

Abstract
HPV16 is the most oncogenic type of human papillomaviruses (HPV). Integration of HPV into the human genome is an important mechanism of carcinogenesis but is absent in at least 30% of HPV16+ tumors. We applied long-read whole-genome sequencing (WGS) to cervical cancer cell lines and tumors to characterize HPV16 carcinogenesis in the absence of integration. Large tandem arrays of full-length and unique truncated viral genomes integrated into multiple chromosomes were identified in two HPV16+ cell lines. The dispersion of characteristic viral variants to multiple integrations sites indicates that viral deletions formed as extrachromosomal DNA (a phenomenon we term HPV superspreading). In addition, we identified an HPV16+ cell line with unintegrated (episomal) DNA that has tandem arrays of full-length, truncated, and rearranged HPV16 genomes (multimer episomes). Cytogenetic analysis of this cell line shows intense extrachromosomal HPV staining, including structures resembling double-minute chromosomes. Whole genome sequencing of HPV16+ cervical tumor samples from Latin America revealed that 11/20 tumors with only episomal HPV (EP) had intact monomer episomes. The remaining nine EP tumors had multimer and rearranged HPV genomes. The majority (80%) HPV rearrangements and deletions disrupted the E1 and E2 genes, and EP tumors overexpressed the E6 and E7 viral oncogenes, a similar profile to tumors with HPV integration. Tumors with putative multimer HPV integrations display HPV multimers and concatemers of human and viral sequences. Our data uncovered a novel mechanism for HPV16 to cause cancer without integration through aberrant episomal replication, forming rearranged, mutated, and multimer episomes.
Keywords: Human Papillomavirus, cervical cancer, head and neck cancer, HPV integration, episomes, extrachromosomal DNA, long-read sequencing
Graphical Abstract
INTRODUCTION
Human Papillomavirus (HPV) causes over 90% of cervical cancer, resulting in at least 300,000 deaths per year, mostly in low-and-middle-income countries (LMICs) (1,2). Two high-risk types, HPV16 and HPV18, are responsible for over 70% of precancerous cervical lesions and advanced cancers (3). However, genetic variation within HPV types is associated with histological type of cervical cancer (adenocarcinoma versus squamous cell carcinoma (SCC)), integration rate, and carcinogenicity (4–6).
The HPV genome replicates inside the nucleus of the host cell as an episome, a form of extrachromosomal DNA (ecDNA). The circular, ~7900 base pair (bp) genome encodes two critical oncoproteins, E6 and E7, which inhibit the tumor suppressors TP53 and RB1 (7–9). E6 and E7 oncogene expression is controlled in part by the E2 protein (10), and E1 and E2 regulate replication of the viral genome (11). In most cancers, the viral genome integrates into the host DNA deleting all or portions of the E1 and E2 genes (12). Thus, most integration events retain a truncated portion of the HPV genome containing the viral upstream regulatory region (URR) and the E6 and E7 genes (type 1 integration), In contrast, a subset of tumors integrates with intact copies of the viral genome (type 2 integration) (13) and extrachromosomal human-HPV hybrid molecules have been proposed (14–17)
Inactivation of E1 and E2 inhibits DNA synthesis at the HPV origin of replication and releases repression of E6 and E7 expression. HPV integration can be associated with amplified viral and flanking cellular genes at the integration site (18–22) and lead to activation of flanking human genes (23). Integration can occur throughout the genome but mainly occurs in transcriptionally active regions. Recurrent sites have been found in or near specific genes (5,21,22,24–26).
Viral integration is not a part of the HPV lifecycle and does not occur in all tumors. While HPV18 and HPV45 integrate into nearly 100% of tumors, the integration rate of HPV16 is only 60–80% (5,16,25,27–30). Some episomal-only HPV-positive tumors have mutations in the URR of HPV (31,32), and HPV16 regulation is altered in cellular models of episomal infection (33). To investigate the mechanism of HPV16 carcinogenesis in the absence of integration, we applied multiple long-read DNA and RNA sequencing strategies to cell lines with and without HPV integration (Supplementary Figure S1A). Finally, we used these methods to study a well-curated set of HPV16 tumors with episomal DNA. Our results provide insight into the extrachromosomal replication of HPV and carcinogenesis.
MATERIALS AND METHODS
Patients/Informed consent
This study was conducted at the Instituto de Cancerología (INCAN) in Guatemala City and the Hospital Central Universitario, Venezuela. The Research Ethical Committees of both institutions approved the protocol, and the study was exempt from institutional review board (IRB) approval by the NIH Office of Human Studies Research. Women gave written informed consent.
Cell culture, DNA and RNA extraction
Cell lines were obtained from American Type Culture Collection (ATCC) and the Korean Cell-Line Bank and Cancer Research Center, Seoul National University (34), verified by Identifiler, checked every 6 months for mycoplasma. Cells were cultured in EMEM or RPMI-640 media with 10% Fetal Bovine Serum 1% Pen Strep (10,000 units/mL of penicillin, 10,000 μg/mL of streptomycin, and 25 μg/mL of Amphotericin B). DNA was extracted using a Gentra Puregene kit from Qiagen or Circulomics Nanobind HMW DNA kit. RNA was prepared from 30 million cells using Trizol (ThermoFisher) and Poly-A+ RNA purified by DYNAL Dynabeads (Invitrogen). Tumor DNA and RNA were simultaneously purified from 1 cm of tumor tissue using the DNeasy Blood & Tissue Kit or RNeasy Mini kit. DNA was quantitated by Nanodrop (Thermo Scientific) and Qubit (Thermo Scientific) and stored at 4°C; RNA was quantitated by Qubit and stored at −80°C.
Determination of HPV integration
For cervical tumors in the TCGA study we used the 169 tumors with detailed information (4) on integration status. For the Guatemalan tumors, 55 HPV+ tumors characterized by HPV capture and deep short-read sequencing were analyzed (27).
Long-read DNA sequencing
For ligation sequencing 1 ug of cell line DNA was sheared to 8–20 kb with a G-tube (Covaris) or used unsheared with the LSK-109 kit. Targeted sequencing of HPV16 was carried out in CaSki and SiHa cells using CRISPR probes to HPV16 probes (Supplementary Table S1). For transposase sequencing of cell lines, 1 ug was used with the Rapid Sequencing kit (SQK-RAD004, Oxford Nanopore). CaSki and SNU-1000 DNA was also prepared using the Ultra-Long protocol of Circulomics and the Ultra-Long DNA Sequencing kit (SQK-ULK001, Oxford Nanopore) with and without adaptive sampling (35,36) using a combined human HG38/high-risk HPV FASTA file selecting for cancer genes, integration loci, and HPV.
Tumor DNAs (0.3 ug) were sequenced using the Rapid Barcoding kit (SQK-RBK004, Oxford Nanopore). A total of 30–50 fM of DNA was loaded onto MinIon R9.4 flow cells (Oxford Nanopore).
Full-length RNA sequencing
Cell line RNA (500ng Poly-A+) was sequencing using the Direct RNA sequencing kit (SQK-RNA002, Oxford Nanopore) and the Direct cDNA kit (DCS109, Oxford Nanopore). Tumor RNA (50ng total RNA) was sequenced with the PCR-cDNA Barcoding kit (SQK-PCB109, Oxford Nanopore) on MinIon R9.4 flow cells. Data for all sequencing runs is displayed in Supplementary Table S2 and coverage for tumors in Supplementary Table S3.
PCR and Sanger sequencing
Primers were designed with Primer3 (37). Overlapping primers spanned the HPV16 and HPV18 genomes. Cell line DNA was amplified using a long-range PCR kit from New England BioLabs. Sanger sequencing was performed using an Applied Biosystems® 3500xL Genetic Analyzer from Thermofisher Scientific.
Cytogenetics and in situ hybridization
Spectral karyotyping (SKY) and fluorescence in situ hybridizations (FISH) (38) protocols can be accessed at https://ccr.cancer.gov/staff-directory/thomas-ried#resources. Metaphase chromosome suspensions were prepared by treating cells with a hypotonic solution (0.075M KCl), followed by fixation using methanol:acetic acid (3:1 [v/v]). Slides were prepared by dropping a small amount of suspension onto slides using a Thermotron chamber (Thermotron) to control humidity. Slides were aged for 1 week at 37°C prior to hybridization. Chromosome preparations were hybridized for 72h with in-house SKY probes and imaged using a Leica DMRXE microscope (Leica) equipped with DAPI and SKY filters (Chroma), a Xenon lamp, and a Spectracube (Applied Spectral Imaging). To perform HPV-FISH, we used the PATHO-GENE® probe HPV screening probe - ENZ-32884 (Enzo) and the tyramide signal amplification kit (Invitrogen). We also performed a non-amplified detection of HPV-FISH after we performed SKY washing off the SKY paints and rehybridized it with the PATHO-GENE® probe HPV screening probe - ENZ-32884. We then detected using our standard FISH detection protocol using BSA blocking followed by incubation with Cy™3 Streptavidin (Jackson ImmunoResearch Laboratories).
For FISH analysis, slides were evaluated using the Leica Thunder Imager microscope with a coolLED lamp and a DMC4500 digital camera (Leica). Leica Application Suite X 3.7.0.20979 software was used to acquire cell images. For each cell line, 18–20 metaphase spreads were acquired for SKY and scored for numerical and structural chromosomal aberrations according to established human chromosome nomenclature rules from ISCN (International Standing Committee on Human Cytogenetic Nomenclature 2009).
Bioinformatics and statistics
The fastq files were aligned to the human (HG38) or HPV genomes using EPI2ME (https://epi2me.nanoporetech.com) Fastq Human Alignment GRCh38 or Fastq Custom Alignment app with a high-risk HPV type fasta file. Reads were merged in Excel or in Filemaker (Claris) and merged with read length data and manually extracted and mapped using BLAT (https://genome.ucsc.edu) or BLAST.
BAM files were merged and indexed using the BamTools Merge and SAMtools Index tools in the Cancer Genomics Cloud (CGC)(39). A ONT WGS Data Processing pipeline was run on align human and HPV reads, produce BAM files.
