

Abstract
Background
An ancillary study to the High-Dose Erythropoietin for Asphyxia and Encephalopathy (HEAL) trial of for neonates with hypoxic-ischemic encephalopathy (HIE) and treated with therapeutic hypothermia examined the hypothesis that neonates randomized to receive erythropoietin (Epo) would have a lower seizure risk and burden compared with neonates who received placebo.
Methods
Electroencephalograms (EEGs) from 7/17 HEAL trial centers were reviewed. Seizure presence was compared across treatment groups using a logistic regression model adjusting for treatment, HIE severity, center, and seizure burden prior to first dose. Among neonates with seizures, differences across treatment groups in median maximal hourly seizure burden were assessed using adjusted quantile regression models.
Results
Forty-six of 150 (31%) of neonates had EEG seizures (31% in Epo vs 30% in placebo, p=0.96). Maximal hourly seizure burden after study drug was not significantly different between groups (median 11.4 for Epo, IQR: 5.6, 18.1 vs median 9.7, IQR: 4.9, 21.0 minutes/hour for placebo).
Conclusion
In neonates with HIE treated with hypothermia who were randomized to Epo or placebo, we found no meaningful between-group difference in seizure risk or burden. These findings are consistent with overall trial results, which do not support Epo use for neonates with HIE undergoing therapeutic hypothermia.
Introduction
Neonatal encephalopathy due to hypoxia-ischemia, or hypoxic-ischemic encephalopathy (HIE), is the most common cause of seizures in neonates.1,2 Erythropoietin (Epo), long been known as a proerythropoietic agent, has more recently been explored as a neuroprotective agent because of its nonhematopoietic functions including neurotrophic and neuroprotective effects.3 Epo has also been shown to reduce the severity of both acute and late seizures in animal models of HIE.4–7 Despite the anti-convulsant effect of Epo in pre-clinical models of HIE,4–7 the United States Food and Drug Administration (FDA) drug label information for Epo warns of a possible pro-convulsant effect based on older trials in adults with renal disease.8–10 It is unclear whether this warning should apply to neonates, as human neonatal studies were previously limited by small cohort sizes and lack of gold standard continuous video-EEG monitoring (cEEG) to diagnose seizures.11,12
The recent High-Dose Erythropoietin for Asphyxia and Encephalopathy (HEAL) multicenter, randomized trial of Epo vs. placebo for neuroprotection in neonates with moderate/severe HIE who receive therapeutic hypothermia showed no meaningful difference between groups in the rate of death or disability at age two to three years.13 However, the primary study did not assess differences in timing and severity of electrographic seizure burden between study groups.
We leveraged the HEAL trial to examine a sub-set of neonates who were evaluated with cEEG throughout cooling and rewarming to examine the hypothesis that neonates who receive Epo have a lower risk and burden of acute provoked seizures after study drug administration as compared to neonates who receive placebo.
Methods
Study Design.
We conducted an ancillary study of the HEAL randomized trial of Epo vs. placebo for neuroprotection in neonates with moderate/severe HIE who received therapeutic hypothermia (NCT02811263)13 to study neurophysiology measures. Epo or placebo treatment was allocated 1:1 and administered at five time points: within 24 hours of birth (day 1), and at days 2, 3, 4, and 7 days after birth at the same time of day. Details of the study protocol have been previously published.14
We included participants enrolled at the seven HEAL enrollment sites (Supplementary Table 1) that performed cEEG throughout cooling and rewarming according to American Clinical Neurophysiology Society (ACNS) guidelines15 as part of routine clinical care. De-identified EEG tracings were collected for central review and inclusion in this ancillary study called HEAL-EEG. The HEAL trial and cEEG data collection were approved by the Institutional Review Board at each participating site and neonates were studied after informed parental consent.
Inclusion and Exclusion Criteria.
Neonates were eligible if they met all four study criteria: 1) born at ≥36 weeks’ gestation; 2) one or more signs of perinatal depression including Apgar score <5 at 10 minutes; cardiorespiratory resuscitation received beyond 10 minutes of age; pH <7.00 or base deficit ≥15 mmol/L in a cord or infant arterial or venous gas obtained within 60 minutes of age; 3) moderate or severe encephalopathy defined as ≥3 of 6 modified Sarnat criteria present at 1 to 6 hours of age; and 4) passive or active therapeutic hypothermia started within 6 hours of birth. Exclusion criteria were birthweight <1800 grams, head circumference <30 cm, genetic or congenital condition affecting neurodevelopment, hematocrit >65.0%, considering redirection of care, encephalopathy attributed to a postnatal event, guardian with diminished capacity, or unlikely to be followed due to unstable social situation.13,14
Additional HEAL-EEG specific inclusion criteria were: 1) cEEG recorded without interruption throughout cooling and rewarming (except for neonates who died during the neonatal admission), and 2) EEG quality sufficient for interpretation by neurophysiologist review.
Measurements.
Maternal and neonatal demographics and clinical characteristics were determined based on medical chart review. Timing and dose of antiseizure medication (ASM) administration were extracted from the medication administration record. Seizure treatment agent and timing were determined by the treating physician(s). Encephalopathy was classified as moderate or severe based on the number of abnormal Sarnat elements in the moderate and severe categories; if equal numbers were present, severity classification was decided based on level of consciousness category.14
EEG Acquisition and Interpretation.
Continuous EEG was recorded using a minimum of 8 electrodes and 10–20 electrode placement modified for the neonate per local clinical practice. Complete recordings were de-identified and stripped of video, then collected for central review by two board-certified clinical neurophysiologists (ALN and CJW). Files were reviewed in the order they were received for centralized review using Persyst software with neurophysiologists able to adjust montages as desired for interpretation. Neurophysiologists were blinded to treatment group, local interpretation, and outcomes. Each neurophysiologist independently reviewed the files; discrepancies were resolved by consensus. Kappa statistic was used to compare agreement in the observed rate of neonates with EEG seizures or status epilepticus and Pearson correlation coefficient was used to assess inter-rater reliability on total minutes of seizure burden. Inter-rater reliability for seizure identification was almost perfect (Kappa=0.82) and for status epilepticus was substantial (Kappa=0.78).16 Inter-rater reliability for overall minutes of seizure burden among neonates with identified seizures was also very high (Pearson r=0.96).
Outcome Measures.
Seizures were defined as a sudden, abnormal EEG event with a repetitive and evolving pattern with a minimum 2μV peak-to-peak voltage and duration of at least 10 seconds.17 Status epilepticus was defined as the summed duration of seizures comprising ≥50% of any 1-hour epoch of recording.17 Only electrographic seizures were considered; clinically-detected seizures were not considered for this ancillary study.
Primary outcome.
EEG maximal hourly seizure burden (in minutes per hour) after Epo administration among neonates with seizures. EEG maximal hourly seizure burden was determined using a sliding one hour time window throughout the course of the EEG recording.
Pre-specified secondary outcomes.
1) Response to initial dose of ASM with complete response defined as no further seizures present >30 minutes after adequate loading dose of an ASM (phenobarbital >20mg/kg, levetiracetam >40mg/kg, or fosphenytoin >20mg/kg) until the end of the recording, 2) Overall seizure burden (minutes of seizure per minutes of cEEG recording) 3) Seizure period (time from the onset of the first seizure to the end of the last seizure), and 4) Presence of status epilepticus.
