

Abstract
Despite the widespread emergence of multidrug-resistant nosocomial Gram-negative bacterial infections and the major public health threat it brings, no new class of antibiotics for Gram-negative pathogens has been approved over the past five decades. Therefore, there is an urgent medical need for developing effective novel antibiotics against multidrug-resistant Gram-negative pathogens by targeting previously unexploited pathways in these bacteria. To fulfill this crucial need, we have been investigating a series of sulfonyl piperazine compounds targeting LpxH, a di-manganese-containing UDP-2,3-diacylglucosamine hydrolase in the lipid A biosynthetic pathway, as novel antibiotics against clinically important Gram-negative pathogens. Inspired by a detailed structural analysis of our previous LpxH inhibitors in complex with K. pneumoniae LpxH (KpLpxH), here we report the development and structural validation of the first-in-class sulfonyl piperazine LpxH inhibitors, JH-LPH-45 (8) and JH-LPH-50 (13), that achieve chelation of the active site di-manganese cluster of KpLpxH. The chelation of the di-manganese cluster significantly improves the potency of JH-LPH-45 (8) and JH-LPH-50 (13). We expect that further optimization of these proof-of-concept di-manganese-chelating LpxH inhibitors will ultimately lead to the development of more potent LpxH inhibitors for targeting multidrug-resistant Gram-negative pathogens.
Keywords: Antibiotics, Gram-negative bacteria, Lipid A, LpxH, Calcineurin-like phosphoesterase, di-Manganese cluster
Graphical Abstract
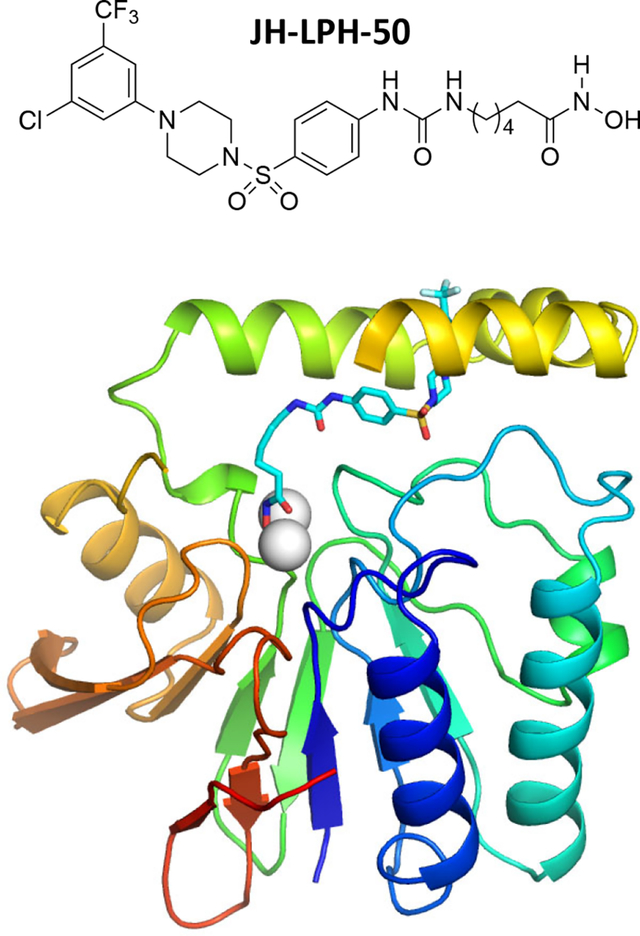
The LpxH enzyme in the lipid A biosynthetic pathway is a promising antibiotic target. In this article, several sulfonyl piperazine LpxH inhibitors are designed and synthesized. Among them, JH-LPH-50 shows improved activity against LpxH by chelating the di-manganese cluster in the active site of LpxH.
Introduction
Gram-negative bacterial pathogens cause many life-threatening illnesses, including pneumonia and bloodstream, wound, and surgical site infections in healthcare and community settings. Antibiotics are essential medications for the treatment of Gram-negative bacterial infection. Unfortunately, as more and more Gram-negative bacteria are becoming resistant to nearly all antibiotics, patients are left with fewer effective treatment options. According to the report by Centers for Disease Control and Prevention on antibiotic resistance threats in the United States, over half of the eighteen antibiotic-resistant threats are from Gram-negative bacteria.[1] In spite of these looming public health concerns, there has been little activity in the antibiotic development pipeline, with no antibiotic with a novel mode of action against Gram-negative bacteria discovered during the past five decades.[2] Hence, there is an urgent need for new antibiotics to treat bacterial infections caused by multidrug-resistant Gram-negative bacteria.
Since the outer membrane of Gram-negative bacteria is required for bacterial viability and serves as a permeability barrier to prevent toxic compounds from entering the cell, disrupting its biogenesis presents an attractive strategy for antibacterial drug discovery.[2,3] A distinctive feature of the outer membrane of Gram-negative pathogens is the presence of lipopolysaccharide (LPS) or lipooligosaccharide (LOS) extruding from the outer monolayer of the outer membrane. Lipid A is the hydrophobic membrane anchor of LPS/LOS and the active component of the bacterial endotoxin that causes Gram-negative septic shock during bacterial infection. Its constitutive biosynthesis via the Raetz pathway is required for bacterial viability and fitness in the human host (Fig. 1A).[4] LpxH is an essential, late-stage lipid A enzyme that hydrolyzes the pyrophosphate group of UDP-2,3-diacylglucosamine (UDP-DAGn) to yield 2,3-diacylglucosamine 1-phosphate (lipid X) and UMP.[5] It is found in ~70% of Gram-negative bacteria and all of the World Health Organization priority Gram-negative pathogens,[6] whereas in the remaining bacterial species, the same chemical reaction is catalyzed by LpxI and LpxG, the functional paralogs of LpxH.[7] Biochemical studies have established LpxH as a member of the calcineurin-like phosphoesterases (CLPs), which requires detergent and a di-manganese cluster for full enzymatic activity.[8] Structural analyses of H. influenzae LpxH,[9] P. aeruginosa LpxH,[10] E. coli LpxH,[11] and most recently K. pneumoniae LpxH[12] have collectively revealed the presence of a conserved core architecture of CLPs with a unique insertion lid above it. The active site, situating between the lid domain and the core CLP architecture, contains a di-manganese cluster that is chelated by residues from the signature metal chelating motifs of CLP enzymes.
