

A versatile synthesis is reported for 5-(arylmethylideneamino)-4-(1H-benzo[d]imidazol-1-yl)pyrimidines and structures are reported for three examples, one of them in two crystal forms, as well as for two intermediates in the synthetic sequence. A diverse range of hydrogen-bonding patterns leads to supramolecular assemblies ranging from finite zero-dimensional aggregates to three-dimensional framework structures.
Keywords: synthesis, pyrimidine, heterocyclic hybrid, NMR spectroscopy, crystal structure, molecular structure, molecular conformation, hydrogen bonding, supramolecular assembly
Abstract
A concise and versatile synthesis of 5-(arylmethylideneamino)-4-(1H-benzo[d]imidazol-1-yl)pyrimidines has been developed, starting from 4-(1H-benzo[d]imidazol-1-yl)pyrimidines, and we report here the synthesis and spectroscopic and structural characterization of three such products, along with those of two intermediates in the reaction pathway. The intermediates 4-[2-(4-chlorophenyl)-1H-benzo[d]imidazol-1-yl]-6-methoxypyrimidine-2,5-diamine, (II), and 4-[2-(4-bromophenyl)-1H-benzo[d]imidazol-1-yl]-6-methoxypyrimidine-2,5-diamine, (III), crystallize as the isostructural monohydrates C18H15ClN5O·H2O and C18H15BrN5O·H2O, respectively, in which the components are linked into complex sheets by O—H⋯N and N—H⋯O hydrogen bonds. In the product (E)-4-methoxy-5-[(4-nitrobenzylidene)amino]-6-[2-(4-nitrophenyl)-1H-benzo[d]imidazol-1-yl]pyrimidin-2-amine, which crystallizes as a 1:1 solvate with dimethyl sulfoxide, C25H18N8O5·C2H6OS, (IV), inversion-related pairs of the pyrimidine component are linked by N—H⋯N hydrogen bonds to form cyclic centrosymmetric R 2 2(8) dimers to which pairs of solvent molecules are linked by N—H⋯O hydrogen bonds. (E)-4-Methoxy-5-[(4-methylbenzylidene)amino]-6-[2-(4-methylphenyl)-1H-benzo[d]imidazol-1-yl]pyrimidin-2-amine, C27H24N6O, (V), crystallizes with Z′ = 2 and the molecules are linked into a three-dimensional framework structure by a combination of N—H⋯N, C—H⋯N and C—H⋯π(arene) hydrogen bonds. The analogous product (E)-4-methoxy-5-[(4-chlorobenzylidene)amino]-6-[2-(4-methylphenyl)-1H-benzo[d]imidazol-1-yl]pyrimidin-2-amine, C26H21ClN6O, (VI), crystallizes from dimethyl sulfoxide in two forms: one, denoted (VIa), is isostructural with (V), and the other, denoted (VIb), crystallizes with Z′ = 1, but as an unknown solvate in which the pyrimidine molecules are linked by N—H⋯N hydrogen bonds to form a ribbon containing two types of centrosymmetric ring.
Introduction
The benzimidazole unit has been shown to be an important heterocyclic fragment present in a large number of compounds with broad biological activity, including antimicrobial and antitumour activity (El-Gohary & Shaaban, 2017 ▸). In addition, aminopyrimidines are important building blocks for the synthesis of new heterocyclic systems (Abdul-Rida et al., 2017 ▸), and they are also considered to constitute an important pharmacophoric fragment (Loving et al., 2009 ▸), because of the wide biological activities that compounds containing this unit have shown, including anti-HIV activity (Al-Masoudi et al., 2016 ▸) and cyclin-dependent kinase 2 (CDK2) inhibitory activity (Cortese et al., 2016 ▸).
Molecules which include both benzimidazole and aminopyrimidine nuclei have been studied against some cancer cell lines, yielding interesting results that motivate the synthesis of this type of hybrid structures. This is the case for a series of novel fused pyrimido–benzimidazole systems reported recently, where one of the structures showed an IC50 value less than 2 µM against the neuroblastoma SK-N-BE(2)-C and Kelly cell lines (Gadde et al., 2023 ▸).
Non-fused pyrimidine–benzimidazole hybrids have also exhibited promising results for antitumour activity in human cancer cell lines (Sana et al., 2021 ▸). A recent report has attributed the cytotoxicity of pyrimidine–benzimidazole hybrids to the presence of methoxy groups on the arene rings linked to the pyrimidine core, while the presence of electron-withdrawing groups seems to eliminate anticancer activity (Ismail et al., 2022 ▸). However, any attempt to predict, prior to experimental evaluation, the effects of substituent variation in the products reported here would, perforce, be largely speculative and thus will not be pursued in this article.
We have recently reported the synthesis and the molecular and supramolecular structures of a set N 5-arylmethyl-6-methoxy-4-(2-aryl-1H-benzo[d]imidazol-1-yl)pyrimidine-2,5-diamines, where the two pendent aryl residues are identical, as they are both introduced in the reaction of N 4-(2-aminophenyl)-6-methoxypyrimidine-2,4,5-triamine with an aryl aldehyde in a 1:2 molar ratio (Vicentes et al., 2019 ▸). Because of the biological importance of both the 2-aminopyrimidine residue (Koroleva et al., 2010 ▸; Jadhav et al., 2021 ▸) and the benzimidazole unit (Singh et al., 2013 ▸; Wu et al., 2022 ▸), whether alone or in combination, in the search for new biological targets (Sana et al., 2021 ▸), we have now explored the combination of different aryl residues linked to the 5-amino group.
We report here an extension of the pyrimidine–benzimidazole hybrid systems reported previously (Vicentes et al., 2019 ▸), in which the N 5-methylaryl-6-methoxy-4-(2-aryl-1H-benzo[d]imidazol-1-yl)pyrimidine-2,5-diamine precusors (A) (see Scheme 1) are subjected to debenzylation effected by ammonium hexanitratocerate(IV) (CAN) to produce the 6-methoxy-4-(2-aryl-1H-benzo[d]imidazol-1-yl)pyrimidine-2,5-diamines (I)–(III) (Scheme 1) for use as intermediates in the derivatization at the 5-amino group. When the corresponding reaction was attempted using the type (A) precursor having X = Y = NO2, no debenzylation was observed, but instead the reaction produced a complex mixture from which only (E)-4-methoxy-5-[(4-nitrobenzylidene)amino]-6-[2-(4-nitrophenyl)-1H-benzo[d]imidazol-1-yl]pyrimidin-2-amine, (IV), could be isolated in pure form as a 1:1 solvate with dimethyl sulfoxide, in a yield of only 15%.