Analysis.
Given this was a select sub study population the baseline characteristics and measurements were first compared between treatment groups using Chi-square tests for categorical variables and Wilcoxon rank sum tests for continuous variables. The maximum hourly seizure burden was defined as the maximum number of seizure minutes over any 60-minute window within time periods (Tj) for j = 0 (before study drug dose 1), j=1 (between study drug doses 1 and 2), or j=2 (between study drug doses 2 and 3). Baseline seizures rates were expected to be similar due to randomization, therefore statistical inference focused on the first and second dose time periods. To estimate the adjusted relative risk (aRR) of seizure burden between groups, we used a Poisson regression model with robust standard errors to allow for overdispersion. The regression model adjusted for sex, recruitment site, HIE severity (moderate or severe), maximum hourly seizure burden observed prior to the first study drug dose, and a log offset log(Tj) to account for variable lengths of cEEG observation time overall or between study drug doses. For secondary binary outcomes measures (any seizures, complete response to initial loading dose of ASM, status epilepticus), we used a log-logistic regression model to calculate relative risks comparing treatment groups adjusting for sex, recruitment site, HIE severity, and any seizure observed prior to the first study drug dose.
Seizure period was defined as the median hours from the first to the last observed seizure and median percentage of total cEEG time with observed seizures were compared between treatment groups using quantile regression (R “quantreg” package). Group differences in medians and 95% confidence intervals (CI) were adjusted for HIE severity and log cEEG observation time.
All analyses were conducted using R software version 4.0.2 (Vienna, Austria).18
Power and Sample Size.
To test the hypothesis that Epo reduces both the proportion of neonates with seizures and seizure burden, we assumed a priori that approximately 50% of placebo-treated neonates would have seizures at a median burden of 4.0 minutes/hour (IQR: 2.0–7.0).19,20 We used simulations to generate data under various scenarios and to assess power for each outcome assuming that Tj is approximately 24 hours. To mimic seizure data, we used a zero inflated model composed of two parts: the probability of no seizures and the seizure burden among those with seizures. For simulation analyses (n=10,000 replications) we used a generalized linear model regression Wald test with alpha=0.05 based on a generalized linear model function assuming a log link and quasi-Poisson family and considered a variety of over-dispersion parameters.
We sought to evaluate n=150 neonates with cEEG, as a sample size of 70 subjects/group provided at least 80% power to detect a treatment effect under three scenarios: 1) a test of whether treatment reduces both the probability of any seizures and the burden of seizures by 25% (a common rate ratio of 0.75), 2) treatment effect associated only with a relative risk of 0.53 for the presence of any seizures, but no reduction in burden among subjects with seizures, and 3) no treatment effect on the percent of subjects with seizures, but the seizure burden is reduced by 40% (rate ratio 0.60) among subjects with seizures.
Results
Patients.
Of 500 neonates included in the modified intention to treat analysis of the HEAL trial, born between January 25, 2017 and October 9, 2019, 235 (47%) participants were enrolled at one of the seven HEAL-EEG centers (Supplementary Table 1). One hundred and eighty-five of 235 (79%) of cEEGs were reviewed for background and seizure burden, to reach the prespecified sample size of 150 recordings with adequate quality for inclusion (37 were excluded for low quality tracing, 15 from the Epo and 22 from the placebo group Figure 1). Twenty-three cEEGs were not reviewed as there were sufficient high-quality tracings to achieve the pre-specified sample size. There were no meaningful differences in characteristics between infants selected for cEEG review compared to those that were not reviewed (Supplemental Table 2). The first dose of Epo was administered at a median 18.5 (IQR 13.1–22.3) hours. Twenty of 150 neonates (13%) died, six of whom had the cEEG removed prior to completing the 72-hour monitoring period.
Figure 1.
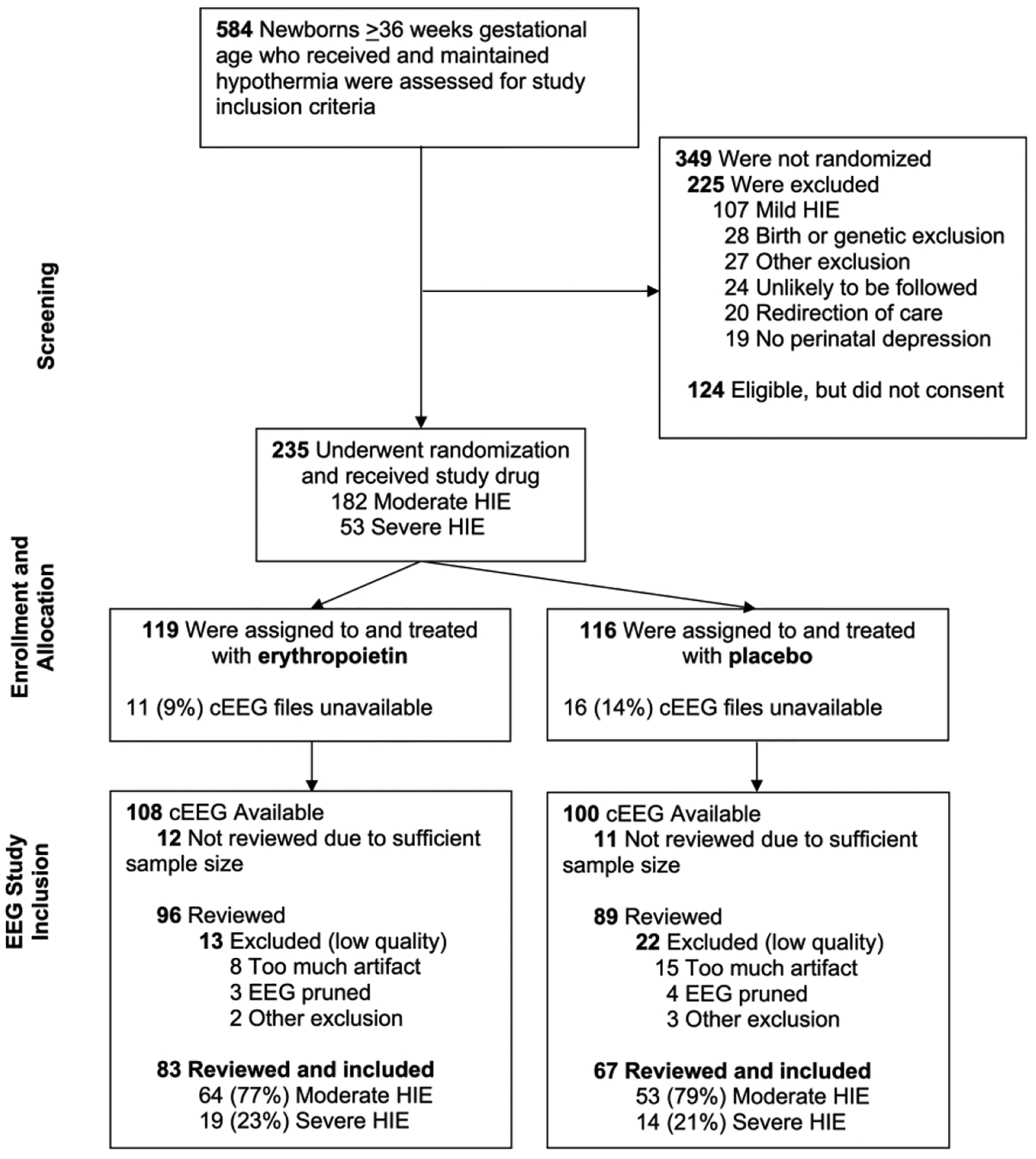
CONSORT diagram for seven sites participating in the HEAL-EEG ancillary study.
