

Abstract
Purpose:
Granulocyte colony stimulating factor (GCSF) enhances colon cancer development. This study defines the prevalence and effects of increased GCSF signaling in human colon cancers and investigates GCSF inhibition as an immunotherapeutic strategy against metastatic colon cancer.
Experimental Design:
Patient samples were used to evaluate GCSF and GCSF receptor (GCSFR) levels by immunohistochemistry with sera used to measure GCSF levels. PBMCs were used to assess the rate of GCSFR+ T cells and interferon γ (IFNγ) responses to chronic ex vivo GCSF. An immune competent mouse model of peritoneal metastasis (MC38 cells in C57Bl/6J) was used to determine the effects of GCSF inhibition (αGCSF) on survival and the tumor microenvironment (TME) with flow and mass cytometry.
Results:
GCSF and GCSFR are increased in human colon cancer samples as compared to patient-matched normal colon. High patient serum GCSF is associated with increases in markers of poor prognosis, (e.g., VEGF, IL6). Circulating T cells from patients express GCSFR at double the rate of T cells from controls. Prolonged GCSF exposure decreases T cell IFNγ production. Treatment with αGCSF shifts both the adaptive and innate compartments of the TME and increases survival (HR=0.46, p=0.0237) and tumor T cell infiltration, activity, and IFNγ response with greater effects in female mice. A negative correlation exists between serum GCSF levels and tumor infiltrating T cells in patient samples from women.
Conclusions:
These findings support GCSF as an immunotherapeutic target against colon cancer with greater potential benefit in women.
Keywords: Colon cancer, immunotherapy, granulocyte colony stimulating factor, tumor immune microenvironment, metastasis
Introduction.
Colorectal cancer is estimated to result in over 52,000 deaths in the United States alone in 2022 (1) and is the second most commonly diagnosed cancer worldwide (2). Colon cancer has a complex tumor immune microenvironment that is strongly associated with patient outcomes. Work from Galon et al has shown that the quantity of T cells infiltrating the tumor positively correlates with improved survival (3–6). Yet, the tumor and patient factors that contribute to shaping the tumor immune microenvironment remain an area of intense study.
While best known for clinical use in mobilizing and maturing neutrophils to support the white blood cell count in patients undergoing cytotoxic chemotherapy, granulocyte colony stimulating factor (GCSF) has complex and less well-characterized effects on T cells (7–10). Mobilization of peripheral blood mononuclear cells (PBMCs) with GCSF for use in stem cell transplantation is associated with short-term adaptive immune suppression as manifested by decreased rates of acute graft vs host disease and increased Th2 and Treg polarization (8, 9, 11–13). The effects of endogenous GCSF signaling on the tumor immune microenvironment of solid tumors such as colon cancer, however, are less clear.
There are multiple case reports of highly aggressive gastrointestinal tumors that secrete GCSF in the literature, as well as a defined role for GCSF in healing mucosal injury by increasing epithelial cell proliferation and modulating the local mucosal cytokine milieu (14–18). Previous work by our group has shown that gene expression of GCSF (CSF3) is elevated in colon cancer as compared to histologically normal colonic tissue and that GCSF stimulates colon cancer epithelial cell proliferation and migration (19). Furthermore, we have shown that inhibition of GCSF with a functional antibody (αGCSF) in a mouse colon cancer development model results in a doubling of T cell infiltration and a shift from alternatively activated macrophages to classically activated. The αGCSF treatment also resulted in a 75% reduction in tumor development (20).
Here we demonstrate that the above data are relevant to human colon cancer and that GCSF inhibition increases survival in an end-stage model of peritoneal carcinomatosis with effects on both the innate and adaptive immune tumor infiltrate. Furthermore, we show that GCSF inhibition as an immunotherapeutic strategy against colon cancer may be more effective in women based on sex-specific differences in T cell responses. This work has rapid translational potential, given the availability of a fully humanized antibody against GCSFR which is already in clinical trials for hidradenitis suppurativa (CSL Behring 324, or Anumigilimab). Finally, this therapeutic strategy is particularly intriguing given that the effects appear to be greater for females, while the majority of currently available anti-cancer immune therapies show greater benefit for male patients (21).
Materials and Methods
All human studies were conducted after obtaining local Institutional Review Board (IRB) approval and in accordance with the ethical principles set forth in the Belmont Report.
Patient selection (University of New Mexico (UNM) cohort)
The study was conducted under approval from the UNM IRB. The surgical pathology database from UNM was queried for patients with colon cancer who underwent resection at UNM from between 1991–2014 and had blocks of tissue available for staining. Sufficient tissue blocks for staining for both GCSF and GCSF receptor (GCSFR) were available for 206 patients. The clinical records were searched for demographic and stage data.
Immunohistochemistry (IHC) and digital analysis (UNM cohort)
Formalin-fixed paraffin embedded (FFPE) surgical specimens were used for the IHC analyses which were performed on the Ventana Discovery platform™. The presence of viable tumor and minimal necrosis in the slides was confirmed by a pathologist (JAH) on hematoxylin and eosin stains. The tissue blocks were sectioned (5 microns), mounted on charged slides and then baked at 60°C for 60 minutes. Prior to application of the antibody, slides were treated with Discovery RiboCC (Ventana, 760-107) for 32 minutes at 91°C (GCSF) or Cell Conditioner #1 (Ventana, 950-500) for 64 minutes at 100°C (GCSFR). Rabbit polyclonal antibody against GCSF (Abcam Cat# ab9691, RRID:AB_308758) was applied and incubated for 60 minutes at 37°C and αGCSFR rabbit polyclonal antibody (Atlas Antibodies Cat# HPA048086, RRID:AB_2680257) applied and incubated for 60 minutes at 36°C respectively. Antibody incubation was followed by anti-rabbit HQ (Roche Cat# 760-4815, RRID:AB_2811171), anti-HQ HRP (Ventana, 760-4820) and DAB CM (Ventana, 760-4304) detection. Slides were counterstained with hematoxylin and bluing solution (Ventana, 760-2012 and 760-2037).
Slides were scanned for high-resolution digital analysis using the Aperio Scan Scope™ (Leica Biosystems) and digital analysis software (HALO™, Indica Labs). The digital analysis software was “trained” to differentiate tumor tissue, positively stained cells, and stromal tissue in an iterative approach by a pathologist (JAH). Total cell counts within tumor were divided by total tumor cells staining positive for GCSF or GCSFR to derive a percentage of ligand or receptor positive cells. This was multiplied by the mean intensity of the staining which was scored on a scale of 0-1 to arrive at GCSF and GCSFR tumor scores. A similar analysis was performed for the histologically normal colonic tissues from the same resection specimens.