DNA reads were also analyzed using Guppy/4.5.4 (https://nanoporetech.com/). Modified base-calling was performed using Megalodon/2.3.3 https://github.com/nanoporetech/megalodon. Structural variation calling was carried out with the Nanopore pipeline-structural-variation. The workflow is available at https://github.com/NCI-CGR/Nanopore_DNA-seq. RNA reads were base-called using BINITO /v0.3.7 and aligned to the HG38 genome using Minimap2/2.17. Isoforms were detected and quantified using Stringtie2/2.1.5 (40) and Freddie (https://www.biorxiv.org/content/10.1101/2021.01.20.427493v1.full) (https://github.com/vpc-ccg/freddie). The Freddie program calls the Gurobi package (www.gurobi.com) to solve optimization problems. The entire workflow is available at https://github.com/NCI-CGR/Nanopore_RNA-seq.
Statistical analysis was performed in GraphPad.
Data availability:
Cell line sequencing data is available at the Sequence Read Archive PRJNA772772 and cervical tumor samples at: dbGaP Study: phs002810.v1. The data analyzed in this study were obtained from TCGA at cbioportal (https://www.cbioportal.org/)and from supplementary data in (4). All other raw data are available upon request from the corresponding author.
Results
HPV type controls integration frequency
HPV types have varying frequencies of integration (5,41). To explore integration frequency by HPV type, we compiled data from The Cancer Genome Atlas (TCGA) and Guatemalan tumors merging type 1 and type 2 integrations into a single ‘integrated’ class (4,27,42). We confirmed that HPV18 and HPV45 integrate into nearly 100% of tumors, while HPV16 and HPV31 integrate into 60–70% of tumors (Figure 1A). HPV18 and 45 are in the alpha-7 clade and HPV16 and 31 the alpha-9 clade (43), indicating that viral genetics in part determines integration rate.
Figure 1. HPV multimers in cancer cell lines.
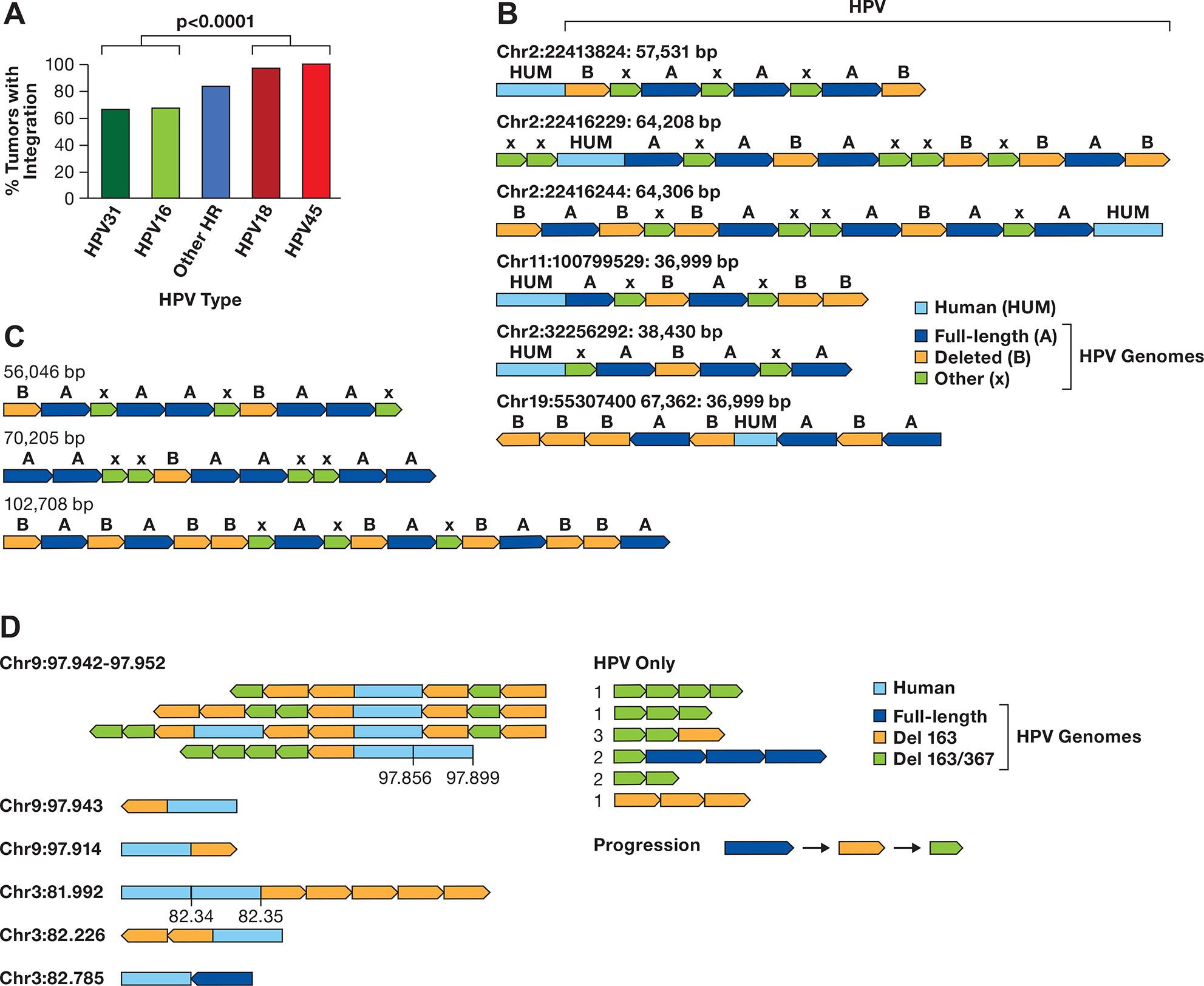
A. The integration frequency of the predominant carcinogenic HPV types is shown using combined data from TCGA and Guatemalan tumors (4,27). The other high-risk (HR) types are combined. The number of samples is HPV31 (N=3), HPV16 (N=170), Other HR (N=43) includes 13 HPV types, HPV18 (N=32), and HPV45 (N=17). The comparision of HPV16/31 to HPV16/45 is significant, P> 0.0001 byt Fishers exact test. B. A diagram of contigs of long DNA sequence reads in CaSki cells aligning to the human genome is shown. Human DNA sequences are in light blue, the full-length HPV16 genome A is dark blue, a 6.5 kb deleted genome derived from genome A is orange, and other smaller HPV16 fragments of varying size are green (X). The arrows show the direction of the HPV genome segments. The location of the human junction and size of the contig is shown. C. HPV only reads from CaSki cells, key as in panel B. D. Contigs of SCC152 HPV16 multimers are shown, with the human/HPV junction location on the left in Mb. For reads with an additional human segment, those coordinates are given above the contig. For HPV-only reads, the number of times that structure was found are shown to the left of the read diagram. A suggested progression in the evolution of the single and double deleted HPV16 genomes is shown.
Three classes of HPV16 tumors have been described: 1) those with only episomal DNA, 2) integrated only tumors (type 1), and 3) tumors with either type 2 integrations or both integrated and episomal DNA (27,44,45). However, hybrid human and HPV extrachromosomal DNA (ecDNA) has been proposed to be present in tumor previously classified as containing both episomal and integrated DNA (14,15). To better understand these different tumor classes, we applied long-range, single-molecule sequencing using methods applicable to linear and circular DNA (Supplementary Figure S1A). We first sequenced SiHa cervical cancer cells (46), known to have a single locus of integration on chromosome 13 (18,47). Using both WGS and CRISPR-targeted sequencing, we constructed a 54 kb contig containing the 7652 bp portion of the integrated HPV16 genome, confirming the rearrangement of flanking human DNA (Supplementary Figure S1B). Therefore, long-read and targeted sequencing can resolve the structure of HPV integration sites.
Long-read DNA sequencing identifies complex integrated HPV multimers
To understand the structure of complex integration events, we performed long-read DNA sequencing of the CaSki cervical cancer cell line containing 800 copies of integrated HPV16 at 30–40 chromosomal sites (18,19). We used sheared, and unsheared DNA to achieve a range of read lengths. Reads of up to 67 kb were obtained containing HPV and human sequences, and 28 out of 35 (80%) recurrent junctions matched those seen previously using short-read WGS(18) (Figure 1B, Supplementary Table S4). The HPV sequences in these reads are concatemers of:
a full-length genome (genome A).
a previously described 6.5 kb genome with a 1.4 kb deletion (genome B)(19).
HPV16 fragments of other sizes.
We obtained reads of up to 102 kb with only HPV16 sequences (Figure 1C) with concatemers of genomes A and B and smaller fragments, some of recurrent structure (Supplementary Figure S2). Reads aligning to the same human-HPV junction assembled into contigs, but there was no identifiable pattern in the order of genomes A and B at different chromosomal locations. We constructed libraries with an ultra-high molecular weight protocol, confirmed many junctions and obtained HPV-human reads of >340 kb and HPV-only reads > 240 kb (Supplementary Figure S2). Therefore, a complex duplication and assembly process likely generated the concatemers.
We also analyzed complex integrations in a head and neck squamous cell carcinoma cell line, SCC152. This line was derived from a relapsed tumor obtained one year after the SCC090 cell line was established from the same patient (48). SCC090 contains 200–500 copies of HPV16 integrated at chromosomes 2, 3, 6, and 9 (18). Our sequencing of SCC152 revealed nine integrated loci, supported by multiple long reads, on chromosomes 2, 3, and 9 (Supplementary Table S5). These integration sites were also identified in SCC090 and, therefore, retained in the relapsed tumor, with the chromosome 9 locus being the only transcribed site. Most integrated loci contained HPV16 arrays composed of 1) full-length genomes, 2) genomes containing a 163 bp deletion, and 3) genomes with both the 163 bp and a 367 bp deletion (Figure 1D).
The 367 bp deletion in SCC152 was observed over 95% of the time with the 163 bp deletion, suggesting that the 163 bp deletion appeared first and the 367 bp deletion occurred on a 163 bp deletion-containing genome. Both deletions are in the URR region, with the 163 bp deletion removing part of the intermediate enhancer region and the 367 bp deletion a portion of the distal region (see below). As with CaSki cells, we observed a random order of the different HPV16 forms integrated into SCC152 cells at distinct genomic locations, although in SCC152, there are very few full-length genomes. The data from both CaSki and SCC152 suggest that deleted forms of HPV formed as HPV ecDNA, present as viral concatemers, before integration. We term this model of episomal amplification, episomal deletion, and rearrangement, followed by integration at multiple chromosomal locations HPV superspreading.