Figure 1.

Disruption of lipid A biosynthesis by LpxH inhibitors. (A) A schematic illustration of the Raetz pathway of lipid A biosynthesis in E. coli. LpxH and its functional orthologs, LpxI and LpxG, are labeled in purple and green, respectively. (B) Chemical structures of the LpxH inhibitors: AZ1 (1), JH-LPH-33 (2), and JH-LPH-41 (3).
LpxH is a uniquely attractive antibiotic target for multidrug-resistant Gram-negative bacteria, as inhibition of LpxH not only disrupts lipid A biosynthesis, but also accumulates toxic lipid A intermediates in the bacterial inner membrane.[13] In 2015, AstraZeneca reported a sulfonyl piperazine compound, dubbed AZ1 (1, Fig. 1B), as the first LpxH inhibitor.[14] AZ1 (1) displays weak antibiotic activity against WT K. pneumoniae and E. coli strains with a compromised outer membrane (yhjD*ΔkdtA)[12] or with an efflux pump deletion (ΔtolC).[14]
Intrigued by the potential of AZ1 as a novel antibiotic targeting LpxH, we analyzed the detailed structure-activity relationship of AZ1 (1)[15] and elucidated the molecular details of the LpxH–AZ1 interaction by X-ray crystallography and NMR.[12] By harnessing the ligand dynamics information and structural insights, we designed JH-LPH-33 (2, Fig. 1B), an AZ1 analog with significantly enhanced potency in enzymatic assays and outstanding antibiotic activities in vitro.[12] Compounds 1 and 2 function by occupying the hydrophobic LpxH substrate chamber that accommodates the 2-N-acyl chain of the substrate, but they do not reach into the active site of LpxH.[12] More recently, we described the development of JH-LPH-41 (3, Fig. 1B) that harbors an extended N-acyl chain reaching into the active site of LpxH.[16] Despite these efforts, no LpxH inhibitor described to date chelates the di-manganese cluster in the active site of LpxH.
Aided by detailed structural analyses of our previous LpxH inhibitors in complex with K. pneumoniae LpxH (KpLpxH), we designed several sulfonyl piperazine LpxH inhibitors with the potential of chelating the di-manganese cluster in the active site of LpxH. Herein, we describe the development of the first-in-class di-manganese-chelating LpxH inhibitors with significantly enhanced potency in suppressing LpxH activity in vitro.
Results and Discussion
Design and synthesis of LpxH inhibitors
As metalloenzymes depend critically on their catalytic metal ion for activity, incorporation of a metal chelating group has been an effective strategy for enhancing the potency of metalloenzyme inhibitors.[17] For example, the FDA-approved histone deacetylase (HDAC) inhibitor Vorinostat has a hydroxamic acid coordinating the catalytic Zn2+ in the HDAC active site as a monoanionic ligand through the carbonyl and hydroxyl donor atoms.[18] Elvitegravir employs a quinolone carboxylic acid metal-binding pharmacophore to inhibit human immunodeficiency virus (HIV) integrase.[19]
As LpxH harbors an active site di-manganese cluster that is required for structural integrity and catalysis, we hypothesized that incorporation of a metal-binding group for the di-manganese cluster into LpxH inhibitors would similarly enhance their potency. We noticed that our most recent LpxH inhibitor JH-LPH-41 (3), containing an aniline core with an N-acyl hydroxamate group extension, unexpectedly occupies the space of the hexose ring shared between the substrate and product as observed in the crystal structures of the lipid X-bound and JH-LPH-41-bound LpxH complexes (Fig. 2A; PDB IDs: 6PH9 and 6WII).[12,16] Careful analysis of the KpLpxH/JH-LPH-41 structure showed that the N-hydroxy-5-ureido-pentanamide linker of JH-LPH-41 not only picks up additional hydrophobic interactions with Y125 of the insertion cap and I171 on the loop connecting the cap back to the CLP core domain, but also forms two hydrogen bonds with the backbone amide and carbonyl group of M172 of the same loop as I171. To achieve these interactions, the N-hydroxy-5-ureido-pentanamide linker must be slightly distorted and adopts a compact conformation. This observation led us to hypothesize that further structural modifications of the linker domain of JH-LPH-41 might prevent the linker from interacting with the insertion cap and the loop connecting the cap back to the CLP core domain and facilitate the chelation to the active site di-manganese cluster of LpxH. To test the hypothesis, we embarked on chemical modifications of the hydroxamate linker of JH-LPH-41 (e.g., aniline vs indoline core, linker length, and rigidity, etc) and assessed the effects of the modifications on the chelation of compounds to the di-manganese cluster of LpxH (Fig. 2B).
