were seeded with the initial density of 5,000 cells per well. (F) Bar plot showing the organoid growth quantified by Celltiter-glo kit at day 7 was normalized to day 0 and presented as fold change (FC). Each bar represents mean ± SD with ten biological replicates. Dunnett’s test was used to compare the expression between control and treatment groups. (** p<0.01)
We sought to evaluate the impact of RXR antagonist using additional ESR1 mutant models. We generated an organoid IPM-PDXO-073 from a patient derived xenograft (CTG-1260) which has an ESR1 D538G mutation. Droplet digital PCR on genomic DNA revealed 50% D538G mutation allele frequency (Supplementary Fig. S8A). E2 stimulation significantly induced two classical downstream genes GREB1 and TFF1, which could be inhibited by ICI 182,780 (ICI) treatment (Fig. 5D). qRT-PCR indicated robust expression of RXRA, but weak or no expression of RXRB and RXRG (Supplementary Fig. S8B). Taken together, these molecular characterizations verified the IPM-PDXO-073 organoid as a heterozygous D538G ESR1 mutant model with estrogen response suitable for our studies. We next exposed IPM-PDXO-073 to RXR agonist and antagonist and consistent with cell line data, the growth of the mutant organoid was promoted by 100nM LG268, whilst 2μM HX531 attenuated growth in the absence and presence of LG268 (Fig. 5E–F). Taken together, these data suggest that RXR antagonists such as HX531 might have therapeutic potential to treat ESR1 mutant breast cancer.
Discussion
Hotspot ESR1 mutations are recognized as frequent mechanisms of resistance to endocrine therapy in recurrent ER+ breast cancers. It is imperative to uncover therapeutic vulnerabilities by deciphering mechanisms of growth and survival endowed by ESR1 mutation. In this study, we identified unique roles of FOXA1 in ESR1 mutant cells including regulation of novel target gene expression and provision of novel RXR binding sites leading to enhanced response to the RXR agonist LG268. In contrast, the role of FOXA1 in canonical ER signaling as seen in WT cells is diminished in ESR1 mutant cells (Fig. 6). It is plausible that ESR1 mutant cells may benefit from RXR activation, which can be inhibited by antagonists such as HX531, providing a novel idea for potential treatment of ESR1 mutant breast cancers.
Figure 6. Graphical presentation of less FOXA1-ER interaction and enhanced response to RXR agonist in ESR1 mutant cells.
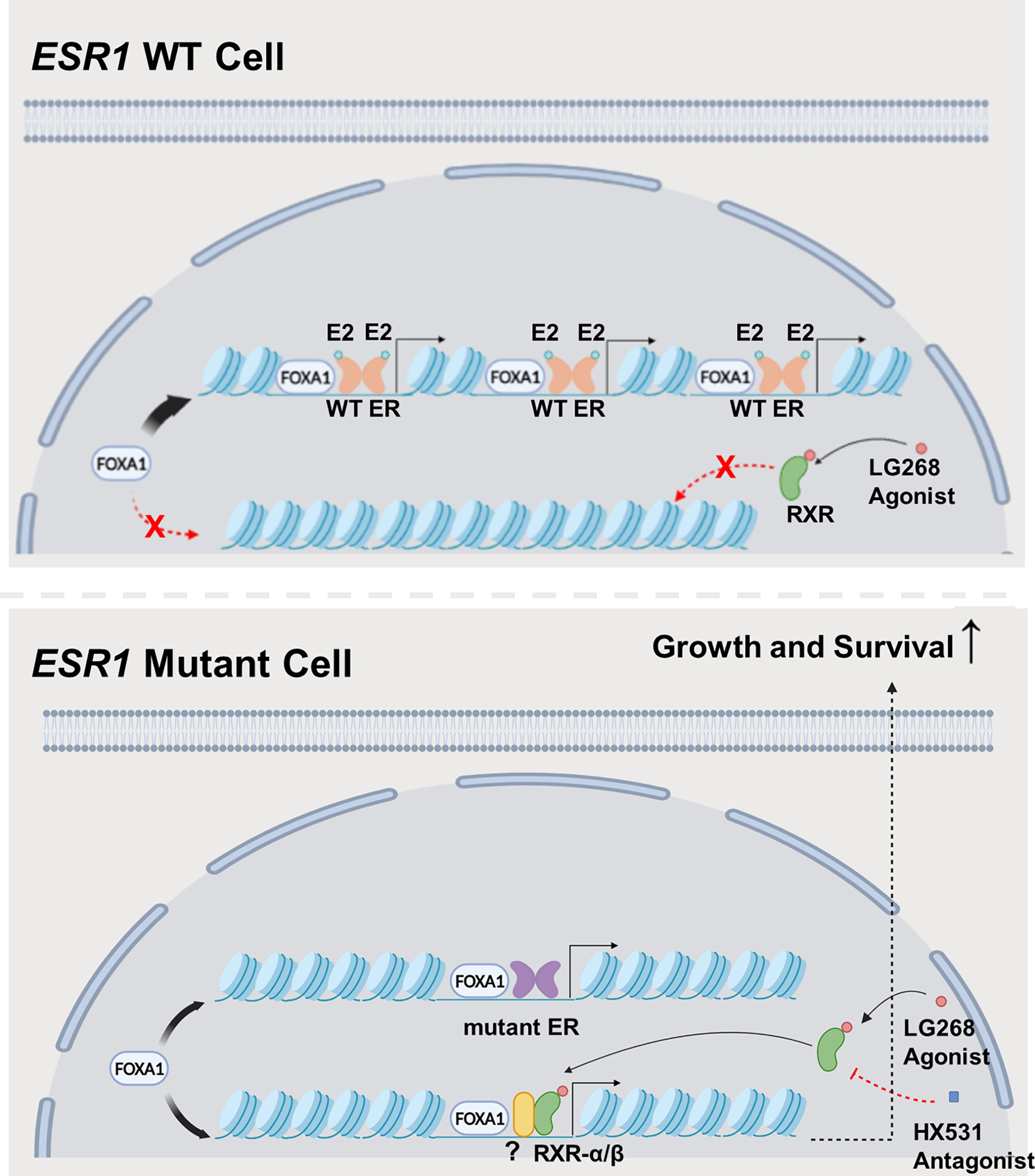