Table 1 describes baseline characteristics of neonates who received Epo (n=83) and placebo (n=67). There were no significant differences between groups in maternal characteristics, pregnancy and delivery complications, infant characteristics (including severity of encephalopathy), or EEG monitoring.
Table 1.
Baseline clinical and electroencephalogram (EEG) characteristic of 150 neonates with hypoxic-ischemic encephalopathy (HIE) undergoing therapeutic hypothermia and treated with erythropoietin or placebo who received continuous video-EEG throughout hypothermia and rewarming.
| Total N=150 | Erythropoietin N=83 | Placebo N=67 | p-value* | |
|---|---|---|---|---|
| Maternal Characteristics, n (%) | ||||
| Race | 0.59 | |||
| White | 104 (69%) | 58 (70%) | 46 (69%) | |
| Black | 18 (12%) | 8 (9.6%) | 10 (15%) | |
| Asian | 15 (10%) | 8 (9.6%) | 7 (10%) | |
| Other | 13 (8.7%) | 9 (11%) | 4 (6.0%) | |
| Hispanic ethnicity | 33 (22%) | 16 (19%) | 17 (25%) | 0.37 |
| Age (years), mean (SD) | 30.1 (6.6) | 30.8 (6.9) | 29.3 (6.3) | 0.16 |
| Education, high school or less | 58 (39%) | 33 (40%) | 25 (37%) | 0.76 |
| Parity = 1 (including subject) | 91 (61%) | 52 (63%) | 39 (58%) | 0.58 |
| Pregnancy and Delivery Complications, n (%) | ||||
| Maternal chorioamnionitis or fever | 28 (19%) | 15 (18%) | 13 (19%) | 0.84 |
| Maternal pre-eclampsia or eclampsia | 16 (11%) | 8 (10%) | 8 (12%) | 0.65 |
| Gestational diabetes | 14 (9.3%) | 6 (7.2%) | 8 (12%) | 0.32 |
| Maternal obesity (BMI >30) | 25 (17%) | 15 (18%) | 10 (15%) | 0.61 |
| Sentinel eventa | 48 (32%) | 26 (31%) | 22 (33%) | 0.85 |
| Cesarean section delivery | 99 (66%) | 57 (69%) | 42 (63%) | 0.33 |
| Infant Characteristics | ||||
| Female, n (%) | 65 (43%) | 33 (40%) | 32 (48%) | 0.44 |
| Birth weight (grams), mean (SD) | 3411 (558) | 3354 (542) | 3481 (573) | 0.14 |
| Gestational age (weeks), mean (SD) | 39.2 (1.5) | 39.1 (1.4) | 39.3 (1.4) | 0.26 |
| 5-minute Apgar score, median (IQR) | 3 (2, 4) | 3 (2, 4) | 4 (2, 5) | 0.26 |
| Lowest pHb, mean (SD) | 6.9 (0.2) | 6.9 (0.2) | 6.9 (0.2) | 0.79 |
| Worst base deficitb, mean (SD) | 17.6 (6.1) | 17.1 (5.6) | 18.2 (6.6) | 0.54 |
| Severe encephalopathyc, n (%) | 33 (22%) | 19 (23%) | 14 (21%) | 0.77 |
| cEEG Monitoring | ||||
| cEEG starting hour after birth, median (IQR) | 8.4 (6.4, 10.3) | 7.9 (6.5, 9.7) | 9.0 (6.3, 10.7) | 0.26 |
| Hours of cEEG, median (IQR) | 89.3 (79.7, 98.0) | 89.7 (77.9, 98.1) | 88.6 (80.5, 97.9) | 0.98 |
| Cannot determine (excess artifact at onset of recording) | 1 (0.7%) | 0 (0%) | 1 (1.5%) | |
| EEG seizures observed prior to 1st dose of study drug, n (%) | 30/140 (21%) | 18/79 (23%) | 12/61 (20%) | 0.66 |
SD standard deviation; BMI body mass index; IQR interquartile range; cEEG continuous video electroencephalogram
P values calculated using Chi-square tests for categorical variables and Wilcoxon rank sum tests for continuous variables.
Sentinel event = placental abruption, shoulder dystocia, uterine rupture, or prolapsed cord.
Lowest pH and worst base deficit among cord arterial, cord venous, and arterial blood gas samples taken before 60 minutes of age.
Severe encephalopathy as defined by modified Sarnat score.
Seizures and seizure treatment.
Electrographic seizures occurred in 46/150 (31%, Table 2). There was no significant difference in the percentage of neonates experiencing seizures between the Epo and placebo groups (26/83, 31% vs. 20/67, 30%; aRR = 1.04; 95% CI = 0.60 – 1.80). Among the 140 neonates who received the first study drug dose after the start of cEEG monitoring, 30 (21%) had seizures before study drug administration, with similar rates comparing the Epo (23%) and placebo (20%) groups. The percentage of neonates with seizures after study drug dosing was also similar across treatment groups. Thirty-four of 150 neonates (27%) had seizures between the first and second doses of study drug, with a similar rate for the Epo (19/83, 23%) and placebo (15/67, 22%) groups, and 16/150 (11%) had seizures after the second dose of study drug, with similar rates across the Epo (11/83, 13%) and placebo (5/67, 7.5%) groups. Anti-seizure medications were administered to 30/83 (36%) of neonates who received Epo and 36/67 (54%) neonates who received placebo (aRR=0.60, 95% CI = 0.40–0.89; p=0.01).
Table 2.
Seizures and anti-seizure medications (ASM) of 150 neonates with hypoxic-ischemic encephalopathy (HIE) undergoing therapeutic hypothermia and treated with erythropoietin or placebo who received continuous video-EEG throughout hypothermia and rewarming.