Figure 2.

Design of new LpxH inhibitors. (A) The N-acyl chain extension of JH-LPH-41 (3) (PDB: 6WII) overlaps with the hexose ring shared by the LpxH product and the substrate (lipid X and UDP-DAGn, respectively) (PDB: 6PH9). (B) Newly designed LpxH inhibitors with an acyl-hydroxamate extension from the indoline and aniline scaffolds.
Indoline and aniline analogs
To develop a di-manganese-chelating LpxH inhibitor, we started with evaluating the effect of acyl chain extension of JH-LPH-33 (2, Fig. 1B), which is the most potent LpxH inhibitor discovered thus far.[12] In addition to the more favorable activity of the indoline core of AZ1 (1) over its corresponding aniline core[15] (Scheme 1A), we hypothesized that switching the aniline core of JH-LPH-41 (3) to the indoline core of JH-LPH-33 (2) might alter the orientation of the N-acyl linker of 3, thus directing the metal binding group, such as a hydroxamic acid, towards the active site di-manganese cluster.
Scheme 1.

The synthesis of indoline and aniline analogs.a
aReagents and conditions: (a) triphosgene, aq. NaHCO3, CH2Cl2, 25 °C, 1 h; (b) NH2(CH2)4CO2Et, DIPEA, CH2Cl2, 25 °C, 1 h, 53% for 2 steps; (c) 1 N NaOH, THF/MeOH (2/1), 25 °C, 30 min, 73%; (d) ClCO2Et, Et3N, THF, 25 °C, 1.5 h; NH2OTBS, MeOH, 25 °C, 1 h, 88%; (e) TFA, CH2Cl2, 25 °C, 25 min, 78%; (f) ClCOCH2(CH2)4CO2Et, Et3N, CH2Cl2, 25 °C, 20 min, 67%; (g) 1 N LiOH, THF/MeOH (2/1), 25 °C, 1.5 h, 99%; (h) ClCO2Et, Et3N, THF, 25 °C, 1 h; NH2OTBS, MeOH, 25 °C, 1 h; (i) TFA, CH2Cl2, 25 °C, 25 min, 21% for 2 steps; (j) OCN(CH2)5CO2Et, CH2Cl2/MeCN (1/1), 25 °C, 18 h, 35%; (k) 1 N NaOH, THF/MeOH (2/1), 25 °C, 3 h, 93%; (l) ClCO2Et, Et3N, THF, 25 °C, 1 h; NH2OTBS, MeOH, 25 °C, 1 h, 94%; (m) TFA, CH2Cl2, 25 °C, 25 min, 81%.
To test this hypothesis, we embarked on the synthesis of the indoline urea analogs 7 and 8 (Scheme 1A). Treatment of the known piperazinyl sulfonyl indoline 6[16] with triphosgene, coupling with ethyl 5-aminopentanoate, and basic hydrolysis afforded the corresponding carboxylic acid 7. Compound 7 was converted to the TBS-protected hydroxamic acid by treatment with ClCO2Et/Et3N/NH2OTBS. Final TBS deprotection by TFA gave the indoline urea analog 8 in 78%. To investigate the effect of the urea linkage of 8 on the orientation of the N-acyl linker, we also prepared the corresponding amide analog 10. Acylation of 6 with ethyl 7-chloro-7-oxoheptanoate followed by hydrolysis proceeded smoothly to give carboxylic acid 9 (Scheme 1A). Treatment of 9 with ClCO2Et/Et3N/NH2OTBS and final TBS deprotection by TFA completed the synthesis of the amide analog 10.
After the successful incorporation of a metal-binding pharmacophore for the di-manganese cluster into the indoline core of JH-LPH-33 (2), we turned our attention to re-examining the design of aniline-based inhibitors such as JH-LPH-41 (3, Fig. 1B). We reasoned that the N-acyl chain of 3 might not be sufficiently long enough to allow the terminal hydroxamate group to reach the di-manganese cluster in LpxH. To test this hypothesis, we designed one-carbon homologated analogs 12 and 13 (Scheme 1B). Coupling of the known piperazinyl sulfonyl aniline 11[16] with ethyl 6-isocyanatohexanoate followed by hydrolysis under basic conditions afforded carboxylic acid 12. To install the hydroxamic acid moiety for metal binding, compound 12 was treated with ClCO2Et/Et3N/NH2OTBS to give the corresponding TBS protected hydroxamic acid. Final TBS deprotection was accomplished by TFA to afford the desired one-carbon homologated analog 13 in 76% over two steps.
Analogs with a cyclic metal-binding group or a rigid linker
Antibiotics against Gram-negative bacteria need to find diffuse through the negatively charged LPS layer present on the surface of the outer membrane or translocate through porins to penetrate the hydrophobic outer membrane.[20] Then, they need to cross the hydrophobic lipid bilayer of the inner membrane before they find their cytosolic targets such as LpxH. The flexible acyl chains of the above-mentioned compounds may affect the membrane permeability of the compounds. To investigate the effect of the flexible acyl chain of 13 on activity, we replaced the acyclic hydroxamic acid moiety of 13 with a more rigid cyclic metal binding group. The 1-hydroxy-2(1H)-pyridinethione metal binding group was originally developed by Cohen and co-workers[21] and has been used in matrix metalloproteinase inhibitors.[22] CDI coupling of 11 to 14 (see the Supporting Information for details) completed the synthesis of the hydroxy pyridinethione analog 15 (Scheme 2).
Scheme 2.

