

Abstract
Predicting in vivo response to antineoplastics remains an elusive challenge. We performed first-of-kind evaluation of two transcriptome-based precision cancer medicine methodologies to predict tumor sensitivity to a comprehensive repertoire of clinically relevant oncology drugs, whose mechanism-of-action we experimentally assessed in cognate cell lines. We enrolled patients with histologically distinct, poor prognosis malignancies who had progressed on multiple therapies, and developed low-passage, patient-derived xenograft models that were used to validate 35 patient-specific drug predictions. Both OncoTarget, which identifies high-affinity inhibitors of individual Master Regulator (MR) proteins, and OncoTreat, which identifies drugs that invert the transcriptional activity of hyper-connected MR modules, produced highly significant 30-day disease control rates (68% and 91%, respectively). Moreover, of 18 OncoTreat-predicted drugs, 15 induced the predicted MR-module activity inversion in vivo. Predicted drugs significantly outperformed antineoplastic drugs selected as unpredicted controls, suggesting these methods may substantively complement existing PCM approaches, as also illustrated by a case study.
Introduction
A major goal of precision cancer medicine (PCM) is to improve clinical outcomes by leveraging the molecular-level properties of a tumor—as encoded by mutational, gene expression, epigenetic modification, and proteomic profiles—to accurately predict sensitivity to candidate therapeutic agents. Application of PCM principles may help generate responder-enriched cohorts for clinical trials when predictions are conserved across a substantial fraction of patients (1, 2), and even help prioritize personalized treatments on an individual patient basis.
Systematic application of the current PCM paradigm is largely predicated on two complementary approaches. The first one (oncogene addiction) aims to identify targeted therapies based on the presence of activating genetic alterations in druggable oncoproteins (3); the second (immunotherapy) is based on the discovery that specific tumor-initiated immunosuppressive programs can be abrogated by pharmacological targeting of immune checkpoints or by sensitizing the immune system to tumor antigens (4).
Despite the remarkable clinical success of these approaches within specific tumor subtypes (5), many tumors may lack actionable genetic alterations, fail to respond to therapy, or develop drug resistance, suggesting an acute need for complementary approaches targeting non-oncogene tumor dependencies (6). In particular, despite its critical role in tumor subtype stratification, use of transcriptome-based approaches in precision medicine has lagged.
We and others have shown that, within each tumor histology, cancer cells adopt only a relatively limited, discrete, and remarkably stable repertoire of molecularly-distinct transcriptional states (7). These states are mechanistically controlled by tightly autoregulated Tumor-Checkpoint Modules (TCMs) (8), comprising a small, highly conserved set of Master Regulator (MR) proteins responsible for canalizing the effect of mutations in their upstream pathways (7–9). About 30% of MR proteins were shown to represent tumor-essential, non-oncogene dependencies, either individually (8) or in combination (10, 11). Indeed, genetic (10–12) or pharmacological (13–15) inhibition of MR proteins has been shown effective in inverting the activity of TCM MRs, resulting in abrogation of tumor viability in vitro and in vivo. TCM-inversion can be effectively achieved due to their hyper-connected, heavily autoregulated nature, supporting their ability to behave as homeostatic on/off control modules (12) (bioRxiv 2020.10.27.357269). As such, individual MRs—and the TCMs they comprise—represent an actionable class of non-oncogene dependencies.
In this study, we test two approaches to leverage these conceptual advances for therapeutic purposes, by targeting either individual, pharmacologically-actionable candidate MRs, with a high-affinity inhibitor (OncoTarget), or the entire TCM (OncoTreat). These methods rely on the ability to accurately measure the transcriptional activity of the regulatory proteins that maintain tumor cell state, by RNASeq profile analysis using the VIPER algorithm (16), which has been shown to compare favorably with antibody-based protein measurements (17).
Specifically, OncoTarget uses VIPER to identify the most aberrantly activated proteins for which a high-affinity inhibitor is already available, thus representing a straightforward, mutation-agnostic extension of the oncogene addiction paradigm. Indeed, aberrant protein activity can result not only from activating mutations in the encoding gene but also by mutations and signals in upstream pathways (7), as well as by autocrine, paracrine, and endocrine signals (18). In contrast, OncoTreat leverages large-scale perturbational profiles—i.e., RNASeq profiles representing the cell’s response to drug treatments—in MR-matched cell lines selected as high-fidelity (cognate) models of a patient tumor. These profiles support direct experimental assessment of context-specific drug Mechanism-of-Action (MoA) and TCM-activity inversion in drug vs. vehicle control-treated tumor cells (13). Compared to other related efforts, such as SynergySeq (19), which predict drug sensitivity based on the greatest divergence between drug perturbation and tumor transcriptomic signatures, our methods capitalize on robust network-based assessment of functional protein activity and are built on a broad mechanistic framework incorporating insights on tumor-specific essentiality of MR proteins. Further, we leverage careful selection of cognate models to assess context-specific drug MoA, and, most critically, we provide in vivo validation.
In this first-in-class application of MR-based tools to predict drug sensitivity, we designed a tumor- and mutation-agnostic non-interventional clinical study to enroll patients with advanced malignancies, across multiple histologies, who had progressed on several lines of therapy (the N of 1 study at Columbia University, IRB-AAAN7562). We present a series of results from treatment of early passages of the first seven patient-derived xenograft (PDX) models established, which were used to assess the overall preclinical efficacy of OncoTarget and OncoTreat-predicted drugs. For the latter, we also performed pharmacodynamic assays to assess in vivo recapitulation of TCM activity inversion, as predicted from in vitro perturbations. Our results demonstrate that OncoTarget and OncoTreat are highly predictive of anti-tumor drug activity in vivo, supporting further development of these clinically actionable tests—both of which are NY and CA Department of Health approved and CLIA compliant (CUIMC pathology department)—for biomarker-driven PCM clinical trials. We discuss the strengths, limitations, and practical challenges encountered in implementing and validating these tools in a clinical context.
Results
Overview of Study Design:
To assess OncoTarget and OncoTreat’s ability to predict drug sensitivity in tumors from pre-treatment RNASeq profile analysis, we designed an innovative, proof-of-concept clinical study with a preclinical end point. The N of 1 study enrolled patients with advanced malignancies refractory or intolerant to standard of care treatment, including several rare tumors (Table S1). Due to trial design, selected participants had generally progressed on most if not all standard of care therapies, by time of enrollment, and lacked actionable genetic alterations suggesting potential targeted drug efficacy.
Clinically indicated biopsies or tumor resections were performed at the request of the treating oncologist; consent was required to allow a portion of the fresh tumor tissue from biopsies with ≥70% cellularity to be processed for RNASeq profiling and transplantation into immunodeficient mice. Here, we report the results of 35 distinct drug arms—including 21 OncoTarget and 22 OncoTreat-predicted drugs, 8 of which were predicted by both methods—in the first seven, consecutively established PDX models that could be expanded for in vivo drug testing. Given its research nature, the protocol did not require patients to be treated with predicted drugs. Instead, subsequent treatment was chosen by the patient’s oncologist using currently available approaches for cancers that have progressed on standard treatments, e.g., mutational profiling, off-label drug use, and referral to therapeutic clinical trials when eligible.
Note that the study was neither designed nor sufficiently powered to assess the efficacy of each individual predicted drug. Rather, our goal was to validate the overall ability of the two methodologies (tests) to predict drugs that elicit in vivo response.
The seven PDX models included three basal-like breast cancers (BLBC) (BC-32398, BC-97359, BC-50291), a pancreatic ductal carcinoma (PDA) (PAC-05647), a colon adenocarcinoma (COAD) (CAR-23659), a KITWT/PDGFRWT gastrointestinal stromal tumor (GIST) harboring both a KRASG12D mutation and a germline SDHB deletion (GIST-81050), and a recurrent WHO grade II anaplastic meningioma (MEN) (CNS-16474). Clinical characteristics of the seven patients are summarized (Figure 1A), including extensive prior therapies and the results of targeted genomic sequencing, when performed at the discretion of the treating oncologist. For two patients (CNS-16474 and PAC-05647), mutational profiling was not available because the oncologist did not expect these tumor types to harbor clinically actionable mutations. Notably, four of the seven subjects had progressed on at least three lines of treatment before enrollment and all seven represented aggressive, drug resistant tumors.
Figure 1.
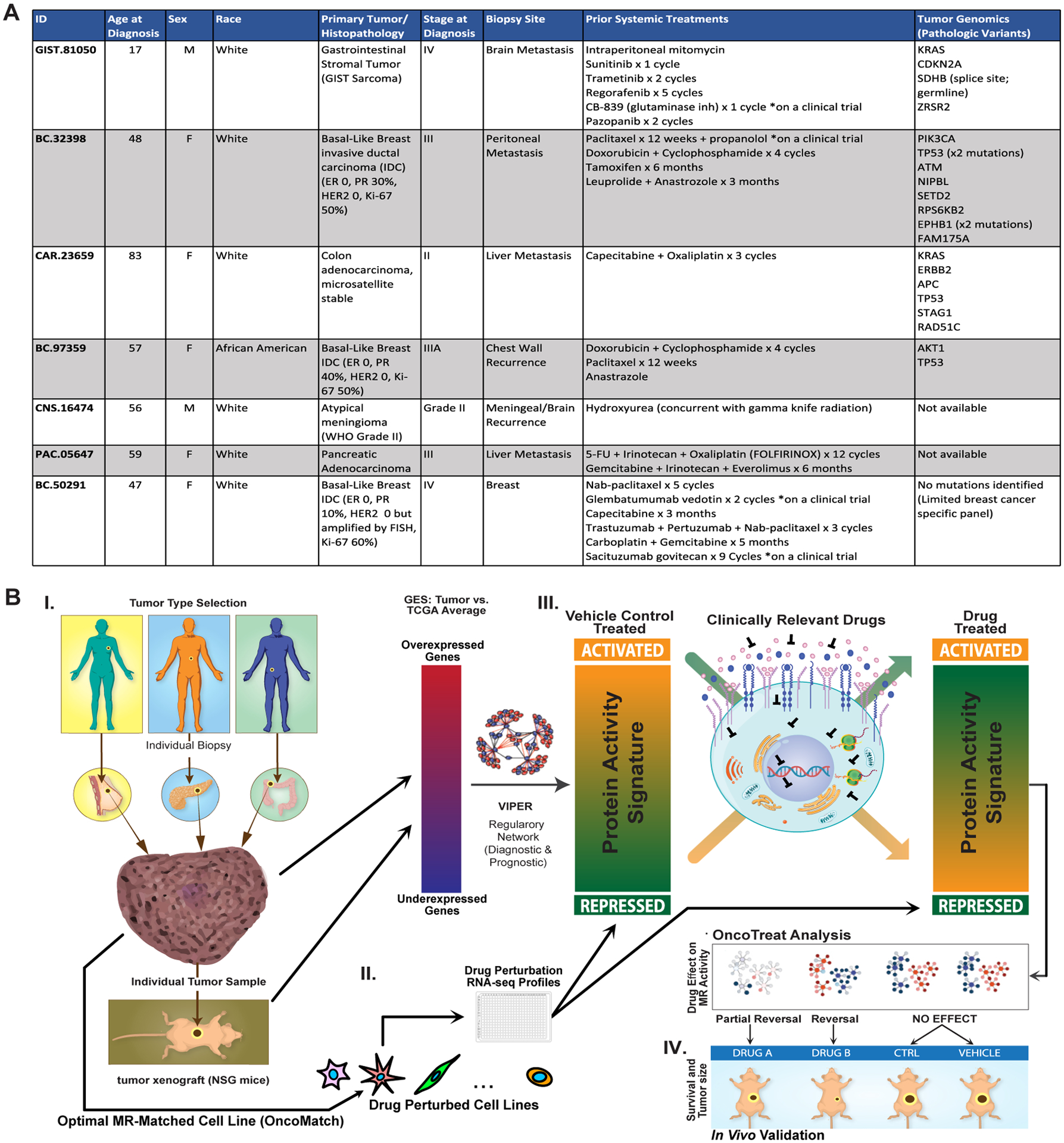
N of 1 study overview. (A) Clinical characteristics, prior systemic treatment, and tumor genomic profiling (if available) for the seven subjects. (B) Study conceptual diagram. I. Adults with advanced solid tumors with progression or intolerance to standard treatments are enrolled. Fresh biopsy tissue is partitioned for clinical pathology review, RNASeq, and xenografting into immunodeficient mice (PDX). Engrafted PDX tumors are also profiled by RNASeq and VIPER to confirm fidelity to the patient-derived tumor (OncoMatch, see Methods). II. High throughput drug screens have been completed in cognate cell lines with high fidelity to distinct cohorts of patient tumors based on recapitulation of Master Regulator (MR) protein activity (Bonferroni p < 10−10 by OncoMatch), collectively comprising the PanACEA database. Cells were perturbed at sub-lethal drug concentrations, and VIPER analysis of post-perturbation RNASeq allows for de novo mechanism inference for each drug in each cellular context. III. VIPER analysis of the patient tumor identifies top MR proteins and drugs are predicted by two methods. First, individual activated druggable MR proteins, e.g. protein kinases and epigenetic regulatory enzymes, are identified (Bonferroni p < 10−5 by OncoTarget). Second, using the best matched cell line(s) in PanACEA, drugs are ranked based on their inverting effect on the top MR proteins, i.e. tumor checkpoint module (TCM)-inverting drugs (Bonferroni p < 10−5 by OncoTreat). IV. Up to six predicted drugs are selected for experimental validation, based on OncoTarget or OncoTreat p-value and a number of practical selection criteria. Mice from early PDX passages (usually P1) are randomized into candidate drug arms, Negative control drug arms, and a Vehicle control arm.
Due to the constraints associated with the expansion and use of early PDX passages for preclinical therapeutic studies, we applied several practical selection criteria to further prioritize predicted drugs for in vivo validation. Specifically: (a) Only drugs classified as antineoplastic agents were considered, (b) Drugs were eliminated if the patient had been previously treated with them, (c) When perturbation profiles were available from suitable models—i.e. for GIST, meningioma and breast cancer—OncoTreat-predicted drugs were selected over OncoTarget-predicted ones with comparable p-values, to ensure an overall balanced number of tested predictions from the two methods, (e) Drugs predicted for the patient but not for the corresponding PDX model were eliminated, and (f) When multiple inhibitors sharing the same canonical mechanism were identified—e.g., multiple HDAC or topoisomerase II inhibitors—only the top FDA approved and/or the most statistically significant one was selected.