| Total N=150 | Erythropoietin N= 83 | Placebo N=67 | aRR (95% CI)* | P value * | |
|---|---|---|---|---|---|
| Seizures and Seizure Timing | |||||
| N (%) with EEG seizures** | 46 (31%) | 26 (31%) | 20 (30%) | 1.04 (0.60, 1.80) | 0.88 |
| With moderate encephalopathy | 29/117 (25%) | 16/64 (23%) | 13/53 (24%) | 0.98 (0.41, 2.34) | |
| With severe encephalopathy | 17/33 (52%) | 10/19 (53%) | 7/14 (50%) | 1.03 (0.62, 1.72) | |
| After 1st dose of study drug | 40 (27%) | 22 (27%) | 18 (27%) | 0.88 (0.44, 1.76) | 0.72 |
| Between 1st and 2nd dose of study drug | 34 (23%) | 19 (23%) | 15 (22%) | 0.88 (0.45, 1.73) | 0.71 |
| After 2nd dose of study drug | 16 (11%) | 11 (13%) | 5 (7.5%) | 1.12 (0.41, 3.07) | 0.83 |
| ASM Administration | |||||
| N (%) administered ASM | 66 (44%) | 30 (36%) | 36 (54%) | 0.60 (0.40, 0.89) | 0.01 |
| Phenobarbital | 64 (43%) | 29 (35%) | 35 (52%) | 0.62 (0.41, 0.93) | 0.02 |
| Levetiracetam | 33 (22%) | 12 (15%) | 11 (16%) | 0.73 (0.34, 1.60) | 0.44 |
| Phenytoin/Fosphenytoin | 13 (8.7%) | 7 (8.4%) | 6 (9.0%) | 1.06 (0.39, 2.88) | 0.91 |
| Other (midazolam, lorazepam, topiramate) | 65 (43%) | 34 (41%) | 31 (46%) | 0.91 (0.61, 1.34) | 0.62 |
aRR adjusted relative risk; EEG electroencephalogram; ASM anti-seizure medication
Adjusted relative risks and P values based upon generalized (binary) logistic regression model and adjusts for treatment, HIE severity, and recruitment site.
Logistic regression models additionally adjusted for the maximum seizure minutes/hour observed prior to first study drug dosing, and a log offset log(Tj) to account for variable lengths of cEEG observation time overall or between study drug doses.
Primary outcome.
Among neonates with seizures, there was no meaningful difference in the median maximal hourly seizure burden after administration of the first dose of study drug between study groups (Epo: 11.4, IQR 5.6, 18.1 minutes per hour compared with placebo: 9.7, IQR 4.9, 21.0 minutes per hour; adjusted difference= −0.2; 95% CI= −6.3 – 5.6; Table 3). Seizure timing by treatment group is presented in Figure 2. In a sensitivity analysis excluding the six children who died and had incomplete cEEG data, median maximal hourly seizure burden was unchanged (median 11.6, IQR 5.6, 17.4 minutes per hour in the Epo group and median 10.4, IQR 4.9, 21.5 minutes per hour in the placebo group).
Table 3.
Seizures and anti-seizure medications 46 neonates with hypoxic-ischemic encephalopathy and confirmed EEG seizures while undergoing therapeutic hypothermia and treated with erythropoietin or placebo who received continuous video-EEG throughout hypothermia and rewarming.
| Total with Seizures N=46 | Erythropoietin and Seizures N=26 | Placebo and Seizures N=20 | Adjusted Group Comparison (95% CI) | |
|---|---|---|---|---|
| Seizures and Seizure Timing | ||||
| Total seizure duration in minutes, median (IQR)* | 51.4 (26.6, 100.3) | 63.8 (28.5, 146.5) | 31.5 (23.4, 77.1) | 34.0 (2.81, 67.4) |
| Prior to the 1st study drug dose | 8.1 (0, 35.3) | 21.0 (0, 70.3) | 3.1 (0, 18.3) | 16.8 (0.6, 30.3) |
| Between 1st and 2nd dose of study drug | 17.4 (1.4, 44.9) | 18.0 (1.4, 44.9) | 17.4 (5.9, 38.8) | 0.2 (−8.2, 15.2) |
| After 2nd dose of study drug | 0 (0, 7.5) | 0 (0, 16.7) | 0 (0, 0.7) | 0 (0, 0.6) |
| Maximal EEG seizure burden (minutes/hour), median (IQR)* | 16.1 (10.3, 32.7) | 19.7 (10.6, 38.1) | 15.0 (10.4, 22.7) | 8.8 (−2.6, 18.6) |
| Prior to the 1st study drug dose | 9.9 (0.0, 25.8) | 16.2 (0.0, 34.4) | 7.7 (0.0, 11.4) | 8.1 (2.5, 17.6) |
| After 1st dose of study drug | 10.8 (5.2, 20.0) | 11.4 (5.6, 18.1) | 9.7 (4.9, 21.0) | −0.2 (−6.3, 5.6) |
| Between 1st and 2nd dose of study drug | 7.9 (0.6, 18.1) | 7.9 (0.6, 15.7) | 8.2 (3.5, 21.0) | 0.3 (−8.9, 4.4) |
| After 2nd dose of study drug | 0.0 (0.0, 3.7) | 0.0 (0.0, 5.7) | 0.0 (0.0, 0.4) | 0 (0, 0.2) |
| Status epilepticus at any time during the recording, n (%)** | 10 (22%) | 9 (35%) | 1 (5.0%) | 2.84 (0.56, 14.39) |
| Hours from start of first seizure to end of last seizure, median (IQR)* | 16.3 (6.2, 25.2) | 20.2 (8.4, 33.1) | 14.9 (3.9, 21.0) | 2.5 (−3.3, 11.6) |
| Percentage of cEEG recording time with observed seizures, median (IQR)* | 1.0 (0.5, 1.7) | 1.2 (0.5, 3.5) | 0.6 (0.4, 1.3) | 0.2 (−0.4, 0.8) |
| Between 1st and 2nd dose of study drug | 0.6 (0.0, 1.3) | 0.5 (0.0, 1.1) | 0.6 (0.2, 1.5) | 0.2 (−0.6, 0.3) |
| After 2nd dose of study drug | 0 (0, 0.1) | 0 (0, 0.4) | 0 (0, 0.0) | 0 (0, 0) |
| Complete response to ASM >=30 minutes after first loading dose of anti-seizure medication, n (%)*** | 13/43 (30%) | 5/24 (21%) | 8/19 (42%) | 0.78 (0.27, 2.26) |
| Received ≥2 ASM to treat neonatal seizures, n (%)*** | 23 (50%) | 12 (46%) | 11 (55%) | 1.18 (0.49, 2.88) |
IQR interquartile range; cEEG continuous video electroencephalogram; ASM anti-seizure medication
Group differences in median hourly seizure burden, percentage, and hours of seizure burden were adjusted for treatment, HIE severity, maximum seizure minutes/hour observed prior to 1st study drug dose, and a log offset log(Tj) to account for variable lengths of cEEG observation time overall or between study drug doses
Relative risk of status epilepticus is based upon generalized (log) logistic regression adjusting for treatment, HIE severity, and maximum seizure minutes/hour observed prior to first study drug dosing.
Relative risks for other dichotomous variables adjust for treatment, HIE severity, recruitment site, maximum seizure minutes/hour observed prior to first study drug dosing, and for a log offset log(Tj) to account for variable lengths of cEEG observation time overall or between study drug doses.
Figure 2.
