

Abstract
This review outlines the effect of disease-causing mutations on proteins’ thermodynamics. Two major thermodynamics quantities, which are essential for structural integrity, the folding and binding free energy changes caused by missense mutations, are considered. It is emphasized that disease effect in case of complex diseases may originate from several mutations over several genes, while monogenic diseases are caused by mutation is a single gene. Nevertheless, in both cases it is shown that pathogenic mutations cause larger perturbations of the above-mentioned thermodynamics quantities as compared with the benign mutations. Recent works demonstrating the effect of pathogenic mutations on the above-mentioned thermodynamics quantities, as well on structural dynamics and allosteric pathways are reviewed.
Keywords: thermodynamics, pathogenicity, folding free energy, binding free energy, missense mutations, diseases
Introduction
With the rapid progress of genomic sequencing and increased number of DNA samples, it became clear that almost all human diseases have genetic component, i.e., diseases are caused in whole or in part by a change(s) in the DNA sequence of an individual away from the normal sequence. Such genetic disorders can be caused by a mutation in one gene (monogenic disorder), by mutations in multiple genes (polygenic/complex disorder), or by a combination of gene mutations, mutations in their regulatory sequence and environmental factors. In the recent years significant progress was made in understanding polygenic and other complex human diseases, due in large part to knowledge of the human genome sequence and the development of new technologies that allow investigators to associate disease phenotypes with genetic loci. However, genetic linkage of complex diseases is much more difficult to assess, compared with monogenic diseases, since the outcome is a result of many small contributions from numerous variants and also depends on the environment (gene by environment (G × E) interactions) [1]. Thus, although polygenic diseases are more common than monogenic disorders, studies of monogenic diseases provide an invaluable opportunity to learn about underlying molecular mechanisms, which is crucial for developing therapeutic solutions [2]. Furthermore, while many genotypes may be associated with a disease, typically the corresponding phenotypes are only few, i.e., numerous genotypes result in only several prominent molecular mechanisms causing the disease. In this review, we will focus on recent works aiming to reveal the effect of pathogenic mutations on thermodynamic properties of the corresponding biological macromolecules. By thermodynamical properties, we refer to folding and binding free energy changes caused by mutations. Figure 1 illustrates such scenarios.
Figure 1.
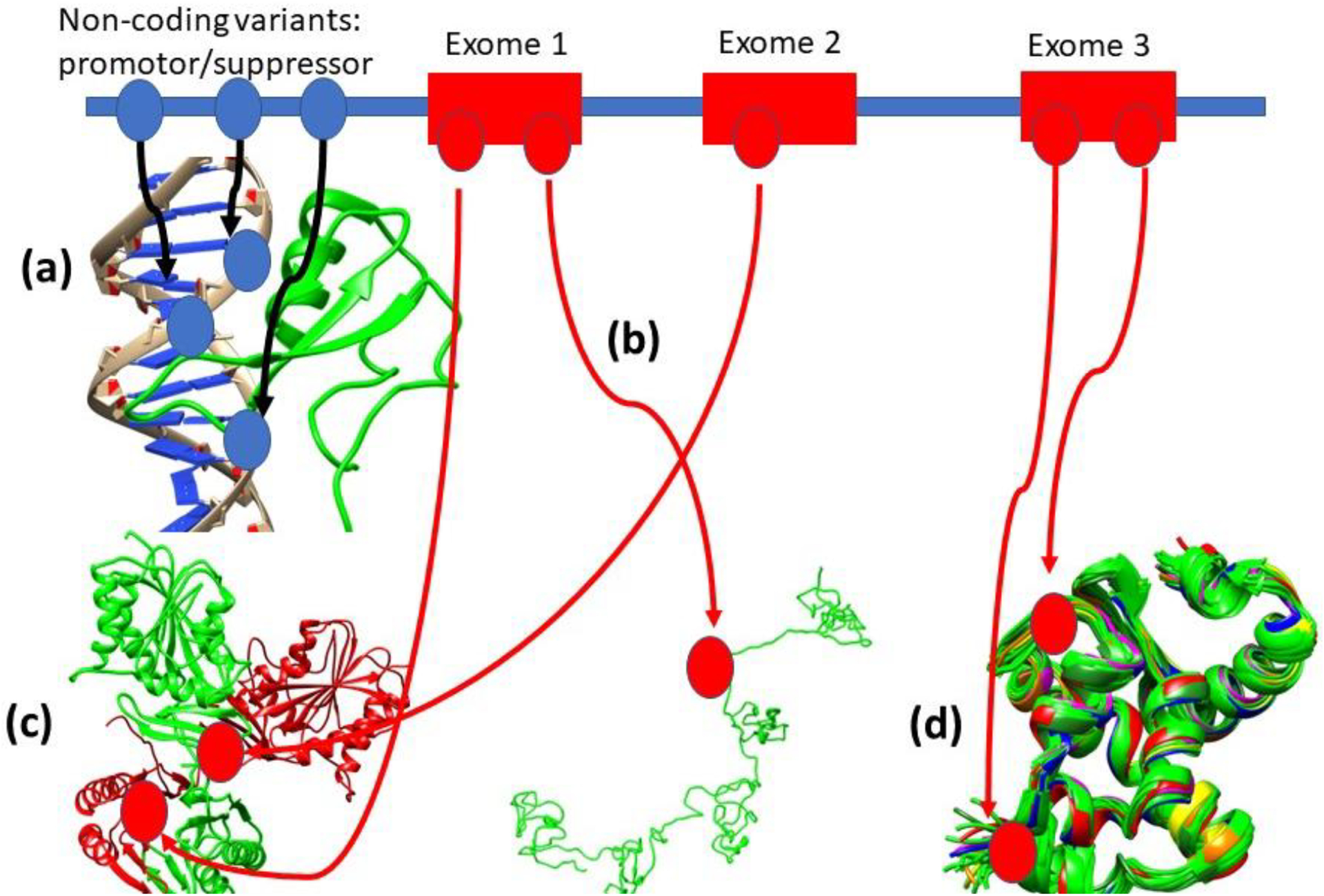
