

Abstract
The design of PROteolysis TArgeting Chimeras (PROTACs) requires bringing an E3 ligase into proximity with a target protein, to modulate the concentration of the latter through its ubiquitination and degradation. Here we present a method for generating high-accuracy structural models of E3 ligase-PROTAC-target protein ternary complexes. The method is dependent on two computational innovations; adding a “silent” convolution term to an efficient protein-protein docking program to eliminate protein poses that do not have acceptable linker conformations, and clustering models of multiple PROTACs that use the same E3 ligase and target the same protein. Results show that the largest consensus clusters always have high predictive accuracy, and that the ensemble of models can be used to predict the dissociation rate and cooperativity of the ternary complex that relate to the degrading activity of the PROTAC. The method is demonstrated by applications to known PROTAC structures, and a blind test involving PROTACs against BRAF mutant V600E. The results confirm that PROTACS function by stabilizing a favorable interaction between the E3 ligase and the target protein, but do not necessarily exploit the most energetically favorable geometry for interaction between the proteins.
Keywords: Protein degradation, protein-protein interaction, conformation generator, linker design, structure prediction, drug design
Graphical Abstract
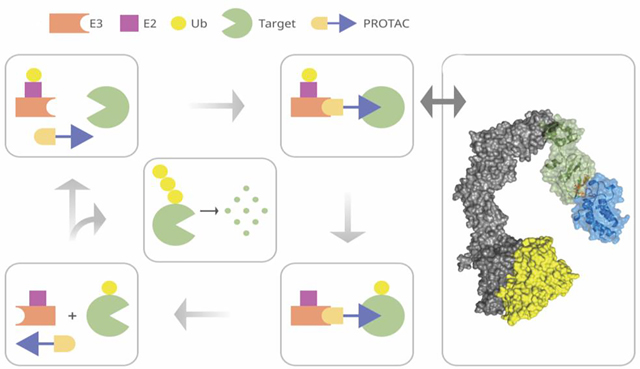
Introduction
Proteolysis-targeting chimeras (PROTACs) represent an emerging therapeutic technology that fundamentally differs from the conventional occupancy-driven pharmacology of traditional small molecule inhibitors, and provides a tool to target proteins that have been considered undruggable. 1, 2 PROTACs are heterobifunctional compounds consisting of two ligands connected by a linker. One of the ligands, sometimes called the “warhead”. 3, 4 binds to the target protein, and the other ligand binds to and recruits an E3 ligase. The goal of this compound is to mediate formation of a ternary complex, leading to the ubiquitination of the target protein. It is expected that such hijacking of the ubiquitin proteasome system, which is a natural pathway for protein degradation in eukaryotic cells, will initiate the degradation of the target protein. It was shown that forming stable, long-lived ternary complexes can be very important to drive faster, more potent degradation.3, 5, 6 The idea is two decades old, 7 but more recently it has been used in an increasing number of applications. 2, 8, 9 To date, there are hundreds of reports describing the use of PROTACs for targeted protein degradation and their utility in chemical biology and drug discovery.10
There are approximately 600 E3 ligases in human cells, although so far only a few have been used in PROTAC implementation studies.4, 10 The two most commonly employed E3 ligases and their substrate recognition domains are the Cullin 2 E3 ligase complex/Von-Hippel Lindau (VHL) pair 11 and the Cullin 4a E3 ligase/Cereblon (CRBN) pair.12 Many PROTAC molecules have been developed to recruit these E3 ligases to a variety of substrates using high-affinity ligands for the target 4, 10. Since in most cases the small-molecule-induced protein degradation benefits from the ligand-mediated binding of two proteins that have not evolved to interact, the design of such compounds is challenging, and remains a largely empirical process in which molecules for new targets frequently fail.13, 14 Factors responsible for this variability in outcome likely include differences in the stability of the E3 ligase-PROTAC-target protein ternary complex, and whether the ternary complex achieves an appropriate relative orientation of the ligase to a site on the target protein that can be ubiquitinated. Thus, while factors such as optimizing the kinetics of degradation may remain empirical, the ability to accurately predict the structure and stability of the ternary complex would be useful for improved PROTAC design.4
The conformation of the ternary complex is described in a high dimensional space, including the rotation and translation of one protein relative to the other, the internal coordinates of the linker, and potential changes in the conformations of the protein side chains upon complex formation. It has been shown that in most cases the interaction between the E3 ligase and the target protein yields major contribution to the binding free energy of the ternary complex.15 and that binding of the three main components is generally cooperative.5 Direct search in this high dimensional space has been considered computationally ineffective, and therefore different ways to partition the space for sampling have been suggested 16. Suggestions included sampling the PROTAC conformations independently, followed by post hoc addition of rigid-body proteins; sampling the PROTAC in the context of one of the proteins, with the second protein added afterward; or sampling PROTAC conformations but adding possible E3 ligase - target protein arrangements via protein−protein docking.16 Recent experience from several groups indicates that a meaningful first step is finding energetically favorable interactions between the target protein and the E3 ligase 15, 17, 18. Providing early insight, Nowak et al. 15 used the RosettaDock 19 program to generate 20,000 docked structures between CRBN and the binding domain 1 (BD1) of the bromodomain BRD4, and among the 200 lowest energy conformations they identified a conformation that closely resembled the one observed in the crystal structure of the complex. The authors also calculated the pairwise shortest distances between the selected solvent-exposed atoms of the BRD4 ligand JQ1 and the CRBN ligand lenalidomide for the top 200 poses, and used the information to design PROTACs with short linkers.15
Recent computational studies have furthered the idea of separately sampling the protein-protein and the linker conformations,17, 18 while restricting the former using some properties of the linker. Zaidman et al.17 generated an ensemble of linker conformations to determine distances to be used as restraints in rigid body docking, applied local docking refinement, and finally concatenated these pieces to form geometrically and energetically acceptable structures for the ternary complex. They applied the method to a set of known PROTAC ternary complexes, in each case starting from the protein structures extracted from the E3 ligase-PROTAC-target protein complex. The authors admitted that the use of such pre-formed protein structures was crucial, and the method did not work when starting from the separately crystallized structures of the component proteins. In contrast, Bai et al.18 considered such separate structures as starting points for model building, oriented the two proteins to position the ligand binding sites toward each other, and used local docking to generate a large number of low energy poses. Following the same approach as Zaidman et al.,17 they generated an ensemble of candidate linker conformations separately, paired the protein-protein poses with compatible linker structures, and refined the resulting models by energy minimization. For any given target and E3 ligase this protocol resulted in many structures that equally satisfied the geometric conditions on the linker and were not distinguishable based on the calculated energy values. Based on this result, Bai et al 18 assumed that predicting a unique structure of a PROTACs may not be an attainable goal, and stated the opinion that the complexes may not even have a unique conformation. Therefore, no attempts were made to model the available X-ray structures of PROTAC-containing ternary complexes. However, they suggested that the population of geometrically and energetically acceptable solutions for a given PROTAC can be used for predicting its effective degradation capability when comparing different linkers for a given E3 ligase and target pair, since larger population generally indicated a more stable ternary complex and a more active PROTAC. While this is a useful result, it appears that none of the methods developed so far is capable of predicting accurate conformations of ternary complexes.
In this paper we go beyond what has been done so far, and show that PROTAC ternary complex structures can in fact be computationally predicted reliably and with high accuracy. This result is based on a new approach that can sample protein-protein and linker conformational space in a single step. The idea involves notionally separating the candidate PROTAC molecule into two pieces, each containing either the warhead or the ligase-binding ligand plus half of the linker atoms. For each protein bound to its cognate ligand we then generate a large ensemble (“cloud”) of half-linker conformations, avoiding clashes with the protein. Using a modified version of an extremely efficient protein-protein docking algorithms based on fast Fourier transforms 20–23 we then directly sample conformations that have a favorable relative orientation of the two proteins and in which the end points of the half-linkers are close to each other. After local refinement, each resulting conformation yields a feasible structure of the ternary complex, including the linker. Our main result is that, for each ternary complex tested, it is always sufficient to consider just a small number of models to identify one or more with an smRMSD of less than 3 Å compared to the corresponding experimental ternary complex structure. Here smRMSD (small molecular RMSD) is defined as the root mean square deviation between the experimental and predicted structures of the protein-bound ligands. We note that smRMSD can be reliably calculated even for low resolution ternary complex structures (see Methods). To reduce the number of potential structures, we use additional information based on the mechanistic knowledge that the ubiquitination reaction requires the E2 Cys and a surface exposed Lys residue of the target protein to be within 50–60 Å to enable the transfer of Ub to the target.24 In addition, we show that the ranking of the models generated by the above algorithm can be much improved if structures are predicted for several PROTACs that connect a given E3 ligase with the same target protein
For the design of a PROTAC it would be important to predict the target degrading capability. We followed the approach suggested by Bai et al.18 and calculated a measure based on the number of acceptable models. As will be described, we were able to predict the dissociation constant Kd and cooperativity α of E3 ligase-PROTAC-target protein ternary complexes. Although these also affect degradation efficiency, the prediction of the later turned out to be qualitative rather than quantitative. The method was tested against retrospective examples of PROTACs for which ternary complex structures or activities have been published, by building structural models and predicting the PROTAC’s degrading activity which we compare against experimentally determined values. We also report results of performing blind prediction for a set of PROTACs targeted against the BRAF V600E mutant.
Results
Overall modeling strategy.
Fig. 1 illustrates the main steps of the protocol. Our modeling of a ternary PROTAC complex is based on the separately crystallized structures of the target protein and of an E3 ubiquitin ligase (E3), both provided as PDB files, together with the chemical structure of the proposed PROTAC provided as a SMILES string. Since PROTACs are most often built using well-characterized warheads, the structure and the position of the ligand bound to the target protein is typically known a priori. Crystal structures of widely used E3-recruiting ligands have also been solved in complex with their cognate E3 ligases. The interactions of each ligand within its respective binding site are unlikely to change in the context of the PROTAC ternary complex. Nevertheless, as shown in Fig. 1A, assuming that future PROTACs may involve novel warheads and warhead-binding sites, for generality the first step of the proposed protocol is docking the small ligands. The next step involves generating 10,000 conformations of the PROTAC linker separately from any protein, and then separating each linker structure into two halves, one attached to the ligase-binding warhead and the other to the to the target-binding ligand. The ensembles of half-linker conformations form two “half-linker clouds” (Fig. 1B). Linker conformations clashing with the protein are removed.