When compound (I) was condensed with 4-methylbenzaldehyde, the product (E)-4-methoxy-5-[(4-methylbenzylidene)amino]-6-[2-(4-methylphenyl)-1H-benzo[d]imidazol-1-yl]pyrimidin-2-amine, (V), was formed in 74% yield and straightforwardly crystallized in the solvent-free form from a mixture of ethyl acetate and hexane. However, the corresponding reaction with 4-chlorobenzaldehyde gave, after crystallization, a mixture of two crystalline forms of (E)-4-chloro-5-[(4-methylbenzylidene)amino]-6-[2-(4-methylphenyl)-1H-benzo[d]imidazol-1-yl]pyrimidin-2-amine, one denoted (VIa), which is isostructural with (V), together with a second form, denoted (VIb), which is a solvate of unknown constitution. The structure of compound (I) has already been reported (Vicentes et al., 2019 ▸) and we report here the molecular and supramolecular structures of compounds (II)–(VI).

Experimental
Synthesis and crystallization
The atom labelling is based throughout on the IUPAC chemical names, with the immediate substituents on the pyrimidine ring labelled according to their location; thus, N21, N41, N51 and O61, with appropriate modifications when Z′ = 2, and with the rest of the substituent labels following the IUPAC name.
All of the signals in the 1H and 13C NMR spectra listed below were assigned using one-dimensional DEPT-135 13C spectra and two-dimensional COSY, HSQC and HMBC spectra.
The precursors of type (A) and the intermediate (I) (see Scheme 1) were prepared using previously described methods (Vicentes et al., 2019 ▸). In the NMR listings given below, the atom labelling for compounds (II)–(IV) and (VI) follows that used in Figs. 1 ▸–3 ▸ ▸ and 6, and the labelling for compound (V) follows that for (VI).
Figure 1.

The two independent components in the structure of compound (II), showing the atom-labelling scheme and the hydrogen bond (drawn as a dashed line) within the selected asymmetric unit. Displacement ellipsoids are drawn at the 50% probability level.
Figure 2.

The two independent components in the structure of compound (III), showing the atom-labelling scheme and the hydrogen bond (drawn as a dashed line) within the selected asymmetric unit. Displacement ellipsoids are drawn at the 50% probability level.
Figure 3.

The two independent components in the structure of compound (IV), showing the atom-labelling scheme and the hydrogen bond (drawn as a dashed line) within the selected asymmetric unit. Displacement ellipsoids are drawn at the 50% probability level.
For the synthesis of compounds (II)–(IV), a solution of ammonium hexanitratocerate(IV) (0.69 g, 1.5 mmol) in a mixture of acetonitrile and water (3:1 v/v, 50 ml) was added to a solution of the appropriate precursor (A) [0.5 mmol; 0.22 g for (II) and 0.26 g for each of (III) and (IV)] in acetonitrile (10 ml); the resulting mixtures were then stirred for 2 h at 273 K. A saturated solution of sodium carbonate (15 ml) was then added and the acetonitrile was removed under reduced pressure. The residue was exhaustively extracted with ethyl acetate and the combined organic extracts were washed with water and then dried over anhydrous sodium sulfate. The solvent was removed under reduced pressure and the crude solid products purified by column chromatography on silica gel (0.040–0.063 mm) using a mixture of ethyl acetate and hexane (3:2 v/v) as the eluent.
Compound (II): colourless solid, yield 54%, m.p. 510 K (decomposition). IR (ATR, cm−1): 3494, 3399, 3303, 3188, 2922, 1606, 1562, 1467, 1450, 1403, 1261, 1241, 1092, 1011, 798, 739. NMR (DMSO-d 6): δ(1H, 400 MHz) 7.79 (ddd, J = 8.0, 1.3, 0.7 Hz, 1H, H44), 7.62 (d, J = 8.8 Hz, 2H, H72, H76), 7.28 (d, J = 8.8 Hz, 2H, H73, H75), 7.25 (dd, J = 4.0, 1.5 Hz, 1H, H45), 7.21 (dd, J = 7.2, 1.3 Hz, 1H, H46), 7.15 (ddd, J = 7.8, 1.4, 0.7 Hz, 1H, H47), 4.56 (s, 2H,NH2), 3.97 (s, 3H, OCH3), 3.06 (s, 2H, NH2); δ(13C, 101 MHz) 162.33 (C6), 155.03 (C2), 150.94 (C42), 143.39 (C43A), 140.29 (C4), 136.25 (C74), 135.24 (C47A), 129.95 (C72, C76), 129.04 (C73, C75), 128.57 (C71), 124.12 (C46), 123.65 (C45), 120.30 (C44), 117.03 (C5), 110.86 (C47), 54.75 (OCH3). HRMS (ESI–QTOF) m/z found 367.1069, [M + H]+ requires for C18H15ClN6O, 367.1069.
Compound (III): colourless solid, yield 48%, m.p. 508 K (decomposition). IR (ATR, cm−1): 3494, 3398, 3302, 3185, 1338, 1609, 1562, 1466, 1450, 1401, 1241, 1053, 1008, 832, 741. NMR (DMSO-d 6): δ(1H, 400 MHz) 7.75 (d, J = 7.3 Hz, 1H, H44), 7.63 (m, 4H, H72, H73, H75, H76), 7.32–7.21 (m, 1H, H45, H46), 7.12 (d, J = 7.4 Hz, 1H, H47), 5.99 (s, 2H, NH2), 4.17 (s, 2H, NH2), 3.96 (s, 3H, OCH3); δ(13C, 101 MHz) 161.49 (C6), 154.72 (C2), 150.65 (C42), 142.74 (C43A), 138.91 (C4), 135.55 (C74A), 131.54 (C73, C75), 130.16 (C72, C76), 129.46 (C71), 123.33 (C46), 123.25 (C45), 122.67 (C74), 119.34 (C44), 116.92 (C5), 111.32 (C47), 54.03 (OCH3). HRMS (ESI–QTOF) m/z found 413.0541, [M + H]+ requires for C18H15BrN6O, 413.0545.
Compound (IV): yellow solid, yield 15%, m.p. 517 K (decomposition). IR (ATR, cm−1): 3449, 3338, 3230, 1638, 1559, 1527, 1448, 1339, 853, 743. NMR (DMSO-d 6): δ(1H, 400 MHz) 8.66 (s, 1H, H57), 8.18 (d, J = 8.8 Hz, 2H, H53, H55), 8.13 (d, J = 9.0 Hz, 2H, H73, H75), 7.88–7.84 (m, 1H, H44), 7.82 (d, J = 9.1 Hz, 2H, H72, H76), 7.59–7.51 (m, 3H, H47, H52, H56), 7.49 (s, 2H, NH2), 7.41–7.27 (m, 2H, H45, H46), 4.01 (s, 3H, OCH3); δ(13C, 101 MHz) 164.07 (C6), 160.63 (C2), 156.79 (C57), 154.28 (C4), 150.82 (C74), 148.31 (C54), 147.57 (C42), 142.59 (C43A), 142.26 (C51), 136.55 (C71), 136.34 (C47A), 128.83 (C72, C73), 128.34 (C53, C56), 124.25 (46), 123.84 (C53, C55), 123.71 (C73, C75), 123.42 (C45), 119.81 (C44), 114.09 (C5), 112.24 (C47), 54.46 (OCH3). HRMS (ESI–QTOF) m/z found 511.1471, [M + H]+ requires for C25H18N8O5, 511.1471.
