

Abstract
Children with treatment-refractory or relapsed (R/R) tumors face poor prognoses. As the genomic underpinnings driving R/R disease are not well defined, we describe here the genomic and transcriptomic landscapes of R/R solid tumors from 202 patients enrolled in Beat Childhood Cancer Consortium clinical trials. Tumor mutational burden (TMB) was elevated relative to untreated tumors at diagnosis, with one-third of tumors classified as having a pediatric high TMB. Prior chemotherapy exposure influenced the mutational landscape of these R/R tumors, with more than 40% of tumors demonstrating mutational signatures associated with platinum or temozolomide chemotherapy and two tumors showing treatment-associated hypermutation. Immunogenomic profiling found a heterogenous pattern of neoantigen and MHC class I expression and a general absence of immune infiltration. Transcriptional analysis and functional gene set enrichment analysis identified cross-pathology clusters associated with development, immune signaling, and cellular signaling pathways. While the landscapes of these R/R tumors reflected those of their corresponding untreated tumors at diagnosis, important exceptions were observed, suggestive of tumor evolution, treatment resistance mechanisms, and mutagenic etiologies of treatment.
Significance:
Tumor heterogeneity, chemotherapy exposure, and tumor evolution contribute to the molecular profiles and increased mutational burden that occur in treatment-refractory and relapsed childhood solid tumors.
Introduction
Cancer represents the primary disease-related cause of death in children in the United States. Though incidence rates have been increasing over the last 45 years, overall survival rates for childhood cancers have significantly improved in recent decades (1). However, patients with relapsed solid tumors still have a poor prognosis despite intensification of therapies. Recent large-scale genomic profiling studies of pediatric cancer have expanded our understanding of the genomic drivers of childhood solid tumors and highlighted the fundamental differences from tumors arising in adults (2, 3). These studies have revealed the influences of cell-of-origin and evolutionary trajectory on the genomic landscapes of individual tumor types, and have implicated the utility for genomic profiling to inform disease classification, prognosis, and treatment. However, these studies have largely focused on untreated tumors at the time of initial diagnosis, and the genomic and transcriptomic landscapes of relapsed or refractory (R/R) childhood solid tumors are less well characterized (4, 5).
For many tumor types, relapsed tumors tend to bear unique molecular features relative to tumors at diagnosis, such as increased mutation burden, while refractory tumors may bear unique resistance drivers. For example, comparison of diagnostic and relapsed neuroblastoma has shown a significantly higher burden of somatic mutations at relapse. Relapsed neuroblastoma tumors frequently retain ALK mutations and MYCN amplification found in initial tumors, but also show enrichment for RAS/MAPK pathway activating events (6). Given intratumoral heterogeneity and tumor evolution, a better understanding of the genomic characteristics of the R/R disease state is needed to inform treatment and ultimately improve clinical outcomes.
Here, we describe the genomic and transcriptomic landscapes of R/R childhood solid tumors [neuroblastoma, sarcomas, central nervous system (CNS) tumors, and other rare tumors] based on tumor-normal whole-exome sequencing (WES) and tumor mRNA-sequencing analysis of 250 tumors from 202 patients profiled within precision medicine studies conducted by the Beat Childhood Cancer (BCC) Consortium.
Materials and Methods
Cohort description
Following written informed patient and/or legal guardian consent and assent, as appropriate, pediatric (1–14 years of age) and adolescent young adult (AYA) patients (15 years and older) with R/R solid tumors were enrolled in one of three Institutional Review Board–approved multi-institutional BCC Consortium precision medicine clinical trials: MGT (NCT01355679), MGT8 (NCT01802567), or MGT9 (NCT02162732). These trials were conducted in accordance with the Declaration of Helsinki and the guidelines of Good Clinical Practice. Written informed consent was obtained for all participants.
Sample processing and analysis
Fresh frozen tumor tissue and whole blood samples were deidentified and shipped to Ashion Analytics (http://www.ashion.com), a College of American Pathologists–accredited, Clinical Laboratory Improvement Amendments–certified laboratory, for tumor-normal WES and tumor mRNA sequencing. Following confirmation of at least 50% viable tumor by local pathology review, tumor DNA and RNA were extracted using the Qiagen AllPrep Kit. Constitutional DNA was extracted using the Qiagen Gentra Puregene Blood Kit. Tumor and constitutional DNA samples were subjected to library construction using Kapa Biosystems’ Kapa Hyper Prep Kit or the Agilent Library Prep Kit, and exome capture performed using a custom Agilent SureSelect target enrichment system with whole-exome and genome-wide copy-number probes. For RNA sequencing (RNA-seq), library construction was performed using Illumina’s TruSeq RNA Sample Preparation V2 Kit. All libraries were sequenced on the Illumina HiSeq2500 with 100 bp paired-end reads. Data were aligned to build 37 of the human reference genome. For the MGT9 cohort, the mean target exome coverage was 455× for mapped tumor reads averaged and 213× for. Constitutional Samples The tumor RNA mapped reads averaged 245M. Samples collected under the MGT and MGT8 studies were sequenced in the research setting at TGen, as described previously (7). Briefly, library construction was performed with the Illumina TruSeq RNA Sample Preparation V2 Kit. Exome sequencing library preparation was performed with the TruSeq DNA Exome V1 kit. Sequencing was performed on the HiSeq2000 with 50 bp paired-end reads. The mean target exome coverage was 54× (tumor) and 71× (constitutional), and the average number of mapped tumor RNA reads was 121M. Sequencing metrics for each biopsy are summarized in Supplementary Table S1.
Somatic and germline variant analysis
Somatic single-nucleotide variants (SNV) and small indels were identified using a consensus approach requiring calls by two of three variant callers: MuTect (RRID:SCR_000559), Strelka (RRID: SCR_005109), and Seurat (8). Filters included a coverage depth of at least 10× in both tumor and normal, an allele frequency of at least 5% in the tumor and less than 2% in the constitutional sample, and a quality score of at least 15. Indels were filtered at a quality score of 25. Germline variant calling was performed using haplotype caller, filtering based on haplotype caller quality score >500, frequencies of 2.5% or less in 1000 Genomes and ExAC, and snpEff annotated impact of significant. Germline variants in 157 genes previously implicated in cancer predisposition were evaluated (2), filtering for variants classified as pathogenic or likely pathogenic based on ClinVar annotations (http://clinvar.com/) or prior clinical germline testing. Copy-number variants were detected using a read depth–based comparative method (https://github.com/tgen/tCoNuT), filtering for copy-number events with a length less than 25 Mb and an absolute log2 fold change greater than 1. Chromosome arm-level events were called using ExomeCNV (RRID:SCR_010815), filtering for regions containing 50 or more genes with a log2 ratio of tumor to matched normal of > = 0.5 (gain) or < = −0.5 (loss). Neighboring events within an arm were combined. Chromosome arm-level events were called if the gain or loss covered > = 50% of the arm. Events seen in > = 15 tumors were included in the oncoprint. The data were also analyzed through a secondary analysis pipeline (4). Results are available on the user-friendly clinomics web interface (https://clinomics.ccr.cancer.gov/clinomics/public/).
GISTIC2.0 (9) was run on the GenePattern server using the following parameters: amplification and deletion thresholds set to 0.1, join segment size of 4, Q value threshold = 0.25, confidence level = 0.99, focal length threshold = 0.5 with values capped at 1.5 and max number of segments capped at 2,500. Arm peel and broad analysis settings were employed and median sample center and extreme gene collapse methods were used. Somatic tumor mutation burden (TMB) was evaluated using an in-house tool to calculate the number of somatic point mutations per callable haploid megabase (Mb). Microsatellite instability (MSI) was assessed using MSIsensor v0.5 (RRID:SCR_006418), run with default parameters on tumor-normal exome binary alignment maps following removal of flagged sequencing duplicates. Tumors with a MSIsensor score of 3.5 or greater were considered positive for MSI. Allele specific copy-number segmentation files and tumor ploidy estimates were generated with Sequenza (RRID:SCR_016662). Genomic scars were assessed using scarHRD (10). A score of 42 or greater was used as the threshold for homologous recombination repair deficiency (HRD; ref. 11). For patients with multiple tumors profiled, the last longitudinal sample, or, in the case of multiple samples from the same timepoint, the sample with the greatest tumor content estimate by Sequenza, was used for cohort level comparisons.
Fusions were called using TopHat (v2.0.8b, RRID:SCR_013035), with a quality score cutoff of 100 or a consensus fusion-calling approach using FusionCatcher (RRID:SCR_000060), STAR-fusion (12), and tophatFusion, filtering for fusions called by at least two callers (4). For gene expression analysis, the conversion of normalized probe set intensities to a relative measure of transcript abundance was performed. Transcripts per million reads (TPM) values were generated using the alignment-free tool Kallisto (RRID:SCR_016582). Cell-type enrichment analysis and immune scoring were performed using xCell (13).
Genes implicated in pediatric cancer were manually curated from the published literature (2, 3, 14), and prioritized based on annotation as a Catalogue of Somatic Mutations in Cancer (COSMIC) cancer gene census gene, prior identification of pathognomonic alterations in pediatric solid tumors, frequency of alterations in pediatric solid tumors, and detection of a hotspot mutation or loss-of-function mutation in a COSMIC cancer oncogene or tumor suppressor gene (TSG), respectively.
Mutational signature analysis
Mutational signature analysis was performed for tumor samples with a minimum of 50 total somatic mutations, using sigProfiler (15, 16) with COSMIC signatures v3.1 under default parameters. The proportion of mutations contributing to each signature was calculated within a sample and then averaged within tumor types.
Neoantigen analysis
Six-digit HLA calls for HLA-A, HLA-B, and HLA-C were determined using bwa-kit (https://github.com/lh3/bwa/tree/master/bwakit), seq2HLA 2.2 (17) and phlat (18) and analysis of HLA expression was performed using seq2HLA 2.2. Mutant peptide sequences corresponding to somatic nonsynonymous coding variants were generated using varcode (19). MHC class I binding affinity predictions of 8- to 11-mer mutant peptides to the HLA genotypes were performed using NetMHCpan 4.0 (20) for each somatic mutation and corresponding wildtype peptide across all HLA genotypes in the cohort. Patient harmonic mean best rank scores were calculated to collapse predictions to a single value per mutation per patient (21). Expression of predicted neoantigens was determined using the detection of the alternate allele expression in the RNA-seq data. Predicted strong-binding neoantigens, defined as a harmonic mean best rank score of <0.5, were used for correlative analysis. Rank order of median HLA-A, HLA-B, and HLA-C expression and number of expressed neoantigens across tumor groups was used to define high (top four tumor groups), moderate (middle five tumor groups), or low (bottom five tumor groups) categories. For cohort analysis, tumors with elevated HLA expression were defined as those in the top 10% of reads per kilobase of transcript, per million mapped reads values for HLA-A, HLA-B, or HLA-C, and low expression was defined as tumors in the bottom quartile in expression for all three genes.