Figure 1.
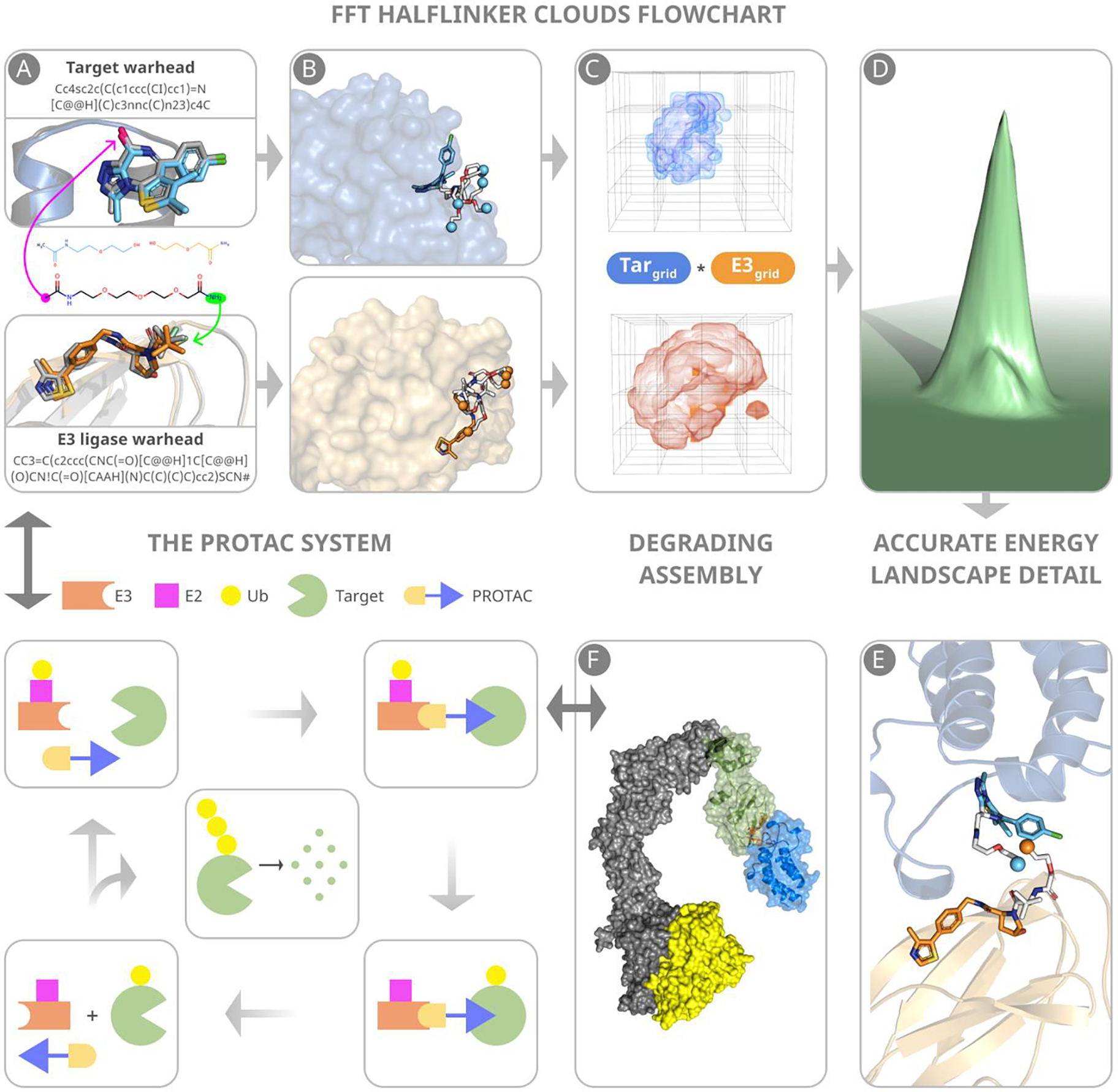
Main steps of predicting PROTAC structures. (A) Warheads are docked to component proteins. Green and magenta arrows indicate attachment points to the E3 ligase and target warheads, respectively. (B) Half-linker conformations are generated and attached to each protein-bound warhead. The small colored spheres represent the half-linker end points. In the VHL:MZ1:BRD4BD2 (PDB ID: 5T35) complex shown, the BRD4 warhead and the end points of the attached half-linkers are depicted in cyan, and the VHL warhead and the end points of the attached half-linkers are in orange. (C) Generating favorable protein-protein poses that have half-linker ends placed sufficiently close to each other. (D) Selection of low energy poses. (E) A resulting pose with half-linker end points in close proximity before connecting the half-linkers. (F) The VHL–ElonginC–ElonginB–Cul2–Rbx1 structural assembly with the CRL2VHL complex shown in blue. The assembly includes a ubiquitin-like protein (ULP), shown in yellow, separated by a favorable distance to facilitate transfer of ubiquitin to the target. The figure also include a schematic outline of the PROTAC system.
The main and most innovative step of the new method is an extension of the fast Fourier based sampling algorithm,20, 22 which uses an additional convolution term to generate only protein-protein complex conformations that also have half-linker ends placed close to each other. Identification of a complex between the E3 ligase and the target protein containing bound ligands with half-linkers that meet at their respective ends indicates that the corresponding PROTAC can productively engage both the target protein and E3 ligase (Fig. 1C). Thus, when used to dock together the two proteins with their bound warheads and half-linkers, this grid-based rigid body search returns ternary complexes with both low energy poses of the two proteins (Fig. 1D) and a potentially viable linker geometry based on the close proximity of the half-linker end points (Fig. 1E). As will be described, the poses generated are then tested to assure ubiquitin accessibility, the half-linker end points are connected, and the retained structures are refined by energy minimization and then clustered to form models of the complete ternary complex.
The flowchart of the protocol in Supplementary Figure 1 shows that the calculations were designed to accomplish two tasks: accurate modeling of a PROTAC ternary complex, and determining the number of sterically and energetically acceptable models. We show that the latter can be used to predict the dissociation kinetics and cooperativity of the target-PROTAC-E3 ligase ternary complex and, to some degree, the expected degradation efficiency of the PROTAC. However, the focus of this paper is the method of predicting the structure of ternary complexes, and the method for the second application needs substantial further development. Note that this latter application does not necessarily require the prediction of a unique structure, but we will argue that information on the likely structure of the protein complex can be useful for improving linker design.
Benchmark set for PROTAC structure prediction.
We explored structure prediction for PROTAC complexes with one or more X-ray structures of the intact E3 ligase-PROTAC-target protein ternary complex available in the PDB, totaling twelve X-ray structures. As shown in Supplementary Table 1, five complexes involve Cereblon (CRBN) as the E3 ligase and the bromodomain-containing protein 4 binding domain 1 (BRD4 BD1) of the Bromodomain and Extra Terminal (BET) family as the target protein. These PROTACs have been named dBET6, dBET23, dBET55, dBET70, and dBET57.15 One complex, with the PROTAC MZ1, uses the von Hippel-Lindau (VHL) E3 ligase to target the binding domain 2 of BRD4 (BRD4 BD2) 5. The next three structures in the table also use VHL to target the BAF ATPase subunits SMARCA2 and SMARCA4, with two compounds, PROTAC1 and PROTAC2, co-crystallized with VHL and SMARCA2, and PROTAC2 also interacting with VHL and SMARCA4 25. The targets in the last four structures in the table are BRD4 BD1,26, 27 focal adhesion kinase (FAK),28 \WD40 repeat domain protein 5 (WDR5),29 and Bcl-xL.30
Fast Fourier transform based sampling with “half-linker clouds”.
As mentioned, the key step in PROTAC design is determining favorable interactions between the E3-warhead complex and the target-warhead complex in the presence of the linker (Fig. 1C). Previous methods developed for the modeling of PROTACs accounted for linker conformation simply as a distance restraint in sampling the protein-protein conformational space 17, or performed local sampling of the latter space starting from a pose with the ligand binding sites facing each other 18. In both cases the sampling had to be followed by searching for compatible members in the set of pre-generated linker conformations for each low energy protein-protein pose. Our new method performs both searches in a single step. We also start the protocol by generating a set of linker conformations, but divide each structure into two halves, attach each half to the respective ligand bound to the E3 ligase and the target protein, and search for favorable poses of the two proteins with their “half-linker clouds” already attached. Supplementary Table 2 shows the PROTACs with the half-linkers developed to target the proteins listed in Supplementary Table 1. This computation is accomplished by a modified version of the program PIPER 23, which is also implemented in our heavily used protein docking server ClusPro.31 PIPER is based on the extremely efficient fast Fourier transform (FFT) correlation approach, which enables dense systematic sampling of the conformational space defined by the relative orientations of the two proteins in the 6D space of rotations and translations. The scoring function of PIPER is a weighted sum of convolutions between protein grids, representing van der Waals, electrostatic, and desolvation energy terms 31. To account for the ‘half-linker clouds” we introduce an additional “silent” convolution, which does not contribute to the energy score, but its values are used for filtering docking poses. The grids of this convolution are indicator functions of the proximity of midway atoms of the non-clashing linker conformations. Namely, if no half-linker conformations in the cloud satisfy the requirement of mid-point proximity, then the corresponding protein poses are removed (see Methods). The 1000 lowest energy docked structures, each with a feasible linker conformation already determined, are retained for further analysis.
In Supplementary Fig. 2 we demonstrate the advantages provided by this approach by comparing the results of numerical experiments using three different methods for the CRBN–dBET6–BRD4 complex. The first method is global sampling of the protein-protein interaction space without any restraints, the second uses the maximum length of the linker as a distance restraint in the sampling, and the third involves the “half-linker cloud” approach which explores the available conformational space accounting for the potential linker geometry. These calculations yield two important results. First, the relative orientation of the two proteins in the PROTAC ternary complex is not at the global minimum of the protein-protein interaction energy. Thus, docking the proteins first and then adding the PROTAC molecule to the model would not have given a correct geometry for the ternary complex as a whole. Second, using only distance as a restraint, the sampling may yield plentiful conformations that satisfy the condition of the linker length. However, as shown in Supplementary Figure 2C, such conformations include false positives, since they cannot be obtained with realistic linker geometry. The proposed half-linker approach eliminates both of these shortcomings. As emphasized by the results shown in Supplementary Figure 2, the relative orientation between the E3 ligase and the target protein in the ternary complex is not at the global energy minimum that the two proteins would adopt when interacting without the PROTAC.