The synthesis of analogs with a cyclic metal bonding group or a rigid linker.a
aReagents and conditions: (a) CDI, DIPEA, THF, 25 °C, 20 h; 14, 25 °C, 20 h, 43%; (b) CDI, DIPEA, THF, 25 °C, 20 h; 16, 25 °C, 20 h, 22%; (c) TFA, CH2Cl2, 25 °C, 30 min, 100%; (d) ClCO2Et, Et3N, THF, 25 °C, 1 h; NH2OTBS, THF, 25 °C, 1 h; (e) TFA, CH2Cl2, 25 °C, 30 min, 74% for 2 steps; (f) OCNCH2CH2Cl, CH2Cl2/MeCN, 40 °C, 20 h, 89%; (g) NaN3, TBAI, DMF, 45 °C, 48 h, 59%; (h) N-hydroxypropiolamide, CuSO4, sodium ascorbate, EtOH/t-BuOH/H2O (2/1/1), 25 °C, 20 h, 28%.
We also made the N-acyl linker of 13 more rigid by incorporating a double bond or a triazole ring. For the synthesis of the (E)-olefinic linker analog 18, we reacted 11 with tert-butyl (E)-6-aminohex-2-enoate (16) (see the Supporting Information for details), CDI, and DIPEA to give the corresponding urea intermediate (Scheme 2). Treatment of the urea intermediate with TFA afforded carboxylic acid 17. Carboxylic acid 17 was converted to the final olefinic linker analog 18 following the same procedure used for 13. Another rigid analog we designed was the triazole linker analog 20 (Scheme 2). To synthesize 20, we started with coupling 11 with 1-chloro-2-isocyanatoethane (89%) followed by treating with NaN3 and TBAI to give azide 19 (59%). The click chemistry[23] of azide 19 and N-hydroxypropiolamide provided the desired triazole analog 20, albeit in low yield (28%).
Evaluation of LpxH inhibition by analogs
After completing the synthesis of sulfonyl piperazine compounds with various forms of linkers, we biochemically characterized the KpLpxH inhibition by these analogs at 0.1 μM using the nonradioactive, colorimetric malachite green assay that we had previously reported.[15] We found that compound 8 was more potent in inhibiting KpLpxH (vi/v0=18 ± 1%) than 2 (vi/v0=21%) (Table 1). Such an enhanced potency requires the presence of the hydroxamate group, as its substitution with a carboxylate group in compound 7 reduced the compound activity (vi/v0=30 ± 6%). This result suggests that the superior inhibitory effect of 8 is likely derived from chelation of the di-manganese cluster.
Table 1.
Percentage activity of KpLpxH in the presence of 0.1 μM LpxH inhibitors
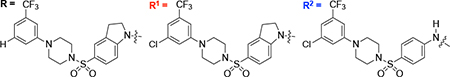
| |||
|---|---|---|---|
| Compounds | Structure | Percentage Activity[a] |
Percentage Inhibition |
|
1 (AZ1) |
|
78[16] | 22[16] |
|
2 (JH-LPH-33) |
|
21[16] | 79[16] |
|
3 (JH-LPH-41) |
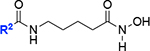
|
36[16] | 64[16] |
| 7 |
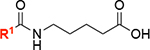
|
30 ± 6 | 70 |
| 8 |
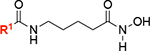
|
18 ± 1 | 82 |
| 10 |
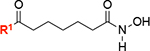
|
93 ± 7 | 7 |
| 12 |
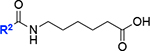
|
57 ± 3 | 43 |
| 13 |
|
3 ± 2 | 97 |
| 15 |
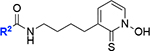
|
39 ± 6 | 61 |
| 18 |
|
34 ± 7 | 66 |
| 20 |
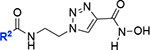
|
59 ± 7 | 41 |
Percentage activity was normalized against the specific activity of KpLpxH measured in the presence of 10% DMSO (309 ± 12 μmol/min/mg). Errors represent the standard percent error.
We also found that compound 13, with a one-carbon extension compared to the acyl hydroxamate group of 3, nearly completely inhibited KpLpxH at 0.1 μM, leaving 3 ± 2% of the enzymatic activity (vi/v0), whereas KpLpxH retained 36% activity in the presence of 0.1 μM of 3 (Table 1). Compound 13 is also a more potent inhibitor than 2 at 0.1 μM (vi/v0=21%). Such a significant improvement in potency was lost when the hydroxamate group was substituted with the carboxylate group (12; vi/v0=57 ± 3%), suggesting that the inhibitory effect similarly requires the hydroxamate group and its ability to chelate the di-manganese cluster.
Unfortunately, neither substituting the hydroxamate group with a cyclic metal binding group as in 15 (vi/v0=39 ± 6% inhibition) nor introducing a double bond as in 18 (vi/v0= 34 ± 7% inhibition) or a cyclic ring as in 20 (vi/v0=59 ± 7% inhibition) next to the hydroxamate group enhanced the compound activity when tested at 0.1 μM concentration (Table 1). It is likely due to the loss of the di-manganese chelation in these analogs.