Hierarchical classification based on genome-wide expression
A SD approach was used to select highly variable genes across the cohort of 245 tumors with RNA-seq data. Highly variable genes were defined as genes with SD ≥1.5 and mean expression of log2(TPM+1) ≥1. Of the 2,199 genes in this group, 95 were COSMIC cancer genes (COSMIC v85) and 11 were housekeeping genes (22). Using this approach, we excluded genes uniformly expressed in a majority of cells while reducing the stochastic noise associated with low copy transcripts to capture the most significant differential gene expression signatures among the individual tumors. Unsupervised hierarchical clustering was performed using pvclust (23) based on TPMs of the 2,199 selected genes using the Spearman correlation distance and average method. DESeq2 analysis (24) was performed on three major groups (one vs. the other two groups) and the significantly differentially expressed genes were selected using log2FC ≥ 1|≤ −1 and Padj < 0.05. For cross-pathology subcluster analysis, DESeq2 analysis was performed on each subcluster (C3a, C3b, C3c) compared with all other samples. Ingenuity Pathway Analysis (RRID:SCR_008653) gene network analysis was performed using highly expressed genes (TPM ≥ 7) as input to identify common shared pathways and functional relationships.
Gene set variation analysis
TPM values from Kallisto were used to evaluate 50 hallmark gene sets from the Molecular Signatures Database (MSigDB) collection (25). The output of gene set variation analysis (GSVA, RRID:SCR_021058) is a projection of each sample with each hallmark set. The resampling-based approach ConsensusClusterPlus (v1.42.0) was used to cluster the samples x projections. Utilizing PAM, a medoid-based clustering approach (26), consensus clustering was run for 1,000 iterations with Pearson correlation as the distance (27). The run computed class assignments for an increasing number of clusters (K), from 2 to 15. Silhouette scores were computed to decide the optimum number of clusters. Specific hallmark markers for each cluster were identified using a generalized linear model with L2 regularization. Hallmark markers with a P < 0.05 were deemed to be predictive for a given class.
Longitudinal analysis
For the patients with longitudinal samples, joint somatic variant and copy-number analysis was performed using lumosVar2 (28), using a tumor-normal mode where the sample fraction of clonal variant groups is set to zero in the constitutional sample. After correcting for copy-number state, somatic variant allele fractions and read depths were imported into CANOPY for phylogenetic analysis (29). The lumosVar2 clonal variant groups were used to seed the variant grouping in CANOPY. TIMESCAPE was used to visualize the resulting trees using the clonal trajectory view (http://bioconductor.org/packages/release/bioc/html/timescape.html).
Data accessibility statement
This study has been deposited in the database of Genotypes and Phenotypes (dbGaP) under accession number phs002238.v1 (http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs002238.v1.p1). Variant calls from secondary analysis with the NCI’s clinomics analysis pipeline are available on the clinomics web interface (https://clinomics.ccr.cancer.gov/clinomics/public/).
Results
Clinical and sequencing characteristics of R/R tumors profiled within the BCC Consortium
To evaluate the genomic and transcriptomic landscape of R/R childhood solid tumors, paired tumor-normal WES and/or tumor mRNA sequencing were performed for 202 pediatric (n = 173) and AYA (n = 29) patients with R/R solid tumors who were enrolled in a BCC Consortium clinical trial (NCT01355679, NCT01802567, NCT02162732) between May 2011 and June 2018. Patients with R/R disease were enrolled following standard-of-care therapy with the exception of patients for whom no established curative standard of care was available (i.e., H3K27M-mutant diffuse midline gliomas, DMG-K27M), who were enrolled at diagnosis (n = 4). Cohort demographics and clinical characteristics are summarized in Table 1 with extended annotation in Supplementary Table S1. The cohort comprised 121 males (60%) and 81 females (40%), with a median age at diagnosis of 6 years (range, <1–20 years). The median time from diagnosis to enrollment on one of these trials was 2 years (range, 0–19 years). Forty-six tumor types are represented, grouped into four general tumor categories: sarcomas (36.1%; n = 73), neuroblastoma (29.2%; n = 59), CNS tumors (23.3%; n = 47), and other rare tumors (11.4%; n = 23). Tumors were grouped into fourteen categories representing the most common tumor types in the cohort: neuroblastoma, ependymoma, glioblastoma (GBM), DMG-K27M, osteosarcoma, Ewing sarcoma, rhabdomyosarcoma (RMS), desmoplastic small round cell tumor (DSRCT), undifferentiated sarcoma, hepatoblastoma, malignant peripheral nerve sheath tumor (MPNST), other CNS tumors, other sarcomas, and other rare tumors, where tumor types with fewer than four tumors are grouped into the other tumor category based on general tumor type (detailed tumor types are listed in Supplementary Table S1). 215 tumors were profiled with both tumor-normal WES and tumor mRNA sequencing, five tumors had tumor-normal WES alone, and 30 tumors had mRNA-sequencing alone. A total of 184 patients had WES data available for at least one tumor and 200 patients had RNA-seq data available for at least one tumor. In total, 250 biopsies from 202 patients were sequenced, including spatial and temporal sampling for a subset of patients.
Table 1.
Relapsed and refractory pediatric and AYA cancer cohort profiled in the BCC Consortium.
| Characteristic | n (%) |
|---|---|
|
| |
| Unique patients | 202 |
| Age at diagnosis (years) | |
| 0–4 | 75 (37.1%) |
| 5–9 | 49 (24.3%) |
| 10–14 | 48 (23.8%) |
| 15–20 | 29 (14.3%) |
| Unknown | 1 (0.5%) |
| Gender | |
| Male | 121 (59.9%) |
| Female | 81 (40.1%) |
| Tumor category | |
| Sarcoma | 73 (36.1%) |
| Neuroblastoma | 59 (29.2%) |
| CNS tumor | 47 (23.3%) |
| Other rare tumor | 23 (11.4%) |
Landscape of somatic mutations
To evaluate the landscape of somatic mutations in R/R childhood tumors, we analyzed mutational features for the cohort of patients with WES data (n = 184). The median somatic TMB was 1.4 mutations per Mb (range: 0.02–56.3; Supplementary Table S1). Mutation burden was highest in neuroblastoma (median: 3.4, range: 1.1–56.3, P < 0.005, Welch t test) followed by sarcomas (median: 1.2, range: 0.02–5.8), other rare tumors (median: 1.0, range: 0.1–9.1), and CNS tumors (median: 0.7, range: 0.1–10.9; Fig. 1A). Within specific tumor type categories, ependymoma bore the lowest and neuroblastoma bore the highest median TMB (0.5 and 3.4, respectively; Fig. 1B). Gröbner and colleagues proposed pediatric mutational burden thresholds, using 2 mutations per Mb to define tumors with high pediatric TMB and 10 mutations per Mb to define hypermutant tumors (2). Applying these thresholds to this R/R cohort, 33.9% of tumors were characterized as having a pediatric high TMB (>2 mutations/Mb). Two additional tumors were classified as hypermutated (TMB >10 mutations/Mb): a neuroblastoma tumor and an anaplastic astrocytoma.
Figure 1.
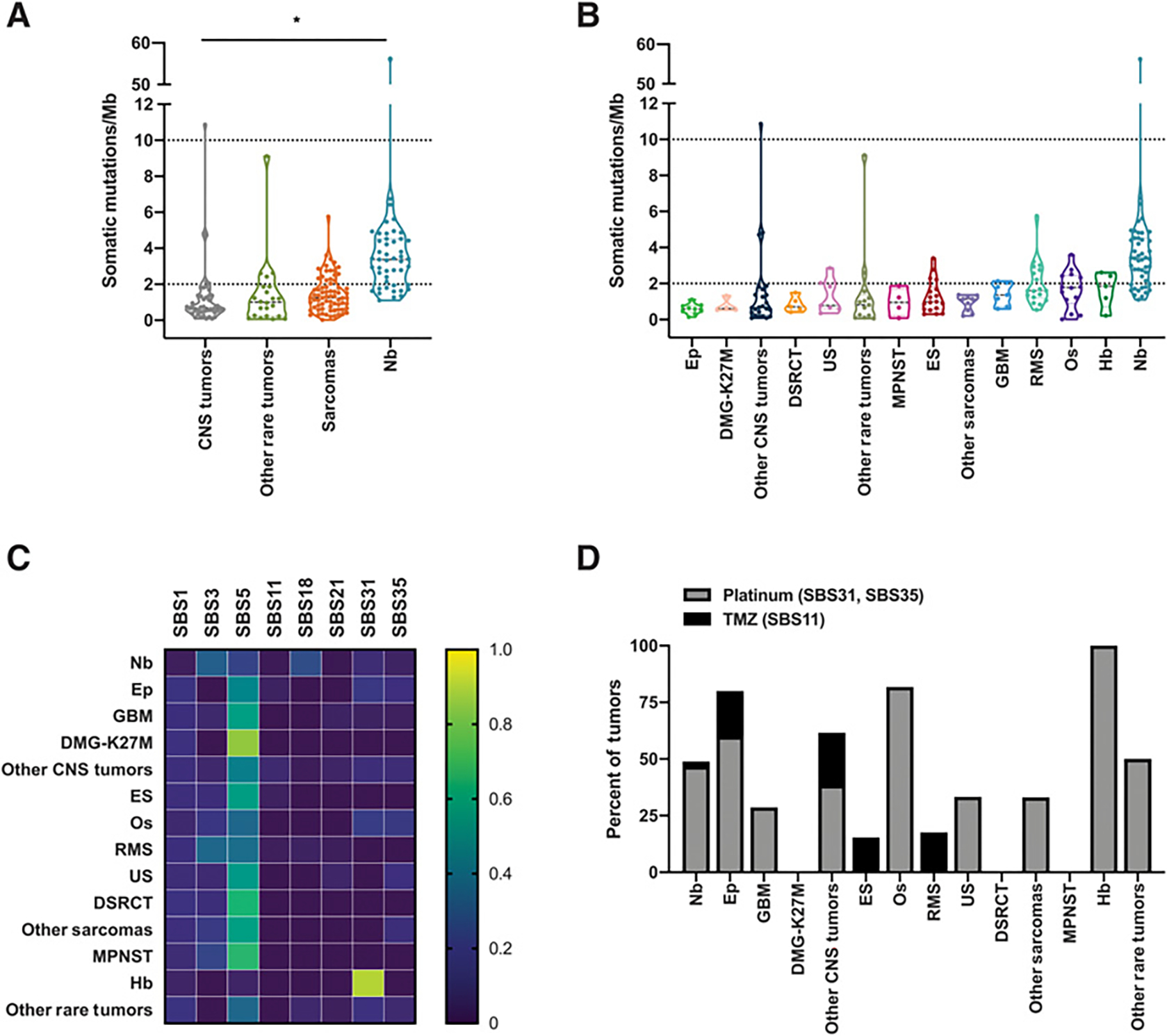
Somatic mutation burden and mutation signatures in relapsed and refractory childhood solid tumors. Somatic mutation burden and mutation signatures for patients with matched tumor and normal WES. The last biopsy is shown for patients with multiple biopsies. A, TMB for general tumor type categories for 184 patients with matched tumor and normal WES. Dashed lines indicate the thresholds for pediatric high TMB (2 or more mutations/Mb) and pediatric hypermutant (10 or more mutations/Mb) tumors. B, TMB for specific tumor type categories for each of 184 patients. Tumor types with at least 4 patients are shown, and tumor types with fewer than 4 distinct patients are grouped into “Other CNS Tumors,” “Other Sarcomas,” and “Other Rare Tumors.” Tumor types are presented in ascending median TMB order. Ep, ependymoma; DMG-K27M, H3K27M-mutant diffuse midline gliomas; DSRCT, desmoplastic small round cell tumor; US, undifferentiated sarcoma; MPNST, malignant peripheral nerve sheath tumor; ES, Ewing sarcoma; GBM, glioblastoma; RMS, rhabdomyosarcoma; Os, osteosarcoma; Hb, hepatoblastoma; Nb, neuroblastoma. Dashed lines indicate the thresholds for pediatric high TMB (2 or more mutations/Mb) and pediatric hypermutant (10 or more mutations/Mb) tumors. C, Mutation signatures reflective of the trinucleotide context of somatic SNVs in the 146 cases with more than 50 somatic SNVs. The proportion of mutations mapping to the indicated COSMIC mutational signatures, averaged within each tumor type, is displayed. D, The frequency of tumors with treatment-associated mutational signatures (SBS11, SBS31, SBS35) is displayed.