In ESR1 WT cells, FOXA1 facilitates ER chromatin binding by increasing chromatin accessibility of ER binding sites. In ESR1 mutant cells, the canonical FOXA1-ER interaction is reduced, but the redistributed FOXA1 remodels chromatin structure to increase accessibility for novel RXR binding sites.
We have comprehensively characterized the impact of FOXA1 reprogramming in ESR1 mutant cells. FOXA1 modulates chromatin structure and increases accessibility of DNA binding elements, augmenting the recruitment of multiple nuclear receptors (62–64). Arnesen et al. have reported the significant enrichment of to FOXA1 motif in D538G-specific open chromatin regions, suggesting the potential role of FOXA1 in remodeling chromatin structure in ESR1 D538G cells (15). We further revealed the impact of FOXA1 on chromatin structure and the binding of other transcriptional factors under ESR1 mutant background, and found in both ESR1 Y537S and D538G cells, a redistributed FOXA1 regulates novel target gene expression at least in part through recruitment of RXR. The mechanisms of how mutant ER alters the FOXA1 cistrome remains unclear. Some studies have suggested that ER could reciprocally act as initiating factors for FOXA1. Caizzi et al. found that transient ER silencing resulted in a significant decrease in FOXA1 binding events in close proximity to ER-bound sites (65). It is plausible that mutant ER serves as an FOXA1 pioneer factor to rewire FOXA1 global binding and thus reshapes a divergent chromatin landscape. Our previous results identified a larger number of FOXA1 differentially binding (DB) peaks in ESR1 mutant cells than WT cells after E2 treatment (34), which supports the concept that the FOXA1 cistrome redistribution in mutant cells is likely an ESR1 mutant-specific gain of function. Some transcription factors have been described as upstream regulators of FOXA1, which may impact FOXA1 binding, such as CTCF and GATA3 (32,66). Notably, we recently reported CTCF cistrome was associated with differential chromatin accessibility and gene expression in ESR1 mutant cells (15,46). Moreover, other epigenetic changes such as histone H3 lysine 4 methylation were also revealed to define FOXA1 binding (67,68). It is likely that mutant ER can impact the FOXA1 cistrome by cooperating with these FOXA1 regulators or by remodeling epigenetic features, another possibility that the activated RXR signaling recruit FOXA1 should not be ignored, but these possibilities require further research. It is necessasy to apply additional biochemical assays like RIME to characterize the FOXA1 and RXR interactions in ESR1 WT and mutant cells.