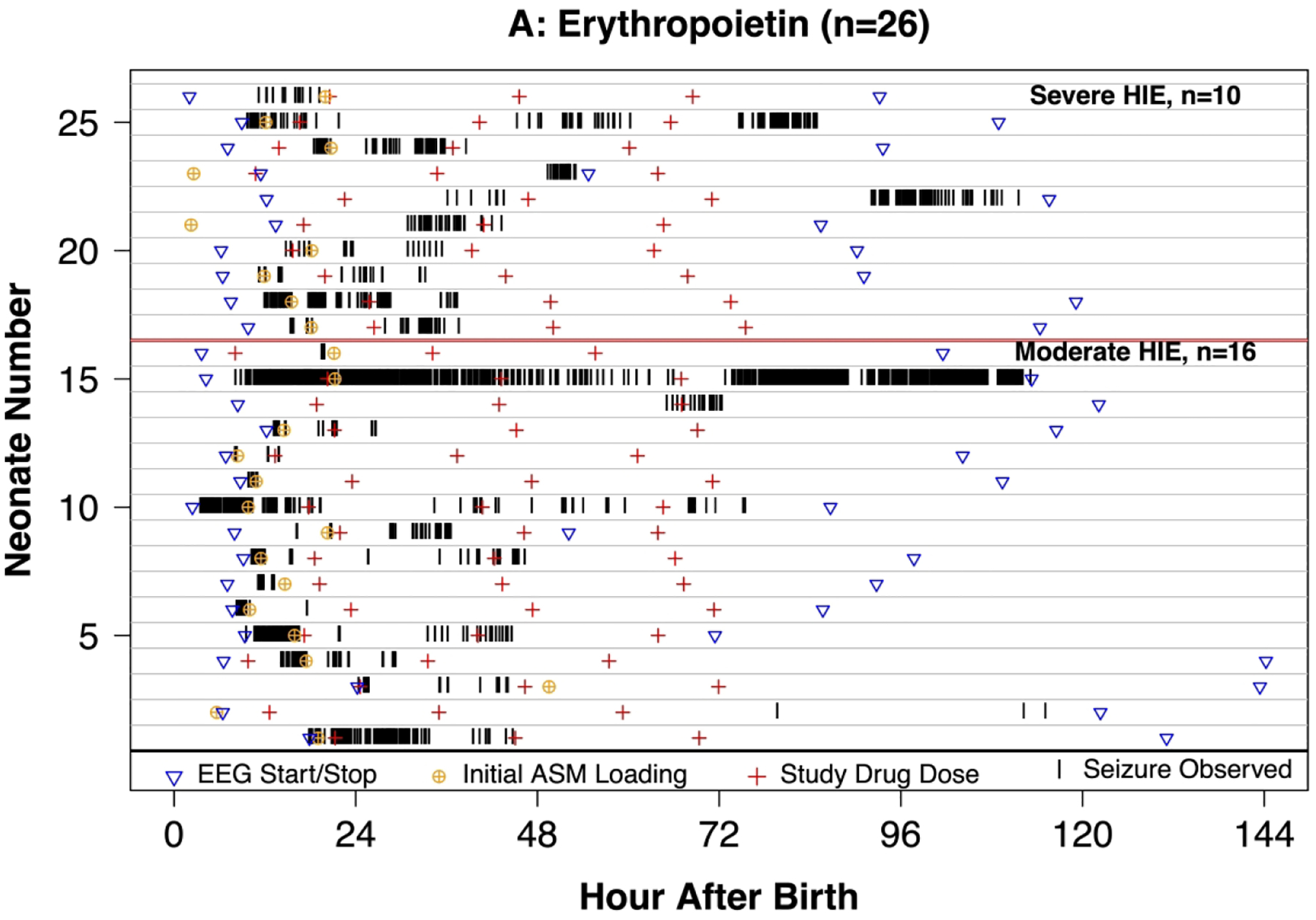
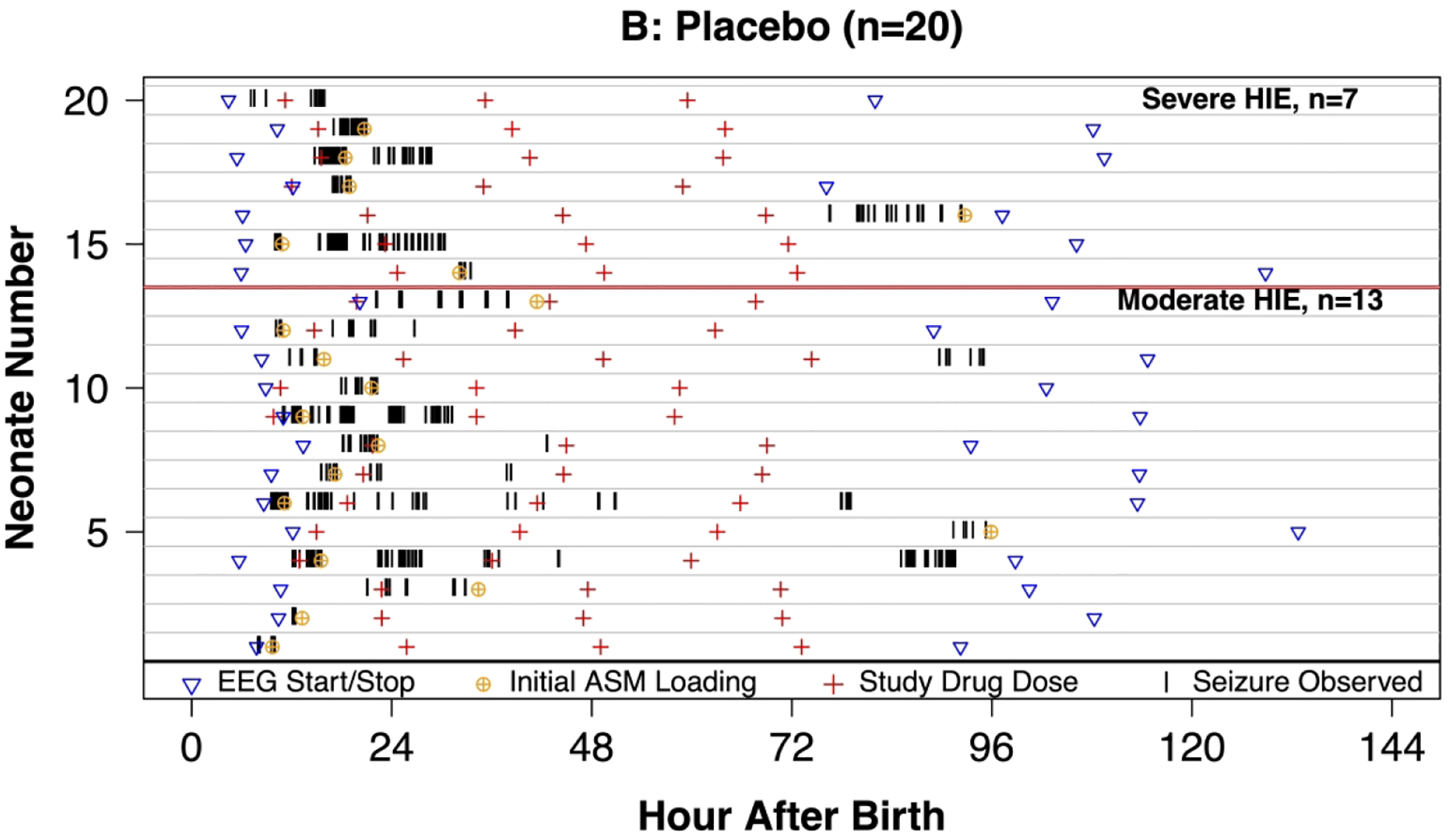
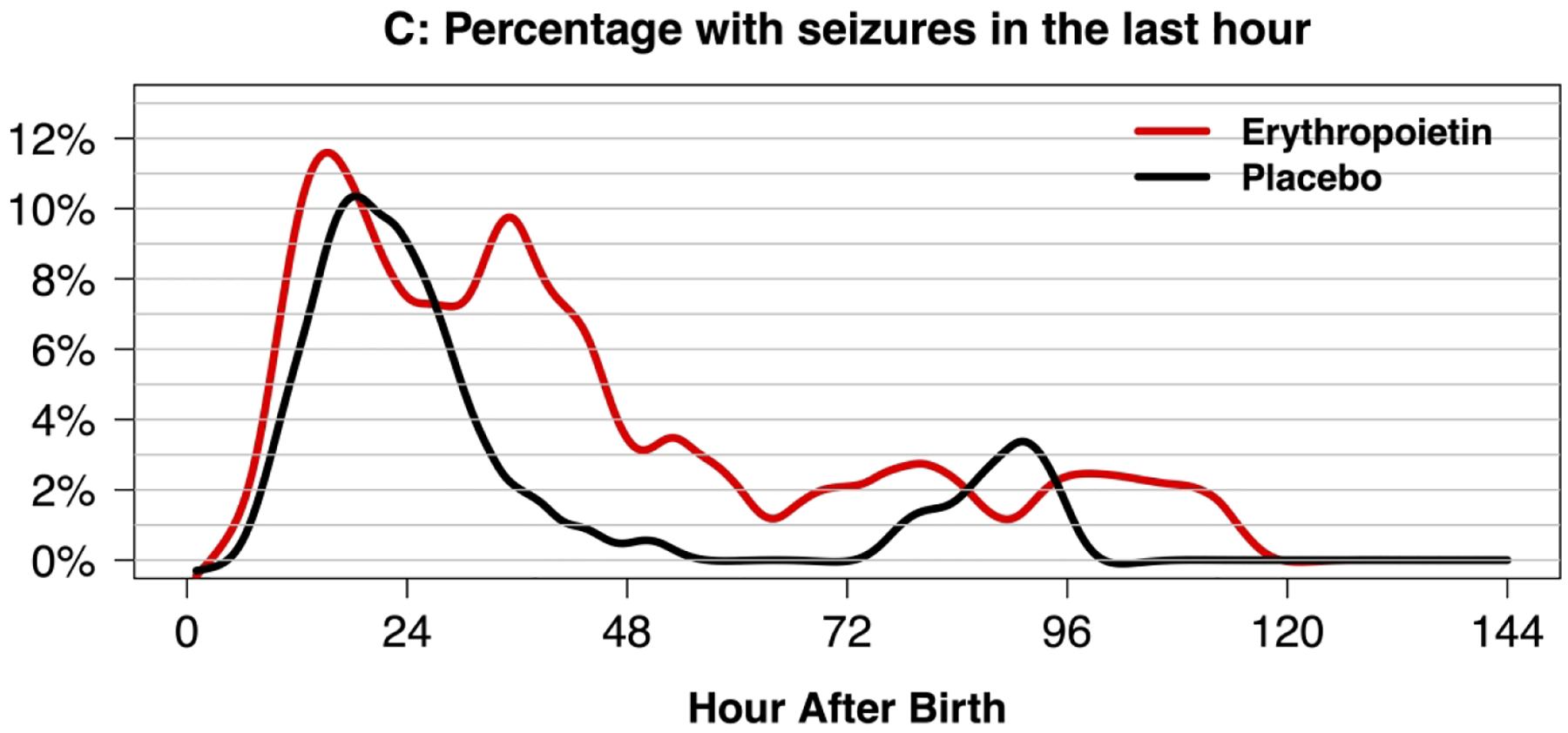
Swimmer plot of 46 neonates with seizures with hypoxic-ischemic encephalopathy (HIE) undergoing therapeutic hypothermia and treated with erythropoietin (Epo, panel A) or placebo (panel B) who received continuous video-EEG throughout hypothermia and rewarming. Vertical lines indicate that a seizure was observed. Panel C: Density plot of neonates with seizure in the previous hour of EEG recording.
Secondary outcomes.
Among the 43 (29%) of neonates that received a loading dose of ASM, 13 (30%) had a complete response. A lower complete response was observed in neonates treated with Epo (5/24, 21%) compared to placebo (8/19, 42%), but the difference was not significant after adjustment for HIE severity and baseline seizure burden prior to first study drug dose (aRR 0.78, 95% CI 0.27 – 2.26).
Among the 46 neonates with seizures, total minutes of observed seizure burden was higher in the Epo group (median=63.8; IQR= 28.5, 146.5) compared to the placebo group (median=31.5; IQR= 23.4, 77.1; adjusted difference=34.0; 95% CI= 2.81 – 67.4; Table 3). However, when considering the total cEEG recording time, the percentage of time with seizures was not significantly different between the Epo group (median=1.2%; IQR= 0.5%, 3.5%) and placebo group (median=0.6%; IQR= 0.4%, 1.3%; adjusted difference= 0.2%; 95% CI= −0.4% - 0.8%). In a sensitivity analysis excluding the six children who died and had incomplete EEG data, the observed seizure burden was similar (median 63.2, IQR 25.5, 81.2) minutes for the Epo group and (median 32.5, IQR 25.5, 81.2) minutes for the placebo group.
The median period over which a neonate had seizures was 16.3 hours (IQR 6.2, 25.2) and was not significantly different between groups (median 20.2 hours, IQR 8.4, 33.1 hours for Epo and median 14.9 hours, IQR=3.9, 21.0 hours for placebo; adjusted difference = 2.5; 95% CI= −3.3 – 11.6).
Status epilepticus was present in 10/46 (22%) of neonates with seizures and occurred more frequently among neonates treated with Epo (9/26, 35%) compared to those treated with placebo (1/20, 5.0%), but was not significantly different after adjustment for pre-treatment seizure burden and HIE severity (aRR = 2.84; 95% CI = 0.56 – 14.39).