The frequency of somatic SNVs in their trinucleotide context can provide clues to mutational etiology (15). Thus, we investigated somatic mutation signatures using the COSMIC reference signatures (SBS, DBS, ID) in tumors with a minimum of 50 somatic mutations (n = 146 tumors; Supplementary Table S2A–S2C). Most tumors (94.5%, 138/146) bore more than one mutation signature (Supplementary Table S2A), suggesting multiple mutagenic processes influenced the tumor mutational landscape, consistent with prior studies (30, 31). The frequency of mutations associated with specific mutational signatures within each tumor type is shown in Fig. 1C. The clock-like age-related signatures (SBS1, SBS5) were the most common signatures across the full cohort, present in 84.9% of tumors (124/146) though less frequently in neuroblastoma compared with other tumors (68.1% vs. 92.9%), consistent with the younger median age of diagnosis of neuroblastoma. Signature SBS3, associated with homologous recombination-based double-stranded DNA break repair defects, was the second most common signature across tumor types (35.6% of all tumors, 11.1% CNS, 59.6% neuroblastoma, 35.1% sarcoma, 6.7% rare tumors). SBS18, linked to reactive oxygen species-induced DNA damage, was seen predominantly in neuroblastoma (72.3%) as well as in a subset of RMS (17.6%) and a Wilms tumor, hepatocellular carcinoma, and a CNS embryonal tumor.
As patients in this cohort had received prior chemotherapy treatment, we evaluated the frequency of therapy-associated mutational signatures. A total of 43.2% of tumors had evidence for mutational signatures associated with prior chemotherapy exposure, a majority of which were associated with platinum chemotherapy (36.3%, SBS31, SBS35) and a smaller subset associated with temozolomide (TMZ) treatment (6.8%, SBS11; Fig. 1D; Supplementary Table S2A). Platinum-associated signatures were most frequently seen in hepatoblastoma, osteosarcoma, and ependymoma. Signature SBS11, a mutational pattern associated with TMZ treatment, was highly enriched in the two hypermutated tumors. Both patients had received prior treatment with TMZ. Nearly half of the mutations in these hypermutant tumors were attributed to the TMZ-associated mutational signature, consistent with TMZ-induced hypermutation.
MSI, reflected in the loss or gain of nucleotides in short repetitive sequences due to defective DNA mismatch repair, was evaluated using MSIsensor (32). Five tumors representing five different tumor types were found to be MSI positive (2.7%, 5/184; Supplementary Table S1). Three of these tumors also showed evidence for mutational signatures associated with mismatch repair deficiency (ID1; Supplementary Table S2C). The sample with the highest number of mutations attributed to ID1 was a choroid plexus carcinoma (CPC). In addition to being characterized as MSI positive with mismatch repair deficiency signature ID1, this CPC tumor had a pediatric high TMB (4.7 mutations/Mb) and a somatic deep deletion in the DNA repair gene PMS2, providing strong support for mismatch repair deficiency in this tumor.
Landscape of somatic copy-number alterations
To characterize the landscape of somatic copy-number alterations in R/R childhood solid tumors, we evaluated large-scale and focal copy-number alterations in the BCC cohort. A total of 27% of samples were characterized as hyperdiploid (ploidy of three or greater; Supplementary Table S1), with enrichment for TP53 mutations in hyperdiploid tumors (P = 0.0025). Genomic scars indicative of genomic instability and HRD were assessed using scarHRD, which combines measures of loss of heterozygosity, telomeric allelic imbalances, and large-scale state transitions (10). A total of 13.6% (25/184) of tumors showed evidence for genomic scars (Supplementary Table S1), with increased frequency in sarcomas (21.9%, 16/73) compared with neuroblastoma, CNS tumors, or other rare tumors (4.3%–8.7%), and an overall association with TP53 alterations (P = 0.0003). Eight tumors showed HR deficiency by both scarHRD assessment and mutational signature analysis: three osteosarcoma, three neuroblastoma, one RMS, and one undifferentiated sarcoma. 11q harbors several DNA repair genes, with 11q deletion occurring in a subset of neuroblastoma. While a majority of neuroblastoma tumors with 11q deletion showed signals of HR deficiency (presence of SBS3 mutational signature or scarHRD scores in the top quartile of all tumors), these associations did not reach statistical significance.
Recurrent chromosomal arm-level events are shown in Fig.2A. The most frequent arm-level event was 17q gain in 20% (36/184) of tumors, predominantly within neuroblastoma (66%, compared with 2%–9% for CNS tumors, sarcomas, and other rare tumors). R/R neuroblastoma also showed enrichment for 3p loss and 11q loss. Gains of 5p and 8q (containing TERT and MYC) showed enrichment within sarcomas and were seen across sarcoma subtypes (osteosarcoma, Ewing sarcoma, DSRCT, RMS, and other sarcomas). After 17q gain, 1q gain was the second most frequent arm-level event (14%), and was seen across multiple tumor types, with similar frequency (11%–18%) within neuroblastoma, CNS tumors, sarcomas, and other rare tumors. Other pan-cohort chromosome arm-level events included 10p, 10q, 17p, and Yp loss. TP53 mutations were enriched in tumors with 17p deletion (P < 0.0001), suggesting biallelic alteration of TP53 in these tumors.
Figure 2.

Genomic landscape of relapsed and refractory childhood solid tumors. The genomic landscape of likely pathogenic driver mutations in 184 patients with BCC with tumor and matched normal WES. A, Somatic coding mutation burden is shown as a per-patient bar graph. Chromosome arm-level gains or losses, present in atleast 15 patients within the cohort, are displayed. Cancer genes bearing likely pathogenic germline SNVs and somatic SNVs, CNVs, SVs, and fusions are ordered according to general tumor type and frequency. Hotspot activating mutations include ACVR1 (R206H), ALK (F1174L, I1171T, R1275Q), BRAF (V600E), CTNNB1 (D32V), EGFR (exon 20 insertion mutation—A767_V769dup, tandem kinase domain duplication), FBXW7 (R465H), FGFR4 (V550L/E), H3F3A (K27M), HRAS/KRAS/NRAS (G12D, G13C/R, Q61K, A146T), MYCN (P44L), PDGFRA (V561D), PIK3CA (E542K, E545K, H1047R), PIK3R1 (N564D), PPM1D (exon 6 truncating mutation, S421*, E423fs, E525*), and PTPN11 (A72D, G503V, T507K). B, Frequency of gene alterations according to general tumor type. C, Venn diagram indicating intersection of cancer gene mutations across general tumor types.
GISTIC2.0 (9) was used to identify statistically significant recurring somatic copy-number events within the cohort. 42 recurrently amplified regions and 71 recurrently deleted regions were identified (Supplementary Fig. S1; Supplementary Table S3A; Supplementary Table S3B). Seven oncogenes contained within six regions of gain were identified: MYCN (2p24.3), TERT (5p15.33), CARD11 (7p22.3), RECQL4 (8q24.3), CCND1 (11q13.3), and MAPK1 and DGCR8 (22q11.23). Twelve TSGs contained in six regions of loss were identified. Three of these regions contained a single TSG, namely TP53 (17p13.1), CDKN2A (9p21.3), and SDHA (5p15.33), whereas other regions contained more than one TSG, including 3p12.3 (ROBO2, FHIT), 4q35.1 (CASP3, FAT1), and 15q25.2 (FES, BLM, CHD2, POLG, PML).
Landscape of somatic alterations in cancer genes
We next evaluated the overall landscape of somatic alterations (SNVs, indels, focal copy-number events, and fusions) within the cohort, focusing on genes previously implicated in pediatric solid tumors (2, 3, 14). 78.3% (n = 144/184) of tumors bore a somatic alteration in at least one pediatric cancer gene, with the highest proportion being sarcomas (59/71; 83.1%), followed by neuroblastoma (39/47; 83.0%), CNS tumors (31/44, 70.5%), and other rare tumors (15/22, 68.2%), (Fig. 2A; Supplementary Table S4).