Additional filtering of poses to assure ubiquitin accessibility.
Each of the top 1000 docking poses produced by the fast Fourier search is evaluated to determine whether productive ubiquitination can occur. Targeted protein degradation involving either the VHL or the CRBN E3 ligases requires that the Cys residue of the E2 subunit of the ligase complex be between 50 and 60 Å of a surface Lys residue on the target protein (12) in order to facilitate the transfer of Ub to the target (9). This condition eliminates some poses that otherwise would be favorable. To check the Cys-to-Lys distances we construct the CRL4CRBN (CRBN–Cul4–Rbx1) (Supplementary Fig. 3A), and the CRL2VHL (VHL–ElonginC–ElonginB–Cul2–Rbx1) (Supplementary Fig. 3B) assemblies and determine the position of the E2 ligase with respect to the target protein (see Methods). For each pose we test whether at least one surface exposed Lys residue on the target protein is located within 60 Å of the Cys residue of the E2 ligase, and remove the structures that fail to meet this condition. We have found that the condition is always satisfied for complexes with CRBN, but only for some of the complexes that have VHL as the E3 ligase.
Refinement by energy minimization, clustering, and ranking.
For each pose retained from the previous stage the two half-linkers are connected by defining the same starting coordinates for some atom in the residue at the middle of the chain (Fig. 1E). Models with a bond angle less than 90° at the connection point are removed. For the remaining poses, the connected halves are clustered with the radius of 3 Å (linker atoms only), and the cluster centroids are used to build models. The models are refined using molecular mechanics energy minimization in which the two protein poses are fixed but the PROTAC is considered flexible. For each minimized model we evaluate the energy of the isolated PROTAC and the RMSDs of the two ligands, bound to their respective proteins in the complex, relative to their coordinates before minimization. Models that result in any of the two ligands shifting by more than 1.5 Å (heavy atom) RMSD during minimization are removed, thereby avoiding models that would produce strained PROTAC conformations. We also take into account the self-energy of the linker by keeping only the lowest energy 25% of the models. The models retained after these filters are clustered with 3 Å pairwise RMSD of the target ligands as the clustering radius (the E3 ligase, with its bound ligand, is kept fixed). The centroids of the 10 largest clusters are selected to form the final models that are ranked based on the energy of the linker. As shown in Supplementary Figure 2C, in the case of Cereblon the thalidomide ligand can flip around a rotatable bond, resulting in two opposite linker attachment locations. Based on this possibility, we generated 10 models for each orientation. The final set was produced by mixing the 20 models, and selecting the 10 with the lowest energy of the linker.
Application to a PROTAC benchmark set.
Supplementary Table 3 shows the smRMSD values of the 10 models generated by the proposed method for the thirteen PROTAC ternary complexes in the benchmark set. As we discussed, the smRMSD values are calculated for the ligands on the two ends of the PROTACs, and the models are ranked on the basis of the energy of the linker. In each case, the RMSD of at least one prediction is less than 3 Å, but the lowest RMSD is not necessarily achieved for the best ranked model. For example, Fig. 2A and Fig. 2B, respectively, show one of the fairly good models (model 3) of the CRBN–dBET23–BRD4 BD1 complex (blue), superimposed on its X-ray structure (orange, PDB ID: 6BN7), and a good model (model 5) of the CRBN–dBET6–BRD4 BD1 complex (blue), superimposed on its X-ray structure (orange, PDB ID: 6BOY). As shown, the predicted linker conformations may deviate from the ones in the X-ray structures, but the bound ligand positions are generally well predicted. For one of the PROTAC complexes, targeting Bcl-xL described by Chung et al.30 our algorithm failed to generate near-native complex conformations. The best compound, PROTAC6, had a very long polyethylene glycol linker that collapsed to a globular structure to bring VHL in unexpected close proximity to Bcl-xL, which is markedly different from the VHL interactions in other PROTAC complexes. VHL – PROTAC6 - Bcl-xL has negative cooperativity, and it has been noted that the two proteins could be brought to interact using a substantially shorter linker.30 It appears that the linker conformational degrees of freedom is too high for our method. In addition, according to our calculations, a large fraction of the binding energy is due to protein-linker rather than protein-protein interactions, and the interface also includes two iodine ions. None of these factors can be taken into account in the current version of our protocol, and possibly should be added in further development. However, we emphasize that this is an unusual complex, and using the very long linker is far from optimal, so it is not clear if such properties should be considered for computational method development.
Figure 2.
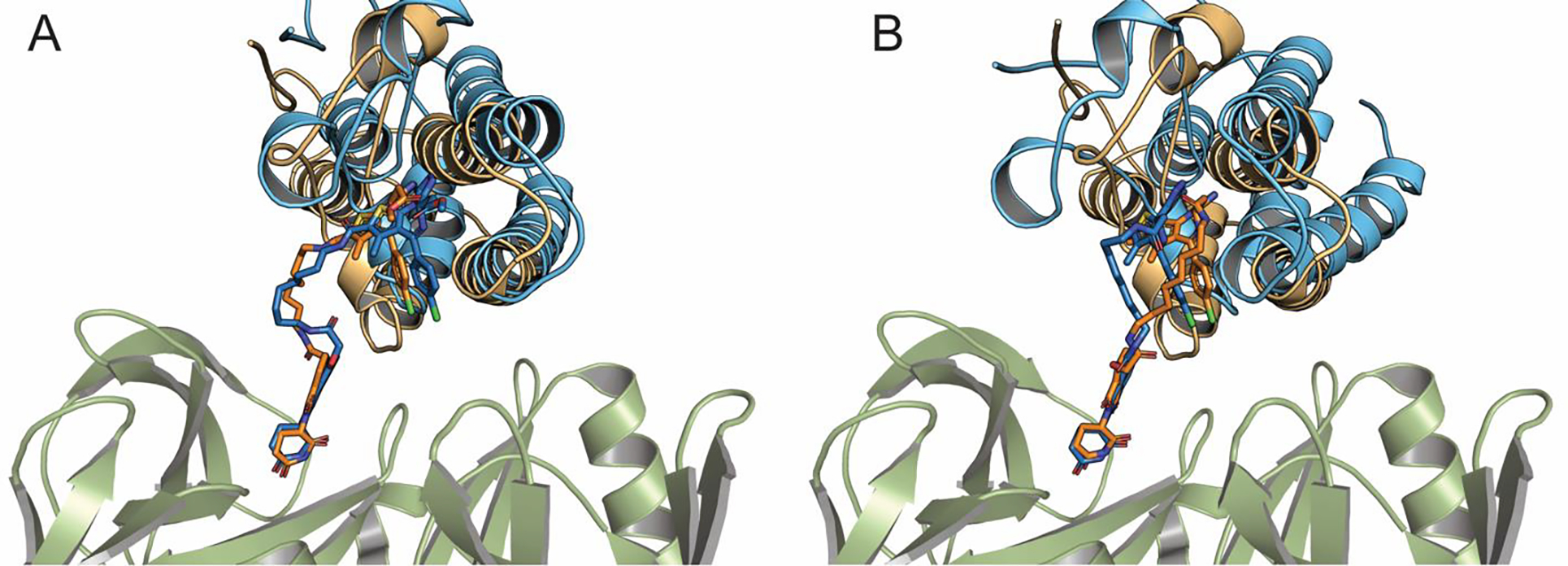
Accuracy of some predicted structures. (A) Model 3 of the CRBN-dBET23-BRD4 BD1 complex (blue), superimposed on its X-ray structure (orange, PDB ID: 6BN7). (B) Model 5 of the CRBN-dBET6-BRD4 BD1 complex (blue), superimposed on its X-ray structure (orange, PDB ID: 6BOY)
Consensus clustering.
We found that the ranking of the best models can be substantially improved if the same E3 ligase and target protein combination is used with multiple PROTACs. Several series of this type can be found in the literature, such as the series of five PROTACs listed in Supplementary Table 1 that target BRD4 using CRBN as the E3 ligase. As shown in Supplementary Table 3, for each PROTAC we generally have multiple acceptable models with favorable interactions between the E3 ligase and the target protein that satisfy the geometric restraints imposed by the linker and also have low PROTAC internal energy. A model is considered acceptable if the two protein have a favorable interaction and the linker is not strained. As described in the Methods, this last condition means that during local minimization of the linker at fixed protein positions the shift in the ligand warheads is less the 1.5 Å RMSD. Superimposing the X-ray structures in such series shows that the same protein poses may occur with several different PROTACs. For example, the superposition of experimental X-ray structures of the BRD4–dBET6–BRD4 and of the BRD4–dBET23–BRD4 complexes in Fig. 3A shows excellent alignment of the corresponding proteins and ligands, in spite of the large difference in the lengths and structures of the two linkers. Thus, we should expect that, apart from the linker conformations, good models of these two ternary complexes will also be very similar to each other. To find such a dominant pose, we performed density-based clustering of all 50 models of the five CRBN–PROTAC–BRD4 complexes (10 models of each), and ranked the resulting clusters based on their population. The clustering radius was 9 Å RMSD, where the pairwise RMSD was calculated for the α-carbon atoms of BRD4 after superimposing the CRBN structures. As shown in Supplementary Table 4 the largest consensus cluster includes three models of dBET6, five models of dBET23, two models of dBET55, and three models of dBET70. The structure closest to the center of the consensus cluster is model 9 of dBET23, which has the smRMSD value of 1.86 Å from the native ligand in the X-ray structure. For dBET6, dBET55, and dBET70 we also selected their models closest to the center of the consensus cluster (Fig. 3B), which resulted in smRMSD values of 1.89 Å, 1.92 Å, and 1.58 Å, respectively, indicating excellent agreement in all cases with the corresponding experimental ternary complex structures. Thus, clustering all models for all tested PROTACs together and selecting the ones closest to the center of the largest consensus cluster solves the ranking problem and leads to very accurate predictions of the ternary complex structure. To demonstrate this quality, in Fig. 3C and Fig. 3D we superimpose the predicted structures of CRBN–dBET70–BRD4 BD1 and CRBN–dBET55–BRD4 BD1 on their X-ray structures.