The 35 unique patient-specific drugs prioritized for in vivo validation, as well as prediction rationale and dosing schedule, are summarized in Table S2 (see Table S3 for further details on drug prediction and selection in each of the seven models).
OncoTarget and OncoTreat Methodology:
We have developed two complementary, RNA-based assays to transform VIPER-based (16) tumor sample-specific protein activity profiles into actionable drug response predictions (see schematics in Figures 1B and S1A-C).
The first one (OncoTarget) identifies aberrantly activated proteins (threshold p ≤ 10−5, Bonferroni corrected, as measured by VIPER) for which a high-affinity inhibitor is currently available, see Methods. To assess target actionability, we analyzed DrugBank (RRID:SCR_002700), the SelleckChem database (RRID:SCR_003823), published literature, and public information from drug development pipelines, resulting in a curated list of 180 proteins representing validated, high-affinity targets of clinically-relevant small molecule compounds (Table S4). These include proteins that are rarely if ever mutated in cancer, such as topoisomerases, chromatin remodeling enzymes, and proteins aberrantly activated by autocrine, paracrine, or endocrine signals.
The second one (OncoTreat) leverages a large compendium of RNASeq profiles, generated to represent the transcriptional response of cell lines to a comprehensive repertoire of antineoplastic agents. These allow identification of compounds capable of inverting the transcriptional activity of a TCM module (TCM-inverters)—as defined by the 25 most activated (25↑) and 25 most inactivated (25↓) candidate MR proteins in a patient tumor—at a conservative statistical significance threshold (p ≤ 10−5, Bonferroni corrected, by 1-tailed analytic-rank based enrichment analysis — aREA), (Figures 1B and S1C) (13, 16), see Methods. As such, rather than using a priori knowledge, OncoTreat predicts TCM-inverter drugs based on drug MoA assessed de novo from experimental perturbational assays—i.e., based on the differential activity of 2,556 regulatory proteins in drug vs. vehicle control-treated cells.
The number of candidate MRs in a TCM was selected based on the average number of MRs necessary to integrate the effect of genetic alterations in their upstream pathways, as assessed in (8). Specifically, we had reported that, for the vast majority of tumor subtypes in The Cancer Genome Atlas (TCGA), there is rapid saturation of mutational events in pathways upstream of the first 1 to 100 candidate MRs of each individual tumor, with 50 MRs sufficient to account for 80% of all mutations in all but 5 of 20 tumor types analyzed (COAD, HNSC, SKCM, STAD, OV). However, we also show that drug prediction is extremely robust and reproducible for any TCM size ranging from n = 10 to 200 activated and inactivated MRs, with mean Spearman’s pairwise correlation ranging from rs = 0.89 in GIST-81050 to rs = 0.98 in BC-32398 (Figure S2A–E).
Cell Line and PDX Models:
Drug MoA and TCM-inversion potential were assessed based on perturbational profiles in selected, high-fidelity (cognate) cell lines. These were identified based on their ability to recapitulate the TCM activity signature—i.e., the activity of the top and bottom 25 most differentially active MRs—of the greatest fraction of a histology-matched patient cohort. Thus, cognate cell lines are meant to represent a biological surrogate of the tumor of interest only in terms of providing an optimal in vitro context to assess tumor-relevant drug mechanisms of action, thus maximizing statistical power. Specifically, assessing MR protein activity decrease or increase following drug perturbation is best accomplished in cells where these are already significantly activated or inactivated, respectively. Likewise, to account for potential tumor drift effects in PDX passaging, we assessed whether the PDX models used in the study also recapitulated their corresponding patients’ TCM-activity signatures. As such, we assess model fidelity to a human tumor based on TCM MR enrichment (i.e., top 25↑+25↓) of the human tumor in differentially active and inactive proteins in the model, respectively (OncoMatch analysis (13, 20) (bioRxiv 2019.677435), threshold Bonferroni p ≤ 10−10, see Methods).
OncoTreat Cell Line Fidelity Assessment:
To illustrate the selection process, in Figure S3A we show the MR-based fidelity of the top 12 breast cancer cell lines identified as candidate cognate models of BLBC tumors in TCGA, as annotated by PAM50 classification (21, 22). Cell lines were selected from a total of 97 profiled breast cancer cell lines, unbiased to receptor status or PAM50 classification, as part of a comprehensive repository that included both the Cancer Cell Line Encyclopedia (CCLE) (23) and the Genentech Cell Line Screening Initiative (gCSI) (24). BT20 emerged among the top five candidates based on the number of patient tumors (78 of 173) whose TCM-activity signature it recapitulated (Bonferroni p ≤ 10−10, by OncoMatch). Since cognate cell lines are identified strictly based on TCM activity recapitulation, we do not expect them to necessarily recapitulate other phenotypic or even transcriptomic characteristics of their matched patients. For instance, while none of the top 12 cell lines identified for BLBC patients would be classified as luminal, several of them were claudin-low or mesenchymal like (e.g., MDAMB231, SUM159PT, BT549, CAL120), unclassifiable (e.g., BT20, HCC1395), or even ER-negative, HER2-amplified/enriched (e.g., JIMT1, HCC1954) by transcriptome-based classifications (25, 26). This is consistent with the fact that BLBC patients in TCGA presented conserved activity of the most differentially active MRs, regardless of claudin or HER2 status (7).
In Figure S3B, we show the fidelity of four cognate cell lines that were used to generate drug perturbation profiles to support OncoTreat analyses for five study patients. While ASPC1 was also identified as a high-fidelity model for the pancreatic tumor in the study, ASPC1-based perturbation profiles were not completed in time for drug prediction and in vivo validation. Nonetheless, for completeness, we are sharing the ASPC1 drug perturbation data as part of this study. As shown, BT20 represents a high-fidelity model for tumor BC-32398 (normalized enrichment score, NES 14.5, p = 10−48) and BC-97359 (NES 8.0, p = 10−15), but not for BC-50291 (NES −3.9, p = 1); both GIST cell lines, GIST430 and GISTT1, were identified as high-fidelity models for GIST-81050 (p < 10−40) despite not harboring the patient SDHBDel/KRASG12D alterations, but rather canonical KIT mutations; finally, the meningioma cell line IOMM was borderline for CNS-16474 (NES 3.3, p = 0.0005).
OncoTreat Perturbation Profile Generation:
On average, ~350 drugs were profiled in each cognate cell line model, including 138 FDA approved antineoplastics, about 170 late-stage experimental drugs in phase II and III oncology clinical trials, as well as a variable number of additional compounds from diverse libraries with cell line-specific EC50 ≤ 2 μM (Table S5), see Methods. Cells were harvested at 24 hours following perturbation with each compound at two sublethal concentrations—the 48-hour EC20 and one tenth of this concentration, as determined by 10-point dose response curves—as well as at 6h (in selected cell lines) to assess early MR activity changes. We used the highest sublethal drug concentration to reveal signatures optimally reflective of the drug’s MoA rather than non-specific effector proteins in stress or death pathways (27, 28). To avoid testing drugs at non-physiologically relevant concentrations, we also capped concentrations at their CMax, defined as the peak serum concentration for the drug’s Maximum Tolerated Dose (MTD), from published pharmacokinetic studies in humans, when available.
Multiplexed, low depth (1–2M reads) RNASeq profiles were generated for each compound using the PLATE-Seq technology (29), which supports low-cost, fully-automated pooled library generation from 96 or 384-well plates. Drug MoA—representing the drug-mediated differential activity of all regulatory proteins—was then assessed by VIPER analysis of drug vs. vehicle-treated (DMSO) samples. In Figure 2A, as a representative example, we show hierarchical clustering of VIPER assessed drug MoA in BT20. Interestingly, while several MoA-related drugs were identified within the same clusters (e.g., topotecan and irinotecan in Cluster 1, sorafenib and vandetanib in cluster 5, and dasatinib, ponatinib, and nilotinib in Cluster 6), several drugs induced similar differential protein activity profiles, despite having distinct high-affinity targets (e.g., thioguanine, a guanine analog, sorafenib, a multi-kinase inhibitor, and vorinostat, a pan-HDAC inhibitor, in Cluster 5). This suggests that cellular networks in BT20 cells may effectively canalize drug MoA towards a small number of relatively distinct cellular responses. Additionally, as shown, several drugs produced highly conserved MoA at multiple concentrations (e.g., topotecan at 0.076 and 0.76 μM, crizotinib at 0.72 and 7.2μM, flubendazole at 1.44 and 14.5 μM, among several such examples, suggesting high reproducibility of these assays, as also previously shown (13, 29)).
Figure 2.
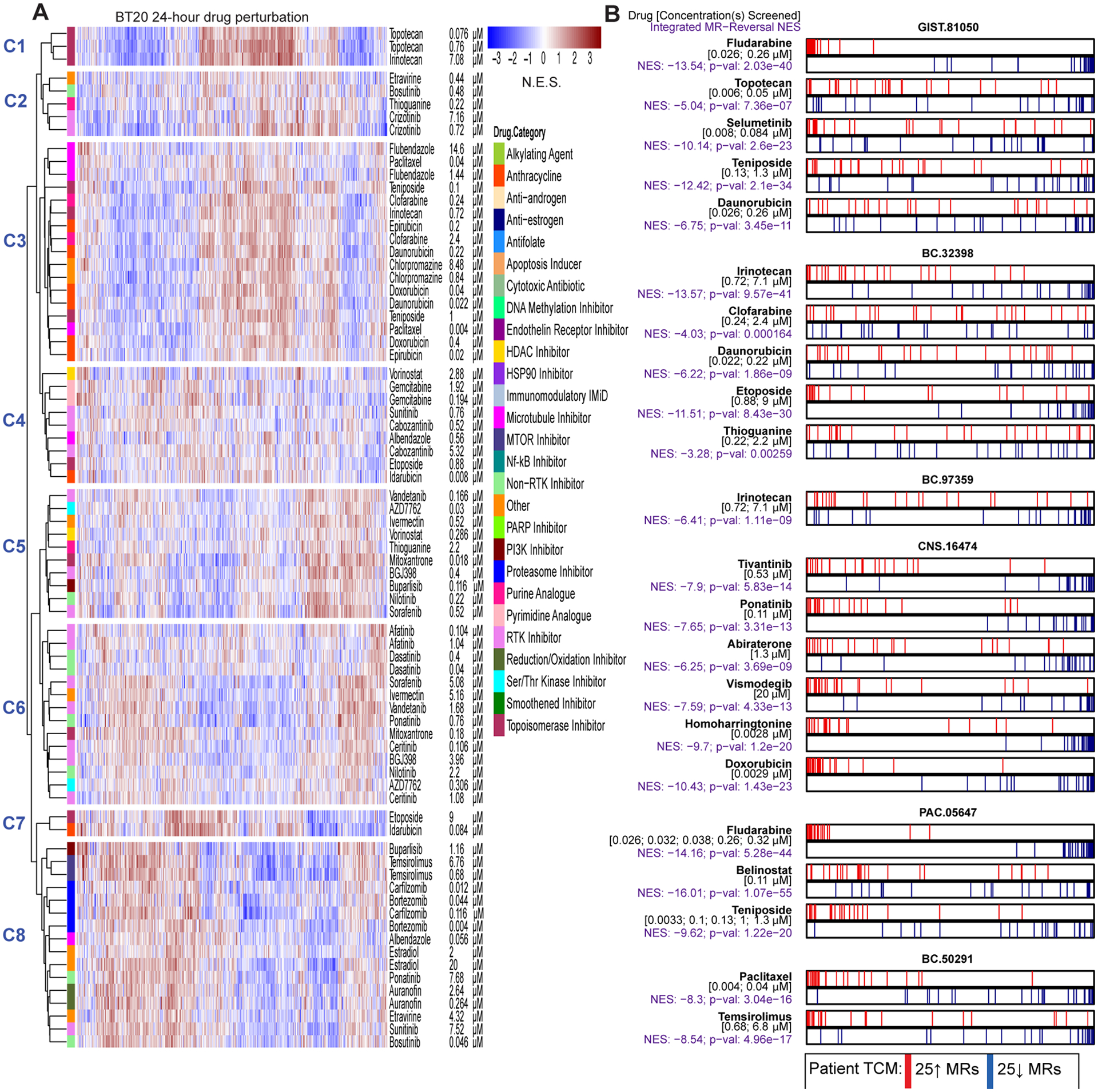
Drug context-specific mechanism and tumor checkpoint module (TCM)-inversion. (A) As an illustrative example, we show a heatmap for the 24-hour drug perturbation in the BT20 breast cancer cell line. The heatmap shows the differential protein activity profile of 38 drugs in BT20 cells compared to vehicle control, annotated by their canonical mechanism and the two sublethal concentrations (EC20 and one-tenth of EC20) screened. VIPER monitored proteins are shown in the columns and we use unsupervised hierarchical clustering to highlight drugs that induce similar transcriptional response, i.e. context-specific observed mechanism of action. (B) Perturbation screens in five relevant cell lines were used to generate the OncoTreat drug predictions we report on here. OncoTreat uses the context-matched de novo drug mechanism information to identify top TCM-inverter drugs. For each patient Id (e.g., GIST-81050) and predicted drug (e.g., fludarabine), we show the placement of the 25 most activated (red bars) and 25 most inactivated (blue bars) patient tumor master regulator (MR) proteins on the drug-induced signature in the cognate cell line(s)—proteins sorted left to right from the most differentially inactivated to the most activated in drug- vs. vehicle control-treated cells. For each model and drug, we report the concentrations whose effect was averaged to rank protein activity, the normalized enrichment score (NES) assessing TCM-inversion in the drug signature, as measured by the aREA algorithm, and the associated p-value. Negative NES indicates TCM-inversion. All but two predictions met the predefined significance threshold (Bonferroni p < 10−5), with clofarabine and thioguanine borderline predictions for BC-32398. For the pancreatic tumor (PAC-05647), drug perturbation profiles in the ASPC1 cell line were not available in time to predict drugs for in vivo testing. Thus, predictions were based on OncoTreat analysis using non-matched cell line models, BT20, GIST430, GISTT1, and IOMM, and integrated using Fisher’s method.
Drug MoA profiles from the appropriate cognate cell line(s) were used to generate OncoTreat predictions for patient and PDX tumors. All regulatory proteins represented in the drug MoA were ranked from most inhibited to most activated, thus providing an optimal reporter assay to assess TCM-inversion. TCM-inversion assessment in vitro for the 22 OncoTreat-predicted drugs that were prioritized for in vivo validation, is shown in Figure 2B.