Structures of K. pneumoniae LpxH in complex with 8 (JH-LPH-45) and 13 (JH-LPH-50)
In order to gain a better understanding of the nature of the interaction of 8 (JH-LPH-45) with K. pneumoniae LpxH (KpLpxH), we determined the co-crystal structure of the KpLpxH/8 complex at 1.74 Å (Fig. 3, and Table S1). The location of 8 is very well defined by the omit map density (Supplementary Fig. S1). Consistent with our design and similar to previously reported LpxH inhibitors, the crystal structure of KpLpxH/8 showed that the 5-((4-(3-chloro-5-(trifluoromethyl)phenyl)piperazin-1-yl)sulfonyl)indoline moiety of 8 occupies the 2-N-acyl chain binding chamber of LpxH defined by a cluster of hydrophobic residues, such as A45, F82, L83, I137, F141, I152, M156, A153, I171 (Fig. 3ABC, Table S1). The sidechain of R80 forms a prototypical cation-p interaction with the indoline group. Additionally, the sulfonyl oxygen atoms form hydrogen bonds with the sidechain of R157 and backbone of W46, and the urea group forms a hydrogen bond with the sidechain of N79 (Fig. 3C). However, compound 8 is distinct from previously reported LpxH inhibitors in that it chelates the di-manganese cluster in the active site of LpxH. It does so with its acyl chain extending towards the di-manganese cluster with the N-hydroxyl group bridging two manganese ions A and B and the carbonyl oxygen of the hydroxamic acid interacting with manganese ion B (Fig. 3D). As a result, manganese ion A is penta-coordinated in a pseudo-square pyramidal configuration with the hydroxamate N-hydroxyl group of 8 occupying a base vertex, whereas manganese ion B is hexa-coordinated in a pseudo-octahedral geometry, with the N-hydroxyl group and carbonyl oxygen atom of the hydroxamic acid group of 8 occupying two vertices. Accordingly, compound 8 displayed a significant reduction of the IC50 value (IC50=18 nM, KI=7.3 nM, Fig. 3E) in comparison with the reported value for 2 (IC50=26 nM, KI=10 nM)[12] which was the most potent LpxH inhibitor discovered prior to this study. Intriguingly, replacing the urea group with the corresponding amide group (compound 10) reduced the compound activity (vi/v0=93 ± 7%) despite the urea nitrogen atoms lacking any interaction with the enzyme, suggesting that the geometric constraints provided by the urea linker are beneficial for activity.
Figure 3.

Inhibition of KpLpxH by 8 (JH-LPH-45). (A) Ribbon diagram of the KpLpxH/JH-LPH-45 complex. KpLpxH is colored in rainbow, with the N-terminus in blue and C-terminus in red. JH-LPH-45 and the di-manganese cluster are shown in the stick and sphere models, respectively. The purple mesh represents the 2mFo-DFc map of JH-LPH-45 at 1s. (B) An enlarged view of JH-LPH-45. Superimposition of the KpLpxH/JH-LPH-45 complex with the previously reported KpLpxH/lipid X complex (PDB: 6PH9) shows that the compound competes with the 2-N-acyl chain of lipid X, but with its acyl-hydroxamate group chelating the active site di-manganese cluster. (C) Interactions between JH-LPH-45 and LpxH residues. The interaction map was generated by LigPlot+.[24] (D) Metal chelation geometry. Distances between the atoms in the hydroxamate group and the di-manganese cluster are labeled with dashed lines. (E) The IC50 curve of the KpLpxH inhibition by JH-LPH-45. Error bars represent the standard error of measurement (SEM, n=3).
To verify that the gain of activity of 13 (JH-LPH-50) was due to chelation of the di-manganese cluster, we additionally analyzed the crystal structure of the KpLpxH/13 complex resolved at 1.73 Å resolution (Fig. 4A; Table S1). The density for compound 13 is also very well defined (Supplementary Fig. S2). Similar to 8 (Fig. 3), compound 13 also has its sulfonyl piperazine tail group occupying the 2-N-acyl chain chamber of LpxH and its acyl hydroxamate reaching into the active site and chelating the di-manganese cluster (Fig. 4ABC). However, there is a major distinction between 8 and 13: although the N-hydroxyl group of 13 similarly bridges both manganese ions, its carbonyl oxygen coordinates manganese ion A instead of B (Fig. 4CD). As a result, manganese ion A is now hexa-coordinated in a pseudo-octahedral geometry, whereas manganese ion B is penta-coordinated in a pseudo square pyramidal geometry. The distance from the carbonyl oxygen of the hydroxamate group is closer for manganese ion A to 13 (2.5 Å) than that of manganese ion B to 8 (2.7 Å), suggesting that it is energetically more favorable. Such an effect may contribute to the lower IC50 value of 13 (IC50=7.7 nM, KI=3.1 nM; Fig. 4E) than that of 8 (IC50=18 nM, KI=7.3 nM; Fig. 3E).
Figure 4.

Inhibition of KpLpxH by 13 (JH-LPH-50). (A) Ribbon diagram of the KpLpxH/JH-LPH-50 complex. KpLpxH is colored in rainbow, with the N-terminus in blue and C-terminus in red. JH-LPH-50 and the di-manganese cluster are shown in the stick and sphere models, respectively. The purple mesh represents the 2mFo-DFc map of JH-LPH-50 at 0.8s with JH-LPH-50 shown in the stick model. (B) An enlarged view of JH-LPH-50. Superimposition of the KpLpxH/JH-LPH-50 complex with the previously reported KpLpxH/lipid X complex (PDB: 6PH9) shows that the compound competes with the 2-N-acyl chain of lipid X, but with its acyl-hydroxamate group chelating the active site di-manganese cluster. (C) Interactions between JH-LPH-50 and LpxH residues. The interaction map was generated by LigPlot+.[24] (D) Metal chelation geometry. Distances between the atoms in the hydroxamate group and the di-manganese cluster are labeled with dashed lines. (E) The IC50 curve of the KpLpxH inhibition by JH-LPH-50. Error bars represent the standard error of measurement (SEM, n=3).