Figure 3.
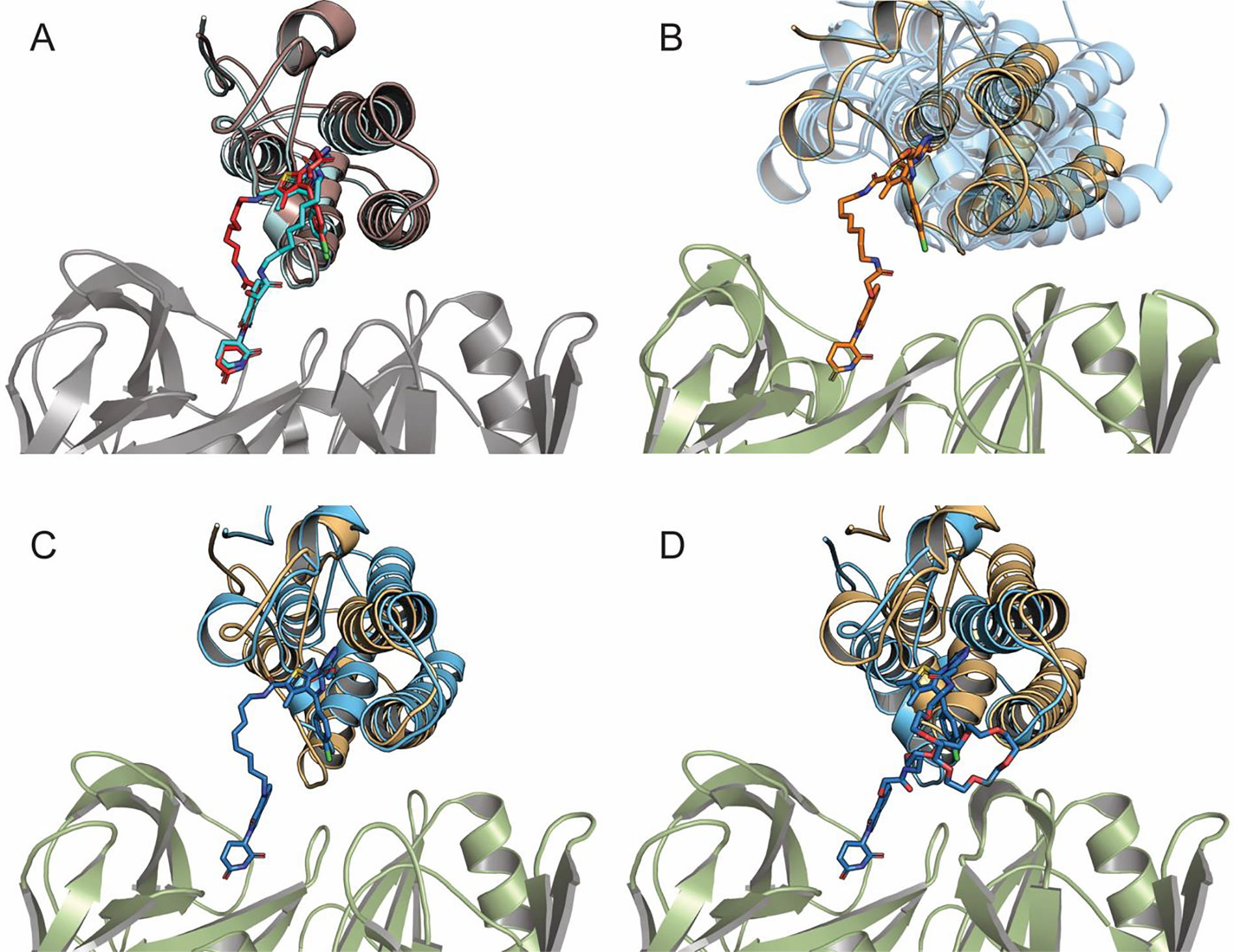
Structures and models of CRBN-PROTAC-BRD4 BD1 ternary complexes. (A) Superimposing the X-ray structures of CRBN-dBET6-BRD4 BD1 (light-blue/cyan, PDB ID: 6BOY) and of CRBN-dBET23-BRD4 BD1 (red/orange, PDB ID: 6BN7). (B) Superimposing the best model CRBN-dBET6-BRD4BD1 at the center of the consensus cluster (orange) and the ternary complexes with dBET23, dBET55, and dBET70 (all shown as transparent blue). (C) Superimposing the consensus model (model 1) of CRBN-dBET70-BRD4 BD1 (blue) and its X-ray structure (light orange, PDB ID: 6BN9). (D) Superimposing the consensus model (model 1) of CRBN-dBET55-BRD4 BD1 (blue) and its X-ray structure (light orange, PDB ID: 6BN8).
For these complexes the PROTACs themselves are not visible in the X-ray structures, but the method generates very good predictions for the relative poses of the BRD4 and CRBN proteins. Similar results for dBET6 and dBET23 are shown in Supplementary Fig. 4A and Supplementary Fig. 4B. We note that the PROTAC dBET57 had no model in the largest consensus cluster. In fact, the linker in dBET57 is much shorter than in the other four PROTACs, and yields substantially different interactions between the BRD4 and CRBN proteins (Fig. 4A). Thus, dBET57 cannot be considered as part of the series, and we cannot use the consensus clustering approach as a tool to select its best model. In fact, three models of dBET57 have very low smRMSD (Fig. 4B), but their ranks are 5, 8, and 9, whereas the top ranked model has the smRMSD of 3.24 Å (Supplementary Table 3).
Figure 4.
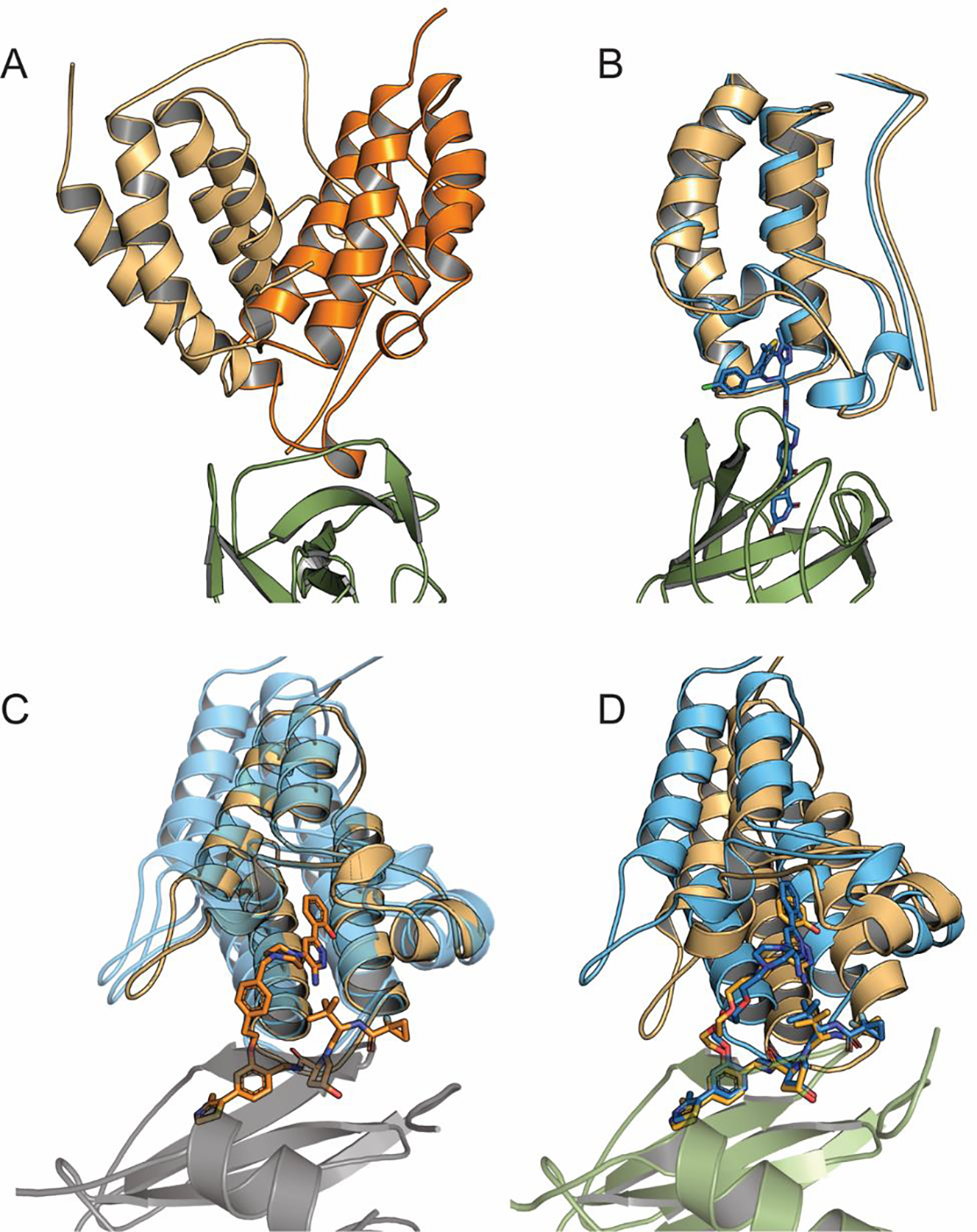
Models of CRBN–dBET57– BRD4BD1 and VHL- PROTAC1/2-SMARCA2/4 complexes. (A) Superposing the best model (model 5) of CRBN–dBET57– BRD4 BD1 (light orange) and the consensus prediction of CRBN–dBET23– BRD4 BD1 (bright orange) to show that they substantially differ. (B) Model 5 of CRBN–dBET57– BRD4 BD1 (blue), superimposed on its X-ray structure (orange, PDB ID: 6BNB). (C) Superimposing the best model (model 2) of VHL- PROTAC2-SMARCA2 (orange) at the center of the consensus cluster and the consensus models of VHL- PROTAC1-SMARCA2 and VHL- PROTAC2-SMARCA4, both shown in transparent blue color. (D) Superimposing the consensus prediction (model 5) of VHL- PROTAC1-SMARCA2 (blue) and its X-ray structure (orange, PDB ID: 5NVX)
We also applied the consensus-based analysis to models of the VHL–PROTAC1/2–SMARCA2/4 series. We superimposed the ten VHL–PROTAC1–SMARCA2 and ten VHL–PROTAC2–SMARCA2 models, and due to the similarity of the targets we added the ten VHL–PROTAC2–SMARCA4 models. As shown in Supplementary Table 4, the largest consensus cluster formed by the 30 models includes one, three, and two models, respectively, of the three ternary complexes. The center of the cluster is located at model 2 of the VHL–PROTAC2–SMARCA2 complex, which has 1.56 Å smRMSD from its PDB structure. Since the ligands are very similar, we find the same smRMSD for the other two complexes. Fig. 4C shows model 2 of VHL-PROTAC2-SMARCA2 (orange) and the models for the other two complexes in the consensus cluster (transparent blue). In Fig. 4D we align model 2 of the VHL-PROTAC2-SMARCA2 to its X-ray structure. Supplementary Fig. 5A and Supplementary Fig. 5B show that the models closest to the center of the consensus cluster for the VHL–PROTAC1–SMARCA2 and the VHL–PROTAC2–SMARCA4 complexes, superimpose with the smRMSD value of 1.56 Å from the respective X-ray structures in both cases (see Supplementary Table 4).