Patient selection (University of Oklahoma Health Sciences Center (OUHSC) cohort)
The study was conducted under approval from the OUHSC Institutional Review Board. Patients undergoing resection of colon cancer were prospectively consented for this study. Formalin-fixed paraffin embedded surgical specimens were retrieved from the Pathology Department.
IHC and digital analysis (OUHSC cohort)
Five-micron thick histological sections, embedded in paraffin and mounted on HistoBond® Plus slides (Statlab Medical Products, 418/45) were rehydrated and washed in Tris Buffered Saline (TBS). For hematoxylin-eosin staining (H&E), tissues were stained according to standard protocols. Antigen retrieval for CD8 antibody (Vector Laboratories, H-3300) or CD4 antibody (Vector Laboratories, H-3301) was accomplished via twenty minutes in a steamer followed by thirty minutes cooling at room temperature. Sections were treated with a peroxidase blocking reagent (Vector Laboratories, SP-6000) to inhibit endogenous peroxidase activity, followed by 2.5% normal horse serum to inhibit nonspecific binding. Mouse αCD8 antibody (C8/144B) (Abcam, #75129) or Rabbit αCD4 antibody (EPR6855) (Abcam Cat# ab133616, RRID:AB_2750883) was applied to each section and incubated for sixty minutes or overnight at 4°C in a humidified chamber. Sections were washed and processed for IHC using the ImmPRESS® Excel Peroxidase Staining kit; Anti-Mouse IgG (Vector Laboratories, MP-7602) or ImmPRESS™ Excel Peroxidase Staining kit; Anti-Rabbit IgG (Vector Laboratories, MP-7601). Slides were incubated with NovaRed® Substrate Kit, Peroxidase (Vector Laboratories, MP-7601) chromogen for visualization. Counterstaining was carried out with Hematoxylin QS Nuclear Counterstain (Vector Laboratories, H-3404). Appropriate positive and negative tissue controls were used.
Slides were scanned and analyzed using Aperio ImageScope Software (Aperio Technologies). Invasive carcinoma was annotated by a pathologist (MK) and the membrane v9 algorithm was used to quantify positive cells for CD4 and CD8. Positivity was assessed using 3+ (high positive), 2+ (medium positive), 1+ (low positive), and 0 (negative) cell counts based on the intensity of Nova Red. Percent positivity was calculated using these values by adding the positive counts together and dividing by the total cell count. The Aperio positive pixel count v9 algorithm was used to quantify positive cells for GCSF and GCSFR (H-score). The algorithm determines the number of pixels with high (Hp), medium (Mp), low (Lp), and negative (Np) intensities of NovaRed. These values were used to calculate the pixel H-score as in Ram et al (22).
Multiplex profiling (OUHSC cohort and mouse studies)
Whole blood was centrifuged to collect serum from prospectively consented colon cancer patients and from mice (see below) injected with 105 MC38 IP. Collected sera were analyzed using a multiplex magnetic bead-based array (Human, Millipore Sigma, Cat: HCYTMAG-60K-PX30 or mouse, MCYTMAG-70K-PX32 Burlington MA, USA) per manufacturer’s protocol as we have done previously (19, 20, 23).
Cell culture
MC38 cells originate from a chemically induced colon cancer in a C57Bl/6 female mouse (24) and were obtained through a kind gift from Dr. Carlton Barnett at the University of Colorado in 2017, validated by short tandem repeat profiling upon arrival and then every 2 years (DDC Medical, Fairfield OH, USA) and tested negative for mycoplasma via MycoAlert Plus Mycoplasma Detection Kit (Lonza, LT07-703) prior to all injections. MC38 cells were cultured in RPMI-1640 (PurMa Biologics, P3S10109-500) with 10% heat inactivated FBS (PurMa Biologics, P3302), 1% penicillin-streptomycin (Gibco, REF:15140-122), and 1% L-glutamine (Corning, 25-005-CI). Cells from passages >15 were not used in these experiments.
Murine models
All procedures were approved by the Institutional Animal Care and Use Committee in accordance with existing national and university policy on humane care and use of laboratory animals.
Defined endpoint model for cytometry time-of-flight (CyTOF)
Five to eight-week-old, male and female C57Bl/6J mice were obtained from Jackson Laboratories (RRID:IMSR_JAX:000664). On Day 0, an intraperitoneal (IP) injection of 105 MC38 cells was performed. Mice were all euthanized on Day 14 after having received 5 total treatments of 25 μg/mouse αGCSF (R&D, MAB414) or 25 μg/mouse IgG1 isotype control (R&D, MAB005) every other day, beginning Day 3.
Survival model
Five to eight-week-old, male and female C57Bl/6J mice were obtained from Jackson Laboratories (000664) and injected with tumor as above. Starting on Day 9, mice were treated with either 25 μg/mouse αGCSF or 25 μg/mouse IgG1 isotype control 3 times a week. Animals were monitored 3 times a week until clinical symptoms developed at which point, they were monitored daily. Physical signs of distress that were evaluated included grooming, lethargy, distress based on a grimace scale and/or a 10% change in weight over 3 days as we have done previously (23). Euthanasia occurred when mice met pre-determined objective criteria as moribund or at Day 63 post cell injection if no symptoms were observed. Results from 2 independent experiments are presented in aggregate.
Tissue preparation for cytometry
Solid tumors ranging from 2-5 mm3 were sharply dissected from the peritoneal surfaces such as the omentum, mesentery, and diaphragm, from animals injected with 105 MC38 IP and treated as described above. The tumors were dissociated using a gentleMACS Dissociator (Miltenyi Biotec, 130-093-235). The dissociation was then incubated with 40 μg/ml Liberase TM (Roche, 05401135001) 25 μg/ml DNase I (Quantabio, 95150-100) in RPMI-1640 supplemented as described above for 40 minutes at 37°C and 5% CO2. 40 μM cell strainers (VWR, 76327-098) were used to filter the sample from each tissue.
Flow cytometry and antibodies
T cells were isolated from peripheral blood mononuclear cells (PBMCs) using a human T cell negative selection kit (STEMCELL, 17951) and cultured in T cell expansion media (Gibco, A1048501) at 37°C with 5% CO2. GCSF or equal volume of vehicle control was added at 10 ng/ml. Stimulation was achieved at 6 hours with 50 ng/ml Phorbol 12-myristate 13-acetate (PMA) and 1 μg/ml ionomycin. Standard protocol was used to perform staining as we have done previously (23). Antibodies used in this study were CD4-BV650 (RM4-5) (BDBiosciences, 9122894), CD8-APC-Cy7 (53-6.7) (Biolegend, B283110), and IFNy-BV421 (XMG1.2) (BDBiosciences, 8144795). Cells were run on a Stratedigm S1400Exi flow cytometer. Data was analyzed using FlowJo V10.8 (FlowJo, RRID:SCR_008520).