A cell line with episomal HPV16 displays multimer and deleted episomes
To further understand the formation of multimer episomes and the mechanisms of HPV superspreading, we searched for cell lines with episomal HPV. The SNU-1000 cell line was established from a cervical squamous cell carcinoma isolated from a 43-year-old Korean patient and published as having episomal and integrated forms of HPV16 (34). Long-read WGS of SNU-1000 identified a 150 bp HPV fragment integrated on chromosome 11q in the intron of the CEP126 gene. This HPV fragment contains a portion of the E7 oncogene but cannot encode a functional E7 protein (Figure 2A). Read count analysis across chromosome 11 identified genes located 5’ to the integration site, such as the progesterone receptor (PGR) amplified 10-fold, and the YAP1, BIRC2, and BIRC3 genes, 3’ to the integration, amplified 25-fold (Figure 2A).
Figure 2. Structure of HPV integration locus in SNU-1000 cells.
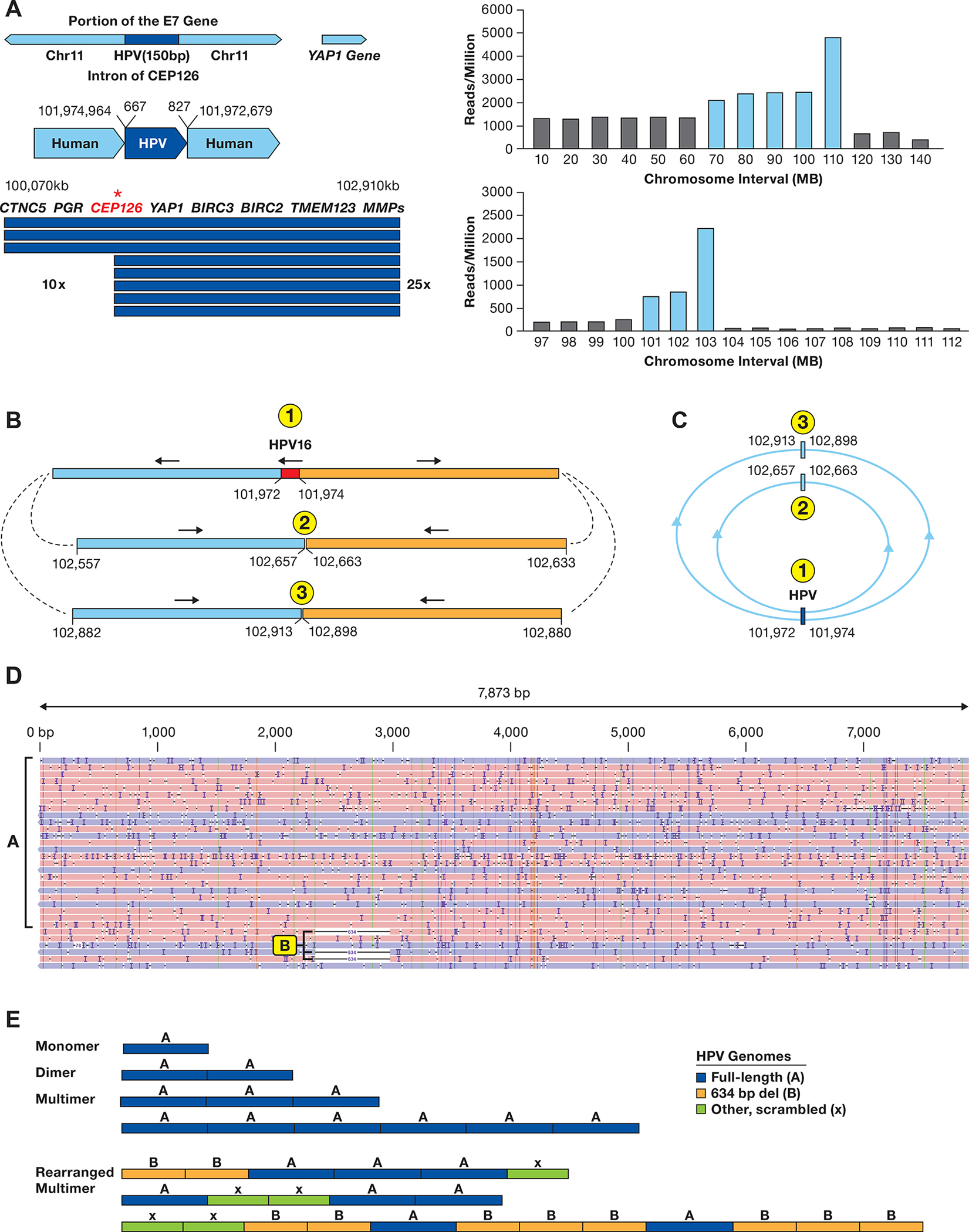
A. The integration locus on chromosome 11 in SNU-1000 is shown along with the fragment of HPV16 from 667–827 bp of the HPV16 genome. The arrow displays the site of integration in the CEP126 gene and the amplified 2.9 Mb region. Below are read counts normalized to reads/million total reads separately in 10 and 1 Mb intervals across chromosome 11 and the 97–112 Mb region. Blue bars represent regions amplified as compared to the rest of the genome. Most other chromosomes do not show such drastic alterations in read depth (Supplementary Figure S9). B. Long-read contigs containing the sequences flanking the integration site are shown along with the direction of the flanking human segments. Also shown are contigs of the two human-human junctions supported by multiple reads joining chr11:102,657 to 102,663 kb and chr11:102,913 to 102,898 kb. C. A model of circular structures that would be consistent with the read junctions, forming 1.4 and 1.9 Mb circles. D. SNU-1000 WGS reads mapped to the HPV16 genome show insertions of multimers of ~7.9 kb, as well as 634 bp deletion genomes and rearranged genomes (arrows). E. Representative SNU-1000 HPV-only reads corresponding to monomer, dimers, and multimer forms of the full-length HPV genome. In addition, multimers containing concatemers of full-length, 634 bp deleted, and other HPV fragments are displayed.
The region distal to 11q22.2 (103–140 Mb) is present at a low copy number from the WGS data, and we identified multiple long reads joining chromosome 11 at 102,657 kb to 102,663 kb and 102,898 kb to 102,913 kb (Figure 2B). Therefore, the chromosome 11 HPV16 integration is accompanied by amplification of the locus and rearrangement of the flanking human DNA. As we did not see chromosome 11q joined to another chromosome, this amplified DNA appears to lack a telomere. Figure 2C presents a model in which these sequences are in circular, extrachromosomal structures.
Sequencing of SNU-1000 DNA was performed with both standard adapter ligation onto linear DNA molecules and insertion of transposase adaptors into linear and circular molecules (Supplemental Figure 1A). In both cases, we identified large HPV16-only reads consistent with episomal DNA. We obtained transposon-tagged, HPV-only reads of 7.9 kb representing monomer episomes (Figure 2D). In addition, there were numerous reads longer than 7.9 kb containing concatemers of full-length genomes, a 634 bp deleted genome, and other truncated genomes (Figure 2E). Some reads contained only full-length genomes consisting of monomer episomes, dimers, and higher order concatemers. Others contained complex patterns of full-length and deleted forms indicating aberrant replication of episomal DNA. The 634 bp deletion removes a portion of the C terminus of the E1 gene and the N-terminus of E2. Therefore, SNU-1000 displays a full spectrum of episomal monomers, multimers, deletions, and complex concatemers as extrachromosomal DNA. Episomal deletion of the E1 and E2 genes provide a mechanism for HPV16 transformation without integration.
To delineate the karyotype and to confirm the presence of extrachromosomal HPV, we performed Spectral Karyotyping (SKY) and HPV-Fluorescence in situ Hybridization (FISH) on metaphase preparations of SNU-1000 and SNU-1245 cells. Figure 3A–B display SKY and HPV-FISH on the same SNU-1000 metaphase. HPV-FISH resulted in very intense hybridization signals outside of the chromosomes, or in some cases adjacent to or on top of chromosomes (Figure 3B, Supplementary Figure S3–S4). In some instances, the extrachromosomal HPV DNA resembled double-minute chromosomes in size and structure (Figure 3B inset). In contrast, a near-tetraploid cell line with a single copy of integrated HPV18 according to our sequencing data, SNU-1245, showed far less intense signals on a specific chromosome (chromosome 1) (Supplementary Figure S3B), in agreement with DNA sequencing data. The karyotype of SNU-1000 revealed extensive diversity between metaphases with a range of 52–76 chromosomes (median 68) exhibiting multiple translocations, and deleted chromosomes (Figure 3C, Supplementary Tables S6–S7–S8). A der(11)t(11;13)(q23;q21) chromosome was observed in all 18 metaphases examined, with an apparent loss of 11q23-ter, consistent with the copy number analysis (Figure 2A). In conclusion, SNU-1000 revealed complex karyotype aberrations and heterogeneity, a 150 bp fragment of integrated HPV16, and a large and variable number of copies of extrachromosomal HPV.
Figure 3. Spectral karyotype and HPV in situ hybridization of SNU-1000 and SNU-1245.