Schematic representation of variants effects: (a) affecting regulation; (b) resulting in unfolded protein: (c) affecting protein-protein binding; and (d) affecting protein dynamics. Circles indicate a variant in DNA and in the corresponding proteins. The figure is only for illustration of genotype-phenotype relations in case of monogenic disorder.
Thermodynamics and pathogenicity
The outcome of disease-causing mutations on structural integrity of biological macromolecules was extensively investigated, both computationally and experimentally [3]. Researchers focused on revealing mutations’ plausible effects on protein stability [3], protein-protein interactions [4] and networks [5], protein-DNA binding [6], characteristics of the active site [7], and many other structural characteristics [8]. However, typically this was done for limited number of cases, either a particular disease or a protein family. A more comprehensive investigation on the linkage between thermodynamics and pathogenicity was reported on a set of thousand mutations and experimentally measured folding and binding free energy changes [9]. It was demonstrated that the Pearson correlation coefficient between the probability of a mutation to be pathogenic and the probability of the same mutation to cause large change of either folding or binding free energies can reach 0.7 [9]. This result was achieved without discriminating complex from monogenic diseases. To eliminate the ambiguity of cases originating from the collective effect of many small contributions, we created a database of monogenic diseases, the corresponding protein sequences, and list of both pathogenic and benign mutations (supplementary material). This resulted in two datasets (available from http://compbio.clemson.edu/lab/downloads/) wherein Dataset 1 contains 686 proteins and 1934 pathogenic and 1405 benign mutations (only mutations that are classified as pathogenic or benign) and Dataset 2 contains 768 proteins and 2559 pathogenic and 1763 benign mutations (Dataset 2 includes the likely benign and likely pathogenic cases as well). The change of the folding free energy caused by mutation was calculated with the leading sequence-based predictors and the ROC curves are shown in Figure 2. It can be seen that there is strong correlation between folding free energy change and a mutation to be pathogenic. The area under curve (AUC) was observed to be 0.71 and Matthew’s correlation coefficient MCC=0.32 for the two datasets when the folding free energy values were averaged using all predictors. An improvement of both, the AUC (AUC=0.77) and MCC (MCC=0.40) was observed by taking the average of folding free energy calculated using SAAFEC-SEQ [10] and INPS-SEQ [11] predictors. It should be clarified that the change of the binding free energy was not considered because it would introduce ambiguity since for most cases in the dataset, the binding partner(s) is unknown or does not exist. Nevertheless, the results indicate strong correlation between folding free energy changes and pathogenicity, which will be further strengthen by outlining resent works on similar topics.
Figure 2.