Predicting properties related to degrader activity and selectivity.
PROTACs exhibit different target degradation efficiencies depending on the linker length and the nature of the ligands. The design of a degrader with high activity is a complicated task normally solved experimentally. However, it has been suggested that the number of docked poses that satisfy both the condition posed by the linker geometry and by ubiquitin accessibility provides information on PROTAC degrading activity.18 To explore this relationship we introduced a measure defined as the weighted sum of the poses that pass all the filtering steps. The weights are assigned according to the solvent accessible surface area (SASA) of non-polar carbons of the linker according to the expression e-(0.1 SASA), where SASA is computed for the non-polar carbon atoms and attached hydrogens of the linker. Thus, this expression reduces the weight of linker conformations that expose substantial hydrophobicity, because linkers that are too hydrophobic tend to collapse, affecting the favorable orientation of the two proteins. A carbon atom is excluded from the summation if the partial charge of any of its neighbors is larger than 0.4 by the absolute value. Partial charges were computed using the AM1-BCC model. This method is implemented as part of our docking algorithm, and due to the exhaustive sampling of the conformational space by the FFT based docking approach we expect fairly good prediction accuracy.
While our plan was to predict degrader activity of PROTACs, we have found that the above measure correlates better with the dissociation constant Kd and the cooperativity α of the E3 ligase – PROTAC – target protein complex. Supplementary Table 5 compares the extensive MZ1 and AT1 thermodynamic and kinetic data from isothermal calorimetry and surface plasmon resonance experiments performed by the Ciulli lab5, 6 to the weighted sum of poses we calculated. Using the MZ1 PROTAC both the highest α and the highest sum of poses occur for BRD4 BD2. In addition, the ternary complex with BRD4 BD2 has the lowest Kd value. The second highest α and sum of poses were obtained for BRD3 BD2. On the other extreme, both α and the sum have the lowest values for BRD4 BD1. The relations between experimental and computed measures are more complex between the two extremes, but the method is clearly able to predict both the most stable and the least stable ternary complexes. To study the specificity of MZ1-induced protein–protein interactions, Gadd et al.5 replaced three residues in the weakly cooperative BRD2 BD1 with the corresponding residues of the highly cooperative BRD4 BD2, producing the variant BRD2 BD1 KEA. As shown in Supplementary Table 5, based on the ITC data the mutations increase the α value to 7.9 from 2.9, and the weighted sum to 18.7 from 10.9. In contrast, replacing three residues in BRD4 BD2 with the residues in BRD2 BD1 to produce the mutant BRD4 BD2 QVK reduces the α value to 4.2 from 17.6, and the sum to 2.5 from 31.1.
Based on the structure of the VHL-MZ1- BRD4 BD2 complex Gadd et al.5 synthesized the PROTAC AT1 to improve selectivity toward BRD4 BD2. As shown in Supplementary Table 5, while all activities were lower than with MZ1, AT1 indeed formed the most cooperative (α = 7) ternary complex with BRD4 BD2 among the BET bromodomains.5 This result was confirmed by surface plasmon resonance experiments.6 Although AT1 is a weaker degrader than MZ1, selectivity toward BRD4 BD2 Improved, as AT1 has almost negligible activity against BRD2 AND BRD3.5 The computed weighted sum values agree well with these observations
The stability and cooperativity of the ternary complex is expected to impact degradation efficiency, and although the latter depends on additional factors, we explored whether the weighted sum of poses can also be used to predict degradation values observed for a number of PROTACs (Figure 5). First we studied a series of PROTACs targeting Bruton’s tyrosine kinase (BTK), a nonreceptor tyrosine kinase essential for B cell maturation. Zorba et al. 32 synthesized and tested a library of 11 PROTACs (P1-P11) of varying linker lengths that engage BTK on one end and CRBN on the other, starting with a chain length of two for P1 (three heavy atoms in total), up to P11 with a chain length of 18 (19 heavy atoms). The BTK binding ligand was a noncovalent analog derived from a previously disclosed covalent phenylpyrazole series 33. They reported that P6 to P11, with linker chain lengths from 11 to 18, potently degraded BTK, but the shorter PROTACs (P1–P4) were largely ineffective. Between these extremes was an intermediate compound, P5 with chain length 8, which demonstrated modest target knockdown. According to our calculations, P1 through P4 yield very small populations of productive docked structures, and the numbers substantially increase for P5 through P8 (Fig. 5A). While this result is in general agreement with the experimental activity data, the observed BTK degradation values are given by low resolution degradation curves, and the paper by Zorba et al.32 discriminates only three different levels of degradation. In view of such data it is difficult to provide a more quantitative relationship between the weighted number of poses and experimental degradation activities. An important observation from the study is that the most potent BTK degradation occurs when steric clashes between BTK and CRBN are alleviated by reaching a critical linker length. In our algorithm, minimizing the energy of the ternary complex removes the steric clashes, but the process stresses the linker, supporting the decision that we should use the energy of the linker as one of the selection criteria.
Figure 5.
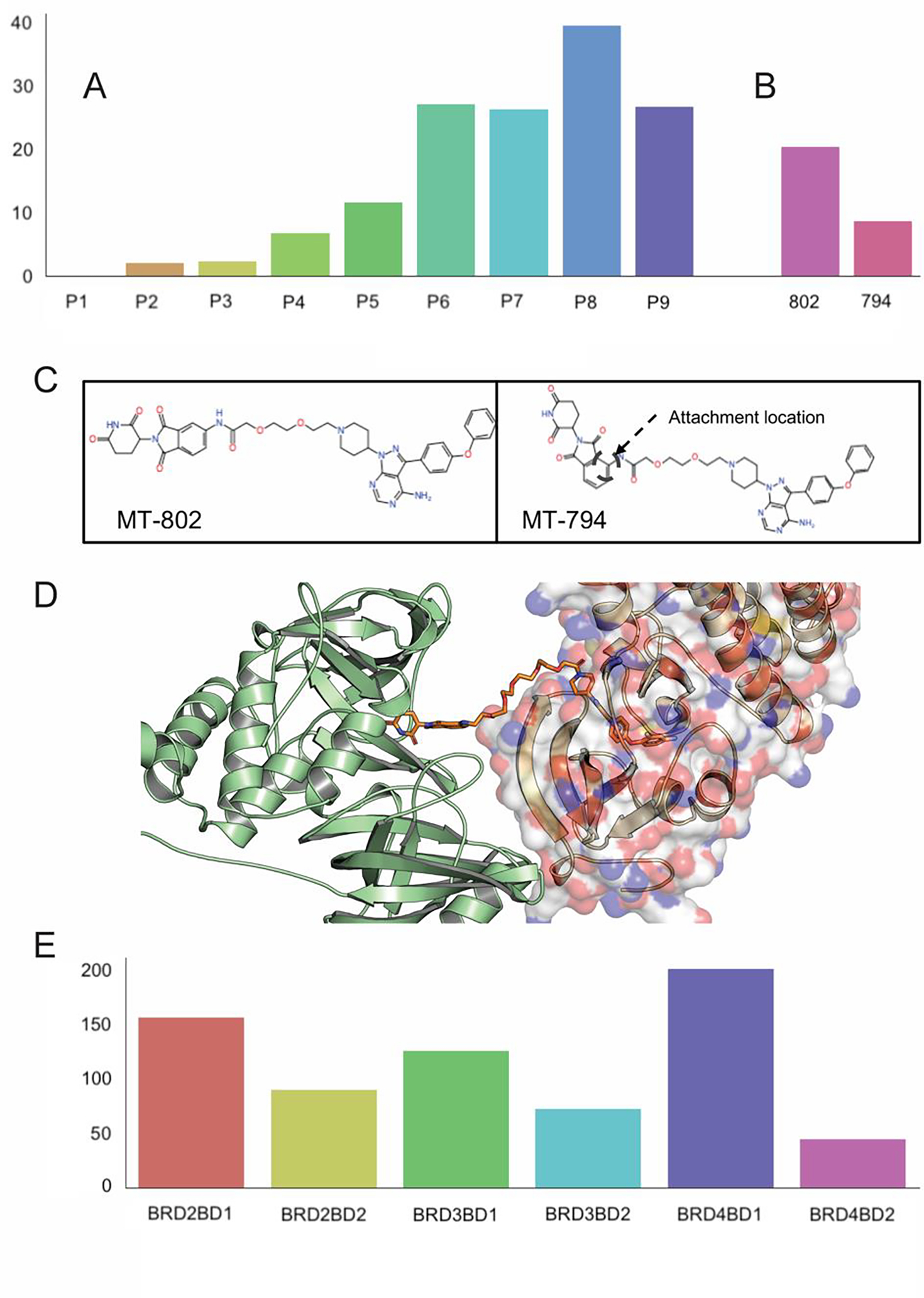
Using the weighted sum of acceptable models for predicting degradation activity and selectivity. (A) Weighted sum values for the series VHL- PROTAC1-BTK (denoted as P1) through VHL- PROTAC10-BTK (denoted as P10), considered here as predictions of degrading activity. (B) Weighted sum of predicted structures that satisfy the restraints posed by the linker for CRBN-MT-802-BTK (denoted as 802) and CRBN-MT-794-BTK (denoted as 794) ternary complexes. (C) Chemical structures of PROTACS MT-802 and MT-794. (D) PROTAC9 (yellow stick model) has limited interactions with CRBN (shown as green cartoon) apart from the ligand binding site, but interacts extensively with the BTK, shown as surface model on the right. (E) Weighted sums of acceptable models for the CRBN-ZXH-3–26-BDR4 BD1, BRD2 BD1, BRD3 BD1, BRD2 BD2, and BRD3 BD2 complexes, demonstrating some level of selective degradation of BRD4 BD1.