PDX Fidelity Assessment:
Several groups have described clonal drift that occurs with sequential passages in PDX models (30). As a result, to minimize drift, we performed all therapeutic studies in the earliest feasible passage, P1 – P5. Additionally, prior to commencing therapeutic testing, we assessed both model fidelity and patient/PDX conservation of drug prediction, as proposed in (31). Following successful engraftment of tumors (P0 passage), we performed RNASeq and subsequent VIPER, OncoTarget, and OncoTreat analyses to determine (a) the MR-based fidelity of the PDX tumor to the patient tumor and (b) conservation of drug predictions. Drugs predicted from patient sample analysis, but not predicted by analysis of the PDX, were only used in the therapeutic study if alternative options were not available.
Six of the seven PDX models, GIST-81050, BC-32398, CAR-23659, BC-97359, CNS-16474, and PAC-05647 met the predefined fidelity threshold (Bonferroni p ≤ 10−10, by OncoMatch), with NES ranging from 13.8 to 17.3 (Figures 3A and S4). In fact, in GIST-81050 and CAR-23659, there was almost perfect fidelity to patient TCM activity, while in BC-32398, BC-97359, CNS-16474, and PAC-05647 there were a handful of MRs with different activity rank between patient tumor and PDX. In BC-50291, however, there was no appreciable fidelity (p = 0.89). Consequently, in BC-50291, we tested drug predictions for the patient tumor that were not conserved in the PDX.
Figure 3.
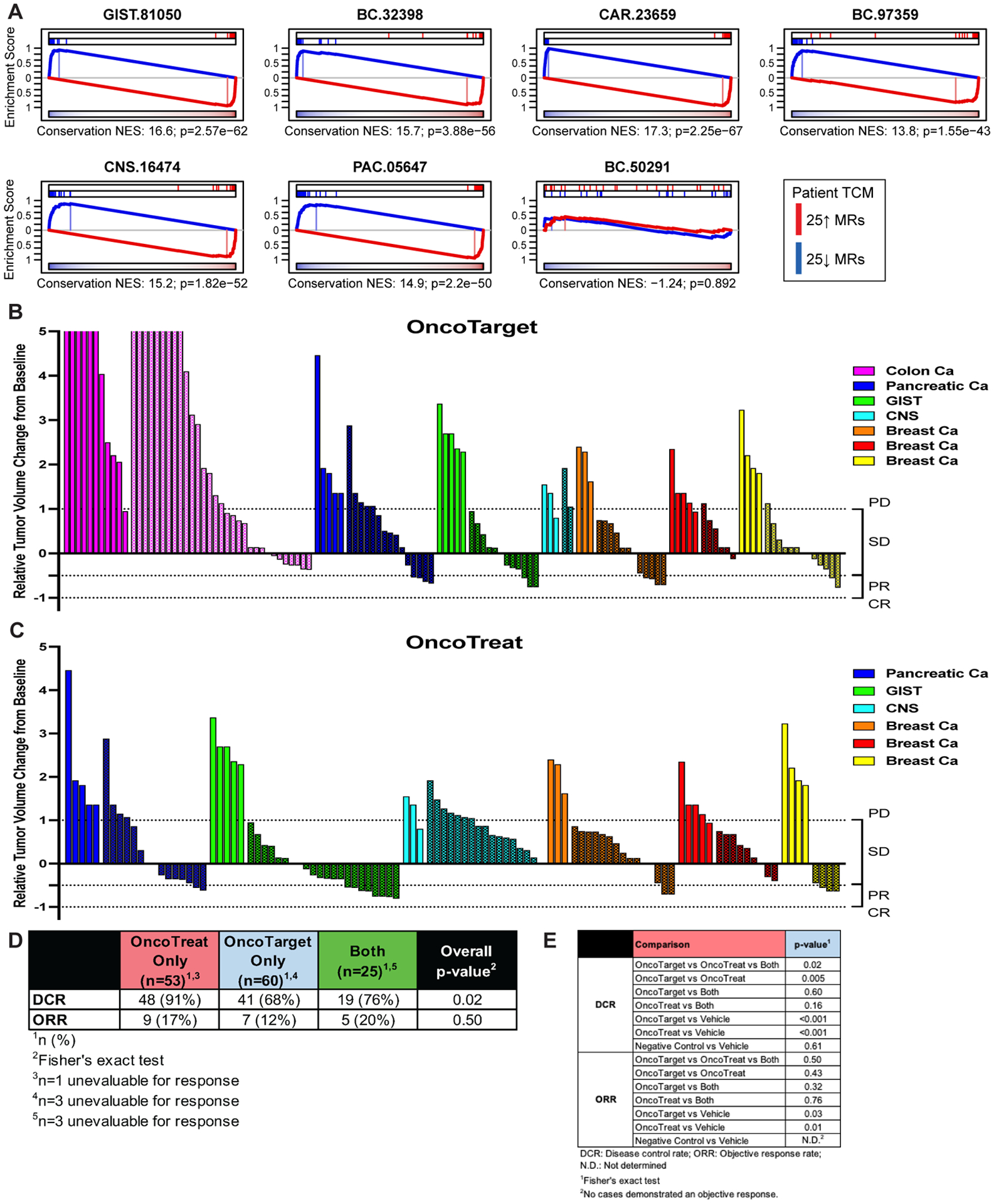
Treatment response in patient-derived xenograft (PDX) models. (A) Fidelity assessment of the seven PDX models. Enrichment of patient tumor master regulators (TCM) in differentially active and inactive proteins in mature P0-passage PDX tumor samples, assessed by OncoMatch. TCM activity was highly conserved in six out of seven models, but not in the BC-50291 breast cancer model, indicating significant early passage drift. (B, C) Waterfall plots for end-of-study time point showing the relative tumor volume change for mice treated for a median of 29 days with OncoTarget-predicted drugs in seven PDX models (B), and OncoTreat-predicted drugs in six PDX models (C). Plots are grouped and color coded by model, with vehicle (solid bars) and drug-treated (textured bars) mice within each PDX presented side by side. OncoTreat predictions were not made for CAR-23659 due to lack of completion of a drug perturbation screen in a cognate colon cancer cell line. (D) Summary of response rates at the end-of-study for each drug prediction category (OncoTreat Only, OncoTarget Only) including a non-overlapping category for drugs predicted by both OncoTarget and OncoTreat (Both). A disease control rate (stable disease + partial response + complete response) of 68% (n = 41/60) and objective response rate (partial + complete response) of 12% (n = 7/60) were observed when treating with OncoTarget-predicted drugs. Responses from OncoTarget [or both]-predicted drugs were primarily stable disease (n = 48) and partial response (n = 12). A disease control rate of 91% (n = 48/53) and objective response rate of 17% (n = 9/53) were observed when treating with OncoTreat-predicted drugs. OncoTreat [or both]-predicted drugs demonstrated stable disease (n = 55) and partial response (n = 14). Overall p-value (Fisher’s exact) is reported for DCR and ORR, assessing for a between groups difference in response rates across all drug prediction groups. (E) Summary statistics of overall and pairwise comparisons of drug prediction groups. Both OncoTarget and OncoTreat were highly accurate in predicting disease control (p < 10−3, 2-tailed U-test) and objective response (OncoTarget p = 0.03; OncoTreat p = 0.01) versus Vehicle control. Note, valid direct comparisons of OncoTarget and OncoTreat are limited by imbalances in number of predictions tested in different models.
OncoTarget and OncoTreat Predict Treatment Response in PDX Models:
A total of 35 individual drugs predicted by the analysis were evaluated in individual PDX therapeutic arms—including 21 OncoTarget-predicted and 22 OncoTreat-predicted, 8 of which were predicted by both methods. In a few cases, the same drug was predicted and tested in more than one model. After expansion of PDX models for the therapeutic study, animals were enrolled once tumor volumes reached ~100 mm3. Models were treated and tumor volume measurements were recorded up to a fixed end-of-study time point with a median of 29 days (range 21 – 30 days). A waterfall plot of the response of each individual mouse, grouped by PDX model, to prioritized and Negative control drugs is shown in Figure 3B–C and comparisons summarized in Figure 3D–E; responses are summarized in Table S6. Overall, across all predictions, Disease Control—comprising stable disease (SD), partial response (PR), or complete response (CR)--was observed for 30 of 35 predictions (85.7%).
We evaluated overall treatment response, at the end of treatment, for OncoTarget-predicted, OncoTreat-predicted, and for drugs predicted by both methods. Compared to Vehicle control, there were significant differences in disease control rate (DCR)—defined as rate of SD + PR + CR—and objective response rate (ORR)—defined as rate of PR + CR—for both OncoTarget (pDCR < 10−3, pORR = 0.03, by Fisher’s exact test) and OncoTreat-predicted drugs (pDCR < 10−3, pORR = 0.01) (Figure 3B–E). Drugs predicted by both had response rates comparable to drugs predicted by just one methodology (DCR = 76%, n = 19/25; ORR = 20%, n = 5/25). Due to unanticipated toxicity possibly related to study treatment (e.g., tumor ulceration in a breast cancer PDX), 3 mice were unevaluable for response in the OncoTarget cohort (BC-97359, n = 2 for MK-2206 arm, n = 1 for panobinostat arm), while 1 mouse was unevaluable in the OncoTreat cohort (BC-97359, n = 1 for irinotecan arm), and 3 mice were unevaluable for response in the Both cohort (GIST-81050, n=2 for daunorubicin arm, n = 1 for topotecan arm). While cautioning that the study was neither designed nor sufficiently powered to evaluate the efficacy of individual drug predictions, for completeness, we provide growth curves for each drug (Figure S5A–C).
As a set of appropriate negative controls, we evaluated response to randomly selecting antineoplastic drugs that were not statistically significant by either OncoTarget or OncoTreat (when available) analysis (i.e., p = 1 from both methods). We note that these are not true random controls but rather potential false negative predictions meant to assess the algorithms’ predictive power. Four PDX models (GIST-81050, CAR-23659, PAC-05647, BC-50291) were thus treated with 13 Negative control drugs (Table S2) and Vehicle control. To avoid bias due to differences in tumor growth rates, these drugs were tested in conjunction with an additional Vehicle control arm in each PDX, thus providing a tumor growth-independent reference. Disease progression was observed across all models treated with Negative control drugs and statistically indistinguishable from concurrent Vehicle control (Fisher’s exact p = 0.6, Table S6).
Cumulative Kaplan-Meier analysis of animals in the OncoTarget, OncoTreat, and OncoTreat + OncoTarget cohorts was performed (Figure 4A). The analysis demonstrates highly statistically significant improvement in disease control using agents predicted by either methodology, compared to Vehicle control (p < 10−4, 2-tailed log-rank test). In contrast, there was no statistically significant difference between animals in the Negative control and passage-matched Vehicle control cohorts (p = 0.38, log-rank) (Figure 4B). Additionally, the treatment-to-control ratio (ΔT/ΔC%: the ratio of change in volumes from baseline in the treatment/control arms), which corrects for baseline differences in model-passage growth rate, was significantly improved in the OncoTarget (mean ΔT/ΔC% = 14.3%, 95% CI: −1.2 – 29.9, p = 0.004, 2-tail Mann-Whitney U-test) and OncoTreat (mean ΔT/ΔC% = 17.3%, 95% CI: −0.8 – 35.3, p = 0.014) treated cohorts, compared to the Negative control cohort (mean ΔT/ΔC% = 46.1%, 95% CI: 25.5 – 61.1), with overall statistical significance (OncoTarget vs OncoTreat vs Negative control, p = 0.002, 2-tail ANOVA) (Figure 4C).
Figure 4.
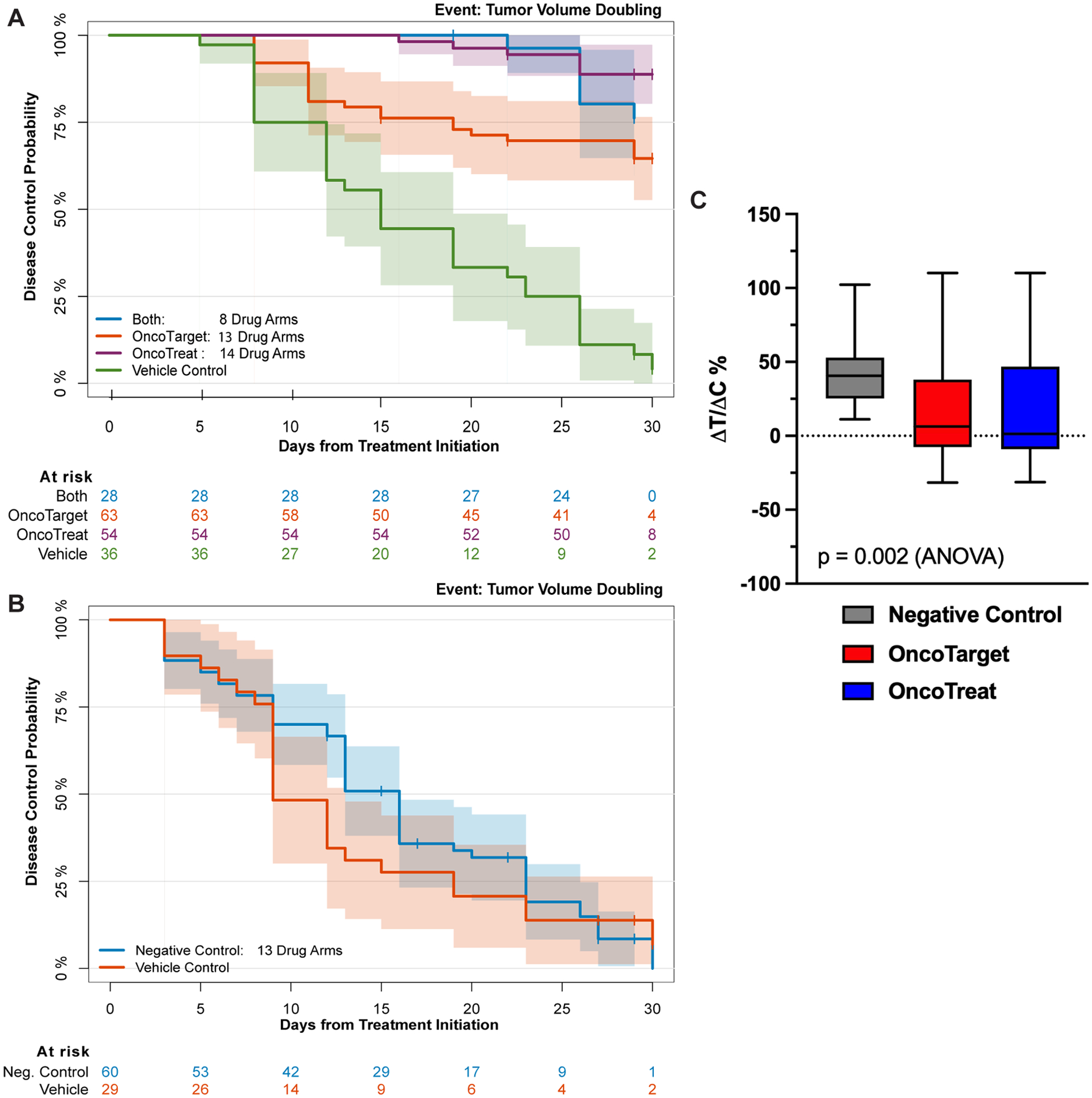
Kaplan-Meier and ΔT/ΔC% Analysis. (A) Kaplan-Meier plot for Disease Control showing significant differences for the arms treated with either OncoTarget-, OncoTreat-, or Both-predicted drugs compared to Vehicle control (p < 10−4, log-rank test). (B) Kaplan-Meier plot for Disease Control showing no difference between the Negative control drugs (not predicted by either OncoTarget or OncoTreat) and matched-Vehicle control (p = 0.38). (C) Boxplots showing the distribution of the treatment-to-control ratio (ΔT/ΔC%: relative change in volumes from baseline in the treatment/control) seen in animals treated with Negative controls, OncoTarget, and OncoTreat-predicted drugs, normalized to matched-Vehicle control. There is a statistically significant difference in mean ΔT/ΔC% in OncoTarget (p = 0.004, Mann-Whitney) and OncoTreat (p = 0.01) treated mice versus Negative controls (overall, p = 0.002, 2-tail ANOVA).