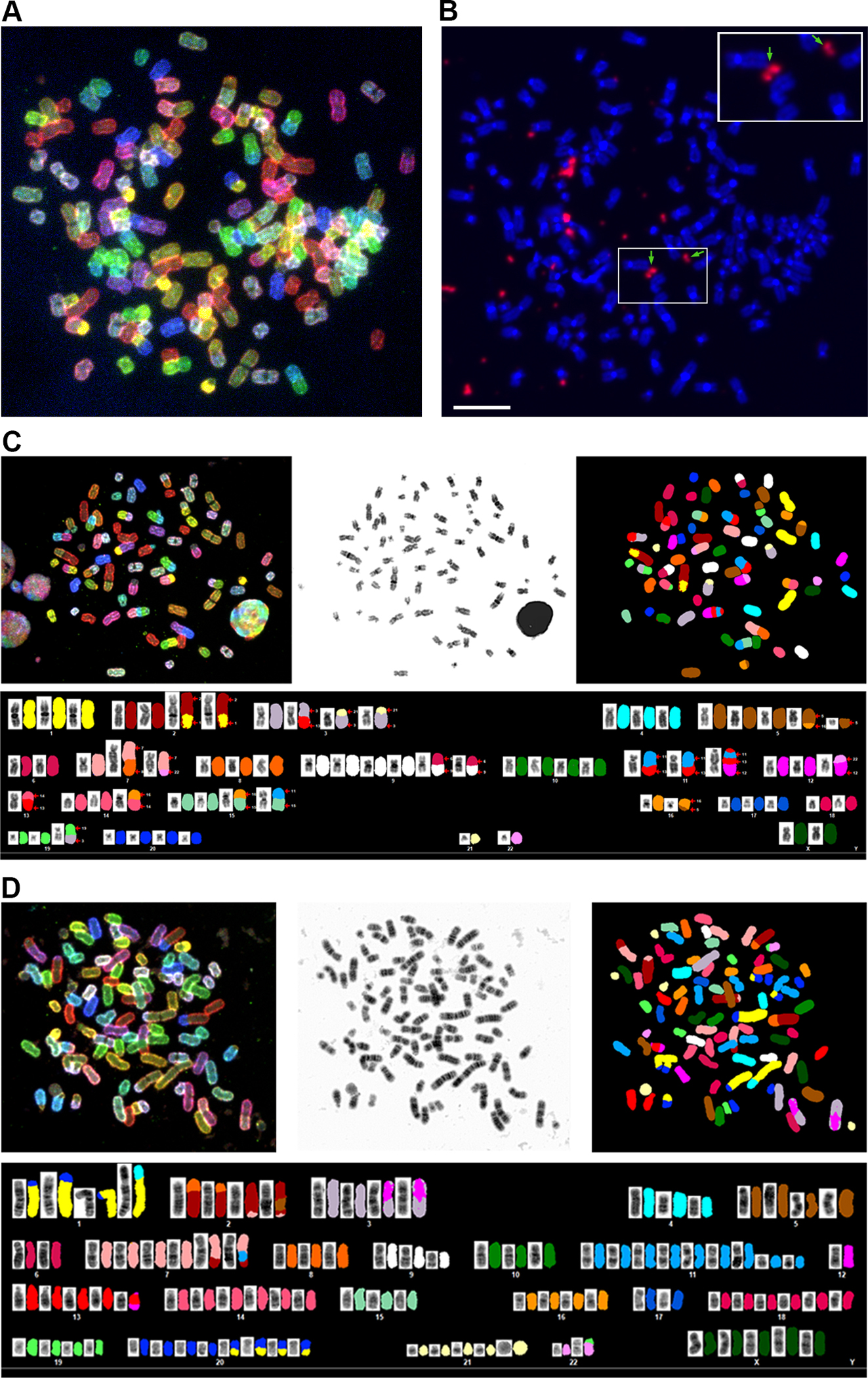
A. A representative spectral karyotype of SNU-1000 cell is shown revealing a near-triploid chromosome content. B. Fluorescent in situ hybridization (FISH) of the same metaphase with a pan-HPV probe reveals intense signals for HPV (in orange) in extrachromosomal regions between or adjacent to chromosomes. Arrows and the inset show HPV signals resembling double minute-like structures. The scale bar is 10 um. C. SKY of SNU-1000 displaying translocation of several chromosomes, including chromosome 11 in the region of the YAP1 amplification. D, SKY of SNU-1245, a near-tetraploid cell line with. Details of metaphases and additional images are in Supplementary Figures S3, S4 and Supplemental Tables 6–8).
Identification of a cell line with integrated HPV18 multimers
Due to the high rate of integration of HPV18 in cancers, we were surprised to find a cell line, SNU-1245, reported to have episomal HPV18 (34). However, in situ hybridization of HPV revealed a signal on chromosome 1 as well as an acrocentric chromosome (Supplementary Figure S3B). SKY analysis of 17 metaphases yielded a median chromosome count of 90 (78–96) with multiple chromosomes showing translocations, deletions, and insertions (Figure 3D Supplementary Table S7–S8). Long-read WGS, and targeted sequencing using computer-guided sequence selection (adaptive sampling) revealed that SNU-1245 has a single multimer integration of HPV18 on chromosome 1q32.2 (Figure 4A). Our sequencing results showed that the locus contains one full-length copy of HPV18 and three HPV18 fragments, a type 2 integration. The HPV sequence block totals 16,615 bp, and this region of chromosome 1 is amplified approximately five times (Figure 4B). In addition, the flanking segments of human DNA are rearranged, consistent with a looping amplification mechanism, as reported previously for HPV16 and HPV18 cell lines and tumors (18). Therefore, the structure of the SNU-1245 locus suggests that HPV18 can also undergo multimer amplification of episomal HPV DNA, followed by integration.
Figure 4. Structure of the SNU-1245 integration locus and HPV expression.
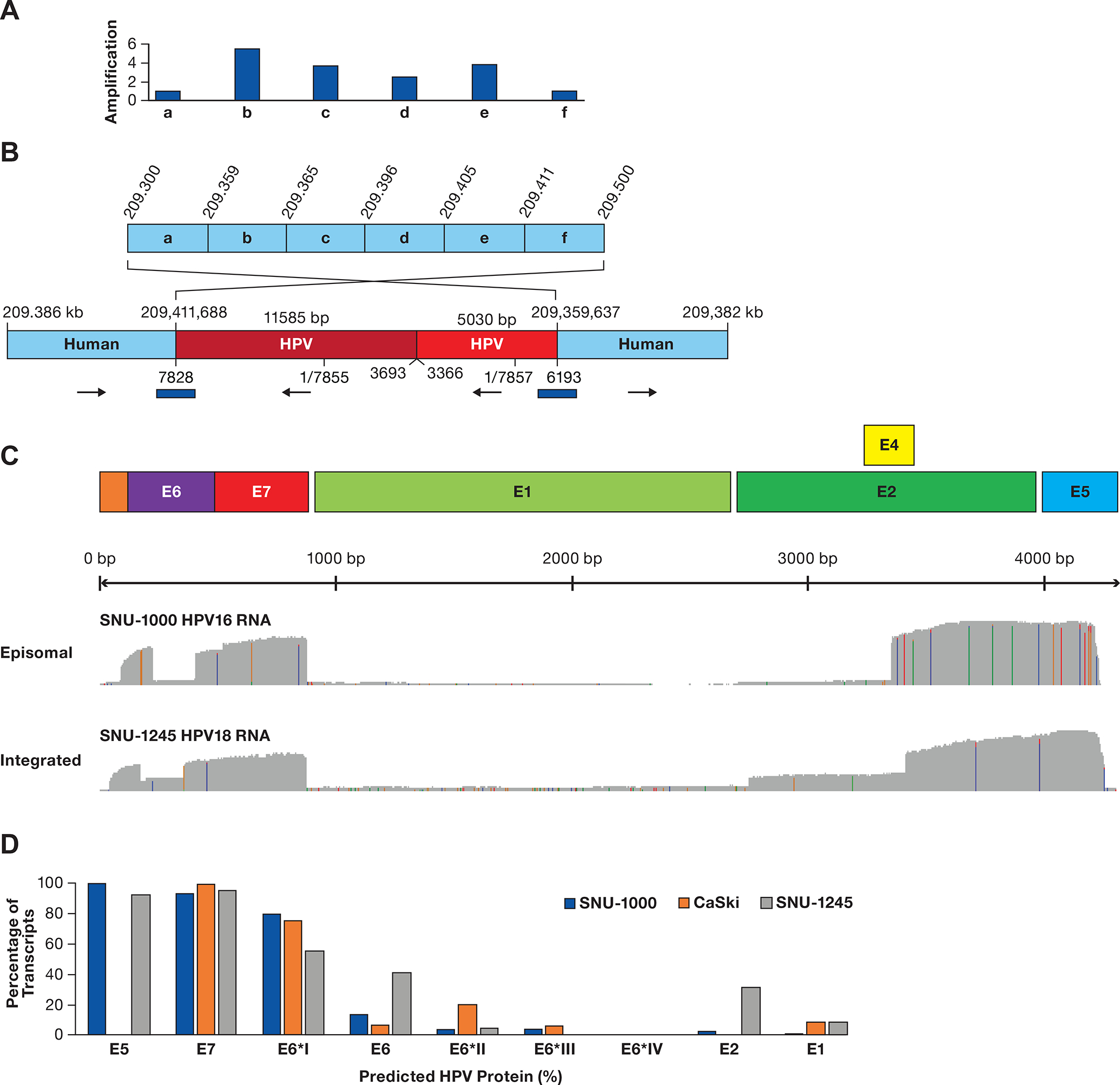
A. A diagram of a region of chromosome 1q24.2 is shown divided into six segments (a-f). Above the diagram are copy number values normalized to segments a and f. B. A contig supported by multiple long reads obtained from ONT WGS, CRISPR targeted sequencing, and adaptive sampling is shown. The two human regions are rearranged, consistent with integration at 209,411,668 bp and looping back to 209,359,637 bp. Human coordinates are above the contig and HPV18 coordinates are below. The total HPV contig is 16.6 kb. The blue bars represent junctions confirmed by PCR and Sanger sequencing. C. Plots of full-length direct cDNA sequencing are shown, displaying an abundant expression of the E6/E7 gene regions with frequent splicing of E6 and deficient expression of the E1/E2 gene region. D. The estimated level of HPV proteins is shown based on content and abundance of transcripts in Supplementary Table S9 and Supplementary Figure S5, assuming transcripts produce equal amounts of protein.
Episomal cervical cancer cells have an HPV expression profile like integrated cells
To study the expression pattern of episomal and integrated HPV, we used full-length direct cDNA and direct RNA sequencing to quantify the HPV16 transcripts in the SNU-1000 cell line and HPV18 transcripts in SNU-1245 (Figure 4C). No HPV-Human hybrid transcripts were observed in either cell line, indicating that HPV expression is within the HPV concatemers. Supplementary Table S9, Supplementary Figure S5 shows that the most abundant HPV16 transcript (transcript B) in SNU-1000 cells encodes the spliced E6*I form of E6, E7, E4, and E5 and accounts for 78% of the transcripts. There is a very low abundance of mRNAs capable of encoding E1 or E2 in SNU-1000, and the percentage of unspliced E6 transcripts in SNU-1000 is in the range of six other cell lines with integrated HPV16 (Supplementary Figure S6, Supplementary Table S10. In the SNU-1245 line, there is a nearly even balance between transcripts encoding the full-length E6 and E6 spliced forms. Furthermore, there is a higher level of transcripts capable of encoding E2 and E1 (transcripts 4, 5, and 6)(Figure 4D). Therefore, the episomal HPV16 in SNU-1000 cells displays an expression profile like that in integrated HPV16 cell lines.