For the synthesis of compounds (V) and (VI) (Scheme 2), a mixture of compound (I) (0.17 g, 0.05 mmol) and the appropriate benzaldehyde (0.7 mmol) [84 mg of 4-methylbenzaldehyde for (V) or 92 mg of 4-chlorobenzaldehyde for (VI)] in acetic acid (3 ml) was stirred at ambient temperature for 1 h. The resulting precipitates were collected by filtration and washed first with an aqueous solution of sodium hydrogen carbonate (10% w/v) and then with water. The crude solid products were then purified by column chromatography on silica gel (0.040–0.063 mm) using a mixture of ethyl acetate and hexane (3:2 v/v) as eluent.

Compound (V): yellow solid, yield 74%, m.p. 491 K (decomposition). IR (ATR, cm−1): 3303, 3157, 1652, 1606, 1576, 1522, 1449, 1345, 1248, 1078, 1041, 817, 738. NMR (DMSO-d 6): δ(1H, 400 MHz) 8.33 (s, 1H, H57), 7.73 (d, J = 6.9 Hz, 1H, H44), 7.45 (d, J = 8.2 Hz, 2H, H72, H76), 7.37 (d, J = 6.7 Hz, 1H, H47), 7.29–7.19 (m, 4H, H45, H46, H52, H56), 7.16–7.07 (m, 6H, H73, H75, H53, H55, NH2), 3.94 (s, 3H, OCH3), 2.26 (s, 3H, C77), 2.25 (s, 3H, C58); δ(13C, 101 MHz) 163.79 (C6), 160.29 (C2), 159.89 (C57), 152.97 (C42), 152.89 (C4), 142.67 (C43A), 140.92 (C34), 139.15 (C74), 136.35 (C47A), 134.01 (C28), 129.06 (C73, C75), 128.88 (C53, C55), 127.87 (C71), 127.73 (C52, C56), 127.66 (C72, C76), 122.92 (C46), 122.55 (C45), 119.03 (C44), 116.05 (C5), 111.45 (C47), 54.18 (OCH3), 21.02 (C77), 20.83 (C58). HRMS (ESI–QTOF) m/z found 449.2084, [M + H]+ requires for C27H24N6O, 449.2084.
Compound (VI): yellow solid, yield 95%, m.p. 493 K (decomposition). IR (ATR, cm−1): 3308, 3138, 1652, 1573, 1520, 1450, 1362, 1080, 1042, 821, 735. NMR (DMSO-d 6): δ(1H, 400 MHz) 8.43 (s, 1H, H57), 7.76 (d, J = 6.8 Hz, 1H, H44), 7.45 (d, J = 8.2 Hz, 2H, H72, H76), 7.44–7.36 (m, 3H, H47, H53, H55), 7.34 (d, J = 8.6 Hz, 2H, H52, H56), 7.31–7.19 (m, 4H, H45, H26, NH2), 7.09 (d, J = 8.0 Hz, 2H, H73, H75), 3.97 (s, 3H, OCH3), 2.25 (s, 3H, CCH3); δ(13C, 101 MHz) 163.85 (C6), 160.18 (C2), 158.37 (C57), 153.78 (C4), 153.01 (C42), 142.70 (C43A), 139.21 (C74), 136.34 (C47A), 135.50 (C54), 135.42 (C51), 129.21 (C52, C56), 128.92 (C73, C75), 128.62 (C53, C55), 127,86 (C71), 127.64 (C72, C76), 123.01 (C46), 122.66 (C45), 119.10 (C44), 115.24 (C5), 111.54 (C47), 54.28 (OCH3), 20.85 (CH3). HRMS (ESI–QTOF) m/z found 469.1538, [M + H]+ requires for C26H21ClN6O, 469.1538.
Crystals of compounds (II)–(V) suitable for single-crystal X-ray diffraction were grown by slow evaporation at ambient temperature and in the presence of air from a solution in dimethyl sulfoxide for (II), (IV) and (V) or from a solution in methanol for (III), providing (II) and (III) as monohydrates, (IV) as a dimethyl sulfoxide (DMSO) solvate and (V) in the solvent-free form. A similar crystallization of (VI) from a solution in DMSO yielded two types of crystal, i.e. the more block-like solvent-free form (VIa) and the more plate-like solvate (VIb); no attempt was made to determine the relative quantities of the two crystalline forms.
Refinement
Crystal data, data collection and refinement details for compounds (II)–(VI) are summarized in Table 1 ▸. For (VIb), one reflection (010), which had been attenuated by the beam stop, and one bad outlier reflection (
 03) were omitted from the data set. All H atoms were located in difference maps. The H atoms bonded to C atoms were then treated as riding atoms in geometrically idealized positions, with C—H = 0.95 (alkenic and aromatic) or 0.98 Å (CH3), and with U
iso(H) = kU
eq(C), where k = 1.5 for the methyl groups, which were permitted to rotate but not to tilt, and 1.2 for all other H atoms. For the H atoms bonded to N or O atoms, the atomic coordinates were refined with U
iso(H) = 1.2U
eq(N) or 1.5U
eq(O), giving the N—H and O—H distances shown in Table 3. For (VIb), conventional refinement converged only to R
1 = 0.146 and wR
2 = 0.3473. Examination of the structure of (VIb) at this point using PLATON (Spek, 2020 ▸) showed that the structure formed by the molecules of (VI) enclosed a void centred at (0,0,
03) were omitted from the data set. All H atoms were located in difference maps. The H atoms bonded to C atoms were then treated as riding atoms in geometrically idealized positions, with C—H = 0.95 (alkenic and aromatic) or 0.98 Å (CH3), and with U
iso(H) = kU
eq(C), where k = 1.5 for the methyl groups, which were permitted to rotate but not to tilt, and 1.2 for all other H atoms. For the H atoms bonded to N or O atoms, the atomic coordinates were refined with U
iso(H) = 1.2U
eq(N) or 1.5U
eq(O), giving the N—H and O—H distances shown in Table 3. For (VIb), conventional refinement converged only to R
1 = 0.146 and wR
2 = 0.3473. Examination of the structure of (VIb) at this point using PLATON (Spek, 2020 ▸) showed that the structure formed by the molecules of (VI) enclosed a void centred at (0,0,
 ), whose volume was ca 166 Å3 in a unit cell of total volume 1272.6 (2) Å3. The void thus occupies ca 13.0% of the total unit-cell volume, and there are a number of significant peaks in the difference map clustered within this void. The largest peak had a magnitude of 4.64 e Å−3 and further examination of this structure using the SQUEEZE procedure (Spek, 2015 ▸) indicated that the void contained around 43 electrons not hitherto accounted for. This number is consistent with the presence of one molecule of dimethyl sulfoxide, but no convincing solvent model could be developed from the difference peaks within the void and hence the reflection data were subjected to the SQUEEZE procedure (Spek, 2015 ▸), and the resultant modified reflection file was used for the refinement reported here. The CIF describing the structure obtained before the SQUEEZE procedure was applied has been included in the supporting information.