TCM-inversion by OncoTreat-predicted Drugs is Conserved In Vivo:
Pharmacodynamic (PD) studies are a critical aspect of drug development to elucidate drug MoA and to characterize primary and acquired drug resistance. PD assessment from early, on-treatment samples help to determine: (a) whether effective, OncoTreat-predicted drugs recapitulate in vivo the TCM-inversion that occurs in the cognate cell line(s) (i.e., MoA conservation) and (b) whether failure of OncoTreat-predicted drugs corresponds to inability to conserve TCM-inversion in vivo, perhaps due to pharmacokinetic factors, or occurs despite TCM-inversion, for instance due to later cell adaptation or clonal selection. The opportunity to investigate the second point was limited in the study, since most OncoTreat-predicted drugs demonstrated strong anti-tumor activity.
Samples for PD assessment were procured from two mice per treatment arm, for the four PDX models treated with at least three OncoTreat-predicted drugs—GIST-81050, BC-32398, CNS-16474, and PAC-05647. Mice were randomly selected for early sacrifice, independent of tumor size, three hours following the third dose, and were excluded from response assessment. The four selected models had the highest patient tumor fidelity (p < 10−10, Figure 3A) and were thus well-suited to evaluate MoA conservation. TCM-inversion was assessed by VIPER analysis of RNASeq signatures comparing drug- vs. Vehicle control-treated PDX tumor samples.
Overall, the vast majority of OncoTreat-predicted drugs with available PD samples—i.e., 15 of 18 (83%)—significantly recapitulated in vivo (p < 10−5, by 1-tailed aREA) the TCM-inversion predicted from cognate cell line perturbational assays, in vitro, with p-values ranging from 10−5 (teniposide in PAC-05647) to 10−40 (daunorubicin in BC-32398) (Figure 5A–D). The three drugs that failed to conserve MoA in vivo included abiraterone in CNS-16474, which was borderline for achieving disease control, belinostat in PAC-05647, which achieved disease control by end-of-study, and daunorubicin in GIST-81050, which also achieved disease control (Table S6). As belinostat was the only HDAC inhibitor evaluated for PD effect, one is tempted to speculate if the early time point was inadequate for an epigenetic modifying drug to fully implement its in vivo effect. Intriguingly, daunorubicin demonstrated strong TCM-inversion in the BC-32398 model but not GIST-81050, where it inverted the 25↓ MRs but failed to invert the 25↑ MRs, and yet had strong anti-tumor activity in both models. Conversely, one drug failed to achieve disease control (homoharringtonine in the CNS-16474 model) despite recapitulating TCM-inversion in vivo.
Figure 5.
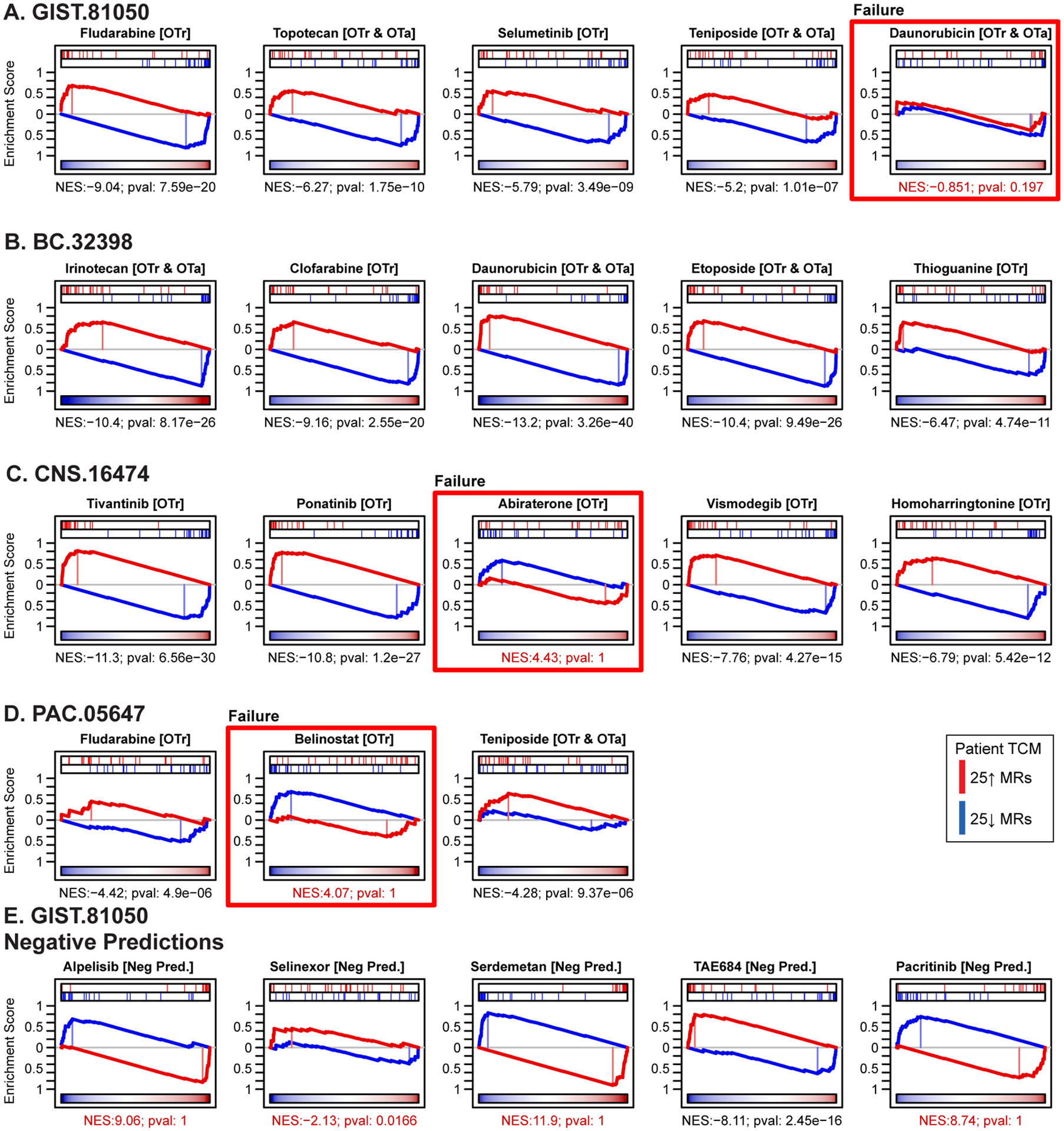
Pharmacodynamic assessment of tumor checkpoint module (TCM)-inversion in vivo, in early on-treatment biopsy samples. In the four PDX models where we tested three or more OncoTreat predicted drugs, two mice from each drug arm were sacrificed after the 3rd dose. VIPER was used to generate a differential protein activity signature for each drug-treated versus Vehicle control-treated arm in the respective PDX model. Enrichment of activated and inactivated patient tumor master regulators in this signature was assessed by aREA. (A – D) Statistically significant TCM-inversion in vivo (Bonferroni p < 10−5), which recapitulated the predictions from cognate cell lines in vitro, was confirmed for 15 of the 18 OncoTreat-predicted drugs for which PD samples were available. Exceptions denoted by red boxes included daunorubicin in GIST-81050, which however achieved disease control, abiraterone in CNS-16474, which induced only modest tumor growth inhibition, and belinostat, an epigenetic modulator, in PAC.05674, which achieved disease control. (E) As expected, four of five Negative control drugs tested in GIST-81050, did not significantly invert TCM activity. TAE684, however, did induce significant TCM-inversion at this early time point. All five drugs failed to induce disease control in this model.
As expected, four of the Negative control drugs—alpelisib, selinexor, serdemetan, and pacritinib—failed to achieve significant TCM-inversion in vivo (Figure 5E) and did not achieve disease control by end-of-study. TAE684 did induce TCM-inversion in vivo (p = 10−16) in the early on-treatment sample, yet still failed to achieve disease control.
In summary, 15 of 18 OncoTreat predicted drugs, including 13 of 16 (81%) that induced disease control, recapitulated significant TCM-inversion in early on-treatment PD samples. This is consistent with our hypothesis that inference of drug induced TCM-inversion in carefully selected models, whether they be cell lines or PDXs, is robust and a feature of the hyper-connected and autoregulated nature of TCMs.
Case Report of N-of-1 Clinical Application:
The proposed PCM framework discussed in this study is uniquely suited to identify therapeutic alternatives in real world scenarios, even for rare cancers lacking actionable mutations and standard of care options. Calcifying Nested Stromal Epithelial Tumor (CNSET) is an exceptionally uncommon primary hepatic tumor that occurs in children and young adults, with only about 40 cases reported in the literature (32). While localized disease is often effectively cured with surgery, recurrent and de novo metastatic disease demonstrates chemotherapy resistance and there are no proven therapeutic options (33–36). We thus report the observed clinical outcome for a CNSET case, where OncoTarget was used off-trial to help guide treatment selection when all other options were exhausted.
A 14-year-old male reported a 1-month history of abdominal pain and fatigue. A computed tomography (CT) scan of the chest and abdomen revealed a large hepatic mass with multiple satellite liver tumors and pulmonary metastases. The mass was biopsied and histopathologic evaluation was consistent with CNSET. The family was initially hesitant to initiate chemotherapy, but the patient developed progressive hepatomegaly, anorexia, weight loss, constipation, and anemia in the subsequent three months. Memorial Sloan Kettering IMPACT (37), a targeted next-generation sequencing panel covering 468 genes, was performed on a biopsy specimen and demonstrated a CTNNB1 hotspot mutation, TERT promoter gain of function mutation, and an NTRK3 point mutation not known to predict response to currently available TRK inhibitors. The estimated tumor mutational burden was only 2.6 per megabase, predicting a low likelihood of response to immune checkpoint inhibitors.
Based on the observation that the tumor shared biological features with Wilms’ Tumor (CTNNB1 and TERT mutations, WT1 and β-catenin expression by immunohistochemistry), the patient was treated with 10 neoadjuvant cycles of a “Wilms’ tumor like-regimen”, including five cycles of vincristine/irinotecan, four cycles of vincristine/dactinomycin/doxorubicin, and one cycle of cyclophosphamide/topotecan (38, 39). There was a partial response to chemotherapy and the patient successfully underwent debulking surgery. Post-operative chemotherapy was complicated by the development of severe colitis, and the family elected to discontinue systemic therapy. Over the next six months, there was evidence of significant disease progression in the liver and lungs (Figure 6A), and the patient developed biliary obstruction and transaminitis that made him ineligible for clinical trials and precluded the use of most chemotherapy agents.
Figure 6.