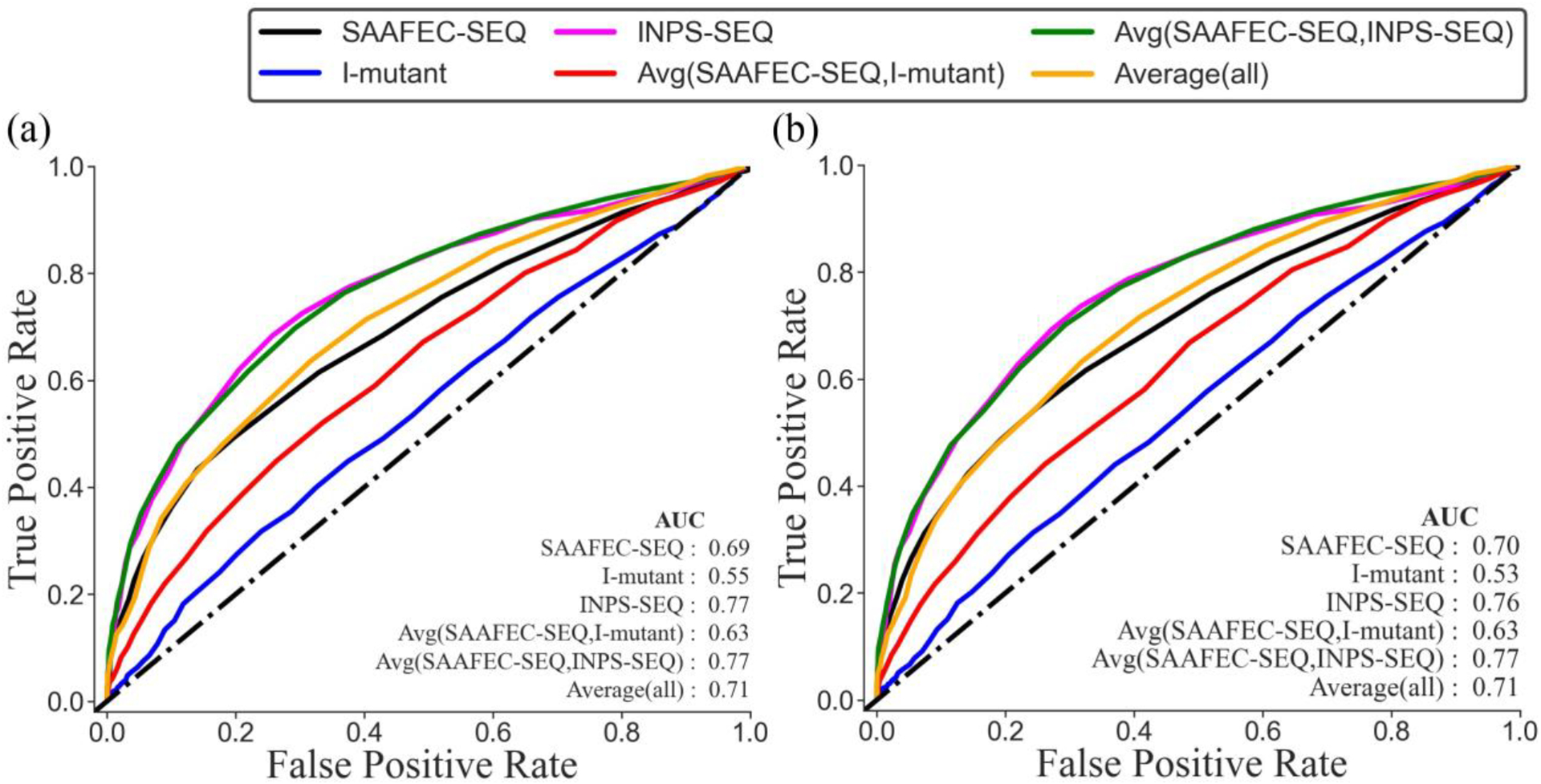
ROC curve for monogenic disorder database. (a) Dataset 1 containing mutations that are listed as pathogenic or benign; (b) Dataset 2 in addition including mutations that are listed as likely pathogenic or likely benign.
Why understanding linkage between pathogenicity and thermodynamics is important?
Understanding the molecular mechanisms that cause disease allows researchers to develop appropriate treatments [12]. For example, if a mutation affects a particular protein–protein interaction, one can target this particular interaction with either appropriate inhibitors (if the mutation makes interaction stronger) [13] or with binding enhancers (if the mutation makes the binding weaker) [14,15]. Similarly, if a pathogenic mutation affects protein conformation, the effect can be mitigated with small molecule which binding to the mutant protein restore the wild-type properties [16]. In terms of pharmacogenetics, one wants to know the effect of amino acid variants on interaction between the target protein and several alternative drugs, thus enabling informative decisions about the suitability of drugs for given patients based on their DNA variants [17].