Multimer Episomes are present in cervical tumors
To analyze the structure of HPV16 episomes in cervical tumors, we performed ONT tagmentation sequencing (Supplementary Figure S1A) on tumors previously classified as episomal (EP) only or episomal and integrated by HPV capture and deep, short-read sequencing (27). In that assay, tumors were classified as EP if they had deep HPV sequence coverage with no gap in HPV coverage and no recurrent human-HPV junction. We have reclassified the episomal and integrated tumors as putative type 2 integrated to reflect current understanding (14). The age range, PIK3CA mutation status, histology, HPV type, and HPV16 sublineage are shown in Figure 5A. From 28 HPV16 EP tumors, we obtained total coverage of at least one HPV16-genome to be able to examine the structure of the 20 episomal tumor genomes. None of these tumors had HPV/human junction reads, or Human-HPV hybrid transcripts consistent with their status as EP. For eight of these tumors, the longest reads began and ended at nearly the same position on the 7906 bp viral genome and had no insertions, deletions, or rearrangements (Supplementary Table S11). This result is consistent with these molecules representing circular episomes, tagged in random positions by the transposon (Figure 5B). These tumors and those with multiple reads all less than 7906 bp were tentatively classified as monomer-only tumors. Therefore, approximately one-half of EP tumors appear to retain a monomer episome.
Figure 5. Diagram displaying data from 62 tumors.
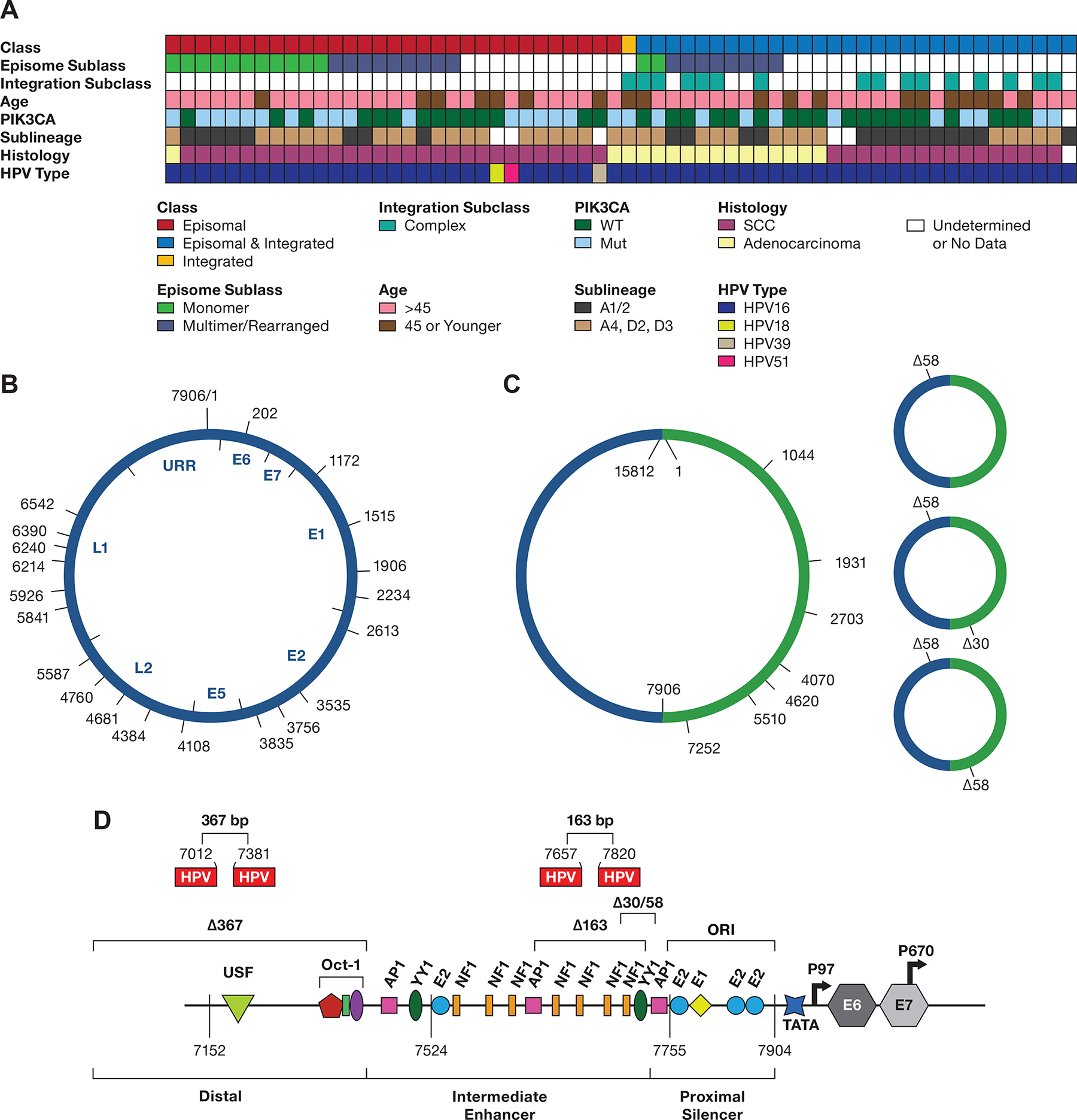
A. The integration class and subclass, episomal subclass, age range, PIK3CA mutation status, HPV type and HPV16 sublineage, and histology are shown. SCC, squamous cell carcinoma; WT, wild type; Int, integration. Blanks represent samples not able to be classified. B. Diagram of the 7906 bp HPV16 genome DNA, displaying the start site of DNA sequence reads from 13 tumors containing a complete or nearly complete monomer genome sequence. The position of the HPV16 genes and URR are shown inside the diagram. C, A diagram of HPV16 dimers from three tumors showing the start position of reads with complete or nearly complete copies of two tandem HPV16 genomes (Supplemental Table 10). D, the location of 30 and 58 bp deletions in the URR occurring in dimer reads from tumor T393.The location of the 163 and 367 bp deletions in the SCC152 cell line is shown, as well as their locations in the HPV16 upstream regulatory region. The 367 bp deletion removes most of the distal region, and the 163 bp deletion removes four NF1 binding sites and two YY1 binding sites (only one is shown) in the Intermediate Enhancer region. Shown are the binding sites for the viral E1 and E2 proteins and the transcription factors OCT1, AP1, YY1, and NF1. ORI, the origin of replication; TATA, TATA-binding site; P97, major promoter; P670, minor promoter. The locations of the coding region of the E6 and E7 genes are indicated
A total of nine additional EP tumors had HPV16 DNA reads larger than 7.9 kb, representing putative multimer episomes, or had rearranged HPV-HPV junctions. Three of these tumors had sequences of 15.8 kb with two complete copies of the HPV16 genome, likely representing HPV dimers (Figure 5C, Supplementary Table S12). As with the monomers, these reads start and stop at nearly the same position on the genome. One tumor, T393, had dimers with both 30 and 58 bp deletions in the URR (Figure 1D). In addition, dimer reads with one or two copies of Δ58 or one copy of Δ30 and Δ58 were observed (Figure 5D). The Δ30 and Δ58 deletions are in overlapping regions of the URR and delete an NF1 binding site and two YY1 binding sites (Figure 1D). These two YY1 binding sites have previously been found deleted in HPV16 isolates from cervical cancer, leading to elevated activity of the P97 promoter (31). Therefore, as seen in the SNU-1000 cell line, deletions affecting key regulatory sites can propagate in episomes replicating in cervical tumors and give rise to multiple aberrant structures, contributing to transformation without integration.
Six other EP tumors had episomal DNA with rearrangement of HPV16 sequences (Figure 6A). Interestingly, 7/8 of these tumors have a breakpoint inside the E1 (914–2666 bp) or E2 genes (2756–3853). These rearrangements could separate the E1 and E2 open reading frames from the P97 promoter and lead to decreased expression of E1 and E2 or disrupt their reading frame. Most of these HPV16 rearrangement junctions were observed in our previously published HPV capture and Ion Torrent sequencing (27), performed on the same DNA samples (Supplementary Figure S7). This data clearly shows that rearrangement of episomal DNA, with frequent inactivation of the E1 and E2 genes, is a common feature of HPV16-driven cervical tumors. In addition, transcriptome analysis of representative EP-only tumors revealed that E6/E7 transcripts are predominant and that nearly all splice inside the E6 gene (Supplementary Figure S8A). Interestingly, integrated tumors had a significantly (P=0.049) higher mean HPV RNA expression than episomal only tumors (Supplementary Figure S8B).
Figure 6. Structure of HPV sequences in Episomal and Integrated tumors.
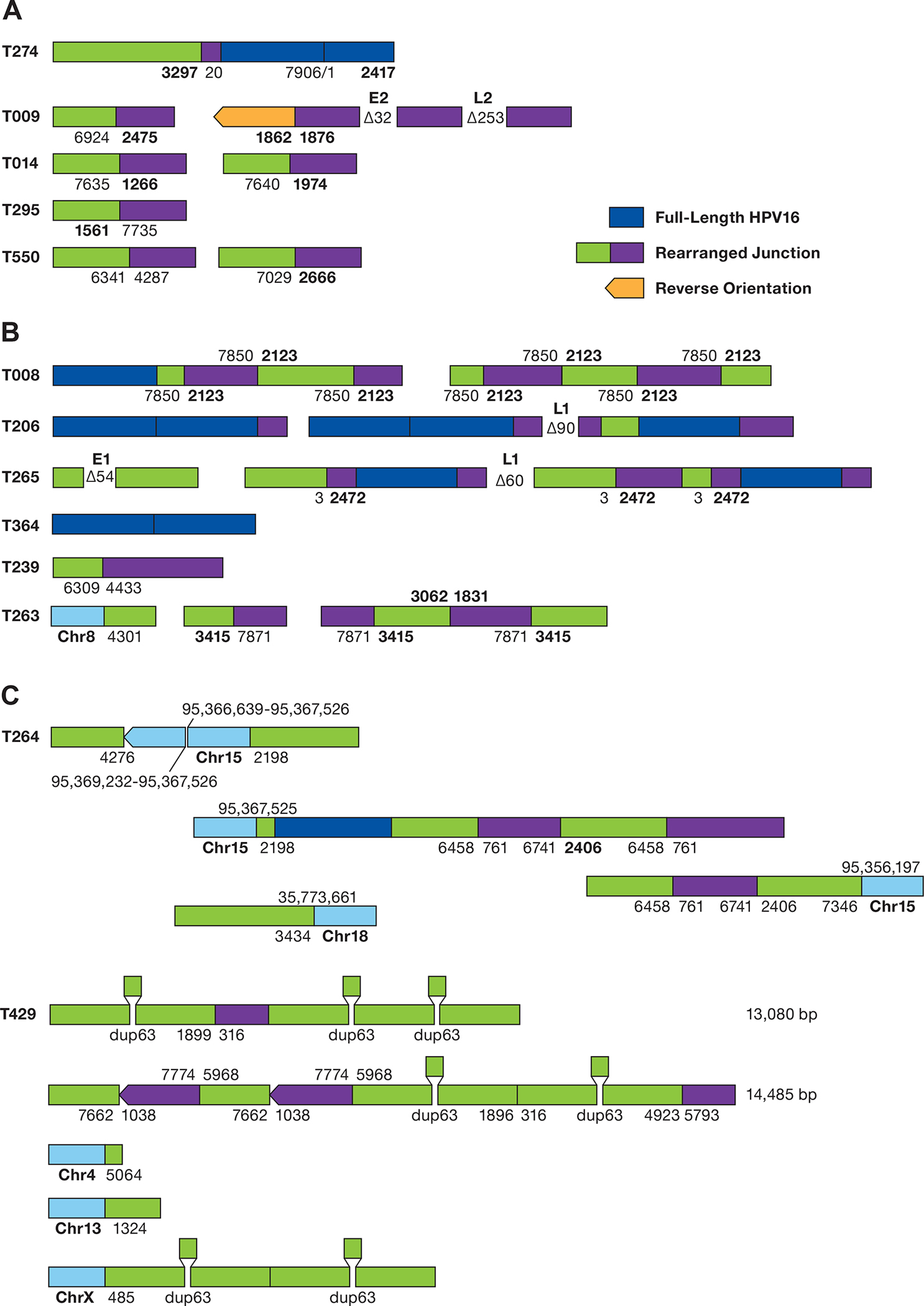
A. The structure of rearranged HPV-only reads in EP tumors is shown. The numbers represent the position of the HPV16 genome at the site of the breakpoint. Deletions are displayed between segments. Unless shown, all HPV sequences are arranged in the same orientation. B. Structure of HPV reads in putative type 2 tumors. Regions of human DNA are shown in light blue. C. The structure of HPV reads in tumor T429 is shown. HPV16 sequence breakpoints are shown joining chromosomes 4, 13, and X to HPV sequences. The position of a 63 bp duplication is shown. The top 2 diagrams represent independent sequence reads of 13,085 and 14,485 bp.