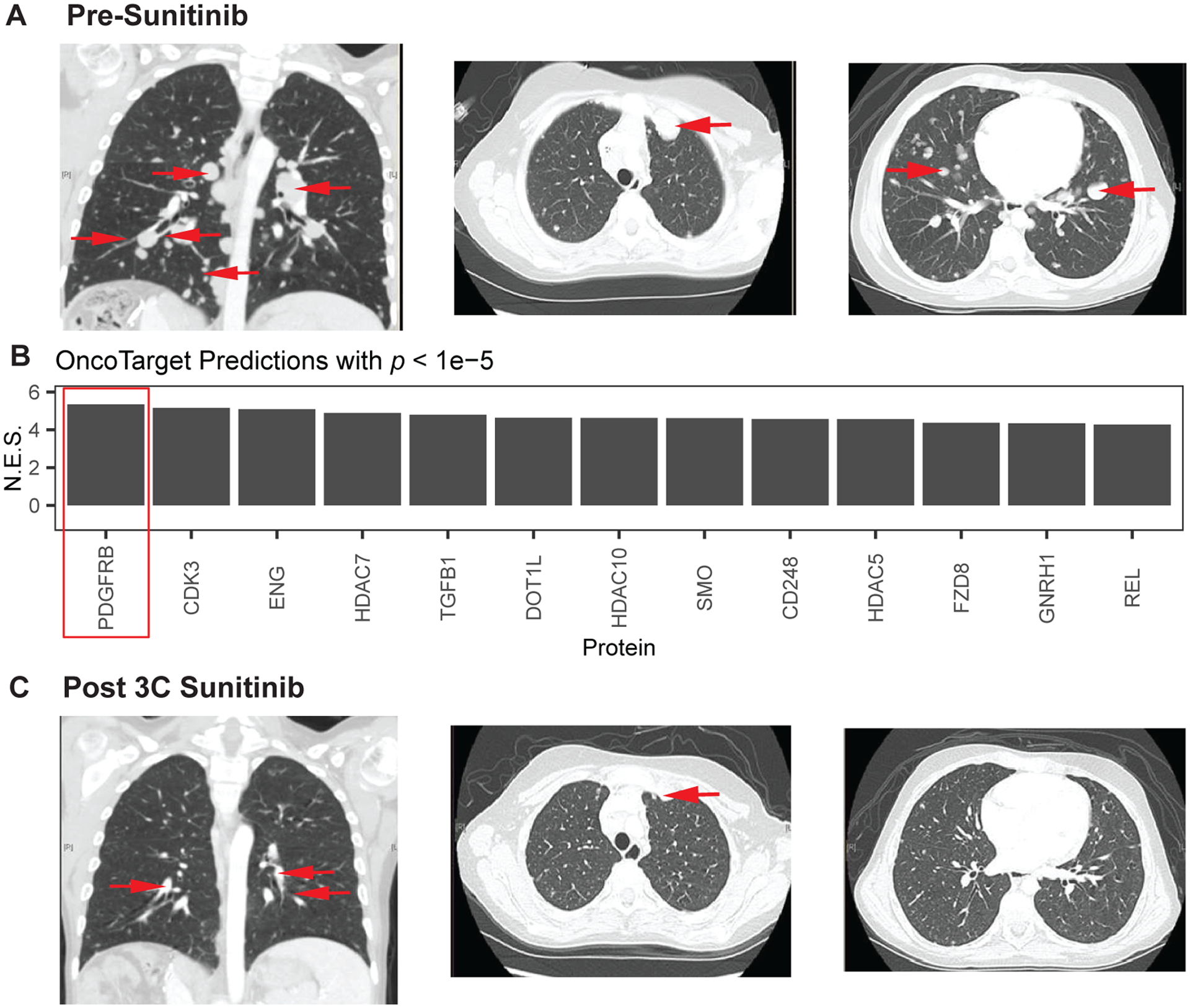
Response to sunitinib in a pediatric patient with Calcifying Nested Stromal Epithelial Tumor (CNSET) with aberrant activation of PDGFR-B, as assessed by OncoTarget analysis. (A) Chest computed tomography (CT) scan pre-sunitinib treatment: coronal section (left) and axial sections (middle and right) demonstrate numerous pulmonary metastases (red arrows) ranging from less than 1 to close to 3 cm in size. Several of the tumors were new or growing on serial scans during the preceding six months. (B) OncoTarget predictions on patient tumor. Multiple proteins were noted to be significantly activated (Bonferroni p < 10−5), but PDGFR-B activation was both the top prediction and also judged to be most actionable by the clinical team. (C) Chest CT following three cycles of sunitinib (six weeks each). Corresponding sections demonstrate that several of the tumors had decreased in size (red arrows) or were no longer radiologically evident.
Given the lack of remaining viable therapeutic options, tumor tissue was sent for the CLIA-compliant OncoTarget test. The most significantly activated targetable protein was PDGFR-B (Bonferroni p = 10−7, Figure 6B). After discussing the results with the family, including the absence of clinical data on targeting PDGFR-B in this exceedingly rare malignancy, we decided sunitinib would be the best candidate drug, given its relative selectivity for PDGFR-B over other kinases (RRID:SCR_003823), accessibility as a drug approved by the FDA in 2006, and safety data in the context of impaired hepatic function and pediatric patients. The patient had a partial response to the first cycle of sunitinib (six weeks) which deepened by the end of cycle 3 (Figure 6C). Remarkably, this patient who had rapidly progressing treatment refractory cancer, has had a durable response and remains on sunitinib, now for two years from his original presentation with only mild side effects such as fatigue. While OncoTarget did predict response, a caveat is that we cannot be certain of mechanism. As pediatric patients do not routinely undergo repeat biopsies when responding to a therapy, we could not confirm treatment effect on PDGFR-B activity or changes in activity of other canonical targets of sunitinib, including KIT and less potently VEGFRs.
Pharmacotype Identification for Clinical Trial Design:
The OncoTarget and OncoTreat approaches can identify multiple candidate drugs for the treatment of most tumors, a majority of which induced disease control in PDX models. Given the inherent conservation of MR and TCM activity within cancer subtypes, as identified by protein activity-based cluster analysis (7), it is reasonable to expect that subsets of patients with predicted sensitivity to the same drugs (pharmacotypes) should emerge from these analyses. Indeed, the majority of TCGA cancer cohorts were effectively stratified into 2 to 7 pharmacotype clusters by joint OncoTreat/OncoTarget analysis.
Figure 7A provides a representative example for the BLBC subtype of breast cancer in TCGA, with the pharmacotype cluster assignment of the three BLBC patients shown. Consistently, tumors are predicted to respond to distinct, pharmacotype-specific drugs. Full pharmacotype stratification of patient cohorts representing the cancers discussed in this study is provided in Figure S6A–D.
Figure 7.
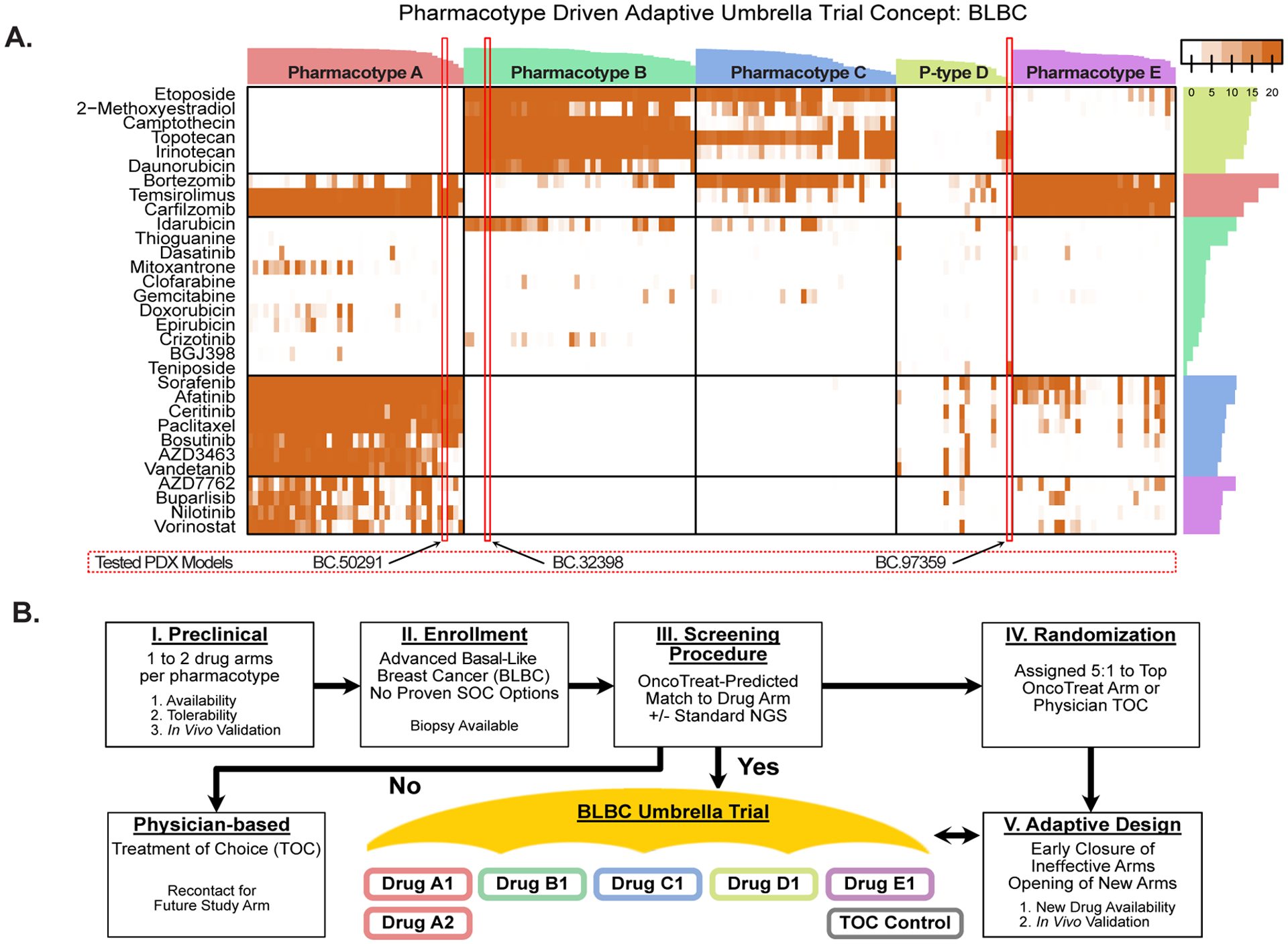
Pharmacotype-based umbrella trial concept. (A) Heatmap showing top OncoTreat predictions (-log10(p)) for 173 basal-like breast cancer (BLBC) samples from The Cancer Genome Atlas (TCGA), as well as for the three BLBC samples from patients enrolled in the study (BC-50291, BC-32398, and BC-97359). Following unsupervised partition-around-medoids-clustering, drug predictions cluster into five main pharmacotypes, with the three N of 1 cases clustering with samples in pharmacotype A, B, and D, respectively. Bars on the top and to the right of the heatmap indicate the cluster reliability score (13) for each tumor sample and drug, respectively. Importantly, at least a few predictions are generated for the majority of tumors, using existing drug perturbation data in PanACEA from one or a few high-fidelity cognate cell lines. (B) Schema for a pharmacotype-based umbrella trial concept in BLBC. One or more drugs per pharmacotype may be prioritized as hypotheses for an umbrella clinical trial, based on availability, tolerability, and preclinical validation in relevant TCM-activity-matched PDX models (I). Patients with advanced BLBC who have exhausted all proven effective treatment options would be enrolled and undergo biopsy (II). An initial screening phase would determine if OncoTreat predicts a match to any of the open drug arms (III). Patients with a statistically significant match to a specific pharmacotype would be randomized 5:1 to OncoTreat-predicted drug versus physician treatment of choice (IV). Using an adaptive design, arms that fail to demonstrate efficacy would be closed early and new arms would be opened, as drugs become available (V).
Discussion
The oncogene addiction (3) and immunotherapy (4) paradigms have illuminated PCM’s potential to meaningfully improve the outcomes of some patients. These approaches are straightforward to implement in the clinic. However, methodologies to predict response to the full repertoire of available antineoplastics, based on rapid and inexpensive RNASeq profiling, are currently underdeveloped and would represent a welcome addition. Here we present a first-in-class application of two complementary and scalable transcriptome-based tests, OncoTarget and OncoTreat, to predict drug sensitivity on an individual patient basis. Critically, both methodologies are NY and CA Department of Health approved and CLIA compliant, and predictions can be generated within 2 to 3 weeks after biopsy, including for OncoTreat if perturbational profiles in a cognate cell line are available.
We encountered a number of practical challenges during this large-scale effort that are worth noting. First, as discussed, both OncoTarget and OncoTreat depend on the VIPER-based measurement of protein activities. We have published extensively on the accuracy and reproducibility of VIPER (8, 16), which compares favorably to gene and protein abundance estimation. However, since, unlike binary determination of genetic alterations, VIPER produces a fully quantitative assessment subject to inherent biological and technical noise, we decided to use a highly conservative statistical significance threshold for predicting drug sensitivity (Bonferroni p ≤ 10−5). As a result, our analyses may likely miss drugs that could have been beneficial (false negatives). In addition, protein activity measurement by VIPER, as reported (16), may be incorrect for 10–20% of proteins, leading to both false positive and false negative drug predictions.
Second, there are several recognized limitations to the use of PDX models. Tumors undergo clonal evolution under various selection pressures, including available nutrients, organ-specific environment, immune editing, pharmacological treatment, and growth kinetics (40). The selection pressures in an immunocompromised PDX may differ from those of the original human host, thus resulting in rapid drift. To minimize this challenge, we restricted therapeutic studies to early passages and confirmed model fidelity on the basis of preserved TCM-activity. Due to cost and constraints in propagating and expanding models at a single early passage, we could not test all statistically significant drugs predicted by OncoTarget and OncoTreat and had to implement practical selection criteria. We also could not test a more comprehensive list of negative control drugs, such as drugs the patient had previously or was currently receiving, or a broader panel of randomly selected drugs, blinded to the OncoTarget and OncoTreat results, across all models. Furthermore, the role of the immune system and thus the effect of immunotherapy is not evaluable in these immunodeficient models.
Additionally, the development of a xenograft from implanted patient tumor tissue may be influenced by sundry factors—including histology, mitotic rate, and fraction of cancer stem cells (41)—while the timeline between implantation and completion of drug testing can vary greatly (e.g., 3 to 18 months in our experience), with some tumor types rarely taking in the xenograft. Consistent with the experience of other groups, less than half of attempted PDXs could be established (Table S1), and there was obvious tumor type bias (42–48). While we would not anticipate these factors to bias comparisons of response between OncoTarget/OncoTreat-predicted and Negative control antineoplastic drugs, it nonetheless limits our ability to confirm the utility of our methodologies in certain tumor types. After a planned interim statistical analysis demonstrated highly significant findings, and given budgetary constraints, we only tested drugs in a fraction (the first seven) of established PDXs. Nonetheless, we believe that our systematic study of 35 algorithmically-predicted drug arms and 13 Negative control drug arms represents the largest such effort to date aimed at validating computational drug sensitivity predictions in vivo.
We further note, that there is no consensus on the optimal PDX end point that best predicts response in patients. While clinical response assessments using RECIST are based on unidimensional measurements, bi-directional caliper measurements to estimate tumor volume are standard practice in mouse studies, but are prone to significant interobserver variability (49, 50). RECIST defined criteria for progressive disease (i.e., >20% increase in the sum of 5 target lesions) translate to a 73% increase in spherical volume (51). When considering the various factors that can contribute to variability in tumor measurements, our definition of progressive disease (100% tumor volume increase relative to baseline) remains within reasonable margins of error (± 1–2 mm) and aligns with volumetric definitions of progression (51–53). Hence, we believe there are advantages to the volumetric end points we present here, and reassuringly, OncoTarget and OncoTreat demonstrated significant benefit using several complementary analytic metrics.
Third, OncoTarget and OncoTreat predictions are unique and vary in number between tumors. In some cases, there are very few predictions by one or both methods and predictions may not be necessarily conserved in the PDX model, while in other cases there are abundant predictions. As such, it is not always possible to test an identical number of predictions of each type across models. This motivated the design of our study to evaluate the efficacy of drugs predicted by the two methods in parallel, and thus we cannot draw any robust conclusions about differences without additional studies. Further, while selecting a conservative statistical threshold generally reduces the number of predicted drugs to a reasonable number (e.g., 5 to 25), the significant variability between tumors is an important consideration for future clinical deployment.