Cytometry Time of Flight (CyTOF)
Preparation of tumor tissue single-cell suspensions
Tumors from animals injected with 105 MC38 IP as described above in the “Defined endpoint model” and treated with either αGCSF or equal amount of isotype control were dissociated using a gentleMACS Dissociator (Miltenyi Biotec, 130-093-235). The dissociation was then incubated with 40 μg/ml Liberase TM (Roche, 05401135001) 25 μg/ml DNase I (Quantabio, 95150-100) in RPMI-1640 supplemented as described above for 40 minutes at 37°C and 5% CO2. 40 μM cell strainers (VWR, 76327-098) were used to filter the sample from each tissue. The cells were then enriched for CD45 using EasySep™ Mouse TIL Positive Selection Kit (STEMCELL, 100-0350).
Single cell mass cytometry sample preparation, data acquisition, and analysis
Cells from control or αGCSF treated mouse tumors were stained for viability with 5 μM cisplatin (Fludigim, 201064). For surface and cytosolic staining, cells were blocked by Human TruStrain FcX (Biolegend, 422301). Antibody cocktail was added to the cells and incubated for 30 minutes at room temperature. Cells were then washed twice with Maxpar Cell Staining Buffer (Fluidigm, 201066). For nuclear staining, cells were incubated with Nuclear Antigen Staining Buffer (Fluidigm, 201063) for 30 minutes at room temperature. Cells were then washed twice with Nuclear Antigen Staining Perm, followed by addition of the antibody cocktail and incubated for 30-45 minutes at room temperature. The Nuclear Antigen Staining Perm washes were repeated, and the cells were fixed using 1.6% formaldehyde for 10 minutes. After fixation, cells were incubated with Intercalator-Ir (Fluidigm, 201192A) in Maxpar Fix and Perm buffer (Fluidigm, 201067) overnight at 4°C. The samples were then suspended in 0.1X EQ beads (Fluidigm, 201078). Data was acquired on the Helios (Fluidigm). The panel of 42 mass-conjugated antibodies used in the study are shown in Supplementary Table S1. Data was uploaded to Cytobank for analysis directly from the core laboratory. The analyses presented here were carried out with a combination of Cytobank (Cytobank, RRID:SCR_014043) and R software v4.0.2. In brief, data were manually gated in Cytobank by event length, live/dead discrimination, and by canonical markers such as CD3. Samples with less than 1000 events were excluded from the analysis. For dimension reduction and cluster analyses, concatenated data with all markers except those used for manual gating (e.g., CD3+) were included. Cytobank’s viSNE dimension reduction algorithm was used with 1000 iterations, a perplexity of 30, and a theta of 0.5. Cytobank’s hierarchical consensus FlowSOM cluster analysis was performed with 10 iterations, 100 clusters, and 15 metaclusters. Density and cluster identification plots were created in R using results of viSNE and FlowSOM analyses. Plots summarizing relative cell abundance in metaclusters (e.g., percentage of CD3+ cells in metacluster #1) were created in R (metaclusters with less than 0.5% of the total cells were excluded). Heatmaps were created in R using Euclidian distance and standard normalization after arc-sin transformation.
Data were generated by the authors with the exception of the CyTOF data which were generated at a core facility.
Data Availability Statement
Data are available upon request to the corresponding author.
Results
GCSF and GCSFR are elevated in the tumor microenvironment of colon cancer as compared to histologically normal colon.
Previously, we demonstrated that the genes for GCSF and GCSFR (CSF3 and CSF3R) were expressed at higher levels in colon cancer samples as compared to histologically normal colon, with node positive tumors having higher expression than node negative tumors (19). To investigate ligand and receptor protein levels across a wider population of colon cancer patients we subjected retrospectively stored samples to IHC with digital segmentation and analysis. A summary of patient demographics and tumor stage is shown in Table 1 (UNM Cohort). Sample staining and digital segmentation are shown in Fig. 1A. Fig. 1B–C show higher expression of both ligand and receptor in tumor tissue as compared to normal tissue in the matched 206 patient samples. Ligand and receptor expression were positively correlated in both cancer and normal colon tissues (Fig. 1D–E) with Spearman’s rho 0.3034 for cancer tissue (p<0.0001) and rho 0.3469 (p<0.0001 for normal tissue). Contrary to our findings with qRT-PCR, in this larger dataset, we did not observe higher GCSF or GCSFR staining at higher stages (Fig. 1F–G). In addition, neither GCSF nor GCSFR levels were different between the sexes (Fig. 1H–I) nor correlated with the age of the patient (Fig. 1J–K). Finally, we anticipated that tumors with higher quantities of GCSF and GCSFR in the tumor microenvironment (TME) would have a stronger tumor associated neutrophil infiltrate, but this was not the case (Supplementary Fig. S1A–B). As expected, the majority of patient samples were available from patients diagnosed at Stage II (57%) or Stage III (31%), thereby limiting the conclusions we can draw from this data regarding a possible relationship between GCSF/R staining and stage. However, the data clearly demonstrate both that there is an elevation in GCSF and GCSFR in the majority of colon cancer tissues as compared to patient matched, histologically normal tissues. In addition, there was a range of levels with a subset of samples showing a greater than 2-fold higher expression of ligand and receptor suggesting a potentially distinct phenotype.