Recent works on folding free energy changes caused by mutations and linkage with diseases
Wild type macromolecules are in their most favorable free energy state and therefore missense mutations are expected to reduce their stability, although examples of opposite do exist [18]. Mutations that shift the equilibrium towards formation of unfolded state are the root cause of several diseases ranging from monogenic to complex diseases. It results in two kinds of problems; first, loss of function caused by scarcity of folded protein and second, formation of supra-molecular assemblies like amyloids that affect series of cellular processes. In a recent study by Aledo et al. [19], the authors developed a neutral continuous fitness-stability model based on Arrhenius law to study the effect of mutation on protein stability, which was validated by performing 137,073,638 mutations in 14,094 proteins. It was shown that most of the mutations destabilize protein structure and their destabilizing effect of amino acid substitution on protein structure correlates positively with the disease-causing potential. Another work focused on the mutational landscape (5187 substitutions) of cancer-associated human NAD(P) H:quinone oxidoreductase 1 (NQO1) [20] and showed that 45% of the mutations affected thermal stability of the protein and 44 % of the mutations resulted in loss of function. Experimental thermal studies for 22 naturally occurring mutants also indicated protein stability to be one of the major reasons for loss of function of NQO1 variants. A recent review article by Gil-Martínez et al. [21] has further outlined proteins’ thermodynamic stability to be a pertinent descriptor in case of rare diseases caused by missense mutations. In parallel, efforts are being made for development of databases documenting diseases resulting from missense mutation that stems from protein stability changes [22,23]. Thus, integration of mutational and stability data can aid in development of methods and diagnostic of clinical relevance. Thus, recent work reported that misfolded organic cation transporter variants can be rescued by chemical chaperone 4-PBA (4-phenyl butyric acid) [24]. More details about the usage of chaperones to assist folding of mutant proteins associated with rare diseases can be found the review article [21]. These limited examples strengthen our claim about the linkage between pathogenicity and macromolecular stability and show that the effect is treatable.
Recent works on binding free energy changes caused by mutations and linkage with diseases
Missense mutations at the surface of the corresponding proteins frequently do not affect protein stability but may affect macromolecular interactions [25]. While unbound proteins might fold to its native functional conformation even in presence of mutation, the affinity and specificity with which proteins interacts with other protein/DNA/RNA could be severely impaired in presence of mutation leading to diseases like cancer, cardiovascular diseases, cystic fibrosis, etc. [26–28]. Specifically, mutation in position located at the core of binding interface compared to the rim of interacting surface contributes towards development of diseases as they contribute significantly to the binding free energy [29,30]. It has been shown in studies that mutations affecting protein-protein interaction are specifically condensed at the binding interface and are evolutionary more conserved compared to other residues [28,31,32]. Considering the importance of protein-protein interaction, several methods have been developed to access binding free energy change caused by mutations [33–35]. As mentioned above, revealing the effect of missense mutations on macromolecular interactions paves the way for development of treatment of treatment which can restore wild type binding affinity and thus to eliminate disease-causing effect.
Recent works on protein dynamics changes caused by mutations and linkage with diseases
Proteins are intrinsically dynamic in nature which play key role in regulating their function. In the earlier sections, we discussed how mutation can affect the protein stability and protein-protein interactions leading to loss of function. Mutations may also affect the conformational dynamics of the protein which is linked to their biological function. Thus, a mutation V486M in dipeptidyl-peptidase 4 (DPP), a multifunctional cell surface glycoprotein, has been shown to disrupt the catalytic activity of inducing conformation collapse of the propeller domain, thus inhibiting DPP4 dimerization [36]. Another example of mutations affecting protein conformation are germline PTEN tumor-suppressor variants associated with autism spectrum disorder (ASD) or cancer display difference in conformational dynamics and influences protein network differently [37,38].