Finally, a requisite component of generating OncoTreat predictions is the completion of one or more perturbation screens in a cognate model (e.g., cell line or organoid). While we do not anticipate post-perturbation cell viability assessments in such models to meaningfully predict clinical response, we do intuitively note that a critical aspect of assessing the ability of a drug to induce patient-specific TCM-inversion is that TCM inversion cannot be assessed in the cell line model if the corresponding MRs are not consistently differentially active. Thus, identifying high-fidelity models with conserved TCM activity was a critical element of the study. In addition to their dependence on model availability, it should be noted that generation of comprehensive, high-throughput perturbation screens is also cost and time prohibitive. As a result, perturbation screens had not yet been completed for all the tumors considered in this study. This is also a real-world consideration, as evidenced for the CNSET case study, where we could only rely on OncoTarget predictions. Fortunately, since completion of this study, we have generated a Pan-cancer Activity by Enrichment Analysis database (PanACEA), comprising perturbational profiles of about 350 drugs in 25 cancer cell lines, each representing a cognate model matched to an MR-based subtype within 15 human malignancies (Table S7). Access to this extensive resource, which greatly extends the generalizability of the proposed approach, will be made available via a companion publication, see also (54).
We also make note of several practical advantages of our methodology. First, these algorithms are tumor- and mutation-agnostic, thus supporting their application to the vast majority of human malignancies. Based on benchmarking of 11,289 primary tumors from TCGA, as well as over 100 prospectively collected samples from patients with metastatic and treatment refractory cancers, these tests can efficiently prioritize multiple candidate drugs for virtually every tumor, thus allowing further drug prioritization based on drug approval status, insurance reimbursement, oncologist experience, prior treatment, and toxicity. We further note that, while our current study was restricted to solid tumors, the approach is equally applicable to hematologic malignancies. Indeed, we have previously reported on several MR-based insights in lymphoma and leukemia (15, 55), including OncoTreat-based prediction of drug synergy (27), and these malignancies will constitute a focus of future studies.
Critically, as the number of validated OncoTreat and OncoTarget predictions increases, including in a clinical setting (56), the need to validate additional predictions in flawed preclinical models will decrease, conceivably leading to direct clinical utilization. This would significantly increase clinical utility especially in the advanced metastatic setting. Indeed, we have already initiated and enrolled subjects to therapeutic trials based on our methodologies, including ricolinostat + nab-paclitaxel in metastatic breast cancer (NCT02632071) (56), entinostat in neuroendocrine tumors (NCT03211988) (13), and the HIPPOCRATES umbrella trial in pancreatic cancer (NCT04476537), without the need for PDX testing.
Second, the ability to stratify patients into a small number of well-defined pharmacotypes—i.e., tumors with shared predicted drug sensitivity—in virtually all analyzed cancer cohorts, supports prioritization and evaluation of drug predictions through standard clinical trial mechanisms. Pharmacotype analysis provides two critical insights: 1) It helps identify drugs consistently predicted across a substantial fraction of patients in a specific tumor type (e.g., BLBC) thus supporting mechanism-based hypotheses for pre-clinical and clinical trials, and 2) It helps disregard singleton drugs—i.e., drugs predicted for a single patient in a cohort—as potential false positives. Basket and umbrella trials may incorporate OncoTarget and OncoTreat as companion diagnostic biomarkers. Lending support to this approach, an OncoTarget-based trial testing the HDAC6 inhibitor ricolinostat in metastatic breast cancer recently concluded with virtually complete validation of sensitivity predictions (AUC = 1) (56), thus paving the road to additional trials with similar design. OncoTarget, in particular, could expand the pool of eligible patients for basket trials using targeted agents, while OncoTreat can be used as the basis of umbrella trials where patients with one or more cancer types can be assigned to preselected drug treatment arms based on pharmacotype classification. To illustrate this concept, we show the pharmacotypes identified by our analysis in BLBC and how this could be leveraged to design an umbrella trial (Figure 7A–B).
Third, the resources required to perform OncoTarget and OncoTreat analysis (i.e., RNASeq and modest computational resources) are relatively affordable and scalable. There is a critical unmet need to diversify enrollment to clinical trials, which further impacts rapid adoption of new technologies in different healthcare settings (56). The ability to perform the analysis from archival FFPE tissue will allow access to clinical trial participation and clinical testing across a broad range of health care facilities. As our methodology identifies candidate drugs for the vast majority of patient tumors, we hope that it will remove at least one roadblock and source of disappointment to patients enrolling on basket/umbrella trials, due to some not matching any drug arm.
Finally, our methodology can capture longitudinal changes, such as we have shown for metastatic progression in breast cancer and neuroendocrine tumors (13, 57), histologic transformation in follicular lymphoma (15), therapy resistance in T-ALL and breast cancer (55, 58), and reprogramming of castrate-resistant prostate adenocarcinoma to a neuroendocrine state (59). OncoTarget and OncoTreat could thus be potentially applied longitudinally to adapt therapy to the dynamic nature of tumor evolution.
There are also potential limitations. Tumor heterogeneity and stroma infiltration are only partially addressed by the current approach—specifically by limiting the analysis to high tumor cellularity samples, a criterion usually met by the mostly metastatic samples collected in our study. This issue will be mitigated in the future by the increasing availability of single cell gene expression profiling technologies. Indeed, due to the robustness of VIPER to low sequencing depth, we have recently shown its applicability to measure protein activity in single cells, comparing favorably with antibody-based measurements and recapitulating bulk measurements (17, 60), thus supporting drug predictions for specific tumor subpopulations (bioRxiv 2022.02.28.482410), including immunosuppressive, non-transformed populations, such as regulatory T cells (bioRxiv 2022.02.22.481404).
We conclude by stating the obvious. It would be naïve to expect that all, or even a majority of predicted drugs will be clinically effective in human patients. Yet, our study and some initial clinical confirmations (56) suggest that these tests may represent a novel PCM option for mechanism-based prioritization of existing, clinically relevant drugs that is not currently available to cancer patients and, thus, potentially complement successful approaches such as targeted therapy and immunotherapy.
Methods
Generation of Gene Regulatory Networks:
To support context-specific regulatory protein activity inference by VIPER, we have generated comprehensive molecular interaction networks (interactomes) using the Algorithm for the Reconstruction of Accurate Cellular Networks (ARACNe) (61, 62), although other suitable algorithms may be used. The networks were reverse engineered by ARACNe from ≥ 100 RNASeq profiles of human cancer tissue from (a) The Cancer Genome Atlas (TCGA) and (b) for meningioma and neuroendocrine tumors, from Columbia University collected datasets (Table S8). TCGA RNASeq level 3 data were downloaded from NCI Genomics Data Commons (63). Raw counts were normalized and variance stabilized by fitting the dispersion to a negative-binomial distribution as implemented in the DESeq2 R-package (RRID: SCR_000154) (64).
ARACNe was run with 100 bootstrap iterations using an input set of candidate regulators including: (a) 1,877 transcription factors annotated in the Gene Ontology (GO) (65) “Molecular Function database” as DNA-binding transcription factor activity (GO:0003700), DNA binding (GO:0003677), Transcription regulator activity (GO:0030528), or Regulation of transcription, DNA-templated (GO:0003677 and GO:0045449); (b) 677 transcriptional co-factors manually curated from genes annotated as Transcription Coregulator Activity (GO:0003712), Plays a Role in Regulating Transcription (GO:0030528), or Regulation of Transcription (GO:0045449); and (c) 3,895 genes encoding for signal transduction proteins, dually annotated in the GO “Biological Process database” as Signal Transduction (GO:0007165) and in the GO “Cellular Component database” as either Intracellular (GO:0005622) or Plasma membrane (GO:0005886). The Data Processing Inequality (DPI) parameter of ARACNe was set to 0 and the Mutual Information (MI) threshold was set to p = 10−8. The mode of regulation was computed based on the correlation between regulator and target gene expression as previously described (16). The version of raw counts and generated networks used in our work are provided (see Key Resources Table).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Abiraterone | Selleck Chemicals | Cat#S122246 |
| Afatinib | Selleck Chemicals | Cat#S1011 |
| Alpelisib | Selleck Chemicals | Cat#S2814 |
| Bardoxolone Methyl | Selleck Chemicals | Cat#S8078 |
| Belinostat | Selleck Chemicals | Cat#S1085 |
| Bosutinib | Selleck Chemicals | Cat#S1014 |
| Celecoxib | Selleck Chemicals | Cat#S1261 |
| Chlorpromazine | Selleck Chemicals | Cat#S5749 |
| Clofarabine | Selleck Chemicals | Cat#S1218 |
| Dabrafenib | Selleck Chemicals | Cat#S2807 |
| Daunorubicin | Selleck Chemicals | Cat#S3035 |
| Doxorubicin | Selleck Chemicals | Cat#S1208 |
| Etoposide | Selleck Chemicals | Cat#S1225 |
| Fludarabine | Selleck Chemicals | Cat#S1491 |
| GSK2606414 | Selleck Chemicals | Cat#S7307 |
| Homoharringtonine | Selleck Chemicals | Cat#S9015 |
| HSP990 | Selleck Chemicals | Cat#S7097 |
| Irinotecan | Selleck Chemicals | Cat#S1198 |
| Lorlatinib | Selleck Chemicals | Cat#S7536 |
| MK-2206 | Selleck Chemicals | Cat#S1078 |
| Obatoclax | Selleck Chemicals | Cat#S6709 |
| Olaparib | Selleck Chemicals | Cat#S1060 |
| Paclitaxel | Selleck Chemicals | Cat#S1150 |
| Pacritinib | Selleck Chemicals | Cat#S8057 |
| Panobinostat | Selleck Chemicals | Cat#S1030 |
| Ponatinib | Selleck Chemicals | Cat#S1490 |
| Prexasertib | Selleck Chemicals | Cat#S6385 |
| Selinexor | Selleck Chemicals | Cat#S7252 |
| Selumetinib | Selleck Chemicals | Cat#S1008 |
| Serdemetan | Selleck Chemicals | Cat#S1172 |
| TAE684 | Selleck Chemicals | Cat#S1108 |
| Temsirolimus | Selleck Chemicals | Cat#S1044 |
| Teniposide | Selleck Chemicals | Cat#S1787 |
| Thioguanine | Selleck Chemicals | Cat#S1774 |
| Tivantinib | Selleck Chemicals | Cat#S2753 |
| Topotecan | Selleck Chemicals | Cat#S9321 |
| Vismodegib | Selleck Chemicals | Cat#S1082 |
| Vorinostat | Selleck Chemicals | Cat#S1047 |
| Critical Commercial Assays | ||
| RNeasy Pure mRNA Bead Kit | Qiagen | Cat#180244 |
| RNeasy Micro Kit | Qiagen | Cat#74004 |
| TruSeq® Stranded mRNA Library Prep | Illumina | Cat#20020594 |
| Deposited Data | ||
| ARACNe generated networks (used for VIPER analysis) | Figshare | https://figshare.com/s/5d1ffd9f8b2e86e37ed6 |
| TCGA processed data (RNASeq counts) | Originally downloaded from genomic data commons; version used uploaded to FigShare | https://figshare.com/s/ad114ea4b274a523bb4a |
| RNASeq from patient and PDX tumors | Gene Expression Omnibus (GEO) | GEO Accession: GSE212854 |
| RNASeq of meningioma tumors sequenced at CUIMC | Gene Expression Omnibus (GEO) | GEO Accession: GSE212377 |
| High throughput drug perturbation expression and VIPER profiles | Figshare | https://figshare.com/s/d77d19eb326b8a8656b8 |
| RNASeq counts and OncoTarget for CNSET case | Figshare | https://figshare.com/s/6327d1857201ce657d47 |
| Experimental Models: Cell Lines | ||
| ASPC1 | ATCC; Cat#ATCC® CRL-1682 | RRID: CVCL_0152 |
| BT20 | ATCC; Cat#ATCC® HTB-19 | RRID: CVCL_0178 |
| GIST430 | Dr. Gary Schwartz | RRID:CVCL_7040 |
| GISTT1 | Dr. Gary Schwartz | RRID:CVCL_4976 |
| IOMM | ATCC; Cat#ATCC® CRL-3370 | RRID:CVCL_5779 |
| Experimental Models: Organisms/Strains | ||
| NOD.Cg-Prkdcscid Il2rgtm1Wjl Hprtb-m3/EshJ | The Jackson Laboratory | RRID: IMSR_JAX:012480 |
| Software and Algorithms | ||
| Prism Software Version 9 | GraphPad | N/A |
| R version 4.2.0 | The R Foundation for Statistical Computing | N/A |
| ARACNe-AP | Margolin, et al., 2006 | http://califano.c2b2.columbia.edu/aracne |
| VIPER R package | Alvarez, et al., 2016 | https://www.bioconductor.org/packages/release/bioc/html/viper.html |
| DESeq2 R package | Love, et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
VIPER Analysis:
We have previously extensively validated the Virtual Proteomics by Enriched Regulon analysis (VIPER) algorithm as a highly robust and specific tool for the accurate inference of regulatory protein activity in tissue context-dependent manner (8, 15, 16). VIPER leverages accurate tissue-specific gene regulatory networks, such as those produced by ARACNe (61, 62), to measure differential protein activity from bulk or single-cell gene expression signatures (GES) (see Key Resources Table). Specifically, akin to a multiplexed gene-reporter assay, VIPER measures a protein’s differential transcriptional activity through a probabilistic enrichment framework that assesses the enrichment of its activated and repressed transcriptional targets (regulon) in genes over and under expressed in a sample of interest compared to a set of control samples (reference model).
We considered multiple options for reference models to apply VIPER to tumor samples for the purposes of predicting drug response. While organ-matched normal tissue expression profiles, such as those available from the Genotype-Tissue Expression (GTEx) resource (66), and cohort-matched tumor and tumor-adjacent profiles were considered, such references would potentially over-emphasize proliferative and cell cycle signals and under-emphasize lineage specific tumor vulnerabilities, respectively. Ultimately, we prospectively opted to use the pan-TCGA tumor dataset as our reference model to run VIPER and the downstream drug prediction algorithms.