In addition to the above set of PROTACs, we estimated the activities of two PROTACs targeting wild-type BTK and the C481S mutant 34. The former can be targeted by the covalent inhibitor ibrutinib, but the C481S mutation, which occurs in >80% chronic lymphocytic leukemia (CLL) patients treated by ibrutinib, eliminates the cysteine that is the site of inhibitor attachment and thus results in resistance to the drug and clinical relapse. Buhimschi et al. 34 reported several PROTACs that have an ibrutinib moiety on one end and the CRBN-binding pomalidomide on the other. The PROTAC MT-802 was found to be the most potent version. It has a linker of length 8, thus similar to PROTAC5 of the series just discussed, but a slightly different BTK ligand. As shown in Fig. 5B, the weighted sum of poses indicated an activity for MT-802 that is somewhere between that of PROTAC5 and PROTAC6, in good agreement with the experimental data.34 Moreover, our calculations indicate that the 8-atom linker is the minimum acceptable length for activity. As shown in Fig. 5B, a modified version of MT-802 with a different attachment of the linker to pomalidomide (MT-794, shown in Fig. 5C) was predicted to result in a substantial drop in degradation potency, as was observed experimentally.34
In addition to estimating the BTK degrading efficiency by the PROTAC1-PROTAC11 series using the weighted sums of acceptable models we also generated predicted structures for these ternary complexes. Although X-ray structures have not been solved for these complexes, our results provide some interesting information. Zorba et al. 32 used solution-phase hydrogen/deuterium exchange coupled with mass spectrometry (HDX–MS) analysis for the assessment of possible long-lived or stable protein–protein interactions within the CRBN–PROTAC–BTK ternary complex. Although CRBN is present in these experiments, they did not observe statistically significant protection of any region of this protein by any ligand, including the PROTAC. They assumed that this result may be due to the low affinity of the CRBN binding ligand, plus the small size of the pomalidomide-binding pocket which provides only a single backbone H-bond donor. In the case of BTK, PROTAC9 led to as much as 23% protection from deuterium exchange, in certain peptide fragments generated in the analysis, compared with the unbound protein. Our top model of the complex fully agrees with these observations. As shown in Fig. 5D, PROTAC9 has limited interactions with CRBN (shown as green cartoon) apart from the ligand binding site. However, the linker interacts with a substantial fraction of the BTK surface, shown on the right side of the figure.
Studying PROTAC selectivity, Nowak et al. 15 found the PROTAC ZXH-3–26 to be active on BDR4 BD1, but inactive on BRD2 BD1, BRD3 BD1, BRD2 BD2, BRD3 BD2, and BRD4 BD2. For generality we predicted the structures of the latter five isoforms by starting from the X-ray structure of BDR4 BD1 and mutating the side chains using the SCWRL 35 program. The sequences between the BD1 and BD2 domains differ significantly, but within the same domain the isoforms differ only by a few residues. Each isoform was docked to CRBN with ZXH-3–26 as degrader.15 As shown in Fig. 5E, BRD4 BD1 demonstrated the largest population among the six BRD types in case of modelled structures, in good agreement with the experimental data.
Our last example shows blind prediction for PROTACs targeting BRAF(V600E). This cancer-causing mutant is a well validated and important target for inhibition of the RAS-ERK signaling pathway. Traditional Inhibitors of BRAF(V600E) are effective, but only for relatively short periods due to the development of resistance. In addition, such inhibitors bind to the active site of BRAF and disable its catalytic output, but do not prevent BRAF dimerization. To overcome these deficiencies, Posternak et al.36 have recently investigated the application of the PROTAC approach to BRAF inhibition. As BRAF binders they used the approved drug dabrafenib or the preclinical inhibitor BI 882370. On the E3 ligase side they used either Cereblon or VHL. Using flexible linkers of various lengths and compositions, 16 different PROTACs were synthesized and tested. Supplementary Table 6 shows DC50 (the concentration at which 50% of maximal degradation was observed) and DCmax (the maximal level of degradation observed) values from cellular degradation experiments for 13 of these complexes, as well as the computed measures of degradation. Based on the experiments, Compound 3, renamed P4B, was selected as the most active PROTAC by Posternak et al.36 P4B used pomalidomide as the E3 ligase binder, BI 882370 as the BRAF binder, and a polyethylene glycol chain with a length of four units (PEG4) as the linker (Fig. 6A). P4B displayed activity in BRAF(V600E) cell lines, with Dmax = 82% (the maximal level of degradation observed) and DC50 =15 nM (the concentration at which 50% of maximal degradation was observed). We applied our prediction algorithm to the BRAF-targeting PROTAC designs, including compounds 28, 29, and 30 that were not yet synthesized and tested in cells at the time of the calculations. Similarly to P4B, all three compounds used pomalidomide and BI 882370, but with different linkers. In 28, the amide group at the attachment of the PEG4 linker to pomalidomide has been N-methylated, whereas 29 and 30 use the same attachment as P4B but have longer and more complex linkers (Fig. 6A). Using the weighted sum of acceptable models the degrading activity of 28 was predicted to be comparable to that of P4B (119.38 versus 108.14), while the activity of 30 was predicted to be ~50% lower, and that of 29 to be lower still, 66.23 and 38.03, respectively (see Supplementary Table 6 and Fig. 6B). To test these predictions, A375 cells were treated with each compounds 28, 29, and 30, plus P4B as a positive control, at concentrations of 40, 200, and 1000 nM for 24 h prior to immunoblot analysis of whole cell lysates. Supplementary Fig. 6A shows the results of these 3-point dose response analyses, showing that 28 is about as effective as P4B at degrading BRAF and suppressing MEK phosphorylation, while 29 and 30 caused much less BRAF degradation. Broader dose response analyses for P4B and 28 confirmed the comparable activity of these two PROTACs (Supplementary Fig. 6B). Moreover, both P4B and 28 showed evidence of a reduction in activity at very high compound concentrations, as expected for a PROTAC mechanism of action due to occupancy of the target and the E3 ligase by separate PROTAC molecules. Compound 28 activity to degrade BRAF is suppressed by pomalidomide, MLN 4924 or MG132 treatment, demonstrating that BRAF degradation is through ubiquitin-proteasome mechanism (Supplementary Fig. 6C). Supplementary Table 6 provides a quantitative analysis of these dose-response curves, showing that 28 and P4B have comparable DC50 and DCmax values, in keeping with their similar linker length and structure, while 29 and 30 have much weaker degrading activity, although they were still able to suppress MEK activation by direct inhibition of BRAF by the BI 882370 warhead. While the computed activities show these differences, the activity of compound 30 is overpredicted, and the predicted activity of compound 22 is a false positive (Supplementary Table 6). Interestingly, compound 22 has a net positive charge unlike the other active PROTACs in this series, which might have contributed to the predicted interaction. We note that no such large deviations between experimental and predicted values are seen for the BET targeting PROTACs MZ1 and AT1 in Supplementary Table 5. However, the latter data are from thermodynamic (ITC) and kinetic (SPR) experiments, whereas Posternak et al.36 measured the level of degradation in A375 melanoma cells, thus in a much more complex system.
Figure 6.
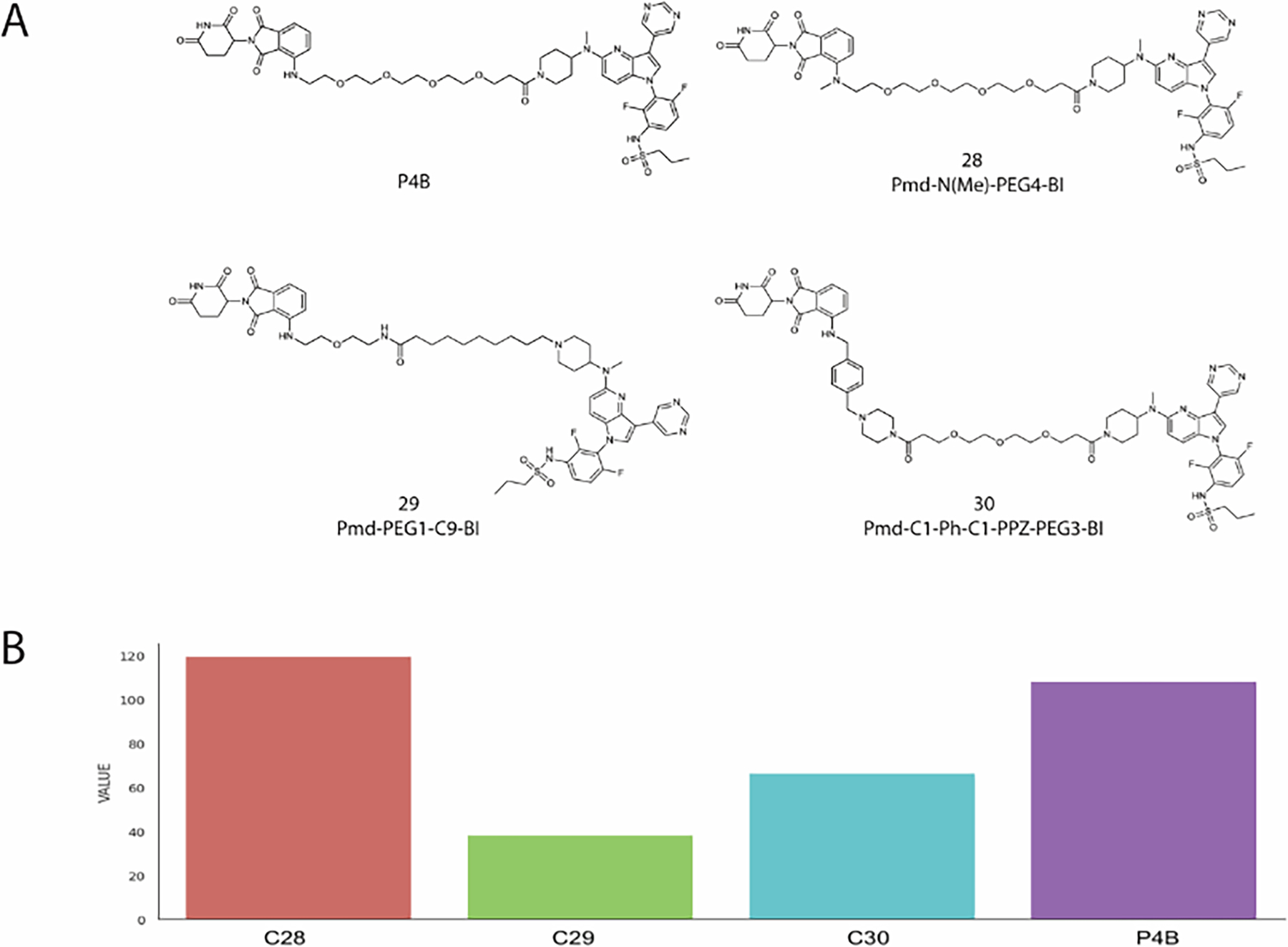
In cell BRAF degradation analysis. (A) Chemical structures of P4B and compounds 28–30. (B) Relative degrading activity of PROTACs 28, 29 and 30 and P4B as predicted using our computational method. Value is the weighted number of predicted structures that satisfy the restraints posed by the linker, considered as the prediction of degrading activity.