Recent works on allosteric changes caused by mutations and linkage with diseases
Many proteins are allosterically regulated, and this has also been used for disease therapy [39,40] specifically with focus on identifying cryptic pockets (allosteric druggable pockets) and allosteric drug discovery [41–44]. Allosteric drugs have several advantages over the typical orthosteric drugs. First, since allosteric drugs binds at allosteric site, i.e., other than the natural substrate site, they don’t compete with natural substrates and therefore, are likely to be more successful and less toxic. Second, most of the active site drugs are inhibitory in nature as they take up the place of natural substrates; however, the allosteric drugs could be both activating and inhibitory in nature [45]. Third, allosteric sites are less conserved as opposed to the active/orthosteric sites and therefore, allosteric drugs are more selective in nature[46–48]. Several allosteric drugs have been approved for example, Cinacalcet, a G-protein-coupled receptors (GPCR) allosteric modulator has been approved for Hyperparathyroidism [49]. Carglumic acid, which binds to carbamoyl phosphate synthetase-I is used for treatment of acute hyperammonaemia [50].
Because of the association between allostery, mutation and diseases, current research in the area has shifted on identification of mutations that causes allosteric dysregulation and causes diseases. In a similar vein, Shen et al. investigated the effect of somatic mutations at the allosteric site on cancer-based regulation [51]. The authors studied the effect of 47000 somatic mutations from 6958 pairwise tumour-normal mismatched pairs distributed across cancer types, out of which they predicted 1990 mutations to be deleterious and mapped 2761 known disease causing mutations of 74 structures. They further identified 20 known and 15 novel cancer associated proteins, which might play important role in tumorigenesis. A recent study by Tang et al. have elucidated the allosteric mechanism of p53 using four mutations (L145Q, P151S, Y220C, and G266R) that leads to loss of function of p53 and cancer development by destabilizing the p53-DNA interaction [52]. Mutations at the distal sites has also been shown to confer drug resistance [53]. The review article by Lu at al. has outlined the reasons that may result in allosteric drug resistance [54]. First, allosteric sites are more likely to be mutated because of low evolutionary pressure and second, resistant mutations can occur on the allosteric transmission pathway, in addition to allosteric sites. A review article by Khamina et al., has outlined the non-canonical allostery in cyclin dependent kinases; PKA (Protein Kinase A) and PKG (Protein Kinase G) caused by disease related mutations [55]. Disease causing mutations may also amplify the allosteric effect by self-associating into amyloid like structures resulting in loss of kinase inhibitory functions [55]. Other efforts towards linking allostery and diseases includes development of database named AlloMAPS, which consists of data of 46 proteins which are involved in allosteric signalling [56].
Supplementary Material
Acknowledgements
The authors are supported by NIH, the grant number R01GM093937.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interests
Authors declare no conflict of interest.
References
- 1.Gianfrancesco O, Bubb VJ, Quinn JP: Treating the “E” in “G × E”: Trauma-Informed Approaches and Psychological Therapy Interventions in Psychosis. Front Psychiatry 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Darden L, Kundu K, Pal LR, Moult J: Harnessing formal concepts of biological mechanism to analyze human disease. PLoS Comput Biol 2018, 14:e1006540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kucukkal TG, Petukh M, Li L, Alexov E: Structural and physico-chemical effects of disease and non-disease nsSNPs on proteins. Curr Opin Struct Biol 2015, 32:18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yates CM, Sternberg MJE: The Effects of Non-Synonymous Single Nucleotide Polymorphisms (nsSNPs) on Protein–Protein Interactions. J Mol Biol 2013, 425:3949–3963. [DOI] [PubMed] [Google Scholar]
- 5.Yates CM, Filippis I, Kelley LA, Sternberg MJE: SuSPect: Enhanced Prediction of Single Amino Acid Variant (SAV) Phenotype Using Network Features. J Mol Biol 2014, 426:2692–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li G, Panday SK, Peng Y, Alexov E: SAMPDI-3D: predicting the effects of protein and DNA mutations on protein–DNA interactions. Bioinformatics 2021, 37:3760–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okerberg ES, Hainley A, Brown H, Aban A, Alemayehu S, Shih A, Wu J, Patricelli MP, Kozarich JW, Nomanbhoy T, et al. : Identification of a Tumor Specific, Active-Site Mutation in Casein Kinase 1α by Chemical Proteomics. PLoS One 2016, 11:e0152934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boccuto L, Aoki K, Flanagan-Steet H, Chen C-F, Fan X, Bartel F, Petukh M, Pittman A, Saul R, Chaubey A, et al. : A mutation in a ganglioside biosynthetic enzyme, ST3GAL5, results in salt & pepper syndrome, a neurocutaneous disorder with altered glycolipid and glycoprotein glycosylation. Hum Mol Genet 2014, 23:418–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peng Y, Alexov E: Investigating the linkage between disease-causing amino acid variants and their effect on protein stability and binding. Proteins: Structure, Function, and Bioinformatics 2016, 84:232–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li G, Panday SK, Alexov E: SAAFEC-SEQ: A Sequence-Based Method for Predicting the Effect of Single Point Mutations on Protein Thermodynamic Stability. Int J Mol Sci 2021, 22:606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Savojardo C, Fariselli P, Martelli PL, Casadio R: INPS-MD: a web server to predict stability of protein variants from sequence and structure. Bioinformatics 2016, 32:2542–2544. [DOI] [PubMed] [Google Scholar]
- 12. **.Fang X, Yeh J-T, Hwang T-C: Pharmacological Responses of the G542X-CFTR to CFTR Modulators. Front Mol Biosci 2022, 9:921680. [DOI] [PMC free article] [PubMed] [Google Scholar]; The manuscript reports development of small-molecule chloride channel cystic fibrosis transmembrane conductance regulator (CFTR) modulators, which facilitate protein folding and expression. It was speculated that such approach can be applied for other cases involving premature termination codon.