Thus, for each cancer sample, we generate a differential gene expression signature (DGES)—computed as the gene-wise relative expression to the distribution of the expression of that gene across 11,289 TCGA samples—and expressed as its quantile relative to the reference model. Genes whose expression is not captured (quantified) in TCGA are excluded from the DGES. Next, VIPER computes enrichment scores for the targets of each regulatory protein in the DGES, using the analytic-rank based enrichment analysis (aREA) test (16), thus determining if the protein’s transcriptional footprint is over-, under-, or normally represented in the tumor’s DGES. Each enrichment score is computed by a 2-tail approach, rank-transforming the DGES for the positively regulated target genes and then inverting the signature to compute enrichment of the repressed target genes. The enrichment score is a weighted average based on targets’ ranks in the signature and the protein-target gene mutual information assignments in the network, with stronger interactions contributing more to the score. Subsequently, statistical significance for the enrichment score is computed by an analytic approach. Under the null hypothesis, the enrichment score is normally distributed with mean of zero and variance proportional to 1/(n−1), where n is the number of targets, scaled to the standard variable. This approximates shuffling the genes in the signature at random (67), outputting a normalized enrichment score (NES). While aREA does not directly account for biological correlation between the expression of various genes (i.e. covariance) in the given biological context, a very conservative NES/p-value threshold is empirically used for downstream analyses. Importantly, due to VIPER’s use of robust reporter sets to compute the activity of each protein, moderate biases in RNA profiles based on RNA extraction method, sequencing instrument, and reference transcriptome version used to map reads, all of which can affect genes captured and abundance estimates, generally have minimal effect on protein activity assessment. Likewise, due to this use of robust reporter sets, for rare cancer types not represented in TCGA, which might have lineage-specific aberrant expression of a subset of genes, we overall do not anticipate a significant impact on protein activity assessment. When cancer type specific networks are not available, we use an integrated network approach as implemented in metaVIPER (60, 68).
The most differentially active proteins identified by VIPER comprise candidate Master Regulator (MR) proteins. Critically, we have shown that MR proteins are ultra-conserved within each of 112 tumor subtypes and play a key role in integrating the effect of genomic alterations to implement a stable tumor cell state, as shown for 20 tumor types in TCGA (7). VIPER reproducibility is extremely high, such that Spearman correlation of activity profiles generated from RNASeq at 30M to as low as 50K read depth is (16) even though correlation of the underlying gene expression profiles is low . While VIPER is uniquely suited for assessing regulatory proteins that directly control gene expression, including transcription factors, co-factors, and chromatin remodeling enzymes, we have shown that the algorithm is equally effective in monitoring activity of signaling proteins (16) and cell surface markers (60).
OncoTarget Analysis:
Through the use of (a) DrugBank (69), (b) the SelleckChem database (RRID:SCR_003823), (c) published literature, and (d) publicly available information on pharmaceutical company drug development pipelines, we have curated a refined list of 180 actionable proteins representing validated targets of high-affinity pharmacological inhibitors, either FDA approved or in clinical trials (Table S4). This manually curated target-drug(s) database is dominated by signaling proteins and established oncoproteins, as expected from the bias in druggability assessment and past focus in drug development efforts. Pharmacological agents with narrow therapeutic indices—such as those targeting neurotransmitters, ion channels, and vasoactive drugs—were purposefully removed from the database as less likely to be successfully repurposed in oncology.
OncoTarget simply analyzes the VIPER outputted protein activity measurements for these 180 actionable proteins, and provides a multiple-testing corrected significance value for the corresponding NES. We used a conservative threshold (Bonferroni p < 10−5) to identify candidate proteins eliciting essentiality when targeted by a pharmacological inhibitor, for in vivo validation. Based on this threshold, the average TCGA tumor yields 15 unique, significant OncoTarget predictions, ranging from an average of n = 4.5 in adrenocortical carcinoma (ACC) to n = 28.7 in renal clear cell carcinoma (KIRC). Tumors representing conserved subtypes tend to have conserved OncoTarget predictions, leading to identification of subsets of patients whose cancer is predicted to respond to the same set of drugs (pharmacotypes) (see Figure 7 and S6A–D for the tumor types discussed herein).
OncoTreat Analysis:
We have previously described OncoTreat (13) as a methodology to systematically elucidate compounds capable of significantly inverting the activity of the 25↑+25↓ MRs comprising the tumor checkpoint module (TCM) that regulates metastatic progression in enteropancreatic and rectal neuroendocrine tumors. In that study, OncoTreat identified entinostat, a class I histone deacetylase inhibitor, among 105 profiled drugs, as a highly effective TCM-inverter drug (13), leading to a clinical trial that is currently accruing patients (NCT03211988).
For the current study, we more broadly adapted OncoTreat to identify TCM-inverter compounds, by comparing individual tumor samples to the entire TCGA repository, as a reference model, as described above. Specifically, for each tumor type considered in the study, the analysis proceeds through the following steps:
Identifying cell lines (typically 1 or 2) jointly representing high-fidelity (i.e., cognate) in vitro models for a majority of samples in the corresponding TCGA cohort (e.g., pancreatic ductal adenocarcinoma, PAAD), based on TCM-recapitulation (Bonferroni p < 10−5), as assessed by enrichment analysis, see next section on OncoMatch. Upon acquisition of cell lines (see Key Resources Table), RNASeq profiles of untreated cell lines were generated at CUIMC to confirm reproducibility of expression and MR/TCM profiles. All cells were inspected for Mycoplasma contamination using the ABM PCR Mycoplasma Detection Kit.
Generating RNASeq profiles of each cognate cell line, from Step 1, at 24h following perturbation with a library of clinically relevant oncology drugs. Drugs were titrated at their maximum sub-lethal concentration (i.e., 48h EC20), as determined by 10-point dose response profiles. Profiled drugs included FDA-approved and late-stage experimental oncology drugs (in Phase II and III clinical trials) (see Table S5). For completed screens, we have attempted to be all inclusive, excluding therapeutic antibodies due to a lack of availability and immunotherapy agents due to lack of appropriateness to screen in vitro. Additional miscellaneous compounds with EC50 ≤ 2μM in the selected cell line were also included. Most compounds were purchased from SelleckChem or Tocris. DMSO was selected as a universal in vitro solvent (vehicle). Multiplexed, low depth (1M to 2M reads) RNASeq profiles were generated using 96-well plates via the PLATE-Seq technology, using fully automated microfluidics for increased throughput and reproducibility [29]. Eight DMSO-treated controls were included in each plate, to avoid plate-dependent batch effects and to mitigate the inherent variability of DMSO treatment.
Generating a subproteome-wide context-specific Drug Mechanism of Action (MoA) for each drug, as represented by the differential activity of each protein in drug- vs. vehicle control (DMSO)-treated cells. Differential protein activity was assessed by VIPER analysis using a tissue-matched gene regulatory network produced by ARACNe, see above.
Identifying sample-specific candidate MRs and the TCMs they comprise, by VIPER analysis of the sample’s DGES, compared to the set of TCGA samples (reference model).
Finally, prioritizing pharmacological agents based on the statistical significance of the enrichment of the tumor sample’s TCM-activity signature (i.e., 25↑+25↓ MRs) in proteins inactivated and activated in drug vs. DMSO-treated cells, respectively, with negative NES indicating TCM-inversion (Bonferroni p < 10−5, 1-tailed aREA). The number of candidate MR proteins (n=50) used to assess TCM-inversion, which for this step was restricted to only transcription factors and co-factors, was selected because we have shown that, on average, across all of TCGA, the vast majority of functionally-relevant genomic events can be found upstream of the top 50 VIPER-inferred candidate MR proteins (7).
OncoMatch, Cell Line and Patient-Derived Xenograft (PDX) Model Fidelity Analysis:
Model fidelity was assessed based on the statistical significance of the TCM-activity conservation between a human-derived sample and a model-derived sample. For computing protein activity in cell line models, we first generated an analogous DGES comparing each cell line against a large repository of cancer cell lines (reference model), which includes both the Cancer Cell Line Encyclopedia (CCLE) (23) and the Genentech Cell Line Screening Initiative (gCSI) (24). Next, we computed the enrichment of the TCM-activity signature (25 most active and 25 most inactive patient tumor-specific MRs) in differentially active and inactive proteins in the model (OncoMatch). The aREA (16) test was again used, but any suitable enrichment analysis algorithm could be substituted. The analysis was used to (a) select optimal cell lines for the generation of perturbational profiles that effectively track the activity of tested drugs on TCM proteins and (b) to assess the fidelity of PDXs prior to validation of drugs predicted from the human sample. All cell lines used in these analyses were re-sequenced on-site, to capture any potential drift effects. A conservative threshold (Bonferroni p < 10−10) was used to identify high-fidelity models.
Establishment of PDX models and therapeutic drug testing:
All animals were maintained under barrier conditions and all experiments were performed in accordance with and approval of the Memorial Sloan Kettering Cancer Center (MSKCC) Institutional Animal Care and Use Committee (IACUC, protocol #16-08-011) and Columbia University Irving Medical Center (CUIMC) IACUC (AAAF5850). Patient tumor tissue was collected under the CUIMC Institutional Review Board (IRB)-approved protocol AAAA7562, with written informed consent provided prospectively by the subject and conducted in accordance with recognized ethical guidelines under the U.S. Common Rule. Generation of PDX models was performed under the MSKCC IRB-approved protocol #17–387 and CUIMC IRB protocol AAAJ5811. PDX models were established as previously described (52). In summary, fresh tumor tissue was fragmented and implanted subcutaneously into nonobese/severe combined immunodeficiency IL2Rγ null, hypoxanthine phosphoribosyltransferase (HPRT)-null (NSGH) mice (Jackson Labs, IMSR catalog no. JAX:012480, RRID: IMSR_JAX:012480) (see Key Resources Table) and tumor engraftment monitored by visual and manual inspection.
Engrafted tumors were measured twice weekly with calipers and drug treatment initiated when tumor volume (TV) reached ~100 mm3 (TV = width2 × ½ length) (see Key Resources Table for sourcing of drugs). Early passage animals (Passage 1 – 5) were used for all therapeutic studies. Aligned with clinical response criteria, treatment response was categorized as complete response (CR, >95% volumetric reduction from baseline or no palpable tumor), partial response (PR, >50% reduction), stable disease (SD, <50% reduction and no more than 100% increase), or progressive disease (PD, >100% increase from baseline). Disease control rate is defined as the sum of CR, PR and SD responses divided by the total responses assessed. Objective response rate is defined as the sum of CR and PR responses divided by the total responses assessed. Tumor responses were assessed at the end of the therapeutic study period. Comparisons of disease control and objective response rates across treatment groups (OncoTarget alone, OncoTreat alone, Both) were performed using a Fisher’s exact test. The Mann-Whitney-Wilcoxon method was used to compare differences in the distribution of relative tumor volume between OncoTarget or OncoTreat cohorts and Vehicle control. Vardi’s test was used to evaluate differences in area under the curve (AUC) between treatment groups across models (70, 71). Curves for Disease Control using the Kaplan-Meier estimator were compared and analyzed using the log-rank test. ΔT/ΔC%, computed as the change in tumor volume from baseline for drug treated mice divided by the change in tumor volume from baseline for Vehicle control treated mice, was determined for OncoTarget, OncoTreat, and Negative control cohorts and differences evaluated using 2-way ANOVA. All relevant statistical analyses were performed using R software (v3.5.0) or GraphPad Prism [v8.4.1 (RRID:SCR_002798)]. Statistical significance was defined as a p < 0.05.
Pharmacodynamic (PD) Assessments of TCM-inversion:
Samples for PD assessment were procured from two mice per treatment arm, for the four PDX models treated with at least three OncoTreat-predicted drugs—GIST-81050, BC-32398, CNS-16474, and PAC-05647. Mice were randomly selected for early sacrifice, independent of tumor size, three hours following the third dose, and were excluded from response assessment. We performed RNASeq and subsequent VIPER on paired drug vs. Vehicle control-treated PDX tumor samples. TCM-inversion was assessed based on the statistical significance of the enrichment of the TCM-activity signature (i.e., 25↑+25↓ MRs of the patient tumor) in proteins inactivated and activated in drug vs. Vehicle control-treated PDX tumors, respectively, again using aREA, although alternative enrichment tests may be used. Negative NES indicates TCM-inversion (Bonferroni p < 10−5).
Supplementary Material
Significance.
Complementary precision cancer medicine paradigms are needed to broaden the clinical benefit realized through genetic profiling and immunotherapy. In this first-in-class application, we introduce two transcriptome-based tumor-agnostic systems biology tools to predict drug response in vivo. OncoTarget and OncoTreat are scalable for the design of basket and umbrella clinical trials.
Acknowledgements:
This work was supported by the NCI Cancer Target Discovery and Development Program (U01 CA217858), the Cancer Systems Biology Consortium (U54 CA209997), and NIH Shared Instrumentation Grants (S10 OD012351 and S10 OD021764), all to A. Califano. Also, this research was funded in part through the NCI Cancer Center Support Grant (P30 CA013696) to CUIMC. Also, this research was funded in part through CureSearch for Children’s Cancer Acceleration Initiative Award to A. Kung and the NCI Cancer Center Support Grants to MSKCC (P30 CA013696 and P30 CA008748).
Footnotes
Conflict of Interest Statement: Dr. Califano is founder, equity holder, and consultant of DarwinHealth Inc., a company that has licensed some of the algorithms used in this manuscript from Columbia University. Dr. Kung is a Medical and Scientific Advisor to DarwinHealth. Columbia University is also an equity holder in DarwinHealth Inc.
Data Availability:
Original tumor bulk RNASeq data generated as part of this work have been deposited to gene expression omnibus (GEO: GSE212854; Key Resources Table). Relevant high throughput drug perturbation data used for de novo assessment of drug MoA is available on Figshare (FS: https://figshare.com/s/d77d19eb326b8a8656b8). RNASeq of a cohort of 102 meningioma tumors sequenced at CUIMC and used to generate a meningioma network have been deposited to GEO (GSE212377). The version of TCGA processed data (RNASeq counts) used to generate networks and perform other analyses presented herein are available on FS (https://figshare.com/s/ad114ea4b274a523bb4a). The ARACNe generated networks used to run VIPER are available on FS (https://figshare.com/s/5d1ffd9f8b2e86e37ed6). The RNASeq counts and OncoTarget results for the CNSET case report are available on FS (https://figshare.com/s/6327d1857201ce657d47). Additional requests should be directed to corresponding author A. Califano.