), whose volume was ca 166 Å3 in a unit cell of total volume 1272.6 (2) Å3. The void thus occupies ca 13.0% of the total unit-cell volume, and there are a number of significant peaks in the difference map clustered within this void. The largest peak had a magnitude of 4.64 e Å−3 and further examination of this structure using the SQUEEZE procedure (Spek, 2015 ▸) indicated that the void contained around 43 electrons not hitherto accounted for. This number is consistent with the presence of one molecule of dimethyl sulfoxide, but no convincing solvent model could be developed from the difference peaks within the void and hence the reflection data were subjected to the SQUEEZE procedure (Spek, 2015 ▸), and the resultant modified reflection file was used for the refinement reported here. The CIF describing the structure obtained before the SQUEEZE procedure was applied has been included in the supporting information.
Table 1. Experimental details.
For all structures: triclinic, P
. Experiments were carried out at 100 K with Mo Kα radiation using a Bruker D8 Venture diffractometer. Absorption was corrected for by multi-scan methods (SADABS; Bruker, 2016 ▸). H atoms were treated by a mixture of independent and constrained refinement.
| (II) | (III) | (IV) | |
|---|---|---|---|
| Crystal data | |||
| Chemical formula | C18H15ClN6O·H2O | C18H15BrN6O·H2O | C25H18N8O5·C2H6OS |
| M r | 384.83 | 429.29 | 588.60 |
| a, b, c (Å) | 8.2156 (6), 11.0343 (7), 11.3968 (8) | 8.1975 (7), 11.1963 (8), 11.3644 (10) | 9.8192 (8), 10.2765 (7), 14.4096 (11) |
| α, β, γ (°) | 107.980 (2), 109.725 (2), 98.541 (2) | 107.938 (2), 109.866 (3), 98.683 (3) | 71.718 (2), 74.872 (3), 88.786 (3) |
| V (Å3) | 887.43 (11) | 894.39 (13) | 1329.87 (18) |
| Z | 2 | 2 | 2 |
| μ (mm−1) | 0.24 | 2.33 | 0.18 |
| Crystal size (mm) | 0.22 × 0.20 × 0.14 | 0.15 × 0.11 × 0.08 | 0.19 × 0.15 × 0.10 |
| Data collection | |||
| T min, T max | 0.907, 0.967 | 0.719, 0.830 | 0.905, 0.982 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 35335, 4071, 3487 | 50505, 4457, 3696 | 66738, 6115, 4641 |
| R int | 0.053 | 0.073 | 0.112 |
| (sin θ/λ)max (Å−1) | 0.650 | 0.667 | 0.649 |
| Refinement | |||
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.036, 0.088, 1.06 | 0.030, 0.072, 1.09 | 0.050, 0.108, 1.05 |
| No. of reflections | 4071 | 4457 | 6115 |
| No. of parameters | 263 | 263 | 388 |
| Δρmax, Δρmin (e Å−3) | 0.34, −0.32 | 0.49, −0.71 | 0.32, −0.35 |
| (V) | (VIa) | (VIb) | |
|---|---|---|---|
| Crystal data | |||
| Chemical formula | C27H24N6O | C26H21ClN6O | C26H21ClN6O |
| M r | 448.52 | 468.94 | 468.94 |
| a, b, c (Å) | 10.2203 (15), 14.821 (2), 16.594 (2) | 10.2298 (7), 14.8344 (9), 16.5321 (10) | 9.6520 (8), 9.7408 (10), 14.1445 (12) |
| α, β, γ (°) | 99.616 (5), 92.153 (6), 106.083 (5) | 99.672 (2), 92.038 (2), 106.704 (2) | 98.183 (4), 104.638 (3), 90.059 (4) |
| V (Å3) | 2371.9 (6) | 2359.4 (3) | 1272.6 (2) |
| Z | 4 | 4 | 2 |
| μ (mm−1) | 0.08 | 0.19 | 0.18 |
| Crystal size (mm) | 0.25 × 0.22 × 0.12 | 0.18 × 0.13 × 0.11 | 0.12 × 0.09 × 0.08 |
| Data collection | |||
| T min, T max | 0.948, 0.990 | 0.926, 0.979 | 0.916, 0.986 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 116861, 10869, 8351 | 126128, 10821, 8240 | 60695, 5849, 4653 |
| R int | 0.074 | 0.079 | 0.074 |
| (sin θ/λ)max (Å−1) | 0.650 | 0.650 | 0.651 |
| Refinement | |||
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.050, 0.127, 1.06 | 0.058, 0.164, 1.04 | 0.050, 0.115, 1.03 |
| No. of reflections | 10869 | 10821 | 5849 |
| No. of parameters | 631 | 629 | 315 |
| Δρmax, Δρmin (e Å−3) | 0.50, −0.23 | 0.74, −0.61 | 0.34, −0.53 |
Results and discussion
Oxidation of the type (A) precursors having X = Cl or Br gave the products (II) and (III) (see Scheme 1) in exactly the same way as reported previously for the formation of (I) (Vicentes et al., 2019 ▸). The formation of (I)–(III) presumably proceeds via the oxidation of the precursors to form the corresponding Schiff bases, which are hydrolysed to (I)–(III) during the subsequent work-up procedures. Accordingly, the formation of (IV), albeit in low yield, when X = NO2, was unexpected, as it might be expected that this Schiff base would be more susceptible to hydrolysis than those having X = Me, Cl or Br. Condensation of (I) with two representative substituted benzaldehydes gave the required hybrid products (V) and (VI) in yields of 74 and 95%, respectively.
The new compounds (II)–(VI) reported here were all fully characterized by high-resolution mass spectrometry, by IR and 1H and 13C NMR spectroscopy, where the NMR spectra exhibited all of the expected signals, and by single-crystal X-ray diffraction. The crystallographic study confirmed fully the constitutions deduced from the spectra and, in addition, demonstrated the E configuration at the exocyclic C=N double bonds in (IV)–(VI), as well as providing information about the molecular conformations in the solid state and about the supramolecular assembly.
In the synthesis of the type (A) precursors (Vicentes et al., 2019 ▸), the benzimidazole unit was constructed during the synthesis by condensation of an aldehyde with a pyrimidine-substituted benzene-1,2-diamine. The ability to incorporate a variety of substituents into both of these components, as well as into the aldehydes used in the formation of the products (V) and (VI), thus offers the possibility of forming a large library of variants containing multiple and varied substituents.