- 13. *.Palanikumar L, Karpauskaite L, Al-Sayegh M, Chehade I, Alam M, Hassan S, Maity D, Ali L, Kalmouni M, Hunashal Y, et al. : Protein mimetic amyloid inhibitor potently abrogates cancer-associated mutant p53 aggregation and restores tumor suppressor function. Nat Commun 2021, 12:3962. [DOI] [PMC free article] [PubMed] [Google Scholar]; The article reports an inhibitor which reduces mutant p53 aggregation and restores tumor suppressor function. The work demonstrated the successful application of a bona fide small-molecule amyloid inhibitor as a potent anticancer agent.
- 14.Kuo S-Y, Castoreno AB, Aldrich LN, Lassen KG, Goel G, Dančík V, Kuballa P, Latorre I, Conway KL, Sarkar S, et al. : Small-molecule enhancers of autophagy modulate cellular disease phenotypes suggested by human genetics. Proceedings of the National Academy of Sciences 2015, 112:E4281–E4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williamson I, Hill RE, Bickmore WA: Enhancers: From Developmental Genetics to the Genetics of Common Human Disease. Dev Cell 2011, 21:17–19. [DOI] [PubMed] [Google Scholar]
- 16. **.Durairaj G, Demir Ö, Lim B, Baronio R, Tifrea D, Hall L v., DeForest JC, Lauinger L, Jebril Fallatah MM, Yu C, et al. : Discovery of compounds that reactivate p53 mutants in vitro and in vivo. Cell Chem Biol 2022, 29:1381–1395.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]; The manuscript describes development of small molecules or potential drugs that restore mutants p53 wild type activity. The outcome demonstrated feasibility of in-silico approaches to identify small molecule corrector drugs for p53 hotspot mutations.
- 17.Reddy A, Kaelin WG: Using cancer genetics to guide the selection of anticancer drug targets. Curr Opin Pharmacol 2002, 2:366–73. [DOI] [PubMed] [Google Scholar]
- 18.Takano K, Liu D, Tarpey P, Gallant E, Lam A, Witham S, Alexov E, Chaubey A, Stevenson RE, Schwartz CE, et al. : An X-linked channelopathy with cardiomegaly due to a CLIC2 mutation enhancing ryanodine receptor channel activity. Hum Mol Genet 2012, 21:4497–4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. *.Aledo P, Aledo JC: Proteome-Wide Structural Computations Provide Insights into Empirical Amino Acid Substitution Matrices. Int J Mol Sci 2023, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]; A study on the linkage between protein stability and pathogenicity by performing 137,073,638 mutations in 14,094 proteins. It was shown that the destabilizing effect of amino acid substitution on protein structure correlates positively with the disease-causing potential.