References
- 1.Le Tourneau C, Delord JP, Goncalves A, Gavoille C, Dubot C, Isambert N, et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015;16(13):1324–34. [DOI] [PubMed] [Google Scholar]
- 2.Tannock IF, Hickman JA. Limits to Personalized Cancer Medicine. N Engl J Med. 2016;375(13):1289–94. [DOI] [PubMed] [Google Scholar]
- 3.Weinstein IB. Cancer. Addiction to oncogenes--the Achilles heal of cancer. Science. 2002;297(5578):63–4. [DOI] [PubMed] [Google Scholar]
- 4.Tang J, Pearce L, O’Donnell-Tormey J, Hubbard-Lucey VM. Trends in the global immuno-oncology landscape. Nat Rev Drug Discov. 2018;17(11):783–4. [DOI] [PubMed] [Google Scholar]
- 5.Zhong L, Li Y, Xiong L, Wang W, Wu M, Yuan T, et al. Small molecules in targeted cancer therapy: advances, challenges, and future perspectives. Signal Transduct Target Ther. 2021;6(1):201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hahn WC, Bader JS, Braun TP, Califano A, Clemons PA, Druker BJ, et al. An expanded universe of cancer targets. Cell. 2021;184(5):1142–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paull EO, Aytes A, Jones SJ, Subramaniam PS, Giorgi FM, Douglass EF, et al. A modular master regulator landscape controls cancer transcriptional identity. Cell. 2021;184(2):334–51e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Califano A, Alvarez MJ. The recurrent architecture of tumour initiation, progression and drug sensitivity. Nat Rev Cancer. 2017;17(2):116–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen JC, Alvarez MJ, Talos F, Dhruv H, Rieckhof GE, Iyer A, et al. Identification of Causal Genetic Drivers of Human Disease through Systems-Level Analysis of Regulatory Networks. Cell. 2014;159(2):402–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463(7279):318–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aytes A, Mitrofanova A, Lefebvre C, Alvarez MJ, Castillo-Martin M, Zheng T, et al. Cross-species regulatory network analysis identifies a synergistic interaction between FOXM1 and CENPF that drives prostate cancer malignancy. Cancer cell. 2014;25(5):638–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rajbhandari P, Lopez G, Capdevila C, Salvatori B, Yu J, Rodriguez-Barrueco R, et al. Cross-Cohort Analysis Identifies a TEAD4-MYCN Positive Feedback Loop as the Core Regulatory Element of High-Risk Neuroblastoma. Cancer Discov. 2018;8(5):582–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alvarez MJ, Subramaniam PS, Tang LH, Grunn A, Aburi M, Rieckhof G, et al. A precision oncology approach to the pharmacological targeting of mechanistic dependencies in neuroendocrine tumors. Nat Genet. 2018;50(7):979–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodriguez-Barrueco R, Yu J, Saucedo-Cuevas LP, Olivan M, Llobet-Navas D, Putcha P, et al. Inhibition of the autocrine IL-6-JAK2-STAT3-calprotectin axis as targeted therapy for HR-/HER2+ breast cancers. Genes & development. 2015;29(15):1631–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bisikirska B, Bansal M, Shen Y, Teruya-Feldstein J, Chaganti R, Califano A. Elucidation and Pharmacological Targeting of Novel Molecular Drivers of Follicular Lymphoma Progression. Cancer Res. 2016;76(3):664–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alvarez MJ, Shen Y, Giorgi FM, Lachmann A, Ding BB, Ye BH, et al. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat Genet. 2016;48(8):838–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Obradovic A, Chowdhury N, Haake SM, Ager C, Wang V, Vlahos L, et al. Single-cell protein activity analysis identifies recurrence-associated renal tumor macrophages. Cell. 2021;184(11):2988–3005 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang VE, Xue JY, Frederick DT, Cao Y, Lin E, Wilson C, et al. Adaptive Resistance to Dual BRAF/MEK Inhibition in BRAF-Driven Tumors through Autocrine FGFR Pathway Activation. Clin Cancer Res. 2019;25(23):7202–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stathias V, Jermakowicz AM, Maloof ME, Forlin M, Walters W, Suter RK, et al. Drug and disease signature integration identifies synergistic combinations in glioblastoma. Nat Commun. 2018;9(1):5315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alvarez MJ, Yan P, Alpaugh ML, Bowden M, Sicinska E, Zhou CW, et al. Reply to ‘ H-STS, L-STS and KRJ-I are not authentic GEPNET cell lines ’. Nat Genet. 2019;51(10):1427–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cancer Genome Atlas N Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–52. [DOI] [PubMed] [Google Scholar]
- 23.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haverty PM, Lin E, Tan J, Yu Y, Lam B, Lianoglou S, et al. Reproducible pharmacogenomic profiling of cancer cell line panels. Nature. 2016;533(7603):333–7. [DOI] [PubMed] [Google Scholar]
- 25.Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007;8(5):R76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bansal M, Yang J, Karan C, Menden MP, Costello JC, Tang H, et al. A community computational challenge to predict the activity of pairs of compounds. Nature Biotechnology. 2014;32(12):1213–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Woo JH, Shimoni Y, Yang WS, Subramaniam P, Iyer A, Nicoletti P, et al. Elucidating Compound Mechanism of Action by Network Perturbation Analysis. Cell. 2015;162(2):441–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bush EC, Ray F, Alvarez MJ, Realubit R, Li H, Karan C, et al. PLATE-Seq for genome-wide regulatory network analysis of high-throughput screens. Nat Commun. 2017;8(1):105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ben-David U, Ha G, Tseng YY, Greenwald NF, Oh C, Shih J, et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat Genet. 2017;49(11):1567–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vasciaveo A, Arriaga JM, de Almeida FN, Zou M, Douglass EF, Picech F, et al. OncoLoop: A Network-Based Precision Cancer Medicine Framework. Cancer Discov. 2023;13(2):386–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benedict M, Zhang X. Calcifying Nested Stromal-Epithelial Tumor of the Liver: An Update and Literature Review. Archives of pathology & laboratory medicine. 2019;143(2):264–8. [DOI] [PubMed] [Google Scholar]
- 33.Heerema-McKenney A, Leuschner I, Smith N, Sennesh J, Finegold MJ. Nested stromal epithelial tumor of the liver: six cases of a distinctive pediatric neoplasm with frequent calcifications and association with cushing syndrome. The American journal of surgical pathology. 2005;29(1):10–20. [DOI] [PubMed] [Google Scholar]
- 34.Assmann G, Kappler R, Zeindl-Eberhart E, Schmid I, Haberle B, Graeb C, et al. beta-Catenin mutations in 2 nested stromal epithelial tumors of the liver--a neoplasia with defective mesenchymal-epithelial transition. Human pathology. 2012;43(11):1815–27. [DOI] [PubMed] [Google Scholar]
- 35.Khoshnam N, Robinson H, Clay MR, Schaffer LR, Gillespie SE, Shehata BM. Calcifying nested stromal-epithelial tumor (CNSET) of the liver in Beckwith-Wiedemann syndrome. European journal of medical genetics. 2017;60(2):136–9. [DOI] [PubMed] [Google Scholar]
- 36.Tehseen S, Rapkin L, Schemankewitz E, Magliocca JF, Romero R. Successful liver transplantation for non-resectable desmoplastic nested spindle cell tumor complicated by Cushing’s syndrome. Pediatric transplantation. 2017;21(6). [DOI] [PubMed] [Google Scholar]
- 37.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn. 2015;17(3):251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daw NC, Chi YY, Kalapurakal JA, Kim Y, Hoffer FA, Geller JI, et al. Activity of Vincristine and Irinotecan in Diffuse Anaplastic Wilms Tumor and Therapy Outcomes of Stage II to IV Disease: Results of the Children’s Oncology Group AREN0321 Study. J Clin Oncol. 2020;38(14):1558–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dix DB, Seibel NL, Chi YY, Khanna G, Gratias E, Anderson JR, et al. Treatment of Stage IV Favorable Histology Wilms Tumor With Lung Metastases: A Report From the Children’s Oncology Group AREN0533 Study. J Clin Oncol. 2018;36(16):1564–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481(7381):306–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hidalgo M, Amant F, Biankin AV, Budinska E, Byrne AT, Caldas C, et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014;4(9):998–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim MP, Evans DB, Wang H, Abbruzzese JL, Fleming JB, Gallick GE. Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat Protoc. 2009;4(11):1670–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sivanand S, Pena-Llopis S, Zhao H, Kucejova B, Spence P, Pavia-Jimenez A, et al. A validated tumorgraft model reveals activity of dovitinib against renal cell carcinoma. Sci Transl Med. 2012;4(137):137ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang X, Claerhout S, Prat A, Dobrolecki LE, Petrovic I, Lai Q, et al. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 2013;73(15):4885–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fichtner I, Rolff J, Soong R, Hoffmann J, Hammer S, Sommer A, et al. Establishment of patient-derived non-small cell lung cancer xenografts as models for the identification of predictive biomarkers. Clin Cancer Res. 2008;14(20):6456–68. [DOI] [PubMed] [Google Scholar]
- 46.Na YS, Ryu MH, Park YS, Lee CW, Lee JK, Park Y, et al. Establishment of patient-derived xenografts from patients with gastrointestinal stromal tumors: analysis of clinicopathological characteristics related to engraftment success. Sci Rep. 2020;10(1):7996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang H, Qi L, Du Y, Huang LF, Braun FK, Kogiso M, et al. Patient-Derived Orthotopic Xenograft (PDOX) Mouse Models of Primary and Recurrent Meningioma. Cancers (Basel). 2020;12(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vaubel RA, Tian S, Remonde D, Schroeder MA, Mladek AC, Kitange GJ, et al. Genomic and Phenotypic Characterization of a Broad Panel of Patient-Derived Xenografts Reflects the Diversity of Glioblastoma. Clin Cancer Res. 2020;26(5):1094–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Euhus DM, Hudd C, LaRegina MC, Johnson FE. Tumor measurement in the nude mouse. J Surg Oncol. 1986;31(4):229–34. [DOI] [PubMed] [Google Scholar]
- 50.Kersemans V, Cornelissen B, Allen PD, Beech JS, Smart SC. Subcutaneous tumor volume measurement in the awake, manually restrained mouse using MRI. J Magn Reson Imaging. 2013;37(6):1499–504. [DOI] [PubMed] [Google Scholar]
- 51.James K, Eisenhauer E, Christian M, Terenziani M, Vena D, Muldal A, et al. Measuring response in solid tumors: unidimensional versus bidimensional measurement. J Natl Cancer Inst. 1999;91(6):523–8. [DOI] [PubMed] [Google Scholar]
- 52.O’Donohue TJ, Ibanez G, Coutinho DF, Mauguen A, Siddiquee A, Rosales N, et al. Translational Strategies for Repotrectinib in Neuroblastoma. Mol Cancer Ther. 2021;20(11):2189–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oxnard GR, Morris MJ, Hodi FS, Baker LH, Kris MG, Venook AP, et al. When progressive disease does not mean treatment failure: reconsidering the criteria for progression. J Natl Cancer Inst. 2012;104(20):1534–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Douglass EF Jr., Allaway RJ, Szalai B, Wang W, Tian T, Fernandez-Torras A, et al. A community challenge for a pancancer drug mechanism of action inference from perturbational profile data. Cell Rep Med. 2022;3(1):100492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Piovan E, Yu J, Tosello V, Herranz D, Ambesi-Impiombato A, Da Silva AC, et al. Direct reversal of glucocorticoid resistance by AKT inhibition in acute lymphoblastic leukemia. Cancer cell. 2013;24(6):766–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zeleke TZ, Pan Q, Chiuzan C, Onishi M, Li Y, Tan H, et al. Network-based assessment of HDAC6 activity predicts preclinical and clinical responses to the HDAC6 inhibitor ricolinostat in breast cancer. Nat Cancer. 2023;4(2):257–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fares J, Fares MY, Khachfe HH, Salhab HA, Fares Y. Molecular principles of metastasis: a hallmark of cancer revisited. Signal Transduct Target Ther. 2020;5(1):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Risom T, Langer EM, Chapman MP, Rantala J, Fields AJ, Boniface C, et al. Differentiation-state plasticity is a targetable resistance mechanism in basal-like breast cancer. Nat Commun. 2018;9(1):3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zou M, Toivanen R, Mitrofanova A, Floch N, Hayati S, Sun Y, et al. Transdifferentiation as a Mechanism of Treatment Resistance in a Mouse Model of Castration-Resistant Prostate Cancer. Cancer Discov. 2017;7(7):736–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ding H, Douglass EF Jr., Sonabend AM, Mela A, Bose S, Gonzalez C, et al. Quantitative assessment of protein activity in orphan tissues and single cells using the metaVIPER algorithm. Nat Commun. 2018;9(1):1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Basso K, Margolin Aa, Stolovitzky G, Klein U, Dalla-Favera R, Califano A. Reverse engineering of regulatory networks in human B cells. Nature genetics. 2005;37:382–90. [DOI] [PubMed] [Google Scholar]
- 62.Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, Dalla Favera R, et al. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC bioinformatics. 2006;7 Suppl 1:S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang Z, Hernandez K, Savage J, Li S, Miller D, Agrawal S, et al. Uniform genomic data analysis in the NCI Genomic Data Commons. Nat Commun. 2021;12(1):1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Consortium GT. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45(6):580–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Coutinho DF, Mundi PS, Marks LJ, Burke C, Ortiz MV, Diolaiti D, et al. Validation of a non-oncogene encoded vulnerability to exportin 1 inhibition in pediatric renal tumors. Med (N Y). 2022;3(11):774–91 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wishart DS, Knox C, Guo AC, Cheng D, Shrivastava S, Tzur D, et al. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic acids research. 2008;36(Database issue):D901–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vardi Y, Ying ZL, Zhang CH. Two-sample tests for growth curves under dependent right censoring. Biometrika. 2001;88(4):949–60. [Google Scholar]
- 71.Wu J, Houghton PJ. Interval approach to assessing antitumor activity for tumor xenograft studies. Pharm Stat. 2010;9(1):46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original tumor bulk RNASeq data generated as part of this work have been deposited to gene expression omnibus (GEO: GSE212854; Key Resources Table). Relevant high throughput drug perturbation data used for de novo assessment of drug MoA is available on Figshare (FS: https://figshare.com/s/d77d19eb326b8a8656b8). RNASeq of a cohort of 102 meningioma tumors sequenced at CUIMC and used to generate a meningioma network have been deposited to GEO (GSE212377). The version of TCGA processed data (RNASeq counts) used to generate networks and perform other analyses presented herein are available on FS (https://figshare.com/s/ad114ea4b274a523bb4a). The ARACNe generated networks used to run VIPER are available on FS (https://figshare.com/s/5d1ffd9f8b2e86e37ed6). The RNASeq counts and OncoTarget results for the CNSET case report are available on FS (https://figshare.com/s/6327d1857201ce657d47). Additional requests should be directed to corresponding author A. Califano.