Over one-third (39.1%; 72/184) of the cohort bore oncogenic fusions and/or oncogenic/likely-oncogenic hotspot mutations in one of these known cancer genes. Pathognomonic fusions were identified in 25% (n = 46/184) of tumors, occurring most frequently in sarcomas. All Ewing sarcoma tumors contained EWSR1-FLI1 (17/18) or EWSR1-ERG (1/18) fusions. EWSR1-WT1 fusions were present in all five DSRCTs. PAX3-FOXO1 fusions were detected in one-third of all RMS (6/18), including five alveolar RMS and one RMS NOS. A PAX7-FOXO1 fusion was detected in one additional RMS sample with RNA-seq. Other pathognomonic fusions in sarcoma samples included the SS18-SSX2 or SS18-SSX1 fusions in spindle cell synovial sarcomas (3/3), an HEY1-NCOA2 fusion in a mesenchymal chondrosarcoma, and a SUZ12-DNAH2 fusion in an undifferentiated sarcoma. In CNS tumors, C11orf95-RELA fusions were identified in ependymoma (3/10, 30%). Over half of the astrocytomas contained receptor tyrosine kinase or MAPK pathway fusions (FGFR1-TACC1 fusion in an anaplastic astrocytoma tumor, EEF1G-ROS1 fusion in a desmoplastic infantile astrocytoma, KIAA1549-BRAF fusion in a juvenile pilocytic astrocytoma, QKI-RAF1 fusion in a pilomyxoid astrocytoma). A TMEM165-PDGFRA fusion was detected in a GBM. In other rare tumors, a pathognomonic PRCC-TFE3 fusion was detected in a renal cell carcinoma. TPM3-NTRK1 and NRBF2-MN1 fusions were iden-tified in tumors labeled as MPNST and hepatocellular carcinoma, respectively.
Recurrent hotspot mutations (33) were identified in 30 of 184 (16.3%) tumors. The majority of these hotspots were detected in tumor types where they had previously been described. For example, in-frame internal tandem duplication of the kinase domain of EGFR was identified in a kidney mesoblastic nephroma from a 1-year-old male. EGFR rearrangements are common events in these soft-tissue tumors in infants (34). In addition to known oncogenic mutations, truncating mutations in TSGs (ARID1A, ARID1B, ATRX, BCOR, BCORL1, NF1, PTEN, RB1, SMARCB1, TP53) were identified in tumors from 24 of 184 (13.0%) patients, with the highest frequency being in CNS tumors (9/44, 20.5%).
The most frequent focally amplified genes, based on sample level copy-number analysis, included MYCN, MYC, CDK4 and PPM1D. The most frequent focally deleted genes included CDKN2A, ATRX, RB1, TP53, and PTEN. Consistent with the known genomic landscape of these tumors, MYCN and PPM1D amplifications and ATRX deletions occurred predominantly in neuroblastoma, MYC amplifications occurred predominantly in osteosarcoma, RB1 deletions occurred primarily in sarcomas (osteosarcoma, RMS, Ewing sarcoma, DSRCT), and PTEN deletions occurred in CNS tumors and sarcomas. TP53 deletions and CDKN2A deletions occurred across multiple tumor types.
The most frequently altered genes across the cohort are shown in Fig. 2B. TP53, CDKN2A, PPM1D, RB1, NF1, and PIK3CA were among the genes altered across all four general tumor types (Fig. 2B and C). TP53 was the most frequently altered gene, with mutations or deletions detected in 11 tumor types.
Oncogenic events were also identified in rare tumors with limited prior genomic characterization and in tumor types where they have not previously been reported. An EGFR exon 20 insertion was identified in a bilateral thalamic grade II glioma in a 4-year-old male. EGFR exon 20 insertions are known oncogenic events that occur in adult non–small cell lung tumors, but are rare events in pediatric glioma, with a single case report to date describing this alteration in another child with bilateral thalamic glioma (35). A known activating mutation in NRAS (G13C) was identified in an MPNST tumor from a 3-year-old female. RAS pathway activation has been implicated in MPNST due to alterations in other MAPK pathway genes (NF1, PTPN11), though NRAS mutations have not been reported previously (36). An exon 6 truncating mutation (E423fs) in PPM1D was identified in an SS18-SSX2 fusion-positive synovial sarcoma tumor from an 18-year-old male. Truncating gain-of-function PPM1D mutations have been reported as oncogenic events that are mutually exclusive with TP53 alterations and implicated in suppression of DNA damage response in brainstem gliomas (37), but have not been previously described in sarcomas. A TPM3-NTRK1 fusion was detected in a tumor labeled as an MPNST. While NTRK fusions have been identified across a wide variety of pediatric and adult tumor types (38), to our knowledge, this represents the first report of an NTRK fusion in pediatric MPNST. An EEF1G-ROS1 fusion was identified in a desmoplastic infantile astrocytoma, adding to the suite of pediatric CNS tumors with reported ROS1 fusions (39).
Putatively pathogenic germline variants
Consistent with previous reports describing pathogenic germline variants in 7%–9% of pediatric cancer (2, 40), 16 of 184 (8.7%) patients in this R/R pediatric cancer cohort were found to have a pathogenic or likely pathogenic germline variant (n = 157 hereditary cancer genes evaluated, Fig.2A; Supplementary Table S5 and S6).The median age at diagnosis for patients with pathogenic germline variants was 12 years (range, 1–17).
Pathogenic germline mutations in TP53 were identified in 3 patients with osteosarcoma, RMS, and medulloblastoma tumors. Osteosarcoma is considered a sentinel cancer associated with TP53-mutant Li-Fraumeni syndrome (LFS), with 3.8% of patients with osteosarcoma younger than 30 years of age found to carry LFS-associated variants (41). In the BCC cohort, one of the 14 patients with osteosarcoma was found to have a known pathogenic germline variant in TP53; this patient was diagnosed with osteosarcoma at age 11. A pathogenic TP53 germline mutation was also identified in 1 patient with RMS (1/18, 5.6%). Consistent with previous reports of TP53 germline mutations occurring in children diagnosed with RMS at a young age (under 3 years at diagnosis; ref. 42), this LFS-associated RMS was diagnosed when the patient was 1 year old. One of the 2 patients with medulloblastoma in this cohort had a known pathogenic germline TP53 variant detected. This medulloblastoma tumor possessed a somatic truncating mutation in SUFU, a regulator of the sonic hedgehog pathway, consistent with previous reports that germline TP53 mutations typically occur within the sonic hedgehog subtype of medulloblastoma (43). However, this medulloblastoma tumor demonstrated 17p loss and a decreased TP53 mutation allele frequency in the tumor (9% compared with 50% in the peripheral blood sample), suggesting loss of the mutant allele in the tumor. Thus, the relevance of this germline TP53 mutation to development of the medulloblastoma tumor is not clear. A pathogenic germline PTPN11 mutation was identified in a patient with neuroblastoma. Germline variants in PTPN11 represent a genetic cause of Noonan syndrome and are associated with increased risk for development of a spectrum of cancers, including neuroblastoma (44). A pathogenic germline NF1 mutation was identified in a patient with MPNST. Patients with neurofibromatosis type 1, an autosomal dominant disorder caused by mutations in the NF1 gene, are at increased risk for developing MPNST (45). Pathogenic germline mutations in DNA repair genes (BRCA2, CHEK2, ERCC2, ERCC4, FANCA, MUTYH) were identified in 9 patients. A second hit in the tumor was not identified for any of these cases, thus the direct contribution of these variants to the development of these cancers is unclear.
Immunogenomic landscape
Given that somatic point mutations can give rise to novel antigens (neoantigens) that, in concert with variation in alleles and expression of HLAs, play a role in shaping immune interactions and immunotherapy responses in cancer, we performed analysis of neoantigen burden leveraging exome and RNA data and in silico predictions of MHC binding affinity. TMB positively correlated with neoantigen burden (Pearson correlation coefficient r = 0.9653; P < 0.0001; Supplementary Fig. S2A). Of the 9,658 nonsynonymous somatic mutations identified in this cohort, 2,756 (28.5%) were predicted to result in strong-binding neoantigens, of which, 55% (1,516/2,756) were detected as expressed in the RNA. Three expressed, strong-binding neoantigens (DMG-K27M, were GBM); DDX11 R186W, n = 2 (RMS, OS); MYCN P44L, n = 2 (neuroblastoma, Wilms tumor)], consistent with previous reports of few recurrent neoantigens across pediatric cancers (46). In the cohort overall, the number of expressed neoantigens increased based on time from initial diagnosis (one-way ANOVA, P = 0.0475; Supplementary Fig. S2B).
Nearly all (94%, 173/184) tumors had at least one predicted strong-binding neoantigen (Supplementary Table S1). The median number of predicted neoantigens in this R/R cohort was 9 (range, 0–400), higher than what has been previously reported for pediatric cancers at the time of diagnosis (46). Consistent with the increased mutational burden in neuroblastoma compared with other tumor type classes in this cohort, neuroblastoma samples showed the highest number of neoantigens (median 21; range, 2–400), followed by sarcomas (median 9; range, 0–51), other rare tumors (median 6; range, 0–107), and CNS tumors (median 4; range, 0–54; Supplementary Fig. S2C).
The median number of neoantigens that were detected as expressed in the RNA-seq data was 5 (range, 0–254), with neuroblastoma bearing the highest number (median neuroblastoma = 10; sarcomas = 4; other rare tumors = 4; CNS tumors = 2; Supplementary Fig. S2D). Eighty-three percent (153/184) of tumors had at least one expressed neoantigen, with 51.6% (95/184) of tumors having five or more expressed neoantigens. Neuroblastoma showed the highest frequency of multiple expressed neoantigens, with 87.2% (41/47) having five or more expressed neoantigens, compared with 47.9% (34/71) of sarcomas, 36.4% (8/22) of other rare tumors, and 27.3% (12/44) of CNS tumors.