Our phenotypic data with cell models from multiple laboratories supports that ESR1 mutant cells with redistributed FOXA1 cistrome can acquire proliferation and survival advantages through RXR activation. RXR agonists are recognized as anti-cancer agents and multiple studies have suggested their potential in breast cancer prevention and treatment (69–72). In our study, we found that RXR agonist stimulation enhanced growth and survival in mutant cells with no or limited agonist activity in WT cells. Indeed, both RXR agonist effects and basal proliferation and colonization were suppressed after RXRB knockdown in both WT and mutant cells. While we find the RXR agonist able to stimulate growth, in other models it has been noted to potentiate apoptotic response to other chemotherapeutic drugs (73,74). It is possible that a novel function gained by ESR1 mutation through FOXA1 reprogramming dictated this RXR growth stimulatory response. Importantly, while some preclinical data suggested anti-cancer efficacy (75), the therapeutic activity of RXR agonists in clinical trials has been minimal. Esteva et al. reported limited efficacy of RXR agonist bexarotene in patients with refractory metastatic breast cancer (76). In addition, some studies reported that high expression of RXR was associated with worse prognosis in breast cancer patients (77,78). Therefore, we surmise that the effects of RXR agonists are likely context-dependent, and that cancer cells with different genetic backgrounds may have different RXR response. For a subset of tumors with an ESR1-mutant background which can hijack RXR response to proliferate, it is plausible to include RXR antagonist as a treatment option, highlighting the need for a precision medicine approach to treatment taking into account ESR1 mutation status.
Notably, although the enhanced responses to RXR agonist were observed in both two ESR1 mutant cells, it was more pronounced in Y537S cells than D538G cells. Under the LG268 treatment, the clonogenic survival ability of Y537S and D538G cells could increase to 5.7 times and 1.9 times respectively, which may attribute to the essential difference between the two mutant subtypes uncovered by multiple studies. Firstly, the crystalized structure analysis revealed that Y537S and D538G mutations could cause different conformational change to the helix 11–12 of ER, and Y537S ER had stronger constitutive activity than D538G ER (79). Secondly, their differences in downstream gene regulation were reflected by distinct ER cistrome, chromatin accessibility and transcriptome (15,18,24). Notably, we have identified mutant ER binding sites were less associated with DE genes, as mutant ER could induce differential gene expression not only through direct ER binding but also through indirect actions (34), additional time course experiments need to be performed for further characterization. Thirdly, some phenotypes were reported to be uniquely acquired by certain mutant, like the Y537S induced epithelial–mesenchymal transition (EMT) (30) and D538G increased migratory ability (34). Indeed, ESR1 mutant breast cancer is a highly heterogenous disease that can have different behaviors conferred by different mutation subtype and background, pressing the need to establish more ESR1 mutant models with extensively different backgrounds for future pre-clinical investigation.
We sought to address the specific molecular mechanism explaining the response to RXR agonist in ESR1 mutant cells. Recognized as the obligate partner for various nuclear receptors, RXR usually forms heterodimers to regulate downstream gene expression in a ligand-dependent manner. Permissive heterodimers can be activated by agonist of either RXR or the other receptors, while nonpermissive heterodimers can be only activated by agonists of the other receptor, but can be enhanced in addition with RXR agonists (53,54,58). From an agonist screen, we found the TR agonist T3 can promote growth of ESR1 mutant cells similar to the RXR agonist, suggesting potential crosstalk between TR and RXR which should be further explored. Intriguingly, T3 stimulated proliferation of ER+ breast cancer cells has been reported by others (80,81). These data reveal the potential to co-target RXR and TR, meanwhile we cannot exclude other possibilities, such as RXR homodimers or other NR-RXR heterodimers requiring higher agonist concentrations than we have explored in this study.
Taken together, our study provides comprehensive analysis of FOXA1 in ESR1 mutant breast cancer cell models, identifying its roles in regulating novel target genes and facilitating RXR binding. With the potential RXR binding sites provided by FOXA1 binding sites redistribution, ESR1 mutant cells gain survival advantage through enhanced RXR response, suggesting that this signaling axis should be further studied with a potential to personalized therapy for patients with ESR1 mutant breast cancer.
Supplementary Material
Implications:
It provides comprehensive characterization of the role of FOXA1 in ESR1 mutant breast cancer and potential therapeutic strategy through blocking RXR activation.