Discussion
Among neonates who received cEEG monitoring within the HEAL randomized, controlled trial of erythropoietin (Epo) vs placebo plus hypothermia for moderate or severe encephalopathy presumed due to hypoxic-ischemic encephalopathy (HIE), 31% had electrographic seizures and there was no significant difference between study groups. Maximal hourly seizure burden, overall timing and duration of seizures, as well as response to antiseizure medication treatment were not significantly different after receiving Epo as compared with placebo.
These findings are not consistent with preclinical studies, where Epo has been shown to reduce the severity of acute and late seizures in animal models of HIE.4–7 Proposed mechanisms for the anti-seizure effects of Epo include microglial activation, reduced inflammation, decreased neuronal death and ectopic granule cell generation, as well as enhanced hippocampal Epo receptor expression (especially following status epilepticus).4–7,21–24 In a study of kainic acid and hypoxia-induced seizures, the latency to seizure onset doubled and duration of seizures dropped by 50% in the Epo-treated animals.6
There are several possible reasons why our data are not aligned with results from animal research. In the HEAL trial, Epo was delivered in combination with hypothermia, while in animal models, it was delivered without cooling. Both Epo and hypothermia may act through similar mechanisms and target similar points in the injury cascade, including reduced apoptotic, inflammatory, and excitotoxic injury.25 Epo may not confer additional benefit beyond hypothermia when used in combination. However, the results of the current study do not exclude the possibility that Epo alone could have anti-seizure properties. Other explanations for our negative findings could include suboptimal timing of administration (specifically administration of Epo early in the injury cascade), or suboptimal dosing of Epo. FDA labeling for Epo warns of a possible pro-convulsant effect based on older trials in adults with renal disease and hypertension,8–10 but there is limited active research in neonates at risk for seizures.