Discussion
PROTACs function by stabilizing a complex between the target protein and an E3 ligase. Thus, methods that can accurately predict the ternary E3 ligase–PROTAC–target protein complex will aid PROTAC design by reducing the need for empirical rounds of synthesis and evaluation. Although a large number of PROTACs have been reported in the last decade, there remain only a few X-ray structures of ternary complexes, suggesting that crystallization of the ternary complex is problematic and emphasizing the need for a reliable computational method to predict these structures. We show here that the protein-protein and linker conformational spaces can be simultaneously sampled by pre-generating half-linker clouds attached to the ligands bound to each of the two proteins, using a modified version of fast Fourier transform based sampling that penalizes protein poses that lack half-linkers positioned so as to give a plausible linker geometry to connect the proteins. After additionally removing poses that do not assure accessibility to ubiquitination sites, the new method always generates high accuracy models among the top ten predictions, though with uncertainty about which of the ten models is nearest to the true geometry. This uncertainty can be eliminated by clustering a set of models generated for different PROTACs with the same E3 ligase and target protein. Results show that beyond a minimum linker length the predicted structures tend to coincide, indicating the dominant mode of E3 ligase–target protein interaction. Shorter linkers may engage the two proteins in a different conformation and generally reduce degradation potency, although they may improve selectivity. 15
The success of our method reveals important features of how PROTACS work, at least in the cases studied. Specifically, the PROTACs function by stabilizing a protein-protein contact that represents an intrinsically favorable mode of interaction between the proteins. If this were not the case, the protein-protein docking component of our approach would be irrelevant, or might even make the predictions less accurate by biasing them towards intrinsically stable protein-protein complex geometries that are unrelated to PROTAC activity. Importantly, however, the geometry observed in the ternary complex is not necessarily the most stable complex the two proteins would otherwise form. Consequently, accurate predictions of the ternary complex may not be achieved simply by docking the proteins in the absence of additional constraints to ensure that the warhead binding sites can be effectively connected by the particular PROTAC. We show that a simple distance constraint relating to the PROTAC linker length is not a good way to ensure a valid prediction, as it is also important to ensure that a feasible linker geometry is possible. A strength of our method is that it searches for suitable protein-protein interaction geometries and suitable linker geometries at the same time, taking advantage of the highly efficient fast Fourier transform sampling algorithm to achieve this in a highly computationally efficient manner.
Following the suggestion by Bai et al.18 we calculated a weighted sum of acceptable models in an attempt to predict the degradation activity of PROTACs. It was observed that the calculated measure provides meaningful prediction of the dissociation constant Kd and the cooperativity α of the E3 ligase – PROTAC – target protein complexes, but predictions of the degrading activity were less accurate. While the stability and cooperativity of the ternary complex clearly impact degrading activity, the latter also depends on other factors, and hence the limited accuracy of predictions is not surprising. Thus, this part of the methodology needs substantial further development. It appears that the predicting the degrading activity has two main shortcomings. First, while the predictions discriminate active degraders from inactive ones in most cases, the numbers do not necessarily correlate with measured degradation activities in the midrange between the two extremes. Second, the relationship between the number of models and predicted degradation values are valid only within particular series of PROTACs with the same target and E3, but the scale is not generally transferable between series with different targets. Comparing Supplementary Tables 5 and 6 shows that the computed values are in better agreement with thermodynamic data obtained by isothermal calorimetry in cell-free system than the results from cellular degradation experiments. In spite of these problems we believe that the accurate prediction of the ternary complex geometry is of high value for the design of effective PROTACs. High potency and degradation efficiency is presumed to benefit from a linker that has the minimum length and flexibility consistent with bringing the E3 ligase and target protein together in an unstrained ternary complex. Knowledge of the structure of the ternary complex can provide important guidance as to how the linker structure might be modified to achieve this end. Similarly, for efficacy certain in vivo properties are also important, such as membrane permeability to allow access to the intracellular target proteins. Knowledge of the linker geometry in the ternary complex can identify which locations might be modified, for example to add lipophilic functionality, to improve membrane permeability. Finally, our approach can indicate which specific lysine residues on the target protein are candidates to be ubiquitinated in the PROTAC mechanism, with possible implications for selectivity of action and robustness to resistance mutations. We therefore believe that our approach is likely to be of high value for future PROTAC design campaigns.
Methods
Ligand docking to the E3 ligase and to the target protein.
Since the protocol is designed to use unbound protein structures, the first step requires modeling of the ligands in the binding site of the E3 ligase and the target protein. The ligands are docked using the recently developed homology based small-molecule docking web-server LigTBM,37 which demonstrated successful performance in blind D3R Grand Challenge 4,38 predicting many compounds with sub-angstrom accuracy. The server accepts the ligand as a SMILES string and the protein as a PDB structure and yields multiple models of the complex. For each ligand we used the first model in the following steps of the prediction algorithm.
Generating the “half-linker clouds” and performing FFT based conformational search.
First, we generate 10000 conformations of the PROTAC’s linker using the ETKDG method from RDKit 39, cut the structures into two halves in the middle, attach each half-linker structure to the E3 ligase- ligand and target protein-ligand complexes, and filter out the clashing conformers using the atom-atom clash cut-off distance of 1.85 Angstrom. The two proteins, together with the generated “clouds” of half-linkers, are docked to each other using a modified version of the PIPER program 23, which performs rigid body docking in the 6D space of rotations and translations. The center of mass of the receptor is fixed at the origin of the coordinate system, and the possible rotational and translational positions of the ligand are evaluated at the given level of discretization. The rotational space is sampled on a sphere-based grid that defines a subdivision of a spherical surface in which each pixel covers the same surface area as every other pixel 40. The 50,000 rotations we consider correspond to about 6 degrees in terms of the Euler angles. The step size of the translational grid is 1 Å, and hence the program evaluates the energy for 109-1010 conformations. The original scoring function of PIPER is a weighted sum of convolutions between protein grids, representing van der Waals, electrostatic, and desolvation energy terms. To account for the ‘half-linker clouds” we introduced an additional “silent” convolution, which does not contribute to the energy score, but its values are used for filtering docking poses. The grids of this convolution are indicator functions of the midway atoms of the non-clashing linker conformations. Namely, target grid values are equal to 1 at the nearest grid points closest to the midway atoms and 0 elsewhere, receptor grid values are equal to 1 at the grid points within 1.5 Å (one and a half of the grid cell size to account for projection error) of the midway atoms and 0 elsewhere. The resulting convolution is 0 if no half-linker conformations satisfy this requirement of mid-point proximity, and the corresponding poses are removed. The 1000 lowest energy docked structures, each with at least one feasible linker conformation, are retained for further analysis. Thus, the modified rigid body sampling generates favorable poses of the two proteins and conformations of their flexible linker in a single and very efficient computational step.
Calculating small molecular RMSD (smRMSD) values.
Some of the ternary complex structures have very low resolution, and hence the linker conformation is uncertaina], and the linker may not be even seen. However, the small molecule RMSD or smRMSD, defined as the RMSD between the X-ray and predicted positions of the warheads, can be still reliably calculated, because high resolution X-ray structures are generally available for both the separate E3 ligase and the target protein, and the ligand binding sites and hence the bound ligand positions and orientations are well defined in both proteins. Thus, to obtain a reliable “experimental” conformations of the two ligands we can superimpose the higher resolution structures of the component proteins with their bound ligand on their structures in the predicted model of the ternary complex, and the low resolution of the latter will have at most moderate impact on the conformations of the ligands. Thus, the RMSD between predicted and “experimental” ligand positions will be almost independent of the resolution of the complex.
Filtering for ubiquitin accessibility.
To test ubiquitin accessibility, we had to construct the CRL4CRBN (CRBN–Cul4–Rbx1) and the CRL2VHL (VHL–ElonginC–ElonginB–Cul2–Rbx1) assemblies. A structural model of the CRL4CRBN (CRBN–Cul4–Rbx1) with bound targets (BRD4 BD1 and BTK) at one end and E2–miUbiquitin at the other end was constructed in PyMOL by aligning the CRBN–DDB1 (PDB entry 6BN7) structure on to the quaternary structure DDB1-DDB2-CUL4A-RBX1 (PDB entry 4A0K). Finally, the Rbx1–E2–Ub arm was modeled based on the crystal structure of Rbx1–Ubc12~NEDD8–Cul1–Dcn1 (PDB entry 4P5O) superposed via the cullin subunit. To account for the flexibility of the DDB1 β-propeller domain, multiple conformations of DDB1 (PDB entries 4A08, 4A09, 3EI1) were aligned to the CRBN–DDB1 structure, followed by subsequent alignment of the quaternary structure DDB1-DDB2-CUL4A- RBX1 (Fig. S2A).