A heterogeneous pattern of expressed neoantigens and MHC class I (HLA-A, HLA-B, HLA-C, B2M) expression was evident across tumor types (Fig. 3A–D; Supplementary Fig. S2E). DSRCT and ependymoma tumors showed low median numbers of expressed neoantigens and low MHC class I expression (relative to the cohort), whereas osteosarcoma, other CNS tumors, and other rare tumors generally showed low number of expressed neoantigens but high MHC class I expression. In contrast, the four tumor types with the highest median number of expressed neoantigens (neuroblastoma, RMS, MPNST, and other sarcomas) showed low to moderate HLA-A, HLA-B, and HLA-C expression. Cohort analysis identified ten tumors with both high neoantigen expression (five or more expressed strong-binding neoantigens) and elevated expression of HLA-A, HLA-B, and/or HLA-C: six neuroblastoma tumors and one each of astrocytoma, GBM, Ewing sarcoma, and ectomesenchymoma. A total of 28.8% (53/184) of this R/R cohort either lacked expression of a strong-binding neoantigen (n = 31/184) or exhibited low expression of HLA-A, HLA-B, and HLA-C (n = 24/184, including two tumors that also lacked neoantigen expression). Tumors lacking neoantigen expression spanned tumor types, including DSRCT (n = 3), Ewing sarcoma (n = 3), osteosarcoma (n = 3), and neuroblastoma (n = 3). Similarly, low MHC class I expression was detected across tumor types, including RMS (n = 7), neuroblastoma (n = 5), synovial sarcoma (n = 3), Wilms tumor (n = 2), and DSRCT (n = 2). Six tumors showed low B2M expression: RMS (n = 3), neuroblastoma (n = 1), embryonal tumor with multilayered rosettes (n= 1), and malignant myoepithelial sarcoma (n= 1; Supplementary Fig. S2E). Elevated RNA expression of the coinhibitory immune checkpoint molecule PDL1 (encoded by CD274) was seen in eight tumors, spanning CNS tumors, sarcomas, and other rare tumors (Supplementary Fig. S2F).
Figure 3.

Immunogenomic landscape of relapsed and refractory childhood solid tumors. A–D, Neoantigen burden and MHC class I expression for patients with tumor-normal WES and tumor mRNA sequencing. The last biopsy is shown for patients with multiple biopsies. Violin plots display the number of expressed, predicted strong-binding neoantigens (A), and HLA-A, HLA-B, and HLA-C expression (B–D) by tumor type. The median value for the entire cohort is indicated by a dotted line. Median values for individual tumor types are shown as a line within the violin plot, and individual values displayed as closed circles. Tumor types with at least 4 patients are shown, with tumor types with less than 4 patients grouped into “Other CNS Tumors,” “Other Sarcomas,” and “Other Rare Tumors.” E, Immune infiltration scores calculated from XCell based on bulk RNA-sequencing data.
Extrinsic factors, such as the tumor immune microenvironment, can influence disease progression and recurrence. To explore the tumor immune microenvironment, we used xCell (13) to infer the immune infiltrate abundance based on deconvolution of bulk tumor transcriptome profiling data. Ependymoma, GBM, DMG-K27M, RMS, DSRCT, and MPNST showed low immune infiltration scores. Other tumor types showed a mixture of immune “hot” and “cold” tumors, with osteosarcoma, neuroblastoma, and other rare tumors representing the highest median immune infiltration scores (Fig. 3E). xCell immune infiltration scores did not correlate with the number of neoantigens.
Gene expression signatures
To investigate gene expression patterns in R/R childhood solid tumors, we performed unsupervised hierarchical clustering analysis of the 2,199 most variable genes across the cohort of tumors with RNA-seq data (n = 245; Supplementary Fig. S3; Supplementary Table S7). Three primary clusters were identified, segregating predominantly according to general tumor types (Fig. 4A), supporting the major role of cell-of-origin and anatomic site in determining gene expression profiles even in the R/R setting.
Figure 4.
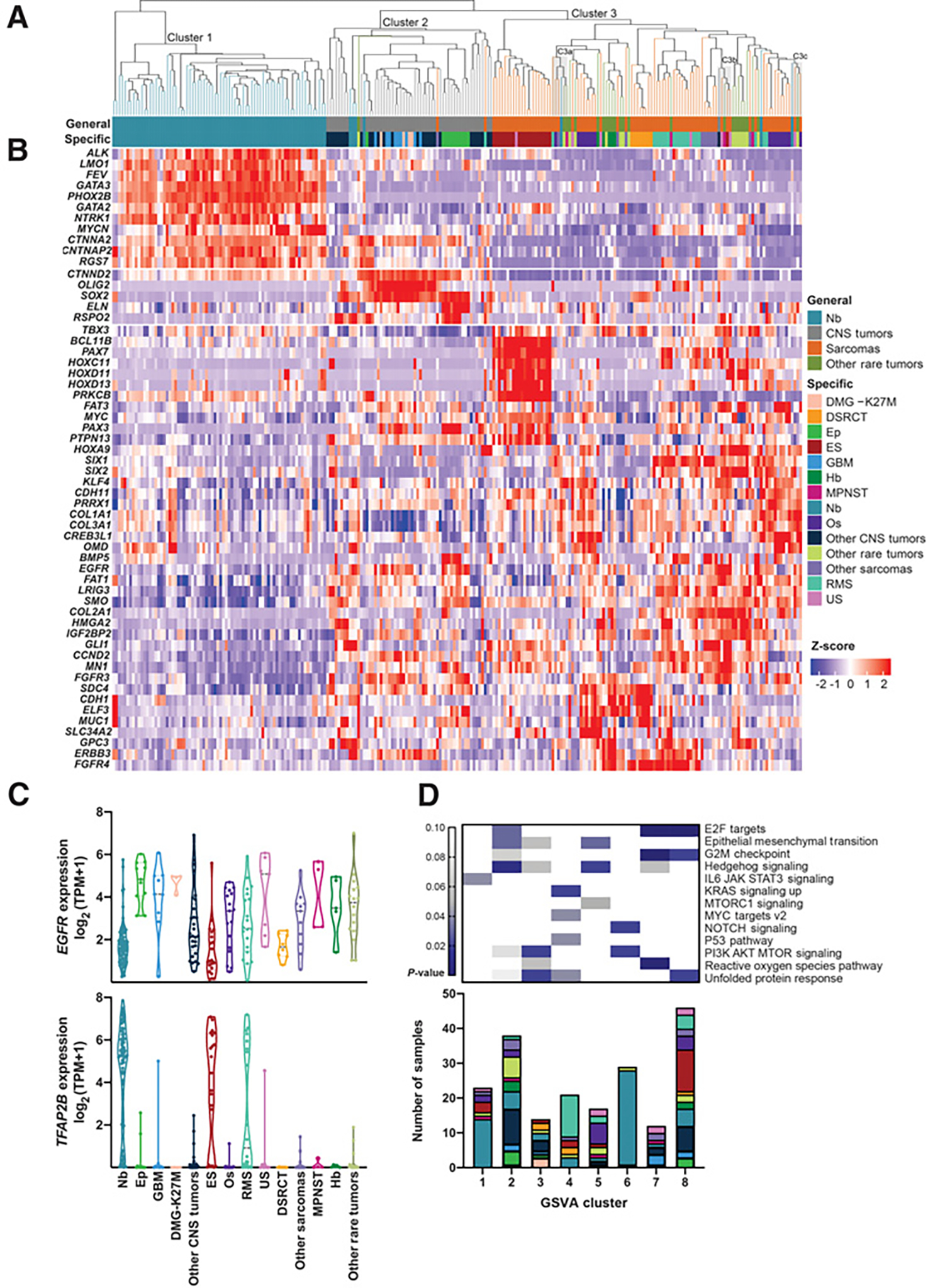
Gene expression patterns in relapsed and refractory childhood solid tumors. A, Unsupervised hierarchical clustering using 2,199 genes. Three main clusters were formed (Cluster 1, Cluster 2, Cluster 3). Three mixed-pathology subclusters within Cluster 3 with P values < 0.05 (C3a, C3b, and C3c) are shaded in gray. B, Heatmap of genes that are significantly differentially expressed in one group versus two others. COSMIC genes are represented here. C, EGFR and TFAP2B as selected examples of genes with cross-pathology expression patterns. D, P values for enrichment of cancer hallmark gene sets within the consensus clustered tumor groups (GSVA clusters 1–8; top). Hallmarks with significant enrichment within at least one group are shown. Tumor types associated with each GSVA cluster (bottom). Colors correspond to the specific tumor types as displayed in the legend for A.
Differential expression analysis was performed using DeSeq2 to identify the genes driving the three main clusters (Supplementary Table S8; Supplementary Table S9), with differentially expressed COSMIC cancer genes (COSMIC v85) shown in the heatmap in Fig. 4B. The most highly significantly differentially expressed gene in Cluster 1 (predominantly neuroblastoma) was PHOX2B (log2FC 8.4), a homeobox transcription factor that plays a role in differentiation of cells in the sympathetic nervous system. PHOX2B has been shown to bear pathogenic variants in hereditary and sporadic neuroblastoma (47, 48). Additional significantly differentially expressed genes in Cluster 1 included MYCN, ALK, GATA3, and LMO1, all of which have been implicated in neuroblastoma (49). Cluster 2 (predominantly CNS tumors) showed high expression of OLIG2 and SOX2, two transcriptional regulators implicated in CNS tumor biology (50). Differentially expressed genes in cluster 3 (predominantly sarcomas and other rare tumors) included HOX genes (HOXD11, HOXD13, HOXC11, and HOXA9), other transcriptional regulators (including PAX3, PAX7, SIX1, SIX2, TBX3, GLI1, and MYC), and receptor tyrosine kinases (FGFR3, FGFR4, EGFR, and ERBB3). Significantly differentially expressed transcription factors and drug targets are shown in Supplementary Fig. S4.
Tumor types largely clustered together within these three main clusters. 76/83 (92%) of the neuroblastoma samples clustered together in the main neuroblastoma group. All GBM, DMG-K27M, and astrocytomas clustered together within the CNS tumor cluster. A total of 20 of the 21 (95%) Ewing sarcoma clustered together within a sarcoma/rare tumor subcluster. A total of 15 of the 20 (75%) RMS clustered within a sarcoma/rare tumor subcluster. All (8/8) of the DSRCTs clustered together within a sarcoma/rare tumor subcluster. Four of the five (80%) hepatoblastoma samples clustered together within a sarcoma/rare tumor subcluster. The ependymoma tumors clustered in two main groups, with all three RELA-fusion positive ependymoma clustered within one CNS tumor subcluster and seven of eight fusion-negative ependymoma in a second CNS tumor subcluster. Similarly, 15 of the 17 osteosarcomas cluster in two main sarcoma/rare tumor subclusters with a trend toward association of the clusters with TP53 mutation status. Osteosarcoma subcluster 1 was predominantly TP53 wildtype whereas osteosarcoma subcluster 2 was predominantly TP53 mutant/deleted (1/7 vs. 6/8; P = 0.10). In contrast to the other tumor types, undifferentiated sarcoma and MPNSTs did not cluster by tumor type.