Although we found no significant increase in seizure risk after Epo administration, several important measures indicated potentially worse seizures in the Epo group (e.g., total seizure duration, overall maximal hourly seizure burden, seizure period, and status epilepticus), and the overall gestalt of the swimmer plots is one of higher seizure burden in the Epo group. Possible explanations for worse seizures in the Epo group include chance (i.e., failure to randomize into two groups with equal likelihood of seizures), less robust ASM treatment in the Epo group, or a true increase in seizure burden related to Epo or its side effects. Importantly, there was no apparent pro-convulsant effect of Epo among neonates with HIE who were treated with hypothermia: differences by treatment group were not significant after adjusting for pre-treatment seizure burden. Prior studies highlight the importance of pre-treatment seizure burden on apparent efficacy of a study drug. For instance, a randomized, controlled trial of bumetanide for acute provoked neonatal seizures had an important imbalance in pre-treatment seizure burden, which limited the study’s ability to detect a treatment effect and led the study authors and an international working group to recommend that future trials of ASMs should take measures to balance baseline seizure severity between the study groups.26,27 Finally, we cannot exclude small differences between groups.
Although we present data from a large, randomized controlled, double-blind trial of Epo vs placebo for neonates with HIE with central review of cEEG recorded throughout cooling and rewarming, our data are not without limitations. First, the rate of EEG-detected seizures (31%) was lower than anticipated and lower than most previously published studies (34% - 65%)20,28,29. The lower seizure rate could have limited the power to detect significant differences between groups. The reason for lower seizure frequency may be related to improved obstetric care and resuscitation, rapid onset of hypothermia, or other neuroprotective measures to prevent secondary brain injury and overall lower frequency of severe encephalopathy at treating sites. Second, seizure identification and treatment were at the discretion of the local care team; the study was not designed to address interactions between Epo treatment and ASM. It is, therefore, possible that the lack of Epo effects were related to unknown confounding interactions.
However, randomization and adjustment for site in our analysis should have mitigated potential confounding. The reasons for differences in ASM use between groups is not known and may relate to treatment of clinically suspected versus electrographic only seizures. Third, we reached our a priori sample size of 150 before reviewing all available cEEGs and before achieving a sample size of 70 in each group. Finally, the parent study excluded families with an unstable social situation, which may limit the generalizability of study results.
Conclusions
In this large cohort of neonates with encephalopathy due to HIE who were randomized to Epo or placebo plus hypothermia and who received cEEG monitoring per ACNS guidelines throughout cooling and rewarming, we found no meaningful differences in key measures of seizure burden or response to treatment in the Epo group. These findings are consistent with overall trial results, which do not support adjunct use of Epo for neonates with HIE undergoing therapeutic hypothermia. The overall proportion of children with seizures was lower that prior studies.
Although Epo combined with hyothermia showed no benefit in seizure reduction, this study provides new important information about the contemporary risk of seizures during hypothermia. We also add to the literature about seizure timing and burden in neonates with HIE. Future studies will address EEG background activity for long-term prognosis in neonates with HIE.
Supplementary Material
Impact:
In the HEAL trial of erythropoietin (Epo) vs placebo for neonates with encephalopathy presumed due to hypoxic-ischemic encephalopathy (HIE) who were also treated with therapeutic hypothermia, electrographic seizures were detected in 31%, which is lower than most prior studies.
Epo did not reduce the proportion of neonates with acute provoked seizures (31% in Epo vs 30% in placebo) or maximal hourly seizure burden after study drug (median Epo: 11.4, IQR 5.6, 18.1 for Epo vs median 9.7, IQR 4.9, 21.0 minutes/hour for placebo).
There was no anti- or pro-convulsant effect of Epo when combined with therapeutic hypothermia for HIE.
Acknowledgements
The authors would like to thank Dr. Taeun Chang (1971-2022) for her tireless contributions to this work, site investigators Drs. John Flibotte and Lori Billinghurst, as well as the Clinical Research Coordinators at each study site.
Funding
The study was funded by NIH/NINDS R01NS104322, U01NS092764, and U01NS092553. Adam L. Numis, MD, received grant support during the study period from NINDS K23NS105918.
Footnotes
Competing Interests
Kaashif A. Ahmad has nothing to disclose.
Sonia L. Bonifacio has nothing to disclose.
Bryan A. Comstock has nothing to disclose.
Hannah C. Glass has nothing to disclose.
Fernando F. Gonzalez has nothing to disclose.
Patrick J. Heagerty has nothing to disclose.
Sandra E. Juul has nothing to disclose.
Nathalie Maitre has nothing to disclose.
Shavonne L. Massey has nothing to disclose.
Dennis E. Mayock has nothing to disclose.
Ulrike Mietzsch has nothing to disclose.
Niranjana Natarajan has nothing to disclose.
Adam L. Numis has nothing to disclose.
Gregory M. Sokol has nothing to disclose.
Cameron Thomas has nothing to disclose.
Krisa P. Van Meurs has nothing to disclose.
Yvonne W. Wu has nothing to disclose.
Consent Statement
Neonates were studied after informed parental consent.
Data Availability Statement
HEAL Trial Data-sharing plan
We will prepare and share a final research data set that the accepted primary pragmatic trial publication is based upon. The final data set will be structured to maximize future scientific value while protecting patient and health system privacy. The UW DCC will remove or de-identify all 18 HIPAA-specified direct identifiers. The aim of our data sharing policy is strive for the least restrictive plan possible while providing appropriate protection for participant privacy, health system privacy, and scientific integrity.
Within 9 months of the end of the final year of funding, a final study data set will be accessible via a supervised private data enclave managed by the National Institute of Neurological Disorder and Stroke (NINDS) at: https://www.ninds.nih.gov/Current-Research/Research-Funded-NINDS/Clinical-Research/Archived-Clinical-Research-Datasets. The shared data set will contain all data collected under both the HEAL Trial protocol and HEAL ancillary studies. Access will be limited to registered users who submit proposed specific questions or analysis plans and sign a data use agreement according to NINDS guidelines. “Supervised” indicates that individual requests are reviewed to protect the intellectual property rights of the project investigative team by restricting external development of manuscripts using the study data that substantially overlap with those that are already in development by study investigators.