Evaluation of antibiotic activity of compounds 8 (JH-LPH-45) and 13 (JH-LPH-50)
To evaluate the effect of di-manganese chelation on antibiotic activity, we tested compounds 8 and 13 against K. pneumoniae (ATCC 10031) in vitro (Table 2). Despite the significant reduction of KI values of 8 (7.3 nM) and 13 (3.1 nM) in comparison with 2 (10 nM) in enzymatic assays, compounds 8 and 13 displayed lower antibiotic activity (MIC=18.7 μg/mL for 8 and MIC=3.3 μg/mL for 13) than the non-metal chelating compound 2 (MIC=0.83 μg/mL). Such a discrepancy was presumably caused by the flexible acyl chain that hindered the membrane permeability of the compounds as suggested by the eNTRY rule.[25]
Table 2.
Antibiotic activity of LpxH inhibitors
| Compounds | 1 (AZ1) | 2 (JH-LPH-33) | 8 (JH-LPH-45) | 13 (JH-LPH-50) |
| MIC (μg/mL) | >64 | 0.83 | 18.7 | 3.3 |
Both JH-LPH-45 and JH-LPH-50 leverage a terminal hydroxamate group to chelate the di-manganese cluster and enhance ligand-LpxH interaction. The hydroxamate group has been used as a metal chelating group in FDA approved histone deacetylase inhibitors, such as Belinostat, Panobinostat, and Vorinostat.[26] Although hydroxamate-containing LpxH inhibitors may inhibit human metalloenzymes, the unique lipid binding feature of LpxH could be leveraged to ensure the specificity of LpxH inhibitors and minimize off-target effects. Furthermore, replacing the hydroxamate group with alternative metal-chelating groups[17] may further enhance the selectivity of LpxH inhibitors containing a metal chelating group, thus mitigating the potential off-target consequence.
Conclusion
The LpxH enzyme in the lipid A biosynthetic pathway is a promising antibiotic target. Building on the sulfonyl piperazine scaffold of AZ1 (1), we previously reported more potent AZ1 analogs, such as JH-LPH-33 (2) with significant improvement of antibiotic activity[12] and JH-LPH-41 (3)[16] that reaches the polar binding pocket near the active site. After careful analyses of the structures of KpLpxH/ JH-LPH-33 (2) and KpLpxH/ JH-LPH-41 (3), we designed JH-LPH-45 (8) and JH-LPH-50 (13) by switching the aniline core to the corresponding indoline core (8) or extending the linker by a methylene unit (13). Consistent with our prediction, both 8 and 13 chelate the di-manganese cluster in the active site of KpLpxH. Most notably, the chelation of the active site di-manganese cluster of KpLpxH significantly improves the potency of compounds 8 and 13, reducing the KI values from 10 nM (JH-LPH-33) to 7.3 nM (8) and 3.1 nM (13). These compounds are the first-in-class LpxH inhibitors that achieve chelation of the di-manganese cluster in LpxH. We anticipate that our work will ultimately contribute to the design and development of potent and selective LpxH inhibitors for multidrug-resistant Gram-negative pathogens.
Experimental Section
Synthesis of sulfonyl piperazine LpxH inhibitors.
See the Supplementary Material for details.
MIC assay.
The MIC assay protocol was adapted from the broth microdilution methods described by the National Committee for Clinical Laboratory Standards[27] using 96-well plates as described previously.[12] In brief, overnight bacterial cultures were diluted to an OD600 of 0.006 in cation-adjusted Mueller-Hinton medium containing 7% DMSO and incubated at 37 °C for 22 h in the presence of varying concentrations of inhibitors. Following overnight incubation, each well is treated with 10 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, 5 mg/mL) for 3 h. Finally, the culture is solubilized by addition of 2-propanol, and the results are read by taking the difference of the UV reading at both 570 and 690 nm. MIC values were reported as the lowest compound concentration that inhibited bacterial growth. The baseline for inhibited cell growth was determined to be < 4× the average control reading due to the baseline optical density of the initial starting bacterial culture.
Enzymatic assay for KpLpxH inhibition.
The LpxE-coupled LpxH activity assay[15] was conducted as described previously using the GB1-K. pneumoniae LpxH-His10 fusion protein.[12,16] Briefly, two reaction mixtures were prepared. Mixture 1 contains 20 mM Tris-HCl (pH 8.0), 0.5 mg/mL BSA, 0.02% Triton X-100, 1 mM MnCl2, 1 mM DTT, 10% DMSO, and 200 μM substrate (UDP-DAGn), and mixture 2 is comprised of the same buffer, but instead of substrate, contains both LpxH (20 ng/mL) and 2× inhibitor. These mixtures were then pre-incubated at 37 °C for 10 min. To initiate the reaction, an equal volume of the LpxH mixture (mixture 2) was added to the substrate mixture (mixture 1) at 37 °C. The final reaction solution contains 100 μM substrate, 10 ng/mL enzyme, and 1× inhibitor. At the desired reaction time points, an aliquot of 20 μL reaction mixture was removed and added to a well in 96-well half-area plate containing 5 mM EDTA (final concentration) to quench the LpxH reaction. Purified Aquifex aeolicus LpxE was then added to a final concentration of 5 μg/mL. The plate was incubated at 37 °C for 30 min followed by addition of formic acid to a final concentration of 3.75 M to quench the LpxE reaction. The malachite green reagent (Sigma Aldrich, catalog MAK307) was diluted 5-fold into the solutions, and the plate was incubated for 30 min at room temperature before the absorbance at 620 nm was measured. All measurements were done in triplicates, and standard error (S.E.) was calculated. Percentage LpxH activities for 1, 2, and 3 at 0.1 μM were previously reported.[12,16] The values of 18 nM for 8 and 7.7 nM for 13 were extracted from fitting of the dose–response curve of . These values correspond to KI values of 7.3 nM for 8 and 3.1 nM for 13 in the competitive binding mode for K. pneumoniae LpxH with a KM value of 68.1 μM.