The intermediates (II) and (III) are isostructural (Table 1 ▸) with the methyl analogue (I) (Vicentes et al., 2019 ▸) and they crystallize as monohydrates (Figs. 1 ▸ and 2 ▸). The product (IV) crystallizes as a stoichiometric solvate with dimethyl sulfoxide (Fig. 3 ▸), but the products (V) and (VIa), which are isostructural, crystallize in the solvent-free form with Z′ = 2 (Figs. 4 ▸ and 5 ▸). The second crystalline form of compound (VI), denoted (VIb) (Fig. 6 ▸), also crystallizes as a solvate, but no coherent model for the disordered solvent could be developed from the peaks in the difference map; accordingly, the SQUEEZE procedure (Spek, 2015 ▸) was applied to the data set for this compound before the final refinements (see Section 2.2). For each of (V) and (VIa), a search for possible additional crystallographic symmetry revealed none; however, the two independent molecules in each of these compounds are related by an approximate, but noncrystallographic, twofold rotation axis (Figs. 4 ▸ and 5 ▸).
Figure 4.

The two independent components in the structure of compound (V), showing the atom-labelling scheme and the hydrogen bonds (drawn as dashed lines) within the selected asymmetric unit. Displacement ellipsoids are drawn at the 50% probability level.
Figure 5.

The two independent components in the structure of the Z′ = 2 form of compound (VI), denoted (VIa), showing the atom-labelling scheme and the hydrogen bonds (drawn as dashed lines) within the selected asymmetric unit. Displacement ellipsoids are drawn at the 50% probability level.
Figure 6.

The molecular structure of the Z′ = 1 form of compound (VI), denoted (VIb), showing the atom-labelling scheme. Displacement ellipsoids are drawn at the 50% probability level.
None of the pyrimidine components in compounds (II)–(VI) exhibits any internal symmetry, as indicated by the key torsion angles (Tables 2 ▸ and 3 ▸), and hence all are conformationally chiral (Moss, 1996 ▸; Flack & Bernardinelli, 1999 ▸), but the space groups (Table 1 ▸) confirm that, in every case, equal numbers of the two conformational enantiomers are present. For each of the products (IV)–(VI), the reference molecules were selected to have a positive sign for the torsion angles Nx3—Cx4—Nx41—Cx42, where x = 1 or 2 for (V) and (VIa), and x = nil for (IV) and (VIb) (Table 3 ▸). On this basis, each product has a negative sign for the torsion angle N3—C4—N41—C47A in (IV) and (VIb) or Nx3—Cx4—Nx41—Cx47 in (V) and (VIa) (see Figs. 3 ▸–6 ▸ ▸ ▸). All of the products have a negative sign for the torsion angle Nx41—Cx42—Cx71—Cx72 and, in each product, the magnitudes of the corresponding torsion angles are very similar (Table 3 ▸). Overall, the products (IV)–(VI) all have very similar molecular structures but their crystallization characteristics are different as noted above and, as discussed below, their supramolecular arrangements are also very different.
Table 2. Selected torsion angles (°) for intermediates (II) and (III).
| Angle | (II) | (III) |
|---|---|---|
| N3—C4—N41—C42 | −59.42 (19) | −59.7 (3) |
| N3—C4—N41—C47A | 104.71 (15) | 103.3 (2) |
| N41—C42—C71—C72 | −34.0 (2) | −33.9 (3) |
| C5—C6—O61—C61 | 177.09 (17) | 177.67 (17) |
Table 3. Selected torsion angles (°) for products (IV)–(VI).
| (IV) | (V) | (V) | (VIa) | (VIa) | (VIb) | |
|---|---|---|---|---|---|---|
| Angle | x = nil | x = 1 | x = 2 | x = 1 | x = 2 | x = nil |
| φ1 | 136.6 (2) | 129.54 (18) | 130.72 (18) | 129.0 (2) | 130.7 (2) | 121.91 (18) |
| φ2 | −58.1 (3) | −68.6 (2) | −70.2 (2) | −69.3 (3) | −72.1 (3) | −62.7 (2) |
| φ3 | −15.6 (3) | −21.6 (3) | −25.7 (2) | −21.4 (3) | −26.0 (3) | −29.4 (3) |
| φ4 | 177.0 (2) | 171.48 (17) | 173.80 (18) | 171.8 (2) | 175.7 (2) | 145.43 (18) |
| φ5 | −4.3 (3) | −2.9 (3) | 3.8 (3) | −4.9 (4) | 0.4 (4) | −3.5 (3) |
| φ6 | −176.38 (18) | −174.4 (2) | 177.12 (17) | 179.8 (3) | 174.8 (2) | −177.71 (17) |
Notes: φ1 represents the torsion angle Nx3—Cx4—Nx41—Cx42; φ2 represents the torsion angles N3—C4—N41—C47A in (IV) and (VIb), and Cx4—Nx41—Nx41—Cx7A in (V) and (VIa) (see Figs. 3 ▸–6 ▸ ▸ ▸); φ3 represents the torsion angle Nx41—Cx42—Cx71—Cx72; φ4 represents the torsion angle Cx4—Cx5—Nx51—Cx57; φ5 represents the torsion angle Nx51—Cx57—Cx51—Cx52; φ6 represents the torsion angle Cx5—Cx6—Ox61—Cx61.
In the intermediates (II) and (III), the signs and magnitudes of the torsion angles N3—C4—N41—C42 and N3—C4—N41—C47A are effectively interchanged compared with the corresponding angles in the products (IV)–(VI) (Tables 2 ▸ and 3 ▸, and Figs. 1 ▸–6 ▸ ▸ ▸ ▸ ▸). In effect, the orientation of the benzimidazole unit in (II) and (III) relative to the pyrimidine differs from that in the products (IV)–(VI) by a rotation of ca 180° about the C—N bond linking these two ring systems. Since the imino atom N43/Nx43 is involved in intermolecular hydrogen bonding in every compound apart from (IV) (Table 4 ▸), it is not easy to understand these orientational differences. In each of compounds (II)–(VI), the methoxy C atom is effectively coplanar with the adjacent pyrimidine ring, as indicated by the torsion angles involving these C atoms (Tables 2 ▸ and 3 ▸).
Table 4. Hydrogen bonds and short intramolecular contacts (Å, °) for compounds (II)–(VI).
Cg1 and Cg2 represent the centroids of the C171–C176 and C271–C276 rings, respectively.