Acknowledgements
This work was in part supported by the National Cancer Institute [R01CA221303 to SO and P30CA047904]; Department of Defense [DOD grant W81XWH1910434 (JG)]; Breast Cancer Research Foundation [AVL and SO]; Magee-Women’s Research Institute and Foundation, Nicole Meloche Foundation, the Pennsylvania Breast Cancer Coalition and the Shear Family Foundation. SO and AVL are Hillman Fellows, Susan G. Komen Scholars [SAC110021 to AVL; SAC160073 to SO]; and ZL was supported by John S. Lazo Cancer Pharmacology Fellowship. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or other Institutes and Foundations.
We would like to thank Dr. Peilu Wang for her contribution to earlier ESR1 mutant-studies and Dr. Rinath Jeselsohn for providing additional information of the Brown FOXA1 ChIP-seq data set. We would also like to thank Champions Oncology for generating CTG-1260 PDX model as the sources of the organoid used in this study.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Conflict of Interest Statement:
SO and AVL receive research support from AstraZeneca PLC. AVL is employee and consultant with UPMC Enterprises, and member of the Scientific Advisory Board, Stockholder and receives compensation from Ocean Genomics. No disclosures were reported by the other authors.
References
- 1.Blows FM, Driver KE, Schmidt MK, Broeks A, van Leeuwen FE, Wesseling J, et al. Subtyping of breast cancer by immunohistochemistry to investigate a relationship between subtype and short and long term survival: a collaborative analysis of data for 10,159 cases from 12 studies. PLoS Med 2010;7(5):e1000279 doi 10.1371/journal.pmed.1000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeSantis CE, Ma J, Goding Sauer A, Newman LA, Jemal A. Breast cancer statistics, 2017, racial disparity in mortality by state. CA Cancer J Clin 2017;67(6):439–48 doi 10.3322/caac.21412. [DOI] [PubMed] [Google Scholar]
- 3.Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, et al. Genome-wide analysis of estrogen receptor binding sites. Nat Genet 2006;38(11):1289–97 doi 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- 4.Burstein HJ. Systemic Therapy for Estrogen Receptor-Positive, HER2-Negative Breast Cancer. N Engl J Med 2020;383(26):2557–70 doi 10.1056/NEJMra1307118. [DOI] [PubMed] [Google Scholar]
- 5.Waks AG, Winer EP. Breast Cancer Treatment: A Review. Jama 2019;321(3):288–300 doi 10.1001/jama.2018.19323. [DOI] [PubMed] [Google Scholar]
- 6.Turner NC, Neven P, Loibl S, Andre F. Advances in the treatment of advanced oestrogen-receptor-positive breast cancer. Lancet 2017;389(10087):2403–14 doi 10.1016/s0140-6736(16)32419-9. [DOI] [PubMed] [Google Scholar]
- 7.Hanker AB, Sudhan DR, Arteaga CL. Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell 2020;37(4):496–513 doi 10.1016/j.ccell.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brufsky AM, Dickler MN. Estrogen Receptor-Positive Breast Cancer: Exploiting Signaling Pathways Implicated in Endocrine Resistance. Oncologist 2018;23(5):528–39 doi 10.1634/theoncologist.2017-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chu D, Paoletti C, Gersch C, VanDenBerg DA, Zabransky DJ, Cochran RL, et al. ESR1 Mutations in Circulating Plasma Tumor DNA from Metastatic Breast Cancer Patients. Clinical cancer research : an official journal of the American Association for Cancer Research 2016;22(4):993–9 doi 10.1158/1078-0432.CCR-15-0943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spoerke JM, Gendreau S, Walter K, Qiu J, Wilson TR, Savage H, et al. Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nature Communications 2016;7(1):11579 doi 10.1038/ncomms11579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guttery DS, Page K, Hills A, Woodley L, Marchese SD, Rghebi B, et al. Noninvasive detection of activating estrogen receptor 1 (ESR1) mutations in estrogen receptor-positive metastatic breast cancer. Clinical chemistry 2015;61(7):974–82 doi 10.1373/clinchem.2015.238717. [DOI] [PubMed] [Google Scholar]