Table 1:
Patient Demographics
| UNM Cohort Total n=206 |
OUHSC Cohort Total n=119 |
OUHSC Cohort (Low serum GCSF) Total n=75 |
OUHSC Cohort (High serum GCSF) Total n=44 |
||
|---|---|---|---|---|---|
| Median age in years (range) | 62 (25-90) | 61 (23-84) | 61 (23-84) | 58 (33-83) | |
| p=0.3054 | |||||
| Sex | |||||
| Women (%) | 105 (51%) | 53 (45%) | 34 (45%) | 19 (43%) | |
| Men (%) | 101 (49%) | 66 (55%) | 41 (55%) | 25 (57%) | |
| p=0.9999 | |||||
| Pathological AJCC Stage | |||||
| Stage I (%) | 14 (7%) | 23 (19%) | 13 (17%) | 10 (23%) | |
| Stage II (%) | 117 (57%) | 36 (30%) | 22 (29%) | 14 (32%) | |
| Stage III (%) | 64 (31% | 36 (30%) | 26 (35%) | 10 (23%) | |
| Stage IV (%) | 11 (5%) | 24 (20%) | 14 (19%) | 10 (23%) | |
| p=0.5709 |
Figure 1:
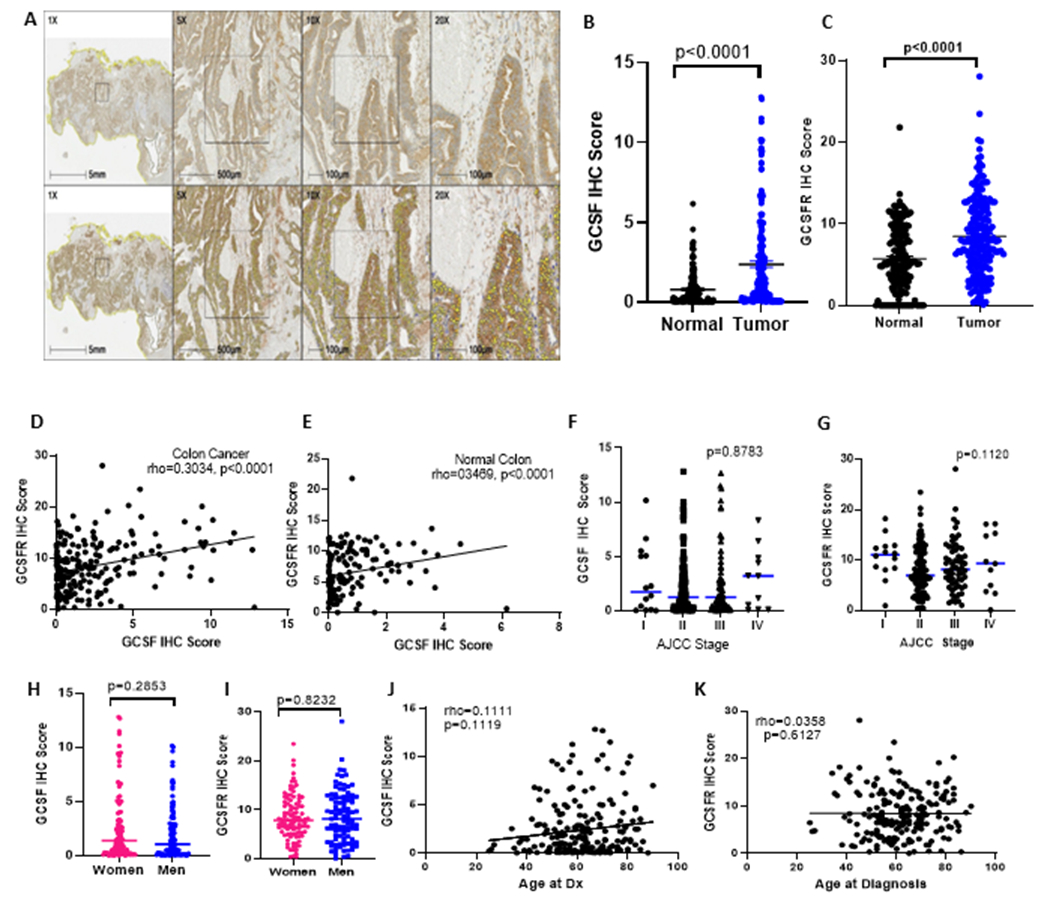
GCSF and GCSFR are elevated in the tumor microenvironment of colon cancer. Sample IHC and digital segmentation are shown in (A). GCSF (B) and GCSFR (C) are elevated in the epithelial compartment of human colon cancer as compared to histologically normal colonic epithelium (n=206). GCSF and GCSFR expression are positively correlated in both tumor bearing (D) and normal (E) epithelium. GCSF (F) and GCSFR (G) levels do not increase with higher stage at diagnosis and are not different between the sexes (H-I), nor do they correlate with age at diagnosis (J-K). Means with SEM and Spearman’s rho are shown.
High serum GCSF signaling is associated with an elevation in established markers of worse prognosis in colon cancer, T cells from colon cancer patients have higher expression of GCSFR than those from healthy controls, and chronic elevations of GCSF decrease interferon γ responses.
To better understand whether higher levels of systemic GCSF were associated with elevations in serum markers of adverse prognosis in colon cancer, we profiled the serum of 119 colon cancer patients drawn immediately prior to resection of their tumors. Demographic and stage data for these patients is shown in Table 1 (OUHSC Cohort). A total of 44 of the 119 (37%) of the patients had serum levels ≥50pg/ml which is 5-fold higher than normal levels in age-matched healthy controls (25). When patients with high serum GCSF (≥50pg/ml GCSF) were compared to those with low GCSF (<50pg/ml GCSF), we observed significantly higher levels of serum analytes associated with worse outcomes in colon cancer. Specifically, VEGF, IL4, IL10, IL6, and IL8 were all higher in patients with serum GCSF≥50pg/ml as compared to those with low serum GCSF (Fig. 2A–E).
Figure 2.

Colon cancer patients with elevated preoperative serum GCSF have elevations in established markers of poor prognosis, T cells from patients with colon cancer have higher GCSFR expression than those from healthy controls, and chronically elevated GCSF decreases interferon γ responses in T cells. Comparison of median serum cytokine levels in patients with elevated vs normal levels of GCSF are shown in (A-E) (Median with 95%CI shown, comparison by Mann-Whitney). Percent of circulating CD8 T cells with GCSFR expression detected by flow cytometry with mean and SEM is shown in (F) (comparison by Student’s t-test). Chronic GCSF (10ng/ml) treatment decreases CD8 interferon γ responses as shown in (G) with mean and SEM (comparison by paired t test).
Because we previously observed significant increases in tumor infiltrating CD8 T cells in mice subjected to the AOM/DSS model and GCSF inhibition (20), we sought to determine whether circulating CD8 T cells from colon cancer patients expressed GCSFR and whether the rate of expression was higher as compared to healthy control donors. Fresh PBMCs were taken from patients and healthy control donors and subjected to negative magnetic bead selection for CD8 T cells. Flow cytometry was used to determine the mean percent of CD8 T cells positive for GCSFR. As shown in Fig. 2F, the rate of GCSFR+ CD8+ T cells in colon cancer patients was double that of normal healthy controls (65% vs 33%, p=0.0097). To determine whether chronically elevated serum GCSF levels would decrease the T cell IFNγ response, we isolated CD8 cells as above from 7 donors, split each sample into 3 wells, rested the cells overnight and then cultured them for 7 days. GCSF (10ng/ml) was added to one well for each sample at Day 1 with the second well receiving GCSF supplementation starting on Day 4. All three samples were assessed for IFNγ expression respectively after 7 days of culture and 0, 3 and 7 days of GCSF treatment. Chronic GCSF treatment significantly decreased the IFNγ expression in the cultured CD8+ T cells as shown in (Fig. 2G).