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A | |
|---|---|---|---|---|---|
| (II) | N21—H21A⋯O81 | 0.90 (2) | 2.10 (2) | 2.989 (2) | 171.1 (18) |
| N21—H21B⋯O81i | 0.86 (2) | 2.094 (19) | 2.8840 (18) | 153.0 (18) | |
| N51—H51A⋯O61 | 0.85 (2) | 2.41 (2) | 2.7059 (17) | 101.1 (17) | |
| N51—H51B⋯N41 | 0.90 (2) | 2.571 (19) | 2.888 (2) | 101.5 (14) | |
| O81—H81A⋯N43ii | 0.87 (2) | 1.98 (2) | 2.8506 (19) | 178 (2) | |
| O81—H81B⋯N51iii | 0.84 (2) | 2.11 (2) | 2.927 (2) | 164 (2) | |
| (III) | N21—H21A⋯O81 | 0.83 (3) | 2.17 (3) | 2.992 (3) | 172 (3) |
| N21—H21B⋯O81i | 0.87 (3) | 2.07 (3) | 2.880 (3) | 154 (2) | |
| N51—H51A⋯O61 | 0.91 (3) | 2.41 (3) | 2.706 (2) | 98 (2) | |
| N51—H51B⋯N41 | 0.88 (3) | 2.56 (3) | 2.885 (3) | 103 (2) | |
| O81—H81A⋯N43ii | 0.83 (3) | 2.03 (3) | 2.862 (3) | 178 (4) | |
| O81—H81B⋯N51iii | 0.86 (3) | 2.11 (3) | 2.932 (3) | 159 (3) | |
| (IV) | N21—H21A⋯O81 | 0.89 (3) | 1.94 (3) | 2.826 (3) | 173 (2) |
| N21—H21B⋯N1iv | 0.86 (3) | 2.32 (3) | 3.161 (3) | 166 (3) | |
| (V) | N121—H12A⋯N23 | 0.90 (2) | 2.13 (2) | 3.022 (2) | 171.5 (19) |
| N121—H12B⋯N243v | 0.92 (2) | 2.18 (2) | 3.062 (2) | 160 (2) | |
| N221—H22A⋯N13 | 0.84 (2) | 2.19 (2) | 3.023 (2) | 173.4 (19) | |
| N221—H22B⋯N143ii | 0.90 (2) | 2.11 (2) | 2.994 (2) | 169 (2) | |
| C146—H146⋯N151ii | 0.95 | 2.57 | 3.390 (3) | 145 | |
| C176—H176⋯N21vi | 0.95 | 2.58 | 3.464 (2) | 154 | |
| C155—H155⋯Cg1iv | 0.95 | 2.60 | 3.465 (2) | 151 | |
| C255—H255⋯Cg2vii | 0.95 | 2.87 | 3.784 (2) | 163 | |
| (VIa) | N121—H12A⋯N23 | 0.86 (3) | 2.18 (3) | 3.016 (3) | 168 (3) |
| N121—H12B⋯N243v | 0.89 (3) | 2.21 (3) | 3.043 (3) | 158 (3) | |
| N221—H22A⋯N13 | 0.78 (3) | 2.23 (3) | 3.012 (3) | 173 (3) | |
| N221—H22B⋯N143ii | 0.82 (3) | 2.18 (3) | 2.988 (3) | 168 (3) | |
| C146—H146⋯N151ii | 0.95 | 2.58 | 3.404 (4) | 146 | |
| C176—H176⋯N21vi | 0.95 | 2.57 | 3.449 (3) | 154 | |
| C155—H155⋯Cg1iv | 0.95 | 2.55 | 3.391 (3) | 147 | |
| C255—H255⋯Cg2vii | 0.95 | 2.85 | 3.754 (3) | 160 | |
| (VIb) | N21—H21A⋯N3vi | 0.85 (2) | 2.35 (2) | 3.196 (2) | 175.9 (19) |
| N21—H21B⋯N43viii | 0.90 (2) | 2.08 (2) | 2.946 (2) | 163.6 (19) |
Symmetry codes: (i) −x + 1, −y + 1, −z; (ii) −x + 1, −y + 1, −z + 1; (iii) −x + 1, −y, −z; (iv) −x + 1, −y + 2, −z + 1; (v) −x + 1, −y + 1, −z + 2; (vi) −x, −y + 1, −z + 1; (vii) −x, −y, −z + 2.
The intermediates (II) and (III) are isostructural with (I) (Vicentes et al., 2019 ▸), and thus exhibit the same pattern of supramolecular assembly, forming complex sheets built from a combination of O—H⋯N and N—H⋯O hydrogen bonds. No additional comment is required except to note that the structure of compound (III) contains a fairly short intermolecular Br⋯O contact whose dimensions are Br74⋯O61i = 3.0972 (16) Å and C74—Br74⋯O61i = 173.70 (9)° [symmetry code: (i) x, y − 1, z], so that the Br⋯O distance is shorter than the sum of the conventional van der Waals radii of 3.41 Å (Rowland & Taylor, 1996 ▸). However, the conventional radii are derived assuming no directional variation in the effective van der Waals radius, but detailed database analysis (Nyburg & Faerman, 1985 ▸) for nonbonded contacts involving halogen atoms bonded to C atoms indicates significant angular variation, with the effective radii diminishing as the contact angle approaches 180°, as here. On this basis, the sum of the effective van der Waals radii, 3.08 Å, differs little from the distance observed here, so that this contact in compound (III) should not be regarded as structurally significant.
For the product (IV), the supramolecular assembly is very simple: inversion-related pyrimidine components are linked by N—H⋯N hydrogen bonds to form a cyclic centrosymmtric
 (8) dimer, to which inversion-related solvent molecules are linked by N—H⋯O hydrogen bonds (Fig. 7 ▸). There are no direction-specific interactions between the four-molecule aggregates of this type.
(8) dimer, to which inversion-related solvent molecules are linked by N—H⋯O hydrogen bonds (Fig. 7 ▸). There are no direction-specific interactions between the four-molecule aggregates of this type.
Figure 7.
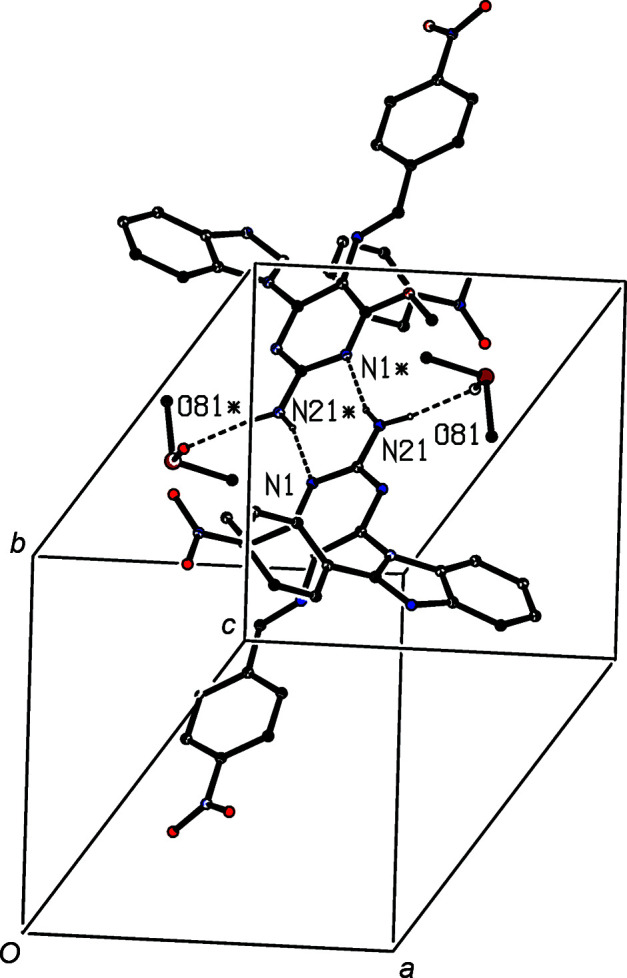
Part of the crystal structure of compound (IV), showing the formation of a centrosymmetric four-molecule aggregate built from N—H⋯O and N—H⋯N hydrogen bonds, which are drawn as dashed lines. For the sake of clarity, H atoms which are not involved in the motifs shown have been omitted. Atoms marked with an asterisk (*) are at the symmetry position (−x + 1, −y + 2, −z + 1).