- 12.Razavi P, Chang MT, Xu G, Bandlamudi C, Ross DS, Vasan N, et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018;34(3):427–38.e6 doi 10.1016/j.ccell.2018.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Katzenellenbogen JA, Mayne CG, Katzenellenbogen BS, Greene GL, Chandarlapaty S. Structural underpinnings of oestrogen receptor mutations in endocrine therapy resistance. Nat Rev Cancer 2018;18(6):377–88 doi 10.1038/s41568-018-0001-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fanning SW, Mayne CG, Dharmarajan V, Carlson KE, Martin TA, Novick SJ, et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. Elife 2016;5 doi 10.7554/eLife.12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arnesen S, Blanchard Z, Williams MM, Berrett KC, Li Z, Oesterreich S, et al. Estrogen Receptor Alpha Mutations in Breast Cancer Cells Cause Gene Expression Changes through Constant Activity and Secondary Effects. Cancer Research 2021;81(3):539 doi 10.1158/0008-5472.CAN-20-1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet 2013;45(12):1439–45 doi 10.1038/ng.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harrod A, Fulton J, Nguyen VT, Periyasamy M, Ramos-Garcia L, Lai C-F, et al. Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene 2017;36(16):2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bahreini A, Li Z, Wang P, Levine KM, Tasdemir N, Cao L, et al. Mutation site and context dependent effects of ESR1 mutation in genome-edited breast cancer cell models. Breast Cancer Research 2017;19(1):60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A, Dvir A, Soussan-Gutman L, Jeselsohn R, et al. D538G mutation in estrogen receptor-α: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer research 2013;73(23):6856–64. [DOI] [PubMed] [Google Scholar]
- 20.Weis KE, Ekena K, Thomas JA, Lazennec G, Katzenellenbogen BS. Constitutively active human estrogen receptors containing amino acid substitutions for tyrosine 537 in the receptor protein. Mol Endocrinol 1996;10(11):1388–98 doi 10.1210/mend.10.11.8923465. [DOI] [PubMed] [Google Scholar]
- 21.Schiavon G, Hrebien S, Garcia-Murillas I, Cutts RJ, Pearson A, Tarazona N, et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Science translational medicine 2015;7(313):313ra182 doi 10.1126/scitranslmed.aac7551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang K, Hong R, Xu F, Xia W, Kaping L, Qin G, et al. Clinical value of circulating ESR1 mutations for patients with metastatic breast cancer: a meta-analysis. Cancer management and research 2018;10:2573–80 doi 10.2147/cmar.S173193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chandarlapaty S, Chen D, He W, Sung P, Samoila A, You D, et al. Prevalence of ESR1 mutations in cell-free DNA and outcomes in metastatic breast cancer: a secondary analysis of the BOLERO-2 clinical trial. JAMA oncology 2016;2(10):1310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jeselsohn R, Bergholz JS, Pun M, Cornwell M, Liu W, Nardone A, et al. Allele-specific chromatin recruitment and therapeutic vulnerabilities of ESR1 activating mutations. Cancer cell 2018;33(2):173–86. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mao C, Livezey M, Kim JE, Shapiro DJ. Antiestrogen Resistant Cell Lines Expressing Estrogen Receptor alpha Mutations Upregulate the Unfolded Protein Response and are Killed by BHPI. Sci Rep 2016;6:34753 doi 10.1038/srep34753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gelsomino L, Gu G, Rechoum Y, Beyer AR, Pejerrey SM, Tsimelzon A, et al. ESR1 mutations affect anti-proliferative responses to tamoxifen through enhanced cross-talk with IGF signaling. Breast cancer research and treatment 2016;157(2):253–65 doi 10.1007/s10549-016-3829-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Z, Levine KM, Bahreini A, Wang P, Chu D, Park BH, et al. Upregulation of IRS1 Enhances IGF1 Response in Y537S and D538G ESR1 Mutant Breast Cancer Cells. Endocrinology 2018;159(1):285–96 doi 10.1210/en.2017-00693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu L, Wang L, Mao C, Duraki D, Kim JE, Huang R, et al. Estrogen-independent Myc overexpression confers endocrine therapy resistance on breast cancer cells expressing ERalphaY537S and ERalphaD538G mutations. Cancer Lett 2019;442:373–82 doi 10.1016/j.canlet.2018.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dustin D, Gu G, Beyer AR, Herzog SK, Edwards DG, Lin H, et al. RON signalling promotes therapeutic resistance in ESR1 mutant breast cancer. Br J Cancer 2021;124(1):191–206 doi 10.1038/s41416-020-01174-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gu G, Tian L, Herzog SK, Rechoum Y, Gelsomino L, Gao M, et al. Hormonal modulation of ESR1 mutant metastasis. Oncogene 2021;40(5):997–1011 doi 10.1038/s41388-020-01563-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 2005;122(1):33–43 doi 10.1016/j.cell.