Although general tumor types largely drove major classifications with the most variably expressed genes likely due to cell-of-origin effects, in some cases a tumor type or set of tumors deviated from pathology boundaries. Eleven individual tumors were identified that broke tumor type boundaries across clusters, including four neuroblastoma tumors that clustered with CNS tumors, three neuroblastoma tumors that clustered with sarcomas and other rare tumors, two sarcomas (RMS and ectomesenchymoma) that clustered with CNS tumors, a neuroglial tumor that clustered with sarcomas/other rare tumors, and a teratoma that clustered with CNS tumors. Interestingly, three mixed pathology subclusters were identified within Cluster 3 (Fig. 4A: C3a, C3b, and C3c; P < 0.05). No genomic alterations were enriched within this mixed pathology subcluster. DESeq2 differential expression analysis was performed to compare each subcluster to all other samples. Significantly differentially expressed genes are represented in Supplementary Table S10, and genes that were uniformly highly expressed (TPM ≥ 7) within a cluster are shown in Supplementary Table S11. C3b, composed of five tumor samples representing four tumor types (glioma, Ewing sarcoma, MPNST, and two undifferentiated sarcoma), showed elevated expression of genes associated with nervous system development and MAPK signaling, including the tyrosine kinase receptors PDGFRA, EGFR, and NTRK3.
To further investigate genes with cross-pathology expression patterns, we focused on the subset of the most variable genes that showed upregulation in more than one main cluster, suggestive of potential cross-pathology oncogenic functions. Ten of these genes are classified as oncogenes in the COSMIC cancer gene census (Supplementary Table S9), including CCND2, EGFR, and FGFR3. EGFR showed a bimodal expression pattern, with elevated expression in ependymoma, GBMs, DMG-K27M gliomas, and in a subset of undifferentiated sarcoma, RMS, osteosarcoma, Ewing sarcoma, and neuroblastoma (Fig. 4C, top). Many genes with cross-pathology expression patterns are not known oncogenes but have been implicated in cancer. For example, TFAP2B, a transcription factor involved in regulating embryonic development, demonstrated bimodal expression within this R/R childhood cancer cohort, with elevated expression in a subset of neuroblastoma, Ewing sarcoma, and RMS tumors, as well as in individual tumors across the cohort, including undifferentiated sarcoma, ependymoma, and GBM tumors (Fig. 4C, bottom). While the function of TFAP2B in childhood cancer is largely undefined, TFAP2B has been identified as a downstream target of the PAX3-FOXO1 and PAX7-FOXO1 fusions in RMS and shown to promote cancer cell survival in this and other cellular contexts (51, 52), suggesting potential relevance for elevated TFAP2B expression in R/R childhood tumors beyond RMS.
GSVA
To evaluate shared functional features and pathway activity across R/R tumors, we performed GSVA utilizing the MSigDB cancer hallmarks gene set. Clustering by cancer hallmark signatures identified eight clusters each composed of multiple tumor types (Fig. 4D; Supplementary Fig. S5A). Neuroblastoma samples clustered predominantly in clusters 1 (not significantly enriched) and 6 (P < 0.001), RMS were enriched in cluster 4 (P < 0.001), and Ewing sarcoma were enriched in cluster 8 (P = 0.035). Clusters 2, 3, 5, and 7 were each composed of multiple tumor types and did not show enrichment for any specific tumor type.
Gene sets predictive of these eight clusters represent developmental, immune signaling, proliferation, and cellular signaling pathways (Fig. 4D). Cluster 1 was associated with immune signaling (IL6 JAK STAT3 signaling). Of note, cluster 1 also had elevated immune infiltration scores by xCell (Supplementary Fig. S5B). Several clusters (2, 4, 7, 8) showed enrichment of various proliferative hallmark gene sets (E2F Targets, G2–M Checkpoint, MYC Targets v2, TP53 Pathway), with cluster 2 also showing activation of hedgehog signaling and developmental signaling [epithelial–mesenchymal transition (EMT)], cluster 4 showing activation of MAPK signaling (KRAS signaling up), cluster 7 showing enrichment for the reactive oxygen species pathway, and cluster 8 showing enrichment for endoplasmic reticulum stress and unfolded protein response. PI3K signaling (PI3K AKT mTOR signaling, mTORC1 signaling) was enriched in clusters 3, 5, and 6, with cluster 3 also showing enrichment for ER stress and unfolded protein response. Cluster 5 was associated with hedgehog signaling and developmental signaling (EMT), while cluster 6 was associated with NOTCH signaling.
Clonal heterogeneity and evolution
We next sought to investigate the genomic evolution of R/R childhood cancers by evaluating 46 longitudinal tumor samples from 20 patients (Supplementary Table S12). The median time between tissue collections was 229 days (range, 70–645 days). TMB was higher in the second R/R sample compared with the initial R/R sample (median 1.11 vs. 2.33; paired t test, P = 0.0187).
Phylogenetic reconstruction of tumor evolution was successfully performed for 19 of the 20 patients with longitudinal samples. Clonal heterogeneity was evident in all patients, and a majority of the longitudinal samples demonstrated predominantly linear or mixed branching evolution (Supplementary Fig. S6; ref. 53). Three patients showed characteristics of collateral branching. Whereas many oncogenic alterations were truncal variants that were maintained over time, 2 of the 3 patients with a collateral branching pattern showed expansion of clones containing oncogenic mutations (Fig. 5). In an RMS tumor, PAX3-FOXO1 fusion and MDM4 amplification were truncal, clonal events present at both timepoints, whereas CDK6 amplification and PIK3CA H1047R hotspot mutation were present at low clonal prevalence in the initial R/R tumor and expanded in clonal prevalence in the recurrent RMS biopsy collected 645 days later (PIK3CA mutation: <0.01% and 26% clonal prevalence, at initial and later R/R biopsy, respectively; Fig. 5A). Similarly, in a neuroblastoma tumor, an activating ALK mutation (I1171T) was detected at a low clonal prevalence (4%) for the initial R/R biopsy and expanded to a clonal prevalence of 35% at the subsequent R/R biopsy 192 days later (Fig. 5B).
Figure 5.
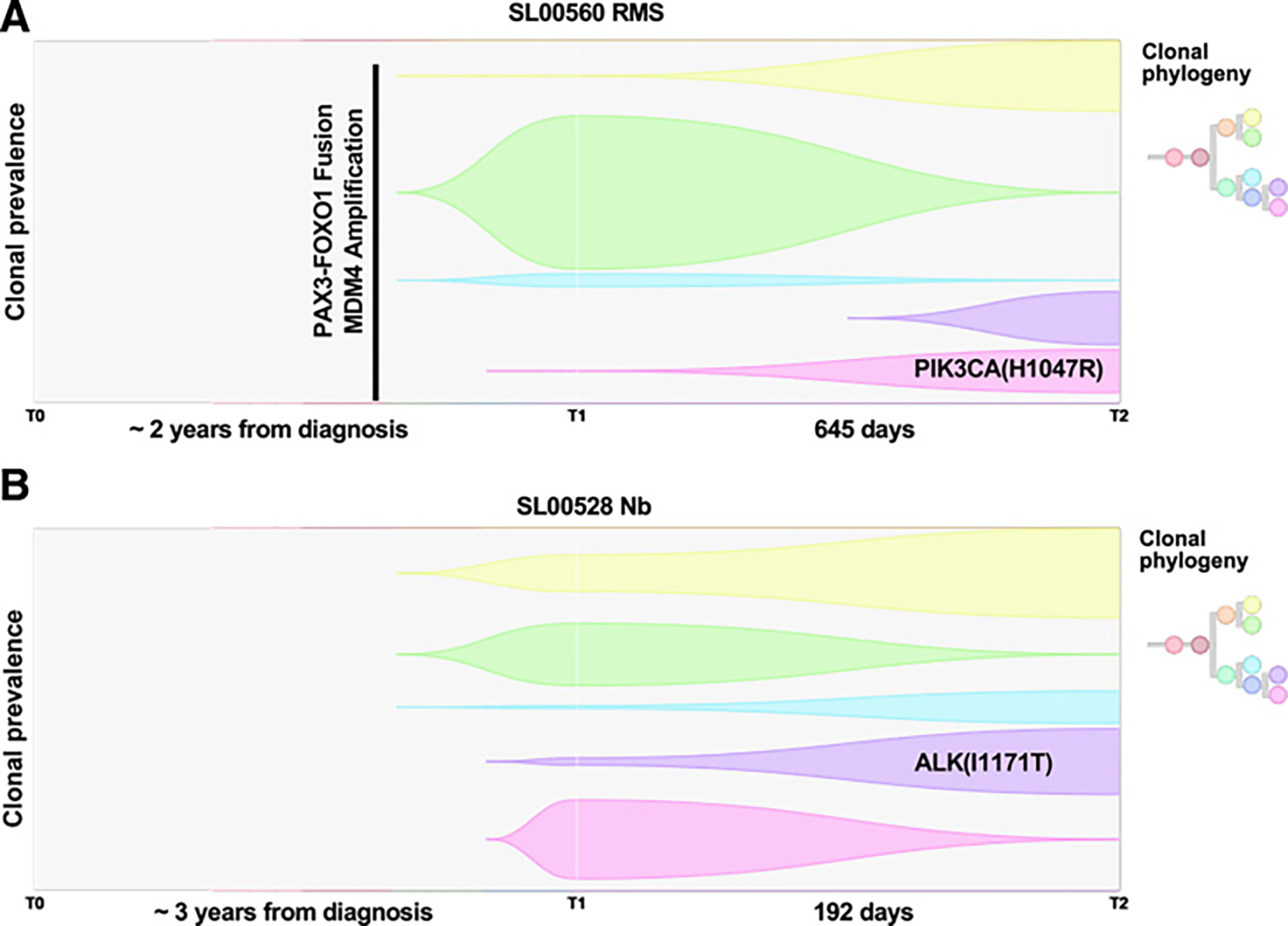
Longitudinal analysis of relapsed and refractory childhood solid tumors. Fish plot and predicted clonal phylogeny for two R/R tumors with longitudinal samples. RMS (A) and neuroblastoma (Nb; B).
Discussion
Here, we present genomic and transcriptomic analysis of 250 R/R tumors from 202 patients representing 46 different tumor types, including longitudinal profiling for 20 patients. Relapsed and treatment-refractory tumors displayed an elevated TMB compared with published cohorts of untreated pediatric cancers at diagnosis, with over one-third (35%) of R/R tumors in this cohort classified as pediatric high TMB or hypermutant compared with only 1.3% of untreated tumors in the Gröbner and colleagues cohort (2). Prior chemotherapy exposure influenced the mutational landscape of these tumors, with 43.2% of tumors showing mutational signatures associated with chemotherapy treatment, predominantly platinum chemotherapy. Two tumors displayed hypermutation, an anaplastic astrocytoma and a neuroblastoma. Both tumors showed enrichment for mutational signature SBS11, a mutational pattern associated with prior treatment with alkylating agents. Both patients had previously been treated with temozolomide chemotherapy, consistent with temozolomide-associated hypermutation previously reported in adult glioblastoma (54). It remains to be determined whether immune checkpoint inhibitors will display clinical activity in pediatric and AYA patients with treatment-induced hypermutant tumors.