Long-read WGS of putative type 2 integrated tumors gave a more complex picture. Despite having only 0.5x genome coverage, we identified the same HPV-human breakpoint in 13/27 tumors found by HPV capture and short-read sequencing (27). Nearly all tumors for which the integration was not confirmed by ONT sequencing yielded less than 10 HPV-containing reads. At least eight type 2 tumors have complex rearrangements of HPV-only sequences (Figure 6B). Several tumors have 50–100 bp deletions internal to the HPV sequence or have multiple rearranged junctions. As with EP tumors, the majority (3/5) of HPV junctions are within the 865–3875 region of the HPV16 genome containing the E1 and E2 genes.
Tumor 429, a type 2 integrated tumor with integration on multiple chromosomes, contains a 63 bp duplication in the E1 gene that is a known viral variant (49). The deletion was seen twice each in single HPV-only molecules of 13 and 14 kb (Figure 6C). In addition, the dup63 mutation was seen twice in a read anchored to chromosome X. Therefore, this tumor appears to have either both episomal and integrated HPV or large HPV multimers.
DISCUSSION
HPVs are among the most oncogenic human cancer viruses, and HPV16 is the most carcinogenic type of HPV (1,6,50). While integration of viral DNA into the human genome is an important mechanism of HPV carcinogenesis, transformation without HPV integration is less well understood. Our data employing long-read and single-molecule sequencing of cell lines and cervical tumors reveal new aspects of HPV oncogenesis. By carrying out long-read WGS on well-characterized cell lines (SiHa, CaSki, SCC152), we established that the methods reliably identify HPV sequences and the complex structure of integrated loci. Using HPV-containing reads of up to 347 kb, we show that all of the integrated loci in the CaSki cell line are composed of complex strings of full-length genomes, a recurrent 6.4 kb truncated genome and other smaller fragments. The finding of concatemers of similar structure and complexity integrated into multiple regions of the genome supports a model where the mixed concatemers of HPV genomes were generated as extrachromosomal DNA and subsequently inserted into the human genome. This model is further supported by the head and neck cancer (HNSCC) cell line, SCC152, which has unique Δ163 bp and Δ163/Δ367 bp HPV16 deletions. In addition, SCC152 has complex concatemers integrated at chromosomes 3 and 9. HPV FISH data for CaSki and SCC-090, a pre-cursor to SCC152, demonstrating that all HPV DNA is integrated (18). To explain the presence of HPV concatemers with unique deleted genomes on multiple chromosomes, we propose an HPV Superspreading model.
To support the model, we analyzed a cell line, SNU-1000, that stably replicates HPV16 episomal DNA (34). SNU-1000 contains only a 150 bp fragment of the HPV16 E7 gene integrated on chromosome 11q22.1, and is truly a cell line with episomal and integrated HPV16. Interestingly, this integration is within a 2.9 Mb amplified region containing the YAP1, BIRC2, and BIRC3 genes. To our knowledge, this is the first time an oncogene amplification has been found at a locus with an incomplete copy of the HPV oncogenes, a new mechanism of HPV carcinogenesis.
Except for the chromosome 11q22.1 integration site, there were no other recurrent human-HPV junction reads in SNU-1000. All other HPV16 sequences in SNU-1000 are HPV-only reads. We used HPV FISH to demonstrate that the HPV concatemers are extrachromosomal and in some cases form structures that resemble double-minute chromsomes.
Using a transposon-based approach that can directly sequence linear and circular DNA, we recovered multiple reads from SNU-1000 that begin and end at nearly the same position on the HPV16 genome. These reads are almost certainly derived from intact monomer episomes. We also identified reads that represent intact dimers and multimers of full-length HPV16 genomes. Therefore, replication of the HPV16 monomer can lead to intact multimer genomes. In SNU-1000 we identified a 634 bp deletion, removing portions of the E1 and E2 genespresent in 15% of viral genomes exclusively in large multimer episomal structures. These multimer episomes are composed of concatemers of full-length HPV16 genomes, Δ634, and rearranged genomes.
Analysis of direct, full-length cDNA and RNA from SNU-1000 demonstrates that the predominant HPV transcript encodes the E6*I, E7, E4, and E5 proteins and low amounts of E1 and E2. Thus, in the absence of integration, rearranged and deleted HPV16 multimers appear to have resulted in the upregulation of E6 and E7 and downregulation of E1 and E2, like tumors with integrated HPV. Therefore, one mechanism of episomal HPV carcinogenesis is the episomal deletion of E1 or E2 (Figure 6).
To understand the mechanisms of carcinogenesis in tumors without HPV16 integration, we studied tumors previously characterized as episomal-only. Nearly one-half of these tumors have intact monomer episomes. Therefore, HPV16 is capable of causing cancer without integration or rearrangement of episomal DNA. Monomer-only tumors were mostly SCC and included HPV16 A1, D2, and D3 sublineages. RNAseq data shows a transcription pattern dominated by the expression of a restricted set of mRNAs containing the E6 and E7 genes. Therefore, even though the entire HPV genome is present in the monomer-only EP tumors they display a viral mRNA expression pattern similar to integrated HPV16 tumors.
We identified a subset of episomal-only tumors with rearranged and deleted episomal HPV and 80% of these deletions and rearrangements have a breakpoint in the E1/E2 genes. Therefore, as multimer episomes expand, they may gain more replication origins, increasing the rate of expansion and deletion, and more carcinogenic episomes are selected. Therefore, HPV16 can cause cervical cancer without integration via a combination of episome deletion, mutation, and rearrangement and favoring E6/E7 oncogene expression.
Integrated forms of HPV nearly always retain the URR, and the origin of replication (ORI). As the E1 and E2 proteins promote replication at the ORI the presence of both episomal and integrated HPV presents a situation where E1 and E2 could promote unscheduled replication at the ORI in integrated HPV. Orav et al. and Peter et al. observed DNA synthesis at integrated ORIs in transfected cells and proposed that this ‘re-replication’ leads to local amplification of HPV and human DNA, the formation of viral/human DNA-containing ecDNA, and chromosome translocations (22,51). Akagi et al. used WGS to characterize HPV integration sites with local amplification and proposed a looping model causing the amplification of viral and flanking human DNA (18). Finally, Kim et al. demonstrated that ecDNA is frequent in solid tumors and 12% of cervical tumors (52). We reanalyzed this data and found that 38% of integrated HPV16 cervical tumors are predicted to have ecDNA (Supplementary Table S13). Several papers have proposed the presence of ecDNA, containing human and HPV sequences, in HPV+ HNSCC tumors (15,53,54). However, our sequencing data does not directly address these post-integration events. We do show that rearrangement and amplification of HPV can occur before integration. Deleting E1 and E2 in episomal DNA, and epigenetic silencing, may permit an integration locus with an ORI to remain stable in the presence of episomal DNA.
There are several methods to identify the presence of HPV integration, either using DNA or RNA-based analyses. We used the presence or absence of an HPV/human DNA junction following HPV capture and deep short-read sequencing to determine integration status. We classified tumors without a detected integration and an intact viral genome as episomal-only. We used overlapping amplicons spanning the HPV genome to detect deletions and classify tumors with complete deletions in the E1/E2 region as type 1 integrated. The classification of tumors as episomal and integrated is more problematic. Tumor cell lines such as CaSki and SCC152 can have full-length copies of HPV16 integrated into the genome (type 2 integration) and therefore could be confused as EP/INT. SNU-1245 was classified as EP/INT based on an intact E2 gene; however, it contains a full-length copy of HPV18 integrated into the genome and no detectable episomal DNA. Morgan et al. argue that, at least for HNSCC, the EP/INT category is mischaracterized and many of these tumors have viral/human ecDNA (14). Our data show that putative type 2 tumors can have apparent monomer, multimer, or rearranged HPV-only sequences, however, we did not obtain deep enough coverage to fully classify these tumors and without very deep, long-read sequencing, it is not possible to determine if these exact state of these sequences.
This study has several limitations, including that some of the analyses involve a small set of cell lines and only one cell line has episomal HPV16. But, there have been few high-risk HPV-containing cell lines with episomal DNA described. SNU-1000 may be unique in having amplified YAP1, a potent cervical cancer oncogene (55).and YAP1 overexpression might aid in the generation of additional episomal HPV models. Due to limitations in DNA quantity and quality, we obtained relatively low coverage WGS of tumors, and did not select for circular DNA. Therefore, we have not detected all HPV-containing species, and our tumor classifications are incomplete. However, to our knowledge, this is the first study to apply long-read sequencing to the study of episomal HPV-containing cervical tumors.