- 20.Pacheco-Garcia JL, Cagiada M, Tienne-Matos K, Salido E, Lindorff-Larsen K,L Pey A: Effect of naturally-occurring mutations on the stability and function of cancer-associated NQO1: Comparison of experiments and computation. Front Mol Biosci 2022, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gil-Martínez J, Bernardo-Seisdedos G, Mato JM, Millet O: The use of pharmacological chaperones in rare diseases caused by reduced protein stability. 2022, doi: 10.1002/pmic.202200222. [DOI] [PubMed] [Google Scholar]
- 22.Woodard J, Zhang C, Zhang Y: ADDRESS: A Database of Disease-associated Human Variants Incorporating Protein Structure and Folding Stabilities. J Mol Biol 2021, 433:166840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, et al. : COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res 2019, 47:D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Angenoorth TJF, Maier J, Stankovic S, Bhat S, Sucic S, Freissmuth M, Sitte HH, Yang J-W: Rescue of Misfolded Organic Cation Transporter 3 Variants. Cells 2022, 12:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Y, Kucukkal TG, Li J, Alexov E, Cao W: Binding Analysis of Methyl-CpG Binding Domain of MeCP2 and Rett Syndrome Mutations. 2016, doi: 10.1021/acschembio.6b00450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jemimah S, Gromiha MM: Insights into changes in binding affinity caused by disease mutations in protein-protein complexes. Comput Biol Med 2020, 123:103829. [DOI] [PubMed] [Google Scholar]
- 27.Zaucha J, Heinzinger M, Kulandaisamy A, Kataka E, Salvádor ÓL, Popov P, Rost B, Gromiha MM, Zhorov BS, Frishman D: Mutations in transmembrane proteins: diseases, evolutionary insights, prediction and comparison with globular proteins. Brief Bioinform 2021, 22. [DOI] [PubMed] [Google Scholar]
- 28.Sahni N, Yi S, Taipale M, Fuxman Bass JI, Coulombe-Huntington J, Yang F, Peng J, Weile J, Karras GI, Wang Y, et al. : Widespread macromolecular interaction perturbations in human genetic disorders. Cell 2015, 161:647–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.David A, Sternberg MJE: The Contribution of Missense Mutations in Core and Rim Residues of Protein–Protein Interfaces to Human Disease. J Mol Biol 2015, 427:2886–2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Navío D, Rosell M, Aguirre J, de la Cruz X, Fernández-Recio J: Structural and Computational Characterization of Disease-Related Mutations Involved in Protein-Protein Interfaces. Int J Mol Sci 2019, 20:1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. *.Xiong D, Lee D, Li L, Zhao Q, Yu H: Implications of disease-related mutations at protein–protein interfaces. Curr Opin Struct Biol 2022, 72:219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]; The article outlines the implication of disrupted protein-protein interaction to human diseases. It reviews works demonstrating cases of disease-related mutations at protein-protein interfaces, mutation effects on protein interactions, and investigation of mutations on specific diseases.
- 32.Meyer MJ, Beltrán JF, Liang S, Fragoza R, Rumack A, Liang J, Wei X, Yu H: Interactome InSIdER: a structural interactome browser for genomic studies. 2018, 15:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiong P, Zhang C, Zheng W, Zhang Y: BindProfX: Assessing Mutation-Induced Binding Affinity Change by Protein Interface Profiles with Pseudo-Counts. J Mol Biol 2017, 429:426–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. **.Wang M, Cang Z, Wei G-W: A topology-based network tree for the prediction of protein–protein binding affinity changes following mutation. Nat Mach Intell 2020, 2:116–123. [DOI] [PMC free article] [PubMed] [Google Scholar]; A novel approach for predicting the changes of binding affinity caused by missense mutation. The article reports deep learning algorithm called NetTree to take advantage of convolutional neural networks and gradient-boosting trees.