A structural model of the CRL2VHL (VHL–ElonginC–ElonginB–Cul2–Rbx1) with bound targets (BRD4BD2, SMARCA2, and SMARCA4) at one end and E2–Ubiquitin at the other end was also constructed in PyMOL by aligning the VHL–EloC–EloB (PDB entry 5T35) structure on to the quaternary structure VHL–EloC–EloB–Cul2NTD (PDB entry 4WQO). Cul2NTD and Cul2CTD were modeled based on the structures of Cul5NTD (PDB entry 2WZK) and Cul1CTD–Rbx1 (PDB entry 3RTR) and superposed onto full-length Cul1 from PDB entry 1LDK. Finally, the Rbx1–E2–Ub arm was modeled based on the crystal structure of Rbx1–Ubc12~NEDD8– Cul1–Dcn1 (PDB entry 4P5O) superposed via the cullin subunit (Fig. S2B).
Energy minimization.
The models retained after checking for ubiquitin accessibility are refined using molecular mechanics energy minimization with fixed proteins and an unconstrained connected PROTAC. Amber 19 41 is used to parameterize the complex, and L-BFGS minimization 42 is carried out using an in-house package.
Synthesis of PROTAC compounds.
For detailed description of the synthetic routes and supporting 1H, 13C and 19F NMR and mass spectrometry information see the Supplementary Notes.
In vitro kinase assays.
BRAF kinase activity (IC50 values in nM) was measured using the KinaseProfiler service (Eurofins Pharma Discovery Services UK). Compounds were sent to Eurofins as dry powders. Individual kinase assay protocols used in the Eurofins KinaseProfiler radiometric protein kinase assays are described in http://www.komabiotech.co.kr/www/product/DD/KinaseProfiler_Assay_Protocol_Guide_Eurofins_v86.pdf.
Methods for in-cell testing.
A375 cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) (GIBCO) supplemented with 10% FBS, penicillin, and streptomycin. At 50–70% confluence, cells were treated for 24 hours with the PROTACS in 0.1% DMSO (final concentration) or with 0.1% DMSO alone. Prior to harvesting, cells were washed, and lifted by scraping into PBS supplemented with 1 mM PMSF. Cell pellets were collected after centrifugation. Cells were lysed in a buffer containing 50 mM HEPES pH 7.4, 150 mM NaCl, 5 mM EDTA, 0.5% NP-40, 5 mM NaF, and 10% glycerol. The buffer was supplemented with fresh Roche protease inhibitor cocktail (5056489001) and phosphatase inhibitor cocktail PhosStop (4906845001). Cell lysates were clarified by centrifugation at 18,000 g for 30 min, the supernatant collected, and the total protein concentration determined by Bradford assay (Bio-Rad). Proteins were resolved by SDS-PAGE, transferred onto nitrocellulose membranes and subjected to standard immunoblot protocol. Western blots were visualized using Bio-Rad Clarity ECL Western Blotting Substrate on a Bio-Rad ChemiDoc MP imaging system. Band intensities were quantified by Bio-Rad Image Lab software.
Supplementary Material
Acknowledgements
The paper is adapted from M.I. thesis.
Funding
This investigation was supported by grants DMS 2054251 and AF 1645512 from the National Science Foundation, and R01GM140098, R35GM118078, RM1135136, R01GM102864, and R01GM140154, from the National Institute of General Medical Sciences. G.P. acknowledges the support of the Ontario Institute for Cancer Research and its funding from the Government of Ontario
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.XXXXX
Block diagram of the method, additional computational results showing some advantages of the proposed approach, dose response analyses of BRAF (V600E) degrading compounds, table of the PROTAC benchmark set of known X-ray structures, table of the PROTAC structures studied with half linkers, predicted structures, dissociation constants, and cooperativity of some of the PROTACs, synthetic methods for the preparation of PROTACs targeting BRAF (V600E).
References
- 1.Neklesa TK, Winkler JD & Crews CM Targeted protein degradation by PROTACs. Pharmacol. Ther. 174, 138–144 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Pettersson M & Crews CM PROteolysis TArgeting Chimeras (PROTACs) - Past, present and future. Drug Discov. Today Technol 31, 15–27 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bondeson DP et al. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem. Biol. 25, 78–87 e75 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fisher SL & Phillips AJ Targeted protein degradation and the enzymology of degraders. Curr. Opin. Chem. Biol. 44, 47–55 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Gadd MS et al. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 13, 514–521 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roy MJ et al. SPR-measured dissociation kinetics of PROTAC ternary complexes influence target degradation rate. ACS Chem. Biol. 14, 361–368 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sakamoto KM et al. Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U S A 98, 8554–8559 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun X et al. PROTACs: great opportunities for academia and industry. Signal Transduct. Target Ther. 4, 64 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Konstantinidou M et al. PROTACs- a game-changing technology. Expert Opin. Drug. Discov. 14, 1255–1268 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schapira M, Calabrese MF, Bullock AN & Crews CM Targeted protein degradation: expanding the toolbox. Nat. Rev. Drug. Discov. 18, 949–963 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Buckley DL et al. Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1alpha interaction. J. Am. Chem. Soc. 134, 4465–4468 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Girardini M, Maniaci C, Hughes SJ, Testa A & Ciulli A Cereblon versus VHL: Hijacking E3 ligases against each other using PROTACs. Bioorg. Med. Chem. 27, 2466–2479 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zoppi V et al. Iterative Design and Optimization of Initially Inactive Proteolysis Targeting Chimeras (PROTACs) Identify VZ185 as a Potent, Fast, and Selective von Hippel-Lindau (VHL) Based Dual Degrader Probe of BRD9 and BRD7. J. Med. Chem. 62, 699–726 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeng M et al. Exploring Targeted Degradation Strategy for Oncogenic KRAS(G12C). Cell Chem. Biol. 27, 19–31 e16 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Nowak RP et al. Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat. Chem. Biol. 14, 706–714 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drummond ML & Williams CI In Silico Modeling of PROTAC-Mediated Ternary Complexes: Validation and Application. J. Chem. Inf. Model. 59, 1634–1644 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Zaidman D, Prilusky J & London N, PRosettaC: Rosetta based modeling of PROTAC mediated ternary complexes. J. Chem. Inf. Model. 60, 4894–4903 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bai N et al. Rationalizing PROTAC-mediated ternary complex formation using Rosetta. J. Chem. Inf. Model. 61, 1368–1382 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gray JJ et al. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J. Mo.l Biol. 331, 281–299 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Katchalski-Katzir E et al. Molecular surface recognition: determination of geometric fit between proteins and their ligands by correlation techniques. Proc. Natl. Acad. Sci.,U S A 89, 2195–2199 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vakser IA Low-resolution docking: prediction of complexes for underdetermined structures. Biopolymers 39, 455–464 (1996). [DOI] [PubMed] [Google Scholar]
- 22.Tovchigrechko A & Vakser IA GRAMM-X public web server for protein-protein docking. Nucleic Acids Res. 34, W310–W314 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kozakov D, Brenke R, Comeau SR & Vajda S PIPER: an FFT-based protein docking program with pairwise potentials. Proteins 65, 392–406 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Duda DM et al. Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell 134, 995–1006 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farnaby W et al. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat. Chem. Biol. 15, 672–680 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dragovich PS et al. Antibody-Mediated Delivery of Chimeric BRD4 Degraders. Part 2: Improvement of In Vitro Antiproliferation Activity and In Vivo Antitumor Efficacy. J. Med. Chem. 64, 2576–2607 (2021). [DOI] [PubMed] [Google Scholar]
- 27.Dragovich PS et al. Antibody-Mediated Delivery of Chimeric BRD4 Degraders. Part 1: Exploration of Antibody Linker, Payload Loading, and Payload Molecular Properties. J. Med. Chem. 64, 2534–2575 (2021). [DOI] [PubMed] [Google Scholar]
- 28.Law RP et al. Discovery and Characterisation of Highly Cooperative FAK-Degrading PROTACs. Angew. Chem. Int. Ed. Engl. 60, 23327–23334 (2021). [DOI] [PubMed] [Google Scholar]
- 29.Yu X et al. A selective WDR5 degrader inhibits acute myeloid leukemia in patient-derived mouse models. Sci. Transl. Med. 13, eabj1578 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chung CW et al. Structural Insights into PROTAC-Mediated Degradation of Bcl-xL. ACS Chem. Biol. 15, 2316–2323 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Kozakov D et al. The ClusPro web server for protein–protein docking. Nat. Protoc. 12, 255–278 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zorba A et al. Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc. Natl. Acad. Sci. U. S. A. 115, E7285–E7292 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rankin AL et al. Selective inhibition of BTK prevents murine lupus and antibody-mediated glomerulonephritis. J. Immunol. 191, 4540–4550 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Buhimschi AD et al. Targeting the C481S Ibrutinib-Resistance Mutation in Bruton’s Tyrosine Kinase Using PROTAC-Mediated Degradation. Biochemistry 57, 3564–3575 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Krivov GG, Shapovalov MV & Dunbrack RL Improved prediction of protein side-chain conformations with SCWRL4. Proteins 77, 778–795 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Posternak G et al. Functional characterization of a PROTAC directed against BRAF mutant V600E. Nat. Chem. Biol. 16, 1170–1178 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alekseenko A et al. ClusPro LigTBM: Automated Template-based Small Molecule Docking. J. Mol. Biol. 432, 3404–3410 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kotelnikov S et al. Sampling and refinement protocols for template-based macrocycle docking: 2018 D3R Grand Challenge 4. J. Comput. Aided Mol. Des. 34, 179–189 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Landrum G RDKit: A software suite for cheminformatics, computational chemistry, and predictive modeling. http://rdkit.sourceforge.net (accessed 2020–09-24).
- 40.Yershova A, Jain S, LaValle SM & Mitchell JC Generating Uniform Incremental Grids on SO(3) Using the Hopf Fibration. Int. J. Robot. Res. 29, 801–812 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Case DA et al. The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chang D, Sun S & Zhang C An Accelerated Linearly Convergent Stochastic L-BFGS Algorithm. IEEE Trans. Neural Netw. Learn. Syst. 30, 3338–3346 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.