References
- 1.Glass HC et al. Contemporary Profile of Seizures in Neonates: A Prospective Cohort Study. J Pediatr 174, 98–103 e101, doi: 10.1016/j.jpeds.2016.03.035 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ronen GM, Penney S & Andrews W The epidemiology of clinical neonatal seizures in Newfoundland: a population-based study. J Pediatr 134, 71–75, doi: 10.1016/s0022-3476(99)70374-4 (1999). [DOI] [PubMed] [Google Scholar]
- 3.Juul S Erythropoietin in the central nervous system, and its use to prevent hypoxic-ischemic brain damage. Acta paediatrica 91, 36–42, doi: 10.1111/j.1651-2227.2002.tb02904.x (2002). [DOI] [PubMed] [Google Scholar]
- 4.Yang J et al. Erythropoietin preconditioning suppresses neuronal death following status epilepticus in rats. Acta Neurobiol Exp (Wars) 67, 141–148 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Nadam J et al. Neuroprotective effects of erythropoietin in the rat hippocampus after pilocarpine-induced status epilepticus. Neurobiol Dis 25, 412–426, doi:S0969–9961(06)00259–2 [pii] 10.1016/j.nbd.2006.10.009 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Mikati MA, El Hokayem JA & El Sabban ME Effects of a single dose of erythropoietin on subsequent seizure susceptibility in rats exposed to acute hypoxia at P10. Epilepsia 48, 175–181, doi: 10.1111/j.1528-1167.2006.00900.x (2007). [DOI] [PubMed] [Google Scholar]
- 7.Chu K et al. Erythropoietin reduces epileptogenic processes following status epilepticus. Epilepsia 49, 1723–1732, doi:EPI1644 [pii] 10.1111/j.1528-1167.2008.01644.x (2008). [DOI] [PubMed] [Google Scholar]
- 8.Brown AL, Tucker B, Baker LR & Raine AE Seizures related to blood transfusion and erythropoietin treatment in patients undergoing dialysis. BMJ 299, 1258–1259, doi: 10.1136/bmj.299.6710.1258 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beccari M, Romagnoni M & Sorgato G Seizures in dialysis patients treated with recombinant erythropoietin. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association 10, 423–424 (1995). [PubMed] [Google Scholar]
- 10.Edmunds ME et al. Seizures in haemodialysis patients treated with recombinant human erythropoietin. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association 4, 1065–1069 (1989). [PubMed] [Google Scholar]
- 11.Wu YW et al. Erythropoietin for neuroprotection in neonatal encephalopathy: safety and pharmacokinetics. Pediatrics 130, 683–691, doi: 10.1542/peds.2012-0498 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baserga MC et al. Darbepoetin administration to neonates undergoing cooling for encephalopathy: a safety and pharmacokinetic trial. Pediatr Res 78, 315–322, doi: 10.1038/pr.2015.101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu YW et al. Trial of Erythropoietin for Hypoxic-Ischemic Encephalopathy in Newborns. N Engl J Med 387, 148–159, doi: 10.1056/NEJMoa2119660 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Juul SE et al. High-Dose Erythropoietin for Asphyxia and Encephalopathy (HEAL): A Randomized Controlled Trial - Background, Aims, and Study Protocol. Neonatology 113, 331–338, doi: 10.1159/000486820 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shellhaas RA et al. The American Clinical Neurophysiology Society’s Guideline on Continuous Electroencephalography Monitoring in Neonates. J Clin Neurophysiol 28, 611–617, doi: 10.1097/WNP.0b013e31823e96d7 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Landis JR & Koch GG The measurement of observer agreement for categorical data. Biometrics 33, 159–174 (1977). [PubMed] [Google Scholar]
- 17.Tsuchida TN et al. American clinical neurophysiology society standardized EEG terminology and categorization for the description of continuous EEG monitoring in neonates: report of the American Clinical Neurophysiology Society critical care monitoring committee. J Clin Neurophysiol 30, 161–173, doi: 10.1097/WNP.0b013e3182872b24 (2013). [DOI] [PubMed] [Google Scholar]
- 18.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, URL https://www.R-project.org/. 2020). [Google Scholar]
- 19.Lynch NE et al. The temporal characteristics of seizures in neonatal hypoxic ischemic encephalopathy treated with hypothermia. Seizure 33, 60–65, doi: 10.1016/j.seizure.2015.10.007 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Glass HC et al. Risk factors for EEG seizures in neonates treated with hypothermia: a multicenter cohort study. Neurology 82, 1239–1244, doi: 10.1212/WNL.0000000000000282 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bahcekapili N et al. Erythropoietin pretreatment suppresses seizures and prevents the increase in inflammatory mediators during pentylenetetrazole-induced generalized seizures. Int J Neurosci 124, 762–770, doi: 10.3109/00207454.2013.878935 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Yoo JY et al. Neuroprotective effects of erythropoietin posttreatment against kainate-induced excitotoxicity in mixed spinal cultures. J Neurosci Res 87, 150–163, doi: 10.1002/jnr.21832 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Montero M et al. Comparison of neuroprotective effects of erythropoietin (EPO) and carbamylerythropoietin (CEPO) against ischemia-like oxygen-glucose deprivation (OGD) and NMDA excitotoxicity in mouse hippocampal slice cultures. Exp Neurol 204, 106–117, doi: 10.1016/j.expneurol.2006.09.026 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Garzon F et al. NeuroEPO Preserves Neurons from Glutamate-Induced Excitotoxicity. J Alzheimers Dis 65, 1469–1483, doi: 10.3233/JAD-180668 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Wassink G et al. Recombinant erythropoietin does not augment hypothermic white matter protection after global cerebral ischaemia in near-term fetal sheep. Brain Commun 3, fcab172, doi: 10.1093/braincomms/fcab172 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soul JS et al. A Pilot Randomized, Controlled, Double-Blind Trial of Bumetanide to Treat Neonatal Seizures. Ann Neurol 89, 327–340, doi: 10.1002/ana.25959 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soul JS et al. Recommendations for the design of therapeutic trials for neonatal seizures. Pediatr Res 85, 943–954, doi: 10.1038/s41390-018-0242-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wusthoff CJ et al. Electrographic seizures during therapeutic hypothermia for neonatal hypoxic-ischemic encephalopathy. J Child Neurol 26, 724–728, doi: 10.1177/0883073810390036 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nash KB et al. Video-EEG monitoring in newborns with hypoxic-ischemic encephalopathy treated with hypothermia. Neurology 76, 556–562, doi: 10.1212/WNL.0b013e31820af91a (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
HEAL Trial Data-sharing plan
We will prepare and share a final research data set that the accepted primary pragmatic trial publication is based upon. The final data set will be structured to maximize future scientific value while protecting patient and health system privacy. The UW DCC will remove or de-identify all 18 HIPAA-specified direct identifiers. The aim of our data sharing policy is strive for the least restrictive plan possible while providing appropriate protection for participant privacy, health system privacy, and scientific integrity.
Within 9 months of the end of the final year of funding, a final study data set will be accessible via a supervised private data enclave managed by the National Institute of Neurological Disorder and Stroke (NINDS) at: https://www.ninds.nih.gov/Current-Research/Research-Funded-NINDS/Clinical-Research/Archived-Clinical-Research-Datasets. The shared data set will contain all data collected under both the HEAL Trial protocol and HEAL ancillary studies. Access will be limited to registered users who submit proposed specific questions or analysis plans and sign a data use agreement according to NINDS guidelines. “Supervised” indicates that individual requests are reviewed to protect the intellectual property rights of the project investigative team by restricting external development of manuscripts using the study data that substantially overlap with those that are already in development by study investigators.