- 35.Pahari S, Li G, Murthy AK, Liang S, Fragoza R, Yu H, Alexov E: SAAMBE-3D: Predicting Effect of Mutations on Protein–Protein Interactions. Int J Mol Sci 2020, 21:2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li T, Peng C, Wang J, Xu Z, Su M, Li J, Zhu W, Li J: Distal mutation V486M disrupts the catalytic activity of DPP4 by affecting the flap of the propeller domain. Acta Pharmacol Sin 2022, 43:2147–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith IN, Thacker S, Seyfi M, Cheng F, Eng C: Conformational Dynamics and Allosteric Regulation Landscapes of Germline PTEN Mutations Associated with Autism Compared to Those Associated with Cancer. Am J Hum Genet 2019, 104:861–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Portelli S, Barr L, de Sá AGC, Pires DEV, Ascher DB: Distinguishing between PTEN clinical phenotypes through mutation analysis. Comput Struct Biotechnol J 2021, 19:3097–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nussinov R, Tsai C-J, Jang H: Dynamic Protein Allosteric Regulation and Disease. In Protein Allostery in Drug Discovery. Edited by Zhang Jian and Nussinov R. Springer; Singapore; 2019:25–43. [DOI] [PubMed] [Google Scholar]
- 40.Wu J, Li D, Liu X, Li Q, He X, Wei J, Li X, Li M, Rehman AU, Xia Y, et al. : IDDB: a comprehensive resource featuring genes, variants and characteristics associated with infertility. Nucleic Acids Res 2021, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu S, He X, Ni D, Zhang J: Allosteric Modulator Discovery: From Serendipity to Structure-Based Design. 2019, doi: 10.1021/acs.jmedchem.8b01749. [DOI] [PubMed] [Google Scholar]
- 42.Kinoshita T: Protein Allostery in Rational Drug Design. In Protein Allostery in Drug Discovery. Edited by Zhang Jian and Nussinov R. Springer; Singapore; 2019:45–64. [DOI] [PubMed] [Google Scholar]
- 43.Qiu Y, Wang Y, Chai Z, Ni D, Li X, Pu J, Chen J, Zhang J, Lu S, Lv C, et al. : Targeting RAS phosphorylation in cancer therapy: Mechanisms and modulators. Acta Pharm Sin B 2021, 11:3433–3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng X, Jiang H: Allostery in Drug Development. In Protein Allostery in Drug Discovery. Edited by Zhang Jian and Nussinov R. Springer; Singapore; 2019:1–23. [Google Scholar]
- 45.Huang Q, Song P, Chen Y, Liu Z, Lai L: Allosteric Type and Pathways Are Governed by the Forces of Protein−Ligand Binding. J Phys Chem Lett 2021 2021, 12:5412. [DOI] [PubMed] [Google Scholar]
- 46.Wu Y, Tong J, Ding K, Zhou Q, Zhao S: GPCR Allosteric Modulator Discovery. In Protein Allostery in Drug Discovery. Edited by Zhang Jian and Nussinov R. Springer; Singapore; 2019:225–251. [DOI] [PubMed] [Google Scholar]
- 47.Guarnera E, Berezovsky IN: Allosteric drugs and mutations: chances, challenges, and necessity. Curr Opin Struct Biol 2020, 62:149–157. [DOI] [PubMed] [Google Scholar]
- 48.Zha J, Li M, Kong R, Lu S, Zhang J: Explaining and Predicting Allostery with Allosteric Database and Modern Analytical Techniques. J Mol Biol 2022, 434:167481. [DOI] [PubMed] [Google Scholar]
- 49.Ng CH, Chin YH, Tan MHQ, Ng JX, Yang SP, Kiew JJ, Khoo CM: Cinacalcet and primary hyperparathyroidism: systematic review and meta regression. Endocr Connect 2020, 9:724–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Filippi L, Fiorini P, la Marca G, Daniotti M: New developments in the treatment of hyperammonemia: emerging use of carglumic acid. Int J Gen Med 2011, doi: 10.2147/IJGM.S10490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shen Q, Cheng F, Song H, Lu W, Zhao J, An X, Liu M, Chen G, Zhao Z, Zhang J: Proteome-Scale Investigation of Protein Allosteric Regulation Perturbed by Somatic Mutations in 7,000 Cancer Genomes. The American Journal of Human Genetics 2017, 100:5–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tang Y, Yao Y, Wei G: Unraveling the Allosteric Mechanism of Four Cancer-related Mutations in the Disruption of p53-DNA Interaction. J Phys Chem B 2021, 125:10138–10148. [DOI] [PubMed] [Google Scholar]
- 53.Yeo JY, Goh G-R, Su CT-T, Gan SK-E: The Determination of HIV-1 RT Mutation Rate, Its Possible Allosteric Effects, and Its Implications on Drug Resistance. Viruses 2020, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lu S, Qiu Y, Ni D, He X, Pu J, Zhang J: Emergence of allosteric drug-resistance mutations: new challenges for allosteric drug discovery. Drug Discov Today 2020, 25:177–184. [DOI] [PubMed] [Google Scholar]
- 55.Khamina M, Martinez Pomier K, Akimoto M, VanSchouwen B, Melacini G: Non-Canonical Allostery in Cyclic Nucleotide Dependent Kinases. J Mol Biol 2022, 434:167584. [DOI] [PubMed] [Google Scholar]
- 56.Tan ZW, Tee W-V, Guarnera E, Booth L, Berezovsky IN: AlloMAPS: allosteric mutation analysis and polymorphism of signaling database. Nucleic Acids Res 2019, 47:D265–D270. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.