In conclusion, we find that multimers of the HPV genome are generated in cervical tumors replicating as extrachromosomal episomes. This HPV replication is associated with deletion and rearrangement of the HPV genome and provides a mechanism for oncogenesis without integration. We provide confirmation by DNA and RNA sequencing that a subset of HPV16-containing tumors has only episomal viral DNA. About half of episomal tumors have intact monomer episomes and an expression pattern dominated by E6/ E7 expression. Another subset of episomal tumors has rearranged episomes, often deleting the E1 and E2 genes. Our data support a model of HPV replicating as ecDNA, accumulating rearrangements leading to the integration of rearranged HPV multimers in the human genome (Figure 7). This process parallels the well-described amplification of oncogenes and drug resistance genes as ecDNA or double minute chromosomes that integrate as homogenously stained regions (HSRs) (56). And we observed HPV16 forming double minute-like structures. Further study of HPV extrachromosomal amplification and integration may provide insight into gene amplification and ecDNA formation across cancer types.
Figure 7. Mechanisms of HPV carcinogenesis.
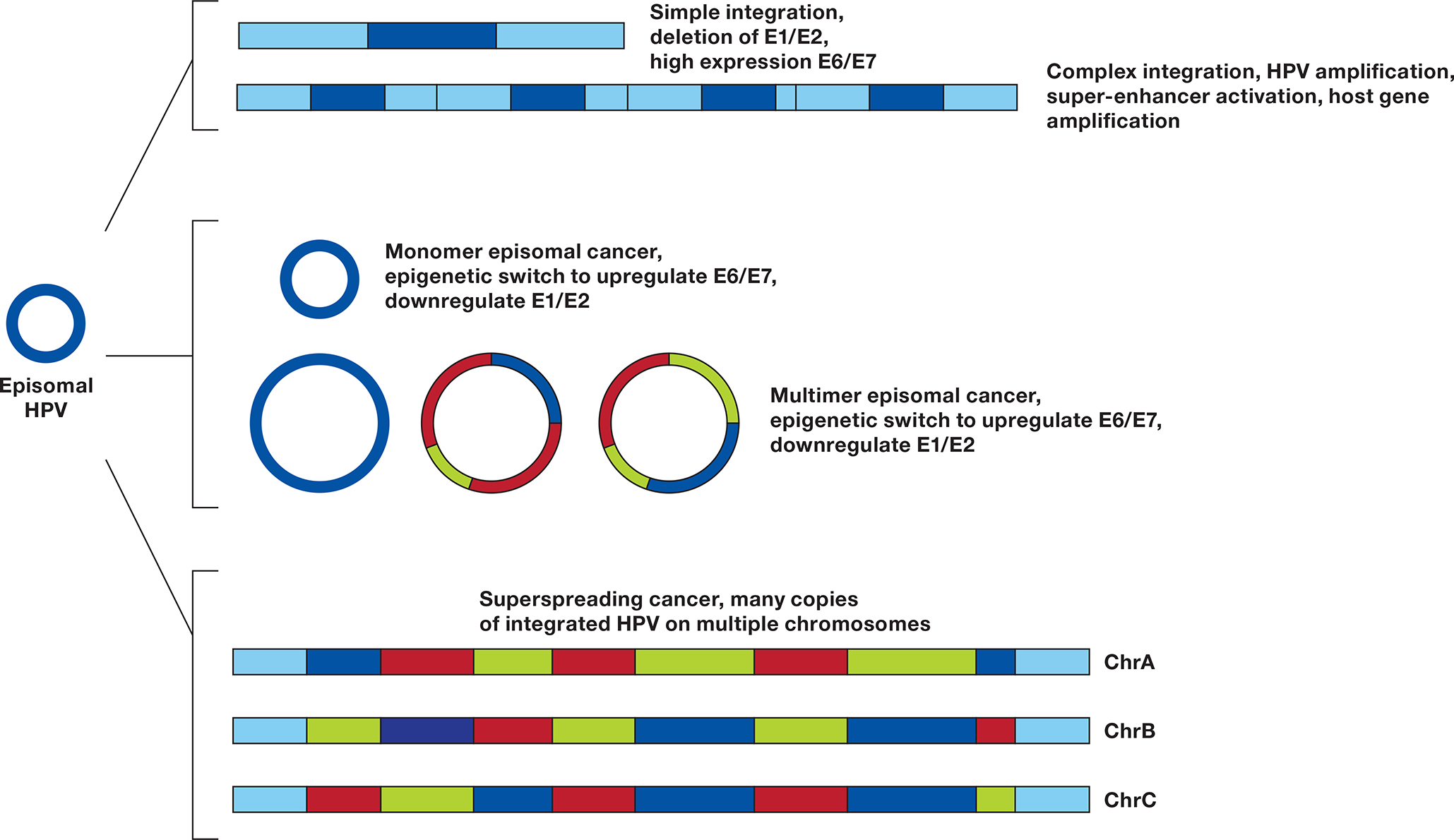
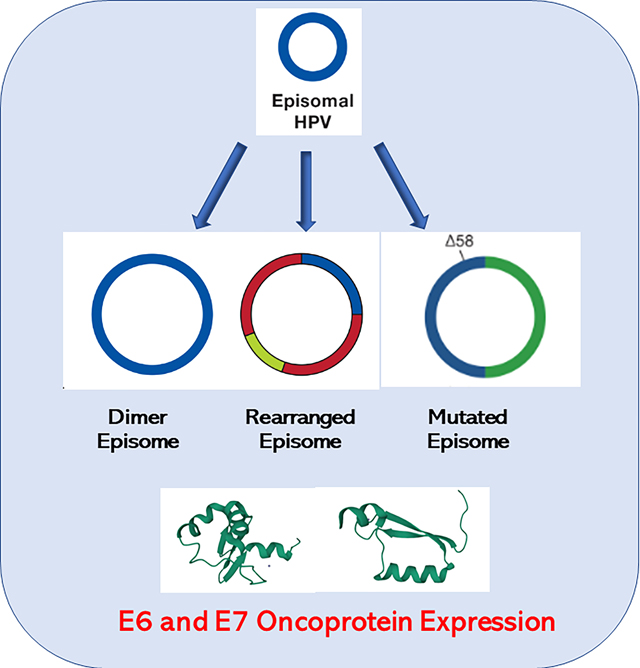
An HPV episome can undergo a simple integration, deleting the E1 and E2 genes, inhibiting DNA synthesis at the HPV origin of replication (ORI) and upregulating E6 and E7. If during integration E1 and E2 are not suppressed, a local amplification can case a complex integration leading to multiple copies of HPV and flanking DNA, resulting in higher expression of E6 and E7, formation of a super-enhancer activating flanking genes (23), and potentially amplifying a carcinogenic host gene. Monomer episomal cancer occurs when the genome remains an episome but undergoes an epigenetic switch to upregulate E6/E7. Aberrant episome replication can lead to Multimer episomal cancer in which deletions and rearrangements in the E1/E2 genes or URR lead to high E6/E7 and low E1/E2 expression. Insertion of multimer episomes into multiple genomic locations give rise to HPV superspreading cancer.
Supplementary Material
Statement of Significance.
Multimers of the HPV genome are generated in cervical tumors replicating as extrachromosomal episomes, which is associated with deletion and rearrangement of the HPV genome and provides a mechanism for oncogenesis without integration.
Acknowledgements
Thanks to Lineth Boror, Ester Avila, and Patricia Zaid for sample collection and Thomas Ried, Monolina Binny, and Dave Roberson for helpful discussions. The authors acknowledge the research contributions of the Cancer Genomics Research Laboratory for their expertise, execution, and support of this research in the areas of project planning, wet laboratory processing of specimens, and bioinformatics analysis of generated data. This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under NCI Contract No. 75N910D00024. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. We are grateful for the use of the NIH Helix Biowulf computing facility. The Seven Bridges Cancer Research Data Commons Cloud Resource has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, Contract No. HHSN261201400008C and ID/IQ Agreement No. 17X146 under Contract No. HHSN261201500003I and 75N91019D00024.
Footnotes
Conflicts of interest
The authors declare no potential conflicts of interest.
Declaration of interests
“The authors declare no competing interests.”