Inhibition of GCSF results in rapid shifts in the tumor immune microenvironment in a mouse model of peritoneal colon cancer carcinomatosis.
Because we saw an increase in T cell infiltration with phenotypic shifts in the innate immune infiltrate within the TME in previous work with single agent αGCSF treatment (20), we sought to gain a more comprehensive understanding of the effects of this treatment in the setting of end-stage, essentially incurable metastatic disease. While patients with solid organ metastasis can undergo combined resection and chemotherapy for long-term survival and even cure in some cases, peritoneal metastases have proven more challenging to treat even with aggressive approaches combining systemic and hyperthermic intraperitoneal chemotherapy. (26, 27) To evaluate the TME at the same timepoint required sacrificing the mice at a defined endpoint (Fig. 3A) to assure the same total treatment exposure. Because preliminary data revealed tumors of approximately 2-4 mm3 throughout the peritoneal surfaces, including visible diaphragmatic and mesenteric implants as early as 9 days post-IP injection, treatments were started on Day 3 with mice (n=6 per group) each receiving a total of 5 treatments (αGCSF vs isotype control) before being sacrificed on Day 14. Tumor samples were processed for mass cytometry with the panel of markers shown in Supplementary Table S1. Unbiased clustering of concatenated data from live CD45+ singlets through Flowsom was performed on Cytobank resulting in 12 metaclusters with >0.5% cells (Fig. 3B). Density plots revealed shifts within one CD8+ metacluster (Fig. 3C) when treatments were compared, leading us to perform separate Flowsom clustering after gating for CD3+ cells (Fig. 3D–E). Both CD3+ and CD19+ cells were gated out to allow assessment of the innate compartment as shown in Fig. 3F–G. Data resulting from gating for CD19+ cells are shown in Supplementary Fig. S2A–B.
Figure 3.

Inhibition of GCSF results in rapid shifts in the tumor immune microenvironment. A total of 12 C57BL/6J mice (5-8-week-old) were injected with 105 MC38 cells on Day 0, with treatments (αGCSF vs isotype control (both 25μg/mouse)) administered 3x/week beginning on Day 3 as shown in (A). Mice received 5 total treatments and were sacrificed on Day 14. Tumors were dissociated and subjected to CyTOF with resulting concatenated data for all cells used to generate 15 metaclusters (B) with treatments compared through creation of density plots with equal numbers of cells/plot (C). Gating concatenated cells for CD3+ cells resulted in a T cell specific two-dimensional visualization (D), with corresponding density plots, cluster jitter plot and heat map shown in (E). Similar plots were created for CD3−CD19− cells (F-G).
Among the CD3+ population, activated CD4 and CD8 T cells increased with αGCSF treatment while CD44-CD4+ cells decreased. Furthermore, while the CD8 cluster that increased upon exposure to αGCSF expressed PD1+ and Lag3+ this cluster did not express Tim3 which has been associated with terminal exhaustion (28).
Plots for cells that did not express either CD3 or CD19 are show in Fig. 3F–G. This revealed a greater diversity in the innate infiltrate as a result of treatment with decreases in a cluster of Ly6C+/Ly6G+/CD11b+/CD44+ cells (cluster 5). In addition, treatment resulted in a modest increase in cluster 3 (F4/80+CD11b+, PDL1med, CD44+).
Inhibition of GCSF improves survival and T cell function within the tumor microenvironment of an advanced model of peritoneal carcinomatosis with female mice receiving greater benefit.
To determine the effects of GCSF inhibition on survival in the setting of advanced metastatic colon cancer, we established peritoneal carcinomatosis by intraperitoneal injection (IP) of 105 MC38 cells in 5–8-week-old C57BL/6J mice as above. Treatment with αGCSF antibody vs isotype control was administered 3 times weekly beginning on Day 9 post tumor injection and continued until mice were sacrificed based on a priori determined criteria (“Survival Model” shown in Fig. 4A). Tumor bearing, treatment naive mice have significant elevations in serum GCSF as compared to controls (Fig. 4B). The efficacy of the GCSF blockade in reducing the systemic GCSF levels to that of healthy mice as compared to isotype control is shown in Fig. 4C. Mice treated with αGCSF antibody had a reduction in the Mantel-Haenszel hazard ratio (HR) to 0.4598 (p=0.0237, n=48) as compared to the control group (Fig. 4D). The survival benefit seen for αGCSF treatment was greater for female mice (HR=0.2923, p=0.0066) than for male mice (HR=0.5398, p=0.3033) (Fig. 4E–F).
Figure 4.

Inhibition of GCSF improves survival in an advanced model of peritoneal carcinomatosis and increases T cell infiltration and activity in the tumor immune microenvironment. A total of 48 C57BL/6J mice (5-8-week-old) were injected with 105 MC38 cells on Day 0, with treatments (αGCSF vs isotype control (both 25μg/mouse)) administered 3x/week beginning on Day 9 as shown in (A). Mice were treated until sacrificed due to reaching a priori humane endpoints. Untreated mice with MC38 IP tumor induction have elevated serum GCSF as compared to healthy controls (B) and αGCSF treatment abrogates this response (C). Treatment with αGCSF resulted in a HR reduction (Mantel-Haenszel) of 0.4598, p=0.0237, as compared to mice treated with isotype control (D). Effect size was sex dependent (E-F). Treatment with αGCSF resulted in a significant increase in total CD8 (G) and CD4 (H) infiltration into the TME in a sex-dependent manner (I and J) as well as increased ex vivo IFNγ responsiveness for both CD8 (K) and CD4 (L) cells that was also sex-specific (M and N). Means with SEM are shown with comparisons made with Mann-Whitney test.
Similar to our previous findings in the AOM/DSS colon cancer development model, we noted a significant increase of tumor infiltrating CD8 and CD4 T cells with αGCSF treatment (Fig. 4G and 4H), with greater effects in female mice as compared to male mice (Fig. 4I and 4J). In addition, the tumor infiltrating CD8 and CD4 cells had a greater IFNγ response to stimulation ex vivo (Fig. 4K and L), suggesting treatment decreased the rate of terminal exhaustion for tumor infiltrating T cells, consistent with the CyTOF findings above. These effects were also greater in the female mice as compared to the male mice (Fig. M and N).
Both increased serum GCSF and a higher gradient between serum GCSF and GCSF levels in the TME correlate with decreased CD4 and CD8 infiltration into colon cancer tumors in women.