Mutational signature analysis also revealed a common theme of DNA repair deficiencies in R/R tumors, with 35.6% of tumors showing evidence for HRD (SBS3). One of these patients had a pathogenic germline BRCA2 mutation and two lines of evidence for HR deficiency in their neuroblastoma tumor (high contribution from SBS3 mutational signature and HR-deficient by scarHRD analysis), suggesting potential activity for a PARP inhibitor. PARP inhibitors are currently being evaluated in the NCI-COG Pediatric MATCH trial in R/R pediatric cancers that harbor a mutation in a DNA repair gene. A subset of R/R childhood tumors were found to be MSI positive, suggesting a potential opportunity for immune checkpoint inhibitors in this subset of R/R tumors. While pan-cancer analyses in adult tumors have revealed MSI across a spectrum of cancers (55), the prevalence in childhood cancer is largely unknown. The MSI-positive tumors in this R/R cohort spanned five tumor types: neuroblastoma, Ewing sarcoma, osteosarcoma, undifferentiated sarcoma, and CPC. None of these patients demonstrated a known pathogenic germline event in a DNA mismatch repair gene. However, three of these tumors showed focal somatic deletion of an MMR gene: MLH1 deletion in the MSI-positive neuroblastoma, PMS2 deletion in the MSI-positive CPC, and EXO1 deletion in the MSI-positive undifferentiated sarcoma, providing molecular support for the MSI phenotype.
Tumor heterogeneity, treatment, and tumor evolution can contribute to the increased TMB and divergent molecular profiles seen in R/R tumors over time. Longitudinal profiling of 20 patients revealed emergence of potentially actionable alterations over time, including a PIK3CA hotspot mutation in an RMS tumor and an ALK mutation in neuroblastoma, illustrating the potential value of rebiopsying when using molecular information from the tumor to guide therapy selection or clinical trial options. This study was limited by the lack of diagnostic biopsies for this R/R cohort. Additional evaluation in a larger longitudinal dataset that includes paired diagnostic and R/R tumors is needed.
A majority (78%) of tumors bore an alteration in a gene previously implicated in pediatric cancer. Genomics events linked to treatment arms of the NCI-COG Pediatric MATCH trial were detected in tumors from 33.7% (62/184) of this BeatCC R/R cohort, a similar rate as has been reported for R/R patients screened through the NCI-COG Pediatric MATCH trial (56). Additional studies are needed to identify and develop additional therapeutic options for patients with R/R cancer.
Though tumor-type-associated genomic features predominated in accordance with differing cells-of-origin and evolutionary trajectories, pan-cancer features were nonetheless evident. TP53 alterations were seen across diverse tumors, reflecting TP53’s global tumor suppressive role spanning cell types. Pathogenic germline TP53 mutations were detected in three patients, and somatic mutations or copy-number alterations in TP53 were identified in 27 of 184 (14.7%) of R/R tumors. MYCN and TERT were identified as recurrently amplified genes and TP53 and CDKN2A were identified as recurrently deleted genes, consistent with reports of newly diagnosed pediatric cancers (2). A 1q gain was detected in 14% of tumors with similar frequency among the tumor groups (neuroblastoma, CNS tumors, sarcomas, and other rare tumors). 1q gain has also been reported in pan-cancer genomic studies of pediatric tumors at diagnosis (2) and has been associated with worse prognosis in various pediatric solid tumors, including neuroblastoma, Ewing sarcoma, and Wilms tumor (57–59). Further studies are needed to evaluate the role of 1q gain as a general marker of poor prognosis across pediatric cancers. Recurrent features reported in this R/R cohort that have not been reported in pan-cancer studies of tumors at diagnosis include gains involving RECQL4, CARD11, CCND1, MAPK1, and DGCR8 and losses involving CASP3, FAT1, SDHA, ROBO2, FHIT, FES, BLM, CHD2, POLG, and PML. RECQL4, a DNA helicase, lies in the region of 8q24.3 identified as recurrently gained in this R/R cohort by GISTIC analysis. RECQL4 overexpression has been linked to poor prognosis in adult cancers and associated with cisplatin resistance (60). It is also one of five gene expression markers associated with poor response to neoadjuvant chemotherapy in pediatric osteosarcoma (61), raising interest into whether gain of this region is contributing to the R/R state. Notably, while this study identified many key driver alterations using clinical tumor-normal WES and tumor RNA-seq, whole-genome sequencing was not performed, limiting the identification and evaluation of certain types of variants such as structural alterations.
Consistent with the increased mutational burden, immunogenomic profiling revealed that most R/R tumors express multiple neoantigens, with more than half of tumors having five or more expressed neoantigens. These R/R tumors showed an elevated number of expressed neoantigens compared with reports in pediatric solid tumors at diagnosis (average 8 expressed neoantigens per tumor for R/R tumors compared with less than 3 for tumors at diagnosis; ref. 46). Our neoantigen analysis focused on tumor-specific somatic mutations and did not explore neoantigens derived from gene fusions, which would be predicted to further increase the potential neoantigen landscape.
However, these tumors also displayed various mechanisms of immune evasion and generally had low levels of immune infiltration. Over 25% of tumors showed low expression of HLA-A, HLA-B, HLA-C, or B2M, lacked expression of a strong-binding neoantigen or showed elevated CD274 expression, and most tumors showed low immune infiltration based on deconvolution of the bulk RNA-seq data, consistent with previous reports that pediatric tumors are largely immunologically cold (reviewed in ref. 62). However, a subset of tumors showed immunogenic potential. Tumors in GSVA cluster 1 were enriched for immune signaling and showed elevated immune infiltration scores based on bulk RNA-seq analysis with xCell. This cross-pathology cluster was composed of multiple tumor types including neuroblastoma, Ewing sarcoma, osteosarcoma, angiosarcoma, MPNST, undifferentiated sarcoma, and a rhabdoid tumor. Two tumors were found to be hypermutated (>10 mutations/Mb). Pan-cancer analysis identified ten tumors with both high neoantigen expression (five or more expressed strong-binding neoantigens) and elevated MHC class I expression, with more than half of these tumors being neuroblastoma. The presence of neoantigens in pediatric cancers is particularly relevant for immunotherapeutic approaches. Understanding the immunogenicity of tumors may help to create immunologic treatments. Tumor specific neoantigens are being studied as novel ways to induce immune responses in tumors (63) and create adoptive T-cell therapies (64). Our results suggest these approaches may be effective in pediatric cancers. R/R neuroblastoma tumors had the highest mutational burden, the highest frequency of expression of multiple neoantigens, and showed elevated immune infiltration compared with R/R tumors overall.
Though transcriptional profiles of R/R childhood solid tumors largely reflect cell lineage, subsets of tumors deviated from these pathology boundaries. Functional grouping of tumors by cancer hallmark gene sets further revealed cross-pathology biological clusters driven by shared biological features of these tumors. Clusters were differentially enriched for various cellular processes (proliferation, developmental EMT signals), immune signaling, and cellular signaling pathways (KRAS, PI3K/AKT/MTOR, Hedgehog, Notch). The early identification of these genomic and/or transcriptomic “outliers” can help inform treatment strategies and advance new drug development for molecular subgroups of patients without curative treatment options.
In this R/R pediatric and AYA cohort, cross-pathology features include gene level events (mutations, copy-number alterations, fusions), large-scale genomic features (TMB, mutational signatures, MSI status, genomic instability), tumor extrinsic features (immune infiltration scoring), and RNA expression features (clustering, cancer hallmark and pathway activity scoring). Sequential tumor sequencing has valuable diagnostic, prognostic, and therapeutic implications in pediatric cancer. In the era of genomic-driven basket trials and immunotherapy, more comprehensive analyses can help identify and inform the design of pathway and biology-driven tumor agnostic therapeutic buckets.
Supplementary Material
Acknowledgments
The authors thank the patients and families for their participation in these studies.
This work was supported by the Dell Inc. Powering the Possible Program, Beat NB Foundation, Meryl and Charles Witmer Foundation, TGen Foundation, Four Diamonds, Hyundai Hope on Wheels, Hartwell Foundation, Padres Pedal the Cause, and the Reid R. Sacco Adolescent and Young Adult Cancer Alliance. This research was supported in part by the NIH, the Intramural Research Program of the NIH, and the NCI Center for Cancer Research.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Authors’ Disclosures
W.P.D. Hendricks reports other support from Vidium Animal Health outside the submitted work. J.E. Oesterheld reports other support from Servier outside the submitted work. G.L. Saulnier Sholler reports grants from Dell Foundation during the conduct of the study. No disclosures were reported by the other authors.
Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Grobner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature 2018;555:321–7. [DOI] [PubMed] [Google Scholar]
- 3.Ma X, Liu Y, Liu Y, Alexandrov LB, Edmonson MN, Gawad C, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 2018;555:371–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang W, Brohl AS, Patidar R, Sindiri S, Shern JF, Wei JS, et al. MultiDimensional ClinOmics for precision therapy of children and adolescent young adults with relapsed and refractory cancer: a report from the center for cancer research. Clin Cancer Res 2016;22:3810–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wong M, Mayoh C, Lau LMS, Khuong-Quang DA, Pinese M, Kumar A, et al. Whole genome, transcriptome and methylome profiling enhances actionable target discovery in high-risk pediatric cancer. Nat Med 2020;26:1742–53. [DOI] [PubMed] [Google Scholar]
- 6.Eleveld TF, Oldridge DA, Bernard V, Koster J, Daage LC, Diskin SJ, et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet 2015;47:864–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saulnier Sholler GL, Bond JP, Bergendahl G, Dutta A, Dragon J, Neville K, et al. Feasibility of implementing molecular-guided therapy for the treatment of patients with relapsed or refractory neuroblastoma. Cancer Med 2015;4:871–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Christoforides A, Carpten JD, Weiss GJ, Demeure MJ, Von Hoff DD, Craig DW. Identification of somatic mutations in cancer through Bayesian-based analysis of sequenced genome pairs. BMC Genomics 2013;14:302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 2011;12:R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sztupinszki Z, Diossy M, Krzystanek M, Reiniger L, Csabai I, Favero F, et al. Migrating the SNP array-based homologous recombination deficiency measures to next generation sequencing data of breast cancer. NPJ Breast Cancer 2018;4: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Telli ML, Timms KM, Reid J, Hennessy B, Mills GB, Jensen KC, et al. Homologous recombination deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clin Cancer Res 2016;22:3764–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haas BJ, Dobin A, Li B, Stransky N, Pochet N, Regev A. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol 2019;20:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 2017;18:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones DTW, Banito A, Grunewald TGP, Haber M, Jager N, Kool M, et al. Molecular characteristics and therapeutic vulnerabilities across paediatric solid tumours. Nat Rev Cancer 2019;19:420–38. [DOI] [PubMed] [Google Scholar]
- 15.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature 2020;578:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bergstrom EN, Huang MN, Mahto U, Barnes M, Stratton MR, Rozen SG, et al. SigProfilerMatrixGenerator: a tool for visualizing and exploring patterns of small mutational events. BMC Genomics 2019;20:685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boegel S, Lower M, Schafer M, Bukur T, de Graaf J, Boisguerin V, et al. HLA typing from RNA-Seq sequence reads. Genome Med 2012;4:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bai Y, Wang D, Fury W. PHLAT: inference of high-resolution HLA types from RNA and whole exome sequencing. Methods Mol Biol 2018;1802:193–201. [DOI] [PubMed] [Google Scholar]
- 19.Rubinsteyn A, Kodysh J, Hodes I, Mondet S, Aksoy BA, Finnigan JP, et al. Computational pipeline for the PGV-001 neoantigen vaccine trial. Front Immunol 2017;8:1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M. NetMHCpan-4.0: improved peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J Immunol 2017;199:3360–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marty R, Kaabinejadian S, Rossell D, Slifker MJ, van de Haar J, Engin HB, et al. MHC-I genotype restricts the oncogenic mutational landscape. Cell 2017;171:1272–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsiao L-L, Dangond F, Yoshida T, Hong R, Jensen RV, Misra J, et al. A compendium of gene expression in normal human tissues. Physiol Genomics 2001;7:97–104. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki R, Shimodaira H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 2006;22:1540–2. [DOI] [PubMed] [Google Scholar]
- 24.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database hallmark gene set collection. Cell Syst 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaufman L, Rousseeuw PJ. Finding groups in data: an introduction to cluster analysis. John Wiley & Sons; 2009. [Google Scholar]
- 27.Monti S, Tamayo P, Mesirov J, Golub T. Consensus clustering: a resampling-based method for class discovery and visualization of gene expression microarray data. Mach Learn 2003;52:91–118. [Google Scholar]
- 28.Halperin RF, Liang WS, Kulkarni S, Tassone EE, Adkins J, Enriquez D, et al. Leveraging spatial variation in tumor purity for improved somatic variant calling of archival tumor only samples. Front Oncol 2019;9:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang Y, Qiu Y, Minn AJ, Zhang NR. Assessing intratumor heterogeneity and tracking longitudinal and spatial clonal evolutionary history by next-generation sequencing. Proc Natl Acad Sci U S A 2016;113:E5528–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Campbell BB, Light N, Fabrizio D, Zatzman M, Fuligni F, de Borja R, et al. Comprehensive analysis of hypermutation in human cancer. Cell 2017;171:1042–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Niu B, Ye K, Zhang Q, Lu C, Xie M, McLellan MD, et al. MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics 2014;30:1015–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang MT, Asthana S, Gao SP, Lee BH, Chapman JS, Kandoth C, et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol 2016;34:155–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wegert J, Vokuhl C, Collord G, Del Castillo Velasco-Herrera M, Farndon SJ, Guzzo C, et al. Recurrent intragenic rearrangements of EGFR and BRAF in soft tissue tumors of infants. Nat Commun 2018;9:2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Antony R, Solomon D, Jin L-W, Perry A, Mueller S, Zwienenberg-Lee M, et al. HGG-12. primary bithalamic glioma with EGFR mutation: a rare case report. Neuro Oncol 2018;20:i91. [Google Scholar]
- 36.Brohl AS, Kahen E, Yoder SJ, Teer JK, Reed DR. The genomic landscape of malignant peripheral nerve sheath tumors: diverse drivers of Ras pathway activation. Sci Rep 2017;7:14992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang L, Chen LH, Wan H, Yang R, Wang Z, Feng J, et al. Exome sequencing identifies somatic gain-of-function PPM1D mutations in brainstem gliomas. Nat Genet 2014;46:726–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol 2018;15:731–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Drilon A, Jenkins C, Iyer S, Schoenfeld A, Keddy C, Davare MA. ROS1-dependent cancers - biology, diagnostics and therapeutics. Nat Rev Clin Oncol 2021;18:35–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med 2015;373:2336–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mirabello L, Yeager M, Mai PL, Gastier-Foster JM, Gorlick R, Khanna C, et al. Germline TP53 variants and susceptibility to osteosarcoma. J Natl Cancer Inst 2015;107:djv101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Diller L, Sexsmith E, Gottlieb A, Li FP, Malkin D. Germline p53 mutations are frequently detected in young children with rhabdomyosarcoma. J Clin Invest 1995;95:1606–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhukova N, Ramaswamy V, Remke M, Pfaff E, Shih DJ, Martin DC, et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J Clin Oncol 2013;31:2927–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bentires-Alj M, Paez JG, David FS, Keilhack H, Halmos B, Naoki K, et al. Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res 2004;64:8816–20. [DOI] [PubMed] [Google Scholar]
- 45.Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet 2002;39:311–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chang TC, Carter RA, Li Y, Li Y, Wang H, Edmonson MN, et al. The neoepitope landscape in pediatric cancers. Genome Med 2017;9:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mosse YP, Laudenslager M, Khazi D, Carlisle AJ, Winter CL, Rappaport E, et al. Germline PHOX2B mutation in hereditary neuroblastoma. Am J Hum Genet 2004;75:727–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Limpt V, Schramm A, Lakeman A, van Sluis P, Chan A, van Noesel M, et al. The Phox2B homeobox gene is mutated in sporadic neuroblastomas. Oncogene 2004;23:9280–8. [DOI] [PubMed] [Google Scholar]
- 49.Bosse KR, Maris JM. Advances in the translational genomics of neuroblastoma: from improving risk stratification and revealing novel biology to identifying actionable genomic alterations. Cancer 2016;122:20–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suva ML, Rheinbay E, Gillespie SM, Patel AP, Wakimoto H, Rabkin SD, et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 2014;157:580–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ebauer M, Wachtel M, Niggli FK, Schafer BW. Comparative expression profiling identifies an in vivo target gene signature with TFAP2B as a mediator of the survival function of PAX3/FKHR. Oncogene 2007;26:7267–81. [DOI] [PubMed] [Google Scholar]
- 52.Wang J, Feng W, Yuan Z, Weber JD, Zhang Y. DHX33 interacts with AP-2beta to regulate Bcl-2 gene expression and promote cancer cell survival. Mol Cell Biol 2019;39:e00017–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Andersson N, Bakker B, Karlsson J, Valind A, Holmquist Mengelbier L, Spierings DCJ, et al. Extensive clonal branching shapes the evolutionary history of high-risk pediatric cancers. Cancer Res 2020;80:1512–23. [DOI] [PubMed] [Google Scholar]
- 54.Yip S, Miao J, Cahill DP, Iafrate AJ, Aldape K, Nutt CL, et al. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin Cancer Res 2009;15:4622–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bonneville R, Krook MA, Kautto EA, Miya J, Wing MR, Chen HZ, et al. Landscape of microsatellite instability across 39 cancer types. JCO Precis Oncol 2017;2017:PO.17.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Parsons DW, Janeway KA, Patton D, Coffey B, Williams PM, Hamilton SR, et al. Identification of targetable molecular alterations in the NCI-COG Pediatric MATCH trial. J Clin Oncol 2019;37:10011-. [Google Scholar]
- 57.Janoueix-Lerosey I, Schleiermacher G, Michels E, Mosseri V, Ribeiro A, Lequin D, et al. Overall genomic pattern is a predictor of outcome in neuroblastoma. J Clin Oncol 2009;27:1026–33. [DOI] [PubMed] [Google Scholar]
- 58.MackintoshC Ordonez JL, Garcia-Dominguez DJ, Sevillano V, Llombart-Bosch A, Szuhai K, et al. 1q gain and CDT2 overexpression underlie an aggressive and highly proliferative form of Ewing sarcoma. Oncogene 2012;31:1287–98. [DOI] [PubMed] [Google Scholar]
- 59.Chagtai T, Zill C, Dainese L, Wegert J, Savola S, Popov S, et al. Gain of 1q as a prognostic biomarker in Wilms Tumors (WTs) treated with preoperative chemotherapy in the international society of paediatric oncology (SIOP) WT 2001 trial: a SIOP renal tumours biology consortium study. J Clin Oncol 2016;34:3195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mo D, Fang H, Niu K, Liu J, Wu M, Li S, et al. Human helicase RECQL4 drives cisplatin resistance in gastric cancer by activating an AKT-YB1-MDR1 signaling pathway. Cancer Res 2016;76:3057–66. [DOI] [PubMed] [Google Scholar]
- 61.Martin JW, Chilton-MacNeill S, Koti M, van Wijnen AJ, Squire JA, Zielenska M. Digital expression profiling identifies RUNX2, CDC5L, MDM2, RECQL4, and CDK4 as potential predictive biomarkers for neo-adjuvant chemotherapy response in paediatric osteosarcoma. PLoS One 2014;9:e95843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Terry RL, Meyran D, Ziegler DS, Haber M, Ekert PG, Trapani JA, et al. Immune profiling of pediatric solid tumors. J Clin Invest 2020;130:3391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Blaeschke F, Paul MC, Schuhmann MU, Rabsteyn A, Schroeder C, Casadei N, et al. Low mutational load in pediatric medulloblastoma still translates into neoantigens as targets for specific T-cell immunotherapy. Cytotherapy 2019;21:973–86. [DOI] [PubMed] [Google Scholar]
- 64.Liu Y Neoantigen: along march toward cancer immunotherapy. Clin Cancer Res 2016;22:2602–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study has been deposited in the database of Genotypes and Phenotypes (dbGaP) under accession number phs002238.v1 (http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs002238.v1.p1). Variant calls from secondary analysis with the NCI’s clinomics analysis pipeline are available on the clinomics web interface (https://clinomics.ccr.cancer.gov/clinomics/public/).