References
- 1.Schiffman M, Doorbar J, Wentzensen N, de Sanjose S, Fakhry C, Monk BJ, et al. Carcinogenic human papillomavirus infection. Nat Rev Dis Primers 2016;2:16086. [DOI] [PubMed] [Google Scholar]
- 2.Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol 1999;189:12–9 [DOI] [PubMed] [Google Scholar]
- 3.Schiffman M, Castle PE. The promise of global cervical-cancer prevention. N Engl J Med 2005;353:2101–4 [DOI] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research N, Albert Einstein College of M, Analytical Biological S, Barretos Cancer H, Baylor College of M, Beckman Research Institute of City of H, et al. Integrated genomic and molecular characterization of cervical cancer. Nature 2017;543:378–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bodelon C, Untereiner ME, Machiela MJ, Vinokurova S, Wentzensen N. Genomic characterization of viral integration sites in HPV-related cancers. International Journal of Cancer 2016;139:2001–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mirabello L, Yeager M, Cullen M, Boland JF, Chen Z, Wentzensen N, et al. HPV16 Sublineage Associations With Histology-Specific Cancer Risk Using HPV Whole-Genome Sequences in 3200 Women. J Natl Cancer Inst 2016;108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990;63:1129–36 [DOI] [PubMed] [Google Scholar]
- 8.Werness BA, Levine AJ, Howley PM. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 1990;248:76–9 [DOI] [PubMed] [Google Scholar]
- 9.Foster SA, Galloway DA. Human papillomavirus type 16 E7 alleviates a proliferation block in early passage human mammary epithelial cells. Oncogene 1996;12:1773–9 [PubMed] [Google Scholar]
- 10.Bellanger S, Tan CL, Xue YZ, Teissier S, Thierry F. Tumor suppressor or oncogene? A critical role of the human papillomavirus (HPV) E2 protein in cervical cancer progression. Am J Cancer Res 2011;1:373–89 [PMC free article] [PubMed] [Google Scholar]
- 11.Dowhanick JJ, McBride AA, Howley PM. Suppression of cellular proliferation by the papillomavirus E2 protein. J Virol 1995;69:7791–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwarz E, Freese UK, Gissmann L, Mayer W, Roggenbuck B, Stremlau A, et al. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature 1985;314:111–4 [DOI] [PubMed] [Google Scholar]
- 13.Jeon S, Allen-Hoffmann BL, Lambert PF. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J Virol 1995;69:2989–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morgan IM, DiNardo LJ, Windle B. Integration of Human Papillomavirus Genomes in Head and Neck Cancer: Is It Time to Consider a Paradigm Shift? Viruses 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou L, Qiu Q, Zhou Q, Li J, Yu M, Li K, et al. Long-read sequencing unveils high-resolution HPV integration and its oncogenic progression in cervical cancer. Nat Commun 2022;13:2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Labarge B, Hennessy M, Zhang L, Goldrich D, Chartrand S, Purnell C, et al. Human Papillomavirus Integration Strictly Correlates with Global Genome Instability in Head and Neck Cancer. Mol Cancer Res 2022;20:1420–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pang J, Nguyen N, Luebeck J, Ball L, Finegersh A, Ren S, et al. Extrachromosomal DNA in HPV-Mediated Oropharyngeal Cancer Drives Diverse Oncogene Transcription. Clin Cancer Res 2021;27:6772–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akagi K, Li J, Broutian TR, Padilla-Nash H, Xiao W, Jiang B, et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res 2014;24:185–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baker CC, Phelps WC, Lindgren V, Braun MJ, Gonda MA, Howley PM. Structural and transcriptional analysis of human papillomavirus type 16 sequences in cervical carcinoma cell lines. J Virol 1987;61:962–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wagatsuma M, Hashimoto K, Matsukura T. Analysis of integrated human papillomavirus type 16 DNA in cervical cancers: amplification of viral sequences together with cellular flanking sequences. J Virol 1990;64:813–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parfenov M, Pedamallu CS, Gehlenborg N, Freeman SS, Danilova L, Bristow CA, et al. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc Natl Acad Sci U S A 2014;111:15544–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peter M, Stransky N, Couturier J, Hupe P, Barillot E, de Cremoux P, et al. Frequent genomic structural alterations at HPV insertion sites in cervical carcinoma. J Pathol 2010;221:320–30 [DOI] [PubMed] [Google Scholar]
- 23.Warburton A, Redmond CJ, Dooley KE, Fu H, Gillison ML, Akagi K, et al. HPV integration hijacks and multimerizes a cellular enhancer to generate a viral-cellular super-enhancer that drives high viral oncogene expression. PLOS Genetics 2018;14:e1007179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Couturier J, Sastre-Garau X, Schneider-Maunoury S, Labib A, Orth G. Integration of papillomavirus DNA near myc genes in genital carcinomas and its consequences for proto-oncogene expression. J Virol 1991;65:4534–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu Z, Zhu D, Wang W, Li W, Jia W, Zeng X, et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat Genet 2015;47:158–63 [DOI] [PubMed] [Google Scholar]
- 26.Tang D, Li B, Xu T, Hu R, Tan D, Song X, et al. VISDB: a manually curated database of viral integration sites in the human genome. Nucleic Acids Res 2020;48:D633–D41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lou H, Boland JF, Torres-Gonzalez E, Albanez A, Zhou W, Steinberg MK, et al. The D2 and D3 Sublineages of Human Papilloma Virus 16-Positive Cervical Cancer in Guatemala Differ in Integration Rate and Age of Diagnosis. Cancer Res 2020;80:3803–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dutta S, Chakraborty C, Dutta AK, Mandal RK, Roychoudhury S, Basu P, et al. Physical and methylation status of human papillomavirus 16 in asymptomatic cervical infections changes with malignant transformation. J Clin Pathol 2015;68:206–11 [DOI] [PubMed] [Google Scholar]
- 29.Vinokurova S, Wentzensen N, Kraus I, Klaes R, Driesch C, Melsheimer P, et al. Type-dependent integration frequency of human papillomavirus genomes in cervical lesions. Cancer Res 2008;68:307–13 [DOI] [PubMed] [Google Scholar]
- 30.Hafner N, Driesch C, Gajda M, Jansen L, Kirchmayr R, Runnebaum IB, et al. Integration of the HPV16 genome does not invariably result in high levels of viral oncogene transcripts. Oncogene 2008;27:1610–7 [DOI] [PubMed] [Google Scholar]
- 31.May M, Dong XP, Beyer-Finkler E, Stubenrauch F, Fuchs PG, Pfister H. The E6/E7 promoter of extrachromosomal HPV16 DNA in cervical cancers escapes from cellular repression by mutation of target sequences for YY1. EMBO J 1994;13:1460–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lace MJ, Isacson C, Anson JR, Lorincz AT, Wilczynski SP, Haugen TH, et al. Upstream regulatory region alterations found in human papillomavirus type 16 (HPV-16) isolates from cervical carcinomas increase transcription, ori function, and HPV immortalization capacity in culture. J Virol 2009;83:7457–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gray E, Pett MR, Ward D, Winder DM, Stanley MA, Roberts I, et al. In vitro progression of human papillomavirus 16 episome-associated cervical neoplasia displays fundamental similarities to integrant-associated carcinogenesis. Cancer Res 2010;70:4081–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ku JL, Kim WH, Park HS, Kang SB, Park JG. Establishment and characterization of 12 uterine cervical-carcinoma cell lines: common sequence variation in the E7 gene of HPV-16-positive cell lines. Int J Cancer 1997;72:313–20 [DOI] [PubMed] [Google Scholar]
- 35.Loose M, Malla S, Stout M. Real-time selective sequencing using nanopore technology. Nat Methods 2016;13:751–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kovaka S, Fan Y, Ni B, Timp W, Schatz MC. Targeted nanopore sequencing by real-time mapping of raw electrical signal with UNCALLED. Nat Biotechnol 2021;39:431–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, et al. Primer3--new capabilities and interfaces. Nucleic Acids Res 2012;40:e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schrock E, Veldman T, Padilla-Nash H, Ning Y, Spurbeck J, Jalal S, et al. Spectral karyotyping refines cytogenetic diagnostics of constitutional chromosomal abnormalities. Hum Genet 1997;101:255–62 [DOI] [PubMed] [Google Scholar]
- 39.Lau JW, Lehnert E, Sethi A, Malhotra R, Kaushik G, Onder Z, et al. The Cancer Genomics Cloud: Collaborative, Reproducible, and Democratized-A New Paradigm in Large-Scale Computational Research. Cancer Res 2017;77:e3–e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kovaka S, Zimin AV, Pertea GM, Razaghi R, Salzberg SL, Pertea M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol 2019;20:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wentzensen N, Vinokurova S, von Knebel Doeberitz M. Systematic review of genomic integration sites of human papillomavirus genomes in epithelial dysplasia and invasive cancer of the female lower genital tract. Cancer Res 2004;64:3878–84 [DOI] [PubMed] [Google Scholar]
- 42.Lou H, Villagran G, Boland JF, Im KM, Polo S, Zhou W, et al. Genome Analysis of Latin American Cervical Cancer: Frequent Activation of the PIK3CA Pathway. Clin Cancer Res 2015;21:5360–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burk RD, Harari A, Chen Z. Human papillomavirus genome variants. Virology 2013;445:232–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu B, Chotewutmontri S, Wolf S, Klos U, Schmitz M, Durst M, et al. Multiplex Identification of Human Papillomavirus 16 DNA Integration Sites in Cervical Carcinomas. PLoS One 2013;8:e66693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheung JL, Cheung TH, Yu MY, Chan PK. Virological characteristics of cervical cancers carrying pure episomal form of HPV16 genome. Gynecol Oncol 2013;131:374–9 [DOI] [PubMed] [Google Scholar]
- 46.Friedl F, Kimura I, Osato T, Ito Y. Studies on a new human cell line (SiHa) derived from carcinoma of uterus. I. Its establishment and morphology. Proc Soc Exp Biol Med 1970;135:543–5 [DOI] [PubMed] [Google Scholar]
- 47.Kalu NN, Mazumdar T, Peng S, Shen L, Sambandam V, Rao X, et al. Genomic characterization of human papillomavirus-positive and -negative human squamous cell cancer cell lines. Oncotarget 2017;8:86369–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.White JS, Weissfeld JL, Ragin CC, Rossie KM, Martin CL, Shuster M, et al. The influence of clinical and demographic risk factors on the establishment of head and neck squamous cell carcinoma cell lines. Oral Oncol 2007;43:701–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sabol I, Matovina M, Gasperov NM, Grce M. Identification of a novel human papillomavirus type 16 E1 gene variant with potentially reduced oncogenicity. J Med Virol 2008;80:2134–40 [DOI] [PubMed] [Google Scholar]
- 50.Mirabello L, Clarke MA, Nelson CW, Dean M, Wentzensen N, Yeager M, et al. The Intersection of HPV Epidemiology, Genomics and Mechanistic Studies of HPV-Mediated Carcinogenesis. Viruses 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Orav M, Geimanen J, Sepp EM, Henno L, Ustav E, Ustav M. Initial amplification of the HPV18 genome proceeds via two distinct replication mechanisms. Sci Rep 2015;5:15952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim H, Nguyen NP, Turner K, Wu S, Gujar AD, Luebeck J, et al. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nat Genet 2020;52:891–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pang J, Nguyen NP, Luebeck J, Ball L, Finegersh A, Ren S, et al. Extrachromosomal DNA in HPV mediated oropharyngeal cancer drives diverse oncogene transcription. Clin Cancer Res 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Akagi K, Symer DE, Mahmoud M, Jiang, Goodwin S, Wangsa D, et al. Intratumoral heterogeneity and clonal evolution induced by HPV integration. Cancer Disc 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lorenzetto E, Brenca M, Boeri M, Verri C, Piccinin E, Gasparini P, et al. YAP1 acts as oncogenic target of 11q22 amplification in multiple cancer subtypes. Oncotarget 2014;5:2608–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carroll SM, DeRose ML, Gaudray P, Moore CM, Needham-Vandevanter DR, Von Hoff DD, et al. Double minute chromosomes can be produced from precursors derived from a chromosomal deletion. Mol Cell Biol 1988;8:1525–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Cell line sequencing data is available at the Sequence Read Archive PRJNA772772 and cervical tumor samples at: dbGaP Study: phs002810.v1. The data analyzed in this study were obtained from TCGA at cbioportal (https://www.cbioportal.org/)and from supplementary data in (4). All other raw data are available upon request from the corresponding author.