Because we observed significant increases in T cell infiltration into the TME that were sex specific with use of αGCSF treatment in the mouse model, we hypothesized that higher serum GCSF levels would inversely correlate with the T cell infiltrate in colon cancer patients. We further hypothesized that a higher GCSF gradient from serum to TME would also be associated with decreased tumor infiltrating T cells. To test this, we used matched serum and tumor resection specimens from prospectively consented colon cancer patients. Serum GCSF levels were measured by bead-based array and IHC with digital analysis (Aperio™) was used to quantify intra-tumoral CD4 and CD8 cell counts as well as GCSF H scores after the tumor containing slide regions were annotated by a pathologist (MK). There was a modest inverse correlation between serum GCSF and CD4 infiltration into the matched tumors (Fig. 5A, Spearman’s rho= −0.3270, p=0.0249). Similarly, while not achieving statistical significance, there was a weak inverse correlation between serum GCSF and CD8 counts (Fig. 5B, Spearman’s rho= −0.2684, p=0.0681). When the gradient between the serum and TME was assessed by division of serum GCSF by tumor GCSF H score (serum:tumor GCSF), the inverse correlation remained for CD4 (Fig. 5C, Spearman’s rho= −0.3602, p=0.0151) and the trend remained for CD8 (Fig. 5D, Spearman’s rho= −0.2813, p=0.0612). However, when we analyzed the relationship by patient sex, it became clear that this inverse correlation was present in tumors taken from women but not men (Fig. 5E–L). The inverse correlations for women were as follows: serum GCSF and CD4, Spearman’s rho= −0.5501, p=0.0044, serum GCSF and CD8, Spearman’s rho= −0.5820, p=0.0023, serum:tumor GCSF and CD4, Spearman’s rho=−0.4923, p=0.0124, and serum:tumor GCSF and CD8, Spearman’s rho= −0.6138, p=0.0011. None of the GCSF/T cell infiltration measures were significantly correlated in the men. Total CD4 and CD8 counts did not differ between the sexes.
Figure 5.

Elevated preoperative serum GCSF and a steep serum to tumor microenvironmental GCSF gradient in CRC patients is associated with decreased CD4 and CD8 (A-D) infiltration into the tumor microenvironment with effects driven by samples from female patients (E-H) and no relationship in samples from male patients (I-L) Serum GCSF was measured in the immediately preoperative period with multiplex bead-based assay, FFPE processed blocks from the resection specimens were stained for CD4 and CD8 with digital analysis (Aperio™) used to assess the percent of nucleated cells that stained for each marker. Sample size, n=47 (25 females, 22 males), Spearman’s rho with best fit linear regression line shown.
Discussion
In this study we demonstrate that GCSF and GCSFR are increased in the tumor microenvironment of colon cancer as compared to histologically normal colon in patient samples as well as reveal a sex-specific survival benefit for use of αGCSF treatment in an end-stage mouse model of peritoneal colon cancer carcinomatosis. Furthermore, elevations of serum GCSF in patients are associated with increases in a pro-angiogenic, immunosuppressive milieu as well as decreased T cell infiltration into colon tumors with greater effects noted in samples from women. Consistent with our previous work in a colon cancer development model, GCSF inhibition increased T cell infiltration and IFNγ responses in an end-stage model of peritoneal colon cancer carcinomatosis, with enhanced anti-tumor effects in female mice. These results suggest that GCSF inhibition is a viable immunotherapeutic target for colon cancer.
The sex-based differences noted in both our human and mouse data suggest that inhibition of GCSF will have a greater efficacy in women which is critical to be aware of for clinical trial considerations of the approach. This could also be of high clinical importance given that the preponderance of available data examining sex-based differences in response to current anti-cancer immunotherapies suggest better results for men even when results from trials for colon cancer alone are considered (21, 29). The mean age of the patients in this study was 61 years-old, a full decade after the mean age of menopause in the United States. In addition, none of the women in the OUHSC cohort were taking hormone replacement therapy at the time of surgery, suggesting the differential effects of GCSF on T cell tumor infiltration between men and women was not due to large differences in circulating estrogen at the time of diagnosis. Indeed, other investigators have found evidence supporting immutable effects of early developmental hormonal exposure on the transcriptome of immune cells (30), suggesting that the immune response to a tumor or to an anti-cancer immunotherapy may be driven by the hormonal milieu of the patient long before the tumor develops.
While recombinant GCSF (rGCSF) has been administered to cancer patients undergoing cytotoxic chemotherapy since FDA approval in 1991, the majority of studies examined the question of whether rGCSF is successful in raising the adjusted neutrophil count or reducing hospitalizations for febrile neutropenia. Recently, however, investigators examined rates of distant organ metastasis following resection and adjuvant chemotherapy for lung cancer in a retrospective study of 307 patients. Receipt of rGCSF was independently associated with a 2.33 HR for metastasis. Furthermore, a dose dependent effect was noted (31). Taken in context with our work, this suggests that the effects of GCSF on solid tumors are complex and potentially supportive of tumor progression.
While our work has focused on colon cancer, other groups have demonstrated similar pro-tumor effects for enhanced GCSF signaling in other solid tumors such as breast cancer, neuroblastoma, head and neck squamous cell cancers, and bladder cancers (32–36). A PubMed search performed on October 18, 2022, for the search term “GCSF producing tumor case report” elicits 238 entries describing exceedingly aggressive courses for patients with GCSF secreting solid tumors including sarcomas, pancreatic cancers, colon cancers and cholangiocarcinomas. While tumors secreting extremely high levels of GCSF are rare, the poor prognosis and sheer variety of solid tumors that have been noted to do so, taken with our data, suggest that tumor production of GCSF supports solid tumor progression and evasion of immune responses.
While the majority of our data supports consideration of GCSF as an anti-colon cancer immunotherapeutic target, it is important to note that there is potential for direct anti-tumor effects on the epithelial compartment as well. We and others have shown that GCSF treatment increases solid tumor proliferation, migration, and is associated with increased stem-like cells in colon cancer, gastric cancer, head and neck cancers, and neuroblastomas (19, 32, 34, 37). Furthermore, in vitro experiments demonstrate a reversal of the pro-proliferation and migration effects with GCSF inhibition (19). The HR reduction of 0.46 with GCSF inhibition in our mouse model suggests that, while single agent treatment may not eliminate widespread metastatic disease, it can slow progression, potentially through multiple different mechanisms.
There are limitations to our study in addition to future directions for this work. While our initial data suggested that higher stage colon cancer was associated with greater gene GCSF (CSF3) expression in the TME, examination of protein expression on a larger scale did not confirm this finding. Given the relatively small numbers of Stage I and IV tumors in our sample, however, this could be due to sample size and will require additional work to confirm or refute the initial findings. Similarly, increased preoperative serum GCSF was not associated with higher stage at presentation, but again, smaller numbers of patients with Stage IV disease were taken for resection. While we suspect that sample size is the issue, it is also possible that patients with high serum and/or tumor GCSF form a distinct subset of patients who may benefit more from GCSF inhibition as a treatment strategy. Indeed, a recent examination of TCGA data reveals that Consensus Molecular Subtype four colon cancers, which exclude immune infiltration, are associated with enhanced TGFβ signaling, dense stroma, and worse overall survival outcomes, have the highest expression of GCSF receptor between the four subtypes (38, 39).