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 32.Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nature genetics 2011;43(1):27–33 doi 10.1038/ng.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinson J, Carroll J. FoxA1 is a Key Mediator of Hormonal Response in Breast and Prostate Cancer. 2012;3(68) doi 10.3389/fendo.2012.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Z, Wu Y, Yates ME, Tasdemir N, Bahreini A, Chen J, et al. Hotspot ESR1 mutations are multimodal and contextual modulators of breast cancer metastasis. Cancer research 2022. doi 10.1158/0008-5472.Can-21-2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sachs N, de Ligt J, Kopper O, Gogola E, Bounova G, Weeber F, et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018;172(1–2):373–86.e10 doi 10.1016/j.cell.2017.11.010. [DOI] [PubMed] [Google Scholar]
- 36.Stark R, Brown G. DiffBind: differential binding analysis of ChIP-Seq peak data. R package version 2011;100:4.3. [Google Scholar]
- 37.Yu G, Wang L-G, He Q-Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015;31(14):2382–3. [DOI] [PubMed] [Google Scholar]
- 38.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, et al. MEME SUITE: tools for motif discovery and searching. Nucleic acids research 2009;37(Web Server issue):W202–8 doi 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buske FA, Bodén M, Bauer DC, Bailey TL. Assigning roles to DNA regulatory motifs using comparative genomics. Bioinformatics (Oxford, England) 2010;26(7):860–6 doi 10.1093/bioinformatics/btq049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinformatics 2011;27(7):1017–8 doi 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anderson AP, Jones AG. erefinder: Genome-wide detection of oestrogen response elements. Molecular ecology resources 2019;19(5):1366–73 doi 10.1111/1755-0998.13046. [DOI] [PubMed] [Google Scholar]
- 42.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics (Oxford, England) 2010;26(6):841–2 doi 10.1093/bioinformatics/btq033 %J Bioinformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krämer A, Green J, Pollard J Jr, Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 2013;30(4):523–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jozwik KM, Chernukhin I, Serandour AA, Nagarajan S, Carroll JS. FOXA1 Directs H3K4 Monomethylation at Enhancers via Recruitment of the Methyltransferase MLL3. Cell Rep 2016;17(10):2715–23 doi 10.1016/j.celrep.2016.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang S, Sun H, Ma J, Zang C, Wang C, Wang J, et al. Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat Protoc 2013;8(12):2502–15 doi 10.1038/nprot.2013.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Z, McGinn O, Wu Y, Bahreini A, Priedigkeit NM, Ding K, et al. ESR1 mutant breast cancers show elevated basal cytokeratins and immune activation. Nature Communications 2022;13(1):2011 doi 10.1038/s41467-022-29498-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Swinstead Erin E, Miranda Tina B, Paakinaho V, Baek S, Goldstein I, Hawkins M, et al. Steroid Receptors Reprogram FoxA1 Occupancy through Dynamic Chromatin Transitions. Cell 2016;165(3):593–605 doi 10.1016/j.cell.2016.02.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Priedigkeit N, Hartmaier RJ, Chen Y, Vareslija D, Basudan A, Watters RJ, et al. Intrinsic subtype switching and acquired ERBB2/HER2 amplifications and mutations in breast cancer brain metastases. JAMA oncology 2017;3(5):666–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Priedigkeit N, Watters RJ, Lucas PC, Basudan A, Bhargava R, Horne W, et al. Exome-capture RNA sequencing of decade-old breast cancers and matched decalcified bone metastases. JCI insight 2017;2(17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Levine KM, Priedigkeit N, Basudan A, Tasdemir N, Sikora MJ, Sokol ES, et al. FGFR4 overexpression and hotspot mutations in metastatic ER+ breast cancer are enriched in the lobular subtype. NPJ breast cancer 2019;5(1):1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robinson DR, Wu Y-M, Lonigro RJ, Vats P, Cobain E, Everett J, et al. Integrative clinical genomics of metastatic cancer. Nature 2017;548(7667):297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pleasance E, Titmuss E, Williamson L, Kwan H, Culibrk L, Zhao EY, et al. Pan-cancer analysis of advanced patient tumors reveals interactions between therapy and genomic landscapes. Nature Cancer 2020;1(4):452–68. [DOI] [PubMed] [Google Scholar]