Cloning and purification of KpLpxH.
Cloning and purification of KpLpxH for crystallography studies were carried out as previously reported.[12,16] Briefly, KpLpxH was cloned into a modified pET21b vector (Novagen/Millipore Sigma), yielding the LpxH fusion protein with a C-terminal TEV protease site (ENLYFQGS) followed by a His10 tag. Vector-transformed BL21 STAR™ (DE3) E. coli cells (Thermo Fisher Scientific) were grown in M9 medium at 37 °C to an OD600 of 0.5, induced with 1 mM IPTG for an additional 5 h at 30 °C, and then harvested by centrifugation. Protein purification was carried out at 4 °C. Cell pellets from 8 L of induced culture were resuspended and lysed in 120 mL of the lysis buffer containing 50 mM phosphate-citrate, 20 mM MES (pH 6.0), 600 mM NaCl, 10% sucrose, 5 mM 2-mercaptoethanol, 10 mM imidazole, and 0.1% Triton X-100 using a French press. Cell debris was removed by centrifugation at 40,000× g for 40 min, and the supernatant was loaded onto a column containing 20 mL of HisPur Ni2+-NTA resin (Thermo Fisher Scientific) pre-equilibrated with 100 mL of the lysis buffer. After extensive column washing with the purification buffer containing 20 mM phosphate-citrate, 20 mM MES (pH 6.0), 300 mM NaCl, 5% glycerol, 5 mM 2-mercaptoethanol, and 40 mM imidazole, LpxH was eluted from the column with a stepwise increase in imidazole concentration from 40 to 400 mM in the purification buffer. The sample was concentrated for further purification with size-exclusion chromatography (Superdex 200; GE Healthcare Life Sciences) in the FPLC buffer containing 20 mM MES (pH 6.0), 800 mM NaCl, 1 mM DTT, and 5% glycerol.
Co-crystallization of KpLpxH with 8 (JH-LPH-45) and 13 (JH-LPH-50).
The FPLC peak fractions containing KpLpxH were exchanged into a buffer containing 20 mM MES (pH 6.0), 200 mM NaCl, 1 mM DTT, and 5% glycerol and then concentrated to 8 mg/mL for crystallization. Compounds 8 and 13 were added to the LpxH solution in two steps to minimize DMSO concentration while still ensuring more than 1:1 concentration of LpxH to compounds. First, 1:1 molar ratio of 8 or 13 was added to the FPLC fraction prior to concentrating them to final concentration. Then, another 1:1 ratio of the compounds was added after concentrating the FPLC fractions to 8 mg/mL. Protein crystals were grown using the sitting-drop vapor diffusion method at 20 °C. Each drop was prepared by mixing 1 μL of the protein solution with 1 μL of the reservoir solution. The final drop solution for 8 contained 4 mg/mL of LpxH, 0.27 mM 8, 10 mM MES (pH 6.0), 100 mM NaCl, 0.5 mM DTT, 2.5% glycerol, 0.625% DMSO, 0.025 M magnesium acetate tetrahydrate, 0.025 M sodium acetate pH 5.4, 12 % v/v PEG 400. The final drop solution for 13 contained 4 mg/mL of LpxH, 0.27 mM 13, 10 mM MES (pH 6.0), 100 mM NaCl, 0.5 mM DTT, and 2.5% glycerol, 0.625% DMSO, 0.05 M calcium chloride dihydrate, 0.05 M Tris pH 6.5, 13 % w/v PEG 2000 MME. Diffraction quality protein crystals were harvested after 2 weeks and soaked for 30 minutes with a cryoprotectant composed of mother liquor diluted 40% additionally containing 20% glycerol, 100 μM MnCl2, 5% DMSO, and 0.27 mM of either 8 or 13.
Structural analysis of KpLpxH complexes with 8 (JH-LPH-45) and 13 (JH-LPH-50).
The X-ray diffraction data of the KpLpxH complex with 8 and 13 were collected at the Northeastern Collaborative Access Team (NECAT) 24-ID-C beamline at the Advanced Photon Source at Argonne National Laboratory. The X-ray diffraction data was processed using XDS.[28] Using molecular replacement with the PHASER in the PHENIX suite, the crystal structure phase information of both the KpLpxH-8 and KpLpxH-13 complexes was obtained using the PDB entry 6PJ3 as the search model. Restraints of the inhibitors were generated using eLBOW and edited manually. Iterative model building and refinement was performed using COOT[29] and PHENIX.
Supplementary Material
Acknowledgments.
This work was supported in part by the grants from the National Institute of Allergy and Infectious Diseases (AI139216) and National Institute of General Medical Sciences (GM115355). X-ray diffraction data were collected at the Northeastern Collaborative Access Team beam lines (24-ID-C), which is funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Pilatus 6 M detector on 24-ID-C beam line is funded by a NIH-ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, Department of Energy Office of Science, United States User Facility operated for the Department of Energy Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. A.F.E was supported by the NIGMS Pharmacological Sciences Training Grant (NIH T32GM133352).