In the isostructural products (V) and (VIa), there are eight independent hydrogen bonds, four of the N—H⋯N type and two each of the C—H⋯N and C—H⋯π(arene) types (Table 4 ▸), which together link the molecules into three-dimensional framework structures. The differences in the details of the C—H⋯N and C—H⋯π(arene) hydrogen bonds involving the two independent molecules confirms the lack of additional crystallographic symmetry. In the selected asymmetric units (Figs. 4 ▸ and 5 ▸), the two molecules are linked by N—H⋯N hydrogen bonds, and these dimeric units can be regarded as the basic building block for the three-dimensional assembly, which is readily analysed in terms of simple one-dimensional substructures (Ferguson et al., 1998a
▸,b
▸; Gregson et al., 2000 ▸). Two N—H⋯N hydrogen bonds, having atoms N143 and N243 as the acceptors (Table 4 ▸), link the basic dimers into a ribbon of alternating
(8) and
 (16) rings running parallel to the [101] direction (Fig. 8 ▸). In the second substructure, the linking of the basic dimeric units by the two C—H⋯N hydrogen bonds generates a ribbon of alternating
(8) and
(16) rings running parallel to the [100] direction (Fig. 9 ▸). In the final substructure, the linkage of the dimers by two C—H⋯π(arene) hydrogen bonds generates a chain of rings running parallel to the [12
] direction (Fig. 10 ▸). The combination of the chain motifs along [100], [101] and [12
] suffices to generate a three-dimensional framework structure.
(16) rings running parallel to the [101] direction (Fig. 8 ▸). In the second substructure, the linking of the basic dimeric units by the two C—H⋯N hydrogen bonds generates a ribbon of alternating
(8) and
(16) rings running parallel to the [100] direction (Fig. 9 ▸). In the final substructure, the linkage of the dimers by two C—H⋯π(arene) hydrogen bonds generates a chain of rings running parallel to the [12
] direction (Fig. 10 ▸). The combination of the chain motifs along [100], [101] and [12
] suffices to generate a three-dimensional framework structure.
Figure 8.

Part of the crystal structure of compound (V), showing the formation of a ribbon of alternating
(8) and
(16) rings running parallel to the [101] direction. Hydrogen bonds are drawn as dashed lines and, for the sake of clarity, H atoms bonded to C atoms have all been omitted.
Figure 9.

Part of the crystal structure of compound (V), showing the formation of a ribbon of alternating
(8) and
(16) rings running parallel to the [100] direction. Hydrogen bonds are drawn as dashed lines and, for the sake of clarity, H atoms bonded to C atoms but not involved in the motif shown have been omitted.
Figure 10.

Part of the crystal structure of compound (V), showing the formation of a chain of rings along [12
] built from N—H⋯N and C—H⋯π(arene) hydrogen bonds, which are drawn as dashed lines. For the sake of clarity, H atoms bonded to C atoms but not involved in the motif shown have been omitted.
Two N—H⋯ N hydrogen bonds link the molecules of (VIb) into a ribbon of edge-fused centrosymmetric rings running parallel to [100], in which
(8) rings (Etter, 1990 ▸; Etter et al., 1990 ▸; Bernstein et al., 1995 ▸) centred at (n,
,
) alternate with
(16) rings centred at (n +
,
,
) (Fig. 11 ▸), where n represents an integer in each case.
Figure 11.

Part of the crystal structure of form (VIb), showing the formation of a ribbon of centrosymmetric
(8) and
(16) rings running parallel to [100]. Hydrogen bonds are drawn as dashed lines and, for the sake of clarity, H atoms bonded to C atoms have all been omitted.
We have previously reported the structures of a wide range of multiply-substituted pyrimidines, but many of these carry either C-nitroso (Quesada et al., 2002 ▸, 2004 ▸; Melguizo et al., 2003 ▸) or C-formyl substituents (Cobo et al., 2008 ▸), whose presence is associated with highly polarized electronic structures.
Summary
We have developed a versatile and efficient synthesis of 5-(arylmethylideneamino)-4-(1H-benzo[d]imidazol-1-yl)pyrimidine hybrids based on simple starting materials and we have characterized three products and two intermediates spectroscopically (IR, 1H and 13C NMR, and HRMS) and have determined their molecular and supramolecular structures.
Supplementary Material
Crystal structure: contains datablock(s) global, II, III, IV, V, VIa, VIb. DOI: 10.1107/S2053229623003728/dv3022sup1.cif
Structure factors: contains datablock(s) II. DOI: 10.1107/S2053229623003728/dv3022IIsup2.hkl
Structure factors: contains datablock(s) III. DOI: 10.1107/S2053229623003728/dv3022IIIsup3.hkl
Structure factors: contains datablock(s) IV. DOI: 10.1107/S2053229623003728/dv3022IVsup4.hkl
Structure factors: contains datablock(s) V. DOI: 10.1107/S2053229623003728/dv3022Vsup5.hkl
Structure factors: contains datablock(s) VIa. DOI: 10.1107/S2053229623003728/dv3022VIasup6.hkl
Structure factors: contains datablock(s) VIb. DOI: 10.1107/S2053229623003728/dv3022VIbsup7.hkl
Supporting information file. DOI: 10.1107/S2053229623003728/dv3022IIsup8.cml
Supporting information file. DOI: 10.1107/S2053229623003728/dv3022IIIsup9.cml
Supporting information file. DOI: 10.1107/S2053229623003728/dv3022IVsup10.cml
Supporting information file. DOI: 10.1107/S2053229623003728/dv3022Vsup11.cml
Supporting information file. DOI: 10.1107/S2053229623003728/dv3022VIasup12.cml
CIF describing the structure of (VI obtained before the SQUEEZE procedure was applied. DOI: 10.1107/S2053229623003728/dv3022sup13.txt
Acknowledgments
The authors thank Centro de Instrumentación Científico-Técnica of Universidad de Jaén for data collection. DV thanks Asociación Universitaria Iberoamericana de Postgrado (AUIP) for a Scholar-Fellowship and acknowledges financial support from the Universidad de Ciencias Aplicadas y Ambientales (UDCA). JC is grateful for financial support from the Spanish Ministerio de Ciencia, Innovacíon, y Universidades, the Universidad de Jaén, Vicerrectorado de Investigación, PAIUJA Acción 1 plan 2019–2020 and 2021–2022, and Consejería de Innovación, Ciencia y Empresa (Junta de Andalucía, Spain).