- 53.Ahuja HS, Szanto A, Nagy L, Davies PJ. The retinoid X receptor and its ligands: versatile regulators of metabolic function, cell differentiation and cell death. Journal of biological regulators and homeostatic agents 2003;17(1):29–45. [PubMed] [Google Scholar]
- 54.Szanto A, Narkar V, Shen Q, Uray IP, Davies PJ, Nagy L. Retinoid X receptors: X-ploring their (patho)physiological functions. Cell Death Differ 2004;11 Suppl 2:S126–43 doi 10.1038/sj.cdd.4401533. [DOI] [PubMed] [Google Scholar]
- 55.Eeckhoute J, Carroll JS, Geistlinger TR, Torres-Arzayus MI, Brown M. A cell-type-specific transcriptional network required for estrogen regulation of cyclin D1 and cell cycle progression in breast cancer. Genes Dev 2006;20(18):2513–26 doi 10.1101/gad.1446006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wright TM, Wardell SE, Jasper JS, Stice JP, Safi R, Nelson ER, et al. Delineation of a FOXA1/ERα/AGR2 regulatory loop that is dysregulated in endocrine therapy-resistant breast cancer. Mol Cancer Res 2014;12(12):1829–39 doi 10.1158/1541-7786.Mcr-14-0195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fu X, Jeselsohn R, Pereira R, Hollingsworth EF, Creighton CJ, Li F, et al. FOXA1 overexpression mediates endocrine resistance by altering the ER transcriptome and IL-8 expression in ER-positive breast cancer. Proc Natl Acad Sci U S A 2016;113(43):E6600–e9 doi 10.1073/pnas.1612835113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Evans RM, Mangelsdorf DJ. Nuclear Receptors, RXR, and the Big Bang. Cell 2014;157(1):255–66 doi 10.1016/j.cell.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lefebvre P, Benomar Y, Staels B. Retinoid X receptors: common heterodimerization partners with distinct functions. Trends in endocrinology and metabolism: TEM 2010;21(11):676–83 doi 10.1016/j.tem.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 60.Rühl R, Krężel W, de Lera AR. 9-Cis-13,14-dihydroretinoic acid, a new endogenous mammalian ligand of retinoid X receptor and the active ligand of a potential new vitamin A category: vitamin A5. Nutrition Reviews 2018;76(12):929–41 doi 10.1093/nutrit/nuy057 %J Nutrition Reviews. [DOI] [PubMed] [Google Scholar]
- 61.Kanayasu-Toyoda T, Fujino T, Oshizawa T, Suzuki T, Nishimaki-Mogami T, Sato Y, et al. HX531, a retinoid X receptor antagonist, inhibited the 9-cis retinoic acid-induced binding with steroid receptor coactivator-1 as detected by surface plasmon resonance. The Journal of steroid biochemistry and molecular biology 2005;94(4):303–9 doi 10.1016/j.jsbmb.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 62.Bernardo GM, Keri RA. FOXA1: a transcription factor with parallel functions in development and cancer. Biosci Rep 2012;32(2):113–30 doi 10.1042/BSR20110046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cirillo LA, McPherson CE, Bossard P, Stevens K, Cherian S, Shim EY, et al. Binding of the winged-helix transcription factor HNF3 to a linker histone site on the nucleosome. EMBO J 1998;17(1):244–54 doi 10.1093/emboj/17.1.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, Zaret KS. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol Cell 2002;9(2):279–89 doi 10.1016/s1097-2765(02)00459-8. [DOI] [PubMed] [Google Scholar]
- 65.Caizzi L, Ferrero G, Cutrupi S, Cordero F, Ballaré C, Miano V, et al. Genome-wide activity of unliganded estrogen receptor-α in breast cancer cells. Proc Natl Acad Sci U S A 2014;111(13):4892–7 doi 10.1073/pnas.1315445111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Theodorou V, Stark R, Menon S, Carroll JS. GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res 2013;23(1):12–22 doi 10.1101/gr.139469.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lupien M, Eeckhoute J, Meyer CA, Wang Q, Zhang Y, Li W, et al. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell 2008;132(6):958–70 doi 10.1016/j.cell.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sérandour AA, Avner S, Percevault F, Demay F, Bizot M, Lucchetti-Miganeh C, et al. Epigenetic switch involved in activation of pioneer factor FOXA1-dependent enhancers. Genome research 2011;21(4):555–65 doi 10.1101/gr.111534.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bischoff ED, Gottardis MM, Moon TE, Heyman RA, Lamph WW. Beyond Tamoxifen: The Retinoid X Receptor-selective Ligand LGD1069 (TARGRETIN) Causes Complete Regression of Mammary Carcinoma. 1998;58(3):479–84. [PubMed] [Google Scholar]
