

To the Editor,
Neurological disorders are considered rare but serious complications after allogeneic hematopoietic stem cell transplantation (allo-HSCT). The presented case report involves a diagnostic challenge for physicians and a great threat for patients.
A 64-year-old patient was admitted to the hospital due to progressive alteration of consciousness for 3 weeks. Sixty days prior, the patient had undergone allo-HSCT due to high-risk acute lymphoblastic leukemia (ALL) with initial infiltration of the cerebrospinal fluid (CSF). At day 36 following allo-HSCT, complete remission with 100% chimerism was confirmed. At day 45, the patient presented with short-term memory loss. Reactivation of cytomegalovirus (CMV) was diagnosed and the patient started therapy with valganciclovir. Despite the decrease of the viral load of CMV, the neurological state of the patient continued to deteriorate. Clinical examination showed psychomotor retardation, moderate dementia, and exaggerated deep tendon reflexes without paresis of the limbs and without meningeal symptoms.
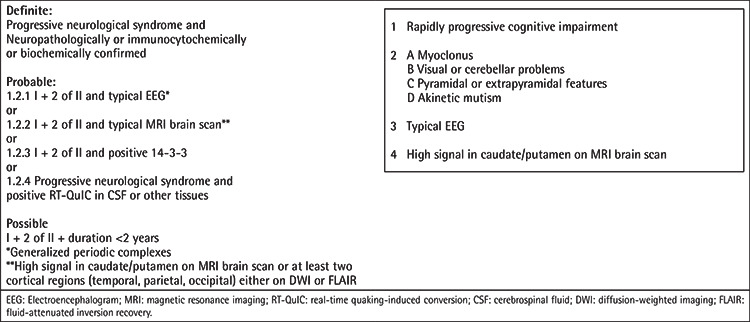
A brain computed tomography scan ruled out ischemic stroke and intracerebral hemorrhage. Bone marrow analysis excluded relapse of ALL. CSF examination showed normal levels of cells, glucose, and protein. CSF and serum analysis also ruled out viral (except for CMV), bacterial, fungal, and parasite infections. Hyponatremia, probably secondary to dehydration, was slowly corrected. All potentially neurotoxic medications were changed or stopped. However, the patient’s clinical condition continued to deteriorate rapidly. Two weeks after admission, the patient was comatose with preserved pain response, and pyramidal and extrapyramidal signs were present. Brain magnetic resonance imaging (MRI) revealed hyperintensity in the left parietal cortex (Figures 1A and 1C), which was initially misinterpreted as encephalitis. With the suspicion of limbic encephalitis, antibody tests of the blood and CSF had negative results. Treatment with plasmapheresis and corticosteroids and subsequently with immunoglobulins, mycophenolate mofetil, and anti-CD20 was administered with no improvement [1]. The patient became deeply comatose with flaccid tetraplegia and continuous myoclonic seizures. In the literature, reports of CNS involvement in chronic graft-versus-host disease are available, but this patient did not meet the relevant criteria [2]. Follow-up MRI presented very suggestive radiological signs of Creutzfeldt-Jackob disease (CJD) (Figures 1B and 1D). EEG results showed slow basal activity of both cerebral hemispheres with generalized synchronic periodic sharp-wave complexes, also suggesting CJD. The CSF analysis indicated the presence of 14-3-3 protein. Finally, the patient was diagnosed with probable sporadic CJD (Table 1) [3].
Figure 1.
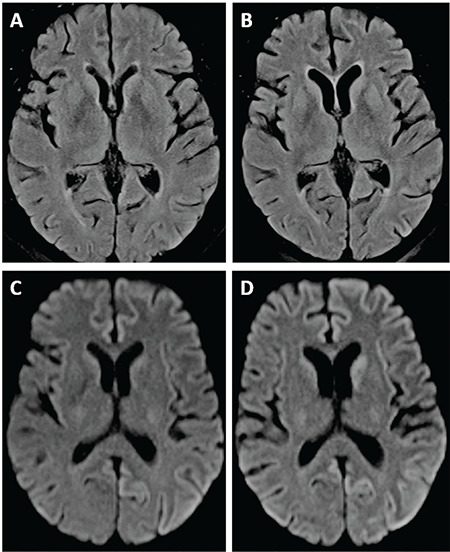
Brain magnetic resonance imaging (MRI) revealed hyperintensity in the left parietal cortex (A, C). Follow-up MRI presented very suggestive radiological signs of Creutzfeldt-Jackob disease (B, D).
Table 1. Diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Adapted from the National Creutzfeldt-Jakob Disease Research & Surveillance Unit [3].
Following symptom onset, the patient’s neurological state deteriorated quickly over the course of 6 weeks and death occurred within 4 months. The patient’s family refused the neuropathological examination required for a definite diagnosis.
CJD is a fatal neurodegenerative disease caused by misfolded prion proteins (PrPSc). The morbidity is approximately one per million per year [5]. Definitive diagnosis of CJD is very difficult as it can occur spontaneously, may be genetically linked, or can be caused by inflammatory or contaminated transplant material, which is not commonly tested for prion diseases [4,5]. While allo-HSCT is a high-risk procedure associated with impaired immunity and serious complications, CJD should also be considered as a potential cause of rapidly progressing dementia.
Footnotes
Ethics
Informed Consent: Obtained.
Authorship Contributions
Surgical and Medical Practices: M.K., M.S., H.B.; Concept: M.K., M.S.; Design: M.K., M.S.; Data Collection or Processing: M.K., M.S., J.B., J.U.; Analysis or Interpretation: M.K., M.S., M.Sz., J.B., J.U.; Literature Search: M.K., M.S., M.Sz., J.B.; Writing: M.K., M.S., M.Sz., J.B., J.U.
Conflict of Interest: No conflict of interest was declared by the authors.
Financial Disclosure: The authors declared that this study received no financial support.
References
- 1.Hermetter C, Fazekas F, Hochmeister S. Systematic Review: Syndromes, early diagnosis, and treatment in autoimmune encephalitis. Front Neurol. 2018;9:706. doi: 10.3389/fneur.2018.00706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pruitt AA, Graus F, Rosenfeld MR. Neurological complications of transplantation: part I: Hematopoietic cell transplantation. Neurohospitalist. 2013;3:24–38. doi: 10.1177/1941874412455338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.National Creutzfeldt-Jakob Disease Surveillance Diagnostic Criteria, 2010. Available online at. [Internet] http://www.cjd.ed.ac.uk.
- 4.Uttley L, Carroll C, Wong R, Hilton DA, Stevenson M. Creutzfeldt-Jakob disease: a systematic review of global incidence, prevalence, infectivity, and incubation. Lancet Infect Dis. 2020;20:e2–e10. doi: 10.1016/S1473-3099(19)30615-2. [DOI] [PubMed] [Google Scholar]
- 5.De Sousa PA, Ritchie D, Green A, Chandran S, Knight R, Head MW. Renewed assessment of the risk of emergent advanced cell therapies to transmit neuroproteinopathies. Acta Neuropathol. 2019;137:363–377. doi: 10.1007/s00401-018-1941-9. [DOI] [PMC free article] [PubMed] [Google Scholar]