Because we found that GCSF inhibition was able to reduce colon cancer development, we sought to determine the potential for anti-tumor effects at the other end of the disease spectrum, using a model of peritoneal metastasis. While patients with limited solid organ metastases from colon cancer are often candidates for resection, sometimes resulting in long-term disease-free survival, patients with peritoneal metastases from colon cancer are generally considered incurable. It is possible, however, that the anti-tumor effects and immune responses to GCSF inhibition may be different in metastases to the liver, suggesting additional avenues for investigation.
In conclusion, GCSF signaling is associated with a pro-angiogenic and immunosuppressive phenotype in colon cancers. Inhibition of GCSF signaling slows tumor progression in an end-stage model of peritoneal carcinomatosis and has potential to synergize with both cytotoxic chemotherapeutic regimens such as oxaliplatin which increase immunogenic cell death and immune checkpoint blockade which requires T cell infiltration into the TME for efficacy against solid tumors such as colon cancer. The anti-tumor and T cell effects of αGCSF appear to be greater in females. While much work remains to be done in understanding the potential for this strategy, the preponderance of available evidence suggests a high likelihood of impact.
Supplementary Material
{kind=link}
{kind=link}
Statement of Translational Relevance:
Here we report that granulocyte colony stimulating factor (GCSF) is increased systemically and in the tumor microenvironment of patients with colon cancer and that inhibition of GCSF increases survival in an end-stage mouse model of peritoneal carcinomatosis from colon cancer. In addition, this single agent treatment results in shifts in the global tumor immune microenvironment with a significant increase in both the number and activity of tumor infiltrating T cells. We further demonstrate that this therapeutic approach has greater benefits in female mice, with corresponding data from human samples suggesting that women would benefit from this approach more than men. This has translational relevance because there is a humanized antibody to the GCSF receptor currently in clinical trials for hidradenitis. Future trials addressing efficacy of this antibody against solid tumors should be stratified by sex based on these data.
Acknowledgements:
The authors would like to thank Dr. Sarah Adams for critical reviews and scientific discussions, Dr. William Berry for collaborations, Drs. Rajagopal Ramesh and Joe Zhao for mentorship, Drs. Caleb Marlin, Carla Guthridge, and Joel Guthridge from the Oklahoma Medical Research Foundation Human Phenotyping Core Facility for processing the CyTOF samples, and Mr. Kurt Scheuermann for statistical advice and support.
Research reported in this publication was supported in part by the following: American Cancer Society Award Number MRSG 15-136-01-CCE (KTM), the National Institutes of General Medical Sciences of the National Institutes of Health under Award Number P20GM103639 (KTM), the National Cancer Institute Cancer Center Support Grant P30CA225520 (KTM, KMF). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Society AC. American Cancer Society. Colorectal Cancer Facts & Figures 2022. 2022. [Google Scholar]
- 2.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians. 2018;68(6):394–424. [DOI] [PubMed] [Google Scholar]
- 3.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–4. [DOI] [PubMed] [Google Scholar]
- 4.Galon J, Pages F, Marincola FM, Angell HK, Thurin M, Lugli A, et al. Cancer classification using the Immunoscore: a worldwide task force. J Transl Med. 2012;10:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pages F, Mlecnik B, Marliot F, Bindea G, Ou FS, Bifulco C, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 2018;391(10135):2128–39. [DOI] [PubMed] [Google Scholar]
- 6.Picard E, Verschoor CP, Ma GW, Pawelec G. Relationships Between Immune Landscapes, Genetic Subtypes and Responses to Immunotherapy in Colorectal Cancer. Front Immunol. 2020;11:369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amariglio N, Jacob-Hirsch J, Shimoni A, Leiba M, Rechavi G, Nagler A. Changes in gene expression pattern following granulocyte colony-stimulating factor administration to normal stem cell sibling donors. Acta Haematol. 2007;117(2):68–73. [DOI] [PubMed] [Google Scholar]
- 8.Bunse CE, Borchers S, Varanasi PR, Tischer S, Figueiredo C, Immenschuh S, et al. Impaired functionality of antiviral T cells in G-CSF mobilized stem cell donors: implications for the selection of CTL donor. PLoS One. 2013;8(12):e77925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bunse CE, Tischer S, Lahrberg J, Oelke M, Figueiredo C, Blasczyk R, et al. Granulocyte colony-stimulating factor impairs CD8(+) T cell functionality by interfering with central activation elements. Clin Exp Immunol. 2016;185(1):107–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franzke A, Piao W, Lauber J, Gatzlaff P, Konecke C, Hansen W, et al. G-CSF as immune regulator in T cells expressing the G-CSF receptor: implications for transplantation and autoimmune diseases. Blood. 2003;102(2):734–9. [DOI] [PubMed] [Google Scholar]
- 11.Ge F, Zhang Z, Hou J, Cao F, Zhang Y, Wang P, et al. Granulocyte colony-stimulating factor decreases the Th1/Th2 ratio in peripheral blood mononuclear cells from patients with chronic immune thrombocytopenic purpura in vitro. Thrombosis research. 2016;148:76–84. [DOI] [PubMed] [Google Scholar]
- 12.Joo YD, Lee WS, Won HJ, Lee SM, Kim HR, Park JK, et al. G-CSF-treated donor CD4+ T cells attenuate acute GVHD through a reduction in Th17 cell differentiation. Cytokine. 2012;60(1):277–83. [DOI] [PubMed] [Google Scholar]
- 13.Malashchenko VV, Meniailo ME, Shmarov VA, Gazatova ND, Melashchenko OB, Goncharov AG, et al. Direct anti-inflammatory effects of granulocyte colony-stimulating factor (G-CSF) on activation and functional properties of human T cell subpopulations in vitro. Cellular immunology. 2018;325:23–32. [DOI] [PubMed] [Google Scholar]
- 14.Fujiwara Y, Yamazaki O, Takatsuka S, Kaizaki R, Inoue T. Granulocyte colony-stimulating factor-producing ascending colon cancer as indicated by histopathological findings: report of a case. Osaka City Med J. 2011;57(2):79–84. [PubMed] [Google Scholar]