- 70.Crowe DL, Chandraratna RAS. A retinoid X receptor (RXR)-selective retinoid reveals that RXR-alpha is potentially a therapeutic target in breast cancer cell lines, and that it potentiates antiproliferative and apoptotic responses to peroxisome proliferator-activated receptor ligands. Breast cancer research : BCR 2004;6(5):R546–R55 doi 10.1186/bcr913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Howe LR. Rexinoids and breast cancer prevention. Clin Cancer Res 2007;13(20):5983–7 doi 10.1158/1078-0432.CCR-07-1065. [DOI] [PubMed] [Google Scholar]
- 72.Suh N, Lamph WW, Glasebrook AL, Grese TA, Palkowitz AD, Williams CR, et al. Prevention and treatment of experimental breast cancer with the combination of a new selective estrogen receptor modulator, arzoxifene, and a new rexinoid, LG 100268. Clin Cancer Res 2002;8(10):3270–5. [PubMed] [Google Scholar]
- 73.Crowe DL, Chandraratna RAS. A retinoid X receptor (RXR)-selective retinoid reveals that RXR-α is potentially a therapeutic target in breast cancer cell lines, and that it potentiates antiproliferative and apoptotic responses to peroxisome proliferator-activated receptor ligands. Breast Cancer Research 2004;6(5):R546 doi 10.1186/bcr913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rendi MH, Suh N, Lamph WW, Krajewski S, Reed JC, Heyman RA, et al. The Selective Estrogen Receptor Modulator Arzoxifene and the Rexinoid LG100268 Cooperate to Promote Transforming Growth Factor β-Dependent Apoptosis in Breast Cancer. 2004;64(10):3566–71 doi 10.1158/0008-5472.CAN-04-0234 %J Cancer Research. [DOI] [PubMed] [Google Scholar]
- 75.Howe LR. Rexinoids and Breast Cancer Prevention. 2007;13(20):5983–7 doi 10.1158/1078-0432.CCR-07-1065 %J Clinical Cancer Research. [DOI] [PubMed] [Google Scholar]
- 76.Esteva FJ, Glaspy J, Baidas S, Laufman L, Hutchins L, Dickler M, et al. Multicenter phase II study of oral bexarotene for patients with metastatic breast cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2003;21(6):999–1006 doi 10.1200/jco.2003.05.068. [DOI] [PubMed] [Google Scholar]
- 77.Lawrence JA, Merino MJ, Simpson JF, Manrow RE, Page DL, Steeg PS. A high-risk lesion for invasive breast cancer, ductal carcinoma in situ, exhibits frequent overexpression of retinoid X receptor. Cancer Epidemiology Biomarkers & Prevention 1998;7(1):29. [PubMed] [Google Scholar]
- 78.Zehni AZ, Batz F, Vattai A, Kaltofen T, Schrader S, Jacob S-N, et al. The Prognostic Impact of Retinoid X Receptor and Thyroid Hormone Receptor alpha in Unifocal vs. Multifocal/Multicentric Breast Cancer. Int J Mol Sci 2021;22(2):957 doi 10.3390/ijms22020957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Katzenellenbogen JA, Mayne CG, Katzenellenbogen BS, Greene GL, Chandarlapaty S. Structural underpinnings of oestrogen receptor mutations in endocrine therapy resistance. Nat Rev Cancer 2018;18(6):377–88 doi 10.1038/s41568-018-0001-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hall LC, Salazar EP, Kane SR, Liu N. Effects of thyroid hormones on human breast cancer cell proliferation. The Journal of steroid biochemistry and molecular biology 2008;109(1–2):57–66 doi 10.1016/j.jsbmb.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 81.Dinda S, Sanchez A, Moudgil V. Estrogen-like effects of thyroid hormone on the regulation of tumor suppressor proteins, p53 and retinoblastoma, in breast cancer cells. Oncogene 2002;21(5):761–8 doi 10.1038/sj.onc.1205136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ER and FOXA1 ChIP-seq data has been deposited onto the Gene Expression Omnibus database (RRID: SCR005012) (GSE125117 and GSE165280). Other data resources used in this study are summarized in Supplementary Table S1. All raw data and scripts are available upon request from the corresponding author.