Funding Statement
Funding for this research was provided by: Spanish Ministerio de Ciencia, Innovacíon, y Universidades (R&D project No. RTI2018-098560-B-C22), co-financed by the FEDER funds of the European Union.
References
- Abdul-Rida, N. A., Mohammed, T. I., Al-Masoudi, N. A. & Frotscher, M. (2017). Med. Chem. Res. 26, 830–840.
- Al-Masoudi, N. A., March, Y. A., Al-Ameri, J. J., Ali, D. S. & Pannecouque, C. (2016). Chem. Biol. Interface, 6, 69–73.
- Bernstein, J., Davis, R. E., Shimoni, L. & Chang, N.-L. (1995). Angew. Chem. Int. Ed. Engl. 34, 1555–1573.
- Bruker (2016). SADABS. Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruker (2017). SAINT. Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruker (2018). APEX3. Bruker AXS Inc., Madison, Wisconsin, USA.
- Cobo, J., Trilleras, J., Quiroga, J., Marchal, A., Nogueras, M., Low, J. N. & Glidewell, C. (2008). Acta Cryst. B64, 596–609. [DOI] [PubMed]
- Cortese, D., Chegaev, K., Guglielmo, S., Wang, L. Z., Golding, B. T., Cano, C. & Fruttero, R. (2016). ChemMedChem, 11, 1705–1708. [DOI] [PubMed]
- El-Gohary, N. S. & Shaaban, M. I. (2017). Eur. J. Med. Chem. 131, 255–262. [DOI] [PubMed]
- Etter, M. C. (1990). Acc. Chem. Res. 23, 120–126.
- Etter, M. C., MacDonald, J. C. & Bernstein, J. (1990). Acta Cryst. B46, 256–262. [DOI] [PubMed]
- Ferguson, G., Glidewell, C., Gregson, R. M. & Meehan, P. R. (1998a). Acta Cryst. B54, 129–138.
- Ferguson, G., Glidewell, C., Gregson, R. M. & Meehan, P. R. (1998b). Acta Cryst. B54, 139–150.
- Flack, H. D. & Bernardinelli, G. (1999). Acta Cryst. A55, 908–915. [DOI] [PubMed]
- Gadde, S., Kleynhans, A., Holien, J. K., Bhadbhade, M., Nguyen, P. L. D., Mittra, R., Yu, T. T., Carter, D. R., Parker, M. W., Marshall, G. M., Cheung, B. B. & Kumar, N. (2023). Bioorg. Chem. 136, 106462. [DOI] [PubMed]
- Gregson, R. M., Glidewell, C., Ferguson, G. & Lough, A. J. (2000). Acta Cryst. B56, 39–57. [DOI] [PubMed]
- Ismail, M. M. F., El-Sehrawi, H., Elzahabi, H. S. A., Shawer, T. & Ammar, Y. A. (2022). Polycyclic Aromat. Compd. 42, 2363–2377.
- Jadhav, M., Sankhe, K., Bhandare, R. R., Edis, Z., Bloukh, S. H. & Khan, T. A. (2021). Molecules, 26, 5170. [DOI] [PMC free article] [PubMed]
- Koroleva, E. V., Gusak, K. N. & Ignatovich, Z. V. (2010). Russ. Chem. Rev. 79, 655–681.
- Loving, K., Salam, N. K. & Sherman, W. (2009). J. Comput. Aided Mol. Des. 23, 541–554. [DOI] [PubMed]
- Melguizo, M., Quesada, A., Low, J. N. & Glidewell, C. (2003). Acta Cryst. B59, 263–276. [DOI] [PubMed]
- Moss, G. P. (1996). Pure Appl. Chem. 68, 2193–2222.
- Nyburg, S. C. & Faerman, C. H. (1985). Acta Cryst. B41, 274–279.
- Quesada, A., Marchal, A., Melguizo, M., Low, J. N. & Glidewell, C. (2004). Acta Cryst. B60, 76–89. [DOI] [PubMed]
- Quesada, A., Marchal, A., Melguizo, M., Nogueras, M., Sánchez, A., Low, J. N., Cannon, D., Farrell, D. M. M. & Glidewell, C. (2002). Acta Cryst. B58, 300–315. [DOI] [PubMed]
- Rowland, R. S. & Taylor, R. (1996). J. Phys. Chem. 100, 7384–7391.
- Sana, S., Reddy, V. G., Srinivasa Reddy, T., Tokala, R., Kumar, R., Bhargava, S. K. & Shankaraiah, N. (2021). Bioorg. Chem. 110, 104765. [DOI] [PubMed]
- Sheldrick, G. M. (2015a). Acta Cryst. A71, 3–8.
- Sheldrick, G. M. (2015b). Acta Cryst. C71, 3–8.
- Singh, G., Kaur, M. & Chander, M. (2013). Int. Res. J. Pharm. 4, 82–87.
- Spek, A. L. (2015). Acta Cryst. C71, 9–18. [DOI] [PubMed]
- Spek, A. L. (2020). Acta Cryst. E76, 1–11. [DOI] [PMC free article] [PubMed]
- Vicentes, D. E., Rodríguez, R., Ochoa, P., Cobo, J. & Glidewell, C. (2019). Acta Cryst. C75, 1405–1416. [DOI] [PubMed]
- Wu, K., Peng, X., Chen, M., Li, Y., Tang, G., Peng, J., Peng, Y. & Cao, X. (2022). Chem. Biol. Drug Des. 99, 736–757. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, II, III, IV, V, VIa, VIb. DOI: 10.1107/S2053229623003728/dv3022sup1.cif
Structure factors: contains datablock(s) II. DOI: 10.1107/S2053229623003728/dv3022IIsup2.hkl
Structure factors: contains datablock(s) III. DOI: 10.1107/S2053229623003728/dv3022IIIsup3.hkl
Structure factors: contains datablock(s) IV. DOI: 10.1107/S2053229623003728/dv3022IVsup4.hkl
Structure factors: contains datablock(s) V. DOI: 10.1107/S2053229623003728/dv3022Vsup5.hkl
Structure factors: contains datablock(s) VIa. DOI: 10.1107/S2053229623003728/dv3022VIasup6.hkl
Structure factors: contains datablock(s) VIb. DOI: 10.1107/S2053229623003728/dv3022VIbsup7.hkl
Supporting information file. DOI: 10.1107/S2053229623003728/dv3022IIsup8.cml
Supporting information file. DOI: 10.1107/S2053229623003728/dv3022IIIsup9.cml
Supporting information file. DOI: 10.1107/S2053229623003728/dv3022IVsup10.cml
Supporting information file. DOI: 10.1107/S2053229623003728/dv3022Vsup11.cml
Supporting information file. DOI: 10.1107/S2053229623003728/dv3022VIasup12.cml
CIF describing the structure of (VI obtained before the SQUEEZE procedure was applied. DOI: 10.1107/S2053229623003728/dv3022sup13.txt