- 15.Joshita S, Nakazawa K, Sugiyama Y, Kamijo A, Matsubayashi K, Miyabayashi H, et al. Granulocyte-colony stimulating factor-producing pancreatic adenosquamous carcinoma showing aggressive clinical course. Intern Med. 2009;48(9):687–91. [DOI] [PubMed] [Google Scholar]
- 16.Kawaguchi M, Asada Y, Terada T, Takehara A, Munemoto Y, Fujisawa K, et al. Aggressive recurrence of gastric cancer as a granulocyte-colony-stimulating factor-producing tumor. Int J Clin Oncol. 2010;15(2):191–5. [DOI] [PubMed] [Google Scholar]
- 17.Tajima S, Waki M, Tsuchiya T, Hoshi S. Granulocyte colony-stimulating factor-producing undifferentiated carcinoma of the colon mimicking a pulmonary giant cell carcinoma: a case showing overexpression of CD44 along with highly proliferating nestin-positive tumor vessels. International journal of clinical and experimental pathology. 2014;7(10):7034–41. [PMC free article] [PubMed] [Google Scholar]
- 18.Kudo T, Matsumoto T, Nakamichi I, Yada S, Esaki M, Jo Y, et al. Recombinant human granulocyte colony-stimulating factor reduces colonic epithelial cell apoptosis and ameliorates murine dextran sulfate sodium-induced colitis. Scand J Gastroenterol. 2008;43(6):689–97. [DOI] [PubMed] [Google Scholar]
- 19.Morris KT, Khan H, Ahmad A, Weston LL, Nofchissey RA, Pinchuk IV, et al. G-CSF and G-CSFR are highly expressed in human gastric and colon cancers and promote carcinoma cell proliferation and migration. Br J Cancer. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morris KT, Castillo EF, Ray AL, Weston LL, Nofchissey RA, Hanson JA, et al. Anti-G-CSF treatment induces protective tumor immunity in mouse colon cancer by promoting protective NK cell, macrophage and T cell responses. Oncotarget. 2015;6(26):22338–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Conforti F, Pala L, Bagnardi V, De Pas T, Martinetti M, Viale G, et al. Cancer immunotherapy efficacy and patients’ sex: a systematic review and meta-analysis. The Lancet Oncology. 2018;19(6):737–46. [DOI] [PubMed] [Google Scholar]
- 22.Ram S, Vizcarra P, Whalen P, Deng S, Painter CL, Jackson-Fisher A, et al. Pixelwise H-score: A novel digital image analysis-based metric to quantify membrane biomarker expression from immunohistochemistry images. PLoS One. 2021;16(9):e0245638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ray AL, Nofchissey RA, Khan MA, Reidy MA, Lerner MR, Wu X, et al. The role of sex in the innate and adaptive immune environment of metastatic colorectal cancer. Br J Cancer. 2020;123(4):624–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Corbett TH, Griswold DP, Roberts BJ Jr., Peckham JC, Schabel FM Jr. Tumor induction relationships in development of transplantable cancers of the colon in mice for chemotherapy assays, with a note on carcinogen structure. Cancer Res. 1975;35(9):2434–9. [PubMed] [Google Scholar]
- 25.Kim HO, Kim H-S, Youn J-C, Shin E-C, Park S. Serum cytokine profiles in healthy young and elderly population assessed using multiplexed bead-based immunoassays. Journal of translational medicine. 2011;9:113-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tomlinson JS, Jarnagin WR, DeMatteo RP, Fong Y, Kornprat P, Gonen M, et al. Actual 10-year survival after resection of colorectal liver metastases defines cure. J Clin Oncol. 2007;25(29):4575–80. [DOI] [PubMed] [Google Scholar]
- 27.Quénet F, Elias D, Roca L, Goéré D, Ghouti L, Pocard M, et al. Cytoreductive surgery plus hyperthermic intraperitoneal chemotherapy versus cytoreductive surgery alone for colorectal peritoneal metastases (PRODIGE 7): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2021;22(2):256–66. [DOI] [PubMed] [Google Scholar]
- 28.Schatton T, Itoh Y, Martins C, Rasbach E, Singh P, Silva M, et al. Inhibition of melanoma cell-intrinsic Tim-3 stimulates MAPK-dependent tumorigenesis. Cancer Res. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ye Y, Jing Y, Li L, Mills GB, Diao L, Liu H, et al. Sex-associated molecular differences for cancer immunotherapy. Nat Commun. 2020;11(1):1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Villa A, Gelosa P, Castiglioni L, Cimino M, Rizzi N, Pepe G, et al. Sex-Specific Features of Microglia from Adult Mice. Cell Rep. 2018;23(12):3501–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, Fang C, Chen R, Yuan S, Chen L, Qiu X, et al. rhG-CSF is associated with an increased risk of metastasis in NSCLC patients following postoperative chemotherapy. BMC Cancer. 2022;22(1):741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsu DM, Agarwal S, Benham A, Coarfa C, Trahan DN, Chen Z, et al. G-CSF receptor positive neuroblastoma subpopulations are enriched in chemotherapy-resistant or relapsed tumors and are highly tumorigenic. Cancer Res. 2013;73(13):4134–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Swierczak A, Cook AD, Lenzo JC, Restall CM, Doherty JP, Anderson RL, et al. The promotion of breast cancer metastasis caused by inhibition of CSF-1R/CSF-1 signaling is blocked by targeting the G-CSF receptor. Cancer Immunol Res. 2014;2(8):765–76. [DOI] [PubMed] [Google Scholar]
- 34.Gutschalk CM, Herold-Mende CC, Fusenig NE, Mueller MM. Granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor promote malignant growth of cells from head and neck squamous cell carcinomas in vivo. Cancer Res. 2006;66(16):8026–36. [DOI] [PubMed] [Google Scholar]
- 35.Aliper AM, Frieden-Korovkina VP, Buzdin A, Roumiantsev SA, Zhavoronkov A. A role for G-CSF and GM-CSF in nonmyeloid cancers. Cancer medicine. 2014;3(4):737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tachibana M, Murai M. G-CSF production in human bladder cancer and its ability to promote autocrine growth: a review. Cytokines Cell Mol Ther. 1998;4(2):113–20. [PubMed] [Google Scholar]
- 37.Sunaga H, Fujieda S, Tsuzuki H, Asamoto K, Fukuda M, Saito H. Expression of granulocyte colony-stimulating factor receptor and platelet-derived endothelial cell growth factor in oral and oropharyngeal precancerous lesions. Anticancer Res. 2001;21(4B):2901–6. [PubMed] [Google Scholar]
- 38.Inamura K. Colorectal Cancers: An Update on Their Molecular Pathology. Cancers. 2018;10(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saunders AS, Bender DE, Ray AL, Wu X, Morris KT. Colony-stimulating factor 3 signaling in colon and rectal cancers: Immune response and CMS classification in TCGA data. PLoS One. 2021;16(2):e0247233. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available upon request to the corresponding author.