Footnotes
P.Z. and J.H. are inventors of a patent covering the designed LpxH inhibitors.
Data Availability.
The crystal structures of the K. pneumoniae LpxH/ JH-LPH-45 (8) complex and the K. pneumoniae LpxH/ JH-LPH-50 (13) complex have been deposited to the Protein Data Bank (www.pdb.org) with access codes of 7SS6 and 7SS7, respectively.
References
- [1].https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf.
- [2].MacNair CR, Tsai CN, Brown ED, Ann. N. Y. Acad. Sci. 2020, 1459, 69–85. [DOI] [PubMed] [Google Scholar]
- [3].Brown DG, Bioorg. Med. Chem. 2016, 24, 6320–6331. [DOI] [PubMed] [Google Scholar]
- [4].a) Raetz CR, Whitfield C, Annu. Rev. Biochem. 2002, 71, 635–700; [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Zhou P, Zhao J, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2017, 1862, 1424–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Babinski KJ, Ribeiro AA, Raetz CR, J. Biol. Chem. 2002, 277, 25937–25946. [DOI] [PubMed] [Google Scholar]
- [6].WHO, Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics, http://www.who.int/medicines/publications/WHO-PPL-Short_Summary_25Feb-ET_NM_WHO.pdf, 2017.
- [7].a) Metzger LE IV, Raetz CR, Biochemistry 2010, 49, 6715–6726; [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Young HE, Zhao J, Barker JR, Guan Z, Valdivia RH, Zhou P, mBio 2016, 7, e00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Young HE, Donohue MP, Smirnova TI, Smirnov AI, Zhou P, J. Biol. Chem. 2013, 288, 26987–27001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cho J, Lee CJ, Zhao J, Young HE, Zhou P, Nat. Microbio. 2016, 1, 16154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Okada C, Wakabayashi H, Kobayashi M, Shinoda A, Tanaka I, Yao M, Sci. Rep. 2016, 6, 32822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bohl TE, Ieong P, Lee JK, Lee T, Kankanala J, Shi K, Demir O, Kurahashi K, Amaro RE, Wang Z, Aihara H, J. Biol. Chem. 2018, 293, 7969–7981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cho J, Lee M, Cochrane CS, Webster CG, Fenton BA, Zhao J, Hong J, Zhou P, Proc. Natl. Acad. Sci. USA 2020, 117, 4109–4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Babinski KJ, Kanjilal SJ, Raetz CR, J. Biol. Chem. 2002, 277, 25947–25956. [DOI] [PubMed] [Google Scholar]
- [14].Nayar AS, Dougherty TJ, Ferguson KE, Granger BA, McWilliams L, Stacey C, Leach LJ, Narita S, Tokuda H, Miller AA, Brown DG, McLeod SM, J. Bacteriol. 2015, 197, 1726–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lee M, Zhao J, Kwak SH, Cho J, Lee M, Gillespie RA, Kwon DY, Lee H, Park HJ, Wu Q, Zhou P, Hong J, ACS Infect. Dis. 2019, 5, 641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kwak SH, Cochrane CS, Ennis AF, Lim WY, Webster CG, Cho J, Fenton BA, Zhou P, Hong J, Bioorg. Chem. 2020, 102, 104055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chen AY, Adamek RN, Dick BL, Credille CV, Morrison CN, Cohen SM, Chem. Rev. 2019, 119, 1323–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Marks PA, Breslow R, Nat. Biotechnol. 2007, 25, 84–90. [DOI] [PubMed] [Google Scholar]
- [19].Hare S, Gupta SS, Valkov E, Engelman A, P. Nature 2010, 464, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a) Cama J, Henney AM, Winterhalter M, J. Mol. Biol. 2019, 431, 3531–3546; [DOI] [PubMed] [Google Scholar]; b)Zgurskaya HI, Lopez CA, Gnanakaran S, ACS Infect. Dis. 2015, 1, 512–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Puerta DT, Cohen SM, Inorg. Chem. 2003, 42, 3423–3430. [DOI] [PubMed] [Google Scholar]
- [22].Martin DP, Blachly PG, McCammon JA, Cohen SM, J. Med. Chem. 2014, 57, 7126–7135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rostovtsev VV, Green LG, Fokin VV, Sharpless KB, Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [DOI] [PubMed] [Google Scholar]
- [24].Laskowski RA, Swindells MB, J. Chem. Inf. Model. 2011, 51, 2778–2786. [DOI] [PubMed] [Google Scholar]
- [25].Richter MF, Hergenrother PJ, Ann. N. Y. Acad. Sci. 2019, 1435, 18–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bondarev AD, Attwood MM, Jonsson J, Chubarev VN, Tarasov VV, Schioth HB, Br. J. Clin. Pharmacol. 2021, 87, 4577–4597. [DOI] [PubMed] [Google Scholar]
- [27].Standards NCfCL, Methods for Dilution Antimicrobial Susceptibility Test for Bacteria that Grow Aerobically (Approved Standard, NCCLS document M7-A1), Wayne, PA, 1997. [Google Scholar]
- [28].Kabsch W, Acta Cryst. 2010, D66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Emsley P, Cowtan K, Acta Cryst. 2004, D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The crystal structures of the K. pneumoniae LpxH/ JH-LPH-45 (8) complex and the K. pneumoniae LpxH/ JH-LPH-50 (13) complex have been deposited to the Protein Data Bank (www.pdb.org) with access codes of 7SS6 and 7SS7, respectively.